

Abstract
Objective:
Native and latent conformers of antithrombin (AT) induce anti-inflammatory and proapoptotic signaling activities, respectively, in vascular endothelial cells by unknown mechanisms. Syndecan-4 (Synd-4) has been identified as a receptor that is involved in transmitting signaling activities of AT in endothelial cells.
Approach and Results:
In this study, we employed flow cytometry, signaling assays, immunoblotting and confocal immunofluorescence microscopy to investigate the mechanism of the paradoxical signaling activities of high-affinity heparin (native) and low-affinity heparin (latent) conformers of AT in endothelial cells. We discovered that native AT binds to glycosaminoglycans on vascular endothelial cells via its heparin-binding D-helix to induce anti-inflammatory signaling responses by recruiting PKC-δ to the plasma membrane and promoting phosphorylation of the Synd-4 cytoplasmic domain at Ser179. By contrast, the binding of latent AT to endothelial cells to a site(s), which is not competed by the native AT, induces a proapoptotic effect by localizing PKC-δ to the perinuclear/nuclear compartment in endothelial cells. Over-expression of a dominant-negative form of PKC-δ resulted in inhibition of anti-inflammatory and proapoptotic signaling activities of both native and latent AT.
Conclusions:
These results indicate that the native and latent conformers of AT may exert their distinct intracellular signaling effects through differentially modulating the subcellular localization of PKC-δ in endothelial cells.
Keywords: antithrombin, HSPG, syndecan-4, PKC, signaling, endothelial cells
Subject codes: Basic, Translational, Clinical Research
Graphical Abstract
Introduction
Antithrombin (AT) is a plasma inhibitor of the serpin superfamily that regulates proteolytic activities of coagulation proteases of the clotting cascade in both intrinsic and extrinsic pathways.1–4 AT is a heparin-binding serpin exhibiting relatively low activity toward coagulation proteases in the absence of heparin. However, the interaction of basic residues of D-helix of AT with a distinct pentasaccharide (H5) sequence of therapeutic heparins containing 3-O-sulfate (3-OS) modification is associated with a conformational change in AT that results in activation and dramatic improvement of its reactivity with coagulation proteases.1,5–7 The vasculature has been reported to be lined with a small population of heparan sulfate proteoglycans (HSPGs) that possess the characteristic 3-OS modified glycosaminoglycans (GAGs) that can bind AT with similar high affinity.7,8 Interestingly, it has been discovered the binding of AT D-helix to vascular GAGs attached to HSPGs not only activates the serpin but also induces an intracellular signaling response in endothelial cells that endows an anti-inflammatory activity for AT that is important for its physiological function.9–12 Results by several groups including ours have indicated that syndecan-4 (Synd-4) is an HSPG receptor that can bind AT to transmit the anti-inflammatory effect of the serpin in endothelial cells.10,13,14 Thus, we have demonstrated genetic knockdown of either Synd-4 or heparan sulfate 3-O-sulfotransferase-1 (the primary isoform involved in 3-OS modification of vascular GAGs)15 by the siRNA approach,10,16 markedly inhibits anti-inflammatory signaling function of AT.
In recent years, there has been a wealth of information relating to identities of anti-inflammatory and proinflammatory signaling molecules that are targets for modulation by AT in endothelial cells. Thus, it has been found that D-helix-dependent interaction of AT with vascular Synd-4 elicits anti-inflammatory responses by inducing prostacyclin (PGI2) production, inhibiting NF-κB activation and downregulating cytokine-mediated expression of cell adhesion molecules.9,10,12,14–16 However, the mechanism by which Synd-4 communicates the protective signaling messages of AT to the relevant second messenger molecules in the signal transduction pathways inside the cell has until now remained unknown. The answer to this question is further confounded by the findings that Synd-4 also functions as an essential co-receptor for receptor tyrosine kinases (in particular the receptor for basic fibroblast growth factor FGF2).13 The participation of Synd-4 in growth factor signaling has been extensively studied and results have indicated that PKC-delta (PKC-δ)-mediated phosphorylation of Ser179 of the cytoplasmic domain of Synd-4 regulates specificity of the receptor signaling.13 Thus, when Ser179 is phosphorylated (Ser183 in rat Synd-4), Synd-4 no longer functions as a co-receptor for the growth factor signaling.13 In light of the basal quiescent phenotype of the vascular endothelial cells under normal physiological conditions, we hypothesized that AT binding to Synd-4 may stabilize the cytoplasmic domain of the receptor in the phosphorylated form, thereby exerting protective effects via synthesis of protective molecules (i.e., PGI2) and inhibition of proinflammatory (i.e., NF-kB) genes. We tested this hypothesis by employing two endothelial cell lines derived from human umbilical vein endothelial cells (EA.hy926 cells) and human dermal microvascular endothelial cells (hTERT-HDMECs) and demonstrate in this study that AT induces the membrane localization of PKC-δ and phosphorylation of Ser179 to exert its anti-inflammatory effect in endothelial cells. We further demonstrate that AT is ineffective in eliciting an anti-inflammatory effect in cells expressing a dominant negative form of PKC-δ. We have demonstrated that latent AT has no anti-inflammatory activity but instead it exerts a potent proapoptotic activity through interaction with vascular HSPGs.10 Interestingly, we discovered that latent AT induces its proapoptotic activity through promoting perinuclear/nuclear localization of PKC-δ in endothelial cells, which is inhibited in cells expressing a dominant negative form of PKC-δ. These results indicate that AT-mediated subcellular localization of PKC-δ modulates signaling specificity of native and latent conformers of AT in vascular endothelial cells.
Materials and methods
The authors declare that all supporting data are available within the article and in online supplementary file.
AT binding to endothelial cell surfaces
AT was conjugated to FITC as described in SureLINK fluorescein (FITC) labeling kit (Seracare Lifesciences; Milford, MA, USA). For staining, EA.hy926 and HDMECs were incubated at room temperature (RT) with 2.5 µM AT-FITC for 45 min and binding was detected using FACSCalibur (BD Biosciences, San Jose, CA, USA) and data was analyzed using Cell Quest software. To determine the specificity of AT-FITC binding, one set of cells was first incubated with a 10-fold molar excess of unlabeled AT (25 µM) followed by staining with AT-FITC.
Detection of inflammatory cell adhesion molecules
EA.hy926 cells and HDMECs were serum deprived when 90% confluent and treated with AT for 3h followed by treatment with TNFα (10 ng/mL) for 3h. Cells were then detached using HBSS containing 10 mM EDTA, washed and resuspended in HBSS containing 2 mM EDTA and 0.1% human serum albumin (HSA). Cells were stained using FITC conjugated anti-ICAM-1 antibody and PE-conjugated anti-VCAM-1 antibody. Cell surface levels of ICAM-1 and VCAM-1 were detected using a FACScan instrument using CellQuest software (BD Life Sciences).
Measurement of prostacyclin synthesis
EA.hy926 and HDMECs were cultured in 60 mm tissue culture plates, serum deprived and stimulated with AT (2.5 μM) for 4h. To measure prostacyclin synthesis, 6-keto-prostaglandin F1 (6-keto-PGF1, a metabolite of PGI2) was purified from 2 mL cell culture media and measured using a commercial prostacyclin competitive ELISA kit (Abcam, Cambridge, MA USA).
Cell permeability assay
Cell permeability in response to TNFα was monitored by spectrophotometric analysis of the leakage of Evans blue-bound albumin across endothelial cell monolayer in a modified two-compartment chamber model as described.18
TUNEL assay
The apoptotic effect of AT-WT and latent AT on EA.hy926 cells and primary HUVECs was analyzed through the TUNEL assay, according to a protocol provided by the manufacturer (Roche, Switzerland).
Confocal immunofluorescence
EAhy.926 cells were cultured on glass coverslips and treated with wild-type AT, latent AT (2.5 μM for 20 min) or TNFα (10 ng/mL for 20 min). After washing with PBS, cells were fixed in 2% paraformaldehyde in PBS for 15 min followed by permeabilization with 0.1% Triton X-100/PBS for 5 min at room temperature. Cells were then washed with PBS and blocked for 1h with 2% bovine serum albumin/PBS. For analysis of PKC-δ localization, cells were incubated with a rabbit polyclonal PKC-δ primary antibody overnight at 4°C. Cells were washed with PBS, incubated with a goat anti-rabbit Alexa Fluor 568-conjugated secondary antibody and counterstained with Hoechst 33342. After washing again with PBS, coverslips were mounted on glass slides with Vecta shield mounting medium. Images were obtained with a Nikon (Melville, NY, USA) C2 Confocal Microscope. For quantitative analysis of translocation, PKC-δ localized to the perinuclear/nuclear compartment was defined as translocation-positive, which were counted for at least 10 randomly selected fields from 3 independent samples. The translocation efficiency was calculated based on the ratio of translocation-positive cells to total cell numbers in these fields.
Statistical analysis
All data are expressed as mean ± SEM. Data sets were tested for normality using Shapiro-Wilk test and equal variance was tested using F test or Brown-Forsythe. Comparisons between 2 groups were performed using 2-tailed Student t- test or Mann-Whitney test for normal and non-normally distributed data, respectively. Comparisons among 3 or more groups were performed using One-way Analysis of Variance (ANOVA) followed by Bonferroni’s multiple comparison test. Values with p<0.05 were considered statistically significant. All analyses were performed using GraphPad Prism 7 software (San Diego, CA). P- values are indicated by asterisks: *P<0.05, **P<0.01, ***P<0.001, and ****P≤0.0001.
Results
Antithrombin binds to endothelial cells and stimulates PGI2 synthesis
We used FITC-conjugated AT to demonstrate that AT can bind two established endothelial cell lines: immortalized HUVECs (EA.hy926 cells) and telomerase reverse transcriptase immortalized human dermal microvascular endothelial cells (hTERT-HDMECs). Analysis by flow cytometry indicated AT-FITC binds both cell lines and an excess of unlabeled AT effectively competes for the binding, suggesting that AT interacts with specific receptor(s) on both cells (Figure 1A,B). The efficiency of AT interaction with HDMECs was significantly higher, suggesting that AT has higher number of specific binding sites on microvascular endothelial cells (Figure 1B), when compared to HUVECs (Figure 1A). AT interacts with cell surface GAGs containing 3-OS modification via D-helix which is also a binding site for the anticoagulant therapeutic drug fondaparinux (a synthetic pentasaccharide, H5).15 To demonstrate if AT interacts with endothelial cells through its basic residues of the D-helix, the binding of AT-FITC to endothelial cell lines were determined in the presence of H5. Results indicated that H5 attenuates or decreases the binding of labeled AT to both cell lines, supporting the hypothesis that AT binds endothelial cells via its D-helix (Figure 1C,D). This is the first direct binding study to the best of our knowledge to demonstrate a D-helix-dependent interaction for AT with vascular endothelial cells. The interaction of AT with cell surface GAGs stimulates PGI2 synthesis in endothelial cells by a time- and concentration-dependent manner.10,14,19 Thus, the effect of AT on stimulation of PGI2 synthesis was analyzed by measuring the concentration of 6-keto-prostaglandin F1 (6-keto-PGF1α, PGI2-metabolite) in the cell culture media. Results demonstrated AT significantly enhances secretion of 6-keto-PGF1αinto the supernatant in both cell lines (Figure 1E,F). The efficiency of AT-mediated PGI2 secretion in HDMECs was significantly higher than EA.hy926 cells, supporting the flow cytometry data that microvascular-derived endothelial cells may have more AT-binding GAGs. We previously demonstrated H5 inhibits the synthesis of 6-keto-PGF1α,20 suggesting the effect is mediated through D-helix interaction with GAGs.
Figure 1.

Antithrombin binds to endothelial cells and induces expression of prostacyclin. FITC-conjugated AT (AT-FITC) (2.5 µM) was used to stain EA.hy926 cells (A) and hTERT-HDMECs (B) in the presence or absence of excess unlabeled AT and analyzed by flow cytometry. C,D) The D-helix binding pentasaccharide (H5) decreases AT binding to EA.hy926 cells and hTERT-HDMECs. AT-FITC (2.5 µM) was incubated with 100-fold molar excess of H5 and then used to stain cells. Histograms shown in A-D are representatives from three independent experiments. The AT-mediated prostacyclin (PGI2) synthesis in EA.hy926 cells (E) and hTERT-HDMECs (F) was analyzed after treating cells with AT (2.5 μM for 4h) and measuring 6-keto-prostaglandin F1α in supernatants using a commercial ELISA kit as described under Materials and methods. Data are presented as mean ± SEM (n=6). Two-tailed Student’s t-test: ***p < 0.001.
Antithrombin attenuates TNFα-induced expression of adhesion molecules
AT exhibits anti-inflammatory activities in response to cytokines in endothelial cells.9–12 We further investigated this question through flow cytometry by measuring expression of cell adhesion molecules in EA.hy926 cells and HDMECs following their treatment with TNFα (10ng/mL) in either the absence or presence of AT. Results demonstrated TNFα promotes cell surface expression of both intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on both cell lines and AT significantly inhibits upregulation of CAMs in response to TNFα on both cell lines (Figure 2).
Figure 2.

Antithrombin attenuates TNFα-induced expression of adhesion molecules. Confluent EA.hy926 cells and hTETR-HDMECs were treated with TNFα (10 ng/mL) with or without prior incubation of cells with AT (2.5 μM for 4h). The cell surface levels of ICAM-1 (A and B) or VCAM-1 (C and D) were measured by flow cytometry. The quantitation of expression levels (mean fluorescence intensity) is presented next to each experiment. Histograms shown in A-D are representative from three independent experiments. Data are mean ± SEM. One-way ANOVA: *p < 0.05, **p < 0.01, and ***p < 0.001.
Antithrombin induces syndecan-4 phosphorylation at Ser179 in endothelial cells
Among HSPGs, syndecan-4 (Synd-4) has been identified as a 3-OS containing receptor that AT binds to exert its anti-inflammatory properties.8,10,13,15 It has been demonstrated that PKC-delta (PKC-δ)-mediated phosphorylation of Ser179 of Synd-4 cytoplasmic domain modulates the signaling specificity of the receptor.13 We hypothesized that AT binding to 3-OS containing Synd-4 may stabilize the Synd-4 cytoplasmic domain in Ser179-phosphorylated form. To test this hypothesis, we analyzed immunoblots of whole cell lysates, with or without treatment with near physiological concentration of AT, employing a specific antibody that recognizes the Ser179-phosphorylated form of Synd-4. Time-course analysis revealed AT increases phosphorylation of Ser179 of Synd-4 by a time-dependent manner with maximal effect occurring at 15min (the earliest time point measured) and remaining stable for 60min (the latest time point measured) (Figure 3A,B). To determine whether increased Synd-4 phosphorylation at Ser179 is mediated through PKC-δ, we generated stable cell lines over-expressing either wild-type (WT) or dominant negative (DN) proteins for PKC-δ by transfecting EA.hy926 cells with expression vectors containing related cDNA constructs. Over-expression of both WT and DN PKC-δ constructs in transfected cells was confirmed by immunoblot analysis of cell lysates, demonstrating that transfected cells express significantly higher levels of PKC-δ proteins when compared with the control vector (Figure 3C). The level of total PKC-α, which was used as a control, was not altered in cells transfected with either control or PKC-δ vectors (Figure 3C). Interestingly, phosphorylation of Synd-4 at Ser179 was found to be significantly downregulated in cells transfected with PKC-δ-DN, but not in cells transfected with PKC-δ-WT (Figure 3D,E). In agreement with the hypothesis that AT promotes Synd-4 phosphorylation at Ser179, AT treatment increased phosphorylation of Synd-4 in cells transfected with PKC-δ-WT (Figure 3F,G), but had an insignificant effect on Synd-4 phosphorylation of cells transfected with PKC-δ-DN (Figure 3H,I).
Figure 3.

Antithrombin treatment of endothelial cells promotes phosphorylation of Synd-4 at Ser179 through PKC-δ. A) Time course of Synd-4 phosphorylation at Ser179 in EA.hy926 cells treated with AT (2.5 μM for 1h) was analyzed by Western-blotting. B) Densitometric analysis of data presented in panel A (n=6). C) Immunoblot analysis of stable EA.hy926 cells transfected with PKC-δ-WT and PKC-δ dominant negative (DN) expression vectors. D) Immunoblot analysis of basal levels of Ser179-phosphorylated Synd-4 in stable EA.hy926 cells transfected with the control expression vector or vectors containing PKC-δ-WT or PKC-δ-DN. E) Densitometric analysis of data in panel D (n=4). F) Immunoblot analysis of AT-mediated Synd-4 phosphorylation (30 min) in EA.hy926 cells over-expressing PKC-δ-WT. G) Densitometric analysis of data in panel F (n=3). H,I) The same as panels F and G except that Synd-4 phosphorylation was determined for cells over-expressing PKC-δ-DN (n=3). GADPH and total Synd-4 were used as loading controls. Data are mean ± SEM. Two-tailed Student’s t-test: *p < 0.05, **p < 0.01, and ***p < 0.001.
Antithrombin has reduced anti-inflammatory function in cells expressing PKC-δ-DN
Anti-inflammatory activity of AT in response to TNFα in stable EA.hy926 cells transfected with either PKC-δ-WT or PKC-δ-DN vectors were assessed. Analysis by flow cytometry indicated AT has a significant inhibitory effect on TNFα-mediated upregulation of CAMs in cells expressing PKC-δ-WT, but not in cells expressing PKC-δ-DN (Figure 4). Results indicated AT inhibits TNFα-mediated upregulation of ICAM-1 (Figure 4A) and VCAM-1 (Figure 4C) in cells transfected with PKC-δ-WT. By contrast, AT was ineffective in inhibiting TNFα-mediated upregulation of either ICAM-1 or VCAM-1 in cells transfected with PKC-δ-DN (Figure 4B,D). The last panels in Figure 4 represent quantitation of expression of CAMs under different conditions. In agreement with these results, AT significantly induced expression of PGI2 in cells expressing PKC-δ-WT (Figure 4E), but not in cells expressing PKC-δ-DN (Figure 4F). Similarly, AT exhibited a significant barrier-protective activity in response to TNFα in cells expressing PKC-δ-WT (Figure 4G), but not in cells expressing PKC-δ-DN (Figure 4H). Taken together, these results provide strong support for the hypothesis that PKC-δ plays a key role in regulating anti-inflammatory function of AT in endothelial cells.
Figure 4.

The inhibitory effect of AT on TNFα-induced expression of cell adhesion molecules and mechanism of anti-inflammatory function of AT in EA.hy926 cells overexpressing PKC-δ. Stable EA.hy926 cells overexpressing PKC-δ-WT or PKC-δ- DN were treated with TNFα (10 ng/mL) with or without prior incubation of cells with AT (2.5 μM for 4h). The cell surface levels of ICAM-1 (A and B) or VCAM-1 (C and D) were measured by flow cytometry. The quantitation of expression levels (mean fluorescence intensity) are presented next to each experiment. Histograms shown in panels A-C are representatives from 3 independent and panel D from 4 independent experiments. Data are mean ± SEM. One-way ANOVA: **p < 0.01, and ***p < 0.001. E-H) Analysis of AT-mediated prostacyclin synthesis and barrier protective function in PKC-δ overexpressing cells. Stable EA.hy926 cells overexpressing PKC-δ-WT (E) or PKC-δ-DN (F) were treated with AT (2.5 μM for 4h) followed by measuring 6-keto-prostaglandin F1α in cell culture supernatants using a commercial ELISA kit as described in Materials and methods. G,H) Confluent PKC-δ-WT and PKC-δ-DN stable EA.hy926 cells were treated with TNFα (10 ng/mL) in the presence or absence of AT (2.5 μM for 4h) and the amount of Evans Blue dye that leaked into the lower chamber in the Trans-well assay plates was measured as described under Materials and methods. Data are mean ± SEM. n=3 for panel E and n=4 for panel F. Two-tailed Student’s t-test. Data in G-H are mean ± SEM. n=5, One-way ANOVA: **p < 0.01, ***p < 0.01 and ****p < 0.0001.
Active and inactive conformers of AT alter subcellular localization of PKC-δ
In light of findings that the signaling function of AT requires PKC-δ-mediated phosphorylation of Synd-4 at Ser179, we investigated the possibility that AT recruits PKC-δ from the cytoplasm to the membrane in order to promote phosphorylation of the receptor. Indeed, Western-blot analysis of fractionated membrane-associated proteins using an anti-PKC-δ antibody revealed that in AT-treated cells, the amount of membrane-associated PKC-δ is significantly higher than untreated cells (Figure 5A). The latent form of AT (AT-Lat.), which lacks anti-inflammatory activity,10,21–23 did not possess this activity and thus exhibited insignificant activity in recruiting PKC-δ to the membrane (Figure 5A). In agreement with these results, native AT, but not latent AT, effectively recruited PKC-δ to the membrane in cells over-expressing wild-type PKC-δ (Figure 5A, middle). This PKC-δ mobilizing function of AT was blunted in cells over-expressing PKC-δ-DN (Figure 5A last panel). Unlike AT-WT, latent AT had no effect on phosphorylation of Synd-4 cytoplasmic domain (Figure 5B). Moreover, analysis by flow cytometry indicated that latent AT also binds to EA.hy926 cells, however, a 20 molar excess of unlabeled AT-WT did not displace the FITC-labeled latent AT from the cell surface, suggesting that AT-WT and latent AT do not interact with the same receptor(s) (Figure 5C).
Figure 5.

AT treatment increases membrane localization of PKC-δ. (A, left panel) Untransfected EA.hy926 endothelial cells were incubated in the presence or absence of wild-type or latent AT (2.5 μM for 30 min). Membrane-associated proteins were isolated and the level of PKC-δ in the membrane fraction was analyzed by Western-blotting. Na-K ATPase was used as a loading control for the membrane fraction. (A, middle panel) The same as above except that PKC-δ-WT cells were treated with AT-WT or AT latent. (A, right panel) The same as middle panel except that cells expressing PKC-δ-DN cells were used in the experiment. Densitometric analysis were performed and represented as fold change. Data are mean ± SEM. n=3, Two-tailed Student’s t-test or ManWhitney test: *p < 0.05 and **p < 0.01. B) Time course of phosphorylation of Synd-4 at Ser179 by latent AT in EA.hy926 cells along with densitometric analysis. C) Latent AT binds to endothelial cells. Latent AT was conjugated to FITC and 2.5 µM of FITC conjugated latent AT was used to stain EA.hy926 cells in the presence or absence of 20-fold molar excess of unlabeled AT-WT and analyzed by flow cytometry.
Unlike AT-WT, which exhibited a significant inhibitory effect on TNFα-mediated upregulation of CAMs in cells expressing PKC-δ-WT, but not in cells expressing PKC-δ-DN (Figure 4), the latent AT had no significant inhibitory effect in response to TNFα in EA.hy926 cells stably expressing either PKC-δ-WT or PKC-δ-DN (Figure 6A–D). Similarly, the latent AT did not exhibit a significant barrier-protective activity in response to TNFα in cells expressing either PKC-δ-WT (Figure 6E) or in cells expressing PKC-δ-DN (Figure 6F). These results support our previous results that the latent AT has no anti-inflammatory activities.10
Figure 6.

The effect of latent AT on TNFα-induced expression of cell adhesion molecules and barrier permeability in PKC-δ overexpressing EA.hy926 cells. EA.hy926 cells stably overexpressing PKC-δ-WT or PKC-δ- DN were treated with TNFα (10 ng/mL for 3h) with or without prior incubation of cells with latent AT (2.5 μM for 2h). The cell surface levels of ICAM-1 (A,B) or VCAM-1 (C,D) were measured by flow cytometry. The quantitation of expression levels (mean fluorescence intensity) are presented next to each experiment. Histograms shown in panels A-D are representatives from 3–4 independent experiments. Data are mean ± SEM. One-way ANOVA: *p<0.05, **p < 0.01, and***p < 0.001. E,F) EA.hy926 cells stably overexpressing PKC-δ-WT and PKC-δ-DN were treated with TNFα (10 ng/mL) in the presence or absence of 2.5 μM latent AT and the amount of Evans Blue dye that leaked into the lower chamber in the Trans-well assay plates was measured as described under Materials and methods. Data are mean ± SEM from 3–5 independent experiments. One-way ANOVA for panels E,F. *p<0.05 and **p < 0.01.
Latent and cleaved forms of AT (both natural constituent of blood plasma)24 are known to have potent antiangiogenic and proapoptotic activities.10,21–23 Since nuclear accumulation of PKC-δ is known to induce apoptosis in endothelial cells,25 we investigated whether latent AT exerts its proapoptotic signaling effect through inducing nuclear localization of PKC-δ. Interestingly, we discovered that latent AT promoted the perinuclear/nuclear localization of PKC-δ in EA.hy926 cells (Figure 7A). In this assay, TNFα (a potent proinflammatory and proapoptotic cytokine) was used as a positive control, which induced the perinuclear/nuclear localization of PKC-δ in EA.hy926 cells. To provide further support for the hypothesis that latent AT exerts its proapoptotic signaling effect through nuclear localization of PKC-δ, nuclear fractions of cells treated with AT derivatives were prepared and analyzed by Western-blotting. The results were essentially mirror images of results presented in Figure 7A since latent AT, but not AT-WT, enhanced perinuclear/nuclear localization of PKC-δ in both EA.hy926 cells and cells over-expressing wild-type PKC-δ and that this effect was minimal in cells over-expressing PKC-δ-DN (Figure 7B).
Figure 7.

Analysis of nuclear localization of PKC-δ and apoptosis in cells treated with wild-type AT or latent AT. A) EA.hy.926 cells were treated with wild-type AT (2.5 μM), latent AT (2.5 μM) or TNFα (10 ng/mL) for 20 min and quantified using confocal fluorescence microscopy for nuclear and/or perinuclear localization of PKC-δ as described under Materials and methods. PKC-δ (red) was stained with rabbit anti-human PKC-δ antibody followed by Phycoerythrin-conjugated anti-rabbit secondary antibody. Hoechst 33342 (blue) was used to stain the nucleus. B) EA.hy926 cells or cells stably overexpressing PKC-δ-WT or PKC-δ-DN were treated with AT (2.5 μM), latent AT (2.5 μM) or TNFα (10ng/mL) for 30 min, the nuclear fractions were isolated and the amount of translocated PKC-δ was determined by Western-blotting. PCNA was used as a loading control for the nuclear fraction. C) Quantification of apoptosis using TUNEL assay after treating EA.hy926 cells with AT-WT or latent AT (2.5 μM each) in the presence or absence of TNFα (10ng/mL) for 16h. D) Quantification of apoptosis using TUNEL assay after treating cells stably overexpressing PKC-δ-WT or PKC-δ-DN with AT-WT (2.5 μM), latent AT (2.5 μM) or TNFα (10ng/mL) for 16h. For quantification of apoptosis, the percentage of TUNEL-positive cells per field of view were determined and expressed as fold over untreated control. Scale bar = 20µm. Data are mean ± SEM. n=3, Two-tailed Student’s t-test for panel A and B. One-way ANOVA for panel C and D. *p < 0.05, **p<0.01 and ***p<0.001.
TUNEL assay was used to determine whether perinuclear/nuclear localization of PKC-δ by latent AT parallels with its proapoptotic function. Analysis of TUNEL positive cells in EA.hy926 cells revealed that latent AT has a significant proapoptotic activity that is completely absent in cells treated with AT-WT (Figure 7C). AT-WT significantly inhibited the proapoptotic effect of TNFα, however, latent AT exhibited similar proapoptotic activity in the absence or presence of TNFα (Figure 7C). Consistent with the hypothesis that latent AT exerts its apoptotic effect through perinuclear/nuclear localization of PKC-δ, markedly higher TUNEL positive cells were detected in treated cells over-expressing wild-type PKC-δ (Figure 7D). By contrast, latent AT, as well as TNFα, had insignificant proapoptotic activity in cells over-expressing the PKC-δ-DN construct (Figure 7D). These results suggest that subcellular localization of PKC-δ by the native and latent AT forms may play a decisive role in initiating distinct signaling responses by the serpin in endothelial cells. The results further suggest that TNFα-mediated nuclear localization of PKC-δ is required for the proapoptotic function of the cytokine. It is worth noting that although similar to TNFα, latent AT was proapoptotic, nevertheless unlike TNFα, which upregulated the surface expression of CAMs, latent AT did not have an effect on expression of either ICAM-1 or VCAM-1 in endothelial cells (Supplementary Figure I).
Finally, some of the key experiments of the study were conducted in primary HUVECs. The inhibitory effect of AT-WT and latent AT on TNFα-induced expression of cell adhesion molecules in primary HUVECs is presented in Figure 8. While latent AT has no significant effect on inhibiting TNFα-induced expression levels of either ICAM-1 (Figure 8A) or VCAM-1 (Figure 8B), AT-WT significantly inhibited their upregulation in primary HUVECs as noted for EA.hy926 cells in Figure 2. Similarly, latent AT, but not AT-WT, induced the nuclear translocation of PKC-δ in primary HUVECs (Figure 8C). Analysis of TUNEL positive cells in primary HUVECs indicated that, similar to EA.hy926 cells, latent AT has a significant proapoptotic activity that is absent in cells treated with AT-WT (Figure 8D). These results suggest that signaling effects of both AT-WT and latent AT are essentially identical in both primary and transformed HUVECs.
Figure 8.

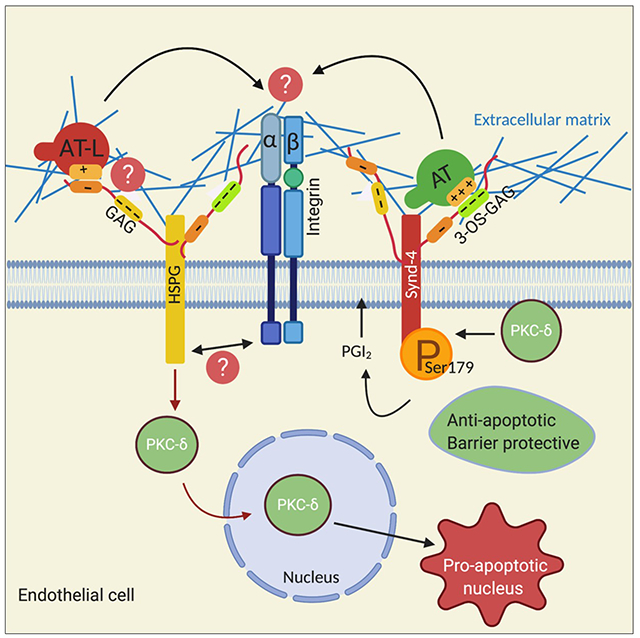
Latent AT lacks cytoprotective effect and induces nuclear translocation of PKC-δ and apoptosis in primary HUVECs. A-B) The inhibitory effect of AT-WT and latent AT on TNFα-induced expression of cell adhesion molecules in primary HUVECs. Cells were treated with TNFα (10ng/mL for 3h) with or without prior incubation of cells with AT or latent AT (2.5 μM for 2h). The cell surface levels of ICAM-1 (A) or VCAM-1 (B) were measured by flow cytometry and represented as mean fluorescence intensity. C) Primary HUVECs were treated with AT-WT (2.5 μM), latent AT (2.5 μM) or TNFα (10ng/mL) for 30 min and the nuclear fractions were isolated and the amount of translocated PKC-δ was determined by Western-blotting. PCNA was used as a loading control for the nuclear fraction. D) Quantification of apoptosis using TUNEL assay after treating primary HUVECs with AT or latent AT (2.5 μM) for 16h. For quantification of apoptosis, the percentage of TUNEL-positive cells per field of view were determined and expressed as fold over untreated control. Data are mean ± SEM from 3 independent experiments except for panel (C) where n=2. One-way ANOVA for panel A and B. Two-tailed Student’s t-test for panel D and *p<0.05, **p < 0.01, and***p < 0.001. E) Hypothetical model of wild-type and latent AT signaling in endothelial cells. The binding of AT to 3-OS containing GAGs on Synd-4 localizes PKC-δ to plasma membrane, thereby mediating the phosphorylation of Ser179 of Synd-4 culminating in synthesis of PGI2 and eliciting a barrier-protective signaling function in endothelial cells. By contrast, the latent AT binds to a low affinity GAG-binding site on an unknown HSPG, thereby leading to perinuclear/nuclear localization of PKC-δ and eliciting a proapoptotic response in endothelial cells. The proapoptotic activity of latent AT is likely mediated though an HSPG receptor crosstalk with integrins interacting with specific extracellular matrix ligands. See the text for more details. AT, antithrombin; AT-L, latent antithrombin; GAG, glycosaminoglycan; HSPG, heparan sulfate proteoglycan. The figure was prepared by software provided by Biorender.com.
Discussion
In addition to its anticoagulant function through direct inhibition of the proteases of the clotting cascade, AT also possesses potent anti-inflammatory signaling activities. The anti-inflammatory activities of AT are mediated via the serpin binding to vascular GAGs and inducing prostacyclin production that culminates in inhibition of NF-κB activation and downregulation of expression of vascular cell adhesion molecules.9–12 Previous results have indicated that interaction of AT with 3-OS modified GAGs on Synd-4 is required for its anti-inflammatory function in endothelial cells.10,13,15,16 In support of this hypothesis, we have demonstrated that genetic knockdown of either Synd-4 or 3-OS-transferase-1 (the enzyme primarily responsible for 3-OS modification in endothelial cells)15 abrogates the barrier-protective activity of AT in cytokine-stimulated cells.10,16 Since Synd-4 is known to function as an essential co-receptor for growth factor signaling by receptor tyrosine kinases (i.e., FGF2 receptor),13 the mechanism through which Synd-4 transmits the intracellular anti-inflammatory signaling activity of AT has not been investigated and remained completely unknown up until now. In this study, we demonstrate for the first time that AT promotes PKC-δ-mediated phosphorylation of the Synd-4 cytoplasmic domain at Ser179, which appears to be required for expression of the anti-inflammatory activity of AT in endothelial cells. This conclusion is based on the observations that AT did not induce PGI2 synthesis in cells expressing PKC-δ-DN nor did it inhibit TNFα-mediated expression of ICAM-1 and VCAM-1 on these cells. By contrast, AT effectively induced synthesis of PGI2 in endothelial cells over-expressing PKC-δ, the extent of which was improved when compared to untransfected cells (Figure 1E). Furthermore, AT did not exhibit a barrier-protective effect in cells expressing PKC-δ-DN, suggesting that the kinase activity of PKC-δ contributes to the protective signaling function of AT in endothelial cells. In support of the hypothesis that Synd-4 is a target receptor for PKC-δ with a key role in the signaling mechanism of AT, phosphorylation of the Synd-4 cytoplasmic domain at Ser179 was significantly downregulated in cells expressing PKC-δ-DN, which correlated with the loss of responsiveness of these cells to the signaling function of AT in all assays employed in this study. These results strongly suggest PKC-δ-mediated phosphorylation of Synd-4 cytoplasmic domain at Ser179 is required for the protective signaling function of AT. Whether in addition to Synd-4, other 3-OS containing HSPGs are involved in transmitting the protective signaling function of AT through a similar or other unknown mechanisms requires further investigation.
Previous studies have indicated nuclear localization of PKC-δ induces a potent proapoptotic effect in endothelial cells.25–28 Intriguingly, unlike AT-WT, latent and cleaved forms of AT, which possess neither protease inhibitory nor anti-inflammatory activities,10 are known to elicit potent antiangiogenic and proapoptotic responses in vascular endothelial cells.21–23 Thus, in light of our results with AT-WT, we speculated that native and inactive conformers of AT may exert their distinct intracellular signaling effects through modulation of subcellular localization of PKC-δ in endothelial cells. Interestingly, Western-blot analysis of isolated membrane and membrane-associated proteins revealed that indeed AT-WT increases the membrane fraction of PKC-δ in EA.hy926 cells. By contrast, no increase in the membrane fraction of PKC-δ could be observed in cells transfected with the PKC-δ-DN construct. A significant increase in AT-mediated membrane localization of PKC-δ in cells transfected with the PKC-δ-WT construct was also observed. These cells also exhibited an increase in PGI2 synthesis (Figure 4E), which was significantly higher than the AT-mediated synthesis of PGI2 by untransfected cells (Figure 1E). On the other hand, AT did not have a significant effect on PGI2 synthesis by cells transfected with the PKC-δ-DN construct (Figure 4F), suggesting that PKC-δ activity is required for the protective signaling function of AT in endothelial cells.
In contrast to native AT, latent AT did not exhibit any effect on the membrane recruitment of PKC-δ in endothelial cells over-expressing either PKC-δ-WT or PKC-δ-DN (Figure 5). Interestingly, however, immunofluorescence analysis indicated that latent AT markedly promotes the perinuclear/nuclear localization of PKC-δ in endothelial cells, a property that is not shared by the native AT (Figure 7A). Interestingly, this property of latent AT was also shared by TNFα, thus inducing the perinuclear/nuclear localization of PKC-δ in endothelial cells (Figure 7A). The TUNEL assay was used to confirm our hypothesis that the latent AT-mediated perinuclear/nuclear localization of PKC-δ accounts for its enigmatic proapoptotic activity in endothelial cells, the mechanism of which has remained unresolved up until now. Results indicated that unlike wild-type AT, latent AT significantly increased the number of TUNEL positive cells (Figure 7C) and this effect of the inactive serpin was also robust in cells over-expressing PKC-δ-WT, but was essentially abrogated in cells over-expressing PKC-δ-DN (Figure 7D). These results strongly suggest that the latent conformer of AT elicits its physiologically important proapoptotic signaling effects through perinuclear/nuclear localization of PKC-δ in vascular endothelial cells. The physiological significance of the latent form of AT, which has been reported to constitute 3% of plasma AT in healthy individuals (~4.8 μg/mL), is not known.29,30 What is known is that distinct variants of AT can readily undergo to a latent transition under stress conditions (i.e., fever and inflammation), thereby causing thrombosis due to their loss of inhibitory functions toward coagulation proteases.31,32 However, because of the GAG-dependent pleiotropic effect of AT in both coagulation and inflammation, further studies will be required to understand the mechanism by which D-helix mutations contribute to defects in these inter-related physiological pathways. It is possible that the latent/cleaved AT may play a role regulating angiogenesis during wound healing after injury (see below).
It has been well-established that apart from its direct signaling effect upon interaction with selected growth factors, Synd-4 functions as a co-receptor for the receptor tyrosine kinases when Synd-4 Ser179 is dephosphorylated.13 In this context, we hypothesize that AT by promoting/stabilizing the Synd-4 cytoplasmic domain in Ser179 phosphorylated state the serpin maintains the growth-factor co-receptor signaling function of Synd-4 at basal physiological conditions under which endothelial cells lining the vasculature can adopt a quiescent phenotype. We believe that with a high plasma concentration of ~2.5 μM, AT through this mechanism makes a key contribution to regulation of the vascular tone and maintenance of the vascular integrity. Similarly, under stimulated conditions (i.e., during injury and/or inflammation) where some amount of inactive loop-inserted AT becomes available as the result of the AT interaction with proteases of the clotting cascade and/or transition to the latent state, the inactive conformers of AT participate in regulation of angiogenesis by interaction with Synd-4 and/or other HSPGs, thereby leading to perinuclear/nuclear localization of PKC-δ and eliciting a potent proapoptotic response in endothelial cells that inhibits angiogenesis, a signaling process associated with wound healing (presented as a hypothetical model in Figure 8E). Unlike the loop-inserted AT, we believe that the covalent protease-AT complexes may not be involved in signaling since they are rapidly removed from the circulation by the low-density lipoprotein receptor-related protein 1 (LRP-1).33 However, this question needs to be further investigated in endothelial cells since interaction of the protease-AT complex with LRP-1 is believed to be involved in monocyte cell signaling.34 The mechanism by which native and latent/cleaved conformers of AT can differentially modulate subcellular localization of PKC-δ by binding to Synd-4 and/or other GAGs is not known. It is however known that synergistic signaling by both syndecans and integrins in the extracellular matrix (ECM) is highly critical for cell survival, proliferation, angiogenesis and many other physiological processes.35,36 It is possible that the occupancy of Synd-4 and/or other GAGs by native and loop-inserted latent/cleaved AT is differentially linked to a receptor crosstalk involving the AT-bound receptor and different combinations of α and β integrins that culminate in transmission of environmental cues from the ECM to the actin cytoskeleton (hypothetical model in Figure 8E). In support of this notion, it has been established that growth factor-induced communication between Synd-4 and integrins (i.e., αvβ3, αvβ5 and α5β1), upon their interaction with the ECM proteins like vitronectin (VN) and fibronectin (FN), plays a key role in cell adhesion, proliferation and migration.35 Thus, it is possible that latent/cleaved conformers of AT are interfering with the binding of αvβ3 and/or α5β1 integrins to the ECM proteins. This is a mechanism through which plasminogen activator inhibitor 1 (PAI-1) inhibits the αvβ3 integrin binding to VN in endothelial cells.37 It is also known that inhibiting αvβ3 and α5β1 integrins interaction with their ECM ligands (VN and FN) induces apoptosis and inhibits angiogenesis in response to FGF2 and VEGF.35 Furthermore, the endogenous proteins tumstatin and endostatin induce apoptosis in the vascular system by inhibiting interaction of integrins with the ECM proteins.38,39 Finally, it has been reported that latent AT is antiangiogenic in wild-type but not in VN-null mice.40 Further investigation is in progress to validate the hypothesis that latent/cleaved conformers of AT induce apoptosis by facilitating a crosstalk between HSPGs and integrins that control the interaction of vascular endothelial cells with the ECM proteins by a PKC-δ-dependent mechanism.
Supplementary Material
Highlights:
In addition to its anticoagulant effect, AT is known to elicit anti-inflammatory signaling responses in endothelial cells.
Unlike active AT, the loop inserted inactive conformers of AT (latent and cleaved forms) are known to elicit proapoptotic and antiangiogenic activities in endothelial cells.
The PKC-δ-dependent phosphorylation of syndecan-4 cytoplasmic domain by AT is required for its anti-inflammatory activity.
The inactive conformer of AT (latent AT) induces its proapoptotic effect by perinuclear/nuclear localization of PKC-δ, a property shared by TNFα.
Acknowledgements
We thank Cindy Carter for technical assistance and Audrey Rezaie for proofreading the manuscript.
Source of funding
This work was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of HL 101917 and HL 62565 to ARR.
Footnotes
Disclosure
The authors declare no conflict of interests.
References
- 1.Olson ST, Richard B, Izaguirre G, Schedin-Weiss S, Gettins PG. Molecular mechanism of antithrombin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie. 2010;92:1587–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gettins PGW. Serpins structure, mechanism, and function. Chem. Rev 2002;102:4751–4803. [DOI] [PubMed] [Google Scholar]
- 3.Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973;246:55–357. [DOI] [PubMed] [Google Scholar]
- 4.Carrell RW, Skinner R, Jin L, Abrahams JP. Structural Mobility of Antithrombin and its Modulation by Heparin. Thromb. Haemost 1997;78:516–519. [PubMed] [Google Scholar]
- 5.Huntington JA, McCoy A, Belzar KJ, Pei XY, Gettins PWG, Carrell RW. The conformational activation of antithrombin. A 2.85-A structure of a fluorescein derivative reveals an electrostatic link between the hinge and heparin binding regions. J. Biol. Chem 2000;275:15377–15383. [DOI] [PubMed] [Google Scholar]
- 6.Danielsson A, Raub E, Lindahl U, Björk I. Role of ternary complexes, in which heparin binds both antithrombin and proteinase, in the acceleration of the reactions between antithrombin and thrombin or factor Xa. J. Biol. Chem 1986;261:15467–15473. [PubMed] [Google Scholar]
- 7.Marcum JA, Rosenberg RD. Anticoagulantly active heparin-like molecules from the vascular tissue. Biochemistry. 1984;23:1730–1737. [DOI] [PubMed] [Google Scholar]
- 8.de Agostini AI, Watkins SC, Slayter HS, Youssoufian H,Rosenberg RD. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: antithrombin binding on cultured endothelial cells and perfused rat aorta. J. Cell. Biol 1990;111:1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oelschläger C, Römisch J,Staubitz A, et al. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood. 2002;99:4015–4020. [DOI] [PubMed] [Google Scholar]
- 10.Bae JS, Rezaie AR. Mutagenesis studies toward understanding the intracellular signaling mechanism of antithrombin. J. Thromb. Haemost 2009;7:803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minnema MC, Chang AC, Jansen PM, et al. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood. 2000;95:1117–1123. [PubMed] [Google Scholar]
- 12.Opal SM. Therapeutic rationale for antithrombin III in sepsis. Crit. Care. Med 2000;28:S34–37. [DOI] [PubMed] [Google Scholar]
- 13.Elfenbein A, Simons M. Syndecan-4 signaling at a glance. J. Cell Sci 2013;126(Pt 17):3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaneider NC, Förster E, Mosheimer B, Sturn DH, Wiedermann CJ. Syndecan-4-dependent signaling in the inhibition of endotoxin-induced endothelial adherence of neutrophils by antithrombin. Thromb. Haemost 2003;90:1150–1157. [DOI] [PubMed] [Google Scholar]
- 15.Shworak NW, Kobayashi T, De Agostini A, Smits NC. Anticoagulant heparan sulfate: To not clot-or not? Prog. Mol. Biol. Transl. Sci 2010;93:153–178. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Wang J, Gao J, et al. Antithrombin up-regulates AMP-activated protein kinase signaling during myocardial ischaemia/reperfusion injury. Thromb. Haemost 2015;113:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezaie AR, Yang L. Probing the molecular basis of factor Xa specificity by mutagenesis of the serpin, antithrombin. Biochim. Biophys. Acta 2001;1528:167–176. [DOI] [PubMed] [Google Scholar]
- 18.Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells, Blood. 2007;110:3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamauchi T, Umeda F, Inoguchi T, Nawata H. Antithrombin III stimulates prostacyclin production by cultured aortic endothelial cells. Biochem. Biophys. Res. Commun 1989;163:1404–1411. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Wang Y, Wang J, et al. Antithrombin is protective against myocardial ischemia and reperfusion injury. J. Thromb. Haemost 2013;11:1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285:1926–1928. [DOI] [PubMed] [Google Scholar]
- 22.Larsson H, Sjöblom T,Dixelius J, et al. Antiangiogenic effects of latent antithrombin through perturbed cell-matrix interactions and apoptosis of endothelial cells. Cancer Res. 2000;60:6723–6729. [PubMed] [Google Scholar]
- 23.Zhang W, Chuang YJ, Swanson R, et al. Antiangiogenic antithrombin down-regulates the expression of the proangiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood. 2004;103:1185–1191. [DOI] [PubMed] [Google Scholar]
- 24.Kjellberg M, Ikonomou T,Stenflo J. The cleaved and latent forms of antithrombin are normal constituents of blood plasma: a quantitative method to measure cleaved antithrombin. J. Thromb. Haemost 2006;4:168–176. [DOI] [PubMed] [Google Scholar]
- 25.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 2002;21:6050–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun F, Zhou B, Lin X, Duan L. Proteomic analysis identifies nuclear protein effectors in PKC-delta signaling under high glucose-induced apoptosis in human umbilical vein endothelial cells. Mol. Med. Rep 2011;4:865–872. [DOI] [PubMed] [Google Scholar]
- 27.Shizukuda Y, Helisch A, Yokota R, Ware JA. Downregulation of protein kinase C delta activity enhances endothelial cell adaptation to hypoxia. Circulation. 1999;100:1909–1916. [DOI] [PubMed] [Google Scholar]
- 28.Niwa K, Inanami O, Yamamori T, et al. Roles of protein kinase C delta in the accumulation of P53 and the induction of apoptosis in H2O2-treated bovine endothelial cells. Free Radic. Res 2002;36:1147–1153. [DOI] [PubMed] [Google Scholar]
- 29.de la Morena-Barrio M, Sandoval E, Llamas P, et al. High levels of latent antithrombin in plasma from patients with antithrombin deficiency. Thromb. Haemost 2017;117:880–888. [DOI] [PubMed] [Google Scholar]
- 30.Kjellberg M, Rimac B,Stenflo J. An immunochemical method for quantitative determination of latent antithrombin, the reactive center loop-inserted uncleaved form of antithrombin. J. Thromb. Haemost 2007;5:127–132. [DOI] [PubMed] [Google Scholar]
- 31.Beauchamp NJ,Pike RN, Daly M, et al. Antithrombins Wibble and Wobble (T85M/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood. 1998;92:2696–2706. [PubMed] [Google Scholar]
- 32.Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn-->Asp). J. Clin. Invest 1994;94:2265–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strickland DK, Muratoglu SC, Antalis TM. Serpin-Enzyme Receptors LDL Receptor-Related Protein 1. Methods Enzymol. 2011;499:17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaultier A, Arandjelovic S, Niessen S et al. Regulation of tumor necrosis factor receptor-1 and the IKK-NF-kappaB pathway by LDL receptor-related protein explains the antiinflammatory activity of this receptor. Blood. 2008;111:5316–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol 2007;8:957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, Strawn TL, Luo M, et al. Plasminogen activator inhibitor-1 inhibits angiogenic signaling by uncoupling vascular endothelial growth factor receptor-2-αVβ3 integrin cross talk. Arterioscler. Thromb. Vasc. Biol 2015;35:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003;100:4766–4771. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 39.Maeshima Y, Sudhakar A, Lively JC, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–143. [DOI] [PubMed] [Google Scholar]
- 40.Yi M, Sakai T, Fassler R,Ruoslahti E. Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. Proc. Natl. Acad. Sci. USA 2003;100:11435–11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.