

Abstract
Intraductal tubulopapillary neoplasm (ITPN) is a distinct precancerous lesion in the pancreas with unique clinical and molecular features. Although in vitro studies in two-dimensional culture have led to numerous important insights in pancreatic cancer, such models are currently lacking for precancerous lesions. In this study, we report the generation and characterization of a cell line from a human pancreatic ITPN. Neoplastic cells were initially cultured in a three-dimensional organoid system, followed by transfer to two-dimensional culture. RNA sequencing revealed a gene expression profile consistent with pancreatic ductal origin, and whole genome sequencing identified many somatic mutations (including in genes involved in DNA repair and WNT signaling) and structural rearrangements. In vitro characterization of the tumorigenic potential demonstrated a phenotype between that of normal pancreatic ductal cells and cancer cell lines. This cell line represents a valuable resource for interrogation of unique ITPN biology, as well as precancerous pancreatic lesions more generally.
INTRODUCTION
Intraductal tubulopapillary neoplasms (ITPNs) are cystic pancreatic intraductal neoplasms characterized by distinct clinical, morphological, and molecular features (1, 2). This lesion was first described in 1992 as “tubular adenoma of the main pancreatic duct”, and in 2010 ITPN was recognized by the World Health Organization as a subtype of premalignant intraductal neoplasm of the pancreas distinct from the more common intraductal papillary mucinous neoplasm (IPMN) (3, 4). ITPNs are uncommon lesions, accounting for less than 3% of all intraductal pancreatic neoplasms. They are morphologically and immunohistochemically distinct from other intraductal neoplasms, with tubulopapillary growth pattern, high-grade cytologic atypia, scarce mucin production, and frequent necrosis (1, 3). The neoplastic cells in ITPNs express cytokeratin as well as MUC1 and MUC6 but typically lack expression of MUC2 and MUC5AC, again highlighting their distinct features compared to IPMNs. Invasive carcinomas co-occur in approximately 40% of ITPNs, and thus like IPMNs, ITPNs are regarded as pancreatic cancer precursor lesions (5). Although examined cohorts are not large, the outcome of carcinoma arising from ITPN seems distinct from that of PDAC, as ITPN-associated carcinomas infrequently metastasize and often show favorable outcomes (1, 6). Genomic analyses have revealed a unique pattern of driver genes in ITPNs, which typically lack somatic alterations in genes commonly associated with ductal pancreatic tumorigenesis, including KRAS, GNAS, TP53, and SMAD4 (7, 8). Candidate drivers suggested in ITPNs include CDKN2A, genes involved in chromatin remodeling (MLL, PBRM1, ATRX), and genes of phosphatidylinositol 3-kinase (PI3K) pathway (PIK3CA, PTEN), among others (7). Recurrent fusions in FGFR2 have also been reported in ITPNs (7). Taken together, these data highlight that ITPNs represent a distinct premalignant pancreatic neoplasm with unique clinical and molecular features.
The development of appropriate disease models is essential for investigating pancreatic tumorigenesis prior to malignant transformation. Unfortunately, there are few cell lines with which to model pancreatic precursor lesions. Human pancreatic duct epithelial (HPDE) cells have been reported as a near-normal pancreatic duct epithelial cell line, but immortalization using HPV E6/E7 proteins leads to perturbations in pathways associated with high-grade pancreatic precursor lesions (p53 and RB pathways) (9). Although HPDE represents an invaluable resource, these alterations call into question how faithfully it can recapitulate “normal” pancreatic duct biology. Previous propagation of human IPMNs has been achieved in murine xenografts, with one IPMN subsequently established as a cell line after in vivo propagation (10). In addition, multiple groups have reported derivation of cell lines from invasive carcinomas arising from IPMNs (11, 12). The scarcity of in vitro models of human premalignant pancreatic neoplasms highlights several difficulties in the establishment of such systems. First, pancreatic precursor lesions are often an incidental finding at time of pancreatic resection for invasive carcinoma and as such are often only identified in examination of fixed tissue, when harvesting of living cells is no longer possible. Second, human pancreatic neoplasms, even invasive carcinomas, are challenging to propagate in vitro in two-dimensional culture (13). Because of these difficulties with two-dimensional cell culture approaches, alternative strategies for propagation of pancreatic precursor lesions (such as murine and chicken egg xenografts as well as three-dimensional culture methods) are currently being explored (14–16). While such strategies may improve our ability to propagate human premalignant neoplasms, they lack the ease of culture and molecular manipulation of two-dimensional cell culture.
In this study, we derived a novel cell line (H58) from a surgically resected, pathologically confirmed pancreatic ITPN. Neoplastic cells were initially cultured in a three-dimensional organoid system and subsequently propagated in two-dimensional culture over the course of >250 days and >30 passages. We report characterization of the new cell line with respect to its pancreatic ductal phenotype, in vitro correlates of malignancy, and molecular signatures. Our ITPN cell line represents a unique resource with which to interrogate this distinct premalignant neoplasm, as well as pancreatic cancer precursor lesions more generally.
MATERIALS AND METHODS
Institutional approval and informed consent
This study was reviewed and approved by the Johns Hopkins University Institutional Review Board. Written informed consent was obtained from the study participant.
Specimen collection and organoid derivation
Neoplastic tissue was collected from a patient with surgically resected ITPN, and organoids were isolated as previously described (17, 18). Briefly, tissue for culture was minced and incubated at 37°C for five hours in an Enviro Genie (Scientific Industries, Inc., NY) with 20 rpm rotation and 40 rpm rocking in advanced DMEM/F12 media (Invitrogen, catalog no. 11320033) containing collagenase type II 5 mg/mL (Life Technology, catalog no. 17101-015), dispase 1.25 mg/mL (Life Technology, catalog no. 17105-041), fetal bovine serum (FBS) (Gibco, catalog no. 26140079) 2.5%. After incubation, dissociated single cells were washed, mixed with Matrigel (BD Bioscience, catalog no. 356231), and seeded in Matrigel domes as described elsewhere (19). Matrigel domes were regularly supplemented with Human Feeding Media (HFM), which is based on AdvDMEM/F12 (Invitrogen, catalog no. 12491-015) supplemented with B27 (Invitrogen, catalog no. 17504044), 1.25 mM N-Acetylcysteine (Sigma, catalog no. A9165), 10 nM gastrin (Sigma, catalog no. G9020) and the growth factors: 50 ng/mL EGF (Peprotech, catalog no. 315-09), 10% RSPO1-conditioned media, 10% Noggin-conditioned media, 100 ng/mL FGF10 (Peprotech, catalog no. 100-26) and 10 mM Nicotinamide (Sigma, catalog no. 1094-61-7) (17, 18). When cell clusters reached confluence, Matrigel domes were broken down with ice-cold media, followed by organoid dissociation and transfer to fresh Matrigel. Passaging was performed in a 1:3–1:6 split ratio approximately once every two weeks.
Two-dimensional cell culture
After seven organoid passages, epithelial cell clusters were dissociated and split into both uncoated culture dishes and dishes coated with rat tail collagen type 1 (BD Bioscience, catalog no. 17100017) – both cultures were supplemented with 100% HFM. Over several passages, cells were gradually weaned from HFM and replaced with Advanced DMEM/F12 up to a ratio of 3:1 (AdvDMEM/F12:HFM). At higher ratios than 3:1, we observed increased vacuolization, cell death, and prolonged doubling times. H58 cells were maintained in two-dimensional culture over 30 passages and >250 days while supplementing AdvDMEM/F12:HFM (3:1) and regularly passaged after application of 0.25% Trypsin/EDTA at 70-90% confluence.
We kindly received HPDE cell line from the laboratory of Ming-Sound Tsao, MD, FRCPC, University Health Network, Canada. PANC-1 cells were purchased from ATCC (CRL-1469). PANC-1 was cultured according to ATCC recommendations with reference to the original article (20). HPDE was cultured as previously described (9). All 2D cell lines were tested before experimental use to confirm identity by short tandem repeat (STR) analysis (GenePrint 10 System, Promega) and negative mycoplasma status by PCR (MycoDetect, Greiner Bio-One) by the Johns Hopkins University Genetics Resources Core Facility.
Mice Xenografts
Five, 6-8 week-old, female nu/nu mice were obtained from Jackson Laboratories (strain no. 002019). 3.5×106 H58 cells suspended in 100 μL of culture media were injected into the flank of each mouse. Tumor volume and weight for each mouse was determined twice weekly for 20 weeks.
DNA and RNA extraction
DNA was extracted from H58 cells in 3D culture and fresh-frozen non-tumor tissue (duodenum) using the PureLink Genomic DNA Mini Extraction Kit (Invitrogen, catalog no. K182000). RNA was extracted from H58 cells in 3D culture using a PureLink RNA mini extraction kit (Invitrogen, catalog no. 12183018A). DNA and RNA were quantified using the Qubit dsDNA HS Assay Kit (Invitrogen, catalog no. Q32851) and Qubit RNA BR Assay Kit (Invitrogen, catalog no. Q10210) respectively, according to manufacturer’s protocols.
Analysis of KRAS and GNAS hotspots
DNA regions containing KRAS exon 2, KRAS exon 3, and GNAS exon 8 were amplified with OneTaq (NEB, catalog no. M0481) according to the manufacturer’s protocol with the following modifications: 1) for GNAS exon 2, an annealing temperature of 62 °C and 30 cycles were used, and 2) for KRAS exon 2 and KRAS exon 3, an annealing temperature of 64 oC and 40 cycles were. Primers for KRAS exon 2 were: 5’-CCCTGACATACTCCCAAGGA-3’ and 5’-TGTAAAACGACGGCCAGTGAGGGTGTGCTACAGGGTGT-3’. Primers for KRAS exon 3 were: 5’-TGTAAAACGACGGCCAGTCCTAGGTTTCAATCCCAGCA-3’ and 5’-CACCAGCAATGCACAAAGAT −3’. Primers for GNAS exon 8 were: 5’-TGTAAAACGACGGCCAGTCACCCCACGTGTCTTTCTTT-3’ and 5’-AAAGAACCACCGCAATGAAC-3’. PCR products were purified with a Qiagen PCR Purification Kit (Qiagen, catalog no.) and Sanger sequenced by Genewiz, Inc. (Plainsfield, NJ).
Whole Genome Sequencing
Whole genome sequencing was performed by the Next Generation Sequencing Core of the Sidney Kimmel Comprehensive Cancer Center of Johns Hopkins University. Libraries for whole genome sequencing were prepared from H58 and matched normal DNA using the TruSeq Nano DNA Kit (Illumina), followed by 2 X 150 bp paired-end sequencing using a HiSeq2500 instrument (Illumina). Bcl2fastq v2.15.0 was used to generate FASTQ files from BCL files. FASTQ files were aligned to the human genome (G) using bwa v.0.7.7 (mem). Read groups were added and duplicate reads removed using Picard-tools v.1.119. Base call recalibration was completed with GATK v.2.6.0 and BAM alignment files generated. Germline variants were called with HaplotypeCaller v.3.6.0. Somatic variants were called with MuTect2 v3.6.0. Structural variants (SVs) were called with lumpy v.0.2.11 and compared to normal using BEDtools. Germline variants and somatic mutations were annotated using ANNOVAR (v.2016-02-01) and FunSeq2 (v2.1.6). Somatic mutations in coding regions and non-coding regions with a FunSeq2 non-coding score > 1.5 were visually inspected in IGV (v2.4.8). SVs from Lumpy software were filtered to include only: 1) SVs not mapping to the mitochondrial genome, 2) SVs supported by at least 2 spanning paired-end reads (PE) and 2 split reads (SR), 3) SVs with an “Evidence Score” 0.0005 or less, and 4) SVs with PE/SR ratio between 1 and 3. Mutation signature and SVs profile were plotted by signeR (v1.4.0) and circlize (v0.4.6) packages, respectively. Default parameters were used unless otherwise specified.
Whole Exome Sequencing
DNA was extracted from archival formalin-fixed paraffin-embedded (FFPE) tissue samples from H58 ITPN (tumor) and duodenum (normal) using the QIAamp DNA FFPE Tissue Kit (Cat No: 56404). Exome capture, library preparation, and sequencing of the paired tumor and normal samples was conducted by the Next Generation Sequencing Core of the Sidney Kimmel Comprehensive Cancer Center of Johns Hopkins University. Briefly, exome capture and library preparation were performed with Agilent SureSelect Target Enrichment V5-post. DNA libraries were sequenced on a Illumina NovoSeq instrument to generate 2 X 150 bp paired-end sequence reads. Sequence reads were aligned to the human genome (hg38) using bwa mem (v0.7.15) with default parameters. Duplicate reads were removed using Picard tools (v1.119). GATK (v3.6) was used to call variants and Mutect2 (v3.6.0) was used to call somatic mutations in tumor compared to normal. Somatic mutations were annotated using ANNOVAR (v.2016-02-01). In order to make the results of the higher coverage FFPE tissue WES comparable to the organoid WGS, we set a threshold mutant allele frequency ≥10% in H58 FFPE WES, followed by visual inspection of each coding mutation identified in WGS or WES using IGV in both samples (v2.4.8).
RT-qPCR
500 ng of RNA from H58 cells in 2D culture was converted to cDNA using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher, catalog no. 4387406) according to the manufacturer’s protocol. Real time PCR for was conducted with the TaqMan Universal Master Mix II with UNG (Thermo Fisher, catalog no. 4440038) and the following gene specific primers according to the manufacturer’s protocol: KRT19 (Thermo Fisher, catalog no. 4331182 Hs01051611_gH), SOX9 (Thermo Fisher, catalog no. 4331182 Hs00165814_m1), NEUROG3 (Thermo Fisher, catalog no 4331182 Hs01875204_s1), VIM (Thermo Fisher, catalog no. 4331182 Hs05024057_m1).
RNA Sequencing
RNA library preparation and sequencing were conducted by Genewiz, Inc. (Plainsfield, NJ). RNA libraries were prepared by mRNA enrichment by polyA selection. mRNA was then fragmented before random priming, cDNA synthesis, A-tailing, adapter ligation, and PCR amplification. RNA libraries were sequenced to generate a 2×150 bp paired-end reads using a HiSeq instrument (Illumina, CA). Adapter sequences were removed from sequence reads using cutadapt (v.1.17). Reads were then mapped to the human genome (GRCh38) using HISAT2 (v2.1.0) before being assembled and gene transcripts per kilobase million (TPM) calculated using StringTie (v1.3.4). Fusion transcripts were detected by STAR-Fusion (v1.5.0). To reduce false positives, fusion events with fusion fragments per million total reads < 0.1 were removed (21). Three or more supporting paired-end reads were required for event detection (22). Default parameters were used unless otherwise specified.
Immunofluorescent staining
5×104 cells of HPDE, PANC-1, and H58 were seeded into chamber slides and cultured for 48 h. Culture media was removed, cells were washed twice with TBST (Tris Buffered Saline + 0.5% Tween20) and fixed with 4% PFA/PBS. For optimal penetration, fixed cells were permeabillized with 0.2% TritonX-100/TBST and subsequently blocked in blocking buffer (5% dry fat milk/TBST). Primary antibody diluted in blocking buffer (1:200 mouse-anti-panCK (Abcam, catalog no. ab7753); 1:500 Dylight594 conjugated rabbit-anti-Vim (Abcam, catalog no. ab154207) was applied and incubated for 2 h. Primary antibody solution was then removed, and cells were washed twice with TBST. Cells were then incubated with secondary antibody diluted in blocking buffer (1:500 Alexa fluor conjugated goat-anti-mouse (Abcam, catalog no. ab150117) for 1 h. Cells were washed with TBST and attachment wells removed. Stained cells were mounted in DAPI solution (Life technologies, catalog no. P36931) and each covered with a cover slip. Images were acquired after 24h incubation in dark at room temperature using a Nikon Eclipse Ti inverted microscope.
Karyotyping
Cytogenetic analysis was performed on cultured cells processed using standard techniques. Briefly, cells were treated with 0.06 µg/ml colcemid for 4 hours, incubated in hypotonic solution (0.075M KCl), and fixed in 3:1 methanol:glacial acetic acid. Slides were prepared and the metaphase chromosomes were treated with trypsin and stained with Leishman for G-banded karyotyping. Fifteen metaphases were analyzed. Chromosomal abnormalities were described based on the 2016 ISCN (International System for Human Cytogenomic Nomenclature).
Soft Agar Assay
2X culture media was prepared for each cell line assayed. PANC-1 received 2X DMEM with 20% FBS and 1% Penicillin/Streptomycin, HPDE received 2X AdvDMEM/F12 with 20% FBS and 1% Penicillin/Streptomycin, and H58 received 2X AdvDMEM/F12/HFM (2:1) with 20% FBS and 1% Penicillin/Streptomycin. We adjusted HPDE to grow in DMEM/F12 through gradual media replacement of SFM-Keratinocyte over several passages. Next, we prepared 0.5% and 1% agar solutions dissolved in cell culture grade water. 0.5% bottom layer agar was obtained by mixing 2X media with pre-warmed 1% Agar solution 1:1 and then 1 mL distributed along nine 12-well plates, considering triplicates for each cell line. 0.5% bottom agar layer solidified at room temperature after 30 min incubation. 0.25% top layer agar was prepared by mixing 2X media with pre-warmed 0.5% Agar solution 1:1. Top layer agar was kept at 42 oC while preparing cells. Cells were washed with PBS, trypsinized and re-diluted in media to calculate cell concentrations with automated cell counter. Appropriate volumes of cell suspensions were added to each 0.25% top layer agar solution to obtain 1×104 cells/mL. 1 mL of warm top layer agar cell suspension was distributed along nine 12-well plates, considering triplicates for each cell line. Top agar layer cell suspension solidified at room temperature after 30 min incubation. 300 μL of respective 2X media was plated on top of agar layers and changed periodically. Cell colonies developed after 4 weeks incubation.
Cell Invasion Assay
Cell conditioned media from respective cell lines were collected and filtered through a 0.45μm membrane (Merck Milipore, catalog no. C3240). Conditioned media was supplemented with 20% FBS to act as a chemoattractant. Culture plates with Matrigel coated inserts from Cell Invasion Assay kit (Cell Biolabs, catalog no. CBA-110) were equilibrated at room temperature and inserts rehydrated with minimum essential media (MEM; Invitrogen, catalog no. 11095080) for 1 h at 37 °C. Cultured cells were washed with PBS, trypsinized, and washed again with PBS to remove any serum-containing media. Cells were diluted in serum-free MEM and cell concentrations calculated with automated cell counter (TC-20, Bio-Rad). 2.5×104 cells for each well were obtained, considering wells in duplicate for each cell line. MEM used for rehydrations was removed from wells and inserts. 500 μL of conditioned media with 20% FBS added to each well. Serum-free cell suspensions containing 2.5×104 cells were added to inserts and inserts placed into wells. Culture plates were incubated in 5% CO2 at 37oC for 48h to 96h. Media in both insert and bottom well were replaced daily to maintain the molecular gradient. After incubation, inserts were washed with PBS and cells from the interior of the insert removed with a Q-tip. Cells on the underside of the insert were washed with PBS and fixed in 4% PFA/PBS. Cells were stained in 0.2% Crystal violet/10%Ethanol/diH20 solution. Insert membranes were removed and mounted on plus microscopy slides with a cover slip and mounting solution. Membranes were imaged on an Olympus BX51 microscope and migrated cells in five distinct 10X high power fields quantified with ImageJ.
Clonogenic assay
Clonogenic assay was conducted as previously described (23). Briefly, cells were washed with PBS, trypsinized, and re-diluted in culture media. Cell concentrations were determined with an automated cell counter (TC-20, BioRad). 2000, 1000, 500, 200, 100 and 50 cells of each cell line were seeded in triplicate into 6-well plates. Cells were incubated for 4 h before receiving either 0 Gy, 1 Gy, 2 Gy, 4 Gy of X-ray radiation using a CIXD Biological Irradiator (Xstrahl Life Sciences). Cells were then incubated for 14 days, when colony formation was visible in all untreated wells. Cells were then washed with PBS and fixed in 4% PFA/PBS. Colonies were stained with 0.5% Crystal violet/10% Ethanol/diH20 solution. Isolated cell colonies were counted for each well at each seeding concentration.
MTT assay
MTT assays were performed to quantify the viability of the cells following treatment with G007-LK (MedChemExpress: Cat. No. HY-12438), a tankyrase inhibitor, and Olaparib (LC Laboratories, Cat. No. O-9201), a poly(ADP-ribose) polymerase inhibitor. H58 cells were seeded into 96-well plates at a concentration of 5000 cells per well and left to attach overnight at 37˚C and 5% CO2. G007-LK was then added to the culture medium to a final concentration of 0, 0.1, 0.5, 1, 2, 4 μmol/L. Olaparib was then added to the culture medium to a final concentration of 0, 1, 5, 10, 20, 40 μmol/L. After 96 hours, MTT (Thermo Fisher Scientific, Cat. No. M6494) was added to the culture media, and the cells were incubated for 4h at 37 ˚C. The culture media was then removed and the formazan crystals in the cells were solubilized using dimethyl sulfoxide (Sigma-Aldrich, Cat. No. 472301), with plate agitation for 10 min. Absorbance at 490 nm was the measure using a Bio-Rad Xmark Microplate Spectrophotometry (Bio-Rad, Hercules, California, USA).
RESULTS
Isolation and culture of neoplastic cells from human ITPN
Neoplastic tissue was harvested from a grossly identified cystic lesion in a pancreatoduodenectomy specimen from a male patient in his 60s. Grossly, the cyst was located in the pancreatic parenchyma and communicated with the duct system. Microscopic examination of the lesion revealed an intraductal neoplasm with minimal mucin and tubulopapillary growth of cuboidal neoplastic cells characteristic of ITPN (Figure 1A). While the lesion was entirely intraductal on frozen section examination at the time of tissue harvesting, a microscopic focus of invasive carcinoma was subsequently identified upon comprehensive review of formalin-fixed paraffin-embedded (FFPE) tissue sections. The harvested fresh tissue was processed to derive organoids embedded in Matrigel and supplemented with human feeding media (HFM). Analysis of oncogenic hotspots in pancreatic driver genes by Sanger sequencing of cultured organoids revealed no mutations in the oncogenic hotspots exons 2 and 3 of KRAS (codons 12, 13, and 61) and exon 8 of GNAS (codon 201). Three-dimensional cell clusters were passaged multiple times before transfer of the neoplastic cells to two-dimensional culture. When primary organoids were transitioned into a two-dimensional culture system, we were able to wean the cells from the growth factor enriched HFM. In two-dimensional culture, the ITPN cell line grew in Advanced DMEM F12/HFM (3:1) with a population doubling time of 36h (Figure 1B, Supplementary Table 1). This cell line, which we labeled H58, was passaged 30 times (including 6 passages in 3D culture and 24 passages in 2D culture) while maintaining cells >250 days in culture. Cells from H58 grow in monolayer without building clusters even at confluence. We observed anchorage dependent growth, as cells adhered solely in collagen type 1 coated flasks.
Figure 1.
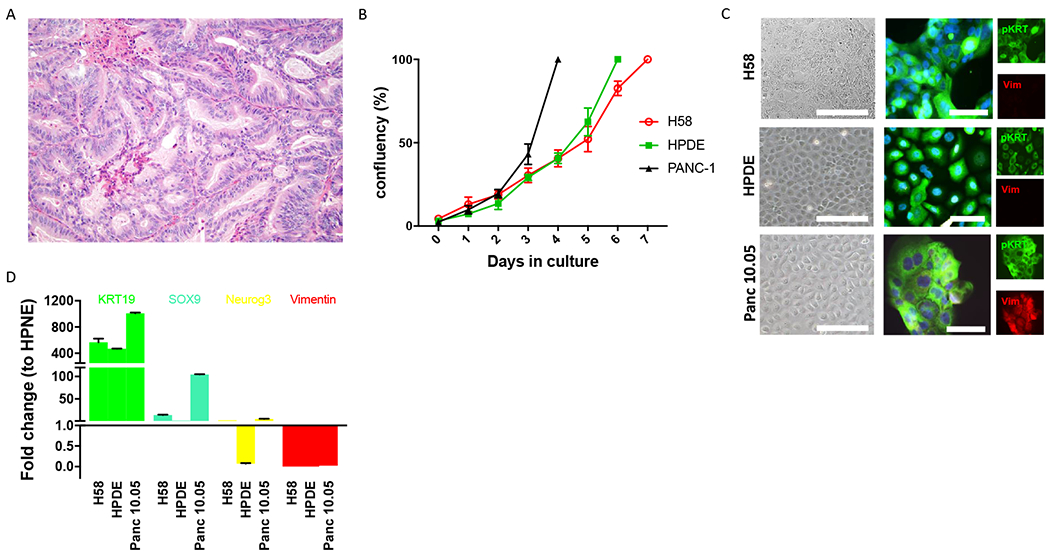
Morphological and molecular features of H58 cell line. A. H&E section (20X) of human pancreatic ITPN from which H58 was derived. The neoplastic cells are organized in closely packed tubules with minimal intracellular mucin. B. The in vitro growth rate of H58 is similar to that of pancreatic ductal cell line HPDE and slower than that of the pancreatic cancer cell line PANC-1. C. Immunofluorescence analysis for pan-cytokeratin (green) and vimentin (red) demonstrates an epithelial phenotype of all three analyzed cell lines. D. RT-PCR analysis confirms the ductal epithelial phenotype of H58, with expression of KRT19 and SOX9.
Thus, we report a new cell line derived from a primary pancreatic ITPN. We next sought to confirm the pancreatic ductal phenotype of H58 and characterize it on the molecular level.
Morphological and molecular characterization of ITPN cell line
Our ITPN cell line exhibited epithelial features both in morphology and protein expression. As is typical for epithelial cell lines, H58 cells adhered and grew in a cobblestone pattern in collagen-coated flasks (Figure 1C). H58 cells labeled strongly with antibodies directed against the epithelial marker cytokeratin (pKRT) by immunofluorescence, and there was no labeling with the mesenchymal marker vimentin (Vim) (Figure 1C). RT-qPCR confirmed ductal phenotype of the H58 cell line, with high expression of cytokeratin 19, modest expression of Sox9, and no expression of neuroendocrine (Neurogenin 3) or mesenchymal (vimentin) markers (Figure 1D). In addition, we validated the origin of our cell line by short tandem repeat (STR) fingerprinting of the H58 cell line and normal tissue from the primary pancreatic resection specimen (Supplementary Figure 1).
RNA-Seq of RNA from H58 in 3D culture generated a total of 45,953,720 sequence reads, representing 13.8 Mb. 89.8% of bases had a quality score ≥ 30. 97.8% of sequence reads mapped to the reference genome (GRCh38), 92.7 % uniquely. Gene counts identified 12,096 expressed genes with tags per million (TPM) ≥ 1 (Supplementary Table 2). Genes associated with epithelial lineage were highly expressed, for example, KRT8, KRT18, and KRT19 (Supplementary Figure 2). Expression of SOX9 and PDX1 was also detected. Expression of genes associated with mesenchymal, neuroendocrine, acinar, and immune cell lineages were not detected or detected low levels (Supplementary Figure 2). Analysis of RNA-Seq data identified 11 fusion genes (Supplementary Table 3), with ZC3H7A-BCAR4 fusion supported by the greatest number of spanning and junction reads.
Whole genome sequencing of DNA from H58 in 3D culture and fresh-frozen matched normal (N58) generated a total of 631,590,513 and 744,400,969 sequence reads with 99.73% and 99.71% mapping to the genome respectively. Across the genome, 13,337 somatic mutations consisting of 11,976 single base substitutions and 1,361 INDELs ≤ 95 nucleotides in length were identified in H58 (Supplementary Table 4). The mutation signature was predominantly C>T and similar to previously described signature 1A related to age (Figure 2) (24). Of these somatic mutations, 129 occurred in coding regions, including 36 synonymous mutations (27.9%), 83 nonsynonymous mutations (64.3%), 7 stop gain mutations (5.4%), 1 frame shift deletion (0.8%), 1 frame shift insertion (0.8%), and 1 in-frame deletion (0.8%) (Supplementary Figure 3; Supplementary Table 5). Of note, somatic mutations were not identified in KRAS, CDKN2A, or SMAD4, confirming the lack of most typical PDAC drivers this ITPN cell line. Somatic mutations were identified in known cancer driver genes, including genes involved in DNA damage response (single base substitutions in BRCA2, MRE11A, and TP53), as well as WNT pathway signaling (oncogenic hotspot mutation in CTNNB1, nonsense mutation in APC). To assess the role of the 13,208 non-coding somatic mutations we annotated mutations with FunSeq2 (25). 76 non-coding somatic mutations had a non-coding score (NCDS) > 1.5 and were predicted to be deleterious. These 76 non-coding somatic mutations were associated with 107 genes (Supplementary Table 6). Predicted deleterious noncoding variants were most frequently found in intronic regions (59.8%), promoter regions (17.8%), and untranslated regions (2.8%). Otherwise, predicted deleterious noncoding variants were found in regions distal to genes (≤10 kb) (Supplementary Figure 4).
Figure 2.
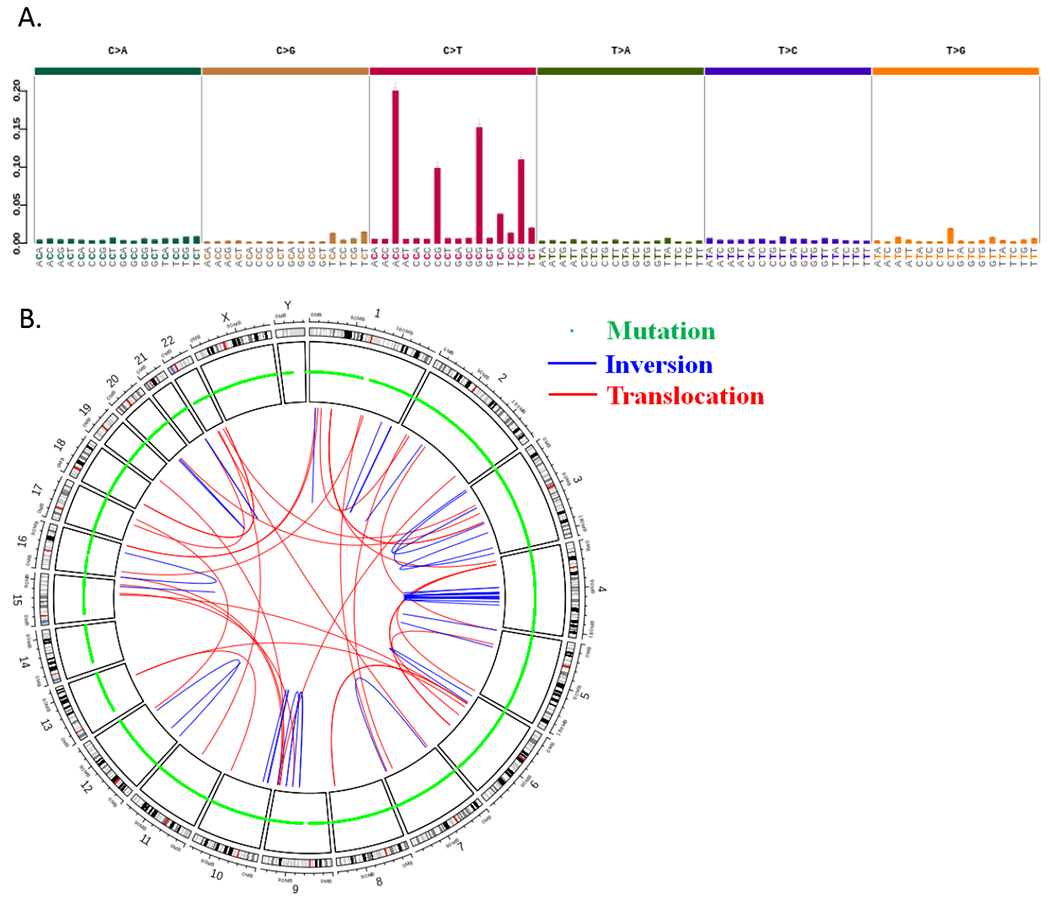
Results of whole genome sequencing of H58 cell line. A. Mutational signature of single base substitutions shows a preponderance of C>T changes. B. CIRCOS plot shows somatic mutations and structural alterations throughout the genome of H58.
To evaluate whether our H58 culture shared the genetic alterations found in bulk ITPN tissue, we compared the coding mutations identified in whole genome sequencing of H58 in culture to whole exome sequencing of H58 from FFPE tissue. Comfortingly, 90 of 129 (69.8%) coding mutations identified in H58 culture by whole genome sequencing were present in whole exome sequenced FFPE tumor tissue. Conversely, 90 of 115 (78.3%) coding mutations identified in whole exome sequenced FFPE tumor tissue were present in whole genome sequenced H58 culture. Importantly, both samples shared somatic mutations in TP53, ERBB2, APC, BRCA2, CTNNB1, MRE11 and MUC4.
Cytogenetic analysis revealed an aneuploid karyotype with multiple clonal structural aberrations (Supplementary Figure 5). Structural variant analysis using whole genome sequencing data identified 173 structural variants including 30 deletions (17.3%), 73 duplications (42.2%), 32 interchromosomal translocations (18.5%), and 38 inversions (30.0%) (Figure 2) (Supplementary Table 7). Interestingly, the structural variant analysis detected a fusion between genes ZC3H7A and BCAR4 that was also detected by RNA-Seq (Supplementary Figure 6; Supplementary Tables 3 and 7). The ZC3H7A-BCAR4 fusion was also validated by PCR amplification and Sanger sequencing.
Together, these results indicate that H58 is a cell line with a pancreatic ductal phenotype, with genomic and transcriptomic alterations characteristic of ITPNs.
Analysis of transformed phenotypes of ITPN cell line
Cancer cell lines frequently exhibit transformed phenotypes with respect to both tumor formation and invasion. As H58 was uniquely derived from a precancerous lesion, we sought to comprehensively assess its phenotype with respect to these features of transformation. H58 cells were implanted bilaterally into flanks of immunodeficient nu/nu mice. No tumors were identified after 6 months of observation, indicating limited tumorigenic potential in this assay. In a soft agar assay, no H58 colonies grew after 6 weeks of culture, confirming the anchorage dependency observed in culture in collagen-coated flasks (Figure 3A). This phenotype was similar to that observed for HPDE but strikingly different from the pancreatic cancer cell line PANC-1, which readily formed colonies in the soft agar assay.
Figure 3.
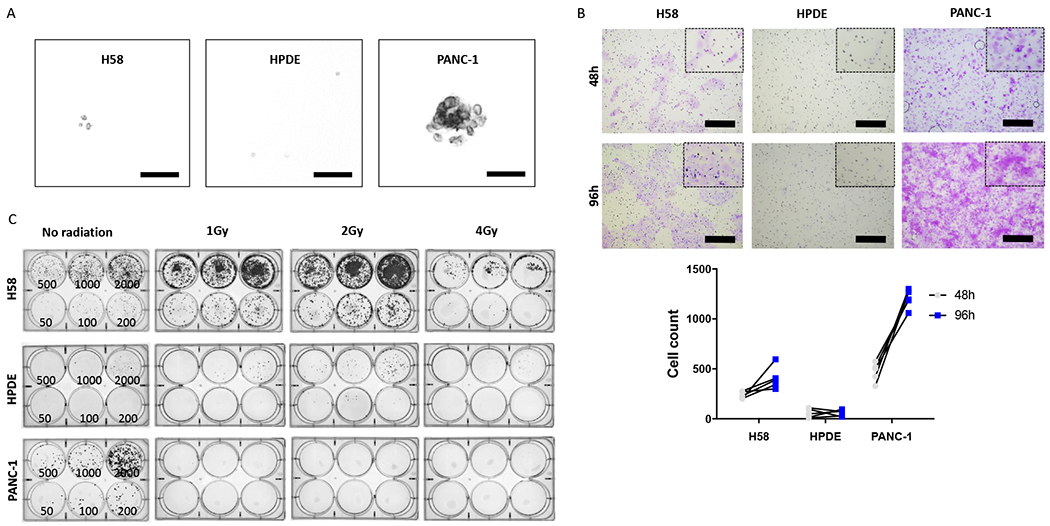
Tumorigenic characteristics of H58 cell line. A. Soft agar assay shows minimal colony formation by H58 cells, in contrast to the pancreatic cancer cell line PANC-1. B. H58 has invasive capability between that of HPDE and PANC-1 in cell invasion assay. C. Clonogenic assay demonstrates enhance colony formation after ionizing radiation in the H58 cell line.
The invasive properties of the H58 cell line were assessed by standardized Cell Invasion Assay 48h and 96h after plating (Figure 3B) (26). Although invasion of the ITPN cell line was observed after 48h, significantly fewer ITPN cells invaded compared to the pancreatic cancer cell line PANC-1 (mean 236 vs 469 cells/10 HPF; p=0.0036; student’s t-test). The difference between the invasive ability of H58 and PANC-1 was even more striking at 96h (mean 406 vs 1202 cells/10 HPF; p<0.001; student’s t-test). By comparison, the normal duct cell line (HPDE) invaded minimally in this assay, with significant differences from H58 at both 48h (236 vs 56 cells/10 HPF; p<0.001; student’s t-test) and 96h (406 vs 58 cells/10 HPF; p<0.001; student’s t-test) (Supplementary Table 8). The results of the Cell Invasion Assay demonstrate that H58 shows an invasive capability between that HPDE and PANC-1, consistent with its origin from a high-grade precancerous lesion.
In order to assess colony formation in cell lines and their replicative activity after radiation we performed a clonogenic assay. We applied either no or low dosages of radiation (1Gy, 2Gy, 4Gy) directly after seeding various cell concentrations (Figure 3C; Supplementary Table 9). In this assay, the H58 cell line was relatively radioresistant after low-dose emittance (surviving fraction 2.28 at 1Gy; 3.74 at 2Gy; 0.47 at 4Gy). This represents greater survival compared to HPDE cells (surviving fraction 0.15 at 1Gy; 0.53 at 2Gy; 0.19 at 4Gy). In contrast, the pancreatic cancer cell line PANC-1 was unable to form colonies after radiation treatment, with no surviving cells after any radiation dose.
The generation of a patient derived cell line with matched genomic data provides a unique opportunity to test the sensitivity of targeted agents based on the identified somatic alterations. As we identified a nonsense somatic mutation in APC and a nonsynonymous somatic mutation in BRCA2 in H58, we tested sensitivity of the H58 cell line to a tankyrase 1/2 specific inhibitor (G007-LK) and a PARP1 inhibitor (olaparib) (27–31). The H58 cell line was not sensitive to tankyrase inhibition, as IC50 was not reached even with a G007-LK concentration of 4 μmol/L (Supplementary Figure 7). This result is in keeping with previous reports that cancer cell lines with APC mutations outside of the mutation cluster region are not sensitive to tankyrase inhibition, as this is the case with the APC somatic mutation identified in H58 (27). Conversely, the H58 cell line was sensitive to PARP1 inhibition as the IC50 for olaparib was 9.23 μmol/L, which is between the IC50 for olaparib in MDA-MB-436 cells (3 μmol/L) and Capan-1 cells (>10 μmol/L), both of which harbor mutations in BRCA1 (Supplementary Figure 7) (29).
DISCUSSION
In this study, we report a novel cell line (H58) derived from a human pancreatic ITPN, a cystic precancerous lesion. Previous reports of derivation of cell lines from precancerous pancreatic lesions have relied on murine xenografts or other growth promoting systems to support early passages in culture (10, 11). In this study, we utilized culture in a three-dimensional organoid system for multiple passages, followed by transfer into a two-dimensional culture system. While in organoid culture, we utilized the previously reported growth factor enriched HFM, which contains factors that promote ductal epithelial growth and stem cell differentiation (EGF, FGF-10, Gastrin I, PGE2), as well as TGFβ inhibition (A83-01, mNoggin), Wnt pathway activation (Wnt3a, R-spondin), and essential vitamin supplementation (Nicotinamide, B27) (17, 18). We cannot determine whether the three-dimensional culture, enriched media, or both were the critical factors that supported the initial propagation of this unique neoplasm. Still, this approach may be useful to establish additional cell lines of precancerous lesions from the pancreas or other organs.
One important caveat of our study is that, although frozen section at the time of initial tissue processing revealed only intraductal neoplasia, a focal invasive carcinoma was identified on comprehensive review of formalin-fixed paraffin-embedded (FFPE) tissue sections. Our method of tissue harvesting, in which small superficial tissue fragments from the cyst wall were selected, should minimize the potential for harvesting carcinoma, which invaded into the underlying stroma. Still, we cannot exclude the possibility that our tissue was harvested from a region with carcinoma. As discussed below, the phenotype of our cell line in culture suggests that it represents premalignant rather than malignant cells. Of note, similar caveats were presented in a previous study of in vitro propagation of IPMNs (10).
Intriguingly, the H58 cell line showed a phenotype intermediate between the pancreatic duct epithelium cell line HPDE and pancreatic cancer cell line PANC-1. H58 demonstrated classic features of primary pancreatic duct epithelial cells in culture: cobblestone appearance, expression of epithelial cytokeratins and other ductal markers, and anchorage dependence (9). H58 continued to show growth factor dependency in two-dimensional culture and required supplementation with HFM even after many passages. The cell line did not produce tumors in nu/nu mice, as is often found with precursor or normal cells (9, 32). However, in the invasion assay, the invasive properties of H58 were between those of HPDE and PANC-1.
In contrast to HPDE, H58 displayed some features typically associated with cancer cell lines, including increased plating efficiency, reduced population doubling time, unlimited population doubling, and aneuploidy – some of these features likely facilitated the cells’ transition to two-dimensional culture. The ITPN cell line displayed resistance to low dose irradiation (1 and 2 Gy) in vitro, in stark contrast to the pancreatic carcinoma cell line PANC-1 which displayed radiosensitivity at all dosages. Of note, PANC-1 has a mutation in TP53, which is known to impact response to radiation, though such response is likely complex and multifactorial (33). The intact and even enhanced colony formation of the H58 cell line after radiation treatment suggests that it has intact DNA repair mechanisms in response to ionizing radiation (33, 34). Thus, the somatic mutations in DNA repair genes in H58, including p.Y88C in TP53, do not affect the cell line’s response to ionizing radiation, at least at the doses used in this assay.
Genomic characterization of ITPNs has previously demonstrated a paucity of somatic alterations in commonly mutated pancreatic driver genes (7). Instead, ITPNs harbor a heterogeneous range of somatic alterations, including mutations in chromatin remodeling genes and components of the PI3K pathway (7). WGS sequencing of the organoids used to derive H58 revealed missense mutations in DNA repair genes (TP53, BRCA2, MRE11), oncogenic hotspot mutation in CTNNB1, nonsense mutation in APC, and multiple putative chromosomal rearrangements, including one in ZC3H7A-BCAR4 that was supported by both WGS and RNA-Seq data. Importantly, we show that the majority of coding somatic mutations identified in H58 in culture were also present in the primary ITPN tumor tissue, confirming the shared clonal origin. There are multiple possible explanations for the discrepant mutations, including genetic heterogeneity in the primary ITPN, differences in sensitivity due to coverage differences in the whole genome and whole exome sequencing approaches, and acquisition of mutations during in vitro culture. Mutations in TP53, BRCA2, and CTNNB1 have been previously reported in ITPNs (7). Intriguingly, the ZC3H7A-BCAR4 fusion has been previously reported in cervical cancer, but to our knowledge this is the first report in pancreatic neoplasia (35).
The somatic mutations in multiple DNA repair genes raise the possibility that our cell line has a homologous recombination deficiency (HDR) phenotype. Though we identified many structural rearrangements, the number (173) is less than the reported threshold for an “unstable” genome (36). Still, the sensitivity of this cell line to the PARP inhibitor olaparib, which is specifically effective in cells with DNA repair defects, suggests that the nonsynonymous BRCA2 mutation has a functional impact on DNA repair (30, 31). Thus, this cell line represents a unique model in which to study DNA repair defects in precancerous lesions.
This is the first report of a cell line derived from a patient with ITPN, an uncommon pancreatic cancer precursor lesion with distinct molecular features. This cell line is a unique resource to study the genetics and biology of pancreatic tumorigenesis prior to the development of invasive carcinoma, offering a disease model for pre-clinical investigation of ITPNs.
Supplementary Material
Supplementary Figure 1. STR analysis of H58 and matched normal tissue.
Supplementary Figure 2. Expression of lineage specific markers in RNA Seq data.
Supplementary Figure 3. Coding mutations identified by whole genome sequencing.
Supplementary Figure 4. Noncoding mutations identified by whole genome sequencing.
Supplementary Figure 5. Metaphase karyotype of H58.
Supplementary Figure 6. Diagrammatic representation of structural variant in BCAR4 – ZC3H7A genomic region.
Supplementary Figure 7. Drug sensitivity of H58 cells to G007-LK and Olaparib by MTT assay.
Supplementary Table 1. Cell line growth characteristics
Supplementary Table 2. Gene expression data from RNA sequencing analysis
Supplementary Table 3. Detection of gene fusions from RNA sequencing data
Supplementary Table 4. Somatic mutations identified by whole genome sequencing
Supplementary Table 5. Somatic mutations in coding regions identified by whole genome sequencing
Supplementary Table 6. Non-coding somatic mutations with FunSeq score >1.5 identified by whole genome sequencing
Supplementary Table 7. Structural variants identified by whole genome sequencing
Supplementary Table 8. Quantification of cell line invasion using Matrigel assay
Supplementary Table 9. Plating efficiency after X-ray irradiation in clonogenic assay
Supplementary Table 10. Coding mutations identified in whole exome sequencing of H58 FFPE tumor tissue and whole genome sequencing H58 cultured cells.
ACKNOWLEDGEMENTS
We thank Dr. James R. Eshleman for helpful discussions.
The authors acknowledge the following sources of support:
Rolfe Pancreatic Cancer Foundation; Susan Wojcicki and Denis Troper; NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; NIH/NCI R00 CA190889; Sol Goldman Pancreatic Cancer Research Center; DFG-German Research Foundation; BIH-Charité Junior Clinician Scientist Program; Buffone Family Gastrointestinal Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; American Cancer Society Research Scholar Grant; Emerson Collective Cancer Research Fund; Joseph C Monastra Foundation; The Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Art Creates Cures Foundation.
REFERENCES
- 1.Basturk O, Adsay V, Askan G, Dhall D, Zamboni G, Shimizu M, et al. Intraductal Tubulopapillary Neoplasm of the Pancreas: A Clinicopathologic and Immunohistochemical Analysis of 33 Cases. The American journal of surgical pathology. 2017;41(3):313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamaguchi H, Shimizu M, Ban S, Koyama I, Hatori T, Fujita I, et al. Intraductal tubulopapillary neoplasms of the pancreas distinct from pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. The American journal of surgical pathology. 2009;33(8):1164–72. [DOI] [PubMed] [Google Scholar]
- 3.Adsay NVF N; Furukawa T; Hruban RH; Klimstra DS; Kloppel G; Offerhaus GJA; Pitman MB; Shimizu M; Zamboni G WHO Classification of Tumours of the Digestive System In: Bosman FTJ ES; Lakhani SR; Ohgaki H, editor. WHO Classification of Tumours. 4th Edition ed. Lyon: International Agency for Research on Cancer; 2010. [Google Scholar]
- 4.Shahinian HK, Sciadini MF, Springer DJ, Reynolds VH, Lennington WJ. Tubular adenoma of the main pancreatic duct. Archives of surgery (Chicago, Ill : 1960). 1992;127(10):1254–5. [DOI] [PubMed] [Google Scholar]
- 5.Basturk O, Hong SM, Wood LD, Adsay NV, Albores-Saavedra J, Biankin AV, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. The American journal of surgical pathology. 2015;39(12):1730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Date K, Okabayashi T, Shima Y, Iwata J, Sumiyoshi T, Kozuki A, et al. Clinicopathological features and surgical outcomes of intraductal tubulopapillary neoplasm of the pancreas: a systematic review. Langenbeck’s archives of surgery. 2016;401(4):439–47. [DOI] [PubMed] [Google Scholar]
- 7.Basturk O, Berger MF, Yamaguchi H, Adsay V, Askan G, Bhanot UK, et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2017;30(12):1760–72. [DOI] [PubMed] [Google Scholar]
- 8.Amato E, Molin MD, Mafficini A, Yu J, Malleo G, Rusev B, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. The Journal of pathology. 2014;233(3):217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ouyang H, Mou L, Luk C, Liu N, Karaskova J, Squire J, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157(5):1623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamiyama H, Kamiyama M, Hong SM, Karikari CA, Lin MT, Borges MW, et al. In vivo and in vitro propagation of intraductal papillary mucinous neoplasms. Laboratory investigation; a journal of technical methods and pathology. 2010;90(5):665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heller A, Angelova AL, Bauer S, Grekova SP, Aprahamian M, Rommelaere J, et al. Establishment and Characterization of a Novel Cell Line, ASAN-PaCa, Derived From Human Adenocarcinoma Arising in Intraductal Papillary Mucinous Neoplasm of the Pancreas. Pancreas. 2016;45(10):1452–60. [DOI] [PubMed] [Google Scholar]
- 12.Fritz S, Fernandez-del Castillo C, Iafrate AJ, Mino-Kenudson M, Neyhard N, LaFemina J, et al. Novel xenograft and cell line derived from an invasive intraductal papillary mucinous neoplasm of the pancreas give new insights into molecular mechanisms. Pancreas. 2010;39(3):308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaffee EM, Schutte M, Gossett J, Morsberger LA, Adler AJ, Thomas M, et al. Development and characterization of a cytokine-secreting pancreatic adenocarcinoma vaccine from primary tumors for use in clinical trials. The cancer journal from Scientific American. 1998;4(3):194–203. [PubMed] [Google Scholar]
- 14.Zhao Z, Bauer N, Aleksandrowicz E, Yin L, Gladkich J, Gross W, et al. Intraductal papillary mucinous neoplasm of the pancreas rapidly xenografts in chicken eggs and predicts aggressiveness. International journal of cancer. 2018;142(7):1440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell stem cell. 2018;22(3):454–67.e6. [DOI] [PubMed] [Google Scholar]
- 16.Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nature medicine. 2015;21(11):1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. The EMBO journal. 2013;32(20):2708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–5. [DOI] [PubMed] [Google Scholar]
- 20.Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int J Cancer. 1975;15(5):741–7. [DOI] [PubMed] [Google Scholar]
- 21.Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, et al. Integrative detection and analysis of structural variation in cancer genomes. Nature genetics. 2018;50(10):1388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell. 2017;32(2):185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature protocols. 2006;1(5):2315–9. [DOI] [PubMed] [Google Scholar]
- 24.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome biology. 2014;15(10):480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall DM, Brooks SA. In vitro invasion assay using matrigel: a reconstituted basement membrane preparation. Methods in molecular biology (Clifton, NJ). 2014;1070:1–11. [DOI] [PubMed] [Google Scholar]
- 27.Schatoff EM, Goswami S, Zafra MP, Foronda M, Shusterman M, Leach BI, et al. Distinct Colorectal Cancer-Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer discovery. 2019;9(10):1358–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka N, Mashima T, Mizutani A, Sato A, Aoyama A, Gong B, et al. APC Mutations as a Potential Biomarker for Sensitivity to Tankyrase Inhibitors in Colorectal Cancer. Molecular cancer therapeutics. 2017;16(4):752–62. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Ndawula C Jr., Zhou H, Gong X, Jin J. JF-305, a pancreatic cancer cell line is highly sensitive to the PARP inhibitor olaparib. Oncology letters. 2015;9(2):757–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. [DOI] [PubMed] [Google Scholar]
- 31.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 32.Soule HD, Maloney TM, Wolman SR, Peterson WD Jr., Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer research. 1990;50(18):6075–86. [PubMed] [Google Scholar]
- 33.Matsui Y, Tsuchida Y, Keng PC. Effects of p53 mutations on cellular sensitivity to ionizing radiation. Am J Clin Oncol. 2001;24(5):486–90. [DOI] [PubMed] [Google Scholar]
- 34.Williams JR, Zhang Y, Zhou H, Gridley DS, Koch CJ, Slater JM, et al. Overview of radiosensitivity of human tumor cells to low-dose-rate irradiation. International journal of radiation oncology, biology, physics. 2008;72(3):909–17. [DOI] [PubMed] [Google Scholar]
- 35.Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. STR analysis of H58 and matched normal tissue.
Supplementary Figure 2. Expression of lineage specific markers in RNA Seq data.
Supplementary Figure 3. Coding mutations identified by whole genome sequencing.
Supplementary Figure 4. Noncoding mutations identified by whole genome sequencing.
Supplementary Figure 5. Metaphase karyotype of H58.
Supplementary Figure 6. Diagrammatic representation of structural variant in BCAR4 – ZC3H7A genomic region.
Supplementary Figure 7. Drug sensitivity of H58 cells to G007-LK and Olaparib by MTT assay.
Supplementary Table 1. Cell line growth characteristics
Supplementary Table 2. Gene expression data from RNA sequencing analysis
Supplementary Table 3. Detection of gene fusions from RNA sequencing data
Supplementary Table 4. Somatic mutations identified by whole genome sequencing
Supplementary Table 5. Somatic mutations in coding regions identified by whole genome sequencing
Supplementary Table 6. Non-coding somatic mutations with FunSeq score >1.5 identified by whole genome sequencing
Supplementary Table 7. Structural variants identified by whole genome sequencing
Supplementary Table 8. Quantification of cell line invasion using Matrigel assay
Supplementary Table 9. Plating efficiency after X-ray irradiation in clonogenic assay
Supplementary Table 10. Coding mutations identified in whole exome sequencing of H58 FFPE tumor tissue and whole genome sequencing H58 cultured cells.