

Abstract
The ubiquitin-proteasome system (UPS) plays a central role in the degradation of cellular proteins. Targeting protein degradation has been validated as an effective strategy for cancer therapy since 2003. Several components of the UPS have been validated as potential anticancer targets, including 20S proteasomes, 19S proteasome-associated deubiquitinases (DUBs) and ubiquitin ligases (E3s). 20S proteasome inhibitors (such as bortezomib/BTZ and carfilzomib/CFZ) have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of multiple myeloma (MM) and some other liquid tumors. Although survival of MM patients has been improved by the introduction of BTZ-based therapies, these clinical 20S proteasome inhibitors have several limitations, including emergence of resistance in MM patients, neuro-toxicities, and little efficacy in solid tumors. One of strategies to improve the current status of cancer treatment is to repurpose old drugs with UPS-inhibitory properties as new anticancer agents. Old drug reposition represents an attractive drug discovery approach compared to the traditional de novo drug discovery process which is time-consuming and costly. In this review, we summarize status of repurposed inhibitors of various UPS components, including 20S proteasomes, 19S-associated DUBs, and ubiquitin ligase E3s. The original and new mechanisms of action, molecular targets, and potential anticancer activities of these repurposed UPS inhibitors are reviewed, and their new uses including combinational therapies for cancer treatment are discussed.
Keywords: Drug reposition, Protein degradation, Post-translational modification, UPS inhibitors, Cancer therapies
1. Introduction
1.1. The ubiquitin-proteasome system (UPS)
The UPS plays an essential role in the degradation of more than 80 % of intracellular proteins. The Nobel Prize in chemistry 2004 was awarded to Avram Hershko, Irwin Rose, and Aaron Ciechanover "for the discovery of ubiquitin-mediated protein degradation". Ubiquitin is a small protein with 76 amino acid residues found universally in eukaryotic cells [1,2]. Ubiquitination, a process of the covalent attachment of ubiquitin to a substrate protein, is highly regulated, involving the sequential action of several enzymes. Initially, ubiquitin activating enzyme (E1) uses energy from ATP hydrolysis to form a thiolester bond between itself and ubiquitin. Following activation, ubiquitin is then transferred to ubiquitin conjugating enzyme (E2) forming an ubiquitin thioester in a similar fashion. In the third step catalyzed by a ubiquitin ligase (E3) an isopeptide is formed between the ubiquitin C-terminus and ε-NH2 group of the substrate protein’s Lys residues [3,4] (Fig. 1).
Fig. 1.

The ubiquitin-proteasome pathway. Ubiquitination, a process of the binding of ubiquitin (Ub) to the substrate, involves the sequential actions of E1, E2 and E3 enzymes in an ATP-dependent manner. Most ubiquitinated substrates are then targeted for degradation by the 26S proteasome, which consists of 20S proteasome and 19S proteasome. The 20S proteasome contains two identical, 7-membered α-rings and two identical, 7-membered β-rings. There are three β-subunits that bear protease active sites. Ubiquitination is a reversible process due to a number of deubiquitinases (DUBs) that catalyze the removal of ubiquitin from target protein.
Once modified by a polyubiquitin chain, most of the target protein is ultimately degraded by 26S proteasome complex (Fig. 1). The 26S proteasome is composed of a core 20S proteasome and one or two regulatory cap 19S proteasomes [5]. The ubiquitinated substrates are recognized by the 19S proteasome, which performs its regulatory functions by binding and removing the polyubiquitin chains. Subsequently, the substrates are unfolded and translocated into the 20S proteasome, where they are degraded into short peptides. The 20S proteasome is a barrel-like complex, which consists of four rings, two identical outer rings being formed by seven α subunits and two identical inner rings formed by seven β-subunits. The proteolytic active sites of 20S proteasome are located on β1-, β2- and β5-subunits, displaying the postglutamyl-peptide-hydrolysis-like (PGPH-L), trypsin-like (T-L) and chymotrypsin-like (CT-L) activities, respectively [5].
Ubiquitination is a dynamic reversible process due to a number of deubiquitinases (DUBs) that catalyze the removal of ubiquitin from the substrate protein (Fig. 1). The human genome encodes approximately one hundred of DUBs which can be divided into six subfamilies [6]. Five of them belong to cysteine DUBs: ubiquitin specifc proteases (USP), ubiquitin C-terminal hydrolase (UCH), Machado-Josephin domain-containing proteases (MJD), ovarian tumor proteases (OTU), and monocyte chemotactic protein-induced protein (MCPIP). The sixth subfamily is the JAB1/MPN/Mov34 metalloenzyme (JAMMs), which are zinc metalloproteases. There are three DUBs associated with the 19S proteasome: USP14/Ubp6, UCHL5/UCH37 and Rpn11/POH1. While Rpn11/POH1 is a permanent 19S proteasomal member, both UCHL5 and USP14 are cysteine DUBs which can physically bind with two 195-base components,ubiquitin receptors Rpn13 and Rpn1, respectivley [7,8].
Targeting protein degradation has been demonstrated as a novel strategy for cancer therapy. Several different components of the UPS have been validated as effective anticancer targets. The first-in-class proteasome inhibitor bortezomib (BTZ) is a dipeptidyl boronic acid that reversibly inhibits the β5 catalytic subunits of the 20S proteasome. BTZ was clinically approved as a therapeutic agent for multiple myeloma (MM) and mantle cell lymphoma (MCL) by the US Food and Drug Administration (FDA) in 2003 and in 2006, respectively [9,10]. In addition, BTZ has shown significant anticancer activity in plasma cell dyscrasias and non-Hodgkin lymphomas, and BTZ has substantial activity in combination with multiple clinically used drugs [11-16]. In addition to BTZ, several second-generation proteasome inhibitors have also been approved by the US FDA, including carfilzomib (CFZ), an irreversible inhibitor, and ixazomib (IXZ), an orally administered reversible inhibitor [17]. The cancer cell selectivity of proteasome inhibition was thought to be due to higher amounts of oncoproteins or aberrant proteins in malignant cells that make the tumor cells more sensitive to the UPS inhibition than normal cells [18,19]. Proteasome inhibition will lead to the buildup of polyubiquitinated proteins in the cancer cells resulting in cell cycle arrest and cell death induction. BTZ-based therapies have become a staple for the treatment of MM; as a result, the survival rate of MM patients has improved significantly since clinical introduction of BTZ and other immunomodulatory drugs. However, BTZ has some shortcomings, such as drug resistance and various toxic side effects [20]. Nevertheless, the clinical success of 20S proteasome inhibitors suggests that the UPS could be considered as a potential target for the development of more specific and potent anticancer agents.
DUBs are an important part of the UPS, and majority of cancer cells have altered expression of DUBs which might act as oncogenes or tumor suppressors or regulators of various essential cancer-associated pathways. Consequently, a number of DUBs are emerging as attractive targets for cancer therapies [21]. Of the three DUBs associated with the 19S proteasome, Rpn11/ POH1 has been suggested as a potential target for drug discovery due to its absolute requirement for cancer cell viability [22]. Overexpression of Rpn11 in mammalian cells can induce the resistance to several cytotoxic drugs [23]. Rpn11 is frequently overexpressed in MM cells and the extent of Rpn11 expression is inversely correlated with the overall patient survival [24]. Knockdown of Rpn11 produces a phenotype similar to that produced by 20S proteasome inhibition, including decreased 20S proteasome activity and buildup of high-molecular-weight ubiquitinated proteins [22]. In addition to Rpn11/POH1, the other two proteasomal DUBs UCHL5 and USP14 have also been shown to be dysregulated in MM cells [25]. UCHL5 can activate the TGF-β signaling through deubiquitinating and stabilizing Smads as well as TGF-β receptor I (TGF-β R1) [26]. UCHL5 was also found to regulate the ubiquitination of an essential RNA splicing factor PRP19, thus promoting the cell migration and invasion of hepatocellular carcinoma (HCC) cells [27]. These observations support the hypothesis that inhibitors of 19S proteasomal DUBs could have potent anticancer properties and potential to treat BTZ-resistant cancer cells [25,28-30].
1.2. Ubiquitin-like systems
Ubiquitin-like proteins (UBLs) are defined as protein families that share structural and evolutionary relationships with ubiquitin as well as the ability to be conjugated to substrates through the concerted action of evolutionarily related E1s, E2s, and E3s. UBLs are conjugated to target proteins or lipids to regulate their activity, stability, subcellular localization, or macromolecular interactions [31].
Several UBLs have been identified, including SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15, all of which share a similar three-dimensional structure to ubiquitin [32]. Most UBLs are conjugated to substrate proteins via an enzymatic cascade that resembles ubiquitylation, but have their own unique enzyme cascades and specific cellular functions [33,34]. The most well characterized UBLs are SUMO and NEDD8. SUMO can regulate most of the nuclear processes including ribosome maturation, DNA repair, transcription and splicing [35]. Many knockouts of SUMO-activating, -conjugating or -deconjugating enzymes in eukaryotic models gave lethal phenotypes, highlighting the essential role of SUMO signal transduction [36]. NEDD8 controls the stability and function of RING-Cullin E3 ligases, thereby regulating the turnover of a subset of substrates involved in cellular processes associated with cancer cell pathways [37].
1.3. Reposition of old drugs and natural compounds
Drug repositioning represents an attractive drug discovery strategy compared with the traditional de novo drug discovery approach, which is a time-consuming and costly process. Drug repositioning refers to the identification of new indications for already approved drugs and the application of the newly identified drugs to the treatment of a different pathological condition from the drug’s intended disease. For some old drugs, their preclinical data and safety information for original indications are known, which enables them to be developed via a cheaper and faster cycle and more efficiently translated from bench to bedside [38]. Drugs, which target the complex diseases (such as cancer) with multiple targets offer a great opportunity to reposition [39]. Drug combination can also be considered as another form of drug repositioning.
Recently, high-throughput screening (HTS) assays have been widely used to identify old drugs/agents or nature compounds as new candidate UPS inhibitors, another type of drug repurposing. HTS allows the testing of large numbers of compounds for their potential inhibitory activity to the UPS. For example, HTS assays can be applied to measure the catalytic activity in a purified or reconstituted enzyme/complex, cell extracts or intact cells [40]. As a result of HTS application, many tool compounds have been identified that have significantly enhanced basic scientific research and identification of lead compounds to be used as repurposed UPS inhibitors (see below and [41]). Application of these technologies has also provided a unique opportunity to re-establish some natural products as new candidate UPS inhibitors.
2. Repurposed drugs as 20S proteasome inhibitors
2.1. Clioquinol and analogs
Through screening National Cancer Institute (NCI)-Diversity Set Library, NSC-109268, an organic copper (Cu) compound (Table 1), was found able to inhibit the purified 20S and the 26S proteasome activities in human prostate cancer cell model [42]. Based on the chemical structure similarity to NSC-109268, the Cu-binding hydroxyquinoline (HQ) family members 8-hydroxyquinol (8-HQ) and clioquinol (5-chloro-7-iodo-8-HQ, CQ) (Table 1) were identified as novel 20S proteasome inhibitors [42].
Table 1.
Repurposed 20S proteasome inhibitors.
| Compounds | Chemical Structure | Proteasome Inhibitory activity | Ref. | |
|---|---|---|---|---|
| HQ | NSC-109268 |  |
Inhibition of the CT-L activity of purified 20S rabbit proteasome (IC50 = 6 μM) | [42] |
| Clioquinol (CQ) |  |
CQ/Cu complex: Inhibition of the CT-L activity of purified 20S proteasome (IC50 = 2.5 μM); ~80% inhibition of proteasomal CT-L activity in prostate cancer cells at 20 μM | [49] | |
| 8-Hydroxyquinoline (8-HQ) |  |
8-HQ/Cu complex: 60 % inhibition of the CT-L activity of purified 20S rabbit proteasome at 10 μM; ~67 % inhibition of proteasomal CT-L activity in DCIS cells. | [42,61] | |
| 5-amino-8-hydroxyquinoline (5AHQ) |  |
Inhibition of the CT-L activity of 20S proteasome with IC50 ranging from 0.30 to 0.51 μM, no significant difference with or without Cu; Inhibition of the purified rabbit proteasome activity with a mean Ki of 2.1 μM (95% CI = 2.08 to 2.33 μM) | [63,64] | |
| 2-amino-8-hydroxyquinoline (2AHQ) |  |
Inhibition of the CT-L activity of 20S proteasome activity with IC50 ranging from 0.30 to 0.51 μM, no significant difference with or without Cu. | [63] | |
| 5-aminomethyl-8-hydroquinoline (AMHQ) |  |
Inhibition of the CT-L activity of 20S proteasome activity with IC50 ranging from 0.30 to 0.51 μM, no significant difference with or without Cu | [63] | |
| 5-chloro-8-hydroxyquinoline (ClHQ) |  |
Inhibition of the CT-L activity of 20S proteasome activity with IC50 about 5 μM, enhanced by Cu | [63] | |
| 5-chloromethyl-8-hydroxyquinoline (ClMHQ) |  |
Inhibition of the CT-L activity of 20S proteasome activity with Cu (IC50 =1.3 μM) | [63] | |
| HIV-PIs | Ritonavir |  |
Inhibition on proteasome activity in mouse model; Inhibition of the CT-L activity of the purified 20S proteasome | [68,69] |
| Saquinavir |  |
Inhibition of the CT-L and PGPH-L activities of the 26S proteasome and purified 20S proteasome | [71] | |
| Nelfinavir |  |
Pan-proteasome inhibition in AMO-1 and U266 myeloma cells; 60 % inhibition of the CT-L activity of 26S proteasome at 5 μM; Inhibition the CT-L activity of the 26S proteasome of CD138-selected plasma cells from MM patients | [72,73] | |
| Indinavir |  |
Inhibition of the CT-L and T-L activities of human 26S proteasome, but not PGPH-L activity of the 26S proteasome and the three peptidase activities of the 20S proteasome | [74] | |
| Tipranavir |  |
Inhibition of the CT-L activity of 26S proteasome | [75] | |
| Lithium | LiCl | Inhibition of the CT-L and PGPH-L activities of both 20S and 26S proteasome | [91] |
CQ has been used for the treatment of neurodegerative disease [43-45]. It acts as a chelator or ionophore of divalent metals, such as Cu and zinc (Zn). X-ray crystallography and X-ray absorption spectroscopy shows that CQ forms complex with Cu through coordination of two CQ molecules to one Cu(II) [46,47]. Further study revealed that CQ could chelate at least some of the Cu from Cu chaperone proteins and oxidizes Cu forming a CQ-Cu(II) complex in the tumors [48].
The complex of CQ and Cu (CQ/Cu) has potent inhibitory effect on the proteasomal activity. It inhibits the purified 20S and the intracellular 26S proteasome activities, leading suppression of cell proliferation and induction of apoptotic cell death in human prostate cancer cells and xenografts [49]. As a Cu chelator, CQ exerts selective anticancer activity toward breast cancer, leukemia, and myeloma with less effect on normal cells [50,51]. Besides acting as the Cu chelator, CQ also works as Zn ionophore [52]. CQ can transport zinc across the plasma membrane [53] and release Zn into the lysosome [54], thereby leading to lysosomal disruption and cell death [53,55]. As cancer cells accumulate more Cu than the normal tissues, it’s reasonable to design a strategy targeting intratumor Cu using CQ to achieve selective anticancer efficacy. However, results from a phase I clinical trial showed that CQ exerted minimal inhibition of the proteasome, and no clinical responses were observed in patients with advanced hematologic malignancies [56]. The possible cause could be the low cellular concentration of CQ in tumor cells. Indeed, one patient with the highest intracellular levels of CQ had inhibition of the proteasome, consistent with the assumption that low cellular CQ was unable to generate biological and clinical effects. Thus it should be taken into consideration, when one plans a future clinical anticancer trial, of improving CQ delivery, increasing CQ cellular levels, and/ or combining CQ with Cu.
8-HQ (Table 1) is an organic compound that is structurally related to CQ. Similar to CQ, 8-HQ and its derivatives can chelate with divalent metal ions, which confers most of their bioactivities [42,57-60]. The anticancer activity of 8-HQ and its derivatives relies on Cu, and they fail to induce cell death if they lose Cu-binding abilities [61]. 8-HQ can inhibit the CT-L activity of purified 20S proteasome in a Cu-dependent manner. The inhibition of CT-L proteasome activity and apoptotic induction by 8-HQ/Cu was not from Cu-mediated oxidative damage to proteins, but from the formation of a proteasome inhibitor inside the tumor cells [42,61]. Based on that finding, several Cu-8-HQ derivatives complexes were developed for cancer treatment, including CuHQTS (Cu 8-HQ-2-carboxaldehyde-thiosemicarbazide) and CuHQDMTS (Cu 8-HQ-2-carboxal dehyde-4,4-dimethyl-3-thiosemicarbazide), two Cu/thiosemicarbazone complexes that showed potent anticancer activity against cisplatin-resistant neuroblastoma cells [62]. In addition, amino-HQ and chloro-HQ derivatives (Table 1) could also inhibit the CT-L activity of the proteasome and induce growth inhibition and apoptosis [63]. Some of the derivatives are FDA-approved drugs, for example, nitroxoline (5-nitro-8-HQ, or 5AHQ; Table 1) is a clinical used urinary antibacterial agent that has a more potent anticancer activity than CQ and 8-HQ [60]. It has been demonstrated that 5AHQ can bind with the α subunits of the 20S proteasome at non-catalytic sites, which results in inhibition of the proteasome and preferable induction of cell death in primary myeloma and leukemia cells over normal hematopoietic cells [64]. Thus, 8-HQ and its derivatives can act as potent 20S proteasome inhibitors with potential anticancer application for various types of cancer.
2.2. Human immunodeficiency virus (HIV)-Protease inhibitors
Since 1996, nine HIV protease inhibitors (HIV-PIs) (ritonavir, saquinavir, nelfinavir, amprenavir, atazanavir, indinavir, tipranavir, darunavir and lopinavir) have been approved by the U.S. FDA and widely used for the treatment of HIV infection [65]. The possible use of HIV-PI as a cancer treatment primarily originated from their success in treating HIV-related Kaposi's sarcoma [66]. As the HIV-I virus coded proteases have similar cleavage activities to the proteasome [67], researchers were interested in knowing whether these HIV-PI could inhibit proteasomes, thus considering to expand their medical implications for the treatment of cancers that are not related to HIV.
Of the nine HIV-PI, five of them are shown to inhibit the proteasome (Table 1). Ritonavir and saquinavir can inhibit the purified 20S proteasome [68-71]. They prefer to inhibit one or two proteasomal proteolytic activities other than the T-L activity [68-71]. The remaining three HIV-PIs can only inhibit the intracellular proteasome. Nelfinavir, is a pan-inhibitor of the 26S proteasome [72], which is a unique property among the HIV-PIs with proteasome inhibitory effects. Also, nelfinavir inhibits the CT-L activity of the 26S proteasome in CD138-selected tumor plasma cells from MM patients [73], while indinavir and tipranavir inhibit CT/T-L and CT-L activities of human 26S proteasome, respectively [74,75].
Similar to specific 20S proteasome inhibitors, inhibition of CT-L activity of proteasome by HIV-PIs leads to accumulation of ubiquinated proteins, inhibition of the NF-κB and induction of ER stress, eventually resulting in growth inhibition and apoptosis in various cancers [70,76-79]. It is well known that upregulation of the unfolded protein response (UPR) by proteasome inhibitors is needed for their anticancer activities and that UPR downregulation is responsible, at least partially for proteasome inhibitor resistance in MM [80]. Ritonavir at clinically relevant concentrations was able to significantly induce UPR; ritonavir-induced UPR can further enhance BTZ’s anticancer effect, resulting in increased levels of cancer cell apoptosis [81,82]. The combination of ritonavir with a second generation proteasome inhibitor such as delanzomib or IXZ also showed synergistical effects [83,84]. These data suggest that HIV-PIs could overcome tumor cell resistance to BTZ or 2nd generation proteasome inhibitors.
The efficacy of HIV-PIs against cancer in preclinical studies has pushed their translation into clinics. Most of the clinical trials are focused on nelfinavir and ritonavir. Nelfinavir can inhibit the intracellular β2 proteasome activity that is not targeted by BTZ or CFZ [72], thus it has been used to overcome 20S proteasome inhibitor resistance in clinical trials. A Phase I trial of nelfinavir-bortezomib-dexamethasone (NeVd) combination from Swiss Group showed promising activity in patients with heavily pretreated, BTZ-lenalidomide doublerefractory MM [85]. Furthermore, NeVd activity was observed in MM refractory to BTZ and more than one immunomodulatory drug in multicenter participated phase II trial, and 65 % objective response rate (ORR) was obtained in this heavily pretreated cohort, similar to that observed with first-line Vd treatment in BTZ-naïive patients [86].
A phase I trial of nelfinavir in combination with chemoradiotherapy for locally advanced pancreatic cancer showed that nelfinavir in combination with radiation and chemotherapeutics (gemcitabine and cisplatin) was well tolerated. Partial chemotherapy responses were observed in 5 of 10 patients who completed chemoradiotherapy, and minor responses were observed in 2 of 10 patients [87], indicating that it can increase the chemoradiotherapy’s effects in pancreatic ductal adenocarcinoma. A phase I trial of nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer (NSCLC) showed that nelfinavir had a positive effect in NSCLC [88].
Most of the completed trials involving ritonavir were set up for HIV-related Kaposi's sarcoma, but the results are currently not available. In a phase II trial, ritonavir/lopinavir was tested in 19 patients with progressive or recurrent high-grade gliomas. This study did not meet its primary efficacy endpoint [89]. Promising antitumour activity was observed using ritonavir in combination with escalating dose of docetaxel (ModraDoc001/r or ModraDoc006/r) for the treatment of solid tumors [90]. Encouraged by this promising result, the same regimen but with variable doses of docetaxel (ModraDoc001-006/r) is currently under investigation in seven prostate and breast cancer clinical trials.
2.3. Lithium
Lithium (Table 1) has been successfully used for the treatment of depression for over 60 years. It has been reinvestigated as an anticancer agent mainly due to its inhibition on GSK3. It should be noted that lithium also has GSK3-independent activities. In addition, it has been found that lithium chloride (LiCl) specifically inhibits the CT-L and PGPH-L activities of both the 20S and 26S proteasome [91,92]. Inhibition of proteasome activity was also observed in LiCl-treated cells prior or post purification, suggesting that LiCl may affect the proteasome structure irreversibly [91]. It possibly occupied the binding site of magnessium, which is required for the activation of CT-L and PGPH-L activities [92]. The effect of LiCl on proteasome enzymatic activity led to stabilization of proteasome substrates such as the CCAAT/enhancer-binding protein alpha (C/EBPα) and the retinoic acid receptor, and potentiating retinoid-induced cell differentiation [91,93].
It has been found that the inactivation rates for the two proteasomal subunits (β5/CT and β1/PGPH) by LiCl are slow, which limits its application as a cancer cell death inducer like BTZ [92]. Clinical studies indicate that LiCl at doses used for bipolar disorder (BD) and depression did not show any anti-tumor effects in low-grade neuroendocrine tumors [94]. However, it can reduce the overall cancer risk in patients with BD [95]. Results from a randomized phase III study using lithium gamolenate in patients with advanced pancreatic adenocarcinoma showed that a low-dose treatment led to survival times similar to those of other treatments, while a high-dose treatment appeared to have an adverse effect. Thus lithium was not recommended for the treatment of advanced pancreatic cancer [96]. However, the lithium salt of gamma-linolenic acid was shown to prolong survival of inoperable pancreatic cancer patients [97].
Since lithium is not a potent proteasome inhibitor and apoptosis inducer, it can not be used as a solo anticancer agent. Consistently, there are some clinical trials currently to investigate lithium’s combinations with clinically used chemotherapeutics in various cancers.
3. Repurposed drugs as 19S-DUB inhibitors
3.1. Old drugs containing α-β-unsaturated ketone as UCHL5/USP14 inhibitors
3.1.1. WP1130
As a repurposed inhibitor of proteasomal cysteine DUBs, UCHL5 and USP14, WP1130 (Degrasyn, Table 2) has an α,β-unsaturated ketone, which could covalently interact with the cysteine sites of these DUBs through the Michael addition. Indeed, it has been found that WP1130 inhibits multiple cysteine DUBs, including proteasomal DUBs (UCHL5 and USP14), USP5, and USP9x [98]. WP1130 can induce proteasomal degradation of the anti-apoptotic protein Mcl-1, which is a substrate of USP9x [99]. In addition, WP1130 has been reported to regulate the deubiquitination of a subset of kinases, such as Bcr-Abl, Janus-activated kinase 2 (Jak2), and unc-51 like autophagy activating kinase 1 (ULK1), thus regulating the signal transduction pathways of these kinases [100-102]. WP1130 was also shown to enhance cytotoxicity of chemotherapeutic drugs such as BTZ, doxorubicin gemcitabine, cisplatin, and spindle poisons in several types of tumor cells [103-106].
Table 2.
Summary of repurposed 19S-deubiquitinating enzyme inhibitors.
| Compounds | Targets | Chemical Structure | DUB Inhibitory activity | Ref. | |
|---|---|---|---|---|---|
| UCHL5i/USP14i | WP1130 | UCHL5, USP14, USP5, USP9x | 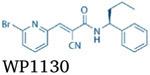 |
WP1130 inhibits the labeling of UCHL5, USP14, USP5 and USP9x in lysates by ≥80 % at 5 μM. | [98] |
| b-AP15/ RA-9/ VLX1570 | UCHL5, USP14 |  |
b-AP15 inhibits the proteasomal DUB activity in vitro at 50 μm. RA-9 inhibits the proteasomal DUB activity in vitro at 20 μM. VLX1570 inhibits proteasomal DUB activity in vitro with IC50 of 10 μM. | [108,113,115] | |
| Curcumin/ AC17 | UCHL5 USP14 |  |
AC17 inhibits the proteasomal DUB activity in vitro with IC50 of 4 μM. | [118,123] | |
| Gambogic acid/dihydrogambogic acid | UCHL5, USP14, 20S |  |
Gambogic acid inhibits the proteasomal DUB activity in vitro at 2 μM and inhibits the CT-like activity of 20 proteasome in vitro with IC50 of 0.5 μM. | [125,126] | |
| Metal-PT (Cu, Zn, Pt, and Ni) | UCHL5, USP14 |  |
Metal-PT inhibits the proteasomal DUB activity in vitro at ~0.5 μM. | [130,136,137,139] | |
| Gold compound (Auranofin, Au(PPh3)PT) | UCHL5, USP14 |  |
Auranofin inhibits the proteasomal DUB activity in vitro at 2 μm. Au(PPh3)PT inhibits the proteasomal DUB activity in vitro at 0.25 μM. | [151, 154] | |
| UCHL5i | RA190 | UCHL5 |  |
RA190 inhibits deconjugation of K48-linked tetraubiquitin by UCHL5 in vitro at 1 μM. | [157] |
| Isothiocyanates | UCHL5, USP9X, USP1 |  |
BITC and PEITC inhibit the labeling of UCHL5 in lysates with IC50 of 22 μM and 13 μM, respectively. | [168] | |
| USP14i | IU1/IU1-248 | USP14 |  |
IU1 and IU1-248 inhibit the USP14 activity in vitro with IC50 of 12.25 and 0.83 μM, respectively. | [170,177] |
| Rpn11i | Capzimin/8TQ | Rpn11 |  |
Capzimin and 8TQ inhibit the Rpn11 activity in vitro with IC50 of 0.34 and 2.4 μM, respectively. | [178] |
| Thiolutin | Rpn11 |  |
Thiolutin inhibits the Rpn11 activity in vitro with IC50 of 0.53 μm. | [187] | |
| ETPs | Rpn11 |  |
SOP11 inhibits the Rpn11 activity in vitro with IC50 of 1.3 μM. | [195] |
3.1.2. b-AP15/RA-9/VLX1570
As another example of drug reposition for 19S-DUB inhibition, b-AP15 (Table 2) has been identified as a specific UCHL5 and USP14 inhibitor through screening a library of potential anticancer drugs that induce the lysosomal membrane permeabilization-mediated apoptosis [107]. b-AP15 induced apoptosis that was independent of p53 and Bcl2 status and displayed antitumor activity in both non-solid and solid tumor models [107-109]. b-AP15 induced accumulation of polyubiquitinated proteins and overcame resistance to 20S proteasome inhibitors in MM cells [109]. Moreover, b-AP15 induced strong apoptotic response through induction of endoplasmic reticulum (ER) stress and production of oxidative stress [110]. b-AP15 also caused atypical cell death by induction of severe mitochondrial damage and proteotoxic stress and inhibition of protein translocation over the ER [111,112].
RA-9 (Table 2), possessing a structure similar to b-AP15, is an inhibitor of UCHL5 and USP14 [113]. However, RA-9 also targets other DUBs such as UCHL1, UCHL3, USP2, USP5, and USP8 [114].
VLX1570 (Table 2) is another chalcone derivative of b-AP15 and prefers to inhibit USP14 over UCHL5. VLX1570 showed increased biological activity and specificity compared to b-AP15, and was advanced to a clinical trial for relapsed MM (now discontinued) [115]. Due to the highly expression of USP14, MM cells were particularly sensitive to VLX1570 treatment [116].
3.1.3. Curcumin/AC17
Curcumin (Table 2) is a natural compound extracted from the golden spice turmeric (Curcuma longa) that has a wide range of beneficial properties, including antitumor, antioxidant, anti-inflammatory, and antiviral [117]. It contains two electrophilic α,β-unsaturated carbonyl groups, which can react with cysteine group of DUBs. Curcumin induces p53-independent cell death partly due to the inhibition of DUBs [118]. Curcumin can also directly inhibit the activity of 20S proteasome, a result from its carbonyl carbons directly interacting with the hydroxyl group of the amino-terminal threonine residue of the 20S proteasomal CT-L subunit [119,120]. Several clinical trials have been carried out to evaluate the beneficial effects of curcumin on multiple types of cancer. It has been confirmed that curcumin can provide symptomatic relief, as well as other parameters of various cancer conditions, including MM, lung, breast, colon, colorectal and prostate cancer [121].
AC17 (Table 2), a 4-arylidene curcumin analog, exhibits improved antitumor activity over curcumin and is initially identified as an inhibitor of IKK (Inhibitory κB Kinase) [122]. AC17 can block the 19S proteasomal DUBs without inhibiting the proteolytic activities of 20S proteasome, resulting in NF-κB inhibition and p53 reactivation. AC17 could be potentially developed into an anticancer agent [123].
3.1.4. Gambogic acid
Gambogic acid (GA) (Table 2) is the principal pigment of gamboge resin of several Garcinia species. The α,β-unsaturated ketone of GA has been found to be essential for growth inhibition and apoptosis induction in cancer cell lines [124]. It was shown that GA and dihydrogambogic acid (Table 2) can inhibit proteasomal DUB activity [125]. GA can also inhibit 20S proteasome function after being metabolized by intracellular CYP2E1, and the C9-C10 double bond of GA is responsible for the proteasome-inhibitory function [126]. Importantly, cancer cell lines have been shown to be more sensitive to GA treatment than normal cell lines [127]. In both animal tumor models and clinical trials, GA shows a favorable safety profile, with minimal side effects [128].
3.2. Metal-based old drugs as UCHL5/USP14 inhibitors
3.2.1. Metal-Pyrithione (M-PT)
Pyrithione (PT) has excellent metal chelating properties that complex with various metals including copper, zinc, platinum, and nickel (Table 2). PT binds with copper which enhances its apoptosis-inducing ability. The pyrithione copper complex, CuPT, has been used as an antifouling ingredient [129]. It was reported that CuPT inhibited the UPS by mainly targeting proteasomal DUBs, UCHL5 and USP14 [130]. Results of silicon docking and active site labeling experiments confirmed that CuPT interacts with these two cysteine DUBs [130]. The pyrithione zinc complex, ZnPT, has been widely used as a booster biocide in antifouling paints and antidandruff shampoos [131-133], and has been shown a significant anticancer potential [134,135]. ZnPT could also target the proteasomal DUBs UCHL5 and USP14, without inhibiting the activities of 20S proteasome [136]. ZnPT efficiently induced apoptosis in cancer cells and significantly suppresses tumor growth in nude mouse xenografts.
Like CuPT and ZnPT, other metal-PT complexes, such as platinum pyrithione (PtPT) and nickel pyrithione (NiPT), can also target the proteasomal DUBs. PtPT dose not bind with DNA, distinct from clinically used platinum-containing drug cisplatin [137]. Cisplatin is highly reactive towards DNA as it has two non-leaving groups (NH3) and two fast-rate leaving groups (Cl) connecting the platinum ion [138]. However, the PT structure in PtPT or NiPT contains two medium-rate leaving groups (S and O) that allows PtPT to react with protein targets such as proteasomal cysteine DUBs over the nuclear DNA. It was found that PtPT and NiPT exhibited better anticancer activities than cisplatin in multiple cancer cell lines and overcome cisplatin resistance in NSCLC cells [137,139]. Both PtPT and NiPT remarkably inhibited tumor growth in nude mice xenografts, without showing any toxic side effects [137,139,140]. Future work should focus on the comprehensive evaluation of the safety of various PT-metal complexes in vivo.
3.2.2. Gold complexes
As early as ancient Chinese days, gold has been used for medicinal purposes towards skin ulcers, infections and inflammations [141]. Moreover, gold complexes were described to exhibit antitubercular activity [142] in 1890 and discovered to be rheumatoid arthritis (RA)-therapeutic in 1929 [143]. Several studies have shown that gold(III) compounds were able to target purified 20S proteasome and 26S proteasome in human breast cancer cells [144-146]. Auranofin (AF, Table 2) is a gold(I)-containing compound approved by the FDA in 1985 for the treatment of rheumatoid arthritis [147]. In recent years, the antitumor activity of AF has been studied by many investigators [148-150], and clinical trials were approved to evaluate the anticancer activity of AF and sirolimus (also called rapamycin, an antifungal agent and an mTOR inhibitor) in ovarian and lung cancer ( NCT03043001; NCT01737502). In 2014, it was reported that AF was a specific inhibitor of the proteasomal DUBs USP14 and UCHL5 [151]. Moreover, in another study, AF showed a potent antitumor activity against imatinib-sensitive and -resistant chronic myeloid leukemia (CML) cells in vitro and in vivo [152]. Notably, DUB inhibition but not ROS induction is required for AF-induced caspase activation and apoptosis [150,152,153]. Recently, it was reported that a new gold(I) complex, Au(PPh3)PT (Table 2), could also suppress tumor growth in vitro and in vivo, and induce apoptosis in cancer cells from acute myeloid leukemia patients [154]. Mechanistically, Au(PPh3)PT impaired UPS function by selectively inhibiting proteasomal DUBs UCHL5 and USP14 as well as other non-proteasomal DUBs, but with minimal effects on the function of 20S proteasome. Importantly, Au(PPh3)PT elicited less cytotoxicity in normal cells than several other metal compounds [154]. In conclusion, repurposing of AF and other gold complexes as effective DUB inhibitors may bring new strategies for cancer treatment.
3.3. UCHL5 inhibitors
3.3.1. RA190
Rpn13 regulates substrate deubiquitinaton by recruiting UCHL5 and activating UCHL5 catalytic activity [155]. Recently, RA190 (Table 2), a bis-benzylidine piperidone compound, was shown to covalently bind to cysteine 88 of Rpn13 in the 19S proteasome, resulting in accumulation of higher-molecular weight polyubiquitylated proteins [156]. The authors also found that the degradation of Ub-AMC by either purified UCHL5 enzyme or purified 19S proteasome was minimally affected by RA190 [156]. More recently, they performed an in vitro assay to directly probe the effects of RA190 on deconjugation of K48-linked tetraubiquitin by UCHL5, and found that RA190 could directly inhibit UCHL5 activity [157]. By using mass spectrometry, RA190 was found to directly react with several cysteines of UCHL5 [157]. Thus, RA190, as a repurposed inhibitor, can inhibit both Rpn13 and UCHL5 through parallel mechanisms. RA190 exerted antitumor effect through induction of caspase-dependent apoptosis and UPR. Moreover, RA190 was found to act synergistically with BTZ, lenalidomide, or pomalidomide against MM [158]. Importantly, RA190 administration retarded growth of MM tumors and ovarian cancer xenografts in mouse models, without affecting body weight or compromising spontaneous antitumor immunity [156]. These findings provide the rationale for novel therapeutics targeting Rpn13 or UCHL5 to treat cancer.
3.3.2. Isothiocyanates (ITCs)
Isothiocyanates (ITCs) (Table 2) are small molecules derived from glucosinolates in edible cruciferous vegetables like broccoli and watercress. The most commonly used ITCs include allyl ITC (AITC), benzyl ITC (BITC), 4-methylthiobutyl-ITC (erucin), sulforaphane (SFN) and phenethyl-ITC (PEITC) [159]. Several clinical trials of ITCs have been approved by the FDA for therapy of malignancies including breast, lung, gastrointestinal tract and prostate cancer [160-162]. Considerable evidence suggests that the possible molecular mechanisms of action by ITCs include: cell cycle arrest, induction of autophagy and apoptosis, and inhibition of inflammatory response [160,161,163-165]. SFN was reported to induce expression of several DUB genes that might be involved in Nrf2-dependent detoxification and antioxidant response [166]. BITC and PEITC have been reported to be strong proteasome inhibitors that suppress the growth of MM cells [167]. Moreover, BITC and PEITC were shown to inhibit DUBs USP9X and UCHL5 through the adduct formation with their active site cysteine residues [168]. USP9X is able to mediate Mcl-1 deubiquitination, and consistently Mcl-1-dependent cancer cells were found to be more sensitive to ITCs than other cells. Using SILAC assisted quantitative mass spectrometry, several other PEITC-targeted DUBs (such as USP1) were identified [169]. PEITC impaired USP1 activity, and thus increased the ubiquitination of PCNA, which functions as an adaptor for recruiting multiple factors to the site of DNA repair [169]. Hence, USP1 inhibition could explain the ability of ITCs to enhance cancer cell sensitivity to the DNA damage inducer cisplatin [169]. Thus, repositioning of ITCs as new potent and selective DUB inhibitors may facilitate their use in clinics.
3.4. USP14 inhibitors
IU1 (Table 2) was identified as a repurposed USP14 inhibitor by HTS from 63,052 compounds using Ub-AMC hydrolysis assay of VS-proteasomes reconstituted with USP14 [170]. IU1 enhanced degradation of proteasomal substrate tau or TDP-43 in murine embryonic fibroblasts (MEFs), and inhibition of USP14 was proposed as a therapeutic strategy for neurodegenerative diseases characterized by tau or TDP-43 aggregation [170]. However, IU1 failed to enhance proteasomal degradation of tau in neurons [171]. In addition, whether IU1 could improve degradation of tau proteins in vivo is still unknown. USP14 inhibition by IU1 induced cell cycle arrest in androgen receptor (AR)-positive prostate and breast cancer cells through inducing the degradation of AR proteins [172,173]. IU1 also enhanced the antiumor effect of enzalutamide through cell growth inhibition and apoptosis induction [174]. USP14 can be phosphorylated and activated by the protein kinase Akt, thus negatively regulating autophagy by removing K63-linked ubiquitination from Beclin 1 [175]. Therefore, USP14 may modulate both UPS and autophagy through regulating K48 and K63 ubiquitination, respectively. The co-crystal structures of USP14 with IU1 showed that IU1 can bind to the thumb-palm cleft region of the USP14 catalytic domain, resulting in steric blockade of the binding of the ubiquitin C-terminus to USP14 [176,177]. Although an IU1 derivative, IU1-248 exhibited a 10-fold higher potency than IU1 under in vitro conditions, there was no in vivo information available about IU1-248 [177].
3.5. Rpn11 inhibitors
3.5.1. Quinoline-8-thiol (8TQ)/Capzimin
Using a fluorescence polarization assay that specifically measures the DUB activity of Rpn11, Li et al. screened hundreds of metal-binding pharmacophores, which yielded quinoline-8-thiol (8TQ) (Table 2) as a Rpn11 inhibitor [178]. They also used HTS assay to screen 330,000 small molecules and identified one compound S-(quinolin-8-yl)-2-bromobenzothioate, a thioester derivative of 8TQ, as a potent Rpn11 inhibitor [178]. These two screenings reveal that 8TQ can serve as a parent scaffold for the further optimization of selective Rpn11 inhibitors. This was consistent with another report in which use of a fragment-based drug discovery approach lead to identification of 8TQ with high affinity for Rpn11 [179]. Inhibition of Rpn11 activity by 8TQ could be through binding to the catalytic Zn2+ ion in the active site of Rpn11. Next, structure-activity relationships (SAR) were explored to improve the potency of 8TQ. A novel compound capzimin (Table 2) showed a marked selectivity for Rpn11 [178]. The computational analysis of binding modes and selectivities of capzimin showed that it binds to the active site Zn2+ of Rpn11 and this interaction extends to the distal ubiquitin binding site [180]. Given that quinoline scaffold is prevalent in several clinical drugs, capzimin may act as a promising proteasome inhibitor for cancer therapy.
3.5.2. Thiolutin
Dithiolopyrrolones (DTPs) are a class of disulfide-containing natural products, which are synthesized by actinomycetes and proteobacteria [181]. The DTP thiolutin (Table 2) was originally thought to inhibit bacterial and fungal RNA polymerases, but no direct data support this hypothesis [182-184]. Thiolutin is also known to inhibit tumor angiogenesis [185]. Moreover, a quantitative mass spectrometric analysis showed that thiolutins stimulated Hsp27 phosphorylation in endothelial cells [186]. The effect of thiolutin on removal of ubiquitin by the proteasomal Rpn11 was tested and Rpn11 was found to be the direct target of thiolutin [187]. Thiolutin inhibits Rpn11 by binding of Zn2+ [187]. Thus thiolutin is a repurposed Rpn11 inhibitor. However, its antitumor potential remains to be further investigated.
3.5.3. Epidithiodiketopiperazines
Epidithiodiketopiperazines (ETPs) (Table 2) are a large group of structurally complex fungal metabolites that exhibit potent antibiotic and antitumor activities [188-190]. Various targets have been proposed to explain the biological activity of ETPs, including histone methyltransferase, HIF-1α/p300 complex, and thioredoxin reductase [191-193]. Previous reports showed that gliotoxin, an ETP, acts as an inhibitor of 20S proteasome depending on its disulfide bond [194]. Using the UbnGST-Wbp2 substrate to monitor 26S proteasome activity, gliotoxin and its core scaffold compound, SOP11 were found to inhibit proteasome function [195]. Furthermore, it was found that SOP11 potently inhibited Rpn11 activity of the proteasome through chelating the catalytic Zn2+ ion [195]. However, further research on the pharmacological antitumor properties of the ETPs is required.
4. Repurposed drugs as ubiquitin ligase inhibitors
An alternative strategy to targeting the UPS is to target enzymes upstream (e.g., E1, E2 or E3) of the 20S proteasome. The success in developing inhibitors of NEDD8-activating enzyme (NAE), an E1-like enzyme for cancer treatment supports this thought [196]. Recently, Piperacillin 1, a semi-synthetic, beta-lactam antibiotic was found to inhibit NAE reversibly. It inhibits Ubc12-NEDD8 formation with high selectivity [197]. Piperacillin 1 has been used for the treatment of febrile and neutropenic types of cancer in combination with a beta-lactamase inhibitor [198]. The discovery of a new therapeutic target may expand its application for other types of cancers that have high levels of NAE. Largazole is another repurposed E1 inhibitor which is also acknowledged as a histone deacetylase (HDAC) inhibitor [199]. It can inhibit the formation of the ubiquitin-adenylate complex without affecting thioester bond formation between ubiquitin and E1 [200].
It has been found that UBE2N (also known as Ubc13; an E2 enzyme) and its partners UBE2V1 and UBE2V2 are highly expressed in some types of cancers such as diffuse large B-cell lymphoma and MM [201,202]. Knockdown of UBE2N induces cell death and decreases cancer cell proliferation as well as tumor growth, suggesting that UBE2N may be an attractive therapeutic target for cancer treatment [201,202]. NSC697923, a novel UBE2N inhibitor repurposed from an existing compound library [201] was shown to decrease melanoma xenograft growth [202], demonstrating anti-cancer effects. NSC697923 exploits a binding groove unique to UBE2N, resulting in covalent adduct formation at the UBE2N active site cysteine. Mutation analysis reveals that the cytotoxic effect of NSC697923 is largely due to specific UBE2N inhibition [203]. Inhibition of UBE2N by NSC697923 in neuroblastoma leads to p53 nuclear accumulation and subsequent tumor suppression. It also exerts antitumor efficacy in neuroblastoma orthotopic xenografts [204]. In addition, NSC697923 was shown to be able to overcome radiation resistance., because this compound causes degradation of hypoxia-inducible factor 1 alpha subunit (HIF1α), a key factor in radiation resistance [205].
Although all three enzymes of the ubiquitination cascade (E1, E2 and E3) are potentially highly druggable, targeting E3 ligase is likely to be much more specific. Due to lacking of clinical data of the repurposed inhibitors of E1 and E2, this section will focus on repurposed drugs as E3 inhibitors.
4.1. IMiDs as repurposed inhibitors of CRL4CRBN E3 Ligase
Thalidomide (Table 3A), an FDA-approved immunomodulatory imid drug (IMiD), was initially developed as a sedative to treat morning sickness in pregnant women, but later repurposed for the treatment of MM, owing to its antiangiogenic, immunomodulatory, and anti-inflammatory properties [206-209]. The use of thalidomide is strictly controlled due to the teratogenicity and adverse side-effects [210]. This led to the development of more potent and less toxic IMiDs, such as lenalidomide (Table 3A) and pomalidomide. All these IMiDs are approved by FDA for the treatment of MM [211]. In addition, lenalidomide has also been approved for the treatment of myelodysplastic syndrome (MDS) with 5q deletion and mantle cell lymphoma (MCL) resistant to BTZ [212,213].
Table 3.
Repurposed E3 inhibitors (3A) and DTC-based proteasome inhibitors (3B).
| Compounds | Targets | Chemical Structure | Inhibitory activity | Ref. | |
|---|---|---|---|---|---|
| 3A | Thalidomide | CRBN |  |
Stabilizes CRBN by inhibiting its autoubiquitination through direct binding, inducing CRBN-mediated ubiquitination and degradation of target proteins | [214] |
| Lenalidomide | CRBN |  |
Same as Thalidomide | [217] | |
| ARV-825 | CRBN, BRD4 | Promotes the CRBN-dependent degradation of BRD4 through dual interactions with both molecules. | [222] | ||
| Lovastatin | Skp2 |  |
Decreases the expression of Skp2 and results in the inhibition of Skp2-mediated ubiquitination and degradation of p27 and p21, leading to cell cycle arrest and apoptosis | [231,232] | |
| Clomipramine | ITCH |  |
Interferes with the HECT Ub transthiolation activity of ITCH in an irreversible manner | [250] | |
| JS-K | Mdm2 |  |
Inhibits Mdm2 E3 ultimately accumulating p53 concentrations leading to inhibition of the ubiquitin proteasome pathway; Also, able to generate DNA damage and increase p53 concentrations | [264] | |
| Suramin | CRL |  |
Targets CRL-mediated K48 polyubiquitination and inhibits Cullin-RING E3 ubiquitin ligase | [267] | |
| 3B | DSF | 20S |  |
DSF-Cu complex: Inhibition of the CT-L activity of the purified 20S proteasome (IC50=~7.5 μM); > 95 % inhibition of proteasomal CT-L activity in MDA-MB-231 cancer cells at 20 μM | [272] |
| DDTC | 20S |  |
DDTC/Cu complex: 35 % inhibition of the CT-L activity of purified 20S proteasome at 50 μM; > 90 % inhibition of cellular 26S proteasome at 20 μM in intact cells | [275] | |
| 19S DUB |  |
AgDT: inhibition of the DUB activities of UCHL5 and USP14 at 0.08 μM. | [283] | ||
| NPL4 | CuET: Inhibition of p97-segregase–dependent protein degradation via interacting with NPL4 and inducing NPL4 immobilization and aggregation. | [290] | |||
| PDTC | 20S |  |
Inhibition of the CT-L activity of purified 20S proteasome with IC50 values of 13.8 μM for PDTC/Zn and 5.3 μM for PDTC/Cu | [287] | |
| Au(DMDT)Br2, | 20S |  |
Inhibition of the CT-L activity of the purified 20S proteasome (IC50 = 7.4 μM) and 26S proteasome in intact MDA-MB-231 cancer cells (10–20 μM) | [144] | |
| AuBr2(ESDT) | 20S | Inhibition the CT-L activity of the purified 20S proteasome (IC50 = 1.13 μM) and 26S proteasome in intact MDA-MB-231 cancer cells | [288] | ||
| [Au(ESDT)]2 | 20S | Inhibition of the CT-L activity of the purified 20S proteasome (IC50 = 17.72 μM) and 26S proteasome in intact MDA-MB-231 cancer cells | [288] |
Mechanically, thalidomide targets the substrate receptor protein cereblon (CRBN) [214], a component of CUL4-RBX1-DDB1-CRBN E3-ligase complex (CRL4CRBN E3 ligase) [215]. As a substrate receptor, CRBN is involved in the ubiquitination of a wide range of substrates, regulating a broad range of essential cellular processes including DNA repair and replication as well as chromatin remodeling [216]. It has been demonstrated that IMiDs binding to CRBN can inhibit auto-ubiquitination of CRBN, but promote CRL4CRBN-mediated ubiquitination and proteasomal degradation of substrate proteins [214,217]. IMiDs promotes myeloma cell death via CRL4CRBN-dependent proteasomal degradation of IKZF1 (Ikaros) and IKZF3 (Aiolos), which are key transcription factors involved in B- and T-cell development [218-220].
Recently, IMiDs were exploited for proteolysis targeting chimeras (PROTACs). PROTACs are bifunctional or hybrid molecules that link the IMiD structure to another small molecule that binds a protein of interest (POI). Such IMiD-based PROTACs are capable of guiding the CRL4CRBN E3 ligase to the POI, resulting in its ubiquitination and degradation [221]. For example, ARV-825 (Table 3A) is a hetero-bifunctional PROTAC that conjugated JQ1, an established chemical inhibitor of bromodomain and extraterminal domain (BET) family members, to a thalidomide analog to promote the CRBN-dependent degradation of Bromodomain-containing protein 4 (BRD4) [222]. BRD4 is a member of the BET family and acts as both a transcriptional/ epigenetic regulator for transcription of oncogenes and apoptosis regulators, such as c-Myc, Bcl-2 and Bcl-xL. Therefore, it has been validated as an important drug target for cancers [223]. It has been demonstrated that ARV-825 has a promising therapeutic potential in various types of cancer, including MM, acute myeloid leukemia, Burkitt's lymphoma, and prostate cancer [222,224-226].
Taken together, the binding of IMiDs or IMiD-based PROTACs to CRBN can induce CRL4CRBN-mediated ubiquitination and degradation of target proteins, leading to the anticancer effect in MM and other types of cancer.
4.2. Statins as repurposed inhibitors of SCFSkp2 E3 ligase
Statins (Table 3A) are a class of small fungal metabolites that inhibit the conversion of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) to mevalonate, a rate-limiting step for the cholesterol biosynthesis [227]. Statins are used as cholesterol-lowering drugs approved by the FDA [228]. It has also been demonstrated that statins are modulators of the UPS [229,230]. Recently, studies indicated that statins could induce the degradation of S-phase kinase-associated protein 2 (SCFSkp2) [231,232]. SCFSkp2 is the substrate recognition factor of Skp2-Skp1/Cullin-1/F-box-protein complex (SCFSkp2 E3 ligase). SCFSkp2 is a promising therapeutic target for cancer treatment since it selectively ubiquitinates and degrades cyclin-dependent kinase inhibitors (CKIs) such as p27 and p21 as well as the G1-/S-specific cyclin E [233]. Notably, previous studies also reported that statins could lead to the induction of the p27 and p21, resulting in decreased cyclin-dependent kinase activity and subsequent cell cycle arrest in G1 phase [234-236]. Clinical studies demonstrated an association of statin use with reduced cancer risk and prolonged survival in patients with certain types of cancer [237-240]. Numerous clinical trials have been performed in cancer patients using statins alone or in combination with other agents (https://clinicaltrials.gov/ct2/results?term=statin+AND+cancer&Search=Search). Although statin monotherapy did not give impressive results [241-243], the combination therapy of statins with other agents have shown more promising results [244-248]. Thus, repurposing statins as anticancer agents provides an encouraging strategy for cancer treatment.
4.3. Clomipramine, a repurposed inhibitor of ITCH E3 ligase
Clomipramine (CMI, brand name Anafranil) is an FDA-approved tricyclic antidepressant. It has been widely used for over 40 years in the treatment of patients with psychiatric disorders [249]. By using an ELISA-based HTS assay, CMI was identified as an ITCH E3 ligase inhibitor that interferes with the HECT Ub transthiolation activity of this enzyme in an irreversible manner (Table 3A) [250]. A number of ITCH substrates that are involved in tumorigenesis and chemosensitivity have been identified, such as p63, p73, Notch1, LATS1 (large tumor suppressor 1) and others [251-255]. Recent study indicated that inhibition of ITCH resulted in the suppression of proliferation and induction of apoptosis in lung cancer cells [256]. These results indicated that ITCH may play a relevant role in cancer biology and inhibition of ITCH is a potential strategy for cancer therapy. CMI has been reported to enhance the anticancer activity of several chemotherapeutic agents since 1980s [257,258]. It also has cytostatic properties and can induce apoptotic cell death [259,260]. As an antidepressant drug, CMI is very well-tolerated and can effectively cross the blood–brain barrier, thus it has potential anticancer activity for glioblastoma [261]. In addition, desmethylclomipramine (DCMI), the active metabolite of CMI, has been reported to inhibit growth of lung cancer stem cells (CSCs), decrease their stemness potential and increase the cytotoxic effect of conventional chemotherapeutic drugs [262]. Future studies should further clarify CMI’s anticancer mechanisms and confirm its anticancer effect in clinical settings.
4.4. Repurposing JS-K as an MDM2 inhibitor
First identified as a NO prodrug used to stabilize NO release, JS-K (Table 3A), a member of the diazeniumdiolate family, was used as a nitric oxide donor, releasing NO in a cell sustainably in conjunction with its reaction with glutathione-S-transferase (GST) [263,264]. Recently, JS-K has been shown to have inhibitory effects on p53 ubiquitylation via inhibiting Mdm2 [264]. Mdm2 is a ubiquitin E3 ligase responsible for p53 ubiquitylation [264]. JS-K-mediated Mdm2 inhibition leads to inhibition of p53 degradation, resulting in accumulation of p53 protein. In addition, by generating DNA damage, JS-K is also able to increase p53 expression [264]. Through this mechanism, JS-K has also been shown to regulate stability and activity of another E3 ligase, Siah2, which may explain how JS-K treatment links to growth inhibition of prostate cancer cells [264]. Other studies also support the idea that JS-K can be repurposed as a novel agent for treating various types of cancer [265].
4.5. Suramin as a repurposed inhibitor of Cullin-RING E3 ligase
Suramin (Table 3A) was first discovered as a remedy for African sleeping sickness and river blindness [266]. More recently, suramin has been recognized as a potent inhibitor of Cullin-RING (CRL) E3 ubiquitin ligase [267]. CRL controls various biological processes by directing several protein substrates for proteasomal degradation. Using HTS, suramin was found to target CRL-mediated K48 polyubiquitination [267]. CRL activity is secondary to the recruitment of the E2 ubiquitin-conjugating enzyme Cdc34, and therefore by targeting E3 cullin’s basic canyon region with suramin, CRL substrates accumulate and the E2-E3 interface is inhibited [267]. Although suramin is toxic and causes a fair number of side effects, it at noncytotoxic doses can sensitize tumor cells to chemotherapeutic agents [268,269]. Therefore, combinations of suramin with chemotherapies have been investigated in various cancer clinical trials (https://clinicaltrials.gov/ct2/results?term=Suramin+AND+cancer&Search=Search).
5. Repurposing disulfiram for cancer therapy
5.1. Dithiocarbamate-based compounds targeting the proteasome
Disulfiram (DSF; Table 3B), also known as Antabuse, is a member of the dithiocarbamate (DTC) family that was approved by the FDA in 1951 as the first drug to treat alcohol dependence. Mechanically, DSF targets and inhibits the enzyme activity of aldehyde dehydrogenase (ALDH), which is a critical enzyme in the metabolism of alcohol. Since the 1970s, scientists have noticed the tumor-suppressive effect of DSF [270], and in the past three decades, increasing evidence indicates that DSF has great potential to treat various human cancers in vitro and in vivo [271]. Inhibition of the UPS has been thought to be the primary mechanism accounting for the anti-cancer effects of DSF, because DSF has been identified as a proteasome inhibitor in a cell-based screening assay that monitors the 26S proteasome activity. Several studies have indicated that DSF has strong inhibitory effect on 26S proteasome activity in various cancer cell lines in a Cu-dependent manner, leading to induction of cancer cell death (Table 3B) [272]. However, several reports found that DSF had no inhibition on purified 20S proteasome [273-276], although one study showed that DSF potently inhibited the CT-L activity of purified human 20S proteasome at low micromolar pharmacological concentrations, via a mechanism different from that of BTZ or MG132 that block the active site of the proteasome [277]. Therefore, DSF may act at some other sites on the proteasome as a slow-binding partial noncompetitive inhibitor.
In acidic or Cu-rich environments, such as the stomach, DSF is reduced to diethyldithiocarbamate (DDTC). DDTC is a very strong chelator of transition divalent metal ions and is an effective drug against nickel carbonyl poisoning [278]. Several metal-DDTC complexes have also been reported to possess both proteasome-inhibitory and apoptosis-inducing activities in cancer cells. Although studies indicated that DDTC/Cu complex inhibits the proteasomal CT-L activity and induces apoptosis in breast, prostate and pancreatic cell lines [276,279], effects of Zn(DDTC)2 and Cu(DDTC)2 against purified 26S proteasome were much stronger than against purified 20S proteasome (Table 3B), indicated that these metal complexes may target 19S proteasome [275]. Moverover, the analysis of electronic density on atoms and bonds within Cu(DDTC)2/Zn(DDTC)2 showed that Cu(DDTC)2/Zn(DDTC)2 should be active against the active site of JAMMs [275], suggesting that Cu(II)-DDTC and Zn(II)-DDTC complexes might also inhibit 19S-JAMMs in tumor cells. Silver ions and silver complex have proven to exhibit excellent anticancer activity in vitro [280-282]. Recently, scientists report that the combination of silver ions and DSF can induce enhanced anticancer activity mainly through inhibition of proteasome function [283]. The silver complex AgDT (Table 3B), synthesized from silver and DDTC, efficiently induced cell death in NSCLC cells by targeting proteasomal DUBs USP14 and UCHL5 [283]. AgDT also induced DUB inhibition and antineoplastic effects in the NSCLC cell xenograft mouse models [283].
Other DTCs, such as pyrrolidine dithiocarbamate (PDTC, Table 3B), is a synthetic analog of DTC that has been reported to inhibit transcription factor nuclear factor-kappaB (NF-κB) [284]. When combined with either Zn(II) or Cu(II) chloride, PDTC was shown to inhibit the UPS [50,285,286]. PDTC/Cu and PDTC/Zn complexes were able to inhibit the CT-L activity of a purified 20S proteasome (Table 3B). These complexes were also able to inhibit proteasome activity and induce apoptosis in prostate and breast cancer cells. However, PDTC alone failed to exhibit similar results in cultured cells [287]. In addition, several gold dithiocarbamates, particularly derivatives of N,N-dimethyldithiocarbamate (DMDT) and ethylsarcosinedithiocarbamate (ESDT), such as Au (DMDT)Br2, AuBr2(ESDT) and Au(ESDT)2 were able to inhibit purified 20S proteasome as well as intact 26S proteasome (Table 3B), resulting in accumulation of ubiquitinated proteins and proteasome target proteins, induction of apoptosis, and inhibition of tumor growth [144,288].
Taken together, all these studies suggest that DTC-based compounds are potent proteasome inhibitors in human cancer cells [289]. Since some of the compounds are clinically used for treatment of other diseases with well-established safety profiles and known side effects, repurposing them as proteasome inhibitors for cancer treatment may reduce the time, cost and risk for the development of novel anticancer drugs.
5.2. CuET targeting p97 segregase adaptor NPL4
Recently, Skrott et al. demonstrated that DDTC/Cu complex (CuET) can target p97 segregase adaptor NPL4 (nuclear protein localization 4) at much lower concentration and suggested that DSF inhibits the processing of ubiquitylated proteins by targeting NPL4-dependent segregase p97 (Table 3B) [290]. p97 is a type II AAA (ATPases associated with various cellular activities) ATPase and acts upstream of the proteasome degradation and downstream of substrate ubiquitination [291,292]. p97 has been reported to use energy from ATP to extract (segregate) polyubiquitinated proteins from different macro-complexes and cellular locations, and then present them to proteasome for degradation, thus functioning as a segregase [293,294]. Furthermore, p97 is also involved in degradation of highly folded ubiquitinated soluble proteasome substrates and thus functions as an unfoldase [295]. The functional diversity of p97 is facilitated by association with many different adaptor proteins that act to recruit or modify substrates. Heterodimer of NPL4 and UFD1 (ubiquitin fusion degradation 1) is the best-characterized p97 adaptor [296]. The NPL4-UFD1 complex binds to the N-terminal domain of p97, forming a p97-UFD1-NPL4 complex, which extracts polyubiquitinated proteins, primarily labeled with Lys48-linked ubiquitin chains, from their holding environment, subcellular membrane or multi-protein complex and delivers them to the proteasome for degradation [296-298]. Given the critical function of p97-UFD1-NPL4 complex in the UPS, it has emerged as an attractive cancer therapeutic target and several inhibitors are under development as anticancer drugs, such as DBeQ and Eeyarestatin I for targeting p97 [299,300].
CuET acts on NPL4 through interacting the putative zinc finger-NPL4 (ZF-NPL4) domain, inducing NPL4 immobilization and aggregation, thereby inhibiting p97-segregase-dependent protein degradation. This is followed by triggering heat-shock response (HSR) and endoplasmic reticulum (ER) stress/UPR and eventually cell death [290]. The authors also detected a higher CuET concentration in tumors than the liver and brain tissues from the same animals after treated with DSF [290]. The preferential accumulation of CuET in tumors may explain the previously finding that the DSF/Cu complex is highly cytotoxic to cancer cells but not damage normal cells [272,301].
5.3. Repurposing DSF in combinational therapy to target cancer stem cells
Recently, accumulated evidence also showed that DSF harbors potent cancer stem cell (CSC)/ tumor initiating cell (TIC)-targeting function based on its irreversible inhibitory effect on aldehyde dehydrogenase (ALDH). The canonical function of ALDH is to convert acetaldehyde to acetate in metabolizing alcohol. In 2007, a pioneer study demonstrated that the tumorigenic cell fraction, identified by high ALDH activity, is capable of self-renewal and of generating tumors that recapitulate the heterogeneity of the parental tumor in breast carcinomas [302]. Later studies further showed that ALDH1A1 and ALDH1A3, two of 19 ALDH isoforms expressed in humans, were generally believed to be responsible for the ALDH activity of CSC or TICs [303,304]. It is worth noting that both normal and cancer human mammary epithelial cells with increased ALDH have stem/progenitor properties [302]. Moreover, the results of our recent study, and several others, also showed an enrichment of ALDH+ cells under therapeutic conditions including chemotherapies and radiation therapy [305-307]. These findings are consistent with the clinical observation that recurrent tumors frequently show more aggressive progression than primary tumors along with resistance to original therapeutic reagents.
As early as 1987, the effect of ALDH inhibitors, including DSF, was studied on the ex vivo sensitivity of human multipotent and committed hematopoietic progenitor cells and malignant blood cells [308]. However, intensive investigation of the effect of DSF on ALDH+ cancer stem cell subpopulation was not started until 2012. So far, significant inhibitory effect of DSF on ALDH+ stem cell has been shown in multiple human cancers including glioblastoma [309], brain cancer, breast cancer [305], NSCLC [310], adult B-lineage acute lymphoblastic leukemia [311], acute myeloid leukemia (AML) [312], MM [313], and pancreatic cancer [314]. The results obtained from these studies indicate that DSF treatment not only prevents tumor recurrence and chemotherapy resistance, but also decreases tumor metastasis. However, the heterogeneity and plasticity of cancer stem cell remains a critical factor that needs to be addressed in order to achieve a better therapeutic efficacy.
Using breast cancer as an example, CD44+/CD24− is has been characterized as another stem cell subpopulation in addition to ALDH+ subpopulation. Both the ALDH+ and CD44+/CD24− sub-populations have strong self-renewal capability and will regenerate tumor mass when they were implanted in mice [315,316]. Multiple research groups, including ours, showed that ALDH+ and CD44+/CD24− populations are not identical in terms of their morphological and oncogenic characteristics [307,317]. Cells co-expressing ALDH+ and CD44+/CD24− may harbor strongest oncogenic and self-renewal capability [318,319]. This notion was confirmed by a transcriptional profiling study based on RNA and single cell sequencing [320]. Aligned with these reports, it has been reproducibly reported that either chemotherapy or radiation therapy could lead to accumulation of CSC cells in vitro and in vivo [321-324]. Due to the heterogeneous and plasticity pattern of CSCs in tumor, it is unlikely that targeting one CSC subpopulation will be therapeutically sufficient to prevent all CSCs’ function. Thus, simultaneously targeting multiple CSC populations or CSC-plasticity related signaling pathways is a more viable alternative.
This speculation was supported by our recent proposed combination therapy, in which DSF was used as an ALDH+-targeting reagent along with BKM120, a pan PI3K inhibitor and a CD44+/CD24−-targeting reagent [307]. Treatment with DSF/Cu or BKM120 induced higher levels of apoptosis in ALDH+ or CD44+/CD24− sub-populations, respectively, than in bulk tumor cells. Combining DSF/Cu and BKM120 treatment simultaneously decreased the ALDH+ and CD44+/CD24− CSCs/TICs. Using a TNBC (triple-negative breast cancer) tumor xenograft mouse model, we found that DSF/BKM in combination with Taxol significantly reduced the tumor burden and delayed tumor recurrence compared to Taxol treatment alone [307]. This study is the first of its kind to use two different drugs to abolish two major TIC subpopulations simultaneously and inhibit tumor recurrence. These results lay a foundation for developing a novel therapy that can improve chemotherapeutic efficacy.
5.4. Current clinical trials of DSF for cancer therapy
As an FDA-approved drug, DSF has a well-studied pharmacokinetics and excellent safety profile. The abilities of DSF to target the UPS and CSC further support it as a repurposed, promising anticancer drug. Hence, numerous clinical trials have been performed in patients with different types of cancer to confirm the anticancer effect of DSF either alone or in combination with other chemotherapeutics (https://clinicaltrials.gov/ct2/results?term=disulfiram+AND+cancer&Search=Search).
Since DSF is capable of penetrating blood-brain barrier, almost half of the trials are aimed to evaluate the effectiveness of DSF/Cu in patients with different stages of glioblastoma (GBM) ( NCT01907165, NCT03034135, NCT02715609, NCT03363659, NCT01777919, NCT02678975, NCT03151772, and NCT02770378). A phase I clinical trial ( NCT01907165) showed that DSF in combination with temozolomide (TMZ) had an acceptable safety profile and produced promising progression free survival (PFS) in GBM patients [325]. A recent phase II study ( NCT03034135) from the same group showed that addition of DSF/Cu to TMZ for TMZ-resistant isocitrate dehydrogenase (IDH)-wild type GBM appears well tolerated but has limited activity for unselected population [326]. A prostate cancer trial ( NCT01118741) demonstrates that DSF has no significant benefit on patients’ survival [327]. An ongoing new phase lb study is to investigate intravenous Cu loading with oral DSF’s effect on prostate cancer ( NCT02963051). Currently, no results are available yet from clinical trials investigating DSF’s effects against other types of tumors, such as melanoma ( NCT02101008, NCT00256230, NCT00571116), pancreatic ( NCT03714555, NCT02671890), liver ( NCT00742911) and breast ( NCT03323346) cancer.
Taken together, the available results from clinical trials indicate that DSF is well tolerated in most cancer patients and it can be combined with Cu or chemotherapeutic agents. All these findings provide promising prospects for repurposing this compound as an effective anticancer drug by further larger scale of trials.
6. Conclusions and perspectives
Since 2003, the 20S proteasome has been validated as an anticancer drug target and several 20S proteasome inhibitors (such as BTZ) have been approved by the FDA for the treatment of MM and some other liquid tumors. BTZ-based therapies have become a staple for the treatment of MM at all stages and the survival rate of MM patients has been increased at least 2-3-folds. However, BTZ has several limitations, such as drug resistance and neuro-toxicities.
Overcoming BTZ resistance can definitely improve the current status of cancer treatment. This could be achieved by, at least, developing new proteasome inhibitor drugs and old drug repositioning. The latter represents an attractive drug discovery strategy compared to the time-consuming, costly traditional de novo drug discovery process.
Literature has presented many examples of discovering old drugs as new inhibitors of the UPS. This review has summarized selected repurposed inhibitors of various UPS components, including 20S proteasomes, 19S proteasome-associated DUBs, and E3 enzymes.
Compared to typical drug discovery and development, old drug repurposing has both advantages and disadvantages. The advantages of drug repositioning include: previously well-known pharmacokinetics and pharmacodynamics in patients; fast drug development cycle; economical, efficient translation from bench to bedside; and easy to get clinical approval. The limitations of drug reposition include low target-specificity and off-target effects.
The fact that the UPS regulates degradation of many substrate proteins supports it as one of the most important drug repurposing targets for cancer treatment. Since an old drug was not designed to treat a new disease, there are often more targets that can be hit by the same old drug. However, anticancer drugs with multiple protein targets could represent excellent candidates for reposition. HTP of old drug libraries and natural product collections could also generate novel leads for drug/agent reposition as cancer treatment. Finally, an old drug, when combined with other compounds/drugs/agents could offer new opportunities to its repositioning.
It is exciting to see that rapidly developed modern technologies have made old drug reposition more feasible, precisional, and systematic. For example, better understanding genetic and epigenetic regulations of tumor formation and progression have increased opportunities of discovering more repurposed anticancer therapeutics. Indeed, recent genome-wide association studies (GWASs) have discovered numberous new potential targets for drug repositioning, which, of course, should be further validated by other functional technologies. It is believed that fast development in modern technologies, including computational, informatics, medicinal chemistry, and clinical profiling and analysis, has offered, and will offer us more oppotunites to reuse approved drugs for treating other human diseases such as cancer.
Acknowledgements
This work was supported by National Cancer Institute grant R21CA184788 (to QP Dou), NCI R01CA172480-01A1 (to GJ Wu), a National Institutes of Health grant P30 CA022453 (to the Karmanos Cancer Institute at Wayne State University), and the National Natural Science Foundation of China (81872439 to H.J. Yang; No. 31700780 to K. Li; 81802405 to X. Chen; 81670156, 81773213, 81772492, and 81600147 to J. Liu). Given the length constraints, we were not able to cite all the papers that are relevant to this topic. We apologize to the scientists whose papers were left out.
Footnotes
Declaration of Competing Interest
The authors declare that there are no conflicts of interest.
References
- [1].Nandi D, Tahiliani P, Kumar A, Chandu D, The ubiquitin-proteasome system, J. Biosci 31 (1) (2006) 137–155. [DOI] [PubMed] [Google Scholar]
- [2].Hershko A, Ciechanover A, The ubiquitin system, Annu. Rev. Biochem 67 (1998) 425–479. [DOI] [PubMed] [Google Scholar]
- [3].Fang S, Weissman AM, A field guide to ubiquitylation, Cell. Mol. Life Sci 61 (13) (2004) 1546–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ciechanover A. The ubiquitin-proteasome proteolytic pathway. In, 79, 1994: 13–21. [DOI] [PubMed] [Google Scholar]
- [5].Adams J, The proteasome: structure, function, and role in the cell, Cancer Treat. Rev 29 (Suppl 1) (2003) 3–9. [DOI] [PubMed] [Google Scholar]
- [6].Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C, Deubiquitinases in cancer: new functions and therapeutic options, Oncogene 31 (19) (2012) 2373–2388. [DOI] [PubMed] [Google Scholar]
- [7].Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Ploegh H, Finley D, Multiple associated proteins regulate proteasome structure and function, Mol. Cell 10 (3) (2002) 495–507. [DOI] [PubMed] [Google Scholar]
- [8].Qiu XB, Ouyang SY, Li CJ, Miao S, Wang L, Goldberg AL, hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37, EMBO J. 25 (24) (2006) 5742–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Richardson PG, Anderson KC, Bortezomib: a novel therapy approved for multiple myeloma, Clin. Adv. Hematol. Oncol 1 (10) (2003) 596–600. [PubMed] [Google Scholar]
- [10].Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ, Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis, J. Immunol 171 (1) (2003) 88–95. [DOI] [PubMed] [Google Scholar]
- [11].Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine L, Porter JB, Schenkein D, Anderson KC, Bortezomib or high-dose dexamethasone for relapsed multiple myeloma, N. Engl. J. Med 352 (24) (2005) 2487–2498. [DOI] [PubMed] [Google Scholar]
- [12].San MJ, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka H, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine L, Liu K, Cakana A, van de Velde H, Richardson PG, Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma, N. Engl. J. Med 359 (9) (2008) 906–917. [DOI] [PubMed] [Google Scholar]
- [13].Rosinol L, Oriol A, Teruel AI, Hernandez D, Lopez-Jimenez J, de la Rubia J, Granell M, Besalduch J, Palomera L, Gonzalez Y, Etxebeste MA, Diaz-Mediavilla J, Hernandez MT, de Arriba F, Gutierrez NC, Martin-Ramos ML, Cibeira MT, Mateos MV, Martinez J, Alegre A, Lahuerta JJ, San MJ, Blade J, Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study, Blood 120 (8) (2012) 1589–1596. [DOI] [PubMed] [Google Scholar]
- [14].Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, Spicka I, Hungria V, Munder M, Mateos MV, Mark TM, Qi M, Schecter J, Amin H, Qin X, Deraedt W, Ahmadi T, Spencer A, Sonneveld P, Daratumumab, bortezomib, and dexamethasone for multiple myeloma, N. Engl. J. Med 375 (8) (2016) 754–766. [DOI] [PubMed] [Google Scholar]
- [15].Bose P, Batalo MS, Holkova B, Grant S, Bortezomib for the treatment of non-Hodgkin’s lymphoma, Expert Opin. Pharmacother 15 (16) (2014) 2443–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Durie BGM, Hoering A, Abidi MH, Rajkumar SV, Epstein J, Kahanic SP, Thakuri M, Reu F, Reynolds CM, Sexton R, Orlowski RZ, Barlogie B, Dispenzieri, Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial, Lancet 389 (10068) (2017) 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG, The proteasome and proteasome inhibitors in multiple myeloma, Cancer Metastasis Rev. 36 (4) (2017) 561–584. [DOI] [PubMed] [Google Scholar]
- [18].Adams J, The proteasome: a suitable antineoplastic target, Nat. Rev. Cancer 4 (5) (2004) 349–360. [DOI] [PubMed] [Google Scholar]
- [19].Almond JB, Cohen GM, The proteasome: a novel target for cancer chemotherapy, Leukemia 16 (4) (2002) 433–443. [DOI] [PubMed] [Google Scholar]
- [20].Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP, Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives, Curr. Cancer Drug Targets 11 (3) (2011) 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Harrigan JA, Jacq X, Martin NM, Jackson SP, Deubiquitylating enzymes and drug discovery: emerging opportunities, Nat. Rev. Drug Discov 17 (1) (2018) 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gallery M, Blank JL, Lin Y, Gutierrez JA, Pulido JC, Rappoli D, Badola S, Rolfe M, Macbeth KJ, The JAMM motif of human deubiquitinase Poh1 is essential for cell viability, Mol. Cancer Ther 6 (1) (2007) 262–268. [DOI] [PubMed] [Google Scholar]
- [23].Spataro V, Simmen K, Realini CA, The essential 26S proteasome subunit Rpn11 confers multidrug resistance to mammalian cells, Anticancer Res. 22 (6C) (2002) 3905–3909. [PubMed] [Google Scholar]
- [24].Yan Song ARDS, Targeting 19S-Proteasome Deubiquitinase Rpn11/POH1/PSMD14 in multiple myeloma, Blood 126 (23) (2015) 1811. [Google Scholar]
- [25].Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, Carrasco RD, Richardson P, Linder S, Chauhan D, Anderson KC, A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance, Blood 123 (5) (2014) 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A, The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-beta signalling, Oncogene 24 (54) (2005) 8080–8084. [DOI] [PubMed] [Google Scholar]
- [27].Fang Y, Fu D, Tang W, Cai Y, Ma D, Wang H, Xue R, Liu T, Huang X, Dong L, Wu H, Shen X, Ubiquitin C-terminal Hydrolase 37, a novel predictor for hepatocellular carcinoma recurrence, promotes cell migration and invasion via interacting and deubiquitinating PRP19, Biochim. Biophys. Acta 1833 (3) (2013) 559–572. [DOI] [PubMed] [Google Scholar]
- [28].D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S, Inhibition of proteasome deubiquitinating activity as a new cancer therapy, Nat. Med 17 (12) (2011) 1636–1640. [DOI] [PubMed] [Google Scholar]
- [29].Song Y, Li S, Ray A, Das DS, Qi J, Samur MK, Tai YT, Munshi N, Carrasco RD, Chauhan D, Anderson KC, Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance, Oncogene 36 (40) (2017) 5631–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liao Y, Liu N, Hua X, Cai J, Xia X, Wang X, Huang H, Liu J, Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor, Cell Death Dis. 8 (2) (2017) e2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cappadocia L, Lima CD, Ubiquitin-like protein conjugation: structures, chemistry, and mechanism, Chem. Rev 118 (3) (2017) 889–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].van der Veen AG, Ploegh HL, Ubiquitin-like proteins, Annu. Rev. Biochem 81 (2012) 323–357. [DOI] [PubMed] [Google Scholar]
- [33].Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE, Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets, Nat. Rev. Drug Discov 10 (1) (2011) 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang DT, Walden H, Duda D, Schulman BA, Ubiquitin-like protein activation, Oncogene 23 (11) (2004) 1958–1971. [DOI] [PubMed] [Google Scholar]
- [35].Flotho A, Melchior F, Sumoylation: a regulatory protein modification in health and disease, Annu. Rev. Biochem 82 (2013) 357–385. [DOI] [PubMed] [Google Scholar]
- [36].Ovaa H, Vertegaal A, Probing ubiquitin and SUMO conjugation and deconjugation, Biochem. Soc. Trans 46 (2) (2018) 423–436. [DOI] [PubMed] [Google Scholar]
- [37].Schwechheimer C, NEDD8-its role in the regulation of Cullin-RING ligases, Curr. Opin. Plant Biol 45 (Pt A) (2018) 112–119. [DOI] [PubMed] [Google Scholar]
- [38].Wurth R, Thellung S, Bajetto A, Mazzanti M, Florio T, Barbieri F, Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds, Drug Discov. Today 21 (1) (2016) 190–199. [DOI] [PubMed] [Google Scholar]
- [39].Sekhon BS, Repositioning drugs and biologics: retargeting old/existing drugs for potential new therapeutic applications, J. Pharm. Educ. Res 4 (1) (2013) 1. [Google Scholar]
- [40].Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS, High-throughput screening assays for the identification of chemical probes, Nat. Chem. Biol 3 (8) (2007) 466–479. [DOI] [PubMed] [Google Scholar]
- [41].Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DV, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam S, Impact of high-throughput screening in biomedical research, Nat. Rev. Drug Discov 10 (3) (2011) 188–195. [DOI] [PubMed] [Google Scholar]
- [42].Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP, Organic copper complexes as a new class of proteasome inhibitors and apoptosis inducers in human cancer cells, Biochem. Pharmacol 67 (2004) 1139–1151. [DOI] [PubMed] [Google Scholar]
- [43].Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL, Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial, Arch. Neurol 60 (2003) 1685–1691. [DOI] [PubMed] [Google Scholar]
- [44].Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG, Treatment of Alzheimer’s disease with clioquinol, Dement. Geriatr. Cogn. Disord 12 (2001) 408–414. [DOI] [PubMed] [Google Scholar]
- [45].Nguyen T, Hamby A, Massa SM, Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 11840–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P, Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc(II) and copper(II) complexes, Inorg. Chem 43 (2004) 3795–3797. [DOI] [PubMed] [Google Scholar]
- [47].Pushie MJ, Nienaber KH, Summers KL, Cotelesage JJ, Ponomarenko O, Nichol K, Pickering IJ, George GN, The solution structure of the copper clioquinol complex, J. Inorg. Biochem 133 (2014) 50–56. [DOI] [PubMed] [Google Scholar]
- [48].Barrea RA, Chen D, Irving TC, Dou QP, Synchrotron X-ray imaging reveals a correlation of tumor copper speciation with Clioquinol’s anticancer activity, J. Cell. Biochem 108 (2009) 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chen D, Cui QC, Yang H, Barrea RA, Sarkar FH, Sheng S, Yan B, Reddy CP, Dou QP, Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts, Cancer Res. 67 (2007) 1636–1644. [DOI] [PubMed] [Google Scholar]
- [50].Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP, Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells, Breast Cancer Res. 7 (2005) R897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mao X, Li X, Sprangers R, Wang X, Venugopal A, Wood T, Zhang Y, Kuntz DA, Coe E, Trudel S, Rose D, Batey RA, Kay LE, Schimmer AD, Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma, Leukemia 23 (2009) 585–590. [DOI] [PubMed] [Google Scholar]
- [52].Ding WQ, Yu HJ, Lind SE, Zinc-binding compounds induce cancer cell death via distinct modes of action, Cancer Lett. 271 (2008) 251–259. [DOI] [PubMed] [Google Scholar]
- [53].Ding WQ, Liu B, Vaught JL, Yamauchi H, Lind SE, Anticancer activity of the antibiotic clioquinol, Cancer Res. 65 (2005) 3389–3395. [DOI] [PubMed] [Google Scholar]
- [54].Yu H, Zhou Y, Lind SE, Ding WQ, Clioquinol targets zinc to lysosomes in human cancer cells, Biochem. J 417 (2009) 133–139. [DOI] [PubMed] [Google Scholar]
- [55].Yu H, Lou JR, Ding WQ, Clioquinol independently targets NF-kappaB and lysosome pathways in human cancer cells, Anticancer Res. 30 (2010) 2087–2092. [PubMed] [Google Scholar]
- [56].Schimmer AD, Jitkova Y, Gronda M, Wang Z, Brandwein J, Chen C, Gupta V, Schuh A, Yee K, Chen J, Ackloo S, Booth T, Keays S, Minden MD, A phase I study of the metal ionophore clioquinol in patients with advanced hematologic malignancies, Clin. Lymphoma Myeloma Leuk 12 (2012) 330–336. [DOI] [PubMed] [Google Scholar]
- [57].Nordenberg J, Novogrodsky A, Beery E, Patia M, Wasserman L, Warshawsky A, Anti-proliferative effects and phenotypic alterations induced by 8-hydroxyquinoline in melanoma cell lines, Eur. J. Cancer 26 (1990) 905–907. [DOI] [PubMed] [Google Scholar]
- [58].Shen AY, Wu SN, Chiu CT, Synthesis and cytotoxicity evaluation of some 8-hydroxyquinoline derivatives, J. Pharm. Pharmacol 51 (1999) 543–548. [DOI] [PubMed] [Google Scholar]
- [59].Chan SH, Chui CH, Chan SW, Kok SH, Chan D, Tsoi MY, Leung PH, Lam AK, Chan AS, Lam KH, Tang JC, Synthesis of 8-hydroxyquinoline derivatives as novel antitumor agents, ACS Med. Chem. Lett 4 (2013) 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jiang H, Taggart JE, Zhang X, Benbrook DM, Lind SE, Ding WQ, Nitroxoline (8-hydroxy-5-nitroquinoline) is more a potent anti-cancer agent than clioquinol (5-chloro-7-iodo-8-quinoline), Cancer Lett. 312 (2011) 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhai S, Yang L, Cui QC, Sun Y, Dou QP, Yan B, Tumor cellular proteasome inhibition and growth suppression by 8-hydroxyquinoline and clioquinol requires their capabilities to bind copper and transport copper into cells, J. Biol. Inorg. Chem 15 (2010) 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang H, Thomas R, Oupicky D, Peng F, Synthesis and characterization of new copper thiosemicarbazone complexes with an ONNS quadridentate system: cell growth inhibition, S-phase cell cycle arrest and proapoptotic activities on cisplatin-resistant neuroblastoma cells, J. Biol. Inorg. Chem 13 (2008) 47–55. [DOI] [PubMed] [Google Scholar]
- [63].Oliveri V, Lanza V, Milardi D, Viale M, Marie I, Sgarlata C, Vecchio G, Amino- and chloro-8-hydroxyquinolines and their copper complexes as proteasome inhibitors and antiproliferative agents, Metallomics 9 (2017) 1439–1446. [DOI] [PubMed] [Google Scholar]
- [64].Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, Wang X, Zhang Y, Verbrugge SE, Adomat H, Li ZH, Trudel S, Chen C, Religa TL, Jamal N, Messner H, Cloos J, Rose DR, Navon A, Guns E, Batey RA, Kay LE, Schimmer AD, Effect of noncompetitive proteasome inhibition on bortezomib resistance, J. Natl. Cancer Inst 102 (2010) 1069–1082. [DOI] [PubMed] [Google Scholar]
- [65].Arts EJ, Hazuda DJ, HIV-1 antiretroviral drug therapy, Cold Spring Harb. Perspect. Med 2 (2012) a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sgadari C, Monini P, Barillari G, Ensoli B, Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumour growth, Lancet Oncol. 4 (2003) 537–547. [DOI] [PubMed] [Google Scholar]
- [67].Flexner C, HIV-protease inhibitors, N. Engl. J. Med 338 (1998) 1281–1292. [DOI] [PubMed] [Google Scholar]
- [68].Andre P, Groettrup M, Klenerman P, de Giuli R, Booth BL Jr, Cerundolo V, Bonneville M, Jotereau F, Zinkernagel RM, Lotteau V, An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 13120–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schmidtke G, Holzhutter HG, Bogyo M, Kairies N, Groll M, de Giuli R, Emch S, Groettrup M, How an inhibitor of the HIV-I protease modulates proteasome activity, J. Biol. Chem 274 (1999) 35734–35740. [DOI] [PubMed] [Google Scholar]
- [70].Pati S, Pelser CB, Dufraine J, Bryant JL, Reitz MS Jr, Weichold FF, Antitumorigenic effects of HIV protease inhibitor ritonavir: inhibition of Kaposi sarcoma, Blood 99 (2002) 3771–3779. [DOI] [PubMed] [Google Scholar]
- [71].Pajonk F, Himmelsbach J, Riess K, Sommer A, McBride WH, The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells, Cancer Res. 62 (2002) 5230–5235. [PubMed] [Google Scholar]
- [72].Kraus M, Bader J, Overkleeft H, Driessen C, Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance, Blood Cancer J. 3 (2013) e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bono C, Karlin L, Harel S, Mouly E, Labaume S, Galicier L, Apcher S, Sauvageon H, Fermand JP, Bories JC, Arnulf B, The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo, Haematologica 97 (2012) 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Piccinini M, Rinaudo MT, Chiapello N, Ricotti E, Baldovino S, Mostert M, Tovo PA, The human 26S proteasome is a target of antiretroviral agents, AIDS 16 (2002) 693–700. [DOI] [PubMed] [Google Scholar]
- [75].Mazza F, Tronconi E, Valerio A, Groettrup M, Kremer M, Tossi A, Benedetti F, Cargnel A, Atzori C, The non-peptidic HIV protease inhibitor tipranavir and two synthetic peptidomimetics (TS98 and TS102) modulate Pneumocystis carinii growth and proteasome activity of HEL299 cell line, J. Eukaryot. Microbiol 53 (Suppl. 1) (2006) SI44–146. [DOI] [PubMed] [Google Scholar]
- [76].Gaedicke S, Firat-Geier E, Constantiniu O, Lucchiari-Hartz M, Freudenberg M, Galanos C, Niedermann G, Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis, Cancer Res. 62 (2002) 6901–6908. [PubMed] [Google Scholar]
- [77].Laurent N, de Bouard S, Guillamo JS, Christov C, Zini R, Jouault H, Andre P, Lotteau V, Peschanski M, Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo, Mol. Cancer Ther 3 (2004) 129–136. [PubMed] [Google Scholar]
- [78].Pyrko P, Kardosh A, Wang W, Xiong W, Schonthal AH, Chen TC, HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress, Cancer Res. 67 (2007) 10920–10928. [DOI] [PubMed] [Google Scholar]
- [79].Bruning A, Burger P, Vogel M, Rahmeh M, Gingelmaiers A, Friese K, Lenhard M, Burges A, Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis, Cancer Biol. Ther 8 (2009) 226–232. [DOI] [PubMed] [Google Scholar]
- [80].Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC, Tiedemann RE, Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma, Cancer Cell 24 (2013) 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kraus M, Malenke E, Gogel J, Muller H, Ruckrich T, Overkleeft H, Ovaa H, Koscielniak D, Hartmann JT, Driessen C, Ritonavir induces endoplasmic reticulum stress and sensitizes sarcoma cells toward bortezomib-induced apoptosis, Mol. Cancer Ther 7 (2008) 1940–1948. [DOI] [PubMed] [Google Scholar]
- [82].Sato A, Asano T, Ito K, Asano T, Ritonavir interacts with bortezomib to enhance protein ubiquitination and histone acetylation synergistically in renal cancer cells, Urology 79 (966) (2012) e913–921. [DOI] [PubMed] [Google Scholar]
- [83].Isono M, Sato A, Asano T, Okubo K, Asano T, Delanzomib interacts with ritonavir synergistically to cause endoplasmic reticulum stress in renal cancer cells, Anticancer Res. 38 (2018) 3493–3500. [DOI] [PubMed] [Google Scholar]
- [84].Sato A, Asano T, Okubo K, Isono M, Asano T, Ritonavir and ixazomib kill bladder cancer cells by causing ubiquitinated protein accumulation, Cancer Sci. 108 (2017) 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Driessen C, Kraus M, Joerger M, Rosing H, Bader J, Hitz F, Berset C, Xyrafas A, Hawle H, Berthod G, Overkleeft HS, Sessa C, Huitema A, Pabst T, von Moos R, Hess D, Mey UJ, Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: a phase I trial (SAKK 65/08), Haematologica 101 (2016) 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Driessen C, Muller R, Novak U, Cantoni N, Betticher D, Mach N, Rufer A, Mey U, Samaras P, Ribi K, Besse L, Besse A, Berset C, Rondeau S, Hawle H, Hitz F, Pabst T, Zander T, Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma, Blood 132 (2018) 2097–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Brunner TB, Geiger M, Grabenbauer GG, Lang-Welzenbach M, Mantoni TS, Cavallaro A, Sauer R, Hohenberger W, McKenna WG, Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer, J. Clin. Oncol 26 (2008) 2699–2706. [DOI] [PubMed] [Google Scholar]
- [88].Rengan R, Mick R, Pryma DA, Lin LL, Christodouleas J, Plastaras JP, Simone CB 2nd, Gupta AK, Evans TL, Stevenson JP, Langer CJ, Kucharczuk J, Friedberg J, Lam S, Patsch D, Hahn SM, Maity A, Clinical outcomes of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a phase 1/2 trial, JAMA Oncol. (2019) [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ahluwalia MS, Patton C, Stevens G, Tekautz T, Angelov L, Vogelbaum MA, Weil RJ, Chao S, Elson P, Suh JH, Barnett GH, Peereboom DM, Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas, J. Neurooncol 102 (2011) 317–321. [DOI] [PubMed] [Google Scholar]
- [90].de Weger VA, Stuurman FE, Hendrikx J, Moes JJ, Sawicki E, Huitema ADR, Nuijen A, Thijssen B, Rosing H, Keessen M, Mergui-Roelvink M, Beijnen JH, Schellens JHM, Marchetti S, A dose-escalation study of bi-daily once weekly oral docetaxel either as ModraDoc001 or ModraDoc006 combined with ritonavir, Eur. J. Cancer 86 (2017) 217–225. [DOI] [PubMed] [Google Scholar]
- [91].Rice AM, Sartorelli AC, Inhibition of 20 S and 26 S proteasome activity by lithium chloride: impact on the differentiation of leukemia cells by all-trans retinoic acid, J. Biol. Chem 276 (2001) 42722–42727. [DOI] [PubMed] [Google Scholar]
- [92].Holtz KM, Rice AM, Sartorelli AC, Lithium chloride inactivates the 20S proteasome from WEHI-3B D+ leukemia cells, Biochem. Biophys. Res. Commun 303 (2003) 1058–1064. [DOI] [PubMed] [Google Scholar]
- [93].Shim M, Smart RC, Lithium stabilizes the CCAAT/enhancer-binding protein alpha (C/EBPalpha) through a glycogen synthase kinase 3 (GSK3)-independent pathway involving direct inhibition of proteasomal activity, J. Biol. Chem 278 (2003) 19674–19681. [DOI] [PubMed] [Google Scholar]
- [94].Lubner SJ, Kunnimalaiyaan M, Holen KD, Ning L, Ndiaye M, Loconte NK, Mulkerin L, Schelman WR, Chen H, A preclinical and clinical study of lithium in low-grade neuroendocrine tumors, Oncologist 16 (2011) 452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Huang RY, Hsieh KP, Huang WW, Yang YH, Use of lithium and cancer risk in patients with bipolar disorder: population-based cohort study, Br. J. Psychiatry 209 (2016) 393–399. [DOI] [PubMed] [Google Scholar]
- [96].Johnson CD, Puntis M, Davidson N, Todd S, Bryce R, Randomized, dosefinding phase III study of lithium gamolenate in patients with advanced pancreatic adenocarcinoma, Br. J. Surg 88 (2001) 662–668. [DOI] [PubMed] [Google Scholar]
- [97].Fearon KC, Falconer JS, Ross JA, Carter DC, Hunter JO, Reynolds PD, Tuffnell Q, An open-label phase I/II dose escalation study of the treatment of pancreatic cancer using lithium gammalinolenate, Anticancer Res. 16 (1996) 867–874. [PubMed] [Google Scholar]
- [98].Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ, Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis, Cancer Res. 70 (22) (2010) 9265–9276. [DOI] [PubMed] [Google Scholar]
- [99].Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP, Jiang W, Rascoe P, Rogers MK, Smythe WR, Cao X, The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition, BMC Cancer 12 (2012) 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, Talpaz M, Donato NJ, Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis, Blood 117 (11) (2011) 3151–3162. [DOI] [PubMed] [Google Scholar]
- [101].Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ, Showalter HD, Talpaz M, Donato NJ, A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination, Cell. Signal 23 (12) (2011) 2076–2085. [DOI] [PubMed] [Google Scholar]
- [102].Driessen S, Berleth N, Friesen O, Loffler AS, Bohler P, Hieke N, Stuhldreier F, Peter A, Schink KO, Schultz SW, Stenmark H, Holland P, Simonsen A, Wesselborg S, Stork B, Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy, Autophagy 11 (9) (2015) 1458–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Pham LV, Tamayo AT, Li C, Bornmann W, Priebe W, Ford RJ, Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications, Mol. Cancer Ther 9 (7) (2010) 2026–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Liu H, Chen W, Liang C, Chen BW, Zhi X, Zhang S, Zheng X, Bai X, Liang T, WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation, Cancer Lett. 361 (2) (2015) 218–225. [DOI] [PubMed] [Google Scholar]
- [105].Fu P, Du F, Liu Y, Yao M, Zhang S, Zheng X, Zheng S, WP1130 increases cisplatin sensitivity through inhibition of usp9x in estrogen receptor-negative breast cancer cells, Am. J. Transl. Res 9 (4) (2017) 1783–1791. [PMC free article] [PubMed] [Google Scholar]
- [106].Ma T, Chen W, Zhi X, Liu H, Zhou Y, Chen BW, Hu L, Shen J, Zheng X, Zhang S, Zhang B, Li H, Liang T, USP9X inhibition improves gemcitabine sensitivity in pancreatic cancer by inhibiting autophagy, Cancer Lett. 436 (2018) 129–138. [DOI] [PubMed] [Google Scholar]
- [107].Erdal H, Berndtsson M, Castro J, Brunk U, Shoshan MC, Linder S, Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis, Proc. Natl. Acad. Sci. U. S. A 102 (1) (2005) 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S, Inhibition of proteasome deubiquitinating activity as a new cancer therapy, Nat. Med 17 (12) (2011) 1636–1640. [DOI] [PubMed] [Google Scholar]
- [109].Wang X, Stafford W, Mazurkiewicz M, Fryknas M, Brjnic S, Zhang X, Gullbo J, Larsson R, Arner ES, D’Arcy P, Linder S, The 19S Deubiquitinase inhibitor b-AP15 is enriched in cells and elicits rapid commitment to cell death, Mol. Pharmacol 85 (6) (2014) 932–945. [DOI] [PubMed] [Google Scholar]
- [110].Brnjic S, Mazurkiewicz M, Fryknas M, Sun C, Zhang X, Larsson R, D’Arcy P, Linder S, Induction of tumor cell apoptosis by a proteasome deubiquitinase inhibitor is associated with oxidative stress, Antioxid. Redox Signal 21 (17) (2014) 2271–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Zhang X, Pellegrini P, Saei AA, Hillert EK, Mazurkiewicz M, Olofsson MH, Zubarev RA, D’Arcy P, Linder S, The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage, Biochem. Pharmacol 156 (2018) 291–301. [DOI] [PubMed] [Google Scholar]
- [112].Hillert EK, Brnjic S, Zhang X, Mazurkiewicz M, Saei AA, Mofers A, Selvaraju K, Zubarev R, Linder S, D’Arcy P, Proteasome inhibitor b-AP15 induces enhanced proteotoxicity by inhibiting cytoprotective aggresome formation, Cancer Lett. 448 (2019) 70–83. [DOI] [PubMed] [Google Scholar]
- [113].Coughlin K, Anchoori R, Iizuka Y, Meints J, MacNeill L, Vogel RI, Orlowski RZ, Lee MK, Roden RB, Bazzaro M, Small-molecule RA-9 inhibits proteasome-associated DUBs and ovarian cancer in vitro and in vivo via exacerbating unfolded protein responses, Clin. Cancer Res 20 (12) (2014) 3174–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Issaenko OA, Amerik AY, Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes, Cell Cycle 11 (9) (2014) 1804–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, Gullbo J, Chanan-Khan A, Linder S, Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15, Chem. Biol. Drug Des 86 (5) (2015) 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, Bossier F, Khan AC, Linder S, D’Arcy P, The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells, Sci. Rep 6 (2016) 26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Maheshwari RK, Singh AK, Gaddipati J, Srimal RC, Multiple biological activities of curcumin: a short review, Life Sci. 78 (18) (2006) 2081–2087. [DOI] [PubMed] [Google Scholar]
- [118].Mullally JE, Fitzpatrick FA, Pharmacophore model for novel inhibitors of ubiquitin isopeptidases that induce p53-independent cell death, Mol. Pharmacol 62 (2) (2002) 351–358. [DOI] [PubMed] [Google Scholar]
- [119].Milacic V, Banerjee S, Landis-Piwowar KR, Sarkar FH, Majumdar AP, Dou QP, Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo, Cancer Res. 68 (18) (2008) 7283–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Hasima N, Aggarwal BB, Targeting proteasomal pathways by dietary curcumin for cancer prevention and treatment, Curr. Med. Chem 21 (14) (2014) 1583–1594. [DOI] [PubMed] [Google Scholar]
- [121].Salehi B, Stojanovic-Radic Z, Matejic J, Sharifi-Rad M, Anil Kumar NV, Martins N, Sharifi-Rad J, The therapeutic potential of curcumin: A review of clinical trials, Eur. J. Med. Chem 163 (2019) 527–545. [DOI] [PubMed] [Google Scholar]
- [122].Qiu X, Du Y, Lou B, Zuo Y, Shao W, Huo Y, Huang J, Yu Y, Zhou B, Du J, Fu H, Bu X, Synthesis and identification of new 4-arylidene curcumin analogues as potential anticancer agents targeting nuclear factor-kappaB signaling pathway, J. Med. Chem 53 (23) (2010) 8260–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhou B, Zuo Y, Li B, Wang H, Liu H, Wang X, Qiu X, Hu Y, Wen S, Du J, Bu X, Deubiquitinase inhibition of 19S regulatory particles by 4-arylidene curcumin analog AC17 causes NF-kappaB inhibition and p53 reactivation in human lung cancer cells, Mol. Cancer Ther 12 (8) (2013) 1381–1392. [DOI] [PubMed] [Google Scholar]
- [124].Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma N, Jiang S, Zhang HZ, Gehlsen KR, Cai SX, Green DR, Tseng B, A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid, Proc. Natl. Acad. Sci. U. S. A 102 (34) (2005) 12095–12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Felth J, Lesiak-Mieczkowska K, D’Arcy P, Haglund C, Gullbo J, Larsson R, Linder S, Bohlin L, Fryknas M, Rickardson L, Gambogic acid is cytotoxic to cancer cells through inhibition of the ubiquitin-proteasome system, Invest. New Drugs 31 (3) (2013) 587–598. [DOI] [PubMed] [Google Scholar]
- [126].Li X, Liu S, Huang H, Liu N, Zhao C, Liao S, Yang C, Liu Y, Zhao C, Li S, Lu X, Liu C, Guan L, Zhao K, Shi X, Song W, Zhou P, Dong X, Guo H, Wen G, Zhang C, Jiang L, Ma N, Li B, Wang S, Tan H, Wang X, Dou QP, Liu J, Gambogic acid is a tissue-specific proteasome inhibitor in vitro and in vivo, Cell Rep. 3 (1) (2013) 211–222. [DOI] [PubMed] [Google Scholar]
- [127].An B, Goldfarb RH, Siman R, Dou QP, Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts, Cell Death Differ. 5 (12) (1998) 1062–1075. [DOI] [PubMed] [Google Scholar]
- [128].Zhao L, Zhen C, Wu Z, Hu R, Zhou C, Guo Q, General pharmacological properties, developmental toxicity, and analgesic activity of gambogic acid, a novel natural anticancer agent, Drug Chem. Toxicol 33 (1) (2010) 88–96. [DOI] [PubMed] [Google Scholar]
- [129].Thomas KV, Brooks S, The environmental fate and effects of antifouling paint biocides, Biofouling 26 (1) (2010) 73–88. [DOI] [PubMed] [Google Scholar]
- [130].Liu N, Liu C, Li X, Liao S, Song W, Yang C, Zhao C, Huang H, Guan L, Zhang P, Liu S, Hua X, Chen X, Zhou P, Lan X, Yi S, Wang S, Wang X, Dou QP, Liu J, A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases, Sci. Rep 4 (2014) 5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Bailey P, Arrowsmith C, Darling K, Dexter J, Eklund J, Lane A, Little C, Murray A, Scott A, Williams A, Wilson D, A double-blind randomized vehicle-controlled clinical trial investigating the effect of ZnPTO dose on the scalp vs. Antidandruff efficacy and antimycotic activity, Int. J. Cosmet. Sci 25 (4) (2003) 183–188. [DOI] [PubMed] [Google Scholar]
- [132].Guthery E, Seal LA, Anderson EL, Zinc pyrithione in alcohol-based products for skin antisepsis: persistence of antimicrobial effects, Am. J. Infect. Control 33 (1) (2005) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Pierard-Franchimont C, Goffin V, Decroix J, Pierard GE, A multicenter randomized trial of ketoconazole 2% and zinc pyrithione 1% shampoos in severe dandruff and seborrheic dermatitis, Skin Pharmacol. Appl. Skin Physiol 15 (6) (2002) 434–441. [DOI] [PubMed] [Google Scholar]
- [134].Magda D, Lecane P, Wang Z, Hu W, Thiemann P, Ma X, Dranchak PK, Wang X, Lynch V, Wei W, Csokai V, Hacia JG, Sessler JL, Synthesis and anticancer properties of water-soluble zinc ionophores, Cancer Res. 68 (13) (2008) 5318–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Rudolf E, Cervinka M, Zinc pyrithione induces cellular stress signaling and apoptosis in Hep-2 cervical tumor cells: the role of mitochondria and lysosomes, Biometals 23 (2) (2010) 339–354. [DOI] [PubMed] [Google Scholar]
- [136].Zhao C, Chen X, Yang C, Zang D, Lan X, Liao S, Zhang P, Wu J, Li X, Liu N, Liao Y, Huang H, Shi X, Jiang L, Liu X, Dou QP, Wang X, Liu J, Repurposing an antidandruff agent to treating cancer: zinc pyrithione inhibits tumor growth via targeting proteasome-associated deubiquitinases, Oncotarget 8 (2017) 13942–13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhao C, Chen X, Zang D, Lan X, Liao S, Yang C, Zhang P, Wu J, Li X, Liu N, Liao Y, Huang H, Shi X, Jiang L, Liu X, He Z, Wang X, Liu J, Platinum-containing compound platinum pyrithione is stronger and safer than cisplatin in cancer therapy, Biochem. Pharmacol 116 (2016) 22–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Marqués-Gallego P, Contaldi S, Dulk HD, Monari M, Brouwer J, Jaehde U, Kalayda GV, Reedijk J, Relevance of the leaving group for antitumor activity of new platinum(II) compounds containing anthracene derivatives as a carrier ligand, J. Inorg. Biochem 103 (12) (2009) 1602–1608. [DOI] [PubMed] [Google Scholar]
- [139].Zhao C, Chen X, Zang D, Lan X, Liao S, Yang C, Zhang P, Wu J, Li X, Liu N, Liao Y, Huang H, Shi X, Jiang L, Liu X, He Z, Dou QP, Wang X, Liu J, A novel nickel complex works as a proteasomal deubiquitinase inhibitor for cancer therapy, Oncogene 35 (45) (2016) 5916–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lan X, Zhao C, Chen X, Zhang P, Zang D, Wu J, Chen J, Long H, Yang L, Huang D, Carter BZ, Wang X, Shi X, Liu J, Nickel pyrithione induces apoptosis in chronic myeloid leukemia cells resistant to imatinib via both Bcr/Abl-dependent and Bcr/Abl-independent mechanisms, J. Hematol. Oncol 9 (1) (2016) 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Huaizhi Z, Yuantao N, China’s ancient gold drugs, Gold Bull. 34 (1) (2001) 24–29. [Google Scholar]
- [142].Koch R, An address on bacteriological research, Br. Med. J 2 (1546) (1890) 380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Forestier J, L’aurothérapie dans les rhumatismes chroniques, Bull Mém Soc Méd Hôp Paris 53 (1929) 323–327. [Google Scholar]
- [144].Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP, A novel anticancer Gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts, Cancer Res. 66 (21) (2006) 10478–10486. [DOI] [PubMed] [Google Scholar]
- [145].Zhang JJ, Ng KM, Lok CN, Sun RW, Che CM, Deubiquitinases as potential anti-cancer targets for gold(III) complexes, Chem. Commun 49 (45) (2013) 5153–5155. [DOI] [PubMed] [Google Scholar]
- [146].Milacic V, Dou QP, The tumor proteasome as a novel target for gold(III) complexes: implications for breast cancer therapy, Coord. Chem. Rev 253 (11-12) (2009) 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Roder C, Thomson MJ, Auranofin: repurposing an old drug for a golden new age, Drugs R&D 15 (1) (2015) 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Hu J, Zhang H, Cao M, Wang L, Wu S, Fang B, Auranofin enhances ibrutinib’s anticancer activity in EGFR-Mutant lung adenocarcinoma, Mol. Cancer Ther 17 (10) (2018) 2156–2163. [DOI] [PubMed] [Google Scholar]
- [149].Hou G, Liu P, Zhang S, Yang M, Liao J, Yang J, Hu Y, Jiang W, Wen S, Huang P, Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis, Cell Death Dis. 9 (2) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chen X, Shi X, Zhao C, Li X, Lan X, Liu S, Huang H, Liu N, Liao S, Zang D, Song W, Liu Q, Carter BZ, Dou QP, Wang X, Liu J, Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms, Oncotarget 5 (19) (2014) 9118–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Liu N, Li X, Huang H, Zhao C, Liao S, Yang C, Liu S, Song W, Lu X, Lan X, Chen X, Yi S, Xu L, Jiang L, Zhao C, Dong X, Zhou P, Li S, Wang S, Shi X, Dou PQ, Wang X, Liu J, Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth, Oncotarget 5 (14) (2014) 5453–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Chen X, Shi X, Wang X, Liu J, Novel use of old drug: anti-rheumatic agent auranofin overcomes imatinib-resistance of chronic myeloid leukemia cells, Cancer Cell Microenviron. 1 (6) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Huang H, Liao Y, Liu N, Hua X, Cai J, Yang C, Long H, Zhao C, Chen X, Lan X, Zang D, Wu J, Li X, Shi X, Wang X, Liu J, Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti-tumor effects in vitro and in vivo, Oncotarget 7 (3) (2016) 2796–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Li X, Huang Q, Long H, Zhang P, Su H, Liu J, A new gold(I) complex-Au(PPh3) PT is a deubiquitinase inhibitor and inhibits tumor growth, EBioMedicine 39 (2019) 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, et al. , Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1, Nat. Cell Biol 8 (2006) 994–1002. [DOI] [PubMed] [Google Scholar]
- [156].Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, et al. , A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer, Cancer Cell 24 (2013) 791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Lu X, Nowicka U, Sridharan V, Liu F, Randles L, Hymel D, et al. , Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets, Nat. Commun 8 (2017) 15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Song Y, Ray A, Li S, Das DS, Tai YT, Carrasco RD, et al. , Targeting proteasome ubiquitin receptor Rpn13 in multiple myeloma, Leukemia 30 (2016) 1877–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Fimognari C, Turrini E, Ferruzzi L, Lenzi M, Hrelia P, Natural isothiocyanates: genotoxic potential versus chemoprevention, Mutat. Res 750 (2) (2012) 107–131. [DOI] [PubMed] [Google Scholar]
- [160].Gründemann C, Huber R, Chemoprevention with isothiocyanates – from bench to bedside, Cancer Lett. 414 (2018) 26–33. [DOI] [PubMed] [Google Scholar]
- [161].Singh SV, Singh K, Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research, Carcinogenesis 33 (10) (2012) 1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Palliyaguru DL, Yuan J, Kensler TW, Fahey JW, Isothiocyanates: translating the power of plants to people, Mol. Nutr. Food Res 62 (18) (2018) 1700965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Choi S, Lew KL, Xiao H, Herman-Antosiewicz A, Xiao D, Brown CK, Singh SV, D,L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1, Carcinogenesis 28 (1) (2007) 151–162. [DOI] [PubMed] [Google Scholar]
- [164].Dinkova-Kostova AT, Rostov RV, Glucosinolates and isothiocyanates in health and disease, Trends Mol. Med 18 (6) (2012) 337–347. [DOI] [PubMed] [Google Scholar]
- [165].Martin SL, Royston KJ, Tollefsbol TO, The role of non-coding RNAs and isothiocyanates in cancer, Mol. Nutr. Food Res 62 (18) (2018) 1700913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Hu R, Xu C, Shen G, Jain M, Khor T, Gopalkrishnan A, Lin W, Reddy B, Chan J, Kong A, Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice, Cancer Lett. 243 (2) (2006) 170–192. [DOI] [PubMed] [Google Scholar]
- [167].Mi L, Gan N, Chung FL, Isothiocyanates inhibit proteasome activity and proliferation of multiple myeloma cells, Carcinogenesis 32 (2) (2011) 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Lawson AP, Long MJC, Coffey RT, Qian Y, Weerapana E, El Oualid F, Hedstrom L, Naturally occurring isothiocyanates exert anticancer effects by inhibiting deubiquitinating enzymes, Cancer Res. 75 (23) (2015) 5130–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Lawson AP, Bak DW, Shannon DA, Long M, Vijaykumar T, Yu R, Oualid FE, Weerapana D, Hedstrom L, Identification of deubiquitinase targets of isothiocyanates using SILAC-assisted quantitative mass spectrometry, Oncotarget 8 (31) (2017) 51296–51316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D, Enhancement of proteasome activity by a small-molecule inhibitor of USP14, Nature 467 (7312) (2010) 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kiprowska MJ, Stepanova A, Todaro DR, Galkin A, Haas A, Wilson SM, Figueiredo-Pereira ME, Neurotoxic mechanisms by which the USP14 inhibitor IU1 depletes ubiquitinated proteins and Tau in rat cerebral cortical neurons: relevance to Alzheimer’s disease, Biochim. Biophys. Acta Mol. Basis Dis 1863 (6) (2017)1157–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Liao Y, Liu N, Hua X, Cai J, Xia X, Wang X, Huang H, Liu J, Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor, Cell Death Dis. 8 (2) (2017) e2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Liao Y, Xia X, Liu N, Cai J, Guo Z, Li Y, Jiang L, Dou QP, Tang D, Huang H, Liu J, Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination, Oncogene 37 (14) (2018) 1896–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Xia X, Huang C, Liao Y, Liu Y, He J, Guo Z, Jiang L, Wang X, Liu J, Huang H, Inhibition of USP14 enhances the sensitivity of breast cancer to enzalutamide, J. Exp. Clin. Cancer Res 38 (1) (2019) 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Xu D, Shan B, Sun H, Xiao J, Zhu K, Xie X, Li X, Liang W, Lu X, Qian L, Yuan J, USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1, Genes Dev. 30 (15) (2016) 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Adelakun N, Obaseki I, Obaseki E, Adeniyi A, Fapohunda O, Omotuyi O, Discovery of new promising USP14 inhibitors: computational evaluation of the thumb-palm pocket, Preprints (2019) 2019120306. [DOI] [PubMed] [Google Scholar]
- [177].Wang Y, Jiang Y, Ding S, Li J, Song N, Ren Y, Hong D, Wu C, Li B, Wang F, He W, Wang J, Mei Z, Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade, Cell Res. 28 (12) (2018) 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, Nguyen K, Vasile S, Suyama E, Smith LH, Sergienko E, Pinkerton AB, Chung T, Palmer AE, Pass I, Hess S, Cohen SM, Deshaies RJ, Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11, Nat. Chem. Biol 13 (5) (2017) 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Perez C, Li J, Parlati F, Rouffet M, Ma Y, Mackinnon AL, Chou TF, Deshaies RJ, Cohen SM, Discovery of an inhibitor of the proteasome subunit Rpn11, J. Med. Chem 60 (4) (2017) 1343–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kumar V, Naumann M, Stein M, Computational studies on the inhibitor selectivity of human JAMM deubiquitinylases Rpn11 and CSN5, Front. Chem 6 (2018) 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Li B, Wever WJ, Walsh CT, Bowers AA, Dithiolopyrrolones: biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics, Nat. Prod. Rep 31 (7) (2014) 905–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Jimenez A, Tipper DJ, Davies J, Mode of action of thiolutin, an inhibitor of macromolecular synthesis in Saccharomyces cerevisiae, Antimicrob. Agents Chemother 3 (6) (1973) 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Khachatourians GG, Tipper DJ, Inhibition of messenger ribonucleic acid synthesis in Escherichia coli by thiolutin, J. Bacteriol 119 (3) (1974) 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Sivasubramanian N, Jayaraman R, Thiolutin resistant mutants of Escherichia coli are they RNA chain initiation mutants? Mol. Gen. Genet 145 (1) (1976) 89–96. [DOI] [PubMed] [Google Scholar]
- [185].Minamiguchi K, Kumagai H, Masuda T, Kawada M, Ishizuka M, Takeuchi T, Thiolutin, an inhibitor of HUVEC adhesion to vitronectin, reduces paxillin in HUVECs and suppresses tumor cell-induced angiogenesis, Int. J. Cancer 93 (3) (2001) 307–316. [DOI] [PubMed] [Google Scholar]
- [186].Dai S, Jia Y, Wu SL, Isenberg JS, Ridnour LA, Bandle RW, Wink DA, Roberts AD, Karger BL, Comprehensive characterization of heat shock protein 27 phosphorylation in human endothelial cells stimulated by the microbial dithiole thiolutin, J. Proteome Res 7 (10) (2008) 4384–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lauinger L, Li J, Shostak A, Cemel IA, Ha N, Zhang Y, Merkl PE, Obermeyer S, Stankovic-Valentin N, Schafmeier T, Wever WJ, Bowers AA, Carter KP, Palmer AE, Tschochner H, Melchior F, Deshaies RJ, Brunner M, Diernfellner A, Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases, Nat. Chem. Biol 13 (7) (2017) 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Gardiner DM, Waring P, Howlett BJ, The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis, Microbiology 151 (Pt 4) (2005) 1021–1032. [DOI] [PubMed] [Google Scholar]
- [189].Reece KM, Richardson ED, Cook KM, Campbell TJ, Pisle ST, Holly AJ, Venzon DJ, Liewehr DJ, Chau CH, Price DK, Figg WD, Epidithiodiketopiperazines (ETPs) exhibit in vitro antiangiogenic and in vivo antitumor activity by disrupting the HIF-1alpha/p300 complex in a preclinical model of prostate cancer, Mol. Cancer 13 (2014) 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kowolik CM, Lin M, Xie J, Overman LE, Horne DA, NT1721, a novel epidithiodiketopiperazine, exhibits potent in vitro and in vivo efficacy against acute myeloid leukemia, Oncotarget 7 (2016) 86186–86197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A, Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9, Nat. Chem. Biol 1 (3) (2005) 143–145. [DOI] [PubMed] [Google Scholar]
- [192].Cook KM, Hilton ST, Mecinovic J, Motherwell WB, Figg WD, Schofield CJ, Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1alpha (HIF-1alpha) and p300 by a zinc ejection mechanism, J. Biol. Chem 284 (39) (2009) 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC, The anticancer agent chaetocin is a competitive substrate and inhibitor of thioredoxin reductase, Antioxid. Redox Signal 11 (5) (2009) 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Kroll M, Arenzana-Seisdedos F, Bachelerie F, Thomas D, Friguet B, Conconi M, The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome, Chem. Biol 6 (10) (1999) 689–698. [DOI] [PubMed] [Google Scholar]
- [195].Li J, Zhang Y, Da SSDS, Wang F, Ma Y, Perez C, Yang Y, Peng J, Cohen SM, Chou TF, Hilton ST, Deshaies RJ, Epidithiodiketopiperazines inhibit protein degradation by targeting proteasome deubiquitinase Rpn11, Cell Chem. Biol 25 (11) (2018) 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Zhou L, Jiang Y, Luo Q, Li L, Jia L, Neddylation: a novel modulator of the tumor microenvironment, Mol. Cancer 18 (2019) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Zhong HJ, Ma VP, Cheng Z, Chan DS, He HZ, Leung KH, Ma DL, Leung CH, Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening, Biochimie 94 (2012) 2457–2460. [DOI] [PubMed] [Google Scholar]
- [198].Li X, Elmira E, Rohondia S, Wang J, Liu J, Dou QP, A patent review of the ubiquitin ligase system: 2015-2018, Expert Opin. Ther. Pat 28 (2018) 919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Hong J, Luesch H, Largazole: from discovery to broad-spectrum therapy, Nat. Prod. Rep 29 (2012) 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Ungermannova D, Parker SJ, Nasveschuk CG, Wang W, Quade B, Zhang G, Kuchta RD, Phillips AJ, Liu X, Largazole and its derivatives selectively inhibit ubiquitin activating enzyme (e1), PLoS One 7 (2012) e29208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J, Sanz I, Chen L, Zhao J, Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A, Blood 120 (2012) 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Dikshit A, Jin YJ, Degan S, Hwang J, Foster MW, Li CY, Zhang JY, UBE2N promotes melanoma growth via MEK/FRA1/SOX10 signaling, Cancer Res. 78 (2018) 6462–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Hodge CD, Edwards RA, Markin CJ, McDonald D, Pulvino M, Huen MS, Zhao J, Spyracopoulos L, Hendzel MJ, Glover JN, Covalent inhibition of Ubc13 affects ubiquitin signaling and reveals active site elements important for targeting, ACS Chem. Biol 10 (2015) 1718–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Cheng J, Fan YH, Xu X, Zhang H, Dou J, Tang Y, Zhong X, Rojas Y, Yu Y, Zhao Y, Vasudevan SA, Zhang H, Nuchtern JG, Kim ES, Chen X, Lu F, Yang J, A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways, Cell Death Dis. 5 (2014) e1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Gombodorj N, Yokobori T, Yoshiyama S, Kawabata-Iwakawa R, Rokudai S, Horikoshi D, Nishiyama M, Nakano T, Inhibition of ubiquitin-conjugating enzyme E2 may activate the degradation of hypoxia-inducible factors and, thus, overcome cellular resistance to radiation in colorectal cancer, Anticancer Res. 37 (2017) 2425–2436. [DOI] [PubMed] [Google Scholar]
- [206].Raje N, Anderson K, Thalidomide–a revival story, N. Engl. J. Med 341 (1999) 1606–1609. [DOI] [PubMed] [Google Scholar]
- [207].Marriott JB, Muller G, Dalgleish AG, Thalidomide as an emerging immunotherapeutic agent, Immunol. Today 20 (1999) 538–540. [DOI] [PubMed] [Google Scholar]
- [208].Stephens TD, Bunde CJ, Fillmore BJ, Mechanism of action in thalidomide teratogenesis, Biochem. Pharmacol 59 (2000) 1489–1499. [DOI] [PubMed] [Google Scholar]
- [209].Bartlett JB, Dredge K, Dalgleish AG, The evolution of thalidomide and its IMiD derivatives as anticancer agents, Nat. Rev. Cancer 4 (2004) 314–322. [DOI] [PubMed] [Google Scholar]
- [210].Svetlana Balkanov K, Sotirova T, Genadieva SS, Cevreska L, Stojanovik A, Balkanov T, Adverse effects of thalidomide administration, in patients with myeloma multiplex? Mater. Sociomed 26 (2014) 134–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Suppiah R, Srkalovic JG, Hussein MA, Immunomodulatory analogues of thalidomide in the treatment of multiple myeloma, Clin. Lymphoma Myeloma 6 (2006) 301–305. [DOI] [PubMed] [Google Scholar]
- [212].Arcioni F, Roncadori A, Di Battista V, Tura S, Covezzoli A, Cundari S, Mecucci C, M.S. Centres, Lenalidomide treatment of myelodysplastic syndromes with chromosome 5q deletion: results from the National Registry of the Italian Drug Agency, Eur. J. Haematol 101 (2018) 78–85. [DOI] [PubMed] [Google Scholar]
- [213].Fuchs O, Treatment of lymphoid and myeloid malignancies by immunomodulatory drugs, Cardiovasc. Hematol. Disord. Drug Targets 19 (2019) 51–78. [DOI] [PubMed] [Google Scholar]
- [214].Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H, Identification of a primary target of thalidomide teratogenicity, Science 327 (2010) 1345–1350. [DOI] [PubMed] [Google Scholar]
- [215].Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca D, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL, Thoma H, Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide, Nature 512 (2014) 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Lee J, Zhou P, DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase, Mol. Cell 26 (2007) 775–780. [DOI] [PubMed] [Google Scholar]
- [217].Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, Karasawa S, Carmel D, Jackson P, Abbasian M, Mahmoudi A, Cathers B, Rychak E, Gaidarova S, Chen R, Schafer PH, Handa H, Daniel TO, Evans JF, Chopra R, Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide, Leukemia 26 (2012) 2326–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, Ito T, Ando H, Waldman MF, Thakurta A, Klippel A, Handa H, Daniel TO, Schafer PH, Chopra R, Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.), Br. J. Haematol 164 (2014) 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL, Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells, Science 343 (2014) 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG Jr, The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins, Science 343 (2014) 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Collins I, Wang H, Caldwell JJ, Chopra R, Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway, Biochem. J 474 (2017) 1127–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM, Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4, Chem. Biol 22 (2015) 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Duan Y, Guan Y, Qin W, Zhai X, Yu B, Liu H, Targeting Brd4 for cancer therapy: inhibitors and degraders, Medchemcomm 9 (2018) 1779–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong D, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG, PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer, Proc Natl Acad Sci U S A 113 (2016) 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Saenz DT, Fiskus W, Qian Y, Manshouri T, Rajapakshe K, Raina K, Coleman DG, Crew AP, Shen A, Mill CP, Sun B, Qiu P, Kadia TM, Pemmaraju N, DiNardo C, Kim MS, Nowak AJ, Coarfa C, Crews CM, Verstovsek S, Bhalla KN, Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells, Leukemia 31 (2017) 1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Lim SL, Damnernsawad A, Shyamsunder P, Chng WJ, Han BC, Xu L, Pan J, Pravin DP, Alkan S, Tyner JW, Koeffler HP, Proteolysis targeting chimeric molecules as therapy for multiple myeloma: efficacy, biomarker and drug combinations, Haematologica (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Geelen MJ, Gibson DM, Rodwell VW, Hydroxymethylglutaryl-CoA reductase-the rate-limiting enzyme of cholesterol biosynthesis. A report of a meeting held at Nijenrode Castle, Breukelen, The Netherlands, August 24, 1985, FEBS Lett. 201 (1986) 183–186. [DOI] [PubMed] [Google Scholar]
- [228].Gupta S, LDL cholesterol, statins and PCSK 9 inhibitors, Indian Heart J. 67 (2015) 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Wojcik C, Bury M, Stoklosa T, Giermasz A, Feleszko W, Mlynarczuk I, Pleban A, Basak G, Omura S, Jakobisiak M, Lovastatin and simvastatin are modulators of the proteasome, Int. J. Biochem. Cell Biol 32 (2000) 957–965. [DOI] [PubMed] [Google Scholar]
- [230].Murray SS, Tu KN, Young KL, Murray EJ, The effects of lovastatin on proteasome activities in highly purified rabbit 20S proteasome preparations and mouse MC3T3-E1 osteoblastic cells, Metabolism 51 (2002) 1153–1160. [DOI] [PubMed] [Google Scholar]
- [231].Vosper J, Masuccio A, Kullmann M, Ploner C, Geley S, Hengst L, Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation, Oncotarget 6 (2015) 2889–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY, Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells, Cell Death Dis. 8 (2017) e2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Frescas D, Pagano M, Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer, Nat. Rev. Cancer 8 (2008) 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI, A cell cycle-regulated inhibitor of cyclin-dependent kinases, Proc. Natl. Acad. Sci. U. S. A 91 (1994) 5291–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Gray-Bablin J, Rao S, Keyomarsi K, Lovastatin induction of cyclin-dependent kinase inhibitors in human breast cells occurs in a cell cycle-independent fashion, Cancer Res. 57 (1997) 604–609. [PubMed] [Google Scholar]
- [236].Danesh FR, Sadeghi MM, Amro N, Philips C, Zeng L, Lin S, Sahai A, Kanwar YS, 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/p21 signaling pathway: implications for diabetic nephropathy, Proc. Natl. Acad. Sci. U. S. A 99 (2002) 8301–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM, Statins and cancer prevention, Nat. Rev. Cancer 5 (2005) 930–942. [DOI] [PubMed] [Google Scholar]
- [238].Undela K, Shah CS, Mothe R.K.J.W.Jo.M.-A.. Statin use and risk of cancer:An overview of meta-analyses, 12 (2017) 590–594. [Google Scholar]
- [239].Lee HS, Lee SH, Lee HJ, Chung MJ, Park JY, Park SW, Song SY, Bang S, Statin use and its impact on survival in pancreatic cancer patients, Medicine (Baltimore) 95 (2016) e3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Lin JJ, Ezer N, Sigel K, Mhango G, Wisnivesky JP, The effect of statins on survival in patients with stage IV lung cancer, Lung Cancer 99 (2016) 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Linden KG, Leachman SA, Zager JS, Jakowatz JG, Viner JL, McLaren CE, Barr RJ, Carpenter PM, Chen WP, Elmets CA, Tangrea JA, Lim SJ, Cochran AJ, Meyskens FL Jr, A randomized, double-blind, placebo-controlled phase II clinical trial of lovastatin for various endpoints of melanoma pathobiology, Cancer Prev. Res. (Phila) 7 (2014) 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Kim WS, Kim MM, Choi HJ, Yoon SS, Lee MH, Park K, Park CH, Kang WK, Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma, Invest. New Drugs 19 (2001) 81–83. [DOI] [PubMed] [Google Scholar]
- [243].Sondergaard TE, Pedersen PT, Andersen TL, Soe K, Lund T, Ostergaard B, Garnero P, Delaisse JM, Plesner T, A phase II clinical trial does not show that high dose simvastatin has beneficial effect on markers of bone turnover in multiple myeloma, Hematol. Oncol 27 (2009) 17–22. [DOI] [PubMed] [Google Scholar]
- [244].Lopez-Aguilar E, Sepulveda-Vildosola AC, Betanzos-Cabrera Y, Rocha-Moreno YG, Gascon-Lastiri G, Rivera-Marquez H, Wanzke-del-Angel V, Cerecedo-Diaz F, de la Cruz-Yanez H, Phase II study of metronomic chemotherapy with thalidomide, carboplatin-vincristine-fluvastatin in the treatment of brain stem tumors in children, Arch. Med. Res 39 (2008) 655–662. [DOI] [PubMed] [Google Scholar]
- [245].Abdel-Rahman O, Statin treatment and outcomes of metastatic pancreatic cancer: a pooled analysis of two phase III studies, Clin. Transl. Oncol (2018). [DOI] [PubMed] [Google Scholar]
- [246].Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, Kim HT, Lee JS, A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer, Clin. Cancer Res 17 (2011) 1553–1560. [DOI] [PubMed] [Google Scholar]
- [247].Lee J, Jung KH, Park YS, Ahn JB, Shin SJ, Im SA, Oh DY, Shin DB, Kim TW, Lee N, Byun JH, Hong YS, Park JO, Park SH, Lim HY, Kang WK, Simvastatin plus irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) as first-line chemotherapy in metastatic colorectal patients: a multicenter phase II study, Cancer Chemother. Pharmacol 64 (2009) 657–663. [DOI] [PubMed] [Google Scholar]
- [248].Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y, Matsuzawa Y, Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial, Br J Cancer 84 (2001) 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Sauter C, Cytostatic activity of commonly used tricyclic antidepressants, Oncology 46 (1989) 155–157. [DOI] [PubMed] [Google Scholar]
- [250].Rossi M, Rotblat B, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, Cavasotto CN, Knight RA, Ciechanover A, Melino G, High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy, Cell Death Dis. 5 (2014) e1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Maillard I, Pear WS, Notch and cancer: best to avoid the ups and downs, Cancer Cell 3 (2003) 203–205. [DOI] [PubMed] [Google Scholar]
- [252].Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G, The ubiquitin-protein ligase Itch regulates p73 stability, EMBO J. 24 (2005) 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, Croce CM, Melino G, The E3 ubiquitin ligase Itch controls the protein stability of p63, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 12753–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Ho KC, Zhou Z, She YM, Chun A, Cyr TD, Yang X, Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected], Proc. Natl. Acad. Sci. U. S. A 108 (2011) 4870–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Suryaraja R, Anitha M, Anbarasu K, Kumari G, Mahalingam S, The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner, Cell Death Dis. 4 (2013) e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Li PF, Zhang QG, Inhibition of ITCH suppresses proliferation and induces apoptosis of lung cancer cells, Cell. Physiol. Biochem 48 (2018) 1703–1709. [DOI] [PubMed] [Google Scholar]
- [257].Tsuruo T, Iida H, Nojiri M, Tsukagoshi S, Sakurai Y, Potentiation of chemotherapeutic effect of vincristine in vincristine resistant tumor bearing mice by calmodulin inhibitor clomipramine, J. Pharmacobio-Dyn 6 (1983) 145–147. [DOI] [PubMed] [Google Scholar]
- [258].Pommerenke EW, Volm M, Reversal of doxorubicin-resistance in solid tumors by clomipramine, In Vivo 9 (1995) 99–101. [PubMed] [Google Scholar]
- [259].Xia Z, Bergstrand A, DePierre JW, Nassberger L, The antidepressants imipramine, clomipramine, and citalopram induce apoptosis in human acute myeloid leukemia HL-60 cells via caspase-3 activation, J. Biochem. Mol. Toxicol 13 (1999) 338–347. [DOI] [PubMed] [Google Scholar]
- [260].Sauter C, Cytostatic activity of commonly used tricyclic antidepressants, Oncology 46 (1989) 155–157. [DOI] [PubMed] [Google Scholar]
- [261].Beaney R, Gullan R, Pilkington G, Therapeutic potential of antidepressants in malignant glioma: clinical experience with clomipramine, J. Clin. Oncol 23 (2005) 1535–1535. [Google Scholar]
- [262].Bongiorno-Borbone L, Giacobbe A, Compagnone M, Eramo A, De Maria R, Peschiaroli A, Melino G, Anti-tumoral effect of desmethylclomipramine in lung cancer stem cells, Oncotarget 6 (2015) 16926–16938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Li X, Elmira E, Rohondia S, Wang J, Liu J, Dou QP, A patent review of the ubiquitin ligase system: 2015-2018, Expert Opin. Ther. Pat 28 (2018) 919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Tan G, Qiu M, Chen L, Zhang S, Ke L, Liu J, JS-K, a nitric oxide pro-drug, regulates growth and apoptosis through the ubiquitin-proteasome pathway in prostate cancer cells, BMC Cancer 17 (2017) 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Maciag AE, Saavedra JE, Chakrapani H, The nitric oxide prodrug JS-K and its structural analogues as cancer therapeutic agents, Anticancer Agents Med. Chem 9 (2009) 798–803. [DOI] [PubMed] [Google Scholar]
- [266].Stevens M, Abdeen S, Salim N, Ray AM, Washburn A, Chitre S, Sivinski J, Park Y, Hoang QQ, Chapman E, Johnson SM, HSP60/10 chaperonin systems are inhibited by a variety of approved drugs, natural products, and known bioactive molecules, Bioorg. Med. Chem. Lett 29 (2019) 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Wu K, Chong RA, Yu Q, Bai J, Spratt DE, Ching K, Lee C, Miao H, Tappin I, Hurwitz J, Zheng N, Shaw GS, Sun Y, Felsenfeld DP, Sanchez R, Zheng JN, Pan ZQ, Suramin inhibits cullin-RING E3 ubiquitin ligases, Proc. Natl. Acad. Sci. U. S. A 113 (2016) E2011–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Xin Y, Lyness G, Chen D, Song S, Wientjes MG, Au JL, Low dose suramin as a chemosensitizer of bladder cancer to mitomycin C, J. Urol 174 (2005) 322–327. [DOI] [PubMed] [Google Scholar]
- [269].Gan Y, Lu J, Yeung BZ, Cottage CT, Wientjes MG, Au JL, Pharmacodynamics of telomerase inhibition and telomere shortening by noncytotoxic suramin, AAPS J. 17 (2015) 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Wattenberg LW, Inhibition of dimethylhydrazine-induced neoplasia of the large intestine by disulfiram, J. Natl. Cancer Inst 54 (1975) 1005–1006. [DOI] [PubMed] [Google Scholar]
- [271].Jiao Y, Hannafon BN, Ding WQ, Disulfiram’s anticancer activity: evidence and mechanisms, Anticancer Agents Med. Chem 16 (2016) 1378–1384. [DOI] [PubMed] [Google Scholar]
- [272].Chen D, Cui QC, Yang H, Dou QP, Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity, Cancer Res. 66 (2006) 10425–10433. [DOI] [PubMed] [Google Scholar]
- [273].Lovborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R, Inhibition of proteasome activity, nuclear factor-KappaB translocation and cell survival by the antialcoholism drug disulfiram, Int. J. Cancer 118 (2006) 1577–1580. [DOI] [PubMed] [Google Scholar]
- [274].Rickardson L, Wickstrom M, Larsson R, Lovborg H, Image-based screening for the identification of novel proteasome inhibitors, J. Biomol. Screen 12 (2007) 203–210. [DOI] [PubMed] [Google Scholar]
- [275].Cvek B, Milacic V, Taraba J, Dou QP, Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells, J. Med. Chem 51 (2008) 6256–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Pang H, Chen D, Cui QC, Dou QP, Sodium diethyldithiocarbamate, an AIDS progression inhibitor and a copper-binding compound, has proteasome-inhibitory and apoptosis-inducing activities in cancer cells, Int. J. Mol. Med 19 (2007) 809–816. [PubMed] [Google Scholar]
- [277].Hasinoff BB, Patel D, Disulfiram is a slow-binding partial noncompetitive inhibitor of 20S proteasome activity, Arch. Biochem. Biophys 633 (2017) 23–28. [DOI] [PubMed] [Google Scholar]
- [278].Sunderman FW, Efficacy of sodium diethyldithiocarbamate (dithiocarb) in acute nickel carbonyl poisoning, Ann. Clin. Lab. Sci 9 (1979) 1–10. [PubMed] [Google Scholar]
- [279].Han J, Liu L, Yue X, Chang J, Shi W, Hua Y, A binuclear complex constituted by diethyldithiocarbamate and copper(I) functions as a proteasome activity inhibitor in pancreatic cancer cultures and xenografts, Toxicol. Appl. Pharmacol 273 (2013) 477–483. [DOI] [PubMed] [Google Scholar]
- [280].Medvetz DA, Hindi KM, Panzner MJ, Ditto AJ, Yun YH, Youngs WJ, Anticancer activity of Ag(I) N-Heterocyclic carbene complexes derived from 4,5-Dichloro-1H-Imidazole, Met. Drugs 2008 (2008) 384010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Kaplan A, Akalin CG, Kutlu HM, Cytotoxic, anti-proliferative and apoptotic effects of silver nitrate against H-ras transformed 5RP7, Cytotechnology 68 (5) (2016) 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Banti CN, Hadjikakou SK, Anti-proliferative and anti-tumor activity of silver(I) compounds, Metallomics 5 (6) (2013) 569–596. [DOI] [PubMed] [Google Scholar]
- [283].Chen X, Yang Q, Chen J, Zhang P, Huang Q, Zhang X, Yang L, Xu D, Zhao C, Wang X, Liu J, Inhibition of proteasomal deubiquitinase by silver complex induces apoptosis in non-small cell lung cancer cells, Cell. Physiol. Biochem 49 (2) (2018) 780–797. [DOI] [PubMed] [Google Scholar]
- [284].Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA, Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells, J. Exp. Med 175 (1992) 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Kim I, Kim CH, Kim JH, Lee J, Choi JJ, Chen ZA, Lee MG, Chung KC, Hsu CY, Ahn YS, Pyrrolidine dithiocarbamate and zinc inhibit proteasome-dependent proteolysis, Exp. Cell Res 298 (2004) 229–238. [DOI] [PubMed] [Google Scholar]
- [286].Chen D, Peng F, Cui QC, Daniel KG, Orlu S, Liu J, Dou QP, Inhibition of prostate cancer cellular proteasome activity by a pyrrolidine dithiocarbamatecopper complex is associated with suppression of proliferation and induction of apoptosis, Front. Biosci 10 (2005) 2932–2939. [DOI] [PubMed] [Google Scholar]
- [287].Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP, Pyrrolidine dithiocarbamate-zinc(II) and -copper(II) complexes induce apoptosis in tumor cells by inhibiting the proteasomal activity, Toxicol. Appl. Pharmacol 231 (2008) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Zhang X, Frezza M, Milacic V, Ronconi L, Fan Y, Bi C, Fregona D, Dou QP, Inhibition of tumor proteasome activity by gold-dithiocarbamato complexes via both redox-dependent and -independent processes, J. Cell. Biochem 109 (2010) 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Buac D, Schmitt S, Ventro G, Kona FR, Dou QP, Dithiocarbamate-based coordination compounds as potent proteasome inhibitors in human cancer cells, Mini Rev. Med. Chem 12 (2012) 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, Kraus M, Michalova M, Vaclavkova J, Dzubak P, Vrobel I, Pouckova P, Sedlacek J, Miklovicova A, Kutt A, Li J, Mattova J, Driessen C, Dou QP, Olsen J, Hajduch M, Cvek B, Deshaies RJ, Bartek J, Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4, Nature 552 (2017) 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Dantuma NP, Hoppe T, Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin-dependent extraction from chromatin, Trends Cell Biol. 22 (2012) 483–491. [DOI] [PubMed] [Google Scholar]
- [292].Bodnar N, Rapoport T, Toward an understanding of the Cdc48/p97 ATPase, F1000Res 6 (2017) 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Torrecilla I, Oehler J, Ramadan K, The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks, Philos. Trans. R. Soc. Lond., B, Biol. Sci 372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Xia D, Tang WK, Ye Y, Structure and function of the AAA+ ATPase p97/Cdc48p, Gene 583 (2016) 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, Young P, A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation, J. Mol. Biol 394 (2009) 732–746. [DOI] [PubMed] [Google Scholar]
- [296].Pye VE, Beuron F, Keetch CA, McKeown C, Robinson CV, Meyer HH, Zhang X, Freemont PS, Structural insights into the p97-Ufd1-Np14 complex, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Ye Y, Meyer HH, Rapoport TA, Function of the p97-Ufd1-Np14 complex in retro translocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains, J. Cell Biol 162 (2003) 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G, A complex of mammalian ufd1 and np14 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways, EMBO J. 19 (2000) 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ, Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Wang Q, Li L, Ye Y, Inhibition of p97-dependent protein degradation by Eeyarestatin I, J. Biol. Chem 283 (2008) 7445–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Safi R, Nelson ER, Chitneni SK, Franz KJ, George DJ, Zalutsky MR, McDonnell DP, Copper signaling axis as a target for prostate cancer therapeutics, Cancer Res. 74 (2014) 5819–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G, ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome, Cell Stem Cell 1 (2007) 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Marcato P, Dean CA, Giacomantonio CA, Lee PW, Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform, Cell Cycle 10 (2011) 1378–1384. [DOI] [PubMed] [Google Scholar]
- [304].Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, Helyer L, Pan L, Leidal A, Gujar S, Giacomantonio CA, Lee PW, Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis, Stem Cells 29 (2011) 32–45. [DOI] [PubMed] [Google Scholar]
- [305].Liu P, Kumar IS, Brown S, Kannappan V, Tawari PE, Tang JZ, Jiang W, Armesilla AL, Darling JL, Wang W, Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells, Br. J. Cancer 109 (2013) 1876–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Wang Y, Li W, Patel SS, Cong J, Zhang N, Sabbatino F, Liu X, Qi Y, Huang P, Lee H, Taghian A, Li JJ, DeLeo AB, Ferrone S, Epperly MW, Ferrone CR, Ly A, Brachtel EF, Wang X, Blocking the formation of radiation-induced breast cancer stem cells, Oncotarget 5 (2014) 3743–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Wu L, Meng F, Dong L, Block CJ, Mitchell AV, Wu J, Jang H, Chen W, Polin L, Yang Q, Dou QP, Wu G, Disulfiram and BKM120 in combination with chemotherapy impede tumor progression and delay tumor recurrence in tumor initiating cell-rich TNBC, Sci. Rep 9 (2019) 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Kohn FR, Landkamer GJ, Manthey CL, Ramsay NK, Sladek NE, Effect of aldehyde dehydrogenase inhibitors on the ex vivo sensitivity of human multipotent and committed hematopoietic progenitor cells and malignant blood cells to oxazaphosphorines, Cancer Res. 47 (1987) 3180–3185. [PubMed] [Google Scholar]
- [309].Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, Guichet PO, Bian X, Armesilla AL, Darling JL, Wang W, Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancerstem-like cells, Br. J. Cancer 107 (2012) 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, Wu C, Yang J, Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells, Oncotarget 7 (2016) 58516–58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Deng M, Jiang Z, Li Y, Zhou Y, Li J, Wang X, Yao Y, Wang W, Li P, Xu B, Effective elimination of adult B-lineage acute lymphoblastic leukemia by disulfiram/copper complex in vitro and in vivo in patient-derived xenograft models, Oncotarget 7 (2016) 82200–82212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Yang X, Yao R, Wang H, Update of ALDH as a potential biomarker and therapeutic target for AML, Biomed Res. Int 2018 (2018) 9192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Morgenroth A, Vogg AT, Zlatopolskiy BD, Siluschek M, Oedekoven C, Mottaghy AM, Breaking the invulnerability of cancer stem cells: two-step strategy to kill the stem-like cell subpopulation of multiple myeloma, Mol. Cancer Ther 13 (2014) 144–153. [DOI] [PubMed] [Google Scholar]
- [314].Cong J, Wang Y, Zhang X, Zhang N, Liu L, Soukup K, Michelakos T, Hong T, DeLeo A, Cai L, Sabbatino F, Ferrone S, Lee H, Levina V, Fuchs B, Tanabe K, Lillemoe K, Ferrone C, Wang X, A novel chemoradiation targeting stem and nonstem pancreatic cancer cells by repurposing disulfiram, Cancer Lett. 409 (2017) 9–19. [DOI] [PubMed] [Google Scholar]
- [315].Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF, Prospective identification of tumorigenic breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H, CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis, Breast Cancer Res. 8 (2006) R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, Hong S, Adams A, D’Angelo R, Ginestier C, Charafe-Jauffret D, Clouthier SG, Birnbaum D, Wong ST, Zhan M, Chang JC, Wicha MS, Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts, Stem Cell Rep. 2 (2014) 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Adams A, Warner K, Pearson AT, Zhang Z, Kim HS, Mochizuki D, Basura G, Helman J, Mantesso A, Castilho RM, Wicha MS, Nor JE, ALDH/CD44 identifies uniquely tumorigenic cancer stem cells in salivary gland mucoepidermoid carcinomas, Oncotarget 6 (2015) 26633–26650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Lin L, Hutzen B, Lee HF, Peng Z, Wang W, Zhao C, Lin HJ, Sun D, Li PK, Li C, Korkaya H, Wicha MS, Lin J, Evaluation of STAT3 signaling in ALDH+ and ALDH+/CD44+/CD24− subpopulations of breast cancer cells, PLoS One 8 (2013) e82821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Colacino JA, Azizi E, Brooks MD, Harouaka R, Fouladdel S, McDermott SP, Lee M, Hill D, Madden J, Boerner J, Cote ML, Sartor MA, Rozek LS, Wicha MS, Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling, Stem Cell Rep. 10 (2018) 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, Thompson EW, Findlay JK, Ahmed N, Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden, Mol. Cancer 12 (2013) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL, Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy, PLoS One 3 (2008) e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM, Therapy-induced enrichment of putative lung cancer stem-like cells, Int. J. Cancer 134 (2014) 1270–1278. [DOI] [PubMed] [Google Scholar]
- [324].Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, Zhang H, Semenza GL, Chemotherapy-induced Ca(2+) release stimulates breast cancer stem cell enrichment, Cell Rep. 18 (2017) 1946–1957. [DOI] [PubMed] [Google Scholar]
- [325].Huang J, Campian JL, Gujar AD, Tran DD, Lockhart AC, DeWees TA, Tsien CI, Kim AH, A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy, J. Neurooncol 128 (2016) 259–266. [DOI] [PubMed] [Google Scholar]
- [326].Huang J, Chaudhary R, Cohen AL, Fink K, Goldlust S, Boockvar J, Chinnaiyan P, Wan L, Marcus S, Campian JL, A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma, J. Neurooncol 142 (2019) 537–544. [DOI] [PubMed] [Google Scholar]
- [327].Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA, Yegnasubramanian S, Carducci MA, Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer, Prostate Cancer Prostatic Dis. 16 (2013) 357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]