

Abstract
Exposure of male C3H mice in utero (from gestational days 8–18) to 85 ppm sodium arsenite via the dams’ drinking water has previously been shown to increase liver tumor incidence by 2 years of age. However, in our companion study (Ahlborn et al., 2009), continuous exposure to 85 ppm sodium arsenic (from gestational day 8 to postnatal day 365) did not result in increased tumor incidence, but rather in a significant reduction (0% tumor incidence). The purpose of the present study was to examine the gene expression responses that may lead to the apparent protective effect of continuous arsenic exposure. Genes in many functional categories including cellular growth and proliferation, gene expression, cell death, oxidative stress, protein ubiquitination, and mitochondrial dysfunction were altered by continuous arsenic treatment. Many of these genes are known to be involved in liver cancer. One such gene associated with rodent hepatocarcinogenesis, Scd1, encodes stearoyl-CoA desaturase and was down-regulated by continuous arsenic treatment. An overlap between the genes in our study affected by continuous arsenic exposure and those from the literature affected by long-term caloric restriction suggests that reduction in the spontaneous tumor incidence under both conditions may involve similar gene pathways such as fatty acid metabolism, apoptosis, and stress response.
Keywords: Sodium arsenite, C3H mouse, In utero, Liver tumor, Gene expression
1. Introduction
Inorganic arsenic is a well-documented human carcinogen, and arsenic-contaminated drinking water is a major concern in many parts of the world (Bates et al., 1995; IARC, 2004; NRC, 1999; Pott et al., 2001; Kitchin, 2001; Simeonova and Luster, 2000). Gestation and early life are periods of high sensitivity to chemical carcinogenesis (Anderson et al., 2000; Barton et al., 2005). Studies from a unique exposure scenario in Chile have shown that arsenic exposure both in utero and during childhood predisposes adults to increased risk of lung and urinary bladder cancer (Marshall et al., 2007; Smith et al., 2006), and children to increased liver cancer mortality (Liaw et al., 2008). Adult mice are typically resistant to experimental induction of tumors with sodium arsenite, and require administration of a co-carcinogen or promoter for tumor formation (Rossman et al., 2004; Morikawa et al., 2000; Mizoi et al., 2005; Motiwale et al., 2005; Uddin et al., 2005). However, studies in C3H and CD-1 mice have found increased liver, lung, adrenal, ovarian and uterine tumor incidence at 2 years following in utero exposure between gestation days 8 and 18 (Waalkes et al., 2003, 2004a,b, 2006), indicating arsenic to be a “complete” transplacental carcinogen in mice. Gene expression changes associated with this exposure included increased expression of genes involved in cell proliferation, stress response, lipid metabolism and cell to cell communication. Altered expression of genes encoding gender-related metabolic enzymes and estrogen-linked genes was also reported (Liu et al., 2006b).
We have recently evaluated age susceptibility to arsenic carcinogenesis by administering inorganic arsenic to C3H mice at three different developmental stages (during gestation, prior to pubescence, and post-pubescence) in order to compare proliferative lesion and tumor outcomes at 1 year (Ahlborn et al., 2009). The C3H mouse strain is known to have a high spontaneous incidence of hepatocellular tumors (Dragani et al., 1995). We observed that urinary bladder hyperplasia incidence was significantly increased in female mice chronically exposed to arsenic from either gestational day (GD) 8 or postnatal day (PD) 21 through 1 year. In contrast, male mice continuously exposed to arsenic from GD8 through 1 year had significantly decreased incidence of liver and adrenal tumors in comparison to both in utero only exposed and untreated control mice. Tumor formation was completely abolished at 1 year in this treatment group (Ahlborn et al., 2009). To elucidate changes at the gene transcript level that may be affecting this unexpected tumor-protective effect, the current study measures global gene expression in liver tissue samples taken from the different treatment regimens.
2. Materials and methods
2.1. In vivo methods
Animal experiments were carried out at the U.S. Environmental Protection Agency, Research Triangle Park, NC in an AAALAC International accredited facility. All procedures involving the care and use of animals were approved by the Institutional Animal Care and Use Committee. The experimental design and procedures are described in detail in our companion study (Ahlborn et al., 2009), from which liver samples were derived for the gene expression analyses reported here. The four treatment groups used for the current study were control (no arsenic exposure), in utero only arsenic exposure (GD8–19), continuous arsenic exposure (GD8 to 1 year), and postnatal arsenic exposure (PD21 to 1 year). Briefly, sodium arsenite was administered in the drinking water at 85 ppm to timed-pregnant C3H dams (Charles River Laboratories, Raleigh, NC). The in utero only treatment group received arsenic entirely via maternal exposure during gestation. For the continuous treatment arsenic was administered maternally from GD8 through weaning of the offspring. At weaning, the offspring were administered 85 ppm sodium arsenite in their own drinking water until 1 year of age. The postnatal exposure group was administered 85 ppm sodium arsenite only in their own drinking water from PD21 until 1 year of age. Mice were maintained on Ralston Purina 5001 Chow, characterized by total background arsenic levels of less than 0.22 ppm. At 1 year of age animals from these four treatment groups were euthanized by CO2 asphyxiation, and their livers flash frozen in liquid nitrogen and stored at −80 ◦C for subsequent analysis.
2.2. Gene expression
Total RNA was isolated from normal appearing tissue using the TriReagent procedure (Molecular Research Center, Cincinnati, OH). The integrity of each RNA sample was confirmed using an Agilent 2100 Bioanalyzer (Agilent, Foster City, CA). Gene expression (four animals chosen randomly per treatment group; one array per animal) was measured using Affymetrix mouse 430 2.0 arrays following the manufacturer’s recommended protocol. Data was normalized using the Rosetta Resolver error-model, and a one-way ANOVA (p ≤ 0.01, with Benjamini Hochberg multiple test correction method) was performed with the four 1-year treatment groups. A Tukey–Kramer post hoc test (p ≤ 0.01) was used to compare the groups of interest (treated vs. untreated control). Lists of probe sets with expression significantly altered due to arsenic were then filtered to include only those probe sets with an absolute fold-change of 1.5 or greater, and exported into Ingenuity Pathways Analysis (IPA). These lists are provided as Supplementary Material.
An additional time course analysis of array results from animals continuously exposed to arsenic in the drinking water from GD8 until euthanasia at various ages (GD19, PD32, PD67, 1 year) was performed using a two-way ANOVA (p ≤ 0.01, with Benjamini Hochberg multiple test correction, Tukey–Kramer post hoc test). The two variables were age when euthanized and treatment (arsenic or control). Genes with expression significantly altered by arsenic treatment were examined at each age and a 1.5-fold-change cutoff applied. These significant gene lists and their functional categorization by IPA are provided as Supplementary Material.
2.3. Quantitative RT-PCR
RNA from each of the four 1-year treatment groups (four samples per group) was used for quantitative reverse transcriptase PCR. One-step qRT-PCR reactions were set up in triplicate for each sample and target gene combination. Eighty-five nanograms of total RNA was loaded into each reaction containing 1× QuantiTect™ Probe RT-PCR master mix [Qiagen, Valencia, CA] and 1× TaqMan® Gene Expression Assays [Applied Biosystems, Foster City, CA] for the desired target. Relative standard curves were generated using serially diluted pooled RNA from the study. Relative expression quantities were generated from the resulting relative standard curves and these values used for comparisons between treatment groups.
3. Results
3.1. Tumor incidence at 1 year
Table 1 summarizes hepatocellular tumor incidence for the different treatment groups of male mice (data from our companion study, Ahlborn et al., 2009). The total tumor incidence for the in utero only treatment group was similar to that of the control group (~30%). Tumor incidence was significantly reduced in the continuous treatment group with no tumors formed by 1 year of age. Tumor incidence for the postnatal exposure group was similar to that of the continuous treatment group, but it was not statistically lower than that of the control or in utero only groups.
Table 1.
Hepatocellular tumor (adenoma/carcinoma) incidence in male C3H mice at 1 year of age (from Ahlborn et al., 2009).
| Treatment | N | No. of animals with tumors | Incidence (%) |
|---|---|---|---|
| Control | 20 | 6 | 30 |
| In utero only | 9 | 3 | 33 |
| Continuous | 28 | 0 | 0* |
| Postnatal | 18 | 1 | 6 |
Statistical significance by the one-sided Fischer’s exact test (p < 0.05).
3.2. Gene expression at 1 year
The current study evaluates gene expression profiles for the three arsenic exposure groups (in utero only, continuous and postnatal) relative to the untreated control group. Numbers of probe sets with significant changes in expression at 1 year due to arsenic are shown in Fig. 1. Because tumor formation and gene expression were affected to the greatest extent in the continuous treatment group that received arsenic from GD8 to 1 year, much of our analysis focused on this group. Table 2 is a summary by functional category of the genes with expression significantly altered by continuous arsenic exposure. Genes involved in cancer and cancer-related functions are highly represented. IPA identified 1407 as cancer genes, with 81 identified as liver cancer genes.
Fig. 1.
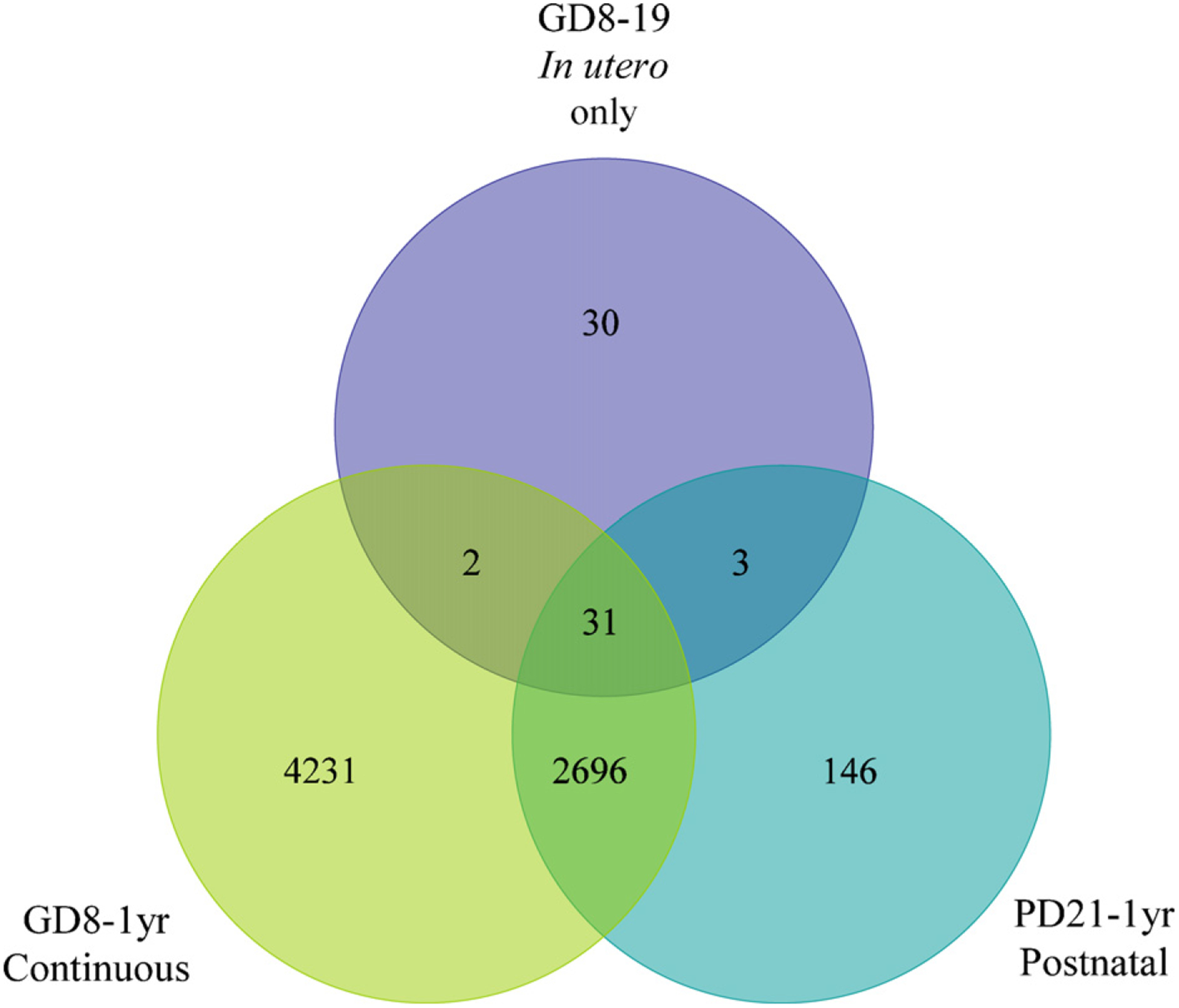
Venn diagram of number of hepatic probe sets significantly altered relative to the untreated control by the arsenic treatment regimens.
Table 2.
IPA functional category analysis of genes whose expression response to arsenic at 1 year was significant for the continuous treatment regimen. Only the top 12 categories are shown for each division, in order of significance.
| No. ofgenes | |
|---|---|
| Molecular and cellular functions | |
| Cellular compromise | 347 |
| Gene expression | 769 |
| Cellular growth and proliferation | 976 |
| Cell death | 951 |
| Post-translational modification | 481 |
| Protein synthesis | 366 |
| Protein degradation | 144 |
| Molecular transport | 354 |
| Protein trafficking | 135 |
| Cell cycle | 448 |
| RNA post-transcriptional modification | 123 |
| Cell morphology | 438 |
| Top canonical pathways | |
| Protein ubiquitination pathway | 96 |
| NRF2-mediated oxidative stress response | 88 |
| PI3/AIKT signaling | 66 |
| Molecular mechanisms of cancer | 142 |
| Purine metabolism | 127 |
| PTEN signaling | 53 |
| Integrin signaling | 86 |
| Mitochondrial dysfunction | 60 |
| Chronic myeloid leukemia signaling | 50 |
| Germ cell-sertoli cell junction signaling | 67 |
| Insulin receptor signaling | 62 |
| B cell receptor signaling | 67 |
| Top Tox lists | |
| Oxidative stress response mediated by NRF2 | 89 |
| Mitochondrial dysfunction | 58 |
| Hypoxia-inducible factor signaling | 40 |
| PPARa/RXR activation | 73 |
| Mechanism ofgene regulation by peroxisome proliferaters via PPARa | 48 |
| Hepatic cholestasis | 58 |
| NFkB signaling | 51 |
| RAR activation | 58 |
| PXR/RXR activation | 29 |
| P53 signaling | 36 |
| Aryl hydrocarbon receptor signaling | 54 |
| FXR/RXR activation | 34 |
Fig. 2 lists expression relative to the untreated control for selected genes from both the chronic treatment groups by functional category. None of the genes in this figure was significantly altered by the in utero only treatment. Arsenic effects on expression were skewed toward down-regulation in both the number of genes and the magnitude of change for all treatment groups, with 67% (in utero only), 71% (postnatal) and 58% (continuous) of the significantly altered genes down-regulated. The magnitude of change ranged from more than −100-fold to approximately +25-fold for all treatment groups.
Fig. 2.
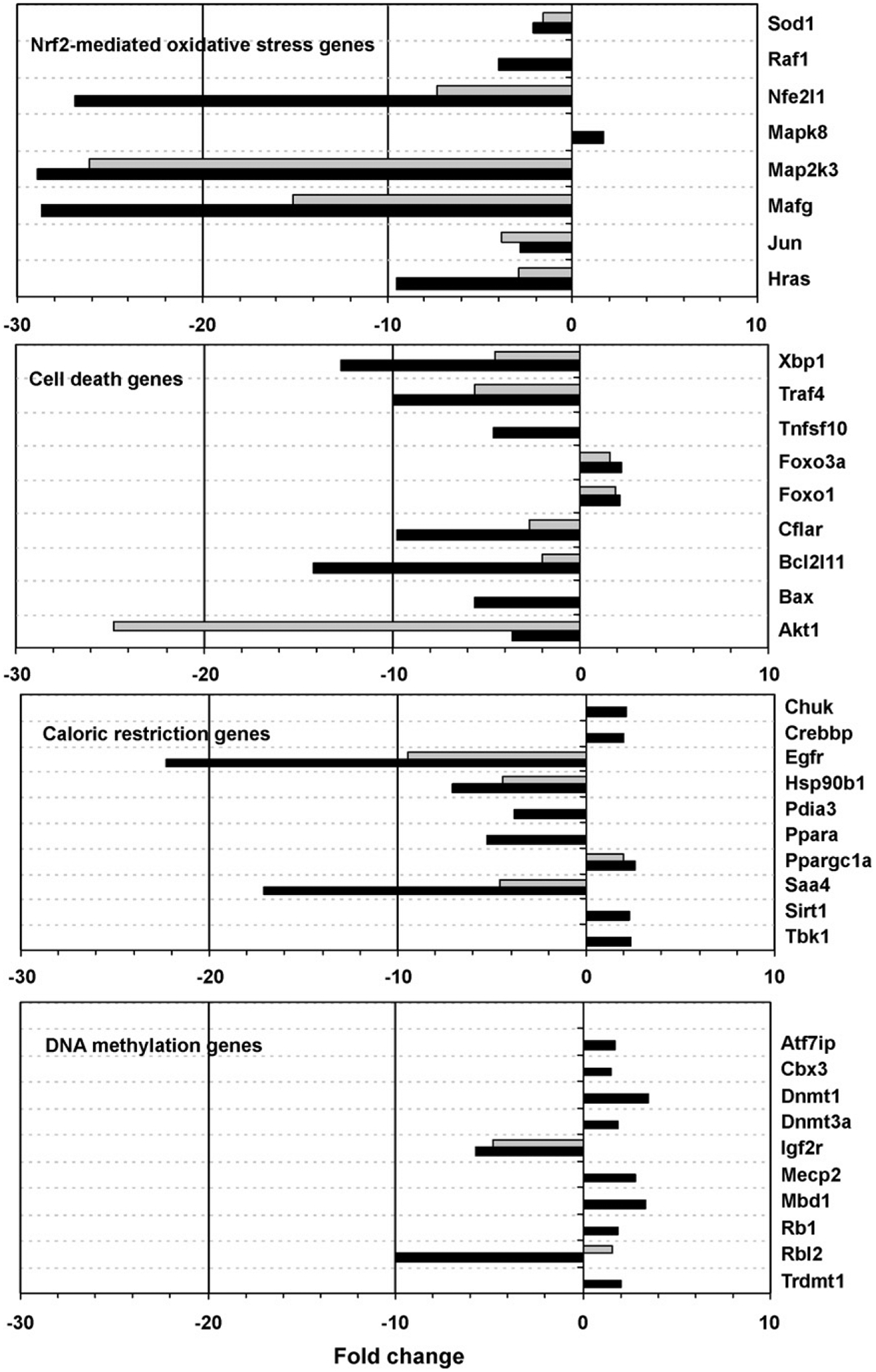
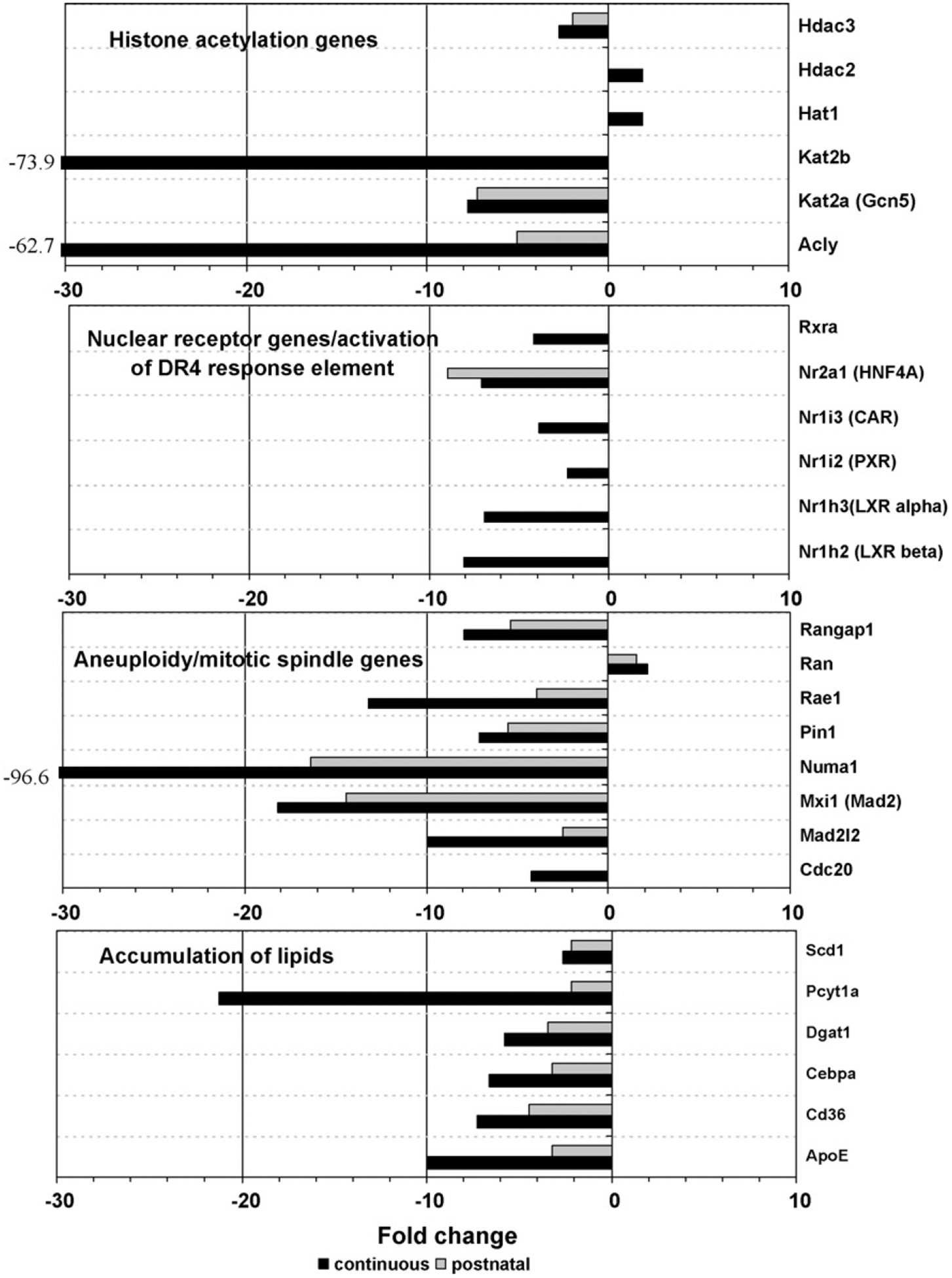
Fold-change expression relative to control for selected genes.
Expression for the insulin receptor signaling pathway genes in the continuous treatment group is shown in Fig. 3. Over half of these genes were similarly affected by the postnatal treatment, with the remainder unaffected. None were significantly altered by the in utero only treatment. A further look at the genes associated with this pathway reveals two areas especially affected by the continuous arsenic treatment. The first is the down-regulation of Akt and resulting upregulation of the Foxo (Fkhr) genes (Figs. 2 and 3), possibly leading to apoptosis. The second is the down-regulation of fatty acid and triglyceride synthesis (lipogenesis) genes through ATP citrate lyase (Acly, Fig. 4). Nearly two-thirds of the genes in Fig. 4 were similarly affected by the postnatal treatment, with the remainder unaffected. None of these genes was significantly altered by the in utero only arsenic treatment. The Scd1 gene shown in Figs. 2 and 4 is of particular interest because of its link to hepatocarcinogenesis in the literature (see Section 4). The differential expression of Scd1 seen with the arrays was confirmed by qRT-PCR, with a significant down-regulation in both chronic exposure (continuous and postnatal) treatment groups (Fig. 5). In time points taken from the continuous treatment group earlier than 1 year (see Supplementary Material) Scd1 expression became significantly reduced (−1.5-fold) at PD32, and expression decreased further from the control level at PD67 (−1.8-fold).
Fig. 3.
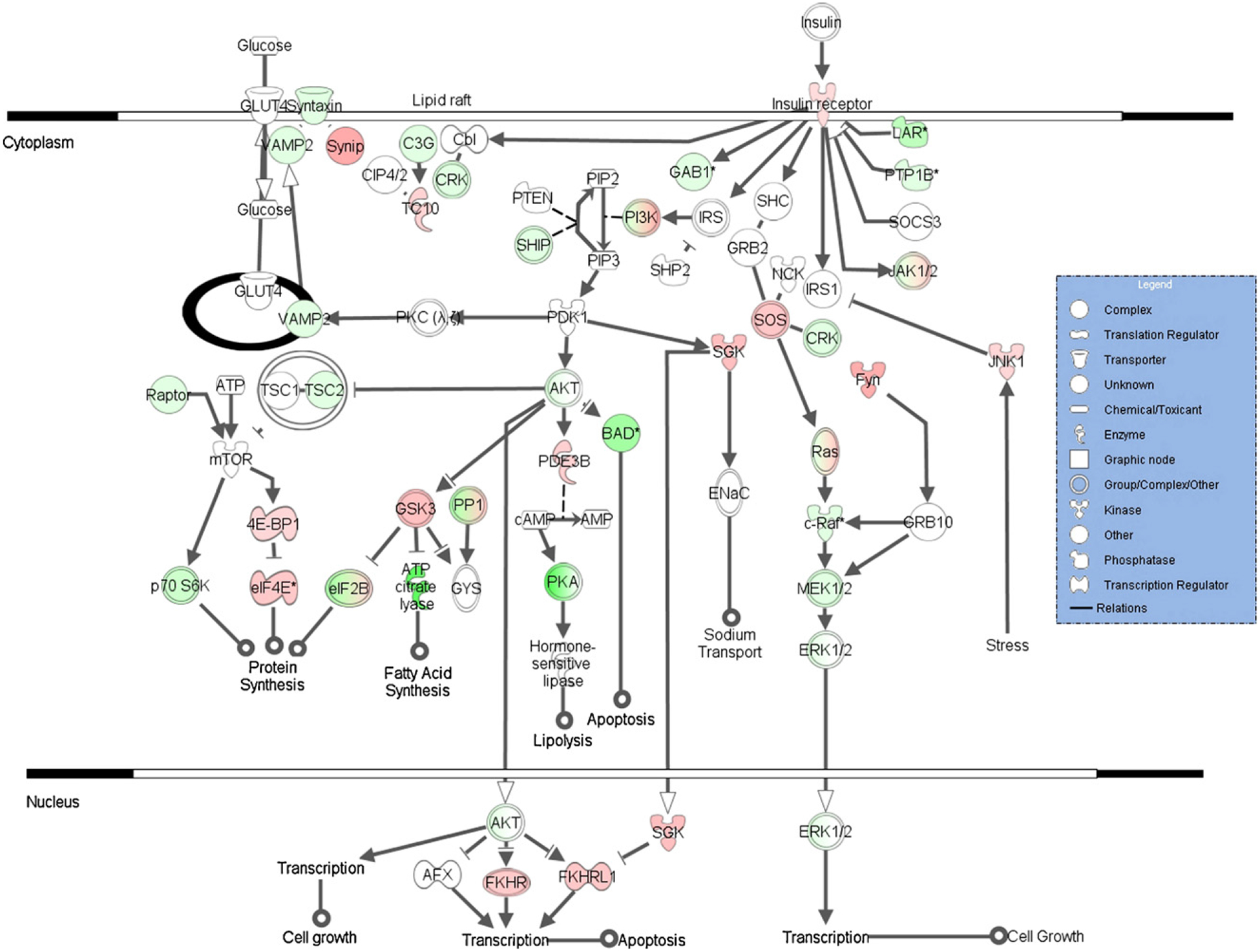
Insulin receptor signaling pathway (from Ingenuity Pathways Analysis) with gene expression significantly different from the control (up: red; down: green) shown for the continuous treatment group. An asterisk denotes multiple identifiers in the dataset mapped to that single gene (replicates). Expression shown (as color intensity) is for the highest fold-change (absolute value) replicate.
Fig. 4.
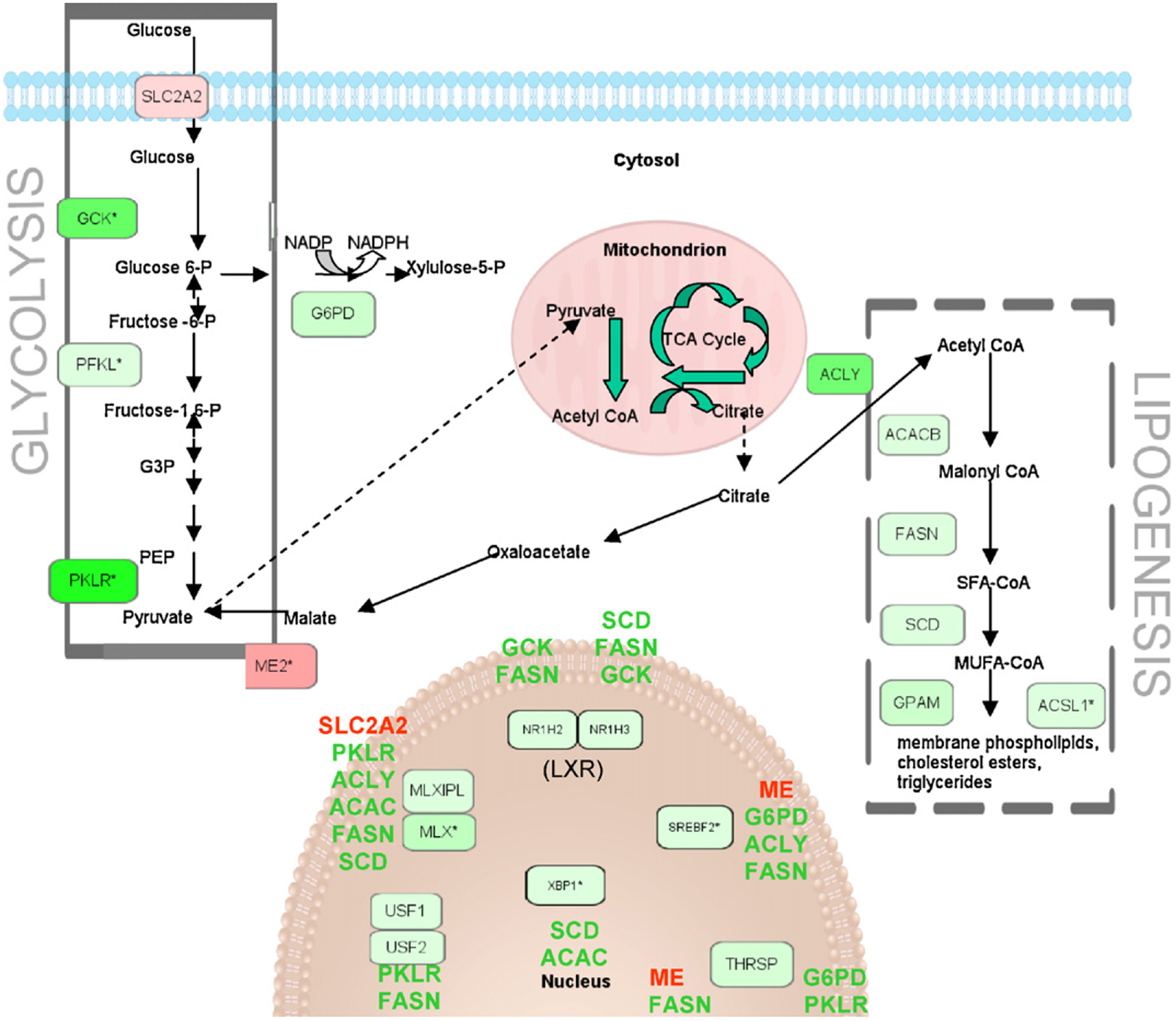
Glycolysis and lipogenesis pathways with gene expression significantly different from the control (up: red; down: green) shown for the continuous treatment group. Regulated genes are listed beside their respective nuclear transcription factors. An asterisk denotes multiple identifiers in the dataset mapped to that single gene (replicates). Expression shown (as color intensity) is for the highest fold-change (absolute value) replicate.
Fig. 5.
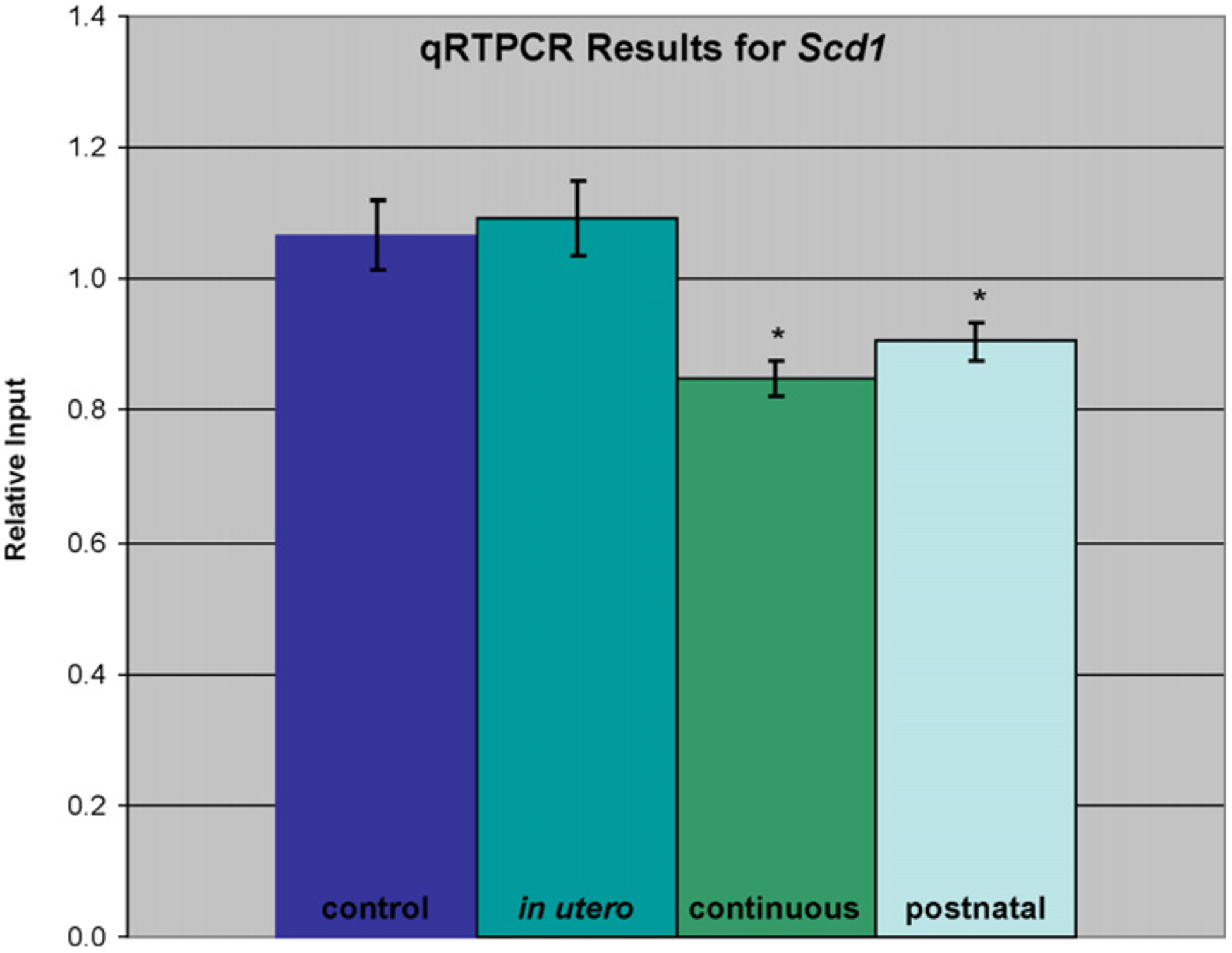
qRT-PCR results for the Scd1 gene. Each value represents the average of three replicate reactions of four unique samples. Values were calculated using a relative standard curve method and normalized to the log of the amount of input RNA. An asterisk indicates statistical significance in comparison to the untreated control sample (t-test, p≤0.05).
4. Discussion
We recently reported that male C3H mice, which exhibit a high background rate of hepatocellular tumors (Dragani et al., 1995), showed a significant reduction in liver tumor incidence compared to control mice when continuously exposed to arsenic for a treatment period beginning in utero and ending at 1 year (Ahlborn et al., 2009). This finding was unexpected, as other mouse studies have reported increased liver tumor incidence at 2 years following an 11 day in utero only exposure (Waalkes et al., 2003, 2004a,b, 2006). We found no increase in liver tumors at 1 year with the same 11 day in utero only treatment regimen (Ahlborn et al., 2009), but it is likely that arsenic tumor induction requires a longer time frame. Our goal in the present study was to identify differential gene expression responses that may lead to the apparent protective effect of continuous arsenic treatment seen at 1 year. It is unknown whether this protective effect would persist beyond 1 year.
Arsenic is a well-documented carcinogen, but is also known to act as an anti-tumor agent. Arsenic trioxide and sodium arsenite have been used for the effective treatment of acute promyelocytic leukemia (APL) and other haematological malignancies, and some cases of prostate cancer (Shen et al., 1997; Lu et al., 2004; Amadori et al., 2005). Interestingly, chronic myeloid leukemia signaling was one of the top canonical pathways for our continuous treatment dataset. Mitochondrial transmembrane potential collapse and retinoic acid signaling are associated with the arsenic trioxide-induced apoptosis and differentiation seen in APL treat ment (Cai et al., 2000). Both mitochondrial dysfunction and RAR activation pathways were indicated to be of significance in our continuous arsenic treatment dataset.
The molecular mechanisms that determine whether arsenic will act as a carcinogen or as an anticarcinogen are not well understood, but are likely related to cell type, arsenic species, and length and dose of exposure. Arsenic’s chemotherapeutic effects have often been attributed to its action as a regulator of gene expression and stimulator of oxidative stress, followed by induction of programmed cell death (Ivanov and Hei, 2006; Bade and Dong, 2002). All three of these categories were highly represented in our IPA analysis of genes significantly altered by continuous arsenic treatment and are discussed below. Increases in cell-cycle G1 arrest and apoptosis presumably eliminate cells damaged by oxidative stress. In addition, many known liver cancer genes fall under one of these categories and were differentially expressed between treatments.
4.1. Gene expression
The propensity toward reduced transcript expression in our dataset was determined to be a treatment effect, not an issue of RNA quality, and may be indicative of generalized toxicity. Other studies have noted a similar trend toward decreased expression with arsenic exposure (Andrew et al., 2007; Rea et al., 2003). Arsenic may decrease gene expression through suppression of key transcription factors such as the nuclear receptors shown in Figs. 2 and 4.
DNA methylation is an important epigenetic regulator of gene expression which is known to be altered by arsenic. In a study by Chen et al. (2004), chronic exposure of adult male mice to 45 ppm arsenic in the drinking water induced hepatic global DNA hypomethylation, which is often postulated as a nongenotoxic mechanism of carcinogenesis. However, in the current study, eighteen genes functioning in DNA methylation were affected by continuous arsenic treatment, with expression elevated for eleven of these, including Dnmt1 and Dnmt3a. This suggests the possibility that DNA hypermethylation plays a role in the tumor-protective effect seen for the continuous treatment group. Hypermethylation could lead to down-regulation of oncogenes and transcription factors involved in the carcinogenic process.
Several genes functioning in another epigenetic mechanism, histone acetylation, were also affected by continuous arsenic exposure, including down-regulation of Kat2a (Gcn5), a gene of known importance for maintenance of global histone acetylation, and down-regulation of the gene encoding ATP citrate lyase (Acly), which generates acetyl-CoA from mitochondria-derived citrate (Fig. 4). Wellen et al. (2009) have shown that ATP citrate lyase-dependent production of acetyl-CoA contributes to histone acetylation during cellular responses to growth factor stimulation, adipocyte differentiation, and the regulation of glucose metabolism genes, providing a link between cellular metabolism, histone acetylation, and gene expression. Reduced histone acetylation generally represses transcription, and has been associated with either the genesis or suppression of cancer depending on the specific target genes involved (Archer and Hodin, 1999).
4.2. Oxidative stress
The antioxidant response element (ARE) mediates transcriptional activation of genes in cells exposed to oxidative stress. ARE-related genes down-regulated by continuous treatment include the ARE regulator Nfe2l1 and its heterodimer Mafg, Jun and Map2k3. ARE-driven genes are induced by toxic metals and metal-loids through the Nrf2-mediated oxidative stress response pathway (He et al., 2006), which was significantly altered in our dataset and may be involved in the differential tumor response observed. However, the mechanism of action of this complex gene system in the transcriptional response to toxins and the carcinogenic response is not well understood.
4.3. Cell death
Arsenic trioxide had dose-dependent opposing effects in a mouse model of hepatocellular carcinoma (Liu et al., 2006a). After 20 days of i.p. treatment, doses greater than 1 mg/kg inhibited tumor growth and angiogenesis while enhancing tumor cell apoptosis, and doses lower than 1 mg/kg promoted tumor growth and angiogenesis. The sodium arsenite dose used in our current study was approximately 10 mg/kg, and total arsenic levels in the male mouse livers were significantly elevated by both chronic arsenic exposure regimens, but not by the short-term in utero only exposure (Ahlborn et al., 2009). Therefore, a similar mechanism of increased apoptosis may be involved in the reduction of liver tumor incidence observed with continuous arsenic exposure in our study.
Arsenic induction of aneuploidy (Ochi et al., 2003; Ramirez et al., 2008) may be a factor in reducing the incidence of liver tumors by increasing tumor cell death. Weaver et al. (2007), working with mice/cells deficient for the centromere protein, CENP-E, observed increases in aneuploidy and a decrease in the incidence of spontaneous liver tumors. They suggested that while lower rates of aneuploidy are potentially tumorigenic, higher rates can cause cell death, thereby suppressing tumor formation. Consistent with this hypothesis, our data shows significant changes with continuous arsenic treatment for a number of aneuploidy and mitotic spindle genes (Fig. 2), including Mad2 (Mxi1). Reduction of this essential component of the mitotic spindle checkpoint has been shown to increase aneuploidy (Weaver et al., 2007).
Apoptosis can be mediated by diverse genes and numerous pathways in the cell. While some pro-apoptotic genes were upregulated by our continuous arsenic treatment, others were down-regulated. The anti-apoptotic genes Xbp1 (also known as the hepatocarcinogenesis-related transcription factor) and Cflar were down-regulated. Jnk (Mapk8, upregulated) has been indicated in arsenic-induced apoptosis (Dong, 2002). Foxo proteins (Foxo1, Foxo3) are regulated by Jnk, CREB binding protein (Crebbp) and sirtuin 1 (Sirt1) (Corton and Brown-Borg, 2005; Yang et al., 2006), all genes with increased expression due to continuous arsenic treatment. However, two pro-apoptotic targets of the Foxo genes, Bim (Bcl2l11) and Trail (Tnfsf10), were down-regulated in our dataset, making interpretation of these gene changes in relation to apoptosis uncertain. Akt, Foxo and other genes in the insulin receptor pathway are also key regulators in caloric restriction and other models of longevity (Corton and Brown-Borg, 2005).
4.4. Insulin signaling and caloric restriction
Caloric restriction (CR) is the most effective means known of reducing cancer incidence and increasing the mean age of onset of age-related diseases and tumors, doing so, in part, by increasing the rate of apoptosis in mitotically competent, and therefore cancer-prone, tissues (Spindler, 2006). Although food consumption was not significantly reduced for either dams or offspring exposed to arsenic at any time throughout our 52-week study, male F1 body weights were significantly lower in both the continuous and post-natal exposure groups beginning 11 days post-weaning (Ahlborn et al., 2009). Because of this reduced body weight, it is possible that chronic arsenite exposure is eliciting responses similar to CR despite an unaltered caloric intake.
CR in rodents dramatically affects insulin signaling by reducing plasma insulin levels, associated with decreased Akt phosphoryla tion and increased expression of Foxo family members (Corton and Brown-Borg, 2005). Sirt1 levels are increased during CR and are negatively regulated by insulin. Pgc-1α (Ppargc1a) is transcriptionally upregulated by Foxo1. Like our continuously exposed arsenite treatment group and CR animals, dwarf mice are protected from spontaneous and chemically induced cancer and exhibit decreased insulin signaling resulting in activation of Foxo1 and Ppargc1a (Corton and Brown-Borg, 2005). Pgc-1 family members mediate effects on gene expression through both ligand-dependent and -independent activation of nuclear receptors (NRs). In our dataset many of the nuclear receptors regulated by Pgc-1α or 1β showed decreased expression by the continuous arsenic treatment rather than the expected increase. These include Car (Nr1i3), Hnf-4a (Nr2a1), Lxr (Nr1h2, h3), Ppara, and Pxr (Nr1i2), all down-regulated, suggesting that Pgc-1 activation of these NRs may be overridden by negative regulation by other transcription factors, ligands, or processes. Reduction of Ppara signaling by the tumor-protective continuous treatment may be particularly relevant as Ppara is activated by a group of rodent hepatocarcinogens known as peroxisome proliferators (Corton and Brown-Borg, 2005; Corton, 2008). ‘Mechanism of gene regulation by peroxisome proliferators via PPARa’ was identified as one of the top toxicity lists in this dataset, and includes many of the genes discussed here.
Dhahbi et al. (2005), working to develop assays to screen for pharamaceuticals capable of reproducing the health- and lifespan-extending effects of long-term caloric restriction, have used Affymetrix arrays to identify gene expression biomarkers useful in evaluating candidate CR mimetics. They have found that the biguanidine antidiabetic drug metaformin (MET) reproduces many of the hepatic gene expression effects of caloric restriction. In addition, MET reduces tumor incidence in both rodents and humans and has been shown to inhibit the development of metabolic syndrome in humans (Spindler, 2006). Common links between CR, MET, and our continuous arsenic treatment include down-regulation of the chaperones TRA1 (Hsp90b1) and GRP58 (Pdia3) and the related transcription factor Xbp1. Chaperone under-expression enhances apoptosis and prevents tumor formation (Dhahbi et al., 2005). In addition, the NFkB-related genes Chuk and Tbk1 are induced by CR, MET and continuous arsenic treatment. CHUK phosphorylates IkB family members, marking them for ubiquitination and degradation, which can lead to apoptosis (Dhahbi et al., 2005). Another proapoptotic effect of CR, MET, and our continuous arsenic treatment is the down-regulation of Traf4. The current study appears to be the first report in the literature of arsenic acting as a CR mimetic. However, it is also the first study to begin arsenic treatment in utero and continue it long-term through adulthood. A few other compounds have been found to reduce spontaneous liver tumor formation in C3H mice (Nishino et al., 1999, 2001). One of them, ginseng, is a herb used for treating type 2 diabetes in Chinese medicine (Park et al., 2008).
Consistent with an apparent negative regulation of the insulin signaling pathway, and in common with CR (Spindler, 2006), expression of many glycolysis and lipogenesis genes was down-regulated by our continuous arsenic exposure (Fig. 4; Cha and Repa, 2007; Dentin et al., 2005; Lee et al., 2008; Towle and Kaytor, 1997; Uyeda and Repa, 2006). In contrast, activation of lipogenic enzymes has been associated with hepatocellular carcinoma (Yahagi et al., 2005). Stearoyl-CoA desaturase (Scd1) is the rate-limiting enzyme that catalyzes the synthesis of monounsatu-rated fatty acids from saturated fatty acids. Scd1 gene expression is correlated with genetic predisposition to hepatocarcinogenesis in mice and rats (Falvella et al., 2002). Scd1 mRNA levels are more than 10-fold higher in the normal liver tissue of C3H mice, which are genetically susceptible to hepatocarcinogenesis, than in BALB/c mice, which are resistant to hepatocarcinogenesis. Similarly, Scd1 mRNA expression is about four-fold higher in the normal liver of F344 rats, which are susceptible to hepatocarcinogenesis, than in Brown Norway rats, which are resistant. In addition to being modulated by dietary and genetic factors, Scd1 expression in mouse liver is induced by peroxisome proliferators, iron overload, and dichloroacetic acid, conditions that induce or promote hepatocarcinogenesis (Falvella et al., 2002). Expression of the Scd1 gene was upregulated by in utero only exposure to arsenite in a previous study by Liu et al. (2006b) that found an increase in liver tumors at 2 years of age. Conversely, down-regulation of this gene in our continuously exposed animals may help explain the reduction in spontaneous liver tumors.
Scd1 deficient mice have increased energy expenditure, reduced body adiposity, increased insulin sensitivity and are resistant to diet-induced obesity and liver steatosis (fatty liver) (Ntambi et al., 2002). Scd1 activity is required for the onset of diet-induced hepatic insulin resistance (Gutierrez-Juarez et al., 2006) and is associated with accumulation of fat in the liver (Dobrzyn et al., 2004). The combination of metabolic disorders known as the metabolic syndrome, including abdominal obesity, fatty liver, insulin resistance, increased serum triglycerides, low HDL cholesterol, hyperglycemia, and hypertension, increases the risk of developing type II diabetes, cardiovascular disease and non-alcoholic fatty liver disease (NAFLD). It has also been identified as a risk factor for hepatocellular carcinoma (Paradis et al., 2008; Watanabe et al., 2008). Inorganic arsenic exposure has been correlated with an increase in multiple metabolic syndrome risk factors, type 2 diabetes, and cardiovascular disease (Wang et al., 2007; Diaz-Villasenor et al., 2007). Therefore, our expression data may help in understanding the link between arsenic’s diverse effects. Although arsenic has been associated with the metabolic syndrome and hepatocellular carcinoma, the liver tumor-protective effect seen with our particular continuous arsenic exposure may be explained by a gene expression profile that is preventative for metabolic syndrome risk factors and mimics that of caloric restriction for genes in key pathways. Multiple genes involved in accumulation of lipids are down-regulated by the continuous arsenic treatment. In addition to gene expression effects and tumor suppressive effects, the reduced serum lipid levels for the continuous arsenic treatment group (Ahlborn et al., 2009) are in agreement with a CR mimetic phenotype.
In conclusion, the unexpected liver tumor-protective effect of continuous arsenic exposure from GD-8 until 1 year in C3H mice can most likely be attributed to gene pathways involving gene expression, oxidative stress, and cell death. The insulin receptor signaling pathway, and in particular fatty acid biosynthesis genes, such as Scd1, likely play an important role. Many of these genes are affected by caloric restriction and the metabolic syndrome, and help provide links between arsenic’s known involvement in cancer, diabetes, and cardiovascular disease. While protective for the liver and adrenals in this study, continuous arsenic exposure did significantly increase the incidence of urinary bladder hyperplasia (Ahlborn et al., 2009). These results highlight the importance of tissue-specificity, life-stage and duration of exposure in arsenic-induced tumorigenicity.
Supplementary Material
Acknowledgements
This research was supported, in part, by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors would like to thank Dr. Julian Preston, Dr. Susan Hester and Dr. Shaeau-Fung Thai for their helpful support, suggestions, and data/manuscript reviews.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The Research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, United States Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views of the Agency, nor does the mention of trade names or commercial products constitute endorsement or recommendation for use.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tox.2009.10.004.
References
- Ahlborn GJ, Nelson GM, Grindstaff RD, Waalkes MP, Diwan BA, Allen JW, Kitchin KT, Preston J, Thomas DJ, Delker DA, 2009. Impact of life stage and duration of exposure on arsenic-induced proliferative lesions and neoplasia in C3H mice. Toxicology 262, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadori S, Fenaux P, Ludwig H, O’dwyer M, Sanz M, 2005. Use of arsenic trioxide in haematological malignancies: insight into the clinical development of a novel agent. Curr. Med. Res. Opin 21 (3), 403–411. [DOI] [PubMed] [Google Scholar]
- Anderson LM, Diwan BA, Fear NT, Roman E, 2000. Critical windows of exposure for children’s health: cancer in human epidemiological studies and neoplasms in experimental animal models. Environ. Health Perspect 108 (Suppl. 3), 573–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew AS, Bernardo V, Warnke LA, Davey JC, Hampton T, Mason RA, Thorpe JE, Ihnnat MA, Hamilton JW, 2007. Exposure to arsenic at levels found in U.S. drinking water modifies expression in the mouse lung. Toxicol. Sci 100, 75–87. [DOI] [PubMed] [Google Scholar]
- Archer SY, Hodin RA, 1999. Histone acetylation and cancer. Curr. Opin. Genet.Dev 9, 171–174. [DOI] [PubMed] [Google Scholar]
- Bade AM, Dong Z, 2002. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit. Rev. Oncol. Hematol 42, 5–24. [DOI] [PubMed] [Google Scholar]
- Barton HA, Cogliano VJ, Flowers L, Valcovic L, Setzer RW, Woodruff TJ, 2005. Assessing susceptibility from early-life exposure to carcinogens. Environ. Health Perspect 113, 1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates MN, Smith AH, Cantor KP, 1995. Case–control study of bladder cancer and arsenic in drinking water. Am. J. Epidemiol 141 (6), 523–530. [DOI] [PubMed] [Google Scholar]
- Cai X, Shen Y-L, Zhu Q, Jia P-M, Yu Y, Zhou L, Huang Y, Zhang J-W, Xiiong S-M, Chen S-J, Wang Z-Y, Chen Z, Chen G-Q, 2000. Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia 14, 262–270. [DOI] [PubMed] [Google Scholar]
- Cha J-Y, Repa JJ, 2007. The liver X receptor (LXR) and hepatic lipogenesis. J. Biol.Chem 282, 743–751. [DOI] [PubMed] [Google Scholar]
- Chen H, Li SF, Liu J, Diwan BA, Barrett JC, Waalkes MP, 2004. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis 25, 1779–1786. [DOI] [PubMed] [Google Scholar]
- Corton JC, Brown-Borg HM, 2005. Peroxisome proliferator-activated receptor λ coactivator 1 in caloric restriction and other models of longevity. J. Gerontol 60A, 1494–1509. [DOI] [PubMed] [Google Scholar]
- Corton JC, 2008. Evaluation of the role of peroxisome proliferator-activated receptor α (PPAR α) in mouse liver tumor induction by trichloroethylene and metabolites. Crit. Rev. Toxicol 38, 857–875. [DOI] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Pegorier J-P, Foufelle F, Viollet B, Vaulont S, Girard J, Postic C, 2005. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Invest 115, 2843–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM, Mote PL, Fahy GM, Spindler SR, 2005. Identification of potential caloric restriction mimetics by microarray profiling. Physiol. Genomics 23, 343–350. [DOI] [PubMed] [Google Scholar]
- Diaz-Villasenor A, Burns AL, Hiriart M, Cebrian ME, Ostrosky-Wegman P, 2007. Arsenic-induced alteration in the expression of genes related to type 2 diabetes mellitus. Toxicol. Appl. Pharmacol 225, 123–133. [DOI] [PubMed] [Google Scholar]
- Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM, 2004. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. PNAS 101, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, 2002. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ. Health Perspect 110, 757–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragani TA, Manenti G, Gariboldi M, De Gregorio L, Pierotti MA, 1995. Genetics of liver tumor susceptibility in mice. Toxicol. Lett 82/83, 613–619. [DOI] [PubMed] [Google Scholar]
- Falvella FS, Pascale RM, Gariboldi M, Manenti G, DeMiglio MR, Simile MM, Dragani TA, Feo F, 2002. Stearoyl-CoA desaturase 1 (Scd1) gene overexpression is associated with genetic predisposition to hepatocarcinogenesis in mice and rats. Carcinogenesis 23, 1933–1936. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Juarez R, Pocai A, Mulas C, Ono H, Bhanot S, Monia BP, Rossetti L, 2006. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest 116, 1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chen MG, Lin GX, Ma Q, 2006. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2-keap1-cul3 complex and recruiting Nrf2-maf to the antioxidant response element enhancer. J. Biol. Chem 281, 23620–23631. [DOI] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer), 2004. Monographs on evaluation of carcinogenic risk to humans Some Drinking Water Disinfectants and Contaminants, Including Arsenic, vol. 84. IARC Press, Lyon, pp. 269–477. [PMC free article] [PubMed] [Google Scholar]
- Ivanov VN, Hei TK, 2006. Sodium arsenite accelerates TRAIL-mediated apoptosis in melanoma cells through upregulation of TRAIL-R1/R2 surface levels and downregulation of cFLIP expression. Exp. Cell Res 312 (20), 4120–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchin KT, 2001. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol 172 (3), 249–261. [DOI] [PubMed] [Google Scholar]
- Lee A-H, Scapa EF, Cohen DE, Glimcher LH, 2008. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw J, Marshall G, Yuan Y, Ferreccio C, Steinmaus C, Smith AH, 2008. Increased childhood liver cancer mortality and arsenic in drinking water in northern Chile. Cancer Epidemiol. Biomarkers Prev 17, 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Pan S, Dong X, Qiao H, Jiang H, Krissansen G, Sun X, 2006a. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci. 97, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ducharme D, Shen J, Diwan BA, Merrick BA, Grissom SF, Tucker CJ, Paules RS, Tennant R, Waalkes MP, 2006b. Global gene expression associated with hepatocarcinogenesis in adult male mice induced by in utero arsenic exposure. Environ. Health Perspect 114, 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Xia L, Luo D, Waxman S, Jing Y, 2004. Dual effects of glutathione-S-transferase pi on As2O3 action in prostate cancer cells: enhancement of growth inhibition and inhibition of apoptosis. Oncogene 23, 3945–3952. [DOI] [PubMed] [Google Scholar]
- Marshall G, Ferreccio C, Yuan Y, Bates MN, Steinmaus C, Selvin S, Liaw J, Smith AH, 2007. Fifty-year study of lung and bladder cancer mortality in Chile related to arsenic in drinking water. J. Natl. Cancer Inst 99 (12), 920–928. [DOI] [PubMed] [Google Scholar]
- Mizoi M, Takabayashi F, Nakano M, An Y, Sagesaka Y, Kato K, Okada S, Yamanaka K, 2005. The role of trivalent dimethylated arsenic in dimethylarsinic acid-promoted skin and lung tumorigenesis in mice: tumor-promoting action through the induction of oxidative stress. Toxicol. Lett 158, 87–94. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Wanibuchi H, Morimura K, Ogawa M, Fukushima Shoji, 2000. Promotion of skin carcinogenesis by dimethylarsinic acid in keratin (K6)/ODC transgenic mice. Jpn. J. Cancer Res 91, 579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiwale L, Ingle A, Rao K, 2005. Mouse skin tumor promotion by sodium arsenate is associated with enhanced PCNA expression. Cancer Lett. 223, 27–35. [DOI] [PubMed] [Google Scholar]
- Nishino H, Murakoshi M, Masuda M, Tokuda N, Satomi Y, Onozuka M, Yamaguchi S, Bu P, Tsuruta A, Nosaka K, Baba M, Takasuka N, 1999. Suppression of lung and liver carcinogenesis in mice by oral administration of myo-inositol. Anticancer Res. 19, 3663–3664. [PubMed] [Google Scholar]
- Nishino H, Tokuda H, Tsunehiro I, Takemura M, Kuchide M, Knazawa M, Mou XY, Bu P, Takayasu J, Onozuka M, Masuda M, Satomi Y, Konoshima T, Kishi N, Baba M, Okada Y, Okuyama T, 2001. Cancer chemoprevention by ginseng in mouse liver and other organs. J. Kor. Med. Sci 16 (suppl), 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC (National Research Concil), 1999. Arsenic in Drinking Water. National Academy Press, Washington, DC. [Google Scholar]
- Ntambi JM, Miyazaki M, Stoehr HP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, Attie AD, 2002. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. PNAS 90, 11482–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Suzuki T, Isono H, Schlagenhaufen C, Goessler W, Tsutsui T, 2003. Induction of structural and numerical changes of chromosome, centrosome abnormality, multipolar spindles and multipolar division in cultured Chinese hamster V79 cells by exposure to a trivalent dimethylarsenic compound. Mutat. Res 530, 59–71. [DOI] [PubMed] [Google Scholar]
- Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, Bedossa P, Belghiti J, 2008. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 49, 851–859. [DOI] [PubMed] [Google Scholar]
- Park SM, Hong SM, Sung SR, Lee JE, Kwon DY, 2008. Extracts of Rehmanniae radix, Ginseng radix and Scutellariae radix improve glucose-stimulated insulin secretion and B-cell proliferation through IRS2 induction. Genes Nutr. 2, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott WA, Benjamin SA, Yang RS, 2001. Pharmacokinetics, metabolism, and carcinogenicity of arsenic. Rev. Environ. Contam. Toxicol 169, 165–214. [DOI] [PubMed] [Google Scholar]
- Rea MA, Gregg JP, Qin Q, Phillips MA, Rice RH, 2003. Global alteration of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis 24, 747–756. [DOI] [PubMed] [Google Scholar]
- Ramirez T, Stopper H, Fischer T, Hock R, Herrera LA, 2008. S-adenosyl-lmethionine counteracts mitotic disturbances and cytostatic effects induced by sodium arsenite in HeLa cells. Mutat. Res 637, 152–160. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, 2004. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol 198, 394–404. [DOI] [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhul, Tang W, Sun GL, Yong KQ, Chen Y, Zhou L, Fong ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY, 1997. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89, 3354–3360. [PubMed] [Google Scholar]
- Simeonova PP, Luster MI, 2000. Mechanisms of arsenic carcinogenicity: genetic or epigenetic mechanisms? J. Environ. Pathol. Toxicol. Oncol 19 (3), 281–286. [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein OS, Steinmaus C, Bates MN, Selvin S, 2006. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ. Health Perspect 114 (8), 1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler SR, 2006. Use of microarray biomarkers to identify longevity therapeutics. Aging Cell 5, 39–50. [DOI] [PubMed] [Google Scholar]
- Towle HC, Kaytor EN, 1997. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu. Rev. Nutr 17, 405–433. [DOI] [PubMed] [Google Scholar]
- Uddin AN, Burns FJ, Rossman TG, 2005. Vitamin E and organoselenium prevent the cocarcinogenic activity of arsenite with solar UVR in mouse skin. Carcinogenesis 26, 2179–2186. [DOI] [PubMed] [Google Scholar]
- Uyeda K, Repa JJ, 2006. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 4, 107–110. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Liu J, Diwan BA, 2003. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol. Appl. Pharmacol 186, 7–17. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Diwan BA, 2004a. Animal models for arsenic carcinogenesis: inorganic arsenic is a transplacental carcinogen in mice. Toxicol. Appl. Pharmacol 198 (3), 377–384. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Diwan BA, 2004b. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis 35, 133–141. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Powell DA, Diwan BA, 2006. Urogenital carcinogenesis in female CD1 mice induced by in utero arsenic exposure is exacerbated by postnatal diethylstilbestrol treatment. Cancer Res. 66 (3), 1337–1345. [DOI] [PubMed] [Google Scholar]
- Wang S-L, Chang F-H, Liou S-H, Wang H-J, Li W-F, Hsieh DPH, 2007. Inorganic arsenic exposure and its relation to metabolic syndrome in an industrial area of Taiwan. Environ. Int 33, 805–811. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Yaginuma R, Ikejima K, Myazaki A, 2008. Liver diseases and metabolic syndrome. J. Gastroenterol 43, 509–518. [DOI] [PubMed] [Google Scholar]
- Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW, 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB, 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Yoshiaki T, Iizuka Y, Ohashi K, Nagai R, Ishibashi S, Kadowaki T, Makuuchi M, Ohnishi S, Osuga J-I, Yamada N, 2005. Coordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur. J. Cancer 41, 1316–1322. [DOI] [PubMed] [Google Scholar]
- Yang J-Y, Xia W, Hu MC-T, 2006. Ionizing radiation activates expression of Foxo3a, Fas ligand, Bim, and induces cell apoptosis. Int. J. Oncol 29, 643–648. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.