

Abstract
Organisms are exposed to fluoride in the air, water, and soil. Yeast and other microbes utilize fluoride channels as a method to prevent intracellular fluoride accumulation and mediate fluoride toxicity. Consequently, deletion of fluoride exporter genes (FEX) in S. cerevisiae resulted in over 1000-fold increased fluoride sensitivity. We used this FEX knockout strain to identify genes, that when overexpressed, are able to partially relieve the toxicity of fluoride exposure. Overexpression of five genes, SSU1, YHB1, IPP1, PHO87, and PHO90, increase fluoride tolerance by two to ten-fold. Overexpression of these genes did not provide improved fluoride resistance in wild type yeast, suggesting that the mechanism is specific to low fluoride toxicity in yeast. Ssu1p and Yhb1p both function in nitrosative stress response, which is induced upon fluoride exposure along with metal influx. Ipp1p, Pho87p, and Pho90p increase intracellular orthophosphate. Consistent with this observation, fluoride toxicity is also partially mitigated by the addition of high levels of phosphate to the growth media. Fluoride inhibits phosphate import upon stress induction and causes nutrient starvation and organelle disruption, as supported by gene induction monitored through RNA-Seq. The combination of observations suggests that transmembrane nutrient transporters are among the most sensitized proteins during fluoride-instigated stress.
Keywords: Fluoride, toxicity, nitrosative stress, phosphate, nutrient starvation, stress response
Graphical Abstract
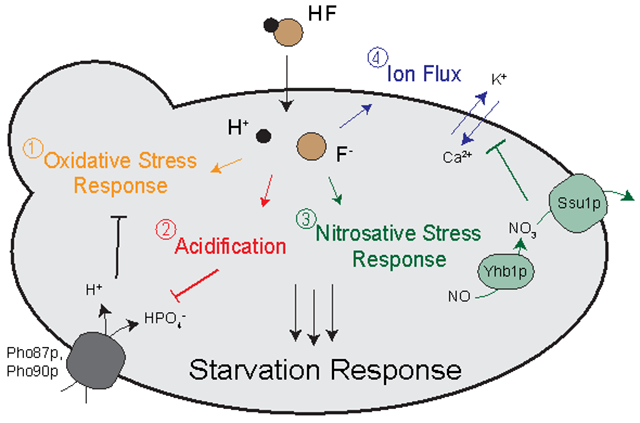
INTRODUCTION.
Fluoride is the thirteenth most abundant element in the Earth’s crust.1 While fluoride is pervasive throughout the environment, it is most highly concentrated in areas with industrial fumes, volcanic activity, or marine sedimentation.2–3 Consequently, the exposure of organisms to fluoride varies with their geographical location. Seawater averages at 1.2-1.5 ppm (60-80 μM), while soil ranges from 100-620 ppm.2 Many countries, including the U.S., add fluoride to drinking water at 0.7-1.5 ppm (40-80 μM) as a method of improving dental hygiene.4 The Center for Disease Control identified water fluoridation as one of the 10 greatest public health achievements of the twentieth century5.
Exposure to high levels of fluoride over a period of weeks to months produce a myriad of stress effects in organisms. High fluoride causes inflammation, lipid peroxidation, and perturbation of the MAPK and NF-κB pathways in multicellular model systems.6–10 At a single-cell level, fluoride activates S-phase cell-cycle arrest and metabolic arrest, and damages both the mitochondria and endoplasmic reticulum.11–16 These effects are linked to the activation by fluoride of oxidative stress and the subsequent generation of free radicals, which causes DNA damage and intracellular acidification.17–22 As the predominant effects of fluoride toxicity are inflammation and free radical damage, it is challenging to distinguish between the specific mechanism of fluoride toxicity and its downstream stress effects.
The mechanism of fluoride toxicity is linked to its size and high affinity for metals. Fluoride is approximately the same size and charge as hydroxide, but much more electronegative (3.98 versus 1.34 on the Pauling scale). Fluoride can displace hydroxide from its coordinating partners, especially metals. In enamel, fluoride displaces the hydroxide in hydroxyapatite to form fluorapatite. Fluorapatite is resistant to demineralization from bacteria, and therefore prevents cavities.23–24 On a molecular level, fluoride has been found to directly modify amino acids, such as Arg-48 of cytochrome c peroxidase, as well as to displace the hydroxides of Ser/Thr phosphatases, which prevents dephosphorylation activity.25–27 More commonly, fluoride binds to metals within metalloprotein active sites, thus inactivating the metalloproteins.28–30 Fluoride-metal complexes can also be a competitive inhibitor of phosphate in phosphoryl transfer enzymes.31–32 Because of its high affinity to metals, fluoride has been shown to inhibit hundreds of proteins at high (millimolar) concentrations in vitro.29, 33
Microorganisms, including bacteria and fungi, have evolved systems to defend themselves against fluoride toxicity. Bacteria utilize riboswitches to activate the transcription of many genes in response to fluoride, including the fluoride exporter Fluc.34–35 Fluc is a four-helix transmembrane protein that forms a homodimer.36–38 It acts as a highly-selective channel to prevent the intracellular accumulation of fluoride.34, 39 Fluc activity is driven by the electrochemical gradient of fluoride across the cell membrane that would otherwise result in the buildup of fluoride in the cytoplasm.39 Bacteria lacking Fluc are 200-fold more fluoride sensitive than wild type.34 Even stronger effects are seen in the eukaryote model organism S. cerevisiae. The constitutively expressed FEX1 and FEX2 (the yeast homologs of Fluc), confer over 1000-fold resistance to fluoride.40 In the absence of a fluoride channel, yeast cannot grow at the concentration of fluoride found in municipal tap water.
The fluoride hypersensitivity of yeast with deletions of both FEX1 and FEX2 (FEX double knockout, or DKO) provides a useful system to investigate fluoride toxicity at low extracellular fluoride. FEX DKO yeast are incapable of removing intracellular fluoride. Consequently, these cells demonstrate signs of toxicity within just a few hours, and at an IC50 of 50 µM NaF, as opposed to 75 mM in wild type.40 The concentration of fluoride needed to cause cell cycle arrest in FEX DKO cells is well below the concentration of fluoride required to inhibit metalloproteins in vitro.29 This observation, combined with the power of yeast genetic manipulation, offers a valuable opportunity to understand intracellular fluoride toxicity in eukaryotes.
An overexpression screen for genes that confirm partial fluoride resistance is a well-documented and high-throughput approach to identify the molecular targets of toxicants and associated pathways.41–42 The screen is comprised of a tiled DNA-fragment library inserted onto high copy plasmids. If the DNA fragment encodes a protein target of the toxicant or a protein that mediates toxicity, increased expression of that protein confers partial resistance to the toxicant. Overexpression screens have been used to elucidate the mechanism of toxicity for many agents, including methylmercury, tunicamycin, methotrexate, and glyphosate.43–46
Transcriptome sequencing (RNA-Seq) serves as a powerful tool for elucidating the response of a cell to a particular stressor. RNA-Seq monitors global RNA expression by reporting the quantity and types of RNA in cells at a given time.47 Consequently, any stress response triggered in cells to toxicants, such as fluoride, can be identified using differential expression analysis. RNA-Seq functions as an unbiased approach for monitoring gene induction and repression. This technique has been used to monitor cellular response to a wide variety of stressors, including nanoparticles, viral infections, and environmental pollutants.48–51
Here we report the results of a gene overexpression screen for fluoride resistance and RNA-Seq to analyze stress pathways during fluoride exposure. We observed patterns consistent with fluoride-induced oxidative stress, nutrient starvation, nitrosative stress, membrane disruption, and metal imbalance. This report furthers our knowledge of fluoride toxicity at the low concentrations that most organisms are exposed to in nature.
EXPERIMENTAL PROCEDURES.
Unless otherwise noted, all experiments were performed in triplicate.
Strains and media:
The wild type yeast used in this study was BY4741, and the FEX DKO strain was SSY3, generated using the hphMX4 and kanMX6 resistance cassettes as reported previously (MATa his3Δ1 leu2Δ0 ura3Δ0 FEX1Δ::kanMX6 FEX2Δ::hphMX).40 Sodium fluoride was purchased from Aldrich Chemistry. YPD media was prepared using 2 grams yeast peptone (Becton, Dickinson and Co.), 1 gram yeast extract (Becton, Dickinson and Co.), 100 μL of 1% adenine (Sigma), and 5 mL of 40% glucose (Sigma) per 100 mL total volume in water. YPD agar was prepared using the aforementioned ingredients plus an additional 2 grams of agar (Becton, Dickinson and Co.). Phosphate concentration of YPD media was verified using a commercially available malachite kit (Sigma) against a standard of increasing phosphate. Yeast media with specific phosphate concentrations were prepared using SD buffer without ammonium or phosphate (Formedium), which was supplemented with ammonium, amino acids, glucose, and adenine, as well as sodium phosphate at the indicated concentrations per experiment (J.T. Baker). Unless otherwise noted, all media were pH adjusted to 6.5-6.54 or as indicated using NaOH (J.T. Baker) and HCl (Macron Fine Chemicals).
Tiling fragments for the overexpression screen are commercially available (Minimal 1588 Plasmid Collection, Open Biosystems/Dharmacon). The fragments were ligated onto pGP564 vectors, although individual genes from tiling hits were testing using pRS426 and pRS426GPD vectors. Plasmids were transformed into yeast using the standard lithium acetate method. pGP564-contining cells were grown and maintained in SD-Leu media/agar, and pRS426-containing cells were grown and maintained in SD-Ura media/agar.
Measurement of intracellular fluoride:
Cells at O.D. 10 were added to 2 mL liquid containing YPD +/− NaF and placed in a shaker at 30°C. At 2, 4, 6, 12, 18, and 24 hours, cells were collected, counted, and harvested. The supernatant was removed via centrifugation and collected for analysis. Cells were washed twice with water before being resuspended in 1 mL lysis buffer, containing 1% Triton X-100 (Sigma), 0.1% SDS (American Bioanalytical), and PBS (American Bioanalytical). Cells were then placed on a shaker overnight at room temperature, and in the following morning sonicated, then measured for fluoride content using a fluoride electrode (Cole-Parmer). The electrode was calibrated used a standard curve of fluoride in lysis buffer. Intracellular fluoride concentration was calculated based on cell count and the corresponding dilution factor.
Measurement of cell viability (Flow Cytometry):
Flow cytometry was performed as outlined in Shen et al., 2014.52 Briefly, yeast were made into spheroplasts by incubating in buffer (1.2 M sorbitol, 0.5 mM MgCl2, and 35 mM potassium phosphate, pH 6.8) with 1 μL zymolyase (Zymo Research, stored in 500 μL of supplied buffer) at 30°C for 1 hour. Yeast were then resuspended in 500 μL binding buffer (10 mM Hepes/NaOH, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4) with 0.5 μL annexin V (Biolegend) and 0.5 μL propidium iodide (MP), placed on ice, and immediately analyzed with flow cytometry using BD FACS-Aria for cell sorting and BD FACS-Verse for analysis.
Measurement of intracellular ATP (Cell Titer Glo):
Intracellular ATP was quantified in yeast starting at O.D. 0.1 using the standard CellTiter-Glo Luminescent Cell Viability kit (Promega), and the concentration was established by comparing samples to a standard curve of increasing dATP.
Measurement of mitochondrial integrity (FUN-1 dye):
FEX DKO yeast were suspended in 3 mL YPD +/− 50 μM NaF at O.D. 0.1 and placed in a 30°C incubator with shaking. At 4 hours, cells at O.D. 0.1 were resuspended in 1 mL PBS with 1 μL FUN-1 and incubated at 30°C water bath for 25 minutes, and then washed twice in PBS. Cells were resuspended in PBS and loaded onto a slide for imaging using a Nikon Eclipse Ti Microscope. Photos of yeast with FUN-1 staining were collected using NIS Elements Software, in which the total fluorescence in the GFP (green) channel was compared with the fluorescence in the TRITC (red) channel.
Measurement of cytoplasmic calcium (Indo-1):
Yeast were grown in 3 mL YPD +/− 50 μM NaF at starting O.D. 0.1 in a 30°C incubator with shaking. At 2, 3, 6, 12, 18, and 24 hours, cells were collected and counted. Cells at O.D. 0.1 were resuspended into 500 μL buffer at pH 5 containing 10 μM Indo-1 AM (Abcam), 10 mM dimethyl glutaric acid, 50 mM KC1, and 100 mM glucose. Cells were placed in a 30°C water bath for 1.5 hours. Cells were washed three times in PBS, then resuspended in 100 μL PBS. Indo-1 fluorescence was recorded at 450 nm emission and 355 nm excitation using a plate reader.
RNA harvesting for RT-qPCR and RNA-Seq:
Yeast were grown in 25 mL YPD at O.D. 0.1. At the indicated time points, cells were spun down twice and resuspended in 1 mL sterile water. RNA was isolated using the protocol by Ares 2012.53 Afterwards, 10 μg of RNA was treated for 20 minutes at 37°C with 1x DNase buffer and 1 uL Turbo DNAse (Invitrogen) before extraction using phenol:chloroform and ethanol. RNA was resuspended in sterile water. For RNA-Seq, samples were sent to the Yale Center for Genome Analysis for quality analysis with a Bioanalyzer and subsequent poly-A sequencing. RT-qPCR was performed using the Luna® Universal One-Step RT-qPCR Kit (NEB #E3005), using the recommended protocol. Background DNA content of each sample was assessed using primers against an intron segment of actin, and relative RNA across samples were compared using primers against an exon segment of actin.
Serial dilutions on YPD-agar:
Yeast were inoculated overnight in 2 mL YPD. The next day, cells were spun down twice and resuspended in 1 mL water at O.D. 1 (denoted as 107 in figures). 1:10 dilutions were conducted in separate tubes of 1 mL total water to generate 106, 105, 104, 103, and 102 of cells/mL. Of these aliquots, 8.75 μL were added sequentially to YPD-agar plates in increasing fluoride. The plates were placed on the counter at room temperature until no liquid was visible, then turned upside-down and placed in a 30°C incubator for two days.
Assessing effects of compounds on growth (liquid growth assay):
Liquid growth assays were conducted as outlined in Li et al., 2013.40 Yeast were added to a 24-well plate at O.D. 0.1. Each well contained 1 mL liquid media of increasing toxin (usually fluoride), as specified per experiment. The plate was shaken at medium speed in a plate reader at 30°C for a period of 24 hours, with the absorbance at 600 nm measured every 3 minutes. The data were plotted on Prism, and IC50 values were calculated by comparing the fold-change in growth by area under the curve (AUC) compared to growth of cells in YPD alone. Unless otherwise noted, media was altered to pH 6.5-6.54 using NaOH and HCl.
Measuring efflux of nitrosative stress substrates (nitric oxide, nitrate, nitrite, and sulfite):
Each substrate was measured using the recommended company protocols. Nitrate and nitrite were assessed using a colorimetric kit (Caymen Chemicals), sulfite using an enzymatic kit (Megazyme), and nitric oxide using DAF-FM diacetate (Caymen Chemicals).
Inductively coupled plasma mass spectrometry (ICP-MS):
Yeast were grown at starting O.D. 10 in 3 mL YPD +/− 50 μM NaF at 30°C for 24 hours. Afterwards, cells were washed three times in water. Yeast at O.D. 20 were suspended in 1 mL of 20% nitric acid. The tubes were sealed and placed in a 95°C heating block overnight. In the morning, tubes were placed on ice to cool. For each sample, three tubes of 1 mL each were combined (3 mL total), and diluted in water to 2% nitric acid. The three tubes combined represented n=1, and this was repeated for three days (n=3). Samples were analyzed using a Perkin Elmer ICP-MS Elan DRC-e. The cell lysate was assessed for concentrations of potassium, phosphorous, oxidized phosphorous, oxidized sulfur, silicon, copper, magnesium, iron, zinc and calcium, then quantified using a standard curve.
Intracellular phosphorous quantification (31P NMR):
Nuclear magnetic resonance (NMR) was conducted based on the requirements outlined by Campbell-Burk and Shulman, 1987.54 Yeast were grown 12 hours, then resuspended at 25% weight/volume in 300 μL water, 150 μL D2O, 100 μL YPD, and 0.75 μL 0.5 M MDP (methylene diphosphonate, or medronic acid). After resuspension, 550 μL was transferred to an NMR tube for immediate analysis. NMR was conducted on a Bruker400 MHz Broadband Probe with 514 scans taken at 1-second intervals.
Monitoring rate of phosphate import (32PO4 influx):
The assay was conducted as in Rothstein and Donovan, 1963, and Wykoff and O’Shea, 2001.55–56 Briefly, cells were grown for three hours at a starting O.D. 0.67 in 5 mL YPD +/− fluoride. O.D. was again measured, and cells were washed three times in SD (synthetic defined, or standard minimal) media lacking phosphate. To cells at O.D. 1, SD media was added containing 10 mM PO4 with 32PO4 tracer +/− fluoride as specified. Cells were then placed on a shaker for the indicated time points. Cells were spun three times and washed with 500 mM PO4 to outcompete import of 32PO4, and the initial supernatant and final pellet were collected and analyzed for radioactivity. All buffers were at pH 6.5 except for the experiments testing pH dependence. In these cases, cells were incubated in increasing concentrations of acetic acid for 30 minutes until they reached the desired pHintra (verified using 5(6)-CFDA dye under its standard protocol), then the cells were quickly washed three times and resuspended in buffer at pH 6.5. For experiments with buffer below pH 6.5, cells were grown for three hours in pH 6.5 buffer, then transferred to buffer acidified by HCl at the same time as 32PO4 exposure. Results were reported as in the Rothstein lab, setting 100% as uptake in control cells at 60 minutes, and comparing phosphate uptake per cell over time.
RNA-Seq analysis:
HiSat2 was used for alignment, HTSeq for generating count data, and DESeq2 for differential expression analysis. The yeast genome from Ensemble was used as a reference. Gene annotation from yeastgenome.org was used for assembling the functional classification chart, and results were plotted using Prism software.
Measurement of plasma membrane electrochemical potential (diS-C3):
Yeast were grown at starting O.D. 0.1 in 2 mL YPD +/− 50 μM NaF at 30°C for 3 hours. Yeast were then washed twice with water, and transferred to 24-well plates at O.D. 0.4 in 1 mL of 10 mM citrate phosphate, pH 6.0 with 2% glucose. 0.5 μL of diS-C3 (3,3’-dipropylthiadicarbocyanine iodide) was added to each well, and the fluorescence emission spectra was measured every 6 minutes on a spectrofluorometer at ℷexcitation = 514, and ℷemission over 543 to 690 nm at a two-step interval. After 30 minutes, either 10 μM FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone), 30 mM HCl, or 50 μM NaF were added to the denoted wells, and reading immediately resumed up to 70 minutes. The reported relative fluorescence units are each the max fluorescence emission, which was ℷ573.
RESULTS.
FEX double-knockout cells are hypersensitive to fluoride exposure
Yeast lacking functional fluoride transporters accumulate intracellular fluoride, resulting in hypersensitivity. We monitored intracellular fluoride at the IC50 of the wildtype and FEX DKO yeast strains. Wildtype yeast exposed to fluoride at its IC50 for 24-hour growth (75 mM NaF) equilibrated external and internal fluoride concentrations at approximately a 1:1 ratio (Fig. 1A). This is consistent with the previous proposal that FEX acts as a channel rather than a pump, and its activity is driven by the fluoride gradient across the membrane.39 Intracellular fluoride in wildtype at the IC50 for FEX DKO (50 μM NaF) was below the detection limit of the electrode. Conversely, FEX DKO yeast exposed to 50 μM NaF accumulated fluoride up to an intracellular concentration of 300 μM within 24 hours (Fig. 1A). This intracellular concentration, while at a higher ratio of inside fluoride to outside fluoride compared with wildtype, is much lower than the total intracellular fluoride concentration of wildtype at their respective IC50’s. This suggests that FEX DKO yeast both accumulates fluoride, and is more sensitive to intracellular fluoride. As the only known role for FEX is to remove fluoride, the enhanced sensitivity to fluoride in the absence of FEX is mostly likely due to increased intracellular fluoride toxicity.
Figure 1:
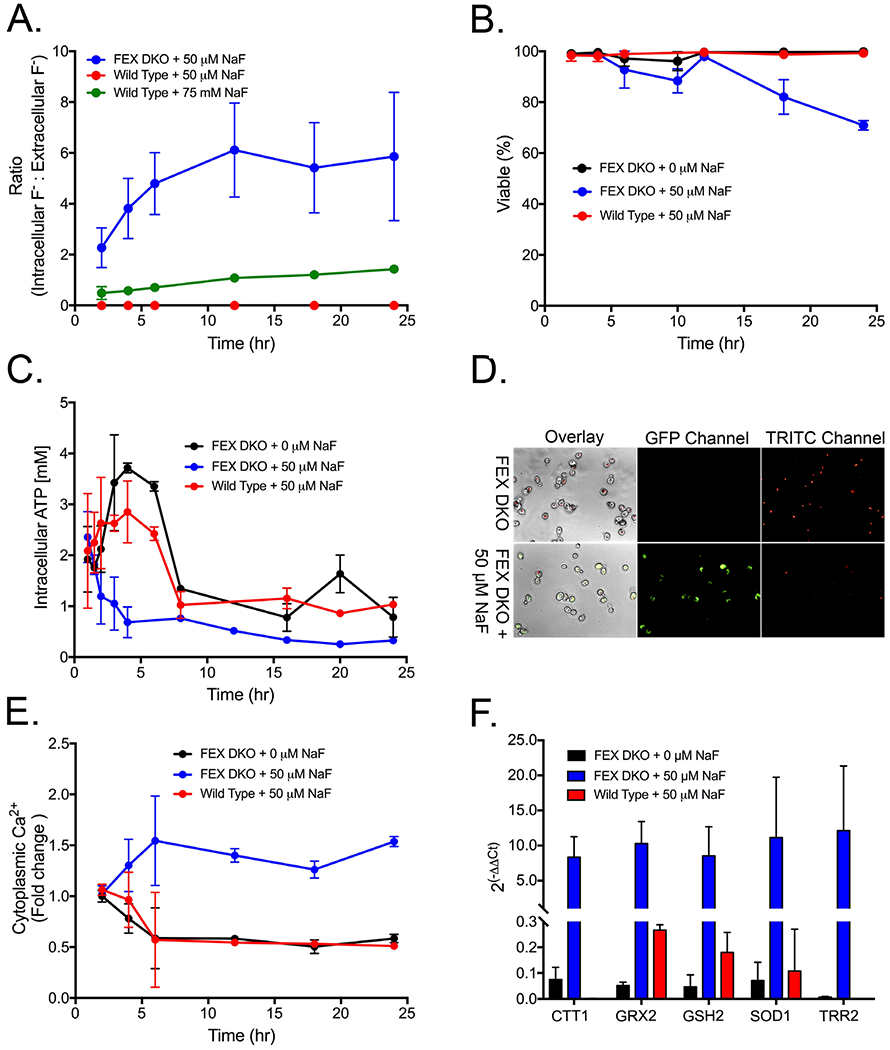
Fluoride toxicity in the fluoride sensitive FEX double knockout yeast. (A) Ratio of intracellular fluoride to extracellular fluoride measured by a fluoride electrode. (B) Cell viability over time, as measured by flow cytometry. Viability was determined as the percent of cells in the lower left gate after staining with propidium iodide and annexin V. (C) Measurement of intracellular ATP using cell titer glo and (D) mitochondrial activity as the incorporation of FUN-1 dye at 6 hours. (E) Fold change in the concentration of cytoplasmic calcium over time, assed using Indo-1. (F) RT-qPCR of oxidative stress response genes at 6 hours of growth. The change is compared with gene expression at 1-hour growth in YPD without fluoride and using actin for normalization.
We hypothesized that removal of FEX would result in a fast onset in hallmarks of fluoride toxicity. Typical markers of fluoride exposure include growth arrest, loss of metabolic activity, and oxidative stress.57 While a few toxicity studies have been conducted in yeast, the vast majority of fluoride studies were conducted in mammalian cell cultures. Mammalian tissue typically take days, or even weeks, to show toxicity phenotypes when exposed to millimolar fluoride.58–60 In contrast to these observations, the FEX DKO yeast display arrested growth at an IC50 of just 50 μM NaF and begin losing viability after just 12 hours of exposure (Fig. 1B).
We tested the onset of stress effects from fluoride in the hypersensitive FEX DKO yeast. Specifically, we investigated the hallmark phenotypes of metabolic arrest and oxidative stress. Intracellular ATP concentration, which provides an assessment of total metabolic activity and cell viability, was measured using a luciferase-based assay. Fluoride decreased intracellular ATP (Fig. 1C). Cells exposed to fluoride did not undergo the three-fold increase in ATP concentration that was observed in untreated cells as they entered log phase. Furthermore, fluoride-treated cells maintained two-to-three-fold less intracellular ATP over 24 hours compared to untreated cells.
Loss of ATP can occur from either glycolytic arrest, or respiratory arrest. We monitored mitochondrial activity using FUN-1 dye, and observed significant reduction of mitochondrial activity after 6 hours of fluoride exposure, as indicated by a shift from red to green fluorescence (Fig. 1D). Decreased mitochondrial activity is generally linked to oxidative stress.61 Cytoplasmic calcium, an early step in oxidative stress signaling, increased within 2 to 4 hours, and peaked at 6 hours of growth (Fig. 1E). As expected, subsequent RT-qPCR indicated increased expression of oxidative stress genes at 6 hours (Fig. 1F). Overall, these data suggest that ATP production is depleted prior to the loss of mitochondrial activity and induction of oxidative stress.
Overexpression of genes involved in nitrosative stress response and orthophosphate accumulation confer fluoride resistance
Fluoride could potentially inhibit hundreds of proteins and activate a myriad of stress pathways. A major challenge in the study of fluoride toxicity is to identify which pathways are directly influenced by fluoride. An overexpression screen using a tiled array provides a high-throughput method to identify important genes that mediate fluoride toxicity. Overexpression screens are composed of DNA-fragment libraries, which are expressed in higher copy numbers within a cell. DNA fragments containing a protein relevant to a mediating toxicity from a particular stressor will confer partial resistant to that stressor when expressed in higher copy numbers.
A commercially-available DNA tiling library was expressed on high-copy plasmids, pooled together, and transformed into FEX DKO yeast. The yeast were screened for their ability to grow on progressively higher concentrations of fluoride. One hundred yeast colonies able to grow in 250 μM NaF were selected, and the plasmids isolated and sequenced. Approximately half of the colonies contained either of the two FEX genes. These were not pursued further. The remaining fluoride resistant colonies contained one of five distinct genome fragments, each with 6-8 genes encoded within the fragments.
To identify which gene was responsible for conferring fluoride resistance within each of the five fragments, we generated high copy plasmids containing just one annotated gene from each fragment. These were expressed under the control of the constitutive promoter GPD. Three genes that conferred resistance were identified using this approach: PHO87, PHO90, and IPP1. All three of these genes have well-established functions. Each corresponding protein increases the total intracellular orthophosphate concentration. Pho87p and Pho90p are both constitutively expressed transmembrane proteins that act to import orthophosphate.56, 62–63 Ipp1p is a cytoplasmic inorganic pyrophosphatase that catalyzes the conversion of pyrophosphate into two orthophosphates.64–65
Two other gene fragments conferred resistance to fluoride, but expression of individual genes within that fragment with a GPD promoter did not confer resistance. We sequentially increased the segment genome fragment onto a plasmid without a GPD promoter, and found that two genes, SSU1 and YHB1, also conferred partial fluoride resistance, but only when controlled by their native promoters (Fig. S2). YHB1 required 1 kilobase of its upstream promoter sequence, while SSU1 required 0.8 kilobase of its promoter. Yhb1p is a flavohemoprotein that acts as a nitric oxide oxidoreductase.66 Ssu1p is a transmembrane exporter of both sulfite and nitrate.67–69
We next tested the extent of fluoride resistance that resulted from the overexpression of individual genes. Yeast grown in liquid culture over 24 hours had two-fold greater fluoride resistance with high-copy plasmids containing IPP1, PHO87, SSU1, or YHB1, and ten-fold greater resistance from the plasmid containing PHO90 (Fig. 2C). With the exception of IPP1, which is a known target for fluoride inhibition, none of these genes have previously been linked to fluoride.
Figure 2:
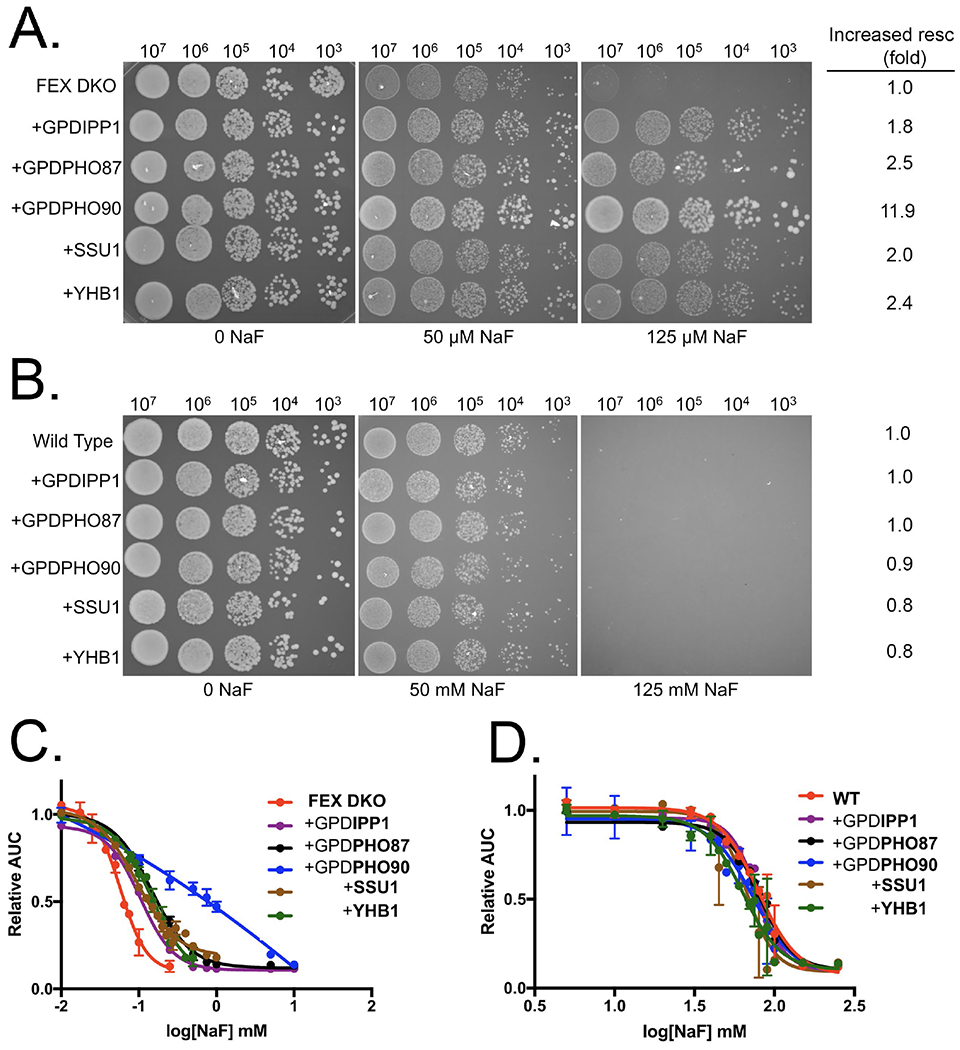
Overexpression of proteins conferring fluoride resistance. Serial dilutions of (A) FEX DKO or (B) wild type cells +/− pRS426 plasmids, tested for growth on YPD-agar supplemented with the designated concentrations of fluoride. On the right of the serial dilutions is the fold-change in fluoride resistance based on the change in IC50’s. Liquid growth assay of (C) FEX DKO and (D) wild type cells +/− pRS426 plasmids over a 24-hour period in increasing fluoride. SSU1 and YHB1 plasmids also contain 1 kb of the corresponding gene’s native promoter.
Given that the wild type and FEX DKO yeast have substantially different sensitivities to fluoride, we tested if the genes that confer resistance in FEX DKO also conferred improved fluoride resistance in the wild type background. Wild type yeast with high-copy plasmids expressing either IPP1, PHO87, PHO90, SSU1, or YHB1 showed no observable increase in fluoride resistance (Fig. 2D). The mechanism by which these genes confer partial fluoride resistance therefore appears to be specific to low fluoride concentrations.
Fluoride is well known to cause oxidative stress and metabolic arrest. Strikingly, no glycolytic enzymes or oxidative stress response proteins were identified in the overexpression screen. As a control, we overexpressed seven genes that were previously reported as in vitro targets of fluoride, including three genes in the glycolytic pathway, and four linked to oxidative stress.29 These genes - ENO1, ENO2, HXK1, SOD1, CTT1, CCP1, and PYK1 - when individually cloned into a high copy plasmid and transformed into FEX DKO, yielded no significant resistance to fluoride toxicity (Fig. S3). While these seven proteins may still be targeted by fluoride in vivo, their individual overexpression does not confer significant rescue to fluoride toxicity. It could be that these genes are not induced to high enough levels to show an effect, or that fluoride has many protein targets. The genes that provide partial rescue from fluoride toxicity (IPP1, PHO87, PHO90, SSU1, and YHB1), are likely to be involved in a broader mechanism of resistance than simply binding to fluoride.
Fluoride induces the nitrosative stress response
We sought to determine how YHB1 and SSU1 confer improved resistance to fluoride. Yhb1p and Ssu1p are both part of the nitrosative stress response pathway that converts the highly toxic nitric oxide into the more chemically inert nitrate, and then exports that nitrate from the cell. However, Ssu1p has a second function; it is also responsible for excreting sulfite. To determine if improved fluoride resistance is associated with nitrate and/or sulfite excretion by Ssu1p, we explored the toxicity of nitrate or sulfite in combination with fluoride. Nitric oxide is a gas, and cannot be readily added to the media at increasing concentrations. At the given IC25 concentrations for sulfite and nitrate (20 mM and 850 mM, respectively), addition of fluoride had contrasting effects (Fig. 3A). Sulfite - a known antioxidant - had a protective effect, shifting the IC50 of fluoride from 50 to 200 μM. Conversely, addition of nitrate shifted the IC50 down to 30 μM. While sulfite protects from fluoride toxicity, nitrate adds to the toxicity. Given that overexpression of SSU1 enhances fluoride resistance, it appears that the benefit of removing nitrate outweighs the cost of excreting sulfite.
Figure 3:
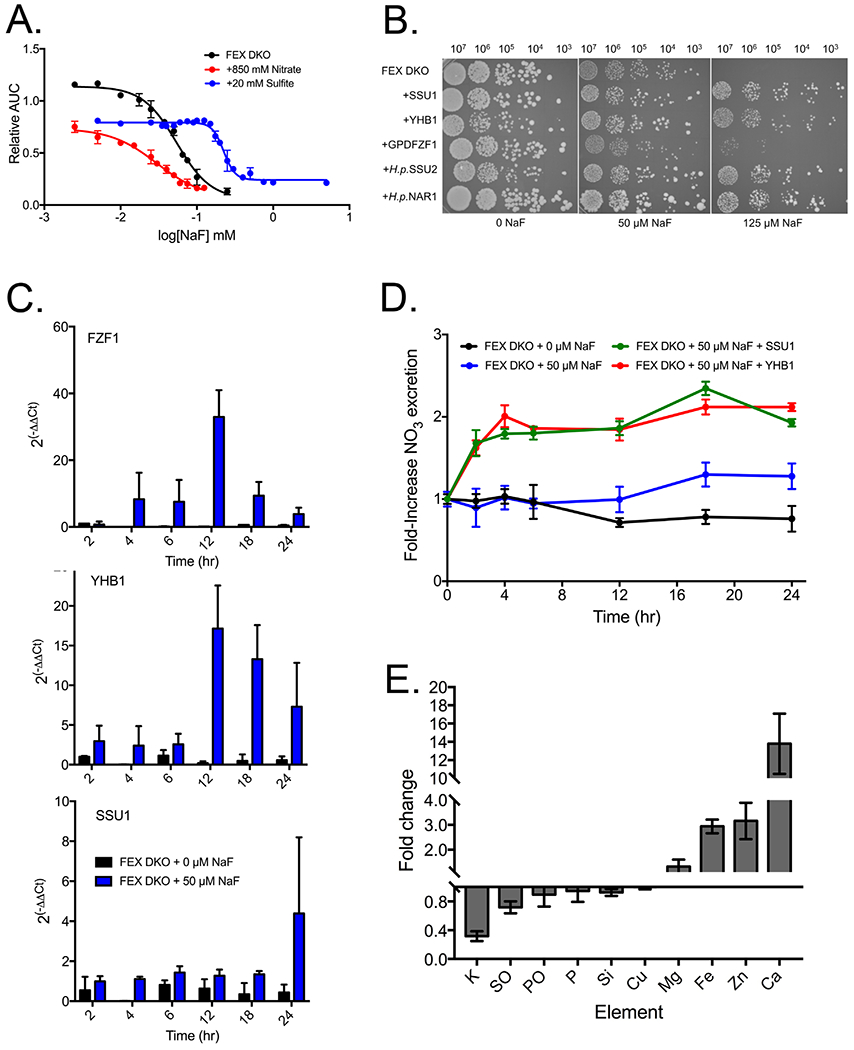
Nitrosative stress in fluoride-treated FEX DKO yeast. (A) Liquid growth assay of the combined effect on growth of yeast with fluoride and the IC25 of nitrate (850 mM) or sulfite (20 mM). (B) Serial dilution of FEX DKO cells +/− pRS426 plasmids over a 24-hour period. For H. polymorpha genes SSU2 and NAR1, both plasmids contain the promoter region upstream of the S. cerevisiae SSU1 gene. (C) RT-qPCR of FZF1, YHB1, and SSU1 gene expression over time, compared with one-hour growth in YPD and using actin as a housekeeping gene. (D) Efflux of extracellular nitrate in cells over time. (E) Fold change in intracellular ions of FEX DKO + 50 μM NaF after 24 hours using ICP-MS. SO and PO are oxidized sulfur and phosphorous, respectively.
To further explore the combined toxicity of fluoride and nitrate, we tested whether other proteins that excrete nitrate also improve fluoride resistance. There are no known yeast channels in S. cerevisiae that excrete only nitrate; however, the yeast strain H. polymorpha expresses two channels that excrete nitrate along with either sulfite or nitrite.69 The H. polymorpha gene SSU2, a sulfite/nitrate exporter, has 44% sequence similarity to S. cerevisiae SSU1, while the H. polymorpha nitrate/nitrite exporter NAR1 has 41% similarity to S. cerevisiae SSU1. Inducing SSU2 or NAR1 in FEX DKO yeast resulted in increased fluoride resistance, depending on the copy number (data not shown). Adding the S. cerevisiae SSU1 promoter to H. polymorpha NAR1 and SSU2 resulted in the same degree of fluoride rescue as overexpression of YHB1 and SSU1 (Fig. 3B). In general, higher expression of any protein that removed intracellular nitrate also provided partial rescue from fluoride toxicity.
The presence of an intact promoter is essential for improved fluoride resistance in both H. polymorpha and S. cerevisiae nitrosative stress response genes. The SSU1 promoter is recognized by the five-zinc finger protein Fzf1p, a transcription factor that induces YHB1 and SSU1 expression during nitrosative stress.70 The necessity of the promoter for fluoride rescue suggests that Fzf1p is activated during fluoride exposure. Overexpression of FZF1 conferred only modest rescue from fluoride, suggesting it may already be induced to optimal levels.
To test whether FZF1, YHB1, and SSU1 are induced under fluoride exposure, expression of each was monitored using RT-qPCR (Fig. 3C). We observed significant induction of FZF1 after 4-6 hours, and further induction after 12 hours. YHB1 was induced after 12 hours. We did not detect significant induction of SSU1 until 24 hours. However, other labs have also reported slight to moderate induction of SSU1 under conditions of nitrosative stress, which indicates that small increases in total SSU1 copy number can have compounding effects on cellular resistance to nitrosative species.70–72
These results suggest that fluoride induces nitrosative stress in FEX DKO yeast. Nitrosative stress is mediated by the conversion of nitric oxide into nitrate, and the subsequent excretion of nitrate from the cell. To determine whether fluoride was activating the nitrosative stress pathway and subsequent excretion of nitrate from cells, we used a colorimetric assay to monitor levels of nitrate in solution. We observed a 1.5-fold increase in extracellular nitrate after fluoride exposure starting at 12 hours, in agreement with the RT-qPCR data that showed induction of nitrosative stress response genes at a similar time (Fig. 3D). Overexpression of either YHB1 or SSU1 resulted in twice the levels of extracellular nitrate. Extracellular nitrite and sulfite concentrations did not change during fluoride exposure (Fig. S4). However, intracellular nitric oxide increased from 6 to 12 hours, before returning to baseline. An initial increase in intracellular nitric oxide and a later increase in extracellular nitrate is consistent with the hypothesis that the nitrosative stress response pathway is activated under fluoride exposure.
Nitrosative stress, while typically reported to correlate with oxidative stress, has also been reported with metal stress.73–74 Fluoride has been shown in mammalian cells to disrupt metal homeostasis.75 To test whether fluoride causes intracellular metal imbalance in FEX DKO yeast, we monitored intracellular the metal concentration using inductively coupled plasma mass spectrometry (ICP-MS). After 24 hours of fluoride exposure, the alkaline earth and transition metals magnesium, iron, zinc, and calcium increased in concentration (Fig. 3E). We also observed a decrease in intracellular potassium, which is a stress-signaling ion. One function of potassium is to regulate the membrane potential required for nutrient transport, and therefore the imbalance in intracellular metals may be linked to loss of potassium.76 The greatest change in concentration upon fluoride exposure was intracellular calcium, which increased by 15-fold. This is consistent with several reports also demonstrating an increase in calcium influx during stress, including hypertonic shock, ethanol, and alkaline stress.77–80 However, it is somewhat inconsistent with our data monitoring of cytoplasmic calcium using the Indo-1 dye, in which we only see a two-fold increase upon fluoride addition (Fig. 1E). This inconsistency is most likely attributed to both the increased sensitivity of ICP-MS, and that Indo-1 only monitors free, cytoplasmic calcium while ICP-MS measures total cellular calcium. ICP-MS does not differentiate between bound and unbound atoms, so we cannot determine if these metals are in complex with fluoride.
As the nitrosative stress response pathway has been shown to occur during metal stress, we hypothesized that YHB1 and SSU1 can reduce metal influx. Therefore, we also monitored intracellular metal ions in FEX DKO yeast in the presence of NaF when either YHB1 or SSU1 was overexpressed (Fig. S5). Magnesium was the only ion whose concentration was not significantly affected by overexpression of SSU1 or YHB1. Overexpressing either SSU1 or YHB1 reduced the efflux of potassium by 3-fold in SSU1, and to baseline in YHB1. Similarly, there were 2-3 fold less influx of transition metals when overexpressing SSU1, and almost no influx when overexpressing YHB1. This rescue could either be direct or indirect; YHB1 and SSU1 could work to excrete metal complexed with nitric oxide, or overexpressing YHB1 or SSU1 could counteract a stress effect upstream of metal imbalance.
Intracellular orthophosphate partially rescues fluoride toxicity in a concentration-dependent manner
The overexpression screen identified PHO87, PHO90, and IPP1 as genes conferring partial rescue to fluoride toxicity. PHO87, PHO90, and IPP1 are all involved in phosphate homeostasis. Pho87p and Pho90p are constitutively expressed transmembrane proteins that act as symporters of orthophosphate and hydrogen.81 Ipp1p is a cytoplasmic protein that converts pyrophosphate into orthophosphate.64 Given these activities, overexpression of IPP1, PHO87 or PHO90 are each predicted to increase the intracellular orthophosphate concentration. To test this hypothesis, 31P NMR was used to monitor total phosphate levels in FEX DKO yeast (Fig. 4A). Overexpression of either PHO87 or PHO90 increased total phosphate levels, by 4- and 6-fold, respectively. Total phosphate levels did not change upon IPP1 overexpression, but a higher fraction of the total cellular phosphate was found as orthophosphate.
Figure 4:
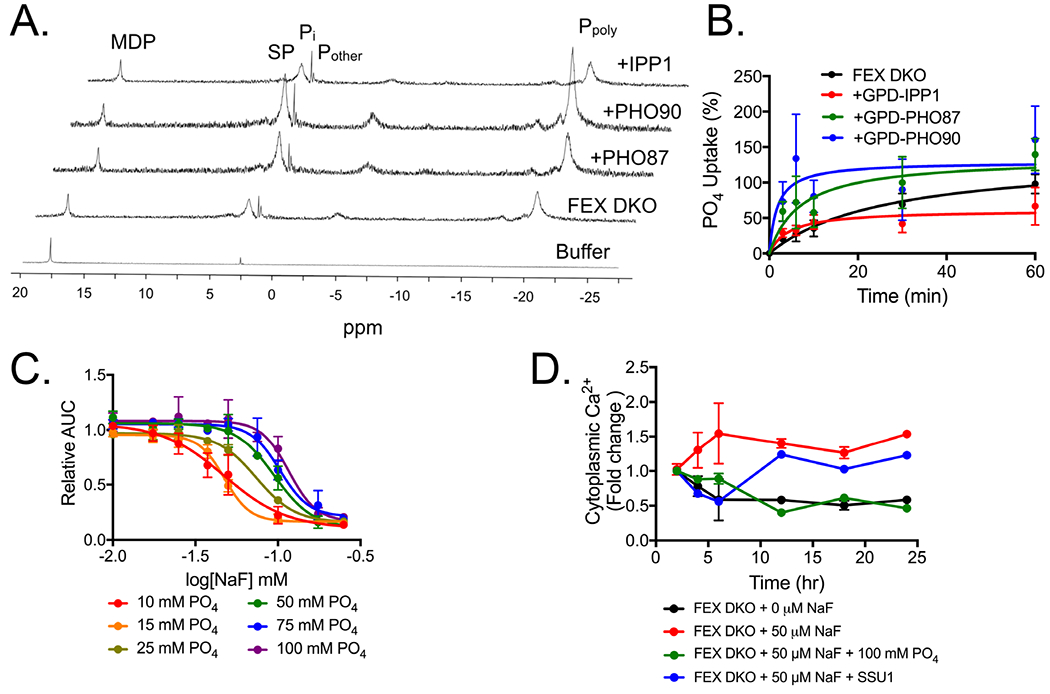
Intracellular phosphate in FEX DKO with high copy plasmids. (A) 31P NMR spectra of yeast after 12 hours growth. The peaks are (from bottom to top) buffer alone, FEX DKO, and FEX DKO with a pRS426GPD promoter expressing PHO87, PHO90, or IPP1. (B) 32PO4 influx assay measuring the rate of phosphate import over 60 minutes. (C) Liquid growth assay of cells grown in synthetic minimal media over 24 hours in increasing fluoride, with the noted final concentration of phosphate present in the media. For all assays, buffer is kept at pH 6.5. (D) Fold change in the concentration of cytoplasmic calcium over time, assed using Indo-1.
The increase in total phosphate observed upon overexpression of PHO87 or PHO90 is most likely due to their function in orthophosphate import. To test this directly, we assessed the rate of phosphate import using radiolabeled phosphate uptake. Over a 1-hour timespan, there was a higher influx of phosphate in cells overexpressing PHO87 or PHO90 compared with IPP1 or FEX DKO alone (Fig. 4B). PHO90 had the highest rate of import, consistent with cells having the largest total phosphate concentrations. As overexpression of PHO90 also resulted in the greatest rescue from fluoride toxicity, we hypothesized that the rescue may directly correlate with the higher orthophosphate concentration.
The connection between greater intracellular orthophosphate and reduced fluoride toxicity suggests that higher orthophosphate alone may provide some rescue from fluoride toxicity. The overexpression screen and concurrent growth assays were conducted in YPD, which contained approximately 10 mM PO4 at pH 6.5, as measured using malachite (data not shown). To assess whether an increase of orthophosphate alone is sufficient to rescue from fluoride toxicity, we performed growth assays in increasing phosphate up to 100 mM while maintaining pH at 6.5. An increase in phosphate from 10 mM to 100 mM shifted the fluoride IC50 from 47 to 116 µM NaF, consistent with a concentration-dependent rescue by phosphate (Fig. 4C). We again used 31P NMR and found that a 10-fold increase in extracellular phosphate led to about a 4-fold increase in intracellular phosphate (Fig. S6). The increase in orthophosphate corresponded with overexpression of PHO87 and IPP1, but was lower than overexpression of PHO90. This is consistent with the level of resistance, in which overexpression of PHO90 had 10-fold enhanced resistance to fluoride toxicity. These results are also consistent with the hypothesis that phosphate rescues fluoride toxicity independently of protein expression.
We next sought to determine whether phosphate alleviated fluoride-induced stress phenotypes. As reported above, overexpressing YHB1 and SSU1 reduced metal influx. Similarly, the addition of phosphate also lessened the efflux of potassium and influx of iron and calcium under fluoride exposure (Fig. S5). We monitored the effects of phosphate on cytoplasmic calcium, which is a signalling ion for oxidative stress (Fig. 4D). Addition of 100 mM PO4 to cells resulted in a depletion of cytoplasmic calcium to the same levels as those without fluoride treatment. In contrast, enhancement of the nitrosative stress response through a high copy plasmid containing SSU1 did not alter cytoplasmic calcium. Together, these data suggest that phosphate is acting to alleviate a general stress phenotype, and is potentially doing so upstream of the onset of oxidative stress.
Fluoride activates the starvation-induced apoptotic pathway
To further understand the mechanism of fluoride toxicity, including how it lowers intracellular orthophosphate and induces nitrosative stress, we examined changes in global gene expression under fluoride exposure using genome-wide RNA-Seq. Prolonged exposure to fluoride leads to eventual oxidative stress and apoptosis.57 Previous studies of RNA-Seq in cells with long exposure to fluoride reported an enrichment for the induction of nonspecific stress, apoptotic and cell cycle arrest signaling genes.60, 82–83 In order to more directly examine the mechanism of fluoride toxicity, we set out to measure gene induction after fluoride had caused toxicity, but before the cells were programmed for death. Oxidative stress signaling does not peak until 6 hours, as determined by cytosolic calcium levels and RT-qPCR, therefore, we measured gene expression of the FEX DKO yeast after 4 hours of exposure to 50 μM NaF.
The results of the RNA-Seq largely complement existing literature on fluoride toxicity (Fig. 5A). Out of 7127 genes monitored, 303 genes had over 1.5-fold change. We noted genes linked to oxidative stress, cell morphology, and general apoptotic signaling. DNA repair genes were induced, as were genes linked to translation, amino acid production and ribosomal maturation were inhibited. Consistent with our findings from the overexpression screen, there was increased expression of the nitrosative stress response transcription factor FZF1. VIP1 was also induced, which produces IP7 as the first step in the phosphate starvation response pathway (Fig. 5C).
Figure 5:
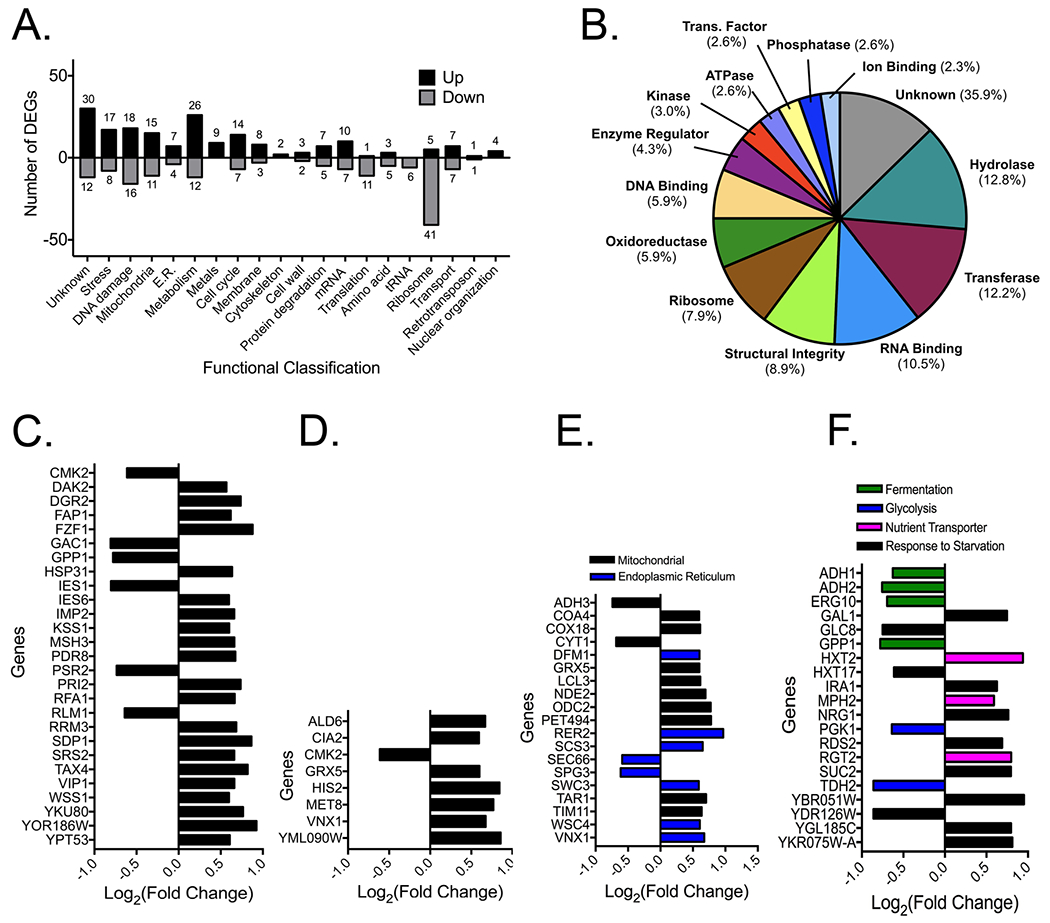
RNA-Seq analysis of FEX DKO treated with 50 μM NaF for four hours. (A) Functional classification graph of genes with more than 1.5-fold difference in expression compared with genes in FEX DKO grown in YPD for four hours. (B) Pie chart of the protein class and function, as mapped using the Saccharomyces Genome Database gene ontology slim mapper. (C) Fold-change in expression of genes from the list of hits linked to stress response and DNA repair, (D) metal homeostasis, (E) mitochondrial and endoplasmic reticulum stress, and (F) glucose starvation.
Included among the induced genes are those linked to metal homeostasis and metalloproteins. A large fraction of the induced proteins are members of classes typically containing metals, including transferases, hydrolases, and oxidoreductases (Fig. 5C). Upregulated genes included those linked to Fe-S protein synthesis, calcium and magnesium regulation, and general metal resistance, as well as mRNA fragments that overlapped with copper transporter CTR1 and iron scavenger ARN2 (Fig. 5D). Upregulated genes also included those linked to mitochondrial and endoplasmic reticulum damage, a widely-reported phenotype in mammalian cells exposed to high doses of fluoride. These included genes involved in replenishing mitochondrial cytochrome c leakage and improving mitochondrial membrane integrity, as well as genes involved in endoplasmic reticulum membrane integrity and calcium storage (Fig. 5E).
Compared to other RNA-Seq experiments from fluoride exposure, a unique observation in this screen was increased expression of a cluster of genes linked to glucose and nutrient starvation (Fig. 5F). Among the genes with significantly altered expression were seven involved in promoting glycolysis, as well as three sugar transporters. Previous studies that tested high extracellular fluoride suggested that fluoride directly inhibits glycolysis.11, 30, 84 This should theoretically lead to a buildup of glucose. Instead, in the model of a FEX double-knockout yeast with low extracellular fluoride, cells appear to be scavenging glucose within four hours of fluoride exposure. This complements the phosphate starvation phenotype, and suggests an overall trend of nutrient starvation under fluoride exposure.
Fluoride-induced stress inhibits phosphate import
The overexpression screen identified two phosphate transporters that confer partial fluoride resistance. Similarly, three sugar transporters are induced in the RNA-Seq experiment. These observations suggest that fluoride inhibits the transport of nutrients. Several previous reports found that fluoride can inhibit glucose import.85–87 We tested for Pho87p/Pho90p inhibition by monitoring phosphate uptake under increasing fluoride (Fig. 6A). Simultaneous addition of fluoride with phosphate showed no detectable difference in phosphate uptake. However, when the cells were pretreated for two doubling times (3 hours) with fluoride, phosphate uptake was inhibited with a KI of 90 μM NaF (Fig. 6B). At this 3-hour timepoint, FEX DKO yeast show slower growth and decreased intracellular ATP (Fig. 1). This suggests that the inhibition of phosphate import by fluoride is indirect.
Figure 6:
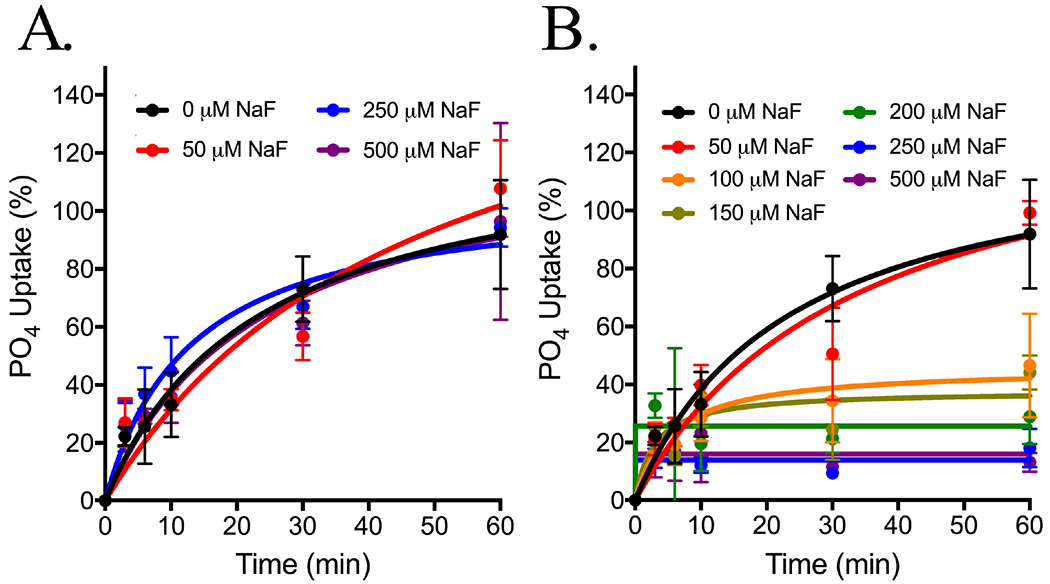
32P influx assay as a measurement of phosphate uptake over 60 minutes in 10 mM PO4 buffer at pH 6.5. FEX DKO yeast were grown three hours in (A) YPD or (B) NaF before transferring to 32PO4 and increasing NaF.
We next sought to determine what phenotype occurs at 3 hours of fluoride exposure that would arrest phosphate import. At this time point, oxidative stress signalling is just beginning to occur (Fig. 6). One major hallmark of oxidative stress is a drop in intracellular pH and consequent disruption of membrane potential.88–90 We directly tested this by monitoring intracellular pH after 3 hours of fluoride exposure, and found that the yeast’s cytoplasmic pH dropped by a full unit (Fig. 7A). We then monitored the plasma membrane electrochemical potential using the dye diS-C3, and found that while immediate exposure to fluoride had no effect on the membrane potential, 3 hours of exposure led to a destabilization consistent with intracellular acidification (Fig. 7B).
Figure 7:
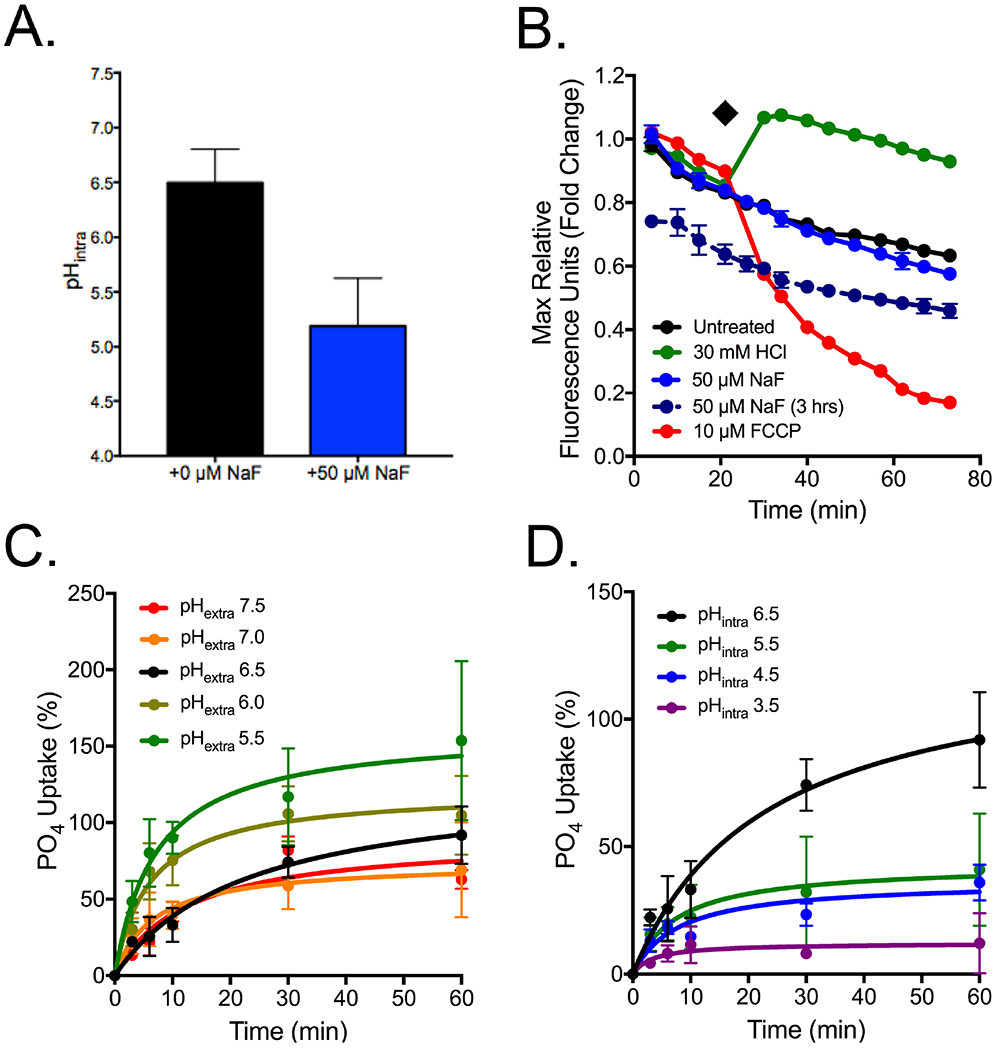
Assessment of phosphate influx as a function of pH. (A) Intracellular pH of FEX DKO cells after three hours growth in YPD or 50 μM NaF, as determined using 5(6)-CFDA. (B) Measurement of electrochemical potential of cells in either 3 hours exposure to fluoride (dashed line), or immediate exposure to NaF, CCCP (an agent that causes a rapid drop in pHintra), or HCl (an agent that causes a rapid drop in pHextra),. The (◆) indicates the point in which compounds were added for immediate exposure. (C) FEX DKO yeast grown for three hours in YPD, before transferring to buffer with pH from 5.5-7.5, using HCl and NaOH. (D) Cells grown in acetic acid for 30 minutes to pHintra of 3.5-6.5, then transferred to buffer at pH 6.5 for measurement of 32PO4 uptake. For further information as to the protocol, see Experimental Procedures.
Although Pho87p and Pho90p are evolved from sodium transporters, they are predicted to be hydrogen symporters, and may be sensitized to changes in membrane potential. To determine whether this model can explain the arrest in phosphate import, we repeated the phosphate influx assay in FEX DKO yeast while changing either extracellular, or intracellular pH. Lowering extracellular pH through the addition of hydrochloric acid increased the amount of orthophosphate able to enter the cells per unit of time (Fig. 7C). Conversely, an increase in intracellular acidity through pre-incubation of cells with acetic acid, then returning cells to a neutral extracellular buffer, arrested phosphate uptake (Fig. 7D). These data collectively suggest that phosphate uptake is highly sensitized to changes in the pH gradient, such as intracellular acidification.
DISCUSSION.
Prolonged exposure to high doses of fluoride is reported to result in many downstream effects, most notably oxidative stress, DNA damage, and metabolic arrest. These phenotypes have been attributed to a wide variety of mechanisms in the literature, most notably metalloprotein inhibition of the glycolytic enzyme enolase as well as mitochondrial respiratory proteins. Here, we report the first investigation of fluoride toxicity in a cell incapable of removing intracellular fluoride. These cells undergo growth arrest at fluoride concentrations well below those required to inhibit metalloprotein in vitro. Fluoride-sensitized yeast showed signs of oxidative stress, nitrosative stress, and nutrient starvation. Both oxidative and nitrosative stress are downstream phenotypes, and correlate with mitochondrial stress signaling and metal imbalance. Nutrient starvation was primarily caused by the inability of yeast to uptake phosphate and glucose upon fluoride exposure. We propose that this effect is due to the acid stress-induced disruption of the plasma membrane electrochemical gradient.
We found that overexpression of the genes IPP1, PHO87, PHO90, YHB1 and SSU1 partially rescue cells from intracellular fluoride toxicity in FEX DKO yeast. None of these proteins provide rescue in wild type background, suggesting that the mechanism of toxicity is different. Of the proteins conferring rescue to FEX DKO yeast, only IPP1p is a known target of fluoride. We found that YHB1 and SSU1 are induced upon fluoride exposure, and import of phosphate by PHO87 and PHO90 is inhibited upon fluoride exposure.
A previous study found that high copies of a genome fragment containing SSU1 rescued selenite toxicity.91 However, when SSU1 was expressed in a plasmid, it did not rescue selenite toxicity. Here we report that the SSU1 gene fragment requires its native promoter to rescue from fluoride toxicity. Likewise, YHB1 also rescues when its native promoter is included. Both SSU1 and YHB1 participate in nitrosative stress response. Nitrosative stress is linked to metal imbalance, oxidative stress, endoplasmic reticulum damage and metalloprotein inhibition. Fluoride is known to inhibit metalloproteins, damage the endoplasmic reticulum and cause oxidative stress. Increased nitric oxide has been found in the plasma of several mammalian models exposed to high levels of fluoride.22, 92–93 Consistent with these reports, we found direct evidence of fluoride activating the nitrosative stress response inside a cell.
PHO90 overexpression provided the most efficient rescue from fluoride toxicity. It was previously shown that cells only expressing PHO90 for phosphate transport survived better in differing phosphate conditions than cells expressing only PHO87, suggesting that PHO90 is more biologically important than PHO87.63 Similarly, our yeast with high copy PHO90 have a higher toxicant resistance than yeast with high copy PHO87. We found that this enhanced survival correlated with greater intracellular orthophosphate concentrations.
The mechanism by which phosphate rescues fluoride toxicity remains unclear. Phosphate has many properties that could contribute. Phosphate drives forward glycolysis, inhibits the mitochondrial pore opening in yeast, chelates metals, increases ATP, and acts as a pH buffer. It is not necessarily the case that only one of these properties is responsible for rescuing from general stress. A study demonstrated that PHO80 mutants – incapable of regulating the PHO pathway – show an influx of intracellular metals similar to what we observe upon fluoride treatment.94 This supports the argument that the metal influx is due to stress and concurrent loss of membrane integrity rather than a specific attribute of fluoride.
Other labs have observed that fluoride causes disruption of mitochondrial membrane potential, the release of cytochrome c into the cytoplasm, and induction of oxidative stress.12–16 Studies have also reported an increase in cytoplasmic calcium and endoplasmic reticulum stress, as well as inhibition of Ca2+-ATPase. 15, 94–97 Our RNA-Seq data complements these findings; however, multiple labs have argued that these effects are all independent from each other and result from the high affinity of fluoride to metalloproteins, which is in the millimolar range. We observed the same pathways are affected at much lower concentrations of fluoride in vivo, suggesting that the inhibition may be indirect. The affected genes can be clustered into the known pathways for starvation-induced apoptosis. Mitochondrial and endoplasmic reticulum stress appear to be downstream stress effects from toxicity and may be a result of calcium influx. Supplementing cells with phosphate reduces stress signalling, further supporting that these phenotypes are downstream of nutrient starvation.
A key finding from this study is the observation that fluoride activated the indirect arrest of nutrient uptake and subsequent starvation pathway. Fluoride is known to inhibit glycolysis and drop ATP concentration. Theoretically, glycolysis inhibition should lead to an accumulation of intracellular glucose. Instead we observed that cells demonstrate a glucose starvation phenotype. Similarly, a recent study in rat liver also showed an induction of glucose scavenger genes during fluoride exposure.60 We also noted an arrest in phosphate uptake upon fluoride exposure. However, RNA-Seq did not show significant induction of the phosphate scavenging proteins in the PHO pathway, with the exception of VIP1. Most likely, metabolic and cell cycle arrest decreased phosphate usage, so total phosphate was not depleted sufficiently within four hours to trigger a substantial phosphate starvation response.
We observed that lowered intracellular pH arrested phosphate import. Similar effects are seen in mammalian cancer cells, where the acidification of the outside of the cells results in a change to membrane potential.98–99 In cancer cases, proteins are upregulated that can function in acidic pH. In yeast, the two inducible high-affinity phosphate transporters, PHO84 and PHO89, have acidic and alkaline pH optima, respectively. This suggests that when the PHO pathway is active, the pH gradient is disrupted. Given that a wide variety of stressors lead to intracellular acidification, phosphate import would be inhibited under many stress conditions.
In summary, yeast lacking fluoride exporters underwent a toxicity phenotype under fluoride exposure distinct from those previously reported at substantially higher fluoride concentrations. These hyper-sensitive yeast underwent metabolic arrest, oxidative stress, and nitrosative stress upon fluoride exposure. The subsequent influx of metals was mitigated by both heightened phosphate and the nitrosative stress response. Fluoride arrested phosphate import and triggered the nutrient starvation apoptotic pathway. Phosphate import was heavily influenced by intracellular acidification, a hallmark of general stress. As such, we hypothesize that the arrest in phosphate import and subsequent nutrient starvation during fluoride stress is caused by the indirect disruption in plasma membrane integrity.
Supplementary Material
ACKNOWLEDGMENTS.
We thank members of the Strobel lab for valuable discussions, as well as David Hiller and Andrew Knappenberger for their critical reading of the manuscript. We also thank the Yale Center for Research Computing, particularly Robert Bjornson and Benjamin Evans, for their insight in bioinformatics.
FUNDING INFORMATION.
This work was supported by funding provided by the N.I.H. Chemical Biology Interface training grant (to N.R.J.). The authors declare no competing financial interests.
ABBREVIATIONS.
- DKO
double knockout
- RT-qPCR
reverse-transcription quantitative polymerase chain reaction
- ICP-MS
inductively coupled plasma mass spectrometry
- YPD
yeast extract peptone dextrose
- SD
synthetic defined
- AUC
area under the curve
Footnotes
SUPPORTING INFORMATION. [Additional flow cytometry graphs, liquid growth assays of YHB1, SSU1, oxidative stress genes, and glycolytic enzymes, concentration changes of sulfite, nitrite, and nitric oxide, additional ICP-MS data, and intracellular phosphate concentrations.]
REFERENCES.
- 1.Smith FA, and Hodge HC (1979) Airborne fluorides and man. CRC Crit. Rev. Envir. Cont 8, 293–271. [Google Scholar]
- 2.Jha SK, Mishra VK, Sharma DK, and Damodaran T (2011) Fluoride in the environment and its metabolism in humans. Rev. Environ. Contam. Toxicol 211, 121–142. [DOI] [PubMed] [Google Scholar]
- 3.Vithanage M, and Bhattacharya P (2015) Fluoride in the environment: sources, distribution and defluoridation. Environ. Chem. Letters 13, 131–147. [Google Scholar]
- 4.U.S. Department of Health and Human Services Federal Panel on Community Water Fluoridation (2015). U.S. Public Health Service recommendation for fluoride concentration in drinking water for the prevention of dental caries. Public health reports (Washington, D.C.: 1974), 130(4), 318–331. DOI: 10.1177/003335491513000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. (1999) Ten great public health achievements: United States, 1900–1999. Morbidity and Mortality Weekly Report 48, 241–243. [PubMed] [Google Scholar]
- 6.Shivarajashankara YM, Shivashankara AR, Bhat PG, and Rao SH (2003) Lipid peroxidation and antioxidant systems in the blood of young rats subjected to chronic fluoride toxicity. Ind. J. Exp. Biol 41, 857–860. [PubMed] [Google Scholar]
- 7.Shanthakumari D, Srinivasalu S, and Subramanian S (2004) Effect of fluoride intoxication on lipid peroxidation and antioxidant status in experimental rats. Toxicology 204, 219–228. [DOI] [PubMed] [Google Scholar]
- 8.Refsnes M, Skuland T, Lag M, Schwarze PE, and Ovrevik J (2014) Differential NF-κB and MAPK activation underlies fluoride- and TPA-mediated CXCL8 (IL-8) induction in lung epithelial cells. J. Inflamm. Res 7, 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo Q, Cui H, Deng H, Kuang P, Liu H, Lu Y, Fang J, Zuo Z, Deng J, Li Y, Wang X, and Zhao L (2017) Sodium fluoride induces renal inflammatory responses by activating NF-κB signaling pathway and reducing anti-inflammatory cytokine expression in mice. Oncotarget 8, 80192–80207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L, Kuang P, Liu H, Wei Q, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, and Zhao L (2019) Sodium fluoride (NaF) induces inflammatory responses via activating MAPKs/NF-κB signaling pathway and reducing anti-inflammatory cytokine expression in the mouse liver. Biol. Trace Elem. Res 189, 157–171. [DOI] [PubMed] [Google Scholar]
- 11.Feig SA, Shohet SB, and Nathan DG (1971) Energy metabolism in human erythrocytes: I. effects of sodium fluoride. J. Clin. Invest 50, 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anuradha CD, Kanno S, and Hirano S (2001) Oxidative damage to mitochondria is a preliminary step to caspase-3 activation in fluoride-induced apoptosis in HL-60 cells. Free Radical Biology and Medicine 31, 367–373. [DOI] [PubMed] [Google Scholar]
- 13.Kubota K, Lee DH, Tsuchiya M, Young CS, Everett ET, Martinez-Mier EA, Snead ML, Nguyen L, Urano F, and Bartlett JD (2005) Fluoride induces endoplasmic reticulum stress in ameloblasts responsible for dental enamel formation. J. Biol. Chem 280, 23194–23202. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Wang A, Xia T, and He P (2008) Effects of fluoride on DNA damage, S-phase cell-cycle arrest and the expression of NF-κB in primary cultured rat hippocampal neurons. Toxicol. Letters 179, 1–5. [DOI] [PubMed] [Google Scholar]
- 15.Sharma R, Tsuchiya M, and Bartlett JD (2008) Fluoride induces endoplasmic reticulum stress and inhibits protein synthesis and secretion. Environ. Health Perspect 116, 1142–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fina BL, Lombarte M, Rigalli JP, and Rigalli A (2014) Fluoride increases superoxide production and impairs the respiratory chain in ROS 17/2.8 osteoblastic cells. PloS One 9, DOI: e100768 10.1371/journal.pone.0100768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamilton IR (1990) Biochemical effects of fluoride on oral bacteria. J. Dent. Res 69, 660–667. [DOI] [PubMed] [Google Scholar]
- 18.Belli E, Buckley DH, and Marquis RE (1995) Weak acid effects and fluoride inhibition of glycolysis by Streptococcus mutans GS-5. Can. J. Microbiol 41, 785–791. [DOI] [PubMed] [Google Scholar]
- 19.Chen Q, Chai YC, Mazumder S, Jiang C, Macklis RM, Chisolm GM, and Almasan A (2003) The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell Death Diff. 10, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He LF, and Chen JG (2006) DNA damage, apoptosis, and cell cycle changes induced by fluoride in rat oral mucosal cells and hepatocytes. World J. Gastroenterol 12, 1144–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gassowska M, Gutowska I, Baranowska-Bosiacka I, and Chlubek D (2013) Effect of fluoride on sodium-proton exchanger activity, intracellular pH and calcium concentration in human non-stimulated platelets. Ann. Acad. Med. Stetin 59, 54–61. [PubMed] [Google Scholar]
- 22.Zhou BH, Zhao J, Liu J, Zhang JL, Li J, and Wang HW (2015) Fluoride-induced oxidative stress is involved in the morphological damage and dysfunction of liver in female mice. Chemosphere 139, 504–511. [DOI] [PubMed] [Google Scholar]
- 23.ten Cate JM, and Featherstone JDB (1991) Mechanistic aspects of the interactions between fluoride and dental enamel. Crit. Rev. Oral Bio. and Med 2, 283–296. [DOI] [PubMed] [Google Scholar]
- 24.Featherstone JDB (1999) Prevention and reversal of dental caries: role of low-level fluoride. Comm. Dent. and Oral Epidem 27, 31–40. [DOI] [PubMed] [Google Scholar]
- 25.Edwards SL, Poulos TL, and Kraut J (1984) The crystal structure of fluoride-inhibited cytochrome c peroxidase. J. Biol. Chem 259, 12984–12988. [PubMed] [Google Scholar]
- 26.Edwards SL, and Poulos TL (1990) Ligand binding and structural perturbations in cytochrome c peroxidase. A crystallographic study. J. Biol. Chem 265, 2588–2595. [PubMed] [Google Scholar]
- 27.Schenk G, Elliott TW, Leung E, Carrington LE, Mitic N, Gahan LR, and Guddat LW (2008) Crystal structures of a purple acid phosphatase, representing different steps of this enzyme’s catalytic cycle. B.M.C. Struct. Biol 8, DOI: 6.10.1186/1472-6807-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baykov AA, Tam-Villoslado JJ, and Aveava SM (1979) Fluoride inhibition of inorganic pyrophosphatase. IV. Evidence for metal participation in the active center and a four-site model of metal effect on catalysis. Biochim. Biophys. Acta 569, 228–238. [DOI] [PubMed] [Google Scholar]
- 29.Adamek E, Palowska-Goral K, and Bober K (2005) In vitro and in vivo effects of fluoride ions on enzyme activity. Ann. Acad. Med. Stetin 51, 69–85. [PubMed] [Google Scholar]
- 30.Qin J, Chai G, Brewer JM, Lovelace LL, and Lebioda L (2006) Fluoride inhibition of enolase: crystal structure and thermodynamics. Biochemistry 45, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang GL, Jiang BH, and Semenza GL (1995) Effect of protein kinase and phosphatase inhibitors on expression of hypoxia-inducible factor 1. Biochem. Biophys. Res. Comm 216, 669–675. [DOI] [PubMed] [Google Scholar]
- 32.Wang P, Verin AD, Birukova A, Gilbert-McClain LI, Jacobs K, and Garcia JG (2001) Mechanisms of sodium fluoride-induced endothelial cell barrier dysfunction: role of MLC phosphorylation. Amer. J. Physiol. Lung Cell Mol. Physiol 281, L1472–1483. [DOI] [PubMed] [Google Scholar]
- 33.The Protein Data Bank Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) Nuc. Acids Res 28, 235–242. DOI: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker JL, Sudarsan N, Weinberg Z, Roth A, Stockbridge RB, and Breaker RR (2012) Widespread genetic switches and toxicity resistance proteins for fluoride. Science 335, 233–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockbridge RB, Robertson JL, Kolmakova-Partensky L, and Miller C (2013) A family of fluoride-specific ion channels with dual-topology architecture. eLife, e01084 DOI: 10.7554/eLife.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rapp M, Granseth E, Seppala S, and von Heijne G (2006) Identification and evolution of dual-topology membrane proteins. Nat. Struc. Molec. Biol 13, 112–116. [DOI] [PubMed] [Google Scholar]
- 37.Stockbridge RB, Koide A, Miller C, and Koide S (2014) Proof of dual-topology architecture of fluc F− channels with monobody blockers. Nat. Comm 5, 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Last NB, Kolmakova-Partensky L, Shane T, and Miller C (2016) Mechanistic signs of double-barreled structure in a fluoride ion channel. eLife, e18767 DOI: 10.7554/eLife.18767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji C, Stockbridge RB, and Miller C (2014) Bacterial fluoride resistance, fluc channels, and the weak acid accumulation effect. J. Gen. Physiol 144, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Smith KD, Davis JH, Gordon PB, Breaker RR, and Strobel SA (2013) Eukaryotic resistance to fluoride toxicity mediated by a widespread family of fluoride export proteins. P. N. A. S 110, 19018–19023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith AM, Ammar R, Nislow C, and Giaever G (2010) A survey of yeast genomic assays for drug and target discovery. Pharmacol, Ther 127, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer AC, and Kishony R (2014) Opposing effects of target overexpression reveal drug mechanisms. Nat. Comm 5, 4296 DOI: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rine J, Hansen W, Hardeman E, Davis RW (1983) Targeted selection of recombinant clones through gene dosage effects. P. N. A. S 80, 6750–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naganuma A, Furuchi T, Miura N, Hwang GH, and Kuge S (2002) Investigation of intracellular factors involved in methylmercury toxicity. Tohoku J. Exp. Med 196, 65–70. [DOI] [PubMed] [Google Scholar]
- 45.Gaines TA, Zhang W, Wang D, Bukun B, Chisholm ST, Shaner DL, Nissen SJ, Patzoldt WL, Tranel PJ, Culpepper AS, Grey TL, Webster TM, Vencill WK, Sammons RD, Jiang J, Preston C, Leach JE, and Westra P (2010) Gene amplification confers glyphosate resistance in Amaranthus palmeri. P. N. A. S 107, 1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnoldo A, Kittanakom S, Heisler LE, Mak AB, Shukalyuk AI, Torti D, Moffat J, Giaever G, and Nislow C (2014) A genome scale overexpression screen to reveal drug activity in human cells. Genome Med. 6, 32 DOI: 10.1186/gm549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Gerstein M, and Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genetics 10, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sessions OM, Tan Y, Goh KC, Liu Y, Tan P, Rozen S, and Ooi EE (2013) Host cell transcriptome profile during wild-type and attenuated dengue virus infection. PLoS Negl. Trop. Dis 7, e2107 DOI: 10.1371/journal.pntd.0002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon DF, Domingos RF, Hauser C, Hutchins CM, Zerges W, and Wilkinson KJ (2013) Transcriptome sequencing (RNA-seq) analysis of the effects of metal nanoparticle exposure on the transcriptome of Chlamydomonas reinhardtii. Appl. Environ. Microbiol 79, 4774–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H, Lam SH, Shen Y, and Gong Z (2013) Genome-wide identification of molecular pathways and biomarkers in response to arsenic exposure in zebrafish liver. PloS One 8, e68737 DOI: 10.1371/journal.pone.0068737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aufauvre J, Misme-Aucouturier B, Vigues B, Texier C, Delbac F, Blot N (2014) Transcriptome analyses of the honeybee response to Nosema ceranae and insecticides. PloS One 9, e91686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen L, Li Y, Jiang L, and Wang X (2014) Response of Saccharomyces cerevisiae to the stimulation of lipopolysaccharide. PloS One 9, e104428 DOI: 10.1371/journal.pone.0104428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ares M (2012) Isolation of total RNA from yeast cell cultures. Cold Spring Harb. Protoc 10, 1082–1086. [DOI] [PubMed] [Google Scholar]
- 54.Campbell-Burk SL, and Shulman RG (1987) High-resolution NMR studies of Saccharomyces cerevisiae. Ann. Rev. Microbiol 41, 595–616. [DOI] [PubMed] [Google Scholar]
- 55.Rothstein A, and Donovan K (1963) Interactions of arsenate with the phosphate-transporting system of yeast. J. Gen. Physiol 46, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wykoff DD, and O’Shea EK (2001) Phosphate transport and sensing in Saccharomyces cerevisiae. Genetics 159, 1491–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbier O, Arreola-Mendoza L, and Del Razo LM (2010) Molecular mechanisms of fluoride toxicity. Chemico-Biol. Inter 188, 319–333. [DOI] [PubMed] [Google Scholar]
- 58.Walton RE, and Eisenmann DR (1974) Ultrastructural examination of various stages of amelogenesis in the rat following parenteral fluoride administration. Arch. Oral Biol 19, 171–178. [DOI] [PubMed] [Google Scholar]
- 59.Deng H, Kuang P, Cui H, Luo Q, Liu H, Lu Y, Fang J, Zuo Z, Deng J, Li Y, Wang X, and Zhao L (2017) Sodium fluoride induces apoptosis in mouse splenocytes by activating ROS-dependent NF-κB signaling. Oncotarget 8, 114428–114441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira HABS, Dionizio AS, Araujo TT, Fernandes MS, Iano FG, and Buzalaf MAR (2018) Proposed mechanism for understanding the dose- and time-dependency of the effects of fluoride in the liver. Toxicol. Appl. Pharmacol 358, 68–75. [DOI] [PubMed] [Google Scholar]
- 61.Ott M, Gogvadze V, Orrenius S, and Zhivotovsky B (2007) Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. [DOI] [PubMed] [Google Scholar]
- 62.Bun-ya M, Shikata K, Nakade S, Yompakdee C, Harashima S, and Oshima Y (1996) Two new genes, PHO86 and PHO87, involved in inorganic phosphate uptake in Saccharomyces cerevisiae. Curr. Genet 29, 344–351. [PubMed] [Google Scholar]
- 63.Ghillebert R, Swinnen E, De Snijder P, Smets B, and Winderickx J (2011) Differential roles for the low-affinity phosphate transporters Pho87 and Pho90 in Saccharomyces cerevisiae. Biochem. J 434, 243–251. [DOI] [PubMed] [Google Scholar]
- 64.Kornberg A (1962) On the metabolic significance of phosphorolytic and pyrophosphorolytic reactions, Academic Press, NY. [Google Scholar]
- 65.Kolakowski LF, Schloesser M, and Cooperman BS (1988) Cloning, molecular characterization and chromosome localization of the inorganic pyrophosphatase (PPA) gene from S. cerevisiae. Nucl. Acids Res 16, 10441–10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cassanova N, O’Brien KM, Stahl BT, McClure T, and Poyton RO (2005) Yeast flavohemoglobin, a nitric oxide oxidoreductase, is located in both the cytosol and the mitochondrial matrix: effects of respiration, anoxia, and the mitochondrial genome on its intracellular level and distribution. J. Biol. Chem 280, 7645–7653. [DOI] [PubMed] [Google Scholar]
- 67.Avram D, and Bakalinsky AT (1999) SSU1 encodes a plasma membrane protein with a central role in a network of proteins conferring sulfite tolerance in Saccharomyces cerevisiae. J. Bacteriology 179, 5971–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park H, and Bakalinsky A (2000) SSU1 mediates sulphite efflux in Saccharomyces cerevisiae. Yeast 16, 881–888. [DOI] [PubMed] [Google Scholar]
- 69.Cabrera E, Gonzalez-Montelongo R, Giraldez T, de la Rosa DA, and Siverio JM (2014) Molecular components of nitrate and nitrite efflux in yeast. Eukaryot. Cell 13, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sarver A, and DeRisi J (2005) Fzf1p regulates an inducible response to nitrosative stress in Saccharomyces cerevisiae. Mol. Biol. Cell 16, 4781–4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nardi T, Corich C, Giacomini A, and Blondin B (2010) A sulphite-inducible form of the sulphite efflux gene SSU1 in a Saccharomyces cerevisiae wine yeast. Microbiol. 156, 1686–1696. [DOI] [PubMed] [Google Scholar]
- 72.Hennicke F, Grumbt M, Lermann U, Ueberschaar N, Palige K, Böttcher B, Jacobsen ID, Staib C, Morschhäuser J, Monod M, Hube B, Hertweck C, and Staib P (2013) Factors supporting cysteine tolerance and sulfite production in Candida albicans. Eukaryot. Cell 12, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wysocki R, and Tamas MJ (2010) How Saccharomyces cerevisiae copes with toxic metals and metalloids. F. E. M. S. Microbiol. Rev 34, 925–951. [DOI] [PubMed] [Google Scholar]
- 74.Sahay S, and Gupta M (2017) An update on nitric oxide and its benign role in plant responses under metal stress. Nitric Oxide 67, 39–52. [DOI] [PubMed] [Google Scholar]
- 75.Zerwekh JE, Morris AC, Padalino PK, Gottshalk F, and Pak CY (1990) Fluoride rapidly and transiently raises intracellular calcium in human osteoblasts. J. Bone Miner. Res 5, S131–136. [DOI] [PubMed] [Google Scholar]
- 76.Navarrete C, Petrezsélyová S, Barreto L, Martínez JL, Zahrádka J, Ariño J, Sychrová H, and Ramos J (2010) Lack of main K+ uptake systems in Saccharomyces cerevisiae cells affects yeast performance in both potassium-sufficient and potassium-limiting conditions. F. E. M. S. Yeast Res 10, 508–517. [DOI] [PubMed] [Google Scholar]
- 77.Bonilla M, and Cunningham KW (2002) Calcium release and influx in yeast: TRPC and VGCC rule another kingdom. Science S. T. K. E 2002, DOI: pe17. 10.1126/stke.2002. [DOI] [PubMed] [Google Scholar]
- 78.Courchesne WE, Vlasek C, Klukovich R, and Coffee S (2011) Ethanol induces calcium influx via the Cch1-Mid1 transporter in Saccharomyces cerevisiae. Arch. Microbiol 193, 323–334. [DOI] [PubMed] [Google Scholar]
- 79.Wang H, Liang Y, Zhang B, Zheng W, Xing L, and Li M (2011) Alkaline stress triggers an immediate calcium fluctuation in Candida albicans mediated by Rim101p and Crz1p transcription factors. F. E. M. S. Yeast Res 11, 430–439. [DOI] [PubMed] [Google Scholar]
- 80.Cyert MS, and Philpott CC (2013) Regulation of cation balance in Saccharomyces cerevisiae. Genetics 193, 677–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Samyn DR, Persson BL (2016) Inorganic phosphate and sulfate transport in S. cerevisiae. Yeast Membrane Transport, Advances in Experimental Medicine and Biology. pp 253–269. [DOI] [PubMed] [Google Scholar]
- 82.Ran S, Sun N, Liu Y, Zhang W, Li Y, Wei L, Wang J, and Liu B (2017) Fluoride resistance capacity in mammalian cells involves complex global gene expression changes. F. E. B. S 7, 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li QS, Li XM, Qiao RY, Shen EH, Lin XM, Lu JL, Ye JH, Liang YR, and Zheng XQ (2018) De novo transcriptome assembly of fluorine accumulator tea plant Camellia sinensis with fluoride treatments. Nat. Sci. Data 5, 180194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shearer TR, and Suttie JW (1970) Effect of fluoride on glycolytic and citric acid cycle metabolites in rat liver. J. Nutrition 100, 749–756. [DOI] [PubMed] [Google Scholar]
- 85.Germaine GR, and Tellefson LM (1986) Role of the cell membrane in pH-dependent fluoride inhibition of glucose uptake by Streptococcus mutans. Antimicrob. Agents Chemother 29, 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iwami Y, Hata S, Schachtele CF, and Yamada T (1995) Simultaneous monitoring of intracellular pH and proton excretion during glycolysis by Streptococcus mutans and Streptococcus sanguis: effect of low pH and fluoride. Mol. Oral Microbiol 10, 355–359. [DOI] [PubMed] [Google Scholar]
- 87.Rogalska A, Kutler K, Zalazko A, Glogowska-Gruszka A, Swietochowska E, and Nowak P (2017) Fluoride alteration of [3H]glucose uptake in Wistar rat brain and peripheral tissues. Neurotox. Res 31, 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai KL, Wang SM, Chen CC, Fong TH, and Wu ML (1997) Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol 502, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Orij R, Brul S, and Smits GJ (2011) Intracellular pH is a tightly controlled signal in yeast. Biochim. Biophys. Acta 1810, 933–944. [DOI] [PubMed] [Google Scholar]
- 90.Birben E, Sahiner UM, Sackesen C, Erzurum S, and Kalayci O (2012) Oxidative stress and antioxidant defense. World Allergy Organ. J 5, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Sampietro M, Serra-Cardona A, Canadell D, Casas C, Arino J, and Herrero E (2016) The yeast Aft2 transcription factor determines selenite toxicity by controlling the low affinity phosphate transport system. Nat. Sci. Rep 6, 32836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krechniak J, and Inkielewicz I (2005) Correlation between fluoride concentrations and free radical parameters in soft tissues of rats. Fluoride 38, 293–296. [Google Scholar]
- 93.Agalakova NI, and Guzev GP (2011) Molecular mechanisms of cytotoxicity and apoptosis induced by inorganic fluoride. Int. Sch. Res. Not 2012, 16. doi: 10.5402/2012/403835. [DOI] [Google Scholar]
- 94.Dominguez JH, Garcia JG, Rothrock JK, English D, and Mann C (1991) Fluoride mobilizes intracellular calcium and promotes Ca2+ influx in rat proximal tubules. Am. J. Physiol 261, F318–327. [DOI] [PubMed] [Google Scholar]
- 95.Boink AB, Wemer J, Meulenbelt J, Vaessen HA, and de Wildt DJ (1994) The mechanism of fluoride-induced hypocalcaemia. Hum. Exp. Toxicol 13, 149–155. [DOI] [PubMed] [Google Scholar]
- 96.Xu H, Zhou YL, Zhang XY, Liu H, Jing L, and Li GS (2007) Effects of fluoride on the intracellular free Ca2+ and Ca2+-ATPase of kidney. Biol. Trace. Elem. Res 116, 279–287. [DOI] [PubMed] [Google Scholar]
- 97.Murphy AJ, and Coll RJ (1992) Fluoride is a slow, tight-binding inhibitor of the calcium ATPase of sarcoplasmic reticulum. J. Biol. Chem 267, 5229–5235. [PubMed] [Google Scholar]
- 98.Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, and Baba Y (2013) Acidic extracellular microenvironment and cancer. Cancer Cell. Int 13, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang M, and Brackenbury WJ (2013) Membrane potential and cancer progression. Front. Physiol 4, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.