

SUMMARY
There has been tremendous insight gained in the last two decades from basic science research. New molecular targets in neoplastic cells are emerging and provide the rationale for clinical development of novel agents in non-Hodgkin lymphoma. These novel agents can be broadly categorized into two groups. The first is by immunotherapy which includes novel monoclonal antibodies and immunomodulating drugs, which takes advantage of or optimizes immune system function. The other group of drugs target small molecules that may play an important role in tumorigenesis. The mechanisms of anti-tumor activity include targeting apoptotic pathways, inhibition of proteasomes, mammalian target of rapamycin (mTOR), cyclin-dependent kinases and histone deacetylases. The purpose of this review is to focus on these novel agents and the various treatment approaches that are currently being evaluated in non-Hodgkin lymphoma.
Keywords: Drug targets, Lymphoma, Novel agents, Small molecules, Immunotherapy
Introduction
The treatment of non-Hodgkin lymphoma (NHL) in the last two decades have heralded an era of increasing exploration of therapies derived from improved biologic understanding of tumors and tumor-host interactions. The focus of drug discovery has moved from identifying classical cytotoxic agents to molecules that target specific pathways involved in signal transduction, apoptosis, and differentiation, to name a few. These efforts have been greatly aided by insights into the structure of proteins and the ability to design specific inhibitors using small molecules or monoclonal antibodies.
Although the introduction of new treatment agents and regimens for NHL has resulted in improved complete response (CR) rates and survival in some settings, the lack of any significant improvement in overall survival in many of the subtypes indicates a clear need for further novel drugs and interventions.1–3 These novel agents can be broadly categorized into two groups. The first is by immunotherapy which includes novel monoclonal antibodies (MAb) and immunomodulating drugs (IMiDs), which take advantage of the immune system and/or optimize tumor cell targeting. The other group of drugs target small molecules that may play an important role in tumorigenesis. The mechanisms of anti-tumor activity include targeting apoptotic pathways, inhibition of proteasomes, mTOR, cyclin-dependent kinases and histone deacetylases. Herein we discuss several of these promising agents and the research that has led several of them into the clinic.
Monoclonal antibody therapy
Until 1980, the molecular architecture of the B-cell surface was known to consist of membrane-bound Ig, complement component receptors, and Fc receptors; beyond that, the molecular constitution of the cell surface was completely uncharacterized. That all changed with the advent of monoclonal antibody (MAb) technology.4 Over the past 25 years, slightly over ten B-cell-specific cell surface molecules have been identified by MAbs. It has been demonstrated that B-cell antigen expression is variable and differs at various stages of B-cell development. However, many of these B-cell antigens are often expressed on malignant B-cells. Most of these antigens are involved in B-cell growth, differentiation, proliferation, and activation or have unknown functions. Several proposed mechanisms by which MAbs appear to produce their cytotoxic effects have been described: (1) Antibody-dependent cellular cytotoxicity (ADCC) is induced through binding of the Fc portion of the antibody to Fc receptors on host effector cells, such as natural killer (NK) cells, granulocytes, and macrophages,5–7 (2) Complement-dependent cytotoxicity (CDC) is induced by promoting complement fixation at the cell surface, leading to complement-dependent lysis of target cells,8,9 and (3) direct cytotoxicity via induction of cellular apoptosis following binding of the target antigen by MAb.10–13 Both ADCC and CDC depend on the interaction of the antibody with the host’s intrinsic immune system, while direct cytotoxicity is independent of it.
Novel anti-CD20 monoclonal antibodies
The CD20 antigen is expressed exclusively on normal and malignant B-cells.14 It has a stable expression and is tightly bound to the membrane with little modulation during maturation. It is neither secreted nor rapidly shed in circulation,15,16 and it likely has an important role in B-cell activation and regulation of cell cycle.17,18 All these features make CD20 an “ideal” anti-B-cell target. Rituximab was the first MAb to be approved by the Food and Drug Administration (FDA) in 1997 for treatment of relapsed or refractory CD20-positive follicular or indolent B-cell NHL. Rituximab is a chimeric human–mouse IgG1 kappa monoclonal antibody that binds to CD20, inducing cell death in both normal and neoplastic B-cells. It has since been used in combination with chemotherapy and is now considered the cornerstone of therapy in both indolent and aggressive B-cell lymphomas. Although rituximab is a valuable addition to the treatment for B-cell NHL, 50% of patients with relapsed or refractory CD20-positive follicular lymphomas do not respond to initial therapy with rituximab19 and close to 60% of patients who were previously treated with rituximab no longer benefit with retreatment.20 Rituximab resistance represents a significant barrier to immunotherapy of B-cell lymphomas.
Newer-generation anti-CD20 antibodies designed to improve on rituximab are currently in development. Several approaches are under evaluation. Modification strategies include humanization of the molecule to decrease infusion reactions and immunogenicity, enhancement of binding affinity, and modification of the Fc portion of the molecule to optimize effector functions, particularly ADCC. Humanized anti-CD20 MAbs are appealing because they avoid antimurine immunogenic response and perhaps have a better side-effect profile. There are three humanized anti-CD20 MAbs undergoing clinical evaluation. One of these is ofatumumab (HuMaxCD20), a fully human IgG1 kappa antibody that binds to a novel CD20 epitope localized in the second extra-cellular loop distinct from that recognized by rituximab.21,22 Compared with rituximab, ofatumumab elicits stronger complement-dependent cytotoxicity (CDC) but induces less apoptosis.23 Preclinical data demonstrates that ofatumumab inhibits the growth of engrafted B-cell tumors in SCID mice more efficiently than rituximab. Ofatumumab was also able to lyse rituximab-resistant Raji Burkitt cells in vitro.23
In a recent phase I/II study, 33 patients with relapsed or refractory CD20-positive chronic lymphocytic leukemia (CLL) were treated with four once-weekly infusions of ofatumumab; 67% of the patients were Binet stage B and median number of previous treatments was 3 (range, 1–9). Three cohorts of patients with the following dosing schedule were used: cohort A, one 100 mg infusion plus three 500 mg infusions (three patients); cohort B, one 300 mg infusion and three 1000 mg infusions (three patients); cohort C, one 500 mg infusion and three 2000 mg infusions (27 patients). All patients had significant reduction in leukemic cells as well as rapid and prolonged depletion of normal B lymphocytes. Their recovery to normal levels was not observed until 5–6 months after completion of therapy. Overall response rate (ORR) in all three cohorts was 44%, and was 50% in cohort C (13 of 26 assessable patients). However, all were partial responses (PR). The majority of related adverse events occurred after the first infusion and these decreased at subsequent administrations. The most common grade 3–4 toxicity reported was myelosuppression (12%), followed by infections (9%). None of the patients developed human anti-human antibodies.24 This phase I/II study indicates that ofatumumab is an active and well tolerated agent in refractory/relapsed CLL in doses up to 2000 mg, with a relatively encouraging objective response. A phase I/II study of 40 patients with follicular lymphoma evaluated 4-weekly infusions of ofatumumab given at different doses (300–1000 mg). Out of 38 patients, 5 CR, 2 CR unconfirmed (CRu), and 9 PR were observed. Based on the median follow-up of 9.2 months, the median time to disease progression was 8.8 months and the median duration of response (DR) was 29.9 months.25 In view of these results, two phase III clinical trials are currently underway. Ofatumumab is being evaluated as a single agent in patients with refractory CLL and in patients with follicular lymphoma that are refractory to rituximab. Also currently under way is a phase II trial of ofatumumab in combination CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) in patients with previously untreated follicular lymphoma.
Veltuzumab (hA20) is another humanized anti-CD20 MAb being evaluated in clinical trials. This MAb is engineered with complementarity-determining regions (CDR) of murine origin and with 90% of the human framework regions identical to epratumumab, a humanized anti-CD22 IgG1 antibody.26,27 The mechanism of cytotoxicity of Veltuzumab is similar to rituximab and also very similar to rituximab in terms of antigen binding and specificity binding avidity. A phase I/II dose escalation study in 82 patients with recurrent B-cell lymphomas demonstrated a 40% ORR, including 17 patients who achieved CR/CRu (21%). In patients with follicular lymphoma (FL) which comprises the largest subgroup in this study, the ORR was 44% (24 of 55 patients) and 27% CR/CRu (15 of 55 patients). The 15 patients with FL who achieved CR/CRu generally had durable responses with a median duration of response (DR) and progression free survival (PFS) of 19.7 and 24.2 months, respectively. The drug was generally well tolerated. All treatment related adverse effects were mild to moderate with the exception of one grade 3 hypoglobulinemia; otherwise, most were transient infusion-related symptoms occurring predominantly at first infusion.28 Another approach being investigated is anti-CD20 MAbs with enhanced binding to FcγRIIIa. Three novel engineered anti-CD20 antibodies, AME-133v, rhuMAb v114, and GA-101, are currently in early phases of clinical development. They are associated with a higher antibody-dependent cytotoxicity as compared with rituximab, and increased direct apoptosis with GA-101 (Table 1).
Table 1.
Comparison of Anti-CD20s.
| Agent | Description | ADCC | CDC | Apoptosis | Binding |
|---|---|---|---|---|---|
| Ofatumumab (HuMax-CD20) | • Human IgG1 kappa backbone • Unique binding site • Phase I/II for NHL and CLL • Kills rituximab-resistant cell lines • 50% B-CLL killing vs. 5% Rituximab (in vitro) |
⬌ | ⬆ | ⬌ | ⬆ |
| Veltuzumab (hA20) | • Humanized • Re-shaped Rituximab binding region with epratuzumab backbone • Phase I/II for NHL • Similar binding affinity, apoptosis and in vitro data as rituximab |
⬌ | ⬌ | ⬌ | ⬌ |
| PRO131921 (rhuMAb v114) | • Humanized • Enhanced binding to FcyRIIIA • B-cell depletion superior to rituximab in murine models |
⬆ | ⬆ | ⬌ | ⬆ |
| AME-133 | • Humanized • Optimized via proprietary AME process (improved FcyRIII binding and ADCC) • 10-fold higher cell killing than rituximab |
⬆ | ⬌ | ⬌ | ⬆ |
| GA101 | • Humanized | ⬆ | ⬇ | ⬆ | ⬌ |
Targeting non-CD20 antigens
Clinical success with anti-CD 20 MAbs has led to further investigation and discovery of other potential targets in B-cell NHL. Some examples include CD22, CD23, CD40, CD8029–32 (Fig. 1, Table 2). These agents have shown promise in early clinical trials and might represent an additional strategy to overcoming rituximab resistance. In this review, only galiximab, a MAb targeting CD80, will be discussed. CD80, a member of the B7 ligand family (B7.1), is a membrane co-stimulatory molecule that is involved in T-cell regulation and in regulation of normal and malignant B-cells.33,34 Preclinical studies demonstrated that cross-linking of CD80 on B-cells resulted in upregulation of proapoptotic proteins such as caspase-3, caspase-8, Fas, Fas ligand, Bak, and Bax and downregulation of anti-apoptotic proteins such as Bcl-XL34 CD80 is a good target antigen because it is constitutively expressed in a variety of B-cell lymphoma cells, including follicular lymphoma and Hodgkin lymphoma.35,36
Fig. 1.
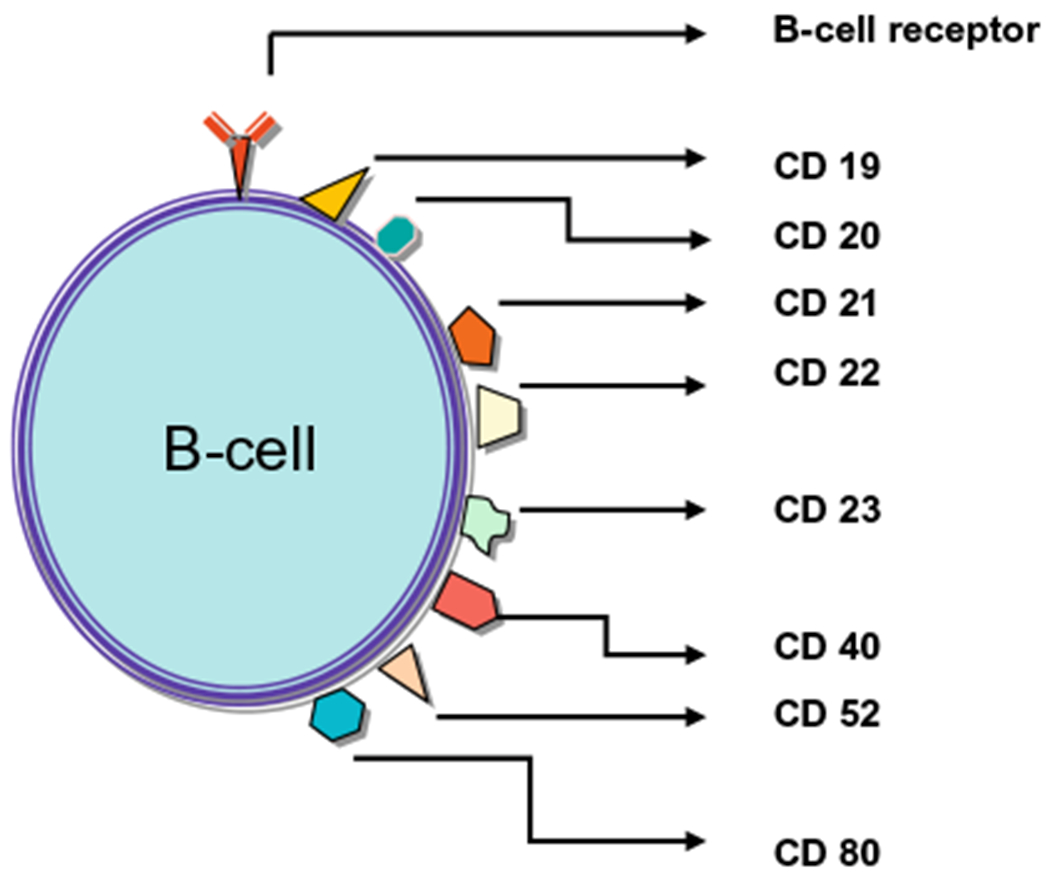
B-cell antigen targets.
Table 2.
Monoclonal antibodies for non-CD20 targets.
| Study | Agent | Target | mAb | Status | Patients | Results (%) |
|
|---|---|---|---|---|---|---|---|
| ORR | CR/CRu | ||||||
| Czuczman et al. [33] | Galiximab | CD80 | Primate-chimeric | Phase I/II | Relapsed/refractory FL (n = 38) | 11 | 5 |
| Leonard et al. [34] | Galiximab Rituximab |
CD80 CD20 |
Phase II | Relapsed/refractory FL (n = 64) | 66 | 33 | |
| Ongoing trial | Galiximab Rituximab |
CD80 CD20 |
Phase II | Untreated FL (n = 61) | 70 | 44 | |
| Leonard et al. [29] | Epratuzumab | CD22 | Humanized | Phase I/II | Relapsed/refractory FL (n = 55) | 18 | 6 |
| Leonard et al. [30] | Epratuzumab | CD22 | Humanized | Phase I/II | Relapsed/refractory aggressive lymphomas (n = 56) | 10 | 5 |
| Strauss et al. [31] | Epratuzumab Rituximab |
CD22 CD20 |
Phase II | Relapsed/refractory NHL (n = 65) | 46 | 22 | |
| Advani et al. [32] | Dacetuzumab (SGN-40) | CD40 | Humanized | Phase I | Relapsed/refractory NHL (n = 50) | 12 | 2 |
CR – complete response; CRu – complete response unconfirmed; FL – follicular lymphoma; MAb – monoclonal antibody; NHL – non-Hodgkin lymphoma; ORR – overall response rate.
Galiximab is a primate–human chimeric anti-CD80 MAb with human IgG1 constant region and macaque variable region, which is structurally indistinguishable from human antibodies. Galiximab was evaluated in a phase I/II study of 38 patients with advanced-stage relapsed or refractory follicular lymphoma. Patients received four infusions of galiximab at doses of 125, 250, 375 or 500 mg/m2, given once weekly for 4 weeks. The overall RR was 11%, with two patients achieving complete response (CR) and two reaching partial response (PR); 12 patients (34%) had stable disease (SD). Both patients with complete response were administered the dose of 375 mg/m2. All enrolled patients were CD80 positive by flow cytometry, but those who responded did not necessarily have greater CD80 density than non-responders. The most common adverse effects reported were grade 1 and 2 fatigue, nausea and headache. There were no grade 4 toxicities. Anti-galiximab antibodies were not observed.37
More recently, a phase II study of galiximab in combination with rituximab in relapsed follicular lymphoma showed better outcomes than with either agent alone. 64 patients received rituximab 375 mg/m2 and galiximab 500 given on a weekly schedule for 4 weeks. The overall RR was 66%, including 19% CR, 14% unconfirmed CR (CRu) and 33% PR. The median progression-free survival was 12.1 months. The addition of galiximab to rituximab did not seem to increase overall incidence of adverse events and appears to be well tolerated. The most common adverse effects were hematologic toxicity followed by fatigue and chills.38 These promising results prompted the initiation of a phase III clinical study of galiximab in combination with rituximab compared with rituximab in combination with placebo for the treatment of patients with relapsed or refractory follicular lymphoma. The study will enroll approximately 700 patients worldwide across 150 cancer centers. This combination is also being evaluated in a phase II trial in previously untreated follicular lymphoma. This study was reported at the International Conference on Malignant Lymphoma, Lugano, Switzerland. The preliminary results for the 61 patients enrolled were encouraging. The ORR was 70% (37.7% CR and 6.6% CRu; 28% PR), and the estimated 1 year disease-free survival probability was 0.87 (95% CI, 0.75–0.93). Treatment was well tolerated, with only 13% experiencing grade 3 adverse events. Potential future directions also include evaluating maintenance therapy with this combination regimen in follicular NHL and galiximab in combination with rituximab, plus chemotherapy for aggressive NHL.
Antibody drug conjugates
Another active area of research has been development of antibody-drug conjugates (ADCs). ADCs comprise an antibody that is conjugated to a cytotoxic drug via a chemical linker. The fundamental principle of the ADC concept is to use an antibody as a vehicle to deliver a cytotoxic drug selectively to a target cell surface antigen. This concept has been around since the 1970s but it is in the last decade that we have witnessed the most significant advances. The development of more target antigens, greater potency drugs used in conjunction with linkers of improved stability, and a better knowledge of ADC cell biology and pharmacology are just some of the key reasons for the development of ADC in the clinical arena. Gemtuzumab ozogamicin (Mylotarg) is currently the sole ADC approved for clinical use. It was approved in 2000 for the treatment of patients with CD33 positive acute myeloid leukemia in first relapse who are at least 60 years of age and considered unsuitable candidates for cytotoxic chemotherapy.39 Since then, many other ADCs have entered clinical trials for use in both solid tumors and hematological malignancies (Table 3).40,41 Currently, the sole ADC being investigated in patients with B-cell NHL is the agent Inotuzumab ozogamicin. It is composed of a humanized CD22 antibody, conjugated to calicheamicin via an acid labile, AcBut, linker. Inotuzumab is a well tolerated drug with significant single agent activity, with a 33% and 69% response rate in patients with relapsed DLBCL and FL, respectively.42 Similarly, the preliminary result from a small Japanese study of 13 patients with relapsed or refractory follicular lymphoma who have been pre-treated with rituximab showed responses in 11 patients.43 The main adverse event in both studies were hematologic, self limiting thrombocytopenia being the most common, and the MTD was established at the dose of 1.8 mg/m2. The encouraging results from the phase I study prompted the initiation of a study using the combination of rituximab with inotuzumab in patients with relapsed FL or DLBCL. The preliminary results of the phase I portion of the study were recently reported. The MTD dose was also confirmed at 1.8 mg/m2. Based on 30 patients that were evaluable for tumor response, the ORR was 80% with a 43% CR rate. Of note, 6 of the 14 patients with relapsed DLBCL achieved a CR with this combination. The safety profile of this combination is similar to that seen in the phase I trials.44 These data support the continuing development of inotuzumab ozogamicin in combination with rituximab in the management of NHL.
Table 3.
Antibody-drug conjugates in lymphomas.
| Study | Status | Agent | Target | Drug | Regimens | Patients | Results (%) |
|
|---|---|---|---|---|---|---|---|---|
| ORR | CR/CRu | |||||||
| Fayad et al. [42] | Phase I | Inotuzumab Ozogamicin | CD22 | Calicheamicin | 1.8 mg/m2 every 28 days | Relapsed/refractory FL or DLBCL (n = 34) | 53 | 24 |
| Tobinai et al. [43] | Phase I | Inotuzumab Ozogamicin | CD22 | Calicheamicin | 1.8 mg/m2 every 28 days | Relapsed/refractory FL (n = 13) | 85 | 54 |
| Fayad et al. [44] | Phase I/II | Inotuzumab Ozogamicin + Rituximab | CD22 CD20 |
Calicheamicin | 1.8 mg/m2 on day 2, every 28 days | Relapsed/refractory FL or DLBCL (n = 30) | 80 | 43 |
| Younes et al. [40] | Phase I | SGN-35 | CD30 | Auristatin | 1.2–2.7 mg/kg every 21 days | Relapsed/refractory CD30+ hematologic malignancies (n = 13) | 54 | 0 |
| Bartlett et al. [41] | Phase I | SGN-35 | CD30 | Auristatin | 0.4–1.0 mg/kg weekly for 3 weeks, every 28 days cycle | Relapsed/refractory HL or ALCL (n = 27) | 48 | 37 |
ALCL – anaplastic large cell lymphoma; CR – complete response; CRu – complete response unconfirmed; DLBCL – diffuse large B-cell lymphoma; FL – follicular lymphoma; HL – Hodgkin lymphoma; NHL – non-Hodgkin lymphoma; ORR – overall response rate.
T-cell engaging antibodies
A number of therapeutic strategies have utilized T-cells in cancer therapy based on their high cytotoxic potential, efficient homing function and high abundance in the host environment. They include diverse vaccination approaches using tumor-associated antigens, blockade of the inhibitory molecule, cytotoxic T-lymphocyte antigen 4 (CTLA-4), on T-cells by monoclonal antibodies, which boost T-cell maturation. Other approaches include the ex vivo expansion and reinfusion of tumor-specific T-cells, a therapy also known as adoptive T-cell transfer. Dramatic tumor responses have been demonstrated in patients with advanced melanoma using this strategy.45,46 However, for most of the other approaches, further development for clinical use have been hampered by rather low response rates. Most of these approaches aim to facilitate tumor antigen presentation and undergo the complicated process of T-cell priming and maturation, resulting in tumor-specific T-cell clonal expansion. The complexity of these processes is frequently the basis for immune escape mechanisms of cancer cells.47
A novel approach that harnesses the enormous cytotoxic potential of T-cell antibodies, independent of peptide antigen presentation by tumor cells or T-cell specificity, are called bispecific T-cell engager (BiTE) antibodies. BiTE antibodies are recombinant protein constructs formed from two single-chain antibodies. One is specific for CD3, a subunit of the T-cell receptor complex, and the other is specific for a selected tumor-associated antigen such as CD19. A unique feature of these antibodies is that bivalent binding is required to trigger its mechanism of action, whereas monovalent binding will not cause T-cell activation. Blinatumomab (MT103) is currently the first BiTE antibody tested clinically in patients with relapsed NHL. A phase I study using Blinatumomab has included 39 patients with relapsed mantle cell lymphoma, follicular lymphoma, immunocytoma, marginal zone lymphoma and small lymphocytic leukemia. Most patients were heavily pre-treated, with a median of three previous regimens. Blinatumomab was administered as a continuous infusion over a period of 4–8 weeks, as one treatment cycle, and with a mean infusion duration of 5.2 weeks per patient. Seven dose levels, ranging from 0.0005 to 0.090 mg/m2/24 h have been tested. No objective responses were seen in patients treated below 0.015 mg/m2/24 h. But responses were observed in 11 out of 27 patients (41%) treated at doses of 0.015 mg/m2/24 h and higher. Five patients (19%) achieved CR and six (22%) reaching PR. At the dose level of 0.060 mg/m2/24 h, all seven patients have shown objective responses. Among patients who were treated at the dose levels of 0.030 and 0.060 mg/m2/24 h, no treatment failures have been observed with the exception of one patient who relapsed at 14 months. Five patients at these dose levels have ongoing responses for more than 6 months. The largest benefit was seen in patients with follicular or mantle cell lymphoma. Three out of five patients with mantle cell lymphoma treated at dose levels of 0.030 and 0.060 mg/m2/24 h all had response duration for more than 1 year. The drug was well tolerated with a manageable toxicity profile, the most common adverse events being lymphopenia, pyrexia and leucopenia. The majority of adverse events improved or resolved while on treatment. Out of 39 patients, treatment was discontinued in eight patients, of which six had fully reversible symptoms of the central nervous system, such as confusion, disorientation, and speech disorder. One patient with a background of hypogammaglobulinemia and bone marrow suppression from prior chemotherapy experienced a fatal sepsis 5 weeks after commencement of treatment.48 On the basis of these encouraging preliminary results, it appears that blinatumomab has activity and potential therapeutic use in an array of CD19-expressing B-cell malignancies.
Another exciting approach is to engineer T-cells to express chimeric antigen receptors (CARs). CARs are made up of an antigenrecognizing receptor coupled to signaling molecules that can activate T-cells expressing the CAR. The antigen-receptors most commonly incorporated into CARs are single chain variable region moieties (scFv) that consist of the light chain and heavy chain variable regions of a monoclonal antibody joined by a peptide linker. Murine models have shown that syngeneic T-cells transduced with retroviruses encoding CARs protected mice from tumor challenges in vivo.49,50 It has also demonstrated that including the signaling domain of CD28 in CARs and increasing the number of CAR-transduced T-cells enhanced tumor protection and persistence of CAR-transduced T-cells.51,52
The only clinical trial of an anti-CD19 CAR that has been completed used electroporation to transfect T-cells prior to adoptive transfer. In this trial, T-cell persistence was limited and an antilymphoma effect was not detected.53 A group from our institution has constructed and extensively tested an anti-CD19 CAR that contains the signaling component of CD28, and using a retroviral vector rather than plasmid transfection to transfer an anti-CD19 receptor to T-cells. They have demonstrated that T-cells that are transduced with gamma-retroviruses encoding the sequence of this CAR can specifically recognize and kill CD19-expressing target cells including primary chronic lymphocytic leukemia cells. These anti-CD19-CAR-transduced T-cells also produce the cytokines interferon and IL-2 specifically in response to CD19.54 There is currently an ongoing clinical trial at our institution in which gamma-retroviruses encoding the anti-CD19 CAR will be used to transduce T-cells from patients with advanced B-cell malignancies. In the phase I portion, patients receive lymphocyte-depleting chemotherapy consisting of fludarabine and cyclophosphamide, anti-CD19-CAR transduced T-cells, and high dose aldesleukin. This is based on murine models which showed that administration of high-dose IL-2 and immunosuppression before adoptive T-cell therapy enhanced anti-tumor activity both in mice and humans.46,52,55 Once the MTD is determined, the study will proceed to the phase II portion. Patients will be randomized to two treatment arms. Patients assigned to treatment arm 1 will receive fludarabine and cyclophosphamide chemotherapy in order to induce lymphocyte depletion prior to infusion of the anti-CD19-CAR-transduced T-cells, and high dose aldesleukin. Patients assigned to treatment arm 2 will receive anti-CD19-CAR-transduced T-cells, and high dose aldesleukin without chemotherapy. This trial design will also assess the effect of immunosuppression before T-cell transfer.
Immunomodulating agents
The immunomodulating drugs (IMiDs) CC-5013 Lenalidomide and CC-4047 Actimid, are structural and functional analogues of thalidomide. These second generation of IMiDs was designed to enhance immunologic and anti-cancer properties while potentially decreasing the neurotoxic and teratogenic adverse effects of the parent compound thalidomide.56 These drugs have been extensively used and have shown clear benefit in other diseases, particularly multiple myeloma and myelodysplastic syndromes. Despite the clinical activity of IMiDs in various malignant diseases, the exact mechanism of their antitumor activity is yet to be elucidated. It has been hypothesized that the mechanism of action includes immunomodulatory and non-immunomodulatory activity. It has been reported to down regulate key prosurvival cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-8 (IL-8), and vascular endothelial growth factor (VEGF).57 Other immunomodulatory activities of IMiDs include inhibition of T regulatory cell function58 and potent T-cell co-stimulatory activity, leading to increased secretion of the T-cell lymphokines interferon (IFN)-γ, and IL-2. This enhances Th1-type cellular immunity and natural killer (NK) T-cell cytotoxicity.57,59–61 IMiDs have also demonstrated direct antiproliferative effects by inhibiting the Akt pathway and increasing the expression of the p21 tumor suppressor protein, leading to G1 cell cycle arrest,62,63 as well as its inhibition of angiogenesis.64
Of this class of drugs, most recent attention has focused on the more active agent, lenalidomide. A phase II study (NHL-002) of 50 patients with relapsed or refractory aggressive NHL showed moderate single-agent activity with lenalidomide. The study included patients with diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle cell lymphoma (MCL), and transformed NHL. Lenalidomide (25 mg/day) was administered on days 1–21, every 28 days for 52 weeks as tolerated or until disease progression. The overall response rate (ORR) was 35% for all patients, 19% for DLBCL patients and 53% in MCL. Clinical responses were also observed in all lymphoma subtypes. The main adverse event was hematologic, severe neutropenia being the most common reason for dose reduction. The result from this study suggests that lenalidomide has potentially important activity in aggressive lymphoma.65 Based on these promising results of the study, an international phase II trial (NHL-003) of single-agent lenalidomide was initiated for patients with relapsed/refractory aggressive NHL. The preliminary results from the 39 MCL and 73 DLBCL patients enrolled in this study were recently reported. The ORR in patients with MCL was 41%, with five patients (13%) achieving CR/CRu and 11 (28%) reaching PR. The response rate was slightly lower in patients with DLBCL, with a ORR of 29%, with three patients (4%) achieving CR and 18 patients (25%) with PR. Eleven patients (15%) had stable disease. The drug was well tolerated with a manageable toxicity profile, similar to that observed in NHL-002. These preliminary results further validate the role of lenalidomide in the treatment of patients with relapsed or refractory MCL and DLBCL.66,67
Clinical data available so far suggest that lenalidomide monotherapy represents a promising new direction for lymphoma therapy. There are at least 10 ongoing clinical trials investigating the efficacy of lenalidomide in different types of lymphoma68–71 (Table 4). The principal challenge facing investigators, given the heterogeneity of the disease and the large number of other active agents, will be the development of rational combinations that will ultimately result in the most clinical benefits for lymphoma patients.
Table 4.
Response rates of single Lenalidomide in NHL.
| Study | Status | Regimens | Patients | Results (%) |
|
|---|---|---|---|---|---|
| ORR | CR/CRu | ||||
| Wiernik et al. [65] | Phase II | 25 mg QD, 21 of 28 days | Relapsed/refractory aggressive NHL (n = 50) | 35 | 6 |
| Zinzani et al. [66] | Phase II | 25 mg QD, 21 of 28 days | Relapsed/refractory indolent NHL (n = 39) | 41 | 13 |
| Chanan-Khan et al. [68] | Phase II | 25 mg QD, 21 of 28 days | Relapsed/refractory CLL (n = 45) | 47 | 9 |
| Ferrajoli et al. [69] | Phase II | 10 mg QD, escalated to maximum of 25 mg QD | Relapsed/refractory CLL (n = 44) | 32 | 7 |
| Chen et al. [70] | Phase I | 2.5 mg QD, escalated to 10 mg QD | Untreated CLL (n = 25) | 65 | 0 |
| Ferrajoli et al. [71] | Phase II | 5 mg QD, escalated to maximum of 25 mg QD | Untreated CLL (n = 35) | 54 | 0 |
CLL – chronic lymphocytic leukemia; CR – complete response; CRu – complete response unconfirmed; FL – follicular lymphoma; MAb – monoclonal antibody; NHL – non-Hodgkin lymphoma; oRr – overall response rate.
Targeting small molecules
The discovery of new signaling pathways that are critical to lymphomagenesis has produced an array of new targets for the treatment of lymphomas. The next section of this review paper will discuss the biologic rationale and clinical trials on several of these small molecules. We will focus on the agents targeting novel pathways regulated by Bcl-2, proteasomes, mTOR inhibitors, cyclin-dependent kinases and histone deacetylases. A number of other drugs that target different pathways such as protein kinase C inhibitors and survivin inhibitors, have also shown promise in early clinical trials and is summarized in (Table 5).72–74
Table 5.
Other targets of small molecules in NHL.
| Study | Status | Agent | Target | Patients | Results (%) |
|
|---|---|---|---|---|---|---|
| ORR | CR/CRu | |||||
| Cheson et al. [72] | Phase I/II | YM155 | Survivin | Relapsed/refractory DLBCL (n = 27) | 11 | 0 |
| Robertson et al. [73] | Phase II | Enzastaurin | Protein kinase C | Relapsed/refractory DLBCL (n = 55) | 6 | 6 |
| Morschhauser et al. [74] | Phase II Ongoing |
Enzastaurin Tanespimycin (17-AAG) |
Protein kinase C HSP 90 |
Relapsed/refractory MCL (n = 60) Relapsed/refractory ALCL, MCL, HL |
0 | 0 |
ALCL – anaplastic large cell lymphoma; CR – complete response; CRu – complete response unconfirmed; DLBCL – diffuse large B-celll lymphoma; HL – Hodgkin lymphoma; MCL – mantle cell lymphoma; NHL – non–Hodgkin lymphoma; ORR – overall response rate.
Proteasome inhibitors
The ubiquitin–proteasome pathway is essential for the degradation of most short- and long-lived intracellular proteins in eukaryotic cells. The 26S proteasome, universally present and abundant in all eukaryotic cells, is an ATP-dependent multicatalytic protease that is central to the degradative pathway. The 26S proteasome functions not only in a housekeeping role to eliminate damaged or misfolded proteins but also as a critical regulator of multiple cellular processes by virtue of the many regulatory proteins governing the cell cycle, transcription factor activation, apoptosis, and cell trafficking that are substrates for proteasome-mediated degradation.75–78 Targeting the proteasome has emerged as a novel approach to cancer therapy. Among the proteins degraded by the ubiquitin–proteasome pathway are cyclins, cyclin-dependent kinase inhibitors such as p21 and p27, the tumor suppressor p53, oncogenes such as c-myc, c-jun and N-myc, and the inhibitory protein IκB, which inhibits nuclear factor kappa-B (NF-κB). Inhibition of the 26S proteasome results in the accumulation of these substrates and therefore causes cell cycle disruption and promotes cell death via multiple pathways.
Bortezomib is the first proteasome inhibitor to be evaluated in human studies. It is a dipeptidyl boronic acid which is a specific and selective inhibitor of multicatalytic sites within the 20S core of the 26S proteasome. The use of bortezomib in NHL was initially based on phase I and II data showing single-agent activity in various NHL subtypes.79,80 The greatest activity was seen in patients with MCL but evidence of activity was also seen in other subtypes, including FL, marginal zone lymphoma (MZL), Waldenstrom’s macroglobulinemia (WM). Based on the results of a phase II study in 155 patients with relapsed or refractory MCL, bortezomib was recently approved for the second-line treatment of MCL. The ORR in the study was 31%, with a CR in 12 patients (7%), two patients (1%) with CRu and PR in 36 patients (23%). The median duration of response (DR) was 9.3 months but was longer at 15.4 months in patients achieving CR or CRu.81 Adverse events were similar to those observed in the trials with multiple myeloma.82
As a single agent, bortezomib is not active in patients with DLBCL.83 But its use as an inhibitor of the NF-κB pathway appears to be important in some DLBCL. Gene expression profiling has revealed that DLBCL consists of at least three distinct diseases. Patients with one DLBCL subtype, termed activated B-cell-like (ABC) DLBCL, have a distinctly inferior prognosis.84 The gene expression profiles of ABC DLBCLs show high expression of target genes of the NF-κB/Rel family of transcription factors, raising the possibility that constitutive activity of the NF-κB pathway may contribute to the poor prognosis. Two cell line models of ABC DLBCL had high nuclear NF-κB DNA binding activity, constitutive IκB kinase (IKK) activity, and rapid IκBα degradation that was not seen in cell lines representing germinal center B-like (GCB) DLBCL. Retroviral transduction of a super-repressor form of IκB or dominant negative forms of IKK killed the ABC but not GCB DLBCL cells.85 These findings establish the NF-κB pathway as a new molecular target in the most clinically intractable subtype of DLBCL and demonstrate that the ABC and GCB DLBCL subtypes defined by gene expression profiling utilize distinct pathogenetic mechanisms. NF-κB dependent transcriptional activity is mediated by dimers of NF-κB family members (p50/105, p52/100, p65/RelA, RelB, or c-Rel), and is regulated by members of the IκB family of inhibitors, principally IκBα, which binds to NF-κB dimers and retains them in the cytoplasm. In response to a wide variety of external signals, IκB is phosphorylated by the IKK complex, IκBα is then targeted for ubiquitination and proteasomal degradation. NF-κB is released and translocates to the nucleus, where it activates antiapoptotic and cell-proliferation genes.86–89 The stabilization of IκB through proteasome inhibition therefore prevents NF-κB activation. This is potentially significant as the inhibition of NF-κB by bortezomib might sensitize DLBCL patients with the ABC subtype to chemotherapy and improve its clinical outcome compared to GCB DLBCL (Fig. 2).
Fig. 2.

Blockade of the NF-κB pathway in ABC DLBCL by bortezomib. NF-κB is regulated by members of the IκB family of inhibitors, principally IκBα, which binds to NF-κB dimers and retains them in the cytoplasm. In response to a wide variety of external signals, IκB is phosphorylated by the IKK complex, IκBα is then targeted for ubiquitination and proteasomal degradation. NF-κB is released and translocates to the nucleus, where it activates antiapoptotic and cell-proliferation genes. The stabilization of IκB through proteosome inhibition therefore prevents NF-κB activation.
To test this hypothesis, we conducted a phase I/II study of bortezomib combined with dose-adjusted-EPOCH chemotherapy (etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin) in 49 patients with relapsed or refractory DLBCL. The study was divided into two parts (A and B). Patients that were clinically stable were started on bortezomib alone 1.3 mg/m2 on days 1, 4, 8 and 11 every 21 days. Treatment was continued until disease progression and at that point, patients received bortezomib in combination with DA-EPOCH (Fig. 3). Patients with serious disease related complications were started on bortezomib and DA-EPOCH (Part B). Bortezomib alone had no activity in DLBCL; out of the 23 patients in part A, only patient had PR. But when combined with chemotherapy, of the 44 patients in part B, 15 (35%) responded, including eight (18%) with CR. In the subset of 27 patients with de novo DLBCL who were classified by gene expression profiling as GCB or ABC DLBCL, the ORR was 13% in GCB compared with 83% in ABC DLBCL. The median overall survival was 10.8 months in ABC compared with 3.4 months in GCB DLBCL. These results supports our hypothesis that bortezomib synergizes with chemotherapy to improve the outcome for patients with ABC DLBCL83 (Fig. 4).
Fig. 3.
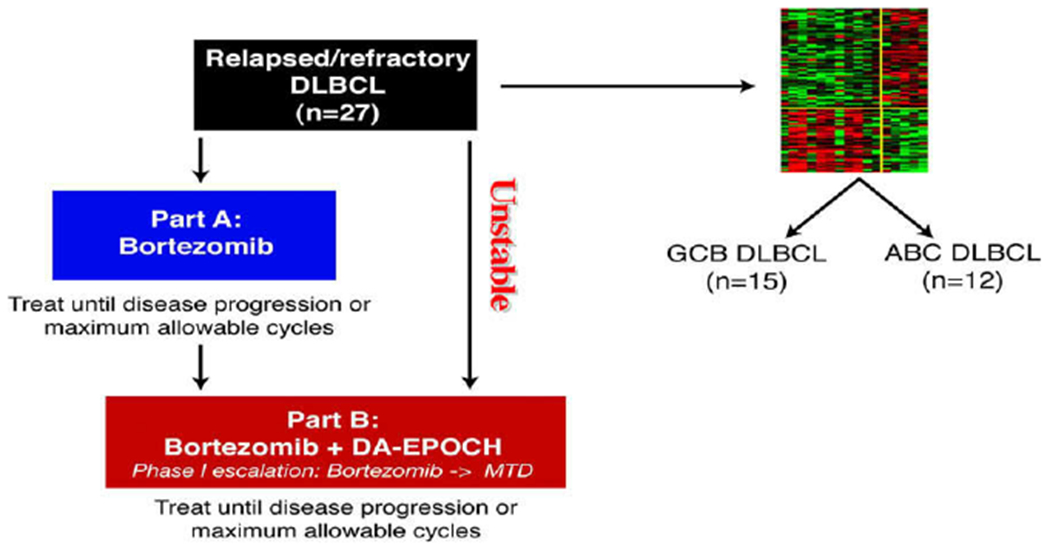
Study schema. Patients initially received bortezomib alone at 1.3 mg/m2 on days 1, 4, 8, and 11 every 21 days (Part A) unless they had disease that the investigators judged to require immediate chemotherapy, as in cases of impending or ongoing organ compromise; these patients received only Part B. Patients with progressive disease in Part A later received bortezomib with DA-EPOCH (Part B). Molecular classification. Of 31 DLBCL cases analyzed by gene expression profiling, 16 were excluded due to ineligible subtype by classification or did not receive Part A, leaving 5 ABC and 10 GCB cases eligible for analysis of outcome. Of 24 paraffin embedded tumor biopsies analyzed by immunohistochemistry, 12 of each were categorized as GCB and ABC type.By combining both methods, cases were identified as GCB in 15 and ABC in 12 and included in the analysis of outcome with Part B.
Fig. 4.
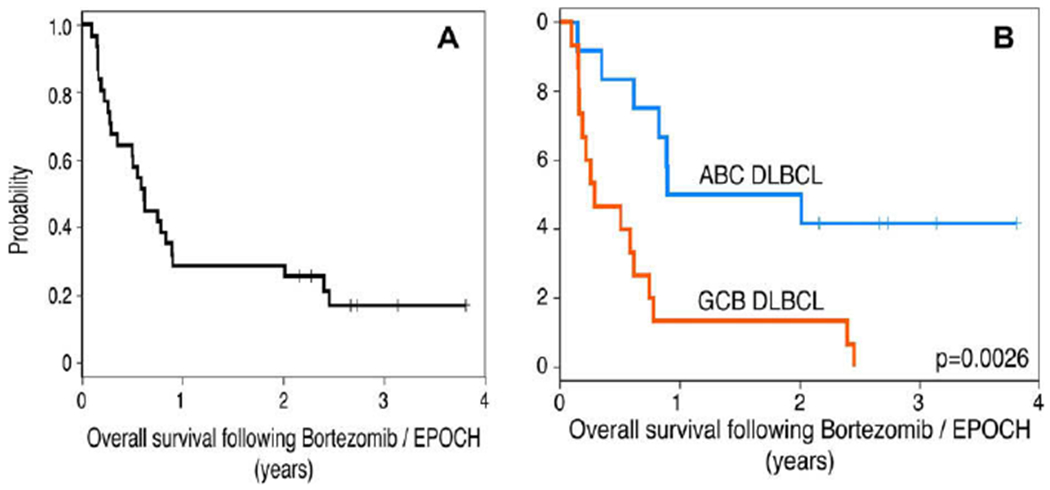
Overall survival in patients with DLBCL. (A) Overall survival of 31 patients with de novo DLBCL who received DA-EPOCH-B. With a median potential follow-up of 49 months, the median survival was 8 months. (B) Overall survival of 27 patients with ABC or GCB DLBCL who received DA-EPOCH-B showed a median survival of 10.8 and 3.4 months, respectively (P = 0.0026).
Targeting apoptotic pathways
Dysregulation of apoptosis is one of the cardinal features in cancer, which may lead to tumor progression and resistance to anticancer therapies. Apoptosis can be triggered either at the cell surface (extrinsic cell-death pathway) or at the mitochondria (intrinsic cell-death pathway). Both pathways eventually converge on the effector caspases, which are key players of the apoptotic cascade, and ultimately leading to cell death. The extrinsic cell-death pathway is activated by cell surface death receptors, such as Fas and tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptors. By contrast, the intrinsic cell-death pathway, also known as the mitochondrial apoptotic pathway, is activated by a wide range of signals, including chemotherapy drugs, irradiation, and growth factor withdrawal.90–92 Members of the Bcl-2 family are important regulators of the intrinsic apoptosis pathway. They control the integrity of the outer mitochondrial membrane through interactions of pro- and anti-apoptotic Bcl-2 family of proteins. The Bcl-2 family comprises 25 pro- and anti-apoptotic members and share up to four Bcl-2 homology (BH) domains. The anti-apoptotic family members (Bcl-XL, Bcl-2, Bcl-w, Bcl-B, A1 and Mcl-1) are characterized by four BH domains that are designated BH1-4. The pro-apoptotic family members can be further subdivided into multidomain proteins (Bax, Bak) and the BH3-only proteins (Bad, Bik, Bid, Bim, Hrk, Bmf, Noxa, Puma).93–95 On receipt of a death signal, the multidomain pro-apoptotic proteins Bax and Bak are oligomerized and activated, leading to mitochondrial outer membrane permeabilization. Once the mitochondrial membranes are permeabilized, cytochrome c is released into the cytoplasm and activates the caspase cascade, resulting in the characteristic biochemical and morphological features associated with apoptosis.96,97 Anti-apoptotic Bcl-2 family proteins inhibit the release of cytochrome c by blocking activation of Bax and Bak. BH3-only proteins, which act upstream of Bax and Bak, selectively bind Bcl-2 family proteins and release Bax and Bad98–101 (Fig. 5).
Fig. 5.

On receipt of a death signal, the multidomain pro-apoptotic proteins Bax and Bak are oligomerized and activated, leading to mitochondrial outer membrane permeabilization. Once the mitochondrial membranes are permeabilized, cytochrome c is released into the cytoplasm and activates the caspase cascade, resulting in apoptosis. Anti-apoptotic Bcl-2 family proteins inhibits the release of cytochrome c by blocking activation of Bax and Bak. BH3-only proteins, which acts upstream of Bax and Bak, selectively binds with anti-apoptotic Bcl-2 family proteins to relieve their inhibition of Bax and Bad98–101 (Used with permission from Abbott Laboratories).
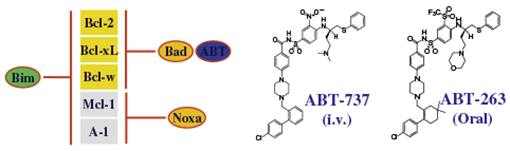
Bcl-2 is expressed by most B-cell lymphomas including FL, MCL, MCL, CLL and DLBCL. However, there is variation in expression of Bcl-2 family proteins amongst the different lymphoma subtypes. Bcl-2 is expressed more in FL, CLL, MCL and MALT than in DLBCL and Burkitt’s lymphoma (BL), whereas Mcl-1 and A1 are expressed more in DLBCL. The first approach targeting Bcl-2 proteins used antisense nucleotides, and subsequently, several small molecules Bcl-2 inhibitors have been developed. By far, the most potent small molecule inhibitors are the Bad-like BH3 mimetics, ABT-737 and its orally active analog ABT-263. However, ABT-737 is not orally bioavailable and its low aqueous solubility makes formulation for intravenous delivery challenging. To overcome this, the oral formulation, ABT-263 was developed. Like its predecessor, ABT-263 binds with very high affinity (Ki ⩽ 1 nM) to Bcl-2, Bcl-XL and Bcl-w but with much lower affinity to Mcl-1 or A1102 (Table 6). By binding to these proteins, ABT-263 releases the pro-apoptotic BCL-2 family members, thus resulting in cell death by apoptosis.
Table 6.
Binding affinities to Bcl-2 family proteins.
 |
||||||
|---|---|---|---|---|---|---|
| Compound | FPA |
|||||
|
Ki (nM) |
TR-FRET | |||||
| Bc1-XL | Bcl-2 | Bcl-w | Mcl-1 | A1 | Bc1-XL | |
| hBad | 0.5 | 15 | 33 | 4800 | >10000 | 0.12 |
| ABT-737 | ⩽0.5 | ⩽1 | 0.9 | >1000 | >1000 | 0.08 |
| ABT-263 | ⩽0.5 | ⩽1 | ⩽1 | 550 | 330 | 0.40 |
Used with permission from Abbott Laboratories.
Pre-clinical studies showed that ABT-737 exhibits potent single agent activity against 10 of 22 cell lines consisting of multiple leukemia and lymphoma types, both B-cell and T-cell malignancies.103 Furthermore, ABT-737 when used in combination with other commonly used therapeutic agents enhanced the treatment outcome in several hematologic malignant xenograft models. This observation was made by Tse and colleagues when they investigated ABT-263 in combination with other agents in several xenograft models. In the DoHH2 DLBCL xenograft, ABT-737 in combination induced complete remission in 70% of the treated tumors, enhancing the efficacy observed in either agent alone. ABT-737 also significantly enhanced the activity of R-CHOP in a mantle cell lymphoma xenograft model, GRANTA-519. The combination resulted in CR in 100% of animals tested with a cure rate of 44%. The activity of bortezomib was also enhanced when used in the combination in an ABT-737 resistant model. The OPM-2 multiple myeloma xenograft model is typically associated with high Mcl-1 expression. This data suggests that the synergy between bortezomib and ABT-737 may be due to neutralization of Bcl-XL/Bcl-2 by ABT-737 and Mcl-1 by bortezomib.103
There are currently three phase I/II studies in patients with lymphoid malignancies, small cell lung cancer and CLL. Our group is currently a site for one of the studies investigating ABT-263 in lymphoid malignancies. The phase I is divided into two parts, each testing a different dosing schedule. The patients in phase Ia will receive the test drug 14 days out of a 21 days cycle and in the phase Ib, this group of patients will receive a continuous dose for 21 days. The phase Ia is completed and the phase Ib with the continuous dosing schedule is still ongoing. The preliminary results of the study were recently reported and demonstrated favorable PK and safety profiles, with anti-tumor activity mostly observed in patients with relapsed or refractory CLL and follicular lymphoma.104 Based on preclinical evidence, observed toxicities are likely to be mechanism based. Potential treatment-related side effects include drug interactions, lymphopenia, testicular effects, and thrombocytopenia.105–107 Bcl-XL is critical in limiting the pro-apoptotic activity of Bak in platelets whereas Bcl-w appears to be essential for spermatogenesis. ABT-263 accelerates apoptosis of circulating mature platelets as older platelets contain less Bcl-XL than younger platelets and are more susceptible to destruction. Hence, the thrombocytopenia from ABT-263 is an acute dose-dependent event, the effects are limited due to release of platelets from the bone marrow.
Cyclin-dependent kinase inhibitors
Interphase cyclin-dependent kinases (CDKs) in concert with regulatory proteins called cyclins orchestrate the complex events that drive the cell cycle. The CDK/cyclin complexes are activated by phosphorylation of the CDK by CDK-activating kinase and the different complexes regulate each step of the cycle.108 The CDKs involved in the cell cycle (CDK2, CDK4, CDK5) are rational targets for cancer therapy because their activity is frequently dysregulated in cancer cells and the overall effect is loss of checkpoint integrity, resulting in uncontrolled proliferation.109 A second group of CDKs (CDKs 7–11), are involved in the regulation of transcription. A key enzyme in the transcription machinery, RNA polymerase II, is phosphorylated by several CDKs.110 The most important regulator is CDK 9/cyclin T which stabilizes the elongation of nascent mRNA transcripts.111 Inhibition of these CDKs has a global impact on cellular transcription, down-regulating the production of anti-apoptotic proteins, cell cycle regulators, NF-κB and p53-responsive gene transcripts.
Flavopiridol is a synthetic flavone which is chemically identical to a natural product obtained Dysoxylum binectariferum. It is pan-CDK inhibitor that blocks the interphase CDKs (CDK2, CDK4, CDK6). Its actions also include the inhibition of CDK7 and cyclin H, and down-regulation of cyclin D1 and D3 expression leading to G1 arrest. In vitro studies demonstrated that exposure of MCL cell lines to flavopiridol, induces apoptosis and decreases cyclin D-1 and Mcl-1 expression in MCL lines.112 Preclinical studies also demonstrated that flavopiridol is significantly toxic for cell lines derived from the ABC-type of DLBCL (OCT-Ly3 cell line) and down-regulates NF-κB pathway which is constitutively activated in the ABC-type of DLBCL.113,114 The results of these preclinical studies have provided the rationale for our study, a phase I/II study of flavopiridol in patients with relapsed or refractory MCL and DLBCL.
One of the great challenges in developing flavopiridol and applying it in clinical trials has been its dose scheduling. Several dosing schedules have been studied and most extensively in CLL patients. Both 24 and 72 h continuous infusion schedules in CLL were associated with 0% response rate. The bolus schedule produced a partial response rate of merely 11%.115 A Phase I/II study using a hybrid schedule was investigated by Byrd and colleagues and was shown to be effective in patients with refractory CLL. Flavopiridol was administered weekly for 4 of 6 weeks, the patients were treated with a 30–40 mg/m2 loading dose followed by 30–50 mg/m2 infusion over 4 h. Twenty one of 52 patients (40%) achieved a PR, and those responses were durable, with a median progression-free survival of 12 months. The dose-limiting toxicity (DLT) was tumor lysis syndrome which occurred in patients with high lymphocyte counts.116,117 Adopting a similar hybrid dosing schedule as mentioned, we recently reported the preliminary results of the phase I portion of our study. Patients (23) were enrolled, 12 patients with MCL and 11 patients with DLBCL. The overall RR was 10%, with two patients achieving PR and five patients (25%) had SD, none achieved CR. The main adverse event was grade 4 neutropenia, observed in 10 (50%) patients. The dose limiting toxicities were tumor lysis syndrome (TLS) and severe vomiting/diarrhea in 2 pts. The MTD and phase II dose has not yet been defined. In this study, we are also assessing multiple biomarkers including effects on proliferation, and phosphorylation of cell cycle associated targets such as cdk2, 4 and 9, p27 and RB. The results are still being analysed.118
mTOR inhibitors
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that regulates translation of key proteins important for cell growth, protein synthesis, and cell cycle progression through interactions with a number of signaling pathways. mTOR inhibitors were originally developed as immunosuppressive agents and is approved for use in both solid organ and allergenic stem cell transplantation. However, preclinical studies have also demonstrated activation of mTOR pathways in a number of hematological malignancies.119–122 Based on these findings, several mTOR inhibitors have been investigated in clinical trials for treatment of various hematological malignancies, including lymphomas, leukemias and multiple myelomas.
Of the novel rapamycin analogs, temsirolimus is the most extensively studied and has the most potential for clinical benefit in patients with mantle cell lymphoma. Two phase II studies have been conducted on patients with relapsed or refractory mantle cell lymphoma, using two different doses. The first phase II study by Witzig et al. reported 35 patients with MCL who had received a median of three prior therapies and investigated a higher dose of IV temsirolimus 250 mg administered once weekly. The overall response rate was 38%, with one patient (3%) achieving complete response and 12 (35%) partial responses.123 The second study enrolled 29 patients but temsirolimus was given at a lower dose of 25 mg weekly. The lower dose was tested in this second trial because 12 patients discontinued treatment for toxicities in the earlier phase II study. The overall response rate was 41%, with one complete remission (4%) and 10 partial responses (37%). The toxicity profile in this study was much improved compared to the higher osing.124 Similar to the earlier study, the main adverse event was still reversible myelosuppression with grade 3 thrombocytopenia and neutropenia occurring in 39% and 18% of patients, respectively. This is in contrast to the higher occurrence of grade 3 or 4 thrombocytopenia and neutropenia seen with the 250 mg dose (66% and 29%, respectively).
A randomized, open label phase III trial of 162 patients with relapsed or refractory MCL was recently reported. Patients were randomized to three arms; two groups received temsirolimus 175 mg weekly for 3 weeks followed by either 75 mg (175/75 mg) or 25 mg (175/25) weekly. The third group received an approved therapy for MCL based on the investigator’s choice. It consisted mostly of monotherapy cytotoxic agents with gemcitabine and fludarabine comprising the majority (42% and 26%, respectively) of the treatment choice. Patients treated with temsirolimus 175/75 mg achieved a significantly longer progression-free survival compared with patients treated with investigator’s choice therapy but this survival benefit was not achieved in the cohort of patients treated with 175/25 mg. The median PFS was 4.8, 3.4, and 1.9 months for the patients treated with temsirolimus 175/75 mg, 175/25 mg, and investigator’s choice, respectively. The objective response rate was significantly higher in the 175/75 mg group compared with the investigator’s choice group (22% vs. 2%; p = 0.019) but the objective response rate in the 175/25 mg group was not (6%; p = 0.6179). Among the responses in the 175/75 mg group, there was one complete remission and 11 partial responses.125 The drug was well tolerated with a manageable toxicity profile, similar to that observed in the phase II trials with myelosuppression being the most common adverse event seen. Grade 3 or 4 thrombocytopenia occurred in 59% of patients treated with 175/75 mg, in 52% of patients in 175/25 mg group, and in 36% of patients receiving investigator’s choice therapy. Both the temsirolimus arms in the phase III trial reported a lower response rate than in the phase II studies. This likely reflects the differences in patient selection between the phase II and the phase III trials. A higher proportion of heavily pre-treated patients were enrolled in the phase III trial, with 50% of the patients receiving four to seven prior therapies. Furthermore, patients in the phase III trial were also required to have had prior treatment with an anthracycline, an alkylating agent, and an anti-CD20 containing regimen. Nonetheless, this is the first phase III study to demonstrate a PFS benefit for heavily pre-treated patients with MCL using the temsirolimus dose of 175/75 mg in comparison with standard chemotherapeutic single agents.
Temsirolimus has also been investigated in clinical trials in patients with non-MCL NHL. A multicenter phase II study of 82 patients with relapsed or refractory NHL showed moderate single-agent activity. The study included patients with diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL) and small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL). Temsirolimus 25 mg IV was administered weekly with a planned 8 weeks of therapy (two cycles) or given until disease progression if tolerated. The overall response rate was 35% for all patients, 9% for DLBCL, 23% for FL, and 3% in SLL/CLL. However, among patients who received at least two cycles of temsirolimus, the objective response rate was 46%. Most non-hematologic toxicities were grade 1 or 2, including stomatitis, rash and metabolic dysregulation but 12 patients were removed from the study due to pneumonia/pneumonitis or stomatitis in seven patients, two patients for infection, and three patients for cytopenia.126 Clinical data available so far suggest that temsirolimus monotherapy represents a promising new direction for lymphoma therapy. Two other mTOR inhibitors, everolimus and deforolimus, have also shown potential in clinical trials in the management of advanced hematological malignancies and their results are summarized in (Table 7).127–129
Table 7.
Response rates of single agent mTOR inhibitors in NHL.
| Study | Status | Agent | Doses | Patients | Results (%) |
|
|---|---|---|---|---|---|---|
| ORR | CR/CRu | |||||
| Witzig et al. [123] | Phase II | Temsirolimus | 250 mg IV weekly | Relapsed/refractory MCL (n = 35) | 38 | 3 |
| Ansell et al. [124] | Phase II | Temsirolimus | 25 mg IV weekly | Relapsed/refractory MCL (n = 29) | 41 | 4 |
| Hess et al. [125] | Phase III | Temsirolimus | 175 mg IV weekly × 3 followed by 75 mg | Relapsed/refractory MCL (n = 54) | 22 | 2 |
| Smith et al. [126] | Phase II | Temsirolimus | 25 mg IV weekly | Relapsed/refractory non-MCL NHL (n = 82) | 35 | 9 |
| Reeder et al. [127] | Phase II | Everolimus | 10 mg/day orally | Relapsed/refractory NHL (n = 37) | 32 | 3 |
| Witzig et al. [128] | Phase II | Everolimus | 10 mg/day orally | Relapsed/refractory aggressive (n = 77) or indolent NHL or HL (n = 17) | 33 | 3 |
| Rizzieri et al. [129] | Phase II | Deforolimus | 12.5 mg/day IV × 5 days every 2 weeks | Relapsed/refractory hematological malignancies (n = 52) | 10 | 0 |
CR – complete response; CRu – complete response unconfirmed; DLBCL – diffuse large B-cell lymphoma; HL – Hodgkin lymphoma; mTOR – mammalian target of rapamycin; MCL – mantle cell lymphoma; NHL – non-Hodgkin lymphoma; ORR – overall response rate.
Histone deacetylase inhibitors
Histones are a family of proteins around which DNA is wound to form the nucleosome, the basic unit of chromatin. Histones can undergo several post-translational modifications, including acetylation, methylation, phosphorylation, and ubiquitylation. These modifications affect the intrinsic chromatin structure and interactions with chromatin-associated proteins, ultimately impacting gene expression.130,131 One of these epigenetic alterations is histone acetylation/deacetylation. Acetylation is regulated by two groups of enzymes with opposite activities, histone acetylase and histone deacetylase (HDAC). When histones are acetylated, the chromatin is relaxed allowing transcriptional activity, while deacetylation is associated with condensed chromatin and transcriptional repression. Alteration of this protein acetylation equilibrium and overexpression of HDAC, has been observed in both hematological and solid malignancies.132–134 Recent data have demonstrated that HDACs regulate an array of proteins that is not limited to histones. A variety of non-histone proteins are regulated by their acetylation level, including transcription factors, chaperones and structural proteins. The gene-silencing activity of DNA methylation also relies on HDAC. As a result of these interactions, control of cell cycle, apoptosis, cellular differentiation, angiogenesis and invasion represents some of the most important pathways affected by HDAC, therefore making HDACs important potential therapeutic targets in anticancer therapy135,136 (Fig. 6). However, it should be emphasized that epigenetic regulation of gene expression is highly complex and the diverse roles of HDAC suggest that the mechanism of cell death resulting from HDAC inhibitors is likely to be multifactorial and not straightforward in predicting its efficacy.
Fig. 6.

Histone deacetylase inhibitors can induce transformed cell growth arrest and death by different pathways. HDACs – histone deacetylases; HIF-1α – hypoxia induced factor-1α; HSP90 – heat shock protein 90; PP1 – protein phosphatase; ROS – reactive oxygen species; TBP2 – thioredoxin binding protein 2; Trx – thioredoxin; VEGF – vascular endothelial growth factor.
HDAC inhibitors (HDACIs) have demonstrated potent anti-tumor activity in vitro with remarkably little toxicity in preclinical studies and are currently in various stages of clinical development. HDACIs are classified according to their chemical structure. The major classes include hydroxamic acid: SAHA (vorinostat), cyclic peptides: depsipeptide (romidepsin), short-chain fatty acids: valproate, benzamides: MS-275, MGCD-0103, thiolate, non-hydroxamic acid, carboxamides, and ketones. Of these, vorinostat, a pan-HDACI is the most advanced of this class of agents in clinical development. It was the first HDACI to be approved by FDA in 2006 for the treatment of progressive, persistent, or recurrent cutaneous T-cell lymphoma (CTCL).137–139 We will now review the clinical progress of HDACIs with respect to peripheral T-cell lymphoma (PTCL) and DLBCL (Table 8).140–142
Table 8.
Histone deacetylase inhibitors in NHL.
| Study | Status | Agent | Regimens | Patients | Results (%) |
|
|---|---|---|---|---|---|---|
| ORR | CR/CRu | |||||
| Duvicet al. [138] | Phase II | Vorinostat | 400 mg QD | Relapsed/refractory CTCL (n = 13) | 31 | 0 |
| Olsen et al. [139] | Phase IIB | Vorinostat | 400 mg QD | Relapsed/refractory CTCL (n = 74) | 30 | 1 |
| Piekarz et al. [143] | Phase II | Romidepsin | 14 mg/m2 on days 1, 8 and 15 of a 28 days cycle | Relapsed/refractory CTCL or PTCL (n = 46) | 28 | 7 |
| Gimsing et al. [140] | Phase I | Belinostat | 1000 mg/m2 on days 1–5 of a 21 days cycle | Relapsed/refractory hematological malignancies (n = 16) | 0 | 0 (5 of 16 patients with SD) |
| Ellis et al. [141] | Phase I | Panobinostat | 20 mg on days 1, 3, 5 weekly, every 28 days cycle | Relapsed/refractory CTCL (n = 10) | 60 | 20 |
| Duvicet al. [142] | Phase II | Panobinostat | 20 mg on days 1, 3, 5 weekly, every 28 days cycle | Relapsed/refractory CTCL (n = 95) | 16 | 2 |
CR – complete response; CRu – complete response unconfirmed; CTCL – cutaneous T-cell lymphoma; NHL – non–Hodgkin lymphoma; ORR – overall response rate; PTCL – peripheral T-cell lymphoma; SD – stable disease.
Romidepsin (Depsipeptide/FK 228) is a cyclic peptide, a selective HDACI, with inhibitory activities against class I and II HDACs (isotype 1, 2, 4 and 6). Romidepsin was first isolated from a broth culture of Chromobacterium violaceum and was observed to be able to reverse the transformed morphology of Ras-1 cells. Early in vitro studies of romidepsin in T-cell lymphomas demonstrated significant apoptosis in the HUT78 human CTCL cell line. Subsequently, a number of phase I and II trials have been conducted in patients with relapsed or refractory CTCL or PTCL. A small phase I trial conducted here at the National Cancer Institute (NCI) identified significant activity with three PRs in CTCL and one CR in PTCL. These encouraging results prompted the initiation of the phase II study. Romidepsin is administered on days 1, 8, and 15 of a 28 days cycle, with a starting dose of 14 mg/m2. Among the 46 patients enrolled, three had CRs, and eight had PRs for an ORR 28%. Overall, the drug was well tolerated with similar adverse effects as in the phase I study, including nausea, vomiting, fatigue, neutropenia, thrombocytopenia and hypocalcemia.143 The results of this study will hopefully be confirmed in a multi-institutional phase II study that was recently initiated. The study is an open-label, single arm, multinational trial that will be conducted at 35 sites in the United States, Europe and Australia.
Conclusion
With the increase in incidence of NHL in recent years, there is a clear clinical need for novel agents to offer new options in resistant disease, and potentially to improve outcomes in curative settings. There has been tremendous insight gained in the last two decades from basic science research, which provided the impetus and rationale for clinical development of novel agents. As a result, there are an unprecedented number of ongoing clinical trials in NHL evaluating various novel targeted therapies, several of which show early evidence of efficacy. However, it is still unclear how to optimally incorporate many of these agents into treatment algorithms for NHL. The principal challenge facing investigators and clinicians, given the heterogeneity of the disease, will be the development of rational combinations that are most effective with limited toxicity, and individualizing treatment algorithms based on the biology of specific NHL.
Footnotes
Conflicts of interest
None declared.
References
- 1.Swenson WT, Wooldridge JE, Lynch CF, Forman-Hoffman VL, Chrischilles E, Link BK. Improved survival of follicular lymphoma patients in the United States. J Clin Oncol 2005;23:5019–26. [DOI] [PubMed] [Google Scholar]
- 2.Feugier P, Van Hoof A, Sebban C, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 2005;23:4117–26. [DOI] [PubMed] [Google Scholar]
- 3.Pfreundschuh M, Trumper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7:379–91. [DOI] [PubMed] [Google Scholar]
- 4.Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol 1980;125:1678–85. [PubMed] [Google Scholar]
- 5.Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002;99:754–8. [DOI] [PubMed] [Google Scholar]
- 6.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 2003;21:3940–7. [DOI] [PubMed] [Google Scholar]
- 7.Kim DH, Jung HD, Kim JG, et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood 2006;108:2720–5. [DOI] [PubMed] [Google Scholar]
- 8.Golay J, Zaffaroni L, Vaccari T, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood 2000;95:3900–8. [PubMed] [Google Scholar]
- 9.van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol 2001;115:807–11. [DOI] [PubMed] [Google Scholar]
- 10.Byrd JC, Kitada S, Flinn IW, et al. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood 2002;99:1038–43. [DOI] [PubMed] [Google Scholar]
- 11.Alas S, Bonavida B. Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res 2001;61:5137–44. [PubMed] [Google Scholar]
- 12.Jazirehi AR, Vega MI, Chatterjee D, Goodglick L, Bonavida B. Inhibition of the Raf-MEK1/2-ERK1/2 signaling pathway, Bcl-xL down-regulation, and chemosensitization of non-Hodgkin’s lymphoma B cells by Rituximab. Cancer Res 2004;64:7117–26. [DOI] [PubMed] [Google Scholar]
- 13.Mathas S, Rickers A, Bommert K, Dorken B, Mapara MY. Anti-CD20- and B-cell receptor-mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer Res 2000;60:7170–6. [PubMed] [Google Scholar]
- 14.Tedder TF, Streuli M, Schlossman SF, Saito H. Isolation and structure of a cDNA encoding the B1 (CD20) cell-surface antigen of human B lymphocytes. Proc Natl Acad Sci USA 1988;85:208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Press OW, Farr AG, Borroz KI, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting human B-cell malignancies. Cancer Res 1989;49:4906–12. [PubMed] [Google Scholar]
- 16.Press OW, Howell-Clark J, Anderson S, Bernstein I. Retention of B-cell-specific monoclonal antibodies by human lymphoma cells. Blood 1994;83:1390–7. [PubMed] [Google Scholar]
- 17.Tedder TF, Boyd AW, Freedman AS, Nadler LM, Schlossman SF. The B cell surface molecule B1 is functionally linked with B cell activation and differentiation. J Immunol 1985;135:973–9. [PubMed] [Google Scholar]
- 18.Jazirehi AR, Bonavida B. Cellular and molecular signal transduction pathways modulated by rituximab (rituxan, anti-CD20 mAb) in non-Hodgkin’s lymphoma: implications in chemosensitization and therapeutic intervention. Oncogene 2005;24:2121–43. [DOI] [PubMed] [Google Scholar]
- 19.McLaughlin P, Grillo-Lopez AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998;16:2825–33. [DOI] [PubMed] [Google Scholar]
- 20.Davis TA, Grillo-Lopez AJ, White CA, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of retreatment. J Clin Oncol 2000;18:3135–43. [DOI] [PubMed] [Google Scholar]
- 21.Cragg MS, Walshe CA, Ivanov AO, Glennie MJ. The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun 2005;8:140–74. [DOI] [PubMed] [Google Scholar]
- 22.Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood 2002;99:3256–62. [DOI] [PubMed] [Google Scholar]
- 23.Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 2004;104:1793–800. [DOI] [PubMed] [Google Scholar]
- 24.Coiffier B, Lepretre S, Pedersen LM, et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1–2 study. Blood 2008;111:1094–100. [DOI] [PubMed] [Google Scholar]
- 25.Hagenbeek A, Gadeberg O, Johnson P, et al. First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: results of a phase 1/2 trial. Blood 2008;111:5486–95. [DOI] [PubMed] [Google Scholar]
- 26.Stein R, Qu Z, Chen S, et al. Characterization of a new humanized anti-CD20 monoclonal antibody, IMMU-106, and Its use in combination with the humanized anti-CD22 antibody, epratuzumab, for the therapy of non-Hodgkin’s lymphoma. Clin Cancer Res 2004;10:2868–78. [DOI] [PubMed] [Google Scholar]
- 27.Goldenberg DM, Rossi EA, Stein R, et al. Properties and structure-function relationships of veltuzumab (hA20), a humanized anti-CD20 monoclonal antibody. Blood 2009;113:1062–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morschhauser F, Leonard JP, Fayad L, et al. Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J Clin Oncol 2009;27:3346–53. [DOI] [PubMed] [Google Scholar]
- 29.Leonard JP, Coleman M, Ketas JC, et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J Clin Oncol 2003;21:3051–9. [DOI] [PubMed] [Google Scholar]
- 30.Leonard JP, Coleman M, Ketas JC, et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin’s lymphoma: phase I/II clinical trial results. Clin Cancer Res 2004;10:5327–34. [DOI] [PubMed] [Google Scholar]
- 31.Strauss SJ, Morschhauser F, Rech J, et al. Multicenter phase II trial of immunotherapy with the humanized anti-CD22 antibody, epratuzumab, in combination with rituximab, in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol 2006;24:3880–6. [DOI] [PubMed] [Google Scholar]
- 32.Advani R, Forero-Torres A, Furman RR, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol 2009;27:4371–7. [DOI] [PubMed] [Google Scholar]
- 33.Kivekas I, Hulkkonen J, Hurme M, Vilpo L, Vilpo J. CD80 antigen expression as a predictor of ex vivo chemosensitivity in chronic lymphocytic leukemia. Leuk Res 2002;26:443–6. [DOI] [PubMed] [Google Scholar]
- 34.Suvas S, Singh V, Sahdev S, Vohra H, Agrewala JN. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J Biol Chem 2002;277:7766–75. [DOI] [PubMed] [Google Scholar]
- 35.Vyth-Dreese FA, Boot H, Dellemijn TA, et al. Localization in situ of costimulatory molecules and cytokines in B-cell non-Hodgkin’s lymphoma. Immunology 1998;94:580–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dorfman DM, Schultze JL, Shahsafaei A, et al. In vivo expression of B7–1 and B7–2 by follicular lymphoma cells can prevent induction of T-cell anergy but is insufficient to induce significant T-cell proliferation. Blood 1997;90:4297–306. [PubMed] [Google Scholar]
- 37.Czuczman MS, Thall A, Witzig TE, et al. Phase I/II study of galiximab, an anti-CD80 antibody, for relapsed or refractory follicular lymphoma. J Clin Oncol 2005;23:4390–8. [DOI] [PubMed] [Google Scholar]
- 38.Leonard JP, Friedberg JW, Younes A, et al. A phase I/II study of galiximab (an anti-CD80 monoclonal antibody) in combination with rituximab for relapsed or refractory, follicular lymphoma. Ann Oncol 2007;18:1216–23. [DOI] [PubMed] [Google Scholar]
- 39.Bross PF, Beitz J, Chen G, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 2001;7:1490–6. [PubMed] [Google Scholar]
- 40.Younes A, Forero-Torres A, Bartlett NL. Objective responses in a phase I dose-escalation study of SGN-35, a novel antibody-drug conjugate (ADC) targeting CD30, in patients with relapsed or refractory Hodgkin lymphoma. J Clin Oncol 2008;26:8526 Abstract. [Google Scholar]
- 41.Bartlett N, Forero-Torres A, Rosenblatt J. Complete remissions with weekly dosing of SGN-35, a novel antibody-drug conjugate (ADC) targeting CD30, in a phase I dose-escalation study in patients with relapsed or refractory Hodgkin lymphoma (HL) or systemic anaplastic large cell lymphoma (sALCL). J Clin Oncol 2009;27:8500 Abstract. [Google Scholar]
- 42.Fayad L, Patel H, Verhoef G. Clinical activity of the immunoconjugate CMC-544 in B-cell malignancies: preliminary report of the expanded maximum tolerated dose (MTD) cohort of a phase 1 study. Blood 2006;108:2711. Abstract. [Google Scholar]
- 43.Tobinai K, Ogura M, Hatake K. Phase I and pharmacokinetic study of inotuzumab ozogamicin (CMC-544) as a single agent in Japanese patients with follicular lymphoma pretreated with rituximab. Blood 2008;112:1565. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fayad L, Patel H, Verhoef G. Safety and clinical activity of the anti-CD22 immunoconjugate inotuzumab ozogamicin (CMC-544) in combination with rituximab in follicular lymphoma or diffuse large B-cell lymphoma: preliminary report of a phase I/II study. Blood 2008;112:266 Abstract. [Google Scholar]
- 45.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005;23:2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002;298:850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 2007;25:267–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bargou R, Kufer P, Goebeler M. Sustained response duration seen after treatment with single agent blinatumomab (MT103/MEDI-538) in the ongoing phase I study MT103- 104 in patients with relapsed NHL. Blood 2008; 112:267. Abstract. [Google Scholar]
- 49.Hwu P, Yang JC, Cowherd R, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res 1995;55:3369–73. [PubMed] [Google Scholar]
- 50.Haynes NM, Trapani JA, Teng MW, et al. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002;100:3155–63. [DOI] [PubMed] [Google Scholar]
- 51.Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res 2006;66:10995–1004. [DOI] [PubMed] [Google Scholar]
- 52.Li S, Yang J, Urban FA, et al. Genetically engineered T cells expressing a HER2-specific chimeric receptor mediate antigen-specific tumor regression. Cancer Gene Ther 2008;15:382–92. [DOI] [PubMed] [Google Scholar]
- 53.Jensen MC, Popplewell L, DiGiusto D. A first-in-human clinical trial of adoptive therapy using CD19-specific chimeric antigen receptor re-directed T-cells for recurrent/refractory follicular lymphoma. Blood 2007;110:288 Abstract. [Google Scholar]
- 54.Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother 2009;32:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 2005;202:907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22. [DOI] [PubMed] [Google Scholar]
- 57.Corral LG, Haslett PA, Muller GW, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol 1999;163:380–6. [PubMed] [Google Scholar]
- 58.Galustian C, Meyer B, Labarthe MC, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother 2009;58:1033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LeBlanc R, Hideshima T, Catley LP, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004;103:1787–90. [DOI] [PubMed] [Google Scholar]
- 60.Chang DH, Liu N, Klimek V, et al. Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood 2006;108:618–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davies FE, Raje N, Hideshima T, et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001;98:210–6. [DOI] [PubMed] [Google Scholar]
- 62.Dredge K, Horsfall R, Robinson SP, et al. Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc Res 2005;69:56–63. [DOI] [PubMed] [Google Scholar]
- 63.Gandhi AK, Kang J, Naziruddin S, Parton A, Schafer PH, Stirling DI. Lenalidomide inhibits proliferation of Namalwa CSN.70 cells and interferes with Gab1 phosphorylation and adaptor protein complex assembly. Leuk Res 2006;30:849–58. [DOI] [PubMed] [Google Scholar]
- 64.Lentzsch S, LeBlanc R, Podar K, et al. Immunomodulatory analogs of thalidomide inhibit growth of Hs Sultan cells and angiogenesis in vivo. Leukemia 2003;17:41–4. [DOI] [PubMed] [Google Scholar]
- 65.Wiernik PH, Lossos IS, Tuscano JM, et al. Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:4952–7. [DOI] [PubMed] [Google Scholar]
- 66.Zinzani PLWT, Vose JM, et al. Confirmation of the efficacy and safety of lenalidomide oral monotherapy in patients with relapsed or refractory mantle-cell lymphoma: results of an international study (NHL-003). Am Soc Hematol Annu Meet 2008;262 Abstract. [Google Scholar]
- 67.Czuczman M, Vose JM, Zinzani PL. Confirmation of the efficacy and safety of lenalidomide oral monotherapy in patients with relapsed or refractory diffuse large-B-cell lymphoma: results of an international study (NHL-003). Blood 2008;112:268 Abstract. [Google Scholar]
- 68.Chanan-Khan A, Miller KC, Musial L, et al. Clinical efficacy of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia: results of a phase II study. J Clin Oncol 2006;24:5343–9. [DOI] [PubMed] [Google Scholar]
- 69.Ferrajoli A, Lee Bn, Schlette EJ, et al. Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood 2008;111:5291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen C, Paul H, Xu W. A phase II study of lenalidomide in previously untreated, symptomatic chronic lymphocytic leukemia. Blood 2008;112:44 Abstract. [Google Scholar]
- 71.Ferrajoli A, O’Brien S, Wierda W. Lenalidomide as initial treatment of elderly patients with chronic lymphocytic leukemia. Blood 2008;112:45 Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheson BD, Vose JM, Bartlett NL. Safety and efficacy of YM155 in diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2009;27:8502 Abstract. [Google Scholar]
- 73.Robertson MJ, Kahl BS, Vose JM, et al. Phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol 2007;25:1741–6. [DOI] [PubMed] [Google Scholar]
- 74.Morschhauser F, Seymour JF, Kluin-Nelemans HC, et al. A phase II study of enzastaurin, a protein kinase C beta inhibitor, in patients with relapsed or refractory mantle cell lymphoma. Ann Oncol 2008;19:247–53. [DOI] [PubMed] [Google Scholar]
- 75.Goldberg AL, Stein R, Adams J. New insights into proteasome function: from archaebacteria to drug development. Chem Biol 1995;2:503–8. [DOI] [PubMed] [Google Scholar]
- 76.Hochstrasser M Protein degradation or regulation: Ub the judge. Cell 1996;84:813–5. [DOI] [PubMed] [Google Scholar]
- 77.King RW, Deshaies RJ, Peters JM, Kirschner MW. How proteolysis drives the cell cycle. Science 1996;274:1652–9. [DOI] [PubMed] [Google Scholar]
- 78.Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res 1996;56:2649–54. [PubMed] [Google Scholar]
- 79.O’Connor OA, Wright J, Moskowitz C, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol 2005;23:676–84. [DOI] [PubMed] [Google Scholar]
- 80.Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75. [DOI] [PubMed] [Google Scholar]
- 81.Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74. [DOI] [PubMed] [Google Scholar]
- 82.Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003;348:2609–17. [DOI] [PubMed] [Google Scholar]
- 83.Dunleavy K, Pittaluga S, Czuczman MS, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009;113:6069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 85.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med 2001;194:1861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649–83. [DOI] [PubMed] [Google Scholar]
- 87.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998;16:225–60. [DOI] [PubMed] [Google Scholar]
- 88.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 2000;18:621–63. [DOI] [PubMed] [Google Scholar]
- 89.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol 1994;10:405–55. [DOI] [PubMed] [Google Scholar]
- 90.Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12. [DOI] [PubMed] [Google Scholar]
- 91.Spierings D, McStay G, Saleh M, et al. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science 2005;310:66–7. [DOI] [PubMed] [Google Scholar]
- 92.Wajant H The Fas signaling pathway: more than a paradigm. Science 2002;296:1635–6. [DOI] [PubMed] [Google Scholar]
- 93.Chittenden T, Harrington EA, O’Connor R, et al. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 1995;374:733–6. [DOI] [PubMed] [Google Scholar]
- 94.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev 1996;10:2859–69. [DOI] [PubMed] [Google Scholar]
- 95.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002;2:183–92. [DOI] [PubMed] [Google Scholar]
- 96.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development ofmultiple tissues. Mol Cell 2000;6:1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006;9:351–65. [DOI] [PubMed] [Google Scholar]
- 99.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007;315:856–9. [DOI] [PubMed] [Google Scholar]
- 100.Kim H, Rafiuddin-Shah M, Tu HC, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 2006;8:1348–58. [DOI] [PubMed] [Google Scholar]
- 101.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005;17:393–403. [DOI] [PubMed] [Google Scholar]
- 102.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677–81. [DOI] [PubMed] [Google Scholar]
- 103.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008;68:3421–8. [DOI] [PubMed] [Google Scholar]
- 104.Wilson WH, O’Connor OO, Roberts AW. ABT-263 activity and safety in patients with relapsed or refractory lymphoid malignancies in particular chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). J Clin Oncol 2009;27(Suppl. 15):8574 Abstract. [Google Scholar]
- 105.Print CG, Loveland KL, Gibson L, et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA 1998;95:12424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2 alpha beta in mice. occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA 1994;91:3700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell 2007;128:1173–86. [DOI] [PubMed] [Google Scholar]
- 108.Kaldis P, Russo AA, Chou HS, Pavletich NP, Solomon MJ. Human and yeast cdk-activating kinases (CAKs) display distinct substrate specificities. Mol Biol Cell 1998;9:2545–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153–66. [DOI] [PubMed] [Google Scholar]
- 110.Oelgeschlager T Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol 2002;190:160–9. [DOI] [PubMed] [Google Scholar]
- 111.Romano G, Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008;7:3664–8. [DOI] [PubMed] [Google Scholar]
- 112.Venkataraman G, Maududi T, Ozpuyan F, et al. Induction of apoptosis and down regulation of cell cycle proteins in mantle cell lymphoma by flavopiridol treatment. Leuk Res 2006;30:1377–84. [DOI] [PubMed] [Google Scholar]
- 113.Lam LT, Pickeral OK, Peng AC, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol 2001;2:0041 Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takada Y, Aggarwal BB. Flavopiridol inhibits NF-kappaB activation induced by various carcinogens and inflammatory agents through inhibition of IkappaBalpha kinase and p65 phosphorylation: abrogation of cyclin D1, cyclooxygenase-2, and matrix metalloprotease-9. J Biol Chem 2004;279:4750–9. [DOI] [PubMed] [Google Scholar]
- 115.Byrd JC, Peterson BL, Gabrilove J, et al. Treatment of relapsed chronic lymphocytic leukemia by 72-hour continuous infusion or 1-hour bolus infusion of flavopiridol: results from Cancer and Leukemia Group B study 19805. Clin Cancer Res 2005;11:4176–81. [DOI] [PubMed] [Google Scholar]
- 116.Byrd JC, Lin TS, Dalton JT, et al. Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 2007;109:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Phelps MA, Lin TS, Johnson AJ, et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009;113:2637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tay K, Shapiro G, Disinski M. Phase I/II study of a hybrid schedule of flavopiridol in relapsed/refractory mantle cell lymphoma (MCL) and diffuse large B-cell lymphoma (DLBCL). J Clin Oncol 2009;27(Suppl. 15):8563. Abstract. [Google Scholar]
- 119.Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood 2006;107:1149–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yazbeck VY, Buglio D, Georgakis GV, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 2008;36:443–50. [DOI] [PubMed] [Google Scholar]
- 121.Costa LJ. Aspects of mTOR biology and the use of mTOR inhibitors in non-Hodgkin’s lymphoma. Cancer Treat Rev 2007;33:78–84. [DOI] [PubMed] [Google Scholar]
- 122.Peponi E, Drakos E, Reyes G, Leventaki V, Rassidakis GZ, Medeiros LJ. Activation of mammalian target of rapamycin signaling promotes cell cycle progression and protects cells from apoptosis in mantle cell lymphoma. Am J Pathol 2006;169:2171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56. [DOI] [PubMed] [Google Scholar]
- 124.Ansell SM, Inwards DJ, Rowland KM Jr, et al. Low-dose, single-agent temsirolimus for relapsed mantle cell lymphoma: a phase 2 trial in the North Central Cancer Treatment Group. Cancer 2008;113:508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hess G, Herbrecht R, Romaguera J, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:3822–9. [DOI] [PubMed] [Google Scholar]
- 126.Smith SM, Pro B, Cisneros A. Activity of single agent temsirolimus (CCI-779) in non-mantle cell non-Hodgkin lymphoma subtypes. J Clin Oncol 2008;26(May 20 suppl):8514. [Google Scholar]
- 127.Reeder CB, Gornet MK, Habermann TM. A phase 2 clinical trial of the oral mTOR inhibitor everolimus (RAD001) in relapsed aggressive non-Hodgkin lymphoma. Blood 2007;110:121 Abstract. [Google Scholar]
- 128.Witzig E, Habermann T, Reeder CB. A phase II trial of the oral mTOR inhibitor everolimus in relapsed non-Hodgkin lymphoma and Hodgkin lymphoma. Haematologica 2009;94(Suppl 2):1081 Abstract. [Google Scholar]
- 129.Rizzieri DA, Feldman E, Dipersio JF, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 2008;14:2756–62. [DOI] [PubMed] [Google Scholar]
- 130.Jenuwein T, Allis CD. Translatingthe histone code. Science 2001;293:1074–80. [DOI] [PubMed] [Google Scholar]
- 131.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009;10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Redner RL, Wang J, Liu JM. Chromatin remodeling and leukemia: new therapeutic paradigms. Blood 1999;94:417–28. [PubMed] [Google Scholar]
- 133.Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell Mol Life Sci 2001;58:728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cress WD, Seto E. Histone deacetylases, transcriptional control, and cancer. J Cell Physiol 2000;184:1–16. [DOI] [PubMed] [Google Scholar]
- 135.Bolden JE, Peart MJ,Johnstone RW. Anticancer activities ofhistone deacetylase inhibitors. Nat Rev Drug Discov 2006;5:769–84. [DOI] [PubMed] [Google Scholar]
- 136.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 2009;107:600–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Invest Drugs 2007;16:1111–20. [DOI] [PubMed] [Google Scholar]
- 138.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007;109:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 2007;25:3109–15. [DOI] [PubMed] [Google Scholar]
- 140.Gimsing P, Hansen M, Knudsen LM, et al. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol 2008;81:170–6. [DOI] [PubMed] [Google Scholar]
- 141.Ellis L, Pan Y, Smyth GK, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res 2008;14:4500–10. [DOI] [PubMed] [Google Scholar]
- 142.Duvic M, Becker JC, Dalle S. Phase II trial of oral panobinostat (LBH589) in patients with refractory cutaneous T-cell lymphoma (CTCL). Blood 2008;112:1005 Abstract.18477770 [Google Scholar]
- 143.Piekarz R, Frye R, Wright J. Update of the NCI multiinstitutional phase II trial of romidepsin, FK228, for patients with cutaneous or peripheral T-cell lymphoma. J Clin Oncol 2007;25(Suppl 18):8027 Abstract. [Google Scholar]