

Abstract
During inner ear development, primary auditory neurons named spiral ganglion neurons (SGNs) are surrounded by otic mesenchyme cells, which express the transcription factor Pou3f4. Mutations in Pou3f4 are associated with DFNX2, the most common form of X-linked deafness, and typically include developmental malformations of the middle and inner ear. It is known that interactions between Pou3f4-expressing mesenchyme cells and SGNs are important for proper axon bundling during development. However, Pou3f4 continues to be expressed through later phases of development, and potential interactions between Pou3f4 and SGNs during this period had not been explored. To address this, we documented Pou3f4 protein expression in the early postnatal mouse cochlea and compared SGNs in Pou3f4 knockout mice and littermate controls. In Pou3f4y/− mice, SGN density begins to decline by the end of the first postnatal week, with approximately 25% of SGNs ultimately lost. This period of SGN loss in Pou3f4y/− cochleae coincides with significant elevations in SGN apoptosis. Interestingly, this period also coincides with the presence of a transient population of Pou3f4-expressing cells around and within the spiral ganglion. To determine if Pou3f4 is normally required for SGN peripheral axon extension into the sensory domain, we used a genetic sparse labeling approach to track SGNs and found no differences compared to controls. We also found that Pou3f4 loss did not lead to changes in the proportions of type I SGN subtypes. Overall, these data suggest that otic mesenchyme cells may play a role in maintaining SGN populations during the early postnatal period.
Keywords: spiral ganglion neuron, mesenchyme, cochlea, Pou3f4, Brn4, RRID:MGI:5909073, RRID:IMSR_JAX:007914, RRID:IMSR_JAX:017593, RRID:MGI:3046172, RRID:SCR_003070, RRID:SCR_014199, RRID: AB_2313552, RRID: AB_10013626, RRID: AB_2814704, RRID: AB_627766, RRID: AB_2313773, RRID:AB_2070042, RRID: AB_442208, RRID:AB_839504, RRID: AB_10013483, RRID:AB_2783873, RRID: AB_2810884, RRID: AB_2552040, RRID: AB_94166, RRID: AB_2687400, RRID: AB_94259, RRID: AB_90725, RRID:SCR_002285
Graphical Abstract
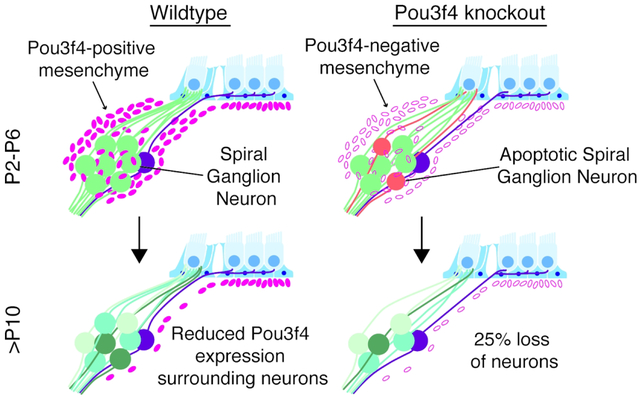
During the first postnatal week of development in the mouse cochlea, Pou3f4 is expressed by mesenchyme cells surrounding the auditory spiral ganglion neurons. In the absence of Pou3f4, approximately 25% of spiral ganglion neurons are lost within this same timeframe.
1. INTRODUCTION
Current therapies to improve hearing, including hearing aids and cochlear implants, require intact and functional auditory neurons. During normal hearing, sound waves from the external environment progress through the cochlea and stimulate mechanosensitive hair cells, which release glutamate onto spiral ganglion neurons (SGNs). Spiral ganglion neurons, so-called because they make up the spiral ganglion, which exists in a spiral within the coiled cochlea, can be subdivided into two major types (Kiang, Rho, Northrop, Liberman, & Ryugo, 1982). Type I SGNs contact inner hair cells, make up 90–95% of the SGN population and transmit auditory information to the brain; type II SGNs contact outer hair cells, make up 5–10% of the neurons and may act as auditory pain sensors (Flores et al., 2015; Liu, Glowatzki, & Fuchs, 2015; Weisz, Glowatzki, & Fuchs, 2009). Recent RNA-seq experiments have additionally identified at least three subclasses of type I SGNs (Petitpré et al., 2018; Shrestha et al., 2018; Sun et al., 2018), and these three subclasses may correspond to previously observed type I populations that are distinguishable in terms of thickness, excitability, and stereotyped synaptic position around the base of inner hair cells (M C Liberman, 1982).
SGNs originate from the cochleovestibular ganglion after it migrates away from the otic epithelium at embryonic day (E) 9 around the time of Neurogenin-1 expression (Ma, Chen, Pompa, & Anderson, 1998; Raft et al., 2007). SGNs then arise from the ventral-medial cochleovestibular ganglion between E10.5 and E12.5, completing the distinction between auditory and vestibular neurons (Koundakjian, Appler, & Goodrich, 2007). The first SGNs to delaminate populate the base of the cochlea, whereas later-born SGNs migrate further and extend along with the coiling cochlear epithelium to ultimately populate the middle and apical cochlear turns (Koundakjian et al., 2007; Matei et al., 2005). This results in a gradient of development along the tonotopic axis, with the base being slightly further developed than the apex at any given developmental time point. By adulthood, there are variations in SGN morphology from the base to the apex, and these variations likely reflect functional differences important in the transmission of different sound frequencies (Adamson, Reid, Mo, Bowne-English, & Davis, 2002; Liberman & Oliver, 1984; Nadol, Burgress, & Reisser, 1990).
As the cochlear epithelium coils during development, the SGNs extend peripheral axons into the cochlear sensory domain where they will eventually form synapses with inner and outer hair cells. Prior to entering the sensory domain, the peripheral axons must navigate through otic mesenchyme cells, which are the most numerous cell type within the cochlea and express the transcription factor Pou3f4, also known as Brn4 (Minowa et al., 1999; Deborah Phippard et al., 1998). Although comparatively little is known about otic mesenchyme cells, they first arise from neural crest cells and paraxial mesoderm and ultimately go on to make up a variety of cochlear structures (Ahn, Passero, & Crenshaw, 2009; Deborah Phippard et al., 1998; Ruben, Van de Water, & Rubel, 1986). These include the vascularized, cartilaginous spiral limbus, the bony spiral lamina surrounding the SGNs, the stria vascularis and lateral wall, which maintain the high potassium levels in the endolymph necessary for hair cell function, as well as Reissner’s membrane, tympanic border cells and the spiral ligament, all of which also line the fluid-filled cochlear ducts (Anniko & Wróblewski, 1986; Jiang et al., 2019; see Figure 1).
Figure 1. Pou3f4 is expressed into adulthood, and loss of Pou3f4 leads to reduced SGN survival.

(a) Section through a P4 cochlea demonstrating the location of spiral ganglion neurons (NF, green) and their peripheral targets, hair cells (MyoVI, white). pa, peripheral axons; ca, central axons; hc, hair cells. The inset is a high-magnification view from the boxed region at the apex. (b-d) Low-magnification micrographs of cross-sectioned P0, P8 and P29 cochleae showing spiral ganglion neuron cell bodies (HuD, green) and the location of Pou3f4-expressing mesenchyme cells (magenta). The insets in b-d are high-magnification views from the boxed regions in each panel and show age-dependent differences in Pou3f4 expression in the spiral lamina. sla, spiral lamina; sg, spiral ganglion; sl, spiral limbus; rm, Reissner’s membrane; tbc, tympanic border cells; sv, stria vascularis. (e) Schematic illustrating the anatomical location of Pou3f4-expressing cells (magenta) relative to the spiral ganglion (green) and hair cells within the sensory epithelium (blue). (f-g) Representative images of the spiral ganglion at the base of the cochlea at P45 in Pou3f4y/+ and Pou3f4y/− cochleae; dotted yellow lines indicate the area of the spiral ganglion used to determine SGN density. (h) At P29 and P45 Pou3f4y/− cochleae show a significant loss of SGN density when measurements from all cochlear turns are compiled; separating out the turns at P45 reveals that SGN loss is only statistically significant at the base of Pou3f4y/− cochleae. Student’s t test; mean ± SEM, *p ≤ 0.05; **p ≤ 0.01; N=3 cochlea Pou3f4y/+, 3 cochlea Pou3f4y/−, 30–56 sections per genotype. Scale = 200 μm (A), 300 μm (B,C), 600 μm (D), 60 μm (E-F).
Previously, otic mesenchyme cells have been shown to influence SGNs in several ways. First, in the spiral limbus, an area that is in close proximity to developing SGNs, mesenchyme cells express the guidance molecule Slit2, which binds Robo1/2 on SGNs to inhibit migration (Wang et al., 2013). Second, otic mesenchyme cells in the spiral lamina express tenascin-C, an extracellular glycoprotein, indicating otic mesenchyme cells may influence SGN peripheral axon organization as they extend toward the sensory epithelium (Whitlon, Zhang, & Kusakabe, 1999). And third, we previously demonstrated that Pou3f4 positively regulates Epha4 expression, and that EphA4 expression on otic mesenchyme cells in the spiral lamina interacts with Ephrin-B2 on SGNs to promote their formation of tight peripheral axon bundles (Coate et al., 2012). In addition, mice lacking Pou3f4 show reduced numbers of presynaptic ribbon synapse contacts on inner hair cells, suggesting reduced hair cell innervation (Coate et al., 2012).
In humans, mutations in the Pou3f4/Brn4 locus account for the most common form of X-linked deafness, DFNX2 (Kok et al., 1995). Hearing loss in humans resulting from Pou3f4 mutations have been attributed to several defects: malformation of the middle ear bones impacting conductive hearing loss (Raft, Coate, Kelley, Crenshaw, & Wu, 2014; Samadi, Saunders, & Crenshaw, 2005), the loss of the high potassium endolymph necessary for proper hair cell function (Kidokoro et al., 2014; Minowa et al., 1999), and issues related to SGN development (Coate et al., 2012). Currently, the majority of studies of Pou3f4 in otic mesenchyme cells have been focused on embryonic stages (Coate et al., 2012), but Pou3f4 expression continues in the ear into adulthood (Ahn et al., 2009). Therefore, what role Pou3f4 plays in the adult cochlea is not well understood. Here, we studied Pou3f4 in the context of SGN and otic mesenchyme interactions in the postnatal mouse cochlea. We found unexpectedly that Pou3f4 is expressed transiently by cells that reside within the region of the SGN cell bodies (“Rosenthal’s canal”), and that Pou3f4 may play a role in SGN survival. These effects occur independently from the impact of Pou3f4 on SGN guidance defects, and impact all three subtypes of type I SGNs equally. Taken together, our data suggest otic mesenchyme cells may provide a previously unappreciated source of trophic support for SGNs in the postnatal cochlea.
2. MATERIALS AND METHODS
2.1. Animal care:
All mice were maintained in accordance with the Georgetown University Institutional Animal Care and Use Committee (protocol# 1147), and the University of Maryland School of Medicine Institutional Animal Care and Use Committee (protocol# 0918005). Pou3f4 null mice (Pou3f4y/−; RRID:MGI:5909073), in which the single exon of the Pou3f4 locus is replaced with the coding region for Cre recombinase, were maintained on a C57BL/6 background (using breeders from Charles River Laboratories). This line was generated in a fashion similar to that of a lacZ insertion line reported previously (D Phippard, Lu, Lee, Saunders, & Crenshaw, 1999). To generate experimental litters, Pou3f4+/− females were crossed with wild type (WT) males to produce males that were Pou3f4y/+ (controls) or Pou3f4y/− (knockouts). Only males were used to avoid any confounds of X-inactivation. PCR genotyping was determined using Direct PCR Lysis reagent (Viagen Biotech) on prepared tail biopsies and the following primer sets: TCCTTGCTTCCTCCAGTCAGAGATC and ACGTCCAGCGGCCAACCCCTCAATG (wild type), and CAATGCTGTTTCACTGGTTATG and CATTGCCCCTGTTTCACTATC (Cre). To generate Pou3f4y/− and WT samples with sparse numbers of fluorescently-labeled SGNs , we cross-bred male mice carrying the R26RtdTomato fluorescent reporter allele (Jackson Laboratories, stock# 007914; RRID:IMSR_JAX:007914) and the Sox2CreERT2 transgene (RRID:IMSR_JAX:017593) to Pou3f4x/lacZ females (RRID:MGI:3046172; D Phippard, Lu, Lee, Saunders, & Crenshaw, 1999). Genotypes were determined by PCR using the following primer sets: CCAAAAACTAATCACAACAATCGC and GGCAAACGGACAGAAGCAT (Sox2CreERT2 mutant); AAGGGAGCTGCAGTGGAGTA and CCGAAAATCTGTGGGAAGTC (R26R wildtype), GGCATTAAAGCAGCGTATCC and CTGTTCCTGTACGGCATGG (R26RtdTomato mutant); CGCCGAAATCCCGAATCTCTA and TCACCGCCGTAAGCCGACCAC (lacZ).
2.2. Immunohistochemistry:
Inner ears were removed from postnatal pups and treated for 0.5–1 hour at room temperature or overnight at 4°C with 4% paraformaldehyde (PFA, Electron Microscopy Sciences) followed by PBS rinses and 48 hrs of 0.125M Ethylenediaminetetraacetic acid (EDTA, Amresco, pH 8.0) at 4°C if pups were P6 or older. For tissue sectioning, samples were equilibrated using 10%, 20% and 30% sucrose solutions followed by cryopreservation in OCT (Optimal Cutting Temperature; Tissue-Tek, Sakura Finetek). Ears were sectioned at 12 μm using a CryoStar NX50 Cryostat (Thermo Fisher Scientific) and stored at −80°C. Whole mount tissue was rinsed following EDTA treatment and stored in PBS at 4°C. Whole mount preparations were generated by removing the cochlear capsule and stria vascularis, revealing the spiral ganglion neurons and the sensory epithelium. Prior to staining, whole mount samples and sections were permeabilized in PBS containing 0.1% Triton X-100 (“PBSTx”) and blocked for two hours at room temperature using either 10% normal horse serum (Jackson ImmunoResearch) or 10% normal donkey serum (Jackson ImmunoResearch), 1:20 BlokhenII (Aves Labs) and either 1:200 AffiniPure Fab Fragment Donkey Anti-Mouse (Jackson ImmunoResearch) or 1:200 Mouse on Mouse blocking reagent (M.O.M.; Vector Laboratories). Primary antibodies were diluted in 0.5% PBSTx, added to tissue samples overnight at 4°C in a humidified chamber, then rinsed extensively in 0.1% PBSTx. Details about primary antibodies are provided in Table 1. Fluorescent secondary antibodies (Jackson ImmunoResearch, 1:1,000) and 4’,6-diamidino-2-phenylindole (DAPI; Roche, 1:1,000) were applied to samples for 30 minutes at room temperature in a humidified chamber.
TABLE 1.
Primary antibodies used.
| Antigen | Characterization, Controls and References | Concentration | ||
|---|---|---|---|---|
| NF-H (200kD) | Raised against NF-H protein purified from adult bovine brains | Anti-NFH immunostaining is neuron specific and is lost in denervated mouse skin (L. Li & Ginty, 2014). | Aves Labs, chicken polyclonal, Lot# NFH-3–1003, RRID: AB_2313552 | 1:2,500 |
| MyoVI | Raised against amino acids 1049–1254 of porcine myosin-VI | On a western blot, anti-MyoVI recognizes a 145kD band (Hasson & Mooseker, 1994). Anti-MyoVI labeling is consistent with hair cell location in the frog saccule and the guinea pig cochlea (Hasson et al., 1997). | Proteus Biosciences, rabbit polyclonal, Cat# 25–6791, RRID: AB_10013626 | 1:1,000 |
| Pou3f4 | Raised against amino acids 158–179 (STQ SLH PVL REP PDH GEL GSH H) (Coate et al., 2012) of mouse Pou3f4 protein | Anti-Pou3f4 immunostaining is not present in Pou3f4y/− mice but is present in littermate controls (Figure 2). | Aves Labs, chicken polyclonal, Lot# 12BG21Y09, RRID: AB_2814704 | 1:30,000 |
| HuD | Raised against amino acids 1–300 of human HuD (ELAVL4) | Immunocytochemistry using anti-HuD shows a reduction in fluorescence when HuD is knocked down in hippocampal cell culture (Vanevski & Xu, 2015). | Santa Cruz Biotechnology, mouse monoclonal, Cat# sc-48421 AF488, RRID: AB_627766 | 1:1,000 |
| Tuj1 | Raised against microtubules from rat brains | On a western blot, anti-Tuj1 recognizes a 50kD band (Lee, Rebhun, & Frankfurter, 1990). This antibody has been well characterized and used previously to identify SGNs. Our staining is consistent with these studies (Coate & Kelley, 2013). | Biolegend, mouse monoclonal, Cat# MMS-435P, RRID: AB_2313773 | 1:1,000 (Figure 1,2) 1:500 (Figure 6,7) |
| CC3 | Raised against a synthetic peptide corresponding to residues adjacent to human caspase-3 Asp175 | On a western blot, anti-CC3 recognizes two cleavage products, 17kD and 19kD, only when cells are treated with staurosporine to induce apoptosis (Cell Signaling data sheet). Anti-CC3 recognizes apoptotic cells in tissue sections and can be blocked using a blocking peptide (Cell Signaling data sheet). | Cell Signaling Technology, rabbit polyclonal, Cat# 9664, RRID:AB_2070042 | 1:500 |
| Sox10 | Raised against E.coli produced recombinant Human Sox10 Met1-Ala118 | Staining patterns of this anti-Sox10 antibody identically match Sox10 gene expression patterns in mouse cochlea described previously (Breuskin et al., 2010). | R&D Systems, goat polyclonal, Cat# AF2864, RRID: AB_442208 | 1:200 |
| Iba1 | Raised against a synthetic peptide corresponding to the mouselratlhuman Iba1 C-terminus | On a western blot, anti-Iba1 recognizes a 17kD band from cultured microglia but not neurons, astrocytes or fibrocytes (Imai, Ibata, Ito, Ohsawa, & Kohsaka, 1996). | FUJIFILM Wako Pure Chemical Corporation, rabbit polyclonal, Cat#019–19741, RRID:AB_839504 | 1:800 |
| dsRed (tdTomato) | Raised against a variant of Discosoma red fluorescent protein | Wildtype zebrafish lacking the tdTomato transgene showed no labeling when treated with anti-dsRed. On a western blot, anti-dsRed only recognizes a 30–38kD band from HEK 239 cells expressing a version of dsRed (Ikenaga et al., 2011) | Clontech, rabbit polyclonal, Cat# 632496, RRID: AB_10013483 | 1:2,000 |
| MyoVI | Raised against amino acids 1049–1254 of porcine myosin-VI (Hasson & Mooseker, 1994) | Staining patterns of this anti-MyoVI antibody match identically the staining patterns of a rabbit polyclonal antibody made from the same immunizing protein (Hasson et al., 1997; Hasson & Mooseker, 1994) | Section of Developmental Neuroscience; NIDCD, goat polyclonal, Cat# TMC_Gt_Myo6, RRID:AB_2783873 | 1:1,000 |
| Calb1 (Figure 7a–i) | Raised against full length recombinant human Calbindin | On a western blot, anti-Calb1 recognizes a 30kD band from rat brain lysate and tagged recombinant calbindin; anti-Calb1 does not recognize tagged recombinant parvalbumin or calretinin (manufacturers technical data). | Abeomics, chicken polyclonal, Cat# 34–1020, RRID: AB_2810884 | 1:500 |
| Calb2 (Figure 7a–i) | Raised against a recombinant fragment corresponding to amino acids 1–271 of Calretinin | On a western blot, anti-Calb2 recognizes a 29kD band from 239T cells transfected with Calb2, but not control cells (manufacturers technical data). | Thermo Fisher Scientific, rabbit polyclonal, Cat# PA5–34688, RRID: AB_2552040 | 1:1,000 |
| Pou4f1 (Brn3a) | Raised against amino acids 186–224 of Brn3a fused to T7 gene 10 protein | On a western blot, anti-Brn3a recognizes a ~56kD band and does not cross react with Brn3b or Brn3c (M. Xiang et al., 1995). Anti-Brn3a does not react with Brn3a−/− tissue in the dorsal root ganglia (Mengqing Xiang, Gan, Zhou, Klein, & Nathans, 1996). | Millipore Sigma, mouse monoclonal, Cat# MAB1585, RRID: AB_94166 | 1:100 (Figure 7a–i) 1:50 (Figure 7j–t) |
| Calb1 (Figure 7j–t) | Raised against recombinant rat Calbindin | On a western blot, anti-Calb1 recognizes a 28kD band from mouse and rat brain (Cell Signaling data sheet) and recognizes a band from Jurkat cells with a Calb1 overexpression vector, but not control vector (Ji et al., 2015). | Cell Signaling Technology, rabbit monoclonal, Cat# 13176, RRID: AB_2687400 | 1:50 |
| Calb2 (Figure 7j–t) | Raised against a recombinant protein specific to the amino terminus of human Calretinin | Anti-Calb2 labels horizontal cells in the mouse retina; Fox4−/− mice lack horizontal cells and show a loss of anti-Calb2 (S. Li et al., 2004). | Millipore Sigma, mouse monoclonal, Cat# MAB1568, RRID: AB_94259 | 1:1,000 |
| Prph | Raised against a trp-E-peripherin fusion protein from rat peripherin | From the manufacturers technical data: “[Anti-Prph] stains a ~57kD band cleanly and specifically and does not stain vimentin, GFAP, alpha-internexin or any of the neurofilament subunits.” Peripherin is specific to type II SGNs in the cochlea (Hafidi, 1998). | Millipore Sigma, rabbit polyclonal, Cat# AB1530, RRID: AB_90725 | 1:200 |
For tissue sections stained with anti-Calb1 (Abeomics, Cat# 34–1020, RRID: AB_2810884), anti-Calb2 (Thermo Fisher Scientific, Cat# PA5–34688, RRID: AB_2552040), anti-Pou4f1 (Millipore Sigma, Cat# MAB1585, RRID: AB_94166) (Figure 7a–i), antigen retrieval was performed. Slides were covered with citrate buffer (10mM citric acid, 0.05% Tween, pH 6.0) and placed on a flat metal rack in an open steamer for 30 minutes. Slides were allowed to cool 5 minutes outside of the steamer before being rinsed in PBS and blocked with 10% normal donkey serum, 1:10 BlokhenII and 1:200 AffiniPure Fab Fragment Donkey Anti-Mouse. Primary antibodies were incubated overnight at 37°C in a humidified chamber and rinsed in 0.1% PBSTx. Fluorescent secondary antibodies (1:1,000) were added to the samples for 1 hour at room temperature in a humidified chamber.
Figure 7. Type I SGN subtype specification is unaltered in Pou3f4 mutants.
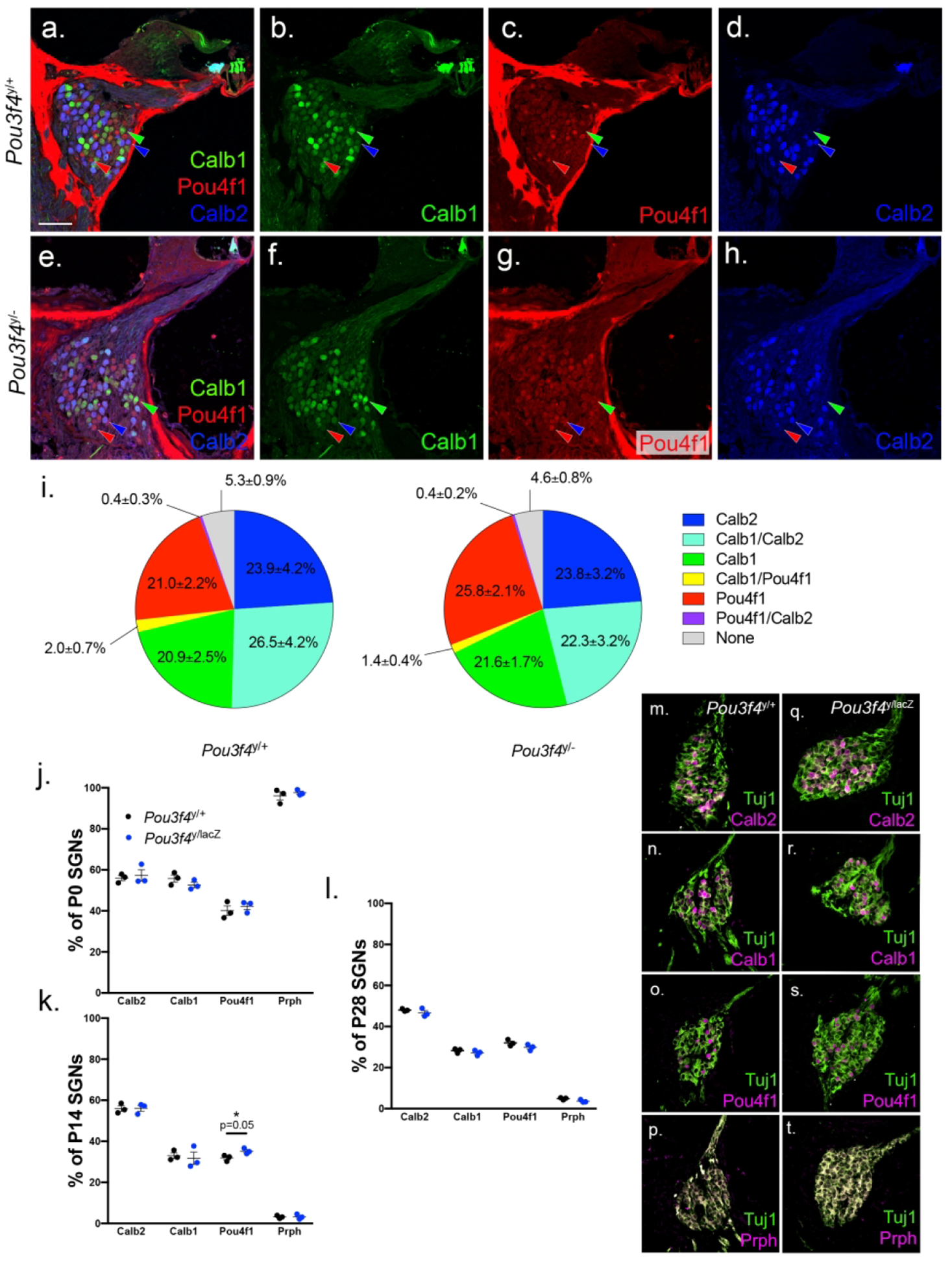
(a-h) 20x images of 4 week old Pou3f4y/+ and Pou3f4y/− cochleae at the base, labeled for the type I spiral ganglion subtype markers Calb1 (green, green arrowhead), Pou4f1 (red, red arrowhead) and Calb2 (blue, blue arrowhead). (i) The percentage of SGNs in Pou3f4y/+ and Pou3f4y/− cochleae positive for one, two or none of the subtype markers. By ordination analysis, differences in proportions of subtype populations were not statistically different; N=5 cochleae Pou3f4y/+, 5 cochleae Pou3f4y/−, minimum 30 sections per genotype. (j-l) The percentage of SGNs in Pou3f4y/+ and Pou3f4y/lacZ cochleae positive for Calb2, Calb1, Pou4f1 or Peripherin at P0, P14 and P28, respectively. (m-t) Representative images of P0 cochleae at the middle turn. Students t test; N=3 cochleae Pou3f4y/+, 3 cochleae Pou3f4y/lacZ, 9 sections per genotype. Scale = 60 μm a-h, 65 μm m-t.
2.3. Image acquisition and processing:
Confocal images were taken using a Zeiss LSM 880 and processed using NIH-ImageJ (https://imagej.net/; RRID:SCR_003070) and Adobe Photoshop (https://www.adobe.com/products/photoshop.html; RRID:SCR_014199).
2.4. Neuron density quantitative analysis:
To quantify SGN density and Pou3f4 expression over time, z-stack images of cochlear cross-sections were taken using a 20x objective. Each optical section within the stack was acquired at 0.79 μm intervals, and 5 optical sections were used for each cross-section counted. SGNs were counted by identifying individual Tuj1- (Biolegend, Cat# MMS-435P, RRID: AB_2313773) or HuD- (Santa Cruz Biotechnology, Cat# sc-48421, RRID: AB_627766) positive neurons within the given z-stack. The number of SGNs was divided by the area occupied by the SGN cell bodies to obtain SGN density values. To determine the number of Pou3f4-positive cells within Rosenthal’s canal, samples were first immunostained with anti-Pou3f4 antibodies (Aves Labs, Lot# 12BG21Y09, RRID: AB_2814704), then imaged by confocal microscopy. For each acquired image, a boundary line was drawn that encircled the HuD-positive SGN cell bodies; any Pou3f4-positive cell within or just on the outside of this boundary line was counted.
2.5. Apoptosis analysis:
To quantify SGN apoptosis, z-stack tile scan images of cochlear whole mount preparations were acquired using a 20x objective. Images include the entire depth of the spiral ganglion as visible by the neuronal cell body marker HuD, and each image of the z-stack was counted individually without being compressed. Cells were deemed “cleaved caspase 3 (CC3)-positive SGNs” if they were (1) anti-CC3 positive (Cell Signaling Technology, Cat# 9664, RRID AB_2070042) and (2) within the spiral ganglion cell body area as defined by HuD. CC3-positive bodies smaller than 7 μm in diameter were considered debris or possibly another cell type (e.g. glia) and were not included in SGN counts. The total number of CC3 positive SGNs was normalized to the length of each cochlea and presented per 500 μm.
2.6. Peripheral axon quantitative analysis:
To quantify the number of peripheral axons in contact with hair cells, z-stack images of individual peripheral fibers in cochlear whole mount samples were acquired using a 40x objective. Each fiber was designated either (1) in contact with hair cells, as defined by overlap with anti-MyoVI (Proteus Biosciences, Cat# 25–6791, RRID: AB_10013626) staining or (2) not in contact with hair cells, as defined by no overlap with MyoVI.
2.7. Neuronal subtype quantitative analyses:
To quantify the percentage of type I SGN subtypes present in a given section (for Figure 7a–i), z-stack images of cochlear cross-sections were taken using a 20x objective and analyzed as maximum intensity projections. Sections were labeled with anti-Calb1 (Abeomics, Cat# 34–1020, RRID: AB_2810884), anti-Calb2 (Thermo Fisher Scientific, Cat# PA5–34688, RRID: AB_2552040), anti-Pou4f1 (Millipore Sigma, Cat# MAB1585, RRID: AB_94166) and DAPI, and SGNs were categorized in the following way: (1) Calb1-positive (2) Calb2-positive (3) Pou4f1-positive (4) Calb1 and Calb2-positive (5) Calb1 and Pou4f1-positive (6) Calb2 and Pouf41-positive and (7) Negative. Neurons were only considered positive for two markers (categories 4–6) if both labels were equally intense. Neurons were only considered positive for Pou4f1 (categories 3,5–6) if the nucleus was brightly positive; nearly all SGNs show faint background-level staining in the cytosol after anti-Pou4f1 immunostaining. Neurons lacking Pou4f1 in the nucleus, Calb1 and Calb2 were considered negative (category 7). For statistical tests, ordination techniques were used (G. Wimp, Georgetown University).
To quantify the percentage of type I SGN subtype change over time (Figure 7j–t; Supplemental Figure 1), cochleae from Pou3f4y/lacZ and littermate controls were embedded in paraffin and sectioned at 7 μm using the Leica RM2135 microtome. Sections were incubated at 37°C overnight and deparaffinized. Antigen retrieval was performed with a 10 mM citric acid solution (0.05% Tween, pH 6.0). Slides were then blocked with 10% fetal bovine serum (Gemini Bio-Products) and 1% bovine serum albumin (Millipore Sigma) in 0.2% PBSTx for 1 hour at room temperature. Sections were labeled with Tuj1 and either anti-Calb1 (Cell Signaling Technology, Cat# 13176, RRID: AB_2687400), anti-Calb2 (Millipore Sigma, Cat# MAB1568, RRID: AB_94259), anti-Pou4f1 (Millipore Sigma, Cat# MAB1585, RRID: AB_94166) or anti-Prph (Millipore Sigma, Cat# AB1530, RRID: AB_90725); all primary antibodies were incubated overnight at 4°C in a humidified chamber and rinsed in 0.2% PBSTx. Fluorescent secondary antibodies (Thermo Fisher Scientific) were added to the samples for 1 hour at room temperature in a humidified chamber. Confocal images were taken using a Nikon 510 spinning disk and processed using Fiji-ImageJ (https://fiji.sc/; RRID:SCR_002285). Z-stack images of cochlear cross-sections were analyzed as maximum intensity projections.
3. RESULTS
3.1. Pou3f4 is expressed in the postnatal cochlea, and loss of Pou3f4 leads to reduced SGN density.
The cochlea is the spiral-shaped organ of the inner ear responsible for converting sound waves into electrical stimuli that can be interpreted as auditory information by the brain. Within the cochlea, mechanosensitive hair cells (MyoVI staining in Figure 1a) respond to changes in fluid pressure created by sound waves, and release glutamate onto primary auditory neurons called spiral ganglion neurons (SGNs). Type I SGNs form individual synaptic contacts with a single inner hair cell, whereas type II SGNs form multiple synaptic contacts with multiple outer hair cells. While type I and type II SGNs perform different functions, both are bipolar neurons with a single peripheral axon (pa) that receives input from hair cell(s) and a single central axon (ca) that projects to the brainstem to send auditory information for higher-order processing (Neurofilament-200 staining in Figure 1a). Pou3f4 immunostaining was used in combination with HuD immunostaining (Figure 1b–d) to label SGN cell bodies. At P0, Pou3f4 was present in the spiral lamina (sla), the area surrounding the SGN cells bodies (sg) as well as cochlear regions further from the SGNs including the mesenchymal layer of Reissner’s membrane (rm), the tympanic border cells (tbc), and the spiral limbus (sl). Along the lateral wall of the cochlear duct, Pou3f4 is expressed at robust levels by spiral ligament cells (see anatomical location in 1e) and fibrocytes within the multi-layered stria vascularis (sv; Figure 1c,e). Consistent with previous reports of Pou3f4 gene enhancer activity (Ahn et al., 2009), Pou3f4 does not appear to be expressed by either marginal or intermediate cells of the stria vascularis.
Within the spiral lamina (sla), at P0 SGNs were surrounded by a scaffold of mesenchyme cells that express Pou3f4 (Figure 1b). This is consistent with the distribution of Pou3f4 and mesenchyme cells reported previously (Coate et al., 2012). Over time, Pou3f4 positive cells were diminished in the spiral lamina, but were retained in other regions of the cochlea (Figure 1c–d). By P29, around the time that SGNs are refined and thought to be fully developed (L. D. Liberman & Liberman, 2016), Pou3f4 is no longer expressed by mesenchyme cells in the vicinity of the SGNs (Figure 1d). The anatomical locations of cochlear structures derived from Pou3f4-expressing cells are illustrated with magenta in Figure 1e. The transient, but continued expression of Pou3f4 in the cochlea through SGN development prompted us to ask whether fully developed SGNs showed any noticeable defects in the absence of Pou3f4. To do this, we used a previously generated Pou3f4 knockout line, in which the Pou3f4 coding region has been replaced by the coding region for Cre recombinase (Deborah Phippard et al., 1998). Using cochlear cross-sections, we calculated the density of neurons within Rosenthal’s canal as an approximation for total SGN number. “Rosenthal’s canal” is the region of the cochlea that houses the SGN cell bodies, but does not include the SGN peripheral or central axons (see outlined region in Figure 1f–g). To measure SGN density, we labeled SGNs with Tuj1, counted the number of neuronal cell bodies, and normalized this number to the total area of the ganglia (dotted line, Figure 1f–g). We examined Pou3f4y/− cochleae and littermate controls in all turns of the cochlea, and first measured density at P29 and P45, two time points when SGNs are fully developed (Coate, Scott, & Gurjar, 2019). At both ages, we found that SGN density was significantly reduced in Pou3f4y/− cochleae compared with controls (Figure 1f–h). Additionally, when the density measurements from the basal, middle and apical turns of P45 cochleae were separated out, the SGN loss in Pou3f4 mutants compared with controls was only statistically significant at the base, although there was a reduction in all turns (Figure 1h). These data suggest that Pou3f4 is necessary for proper SGN density, especially at the cochlear base.
3.2. SGN loss in Pou3f4 mutants occurs shortly after birth
Given we detected fewer SGNs at P29 and P45 in Pou3f4y/− cochleae, we next wanted to ask whether this represented a defect in SGN development, or a defect in SGN survival after development occurred. Thus, we repeated the SGN density measurements described (see 3.1) ina series of postnatal ages. At P0, there was no significant difference in SGN density between Pou3f4y/− cochleae and littermate controls (Figure 2 a–b and g). From P2 to P6, SGN density was also not significantly different in either genotype, although there was a trend toward reduced SGN density in Pou3f4y/− cochleae (Figure 2c–d and g). By P8 and P10, SGN density decreased in Pou3f4y/− cochleae by approximately 15–20% (Figure 2e–f and g). Beyond P10, neuron loss did not surpass 28% at any time point examined (Figure 2g). These data suggest that Pou3f4 is needed for SGN survival during the first two postnatal weeks in mouse.
Figure 2. Loss of Pou3f4 leads to reduced spiral ganglion neuron density.
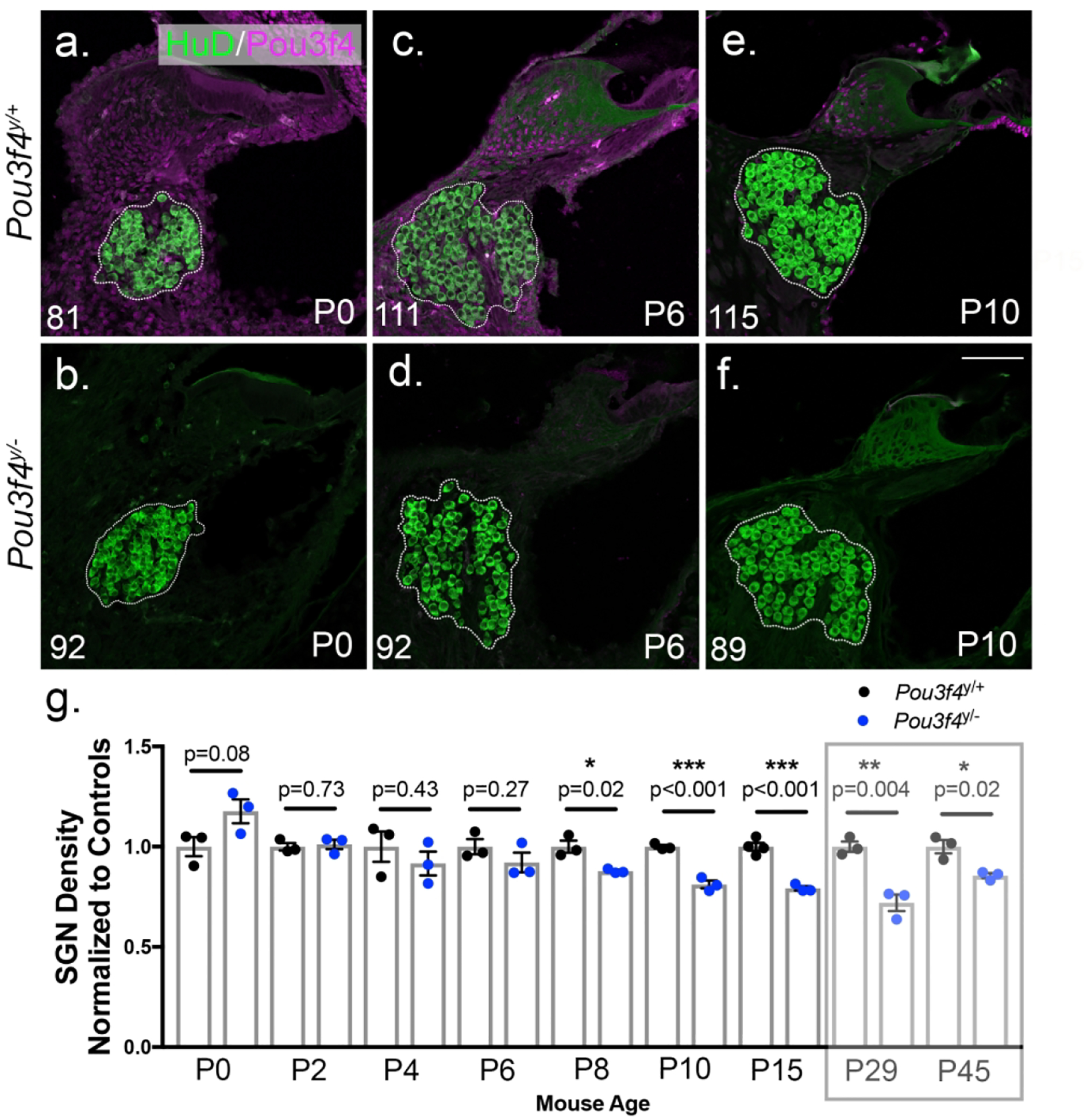
(Top row; a,c,e) Representative z-stack projections of SGN cross-sections from the base of Pou3f4y/+ cochleae at P0, P6 and P10 respectively. (Bottom row; b,d,f) Same as a,c,e but from Pou3f4y/− cochleae. SGN cell bodies are labeled with anti-HuD (green); Pou3f4 is labeled in magenta. Dotted lines indicate the area of the spiral ganglion used to determine SGN density. Number in the left lower corner indicates the number of SGNs in the image, and images show ganglia with similar areas. (g) Quantification of SGN density in Pou3f4y/− cochleae (including all turns of the cochlea) normalized to littermate controls. Semitransparent box indicates data presented in Figure 1. Approximately 25% of spiral ganglion neurons are lost in the early postnatal period. Student’s t test; mean ± SEM, *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; N=3–4 cochleae Pou3f4y/+, 3 cochleae Pou3f4y/−, 30–66 sections per genotype. Scale = 100 μm.
3.3. Pou3f4 is expressed dynamically around and among the SGNs during the first postnatal week.
Pou3f4 is first expressed in the mouse cochlea at embryonic day 10.5 (E10.5) specifically in mesenchyme cells, and remains expressed in mesenchyme cells throughout development (Coate et al., 2012; Deborah Phippard et al., 1998). A previous study using a mouse line that reports Pou3f4 enhancer activity also suggested Pou3f4 expression likely persists into adulthood (Ahn et al., 2009), but a detailed understanding of Pou3f4 protein distribution in the early postnatal cochlea was lacking. Given the reduced survival of SGNs in the Pou3f4y/− cochleae during the first two postnatal weeks, we next asked when and where Pou3f4 protein is expressed in the cochlea during this time frame. To do this, we used previously generated anti-Pou3f4 antibodies (Coate et al., 2012) on cochlear cross-sections from mice ages P0 through P10. Throughout this time, cells of the spiral limbus (sl), stria vascularis (sv), tympanic border (tbc) and Reissner’s membrane (rm) show consistent levels of Pou3f4 . However, in regions of the cochlea closer to the SGN cell bodies, Pou3f4 density was reduced.
During the course of these studies, we were surprised to find the presence of Pou3f4-positive cells near SGN cell bodies within Rosenthal’s canal (Figure 3a–g). In Rosenthal’s canal (rc), the Pou3f4-positive cells are present starting at P0, but become more abundant as development progresses, peaking at P6 (Figure 3b–d and i; see outlined region). However, at P8 there is a sharp decline in the number of Pou3f4-positive cells in Rosenthal’s canal, and this decline is particularly evident in the base compared to the apex (Figure 3e–f and i; see outlined region). In fact, we can see that, at P8, mesenchyme cells at the base of Rosenthal’s canal are faintly positive for Pou3f4 compared with the apex, suggesting that Pou3f4 is being shut down first at the base (Fig 3e–f; yellow arrowheads).
Figure 3. Pou3f4 is expressed transiently within and surrounding Rosenthal’s canal.
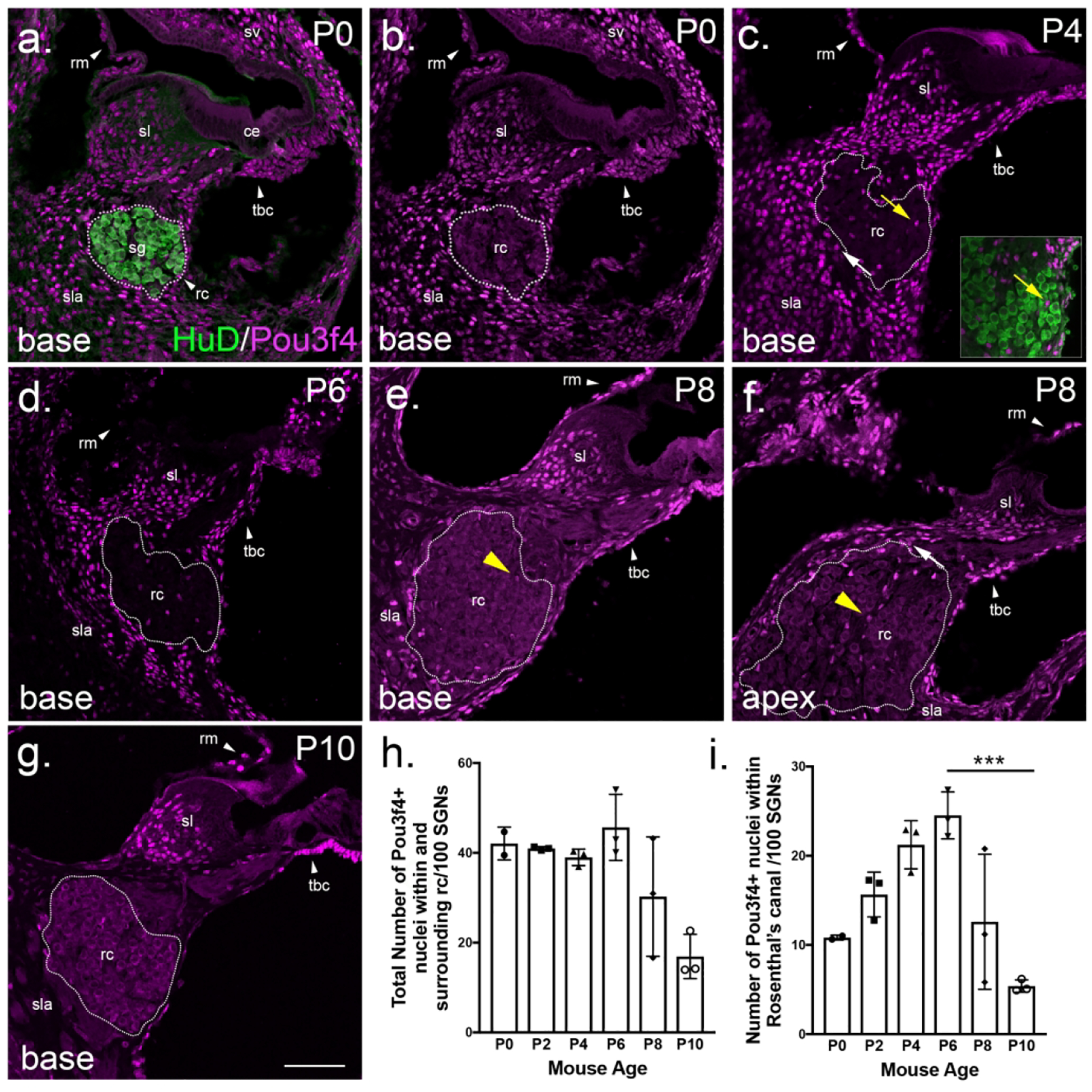
(a-g) Cross-sections at the base of the cochlea (except f at the apex) from P0–P10. Dotted lines represent Rosenthal’s canal, the location of SGN cell bodies; the yellow arrow in c points to Pou3f4-expressing cells within Rosenthal’s canal; the white arrow points to Pou3f4-expressing cells directly surrounding Rosenthal’s canal in the spiral lamina. In e and f, the yellow arrowheads illustrate different intensities of Pou3f4 expression at P8 between the base and the apex. sg, spiral ganglion; sl, spiral limbus; ce, cochlear epithelium; rm, Reissner’s membrane; sv, stria vascularis; rc, Rosenthal’s canal; sla, spiral lamina; tbc, tympanic border cells. (h-i) Quantification of numbers of Pou3f4-positive cells in Rosenthal’s canal from P0–P10 normalized to SGN number. Panel h includes Pou3f4-positive cells both within and directly surrounding Rosenthal’s canal (rc); Panel i represents Pou3f4-positive cells only within Rosenthal’s canal. Data expressed as mean ± SEM. Student’s t test; ***p ≤ 0.001; N=3 cochleae for Pou3f4y/+, 3 cochleae for Pou3f4y/−; 45 sections P6, 34 sections P10. Scale = 100 μm.
In addition to Rosenthal’s canal, Pou3f4 expression was also dynamic in the spiral lamina, the region surrounding the SGN cell bodies (Figure 3b–h; see “sla”). Starting at P0 and continuing through P6, Pou3f4-positive cells in the spiral lamina were abundant (Figure 3b–d and h; see “sla”). Like in Rosenthal’s canal, the number of Pou3f4-positive cells declined at P8 and was minimal by P10 (Figure 3e–h; see “sla”). It is important to note that the histogram in Figure 3h includes Pou3f4-positive cells within the spiral lamina that are in close proximity to the SGN cells bodies in addition to those within Rosenthal’s canal. By contrast, the histogram in Figure 3i exclusively represents Pou3f4-positive cells within Rosenthal’s canal (Figure 3c; see yellow vs. white arrows). By separating out these two populations, we found that the number of Pou3f4-expressing cells in the spiral lamina was stable from P0 to P6 and dropped off around P8 (Figure 3h). In contrast, the population of Pou3f4-expressing cells within Rosenthal’s canal slowly increased following birth, peaked at P6, and then sharply declined around P8 (Figure 3i). Interestingly, the peak of Pou3f4 expression among the SGN cell bodies aligns with the period when SGN loss is observed (Figure 2g).
3.4. Pou3f4-positive cells within Rosenthal’s canal are not Sox10-positive glia or macrophages
Given the position of the Pou3f4-expressing cells within Rosenthal’s canal, we next wanted to determine whether these cells were otic mesenchyme, or possibly one of the nonmesenchyme cell types present in Rosenthal’s canal: Schwann cells, satellite cells or macrophages. To do this, we first examined cross-sections for overlap between Pou3f4 and the glial lineage marker Sox10. Previous work has shown that Sox10 is necessary for the generation of Schwann cells and satellite cells in the periphery, and that all Schwann and satellite cells are Sox10-positive (Britsch et al., 2001; Jessen & Mirsky, 2005; Kuhlbrodt, Herbarth, Sock, Hermans-Borgmeyer, & Wegner, 1998). Consistent with observations in the developing cochlea (Coate et al., 2012), Pou3f4-expressing cells in the postnatal cochlea were distinct from Sox10-positive Schwann or satellite cells (Figure 4a). High-magnification images of nuclei within Rosenthal’s canal revealed no overlap between Pou3f4 and Sox10 (Figure 4b–d). Furthermore, the overall distribution of glia appeared unaffected by the loss of Pou3f4 (Figure 4e–h), suggesting the Pou3f4 cell population in Rosenthal’s canal does not alter glia numbers. We also wanted to determine if the Pou3f4-expressing cells in Rosenthal’s canal were macrophages, and whether Pou3f4 loss altered macrophage distribution. High-magnification images reveal that cells positive for Pou3f4 were clearly distinct from Iba1-expressing macrophages (Figure 4i–j). In addition, Iba1 distribution was similar between Pou3f4y/+ and Pou3f4y/− cochleae (Figure 4i vs. Figure 4k), thus Pou3f4 does not appear to affect macrophage numbers. However, we did notice that some of the macrophages in the Pou3f4y/− cochleae appeared more amoeboid (i.e. rounded) compared to those in the control samples, where most appeared more ramified (i.e. protrusive) (Figure 4j vs. Figure 4l). This could reflect an increase in activity and phagocytosis of SGNs following apoptosis (Parakalan et al., 2012). Although glia and macrophages were present in the spiral ganglion at early postnatal ages (Dong et al., 2018; Romand & Romand, 1985), neither of these cell types stained positively for Pou3f4 and their numbers were not affected by Pou3f4 loss. Overall, the cells in Rosenthal’s canal that express Pou3f4 appear to be otic mesenchyme cells.
Figure 4. Pou3f4-positive cells within Rosenthal’s canal are not Sox10-positive glia or macrophages.
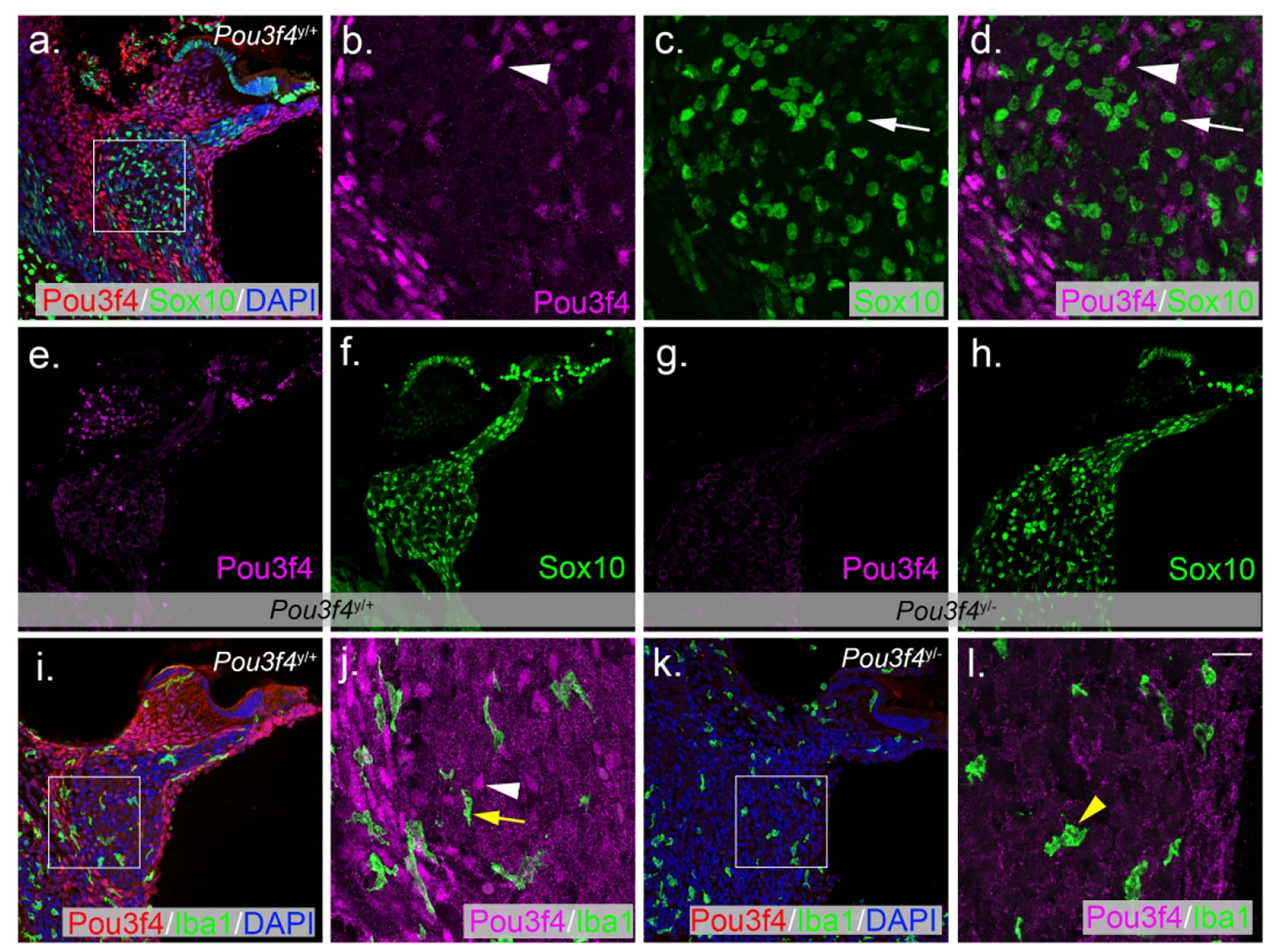
(a) P6 cross-section of the cochlear base labeled with Pou3f4 (red) and Sox10 (green) immunostaining, and DAPI (blue). (b-d) High-magnification views of the boxed region of A reveals that Pou3f4-positive mesenchyme (magenta) and Sox10 positive glia (green) are distinct cell populations within the ganglia. (e-h) P10 sections from Pou3f4y/+ (e-f) and Pou3f4y/− (g-h) cochleae show no change in overall Schwann cell distribution between genotypes. (i) P6 section at the cochlear base labeled with Pou3f4 (red) and Iba1 (green) immunostaining, and DAPI (blue). (j) A high-magnification view from i shows no overlap between Pou3f4-expressing mesenchyme (magenta) and Iba1-positive macrophages (green). Arrowheads indicate Pou3f4 positive nuclei; white arrow indicates a Sox10 expressing cell, yellow arrow indicates an Iba1 expressing cell. (k) There is no difference in the distribution of Iba1-positive cells in Pou3f4y/− cochleae. (l) High-magnification view of Pou3f4y/− cochleae show macrophages appear more amoeboid compared to controls (see yellow arrowhead). Scale = 60 μm (a, e-i,k), 16 μm (b-d,j,l).
3.5. Loss of Pou3f4 leads to increased SGN apoptosis
Next, we considered whether the decrease in SGN density in the absence of Pou3f4 could be due to an increase in SGN apoptosis. We first looked for signs of SGN apoptosis by looking for the apoptosis marker cleaved-caspase 3 (CC3) at P2 and P8. We surmised that, although neuronal loss is not significant until P8, neurons likely initiate apoptosis and are cleared from the ganglion prior to this period. Indeed, we found evidence of CC3-positive neurons in sections at the base of Pou3f4y/− cochleae at both P2 and P8 (Figure 5a–d). While cross-sections such as these provide excellent cell-type resolution, they also only show a small portion of the total SGN population. Therefore, we also utilized whole mount preparations to view the entirety of the spiral ganglion and compared the number of neurons entering apoptosis between Pou3f4y/− and Pou3f4y/+ cochleae. For this quantification, we chose to examine cochleae at P2, which is the time point just prior to the clear onset of SGN loss in the Pou3f4y/− cochleae (Figure 2). We considered apoptotic SGNs to be any CC3-positive cell that was larger than 7 μm and overlapped with the neuronal marker HuD (see Materials and Methods 2.5). Compared with controls, Pou3f4y/− cochleae showed significant increases in SGN apoptosis in both the basal and apical halves (Figure 5e–i). Interestingly, the distribution of CC3-positive SGNs in the Pou3f4y/− cochleae varied by location along the frequency axis: whereas the cochlear apex showed just a 1.85-fold increase, the base showed a 7.34-fold increase (Figure 5j). These data are consistent with the statistically significant loss of SGNs in the base compared to the apex in Pou3f4y/− cochleae at P45 (Figure 1h).
Figure 5. Pou3f4y/− cochleae show increased SGN apoptosis.

(a-d) Cross-sections at P2 (a-b) and P8 (c-d) reveal evidence of the apoptosis marker cleaved caspase 3 (CC3) in Pou3f4y/− spiral ganglion neurons. (e-h) Tile scan images of P2 Pou3f4y/+ and Pou3f4y/− cochlear whole mounts show an increase in apoptotic SGNs in both the apex and the base of the Pou3f4 mutant cochlea. Arrowheads indicate CC3-positive cells; insets in e and f show higher magnification views of apoptotic cells within the spiral ganglion. (i) Quantification of apoptotic cells in Pou3f4y/+ and Pou3f4y/− cochleae per 500 μm of ganglia. Student’s t test; mean ± SEM, *p ≤ 0.05; **p ≤ 0.01; N=6 cochleae for Pou3f4y/+, 6 cochleae for Pou3f4y/−. (j) Pou3f4y/− cochleae show a 7.34-fold increase in CC3-positive cells at the base of the cochlea and a 1.85-fold increase at the apex of the cochlea compared to Pou3f4y/+. Scale = 60 μm (a-d), 100 μm (e-h).
3.6. SGNs from Pou3f4y/− cochleae do not show defects in peripheral axon extension into the cochlear sensory domain.
Numerous studies from the past have highlighted the importance of trophic cues like brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) on SGN survival (Green, Bailey, Wang, & Davis, 2012). BDNF and NT-3 are expressed by hair and supporting cells during SGN peripheral axon extension, and removal of either of these factors, or their receptors, leads to significant SGN loss (Ernfors, Kucera, Lee, Loring, & Jaenisch, 1995; Flores-Otero & Davis, 2011; Fritzsch, Barbacid, & Silos-Santiago, 1998; Fritzsch, Fariñas, & Reichardt, 1997; SUGAWARA, MURTIE, STANKOVIC, LIBERMAN, & CORFAS, 2007). Because SGNs from Pou3f4y/− cochleae show significant fasciculation defects (Coate et al., 2012) we reasoned that these defects may lead to reduced contact with the sensory epithelium, less neurotrophin exposure and ultimately SGN loss. To determine if postnatal SGN loss could be due to hair cell targeting defects, we compared the number of individually-labeled peripheral axons (pa) overlapping with the hair cell marker myosin-VI (MyoVI) in Pou3f4 mutants and littermate controls at P0. P0 is an ideal stage for this analysis because all SGN peripheral axons have reached the sensory epithelium by this stage (Coate, Spita, Zhang, Isgrig, & Kelley, 2015; Druckenbrod & Goodrich, 2015; Koundakjian et al., 2007), and aberrant SGN death in the Pou3f4y/− cochleae has not yet begun (Figure 2g). In order to visualize individual axons, we bred Pou3f4x/lacZ females to males carrying Sox2CreERT2 (Arnold et al., 2011) and R26RtdTomato and examined the cochleae of resulting progeny. Sox2CreERT2 combined with R26RtdTomato led to beautiful sparse SGN labeling in the absence of tamoxifen, as well as some sparse labeling of non-neuronal cells that are also derived from the Sox2 lineage including hair and supporting cells in the sensory epithelium (Figure 6a–c). Because this sparse labeling method relies on expression of Cre recombinase, we were unable to use the Pou3f4 mutant line used in our other analyses because it has Cre in the Pou3f4 coding region. The Pou3f4lacZ line, in which Pou3f4 is removed and lacZ is inserted in its place, was used instead (Heydemann, Nguyen, & Crenshaw, 2001). In both Pou3f4y/+ and Pou3f4y/lacZ cochleae, the majority of axons (96.9% and 98.9%, respectively) overlap with the hair cell marker MyoVI, indicating that SGN loss is most likely not secondaryto a hair cell targeting defect in Pou3f4 mutants (Figure 6d–f).
Figure 6. Loss of Pou3f4 does not prevent spiral ganglion neurons from reaching the sensory epithelium.
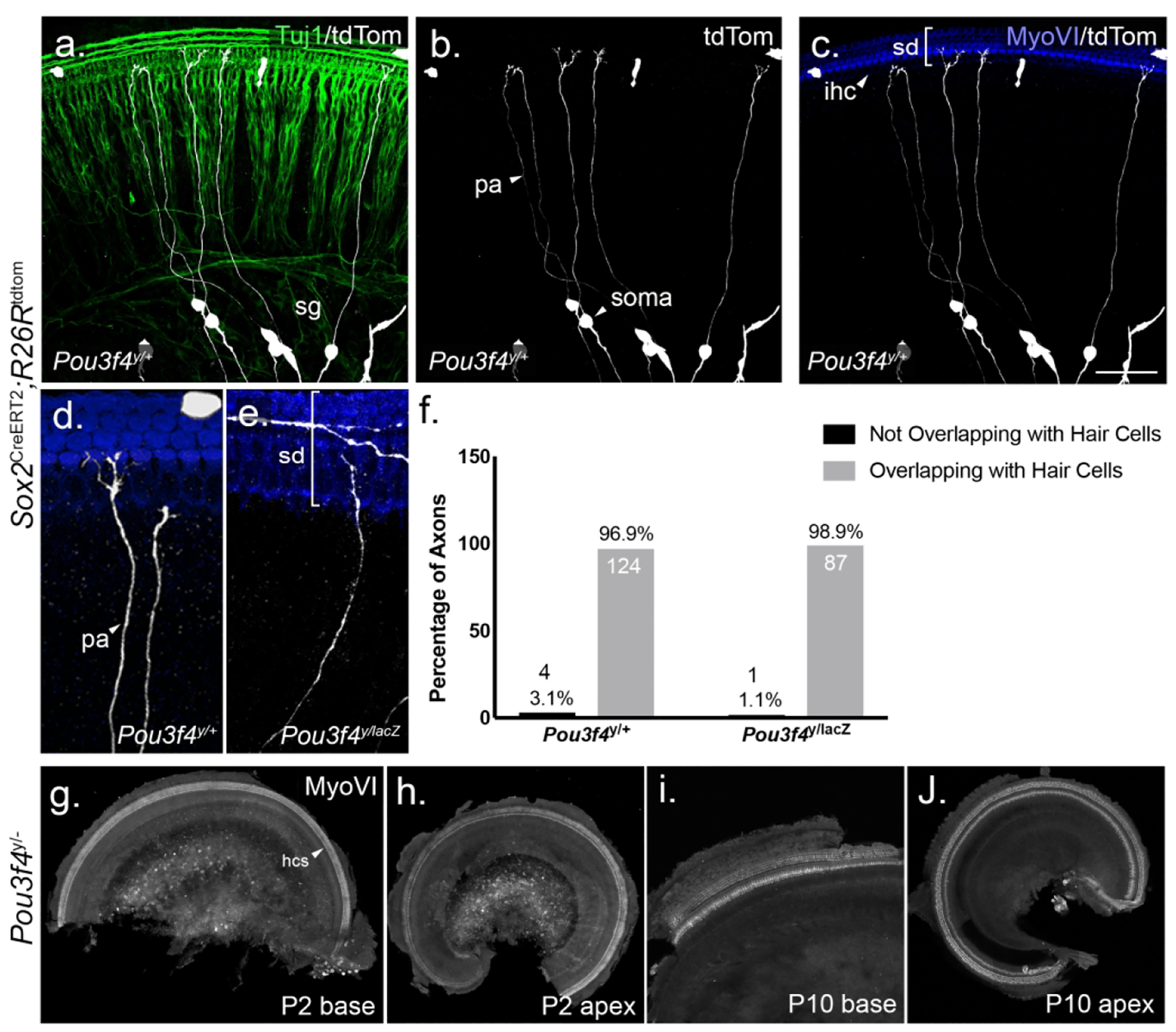
(a-c) A wholemount view of a P0 cochlea demonstrating the advantage of the Sox2CreERT2;R26Rtdtomato sparse label (tdTom, white) over the pan-neuronal marker (Tuj1, green) for visualizing individual peripheral axons. sg, spiral ganglion; pa, peripheral axon. Note in c how the SGNs are visualized overlapping with hair cells (MyoVI, blue). sd, sensory domain; ihc, inner hair cell. (d-e) Representative images of SGN peripheral axons (white) and hair cells (blue) in Pou3f4y/+and Pou3f4y/lacZ cochleae. (f). Quantification showing how the majority of SGN peripheral axons in both genotypes (96.9% in Pou3f4y/+; 98.9% in Pou3f4y/lacZ) contact hair cells by P0. N=5 cochleae, 128 axons Pou3f4y/+; 5 cochleae, 88 axons Pou3f4y/lacZ. (g-j) Representative images from Pou3f4y/− cochleae demonstrating no loss of hair cells (MyoVI, white) throughout the cochlea at P2 and P10. hcs, hair cells. Scale = 60 μm (a-c), 32 μm (d-e), 480 μm (g-h,j), 240 μm (i).
SGN death has also been noted following hair cell loss in multiple mammalian species (Dupont & Guilhau, 1993; Mcfadden, Ding, Jiang, & Salvi, 2004). Previous work indicated that hair cells appeared healthy and were present in normal numbers in a different Pou3f4 null strain (Minowa et al., 1999). However, we wanted to check for any changes in hair cell survival in our Pou3f4 mutants. At both P2 (Figure 6g–h), when we noted an increase in SGN apoptosis (Figure 5i), and P10 (Figure 6i–j), when about 1/5 of neurons are lost (Figure 2g), we did not observe any missing hair cells in either the basal or apical turns of Pou3f4y/− cochleae (Figure 6). Taken together, these data suggest that Pou3f4 does not indirectly impact SGN survival via hair cell targeting defects or hair cell loss.
3.7. The proportion of type I SGN subtypes are unaltered by the loss of Pou3f4
Recent single cell RNA-seq work has identified three subtypes of type I SGNs (Petitpré et al., 2018; Shrestha et al., 2018; Sun et al., 2018), which correspond to previous observations that SGNs vary in thickness and excitability, and organize in a stereotyped way around the base of the inner hair cell (M C Liberman, 1982). As a result of the extended period of SGN refinement, these subtypes are not clearly defined until about P28. At this point, type I SGNs can be classified by the expression of Calb1, Calb2 and Pou4f1, although some SGNs are positive for two of these factors (Shrestha et al., 2018). Given that approximately 25% of SGNs die in Pou3f4 mutants, we wanted to determine if one particular subtype was more susceptible to dying in the absence of Pou3f4, or if all subtypes were equally susceptible. To determine this, we labeled cross-sections of 4 week old cochleae from both Pou3f4y/+ and Pou3f4y/− mice with anti-Calb1, -Calb2, and -Pou4f1 antibodies, counted how many of each type were present in each section and converted this into a percentage. Since some SGNs are positive for two of these factors, we labeled for all three types in the same section and came up with seven separate categories (Figure 7; see Materials and Methods 2.7). Given that SGNs at the base of the cochlea are most sensitive to Pou3f4 loss (Figure 5j), we limited this analysis to the cochlear base.
Overall, we found type I SGN subtypes in proportions similar to what was described previously (Petitpré et al., 2018; Shrestha et al., 2018; Sun et al., 2018). First, slightly less than a quarter of SGNs were positive for only Calb1 or only Pou4f1 (Figure 7b–c, i; 20.9±2.5% and 21.0±2.2%, respectively), with a small portion of overlap between the two (Figure 7i; 2.0±0.7%). Second, another 20–25% of neurons were positive only for Calb2 (Figure 7d,i; 23.9±4.2%), with a large overlap between the Calb1 and Calb2 populations (Figure 7i; 26.5±4.2%). A very small portion of the neurons also overlapped between Pou4f1 and Calb2 (Figure 7i; 0.4±0.3%). Finally, neurons positive for none of the markers are classified as “none” and make up around 5% of the neurons in each section, indicating that this population is most likely type II SGNs (Figure 7i; 5.3±0.9%). Surprisingly, our Pou3f4y/− samples showed almost no significant change in the distribution of any of these subtypes (Figure 7e–i), except for a slight increase in the percentage of Pou4f1 positive SGNs in the Pou3f4 mutants (Figure 7i; 25.8±2.1%). For statistical tests, we examined possible shifts in these populations using ordination techniques and found no significant differences. Given that no single population of type I SGN is absent or greatly reduced in the Pou3f4 mutants, Pou3f4-mediated neuronal survival appears to be independent from SGN subtype specification.
In a separate study using Pou3f4lacZ mutants and littermate controls, we asked if there were any changes in the overall percentage of neurons positive for Calb2, Calb1, Pou4f1 or the type II SGN marker Prph (Shrestha et al., 2018) at P0, P14 and P28. Each section was labeled with one subtype marker and anti-Tuj1, and the number of positive SGNs was calculated as a percentage of total neurons labeled by Tuj1. Even at earlier ages, we found little difference between the Pou3f4y/+ and Pou3f4y/lacZ type I SGN subtypes (Figure 7j–l). At P0, there were no notable differences in any of the type I subtypes (Figure 7j, m-t) and at P14 only a slight uptick in the Pou4f1 population was noted (Figure 7k; 31.90% vs. 35.21%, p=0.05; Supplemental Figure 1a–h). However, this difference was not seen at P28 between Pou3f4y/+ and Pou3f4y/lacZ cochleae (Figure 7l; 32.05% vs. 29.87%, p=0.15; Supplemental Figure 1i–p) indicating that Pou3f4 is dispensable for the development of proper type I SGN subtype proportions.
4. DISCUSSION
Numerous studies have shown that SGNs are dependent on multiple trophic cues to survive during and after development (Green et al., 2012). Our data suggest otic mesenchyme cells may provide an additional, previously unknown, source of support to SGNs in the early postnatal cochlea. In the absence of the mesenchyme-specific transcription factor Pou3f4, a portion of SGNs undergo apoptosis shortly after birth resulting in about a 25% decrease in the overall SGN population (Figures 2 and 5). Given how the proportion of SGN loss in Pou3f4 mutants is relatively small, it is likely that trophic factors, such as BDNF or NT3 (known in SGN survival and discussed below), are still present. This SGN loss is not due to a lack of nearby glia (Figure 4) and is likely not a result of the axon guidance defects described previously (Figure 6). One possibility is that Pou3f4-expressing mesenchyme cells provide a direct source of trophic support to the SGNs (Summary Figure 8a; see “1”). This idea is based on the location and timing of Pou3f4 expression: Pou3f4-expressing mesenchyme cells are most abundant surrounding the SGN cell bodies within the same developmental time frame (P2–P8) that Pou3f4y/− cochleae show a loss of SGNs (Figures 2 and 3). The transient expression of Pou3f4 by mesenchyme cells in the vicinity of the SGN cell bodies is in contrast to Pou3f4 expression in cochlear regions further from the SGNs, like the stria vascularis, which maintains Pou3f4 expression throughout adulthood. The continued expression of Pou3f4 in other areas of the cochlea, but not in proximity to the SGNs, underscores the possibility that these mesenchyme cells could be providing some form of direct trophic support. Additionally, if this interaction were dependent upon direct cell to cell contact, it could account for why only a portion of SGNs are lost in Pou3f4 mutants.
Figure 8. Pou3f4-expressing mesenchyme cells promote early postnatal spiral ganglion neuron survival via an unknown mechanism.
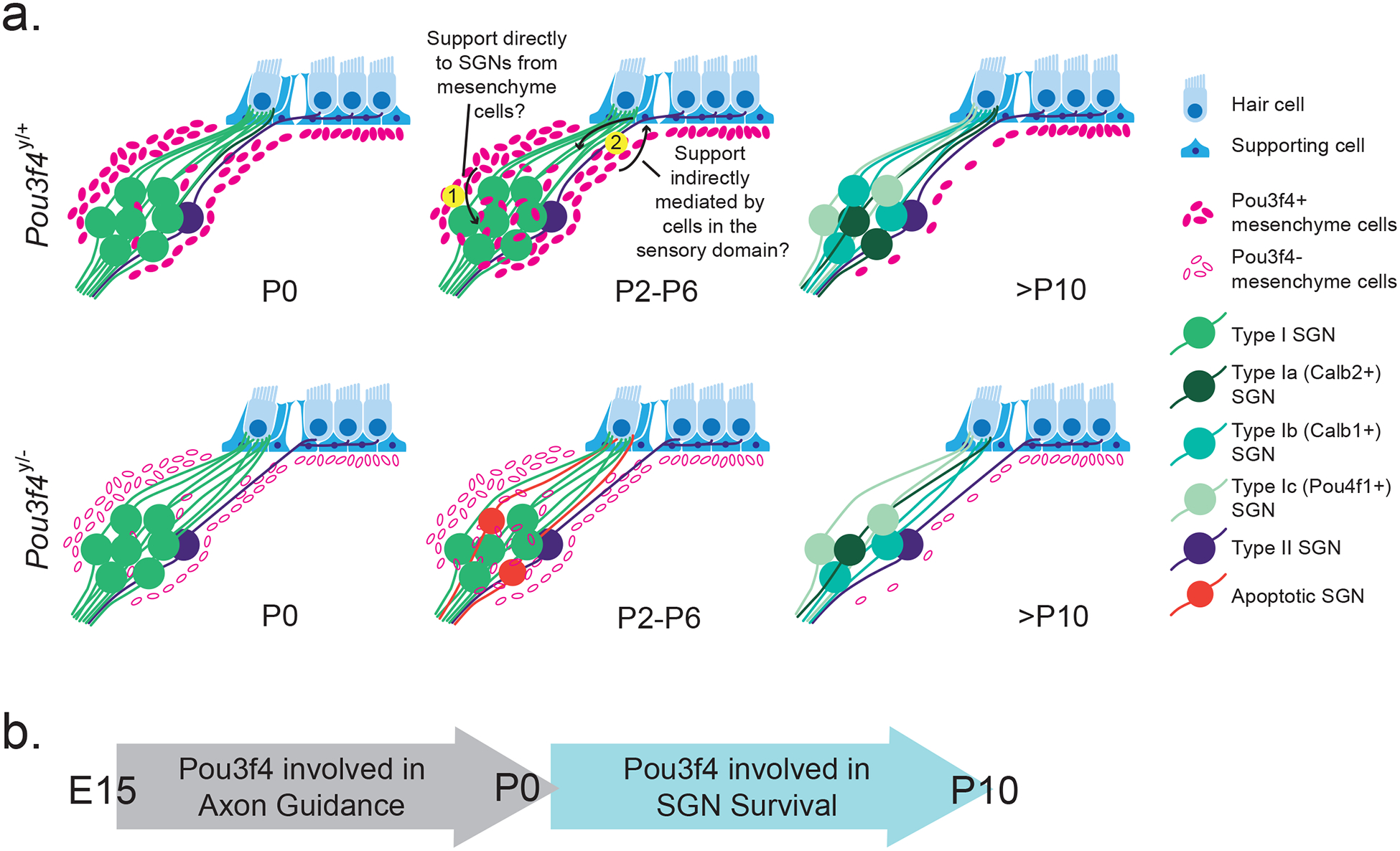
(a) Schematic showing normal Pou3f4 expression within and surrounding Rosenthal’s canal within the first 10 postnatal days (top row) and the impact of Pou3f4 loss on SGNs (bottom row). At P0, Pou3f4y/+ and Pou3f4y/− cochleae have comparable numbers of SGNs, and SGNs in both genotypes reach the sensory domain and contact hair cells. Starting at P2, a portion of SGNs in Pou3f4y/− cochleae enter apoptosis; by P10 about 25% of SGNs are lost. Within the same time frame, Pou3f4-expressing mesenchyme cells peak in number and expression near the SGNs in Pou3f4y/+ cochleae. It is possible that (1) Pou3f4-expressing mesenchyme cells provide an unknown trophic factor directly to SGNs or (2) Pou3f4-expressing mesenchyme cells signal to the cells in the sensory domain to provide trophic support to SGNs. (b) At embryonic stages, Pou3f4 is involved in SGN axon guidance (Coate et al., 2012); whereas Pou3f4 has a role in SGN survival during postnatal stages.
Another possibility is that Pou3f4-expressing mesenchyme cells interact with SGNs indirectly by signaling to cells in the sensory epithelium, which then provide trophic support to the SGNs (Summary Figure 8a; see “2”). In this scenario, factors like BDNF and NT-3 could be examined given their importance in SGN development and survival (Ernfors, Van De Water, Loring, & Jaenisch, 1995; Schimmang et al., 2003; Stankovic & Corfas, 2003). Moreover, BDNF and NT-3 are expressed in basal-to-apical and apical-to-basal gradients, respectively, thus any changes in their expression in Pou3f4 mutants could result in SGN loss in a spatially biased manner. Members of the TGFbeta family of growth factors, including glial cell-line-derived neurotrophic factor (GDNF), neurturin, artemin and persephin are also expressed in the postnatal cochlea (Stankovic & Corfas, 2003; Stöver, Gong, Cho, Altschuler, & Lomax, 2000). All four have been localized to the spiral ganglion via immunohistochemistry (Stöver, Nam, Gong, Lomax, & Altschuler, 2001; Wefstaedt, Scheper, Rieger, Lenarz, & Stöver, 2006). In addition, mRNA of these four trophic factors were detectable in cochlear tissue even when the SGNs and sensory domain were removed (Stöver et al., 2000), suggesting the area surrounding the SGNs may also express these factors. Many other families of growth factors and their receptors have also been localized to the cochlear organ of Corti, stria vascularis and spiral ganglion (Malgrange et al., 1998) such as ciliary neurotrophic factor (CNTF), which improves SGN survival in culture (D. S. Whitlon, Grover, Tristano, Williams, & Coulson, 2007). Indeed, in situ data from online databases show that otic mesenchyme cells express members of the platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) families (Allen Institute for Brain Science, 2015; Visel, Thaller, & Eichele, 2004). In addition, Eph/Ephrin signaling is known to regulate neuron numbers (Depaepe et al., 2005), and Pou3f4 is known to transcriptionally regulate levels of these factors (Coate et al., 2012; Raft et al., 2014). In future studies, it will be important to determine how possible trophic cues downstream of Pou3f4 control SGN survival.
The mechanism of Pou3f4-mediated neuronal support is still unclear, as it does not seem that Pou3f4 controls the distribution of glia and macrophages, or hair cell survival. As shown in Figure 4, the overall distribution of the glial marker Sox10 and the macrophage marker Iba1 do not change in the absence of Pou3f4. However, we did notice an increase in amoeboid macrophages in the Pou3f4y/− cochleae at P6. This change in morphology may reflect more active, phagocytic macrophages (Parakalan et al., 2012) responding to the increase in apoptotic neurons shown in Figure 5. Future studies should include a more in-depth look at activity of macrophages in Pou3f4y/− cochleae.
The data reported here offer clarity on the previous observation that Pou3f4y/− cochleae show a decline in inner hair cell synapse numbers (Coate et al., 2012). This decline was originally attributed to impaired hair cell targeting resulting from axon fasciculation deficits during development (Coate et al., 2012). However, these synapse counts were taken at P8, an age at which we already see a loss of SGNs (Figure 2). Instead, it is more likely that ribbon synapse loss in Pou3f4y/− cochleae results from SGN death rather than axon guidance defects. In support of this, we found that SGNs from WT and Pou3f4y/− cochleae at P0 show comparable percentages of fibers having arrived at the cochlear epithelium (Figure 6). We also detected no deficiencies in hair cell (Figure 6) or supporting cell (not shown) numbers in the in Pou3f4y/− cochleae, indicating that SGN loss is likely not due to defects in organ of Corti formation. We do note that Pou3f4 mutants are reported to show reduced endocochlear potential (Minowa et al., 1999), which is necessary for normal hair cell function. While this likely causes the hearing loss in Pou3f4 mutant adults, normal endocochlear potential does not reach adult levels until around P17 (Sadanaga & Morimitsu, 1995). Given that we detected SGN apoptosis in Pou3f4y/− cochleae starting at P2 (Figure 5), we predict that loss of endocochlear potential likely does not contribute to SGN death.
Several studies both in the cochlea and in other systems have illustrated the importance of programmed cell death for normal tissue structures to develop (Fuchs & Steller, 2011; Mammano & Bortolozzi, 2017). For example, in the superior cervical ganglion, neuron number is regulated by nerve growth factor (NGF), and neurons lacking NGF undergo programmed apoptosis (Kristiansen & Ham, 2014). A thorough analysis of early postnatal apoptosis in the gerbil cochlea revealed that programmed cell death also contributes to final numbers of SGNs. The majority of programed SGN death occurs within the apical and middle turns of the cochlea between P4 and P6, although some SGN death was also noted at the base. Interestingly, even at its peak, only about 2% of SGNs were apoptotic at any time (Echteler, Magardino, & Rontal, 2005). This is consistent with our observation that apical neurons are more likely to be apoptotic in both Pou3f4y/+ and Pou3f4y/− cochleae (Figure 5), and emphasizes that the increase in CC3-positive neurons at the base of Pou3f4y/− cochleae represents elevated SGN apoptosis. It is possible that programmed SGN death is involved in the careful refinement of SGN-hair cell contacts that continues well into the first month of cochlear development (Coate et al., 2019). So, given that normal SGN apoptosis coincides with the peak of Pou3f4 expression surrounding the spiral ganglion, and that loss of Pou3f4 results in elevated SGN death, it is possible that Pou3f4 and mesenchyme cells play a role in regulating the normal phase of programmed SGN death.
Finally, we found that Pou3f4y/+ and Pou3f4y/− cochleae did not differ in proportions of type I SGN subtypes despite the loss of SGNs in Pou3f4 mutants. This presents two interesting possibilities. First, Pou3f4 possibly regulates SGN survival prior to the normal period of type I SGN differentiation. It has been shown that the appearance of well-defined subtypes occurs gradually after birth: during the first postnatal week, most SGNs are positive for multiple subtype markers compared to the fourth postnatal week, when SGNs have differentiated (Shrestha et al., 2018). Pou3f4y/− SGN death could occur prior to this period of specification, resulting in the development of normal SGN subtype proportions despite the loss of SGN numbers. Second, it is possible that Pou3f4 mutants escape defects in SGN differentiation that result from activity deficits. Previously, normal patterns of SGN subtype specification were shown to be altered in two other hearing loss models, Tmie−/− and Vglut3−/− mice (Shrestha et al., 2018; Sun et al., 2018), and the defects were attributable to a loss of afferent synaptic activity. To date, activity has not been investigated in Pou3f4 mutants aside from measurements of endocochlear potential in adult mice (Minowa et al., 1999), and it is likely that spontaneous activity (Tritsch, Zhang, Ellis-Davies, & Bergles, 2010) is intact in Pou3f4 mutants. Spontaneous activity in SGNs occurs prior to hearing onset and the development of endocochlear potential, and this likely allows normal proportions of type I SGNs to develop in Pou3f4 mutants. Ultimately, it seems that Pou3f4 is dispensable with regard to SGN subtype specification.
One idea that has emerged from this work is that mesenchyme cells may have potential for supporting SGNs in adult cochleae following trauma. Great progress has been made in the field of SGN survival following trauma including increasing the epithelial factors BDNF and NT-3 either via intracochlear infusion (Sly et al., 2016) or specially modified viral vectors (Wise et al., 2011). Intracochlear infusions can help mediate SGN loss following aminoglycoside-induced hair cell death by temporarily increasing BDNF or NT-3 levels (Sly et al., 2016). Additionally, modified viral vectors have proven effective at restoring hearing loss in mice with particular genetic hair cell deficits (Chien et al., 2016), and viral delivery of neurotrophin genes to supporting cells can support SGN survival even multiple weeks following acoustic trauma, although there are still some challenges (Wise et al., 2011). Given that mesenchyme cells are abundant and in close proximity to SGNs, they could potentially offer another abundant, targetable cell for viral-mediated trophic SGN support. Moving forward it will be important to identify the mechanism(s) by which Pou3f4 and mesenchyme cells provide trophic support to early postnatal SGNs.
Supplementary Material
Supplemental Figure 1. Type I SGN subtype specification of Pou3f4 mutants at P14 and P28. (a-h) Images of P14 Pou3f4y/+ and Pou3f4y/lacZ cochleae at the middle turn, labeled with anti-Tuj1 to label all SGNs and either anti-Calb2, -Calb1, -Pou4f1 or -Prph. (i-p) Images of P28 Pou3f4y/+ and Pou3f4y/lacZ cochleae at the middle turn, labeled with anti-Tuj1 to label all SGNs and either anti-Calb2, -Calb1, -Pou4f1 or -Prph. Scale = 50 μm.
Acknowledgements:
This work is supported by the National Institutes of Health R01 DC016595 (to TMC and RH). We thank the members of the Coate laboratory (Georgetown University) for their thoughtful discussions and technical assistance. We also thank Dr. Katie Kindt (National Institute on Deafness and other Communication Disorders) for critical reading of the manuscript and Dr. Gina Wimp for her statistics expertise. Talya Inbar (Georgetown University) was instrumental in optimizing the staining protocol used for the experiments in Figure 7a–i.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Footnotes
Conflicts of Interest: none.
REFERENCES
- Adamson CL, Reid MA, Mo ZL, Bowne-English J, & Davis RL (2002). Firing features and potassium channel content of murine spiral ganglion neurons vary with cochlear location. Journal of Comparative Neurology, 447(4), 331–350. 10.1002/cne.10244 [DOI] [PubMed] [Google Scholar]
- Ahn KJ, Passero F, & Crenshaw EB (2009). Otic mesenchyme expression of Cre recombinase directed by the inner ear enhancer of the Brn4/Pou3f4 gene. Genesis, 47, 137–141. 10.1002/dvg.20454 [DOI] [PubMed] [Google Scholar]
- Allen Institute for Brain Science. (2015). Allen Mouse Brain Atlas. Allen Mouse Brain Atlas. Retrieved from hhtp://mouse.brain-map.org [Google Scholar]
- Anniko M, & Wróblewski R (1986). Ionic environment of cochlear hair cells. Hearing Research, 22(1–3), 279–293. 10.1016/0378-5955(86)90104-8 [DOI] [PubMed] [Google Scholar]
- Arnold K, Sarkar A, Yram MA, Polo JM, Bronson R, Sengupta S, … Hochedlinger K (2011). Sox2 + adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell, 9(4), 317–329. 10.1016/j.stem.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuskin I, Bodson M, Thelen N, Thiry M, Borgs L, Nguyen L, … Malgrange B (2010). Glial but not neuronal development in the cochleo-vestibular ganglion requires Sox10. Journal of Neurochemistry, 114, 1827–1839. 10.1111/j.1471-4159.2010.06897.x [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, … Wegner M (2001). The transcription factor Sox10 is a key regulator of peripheral glial development. Genes and Development, 15(1), 66–78. 10.1101/gad.186601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien WW, Isgrig K, Roy S, Belyantseva IA, Drummond MC, May LA, … Cunningham LL (2016). Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Molecular Therapy, 24(1), 17–25. 10.1038/mt.2015.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate TM, & Kelley MW (2013). Making connections in the inner ear: Recent insights into the development of spiral ganglion neurons and their connectivity with sensory hair cells. Seminars in Cell and Developmental Biology, 24(5), 460–469. 10.1016/j.semcdb.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate TM, Raft S, Zhao X, Ryan A, Crenshaw EB, & Kelley MW (2012). Otic Mesenchyme Cells Regulate Spiral Ganglion Axon Fasciculation through a Pou3f4/EphA4 Signaling Pathway. Neuron, 73(1), 49–63. 10.1016/j.neuron.2011.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate TM, Scott MK, & Gurjar MC (2019). Current Concepts in Cochlear Ribbon Synapse Formation. Synapse, 73(5), e22087 10.1002/syn.22087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate TM, Spita NA, Zhang KD, Isgrig KT, & Kelley MW (2015). Neuropilin-2/semaphorin-3F-mediated repulsion promotes inner hair cell innervation by spiral ganglion neurons. ELife, 4(AUGUST2015), 1–24. 10.7554/eLife.07830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, … Vanderhaeghen P (2005). Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 10.1038/nature03651 [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang C, Frye M, Yang W, Ding D, Sharma A, … Hu BH (2018). Differential fates of tissue macrophages in the cochlea during postnatal development. Hearing Research. 10.1016/j.heares.2018.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druckenbrod NR, & Goodrich LV (2015). Sequential Retraction Segregates SGN Processes during Target Selection in the Cochlea. Journal of Neuroscience, 35(49), 16221–16235. 10.1523/JNEUROSCI.2236-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont J, & Guilhau A (1993). Neuronal degeneration of primary cochlear and vestibular innervations after local injection of sisomicin in the guinea pig. [DOI] [PubMed]
- Echteler SM, Magardino T, & Rontal M (2005). Spatiotemporal patterns of neuronal programmed cell death during postnatal development of the gerbil cochlea. Developmental Brain Research, 157, 192–200. 10.1016/j.devbrainres.2005.04.004 [DOI] [PubMed] [Google Scholar]
- Ernfors P, Kucera J, Lee K, Loring J, & Jaenisch R (1995). Int J Dev Biol - Studies on the physiological role of brain-derived neurotrophic factor and neurotrophin-3 in knockout mice. International Journal of Developmental Biology, 807, 799–807. [PubMed] [Google Scholar]
- Ernfors P, Van De Water T, Loring J, & Jaenisch R (1995). Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron, 14(6), 1153–1164. 10.1016/0896-6273(95)90263-5 [DOI] [PubMed] [Google Scholar]
- Flores-Otero J, & Davis RL (2011). Synaptic proteins are tonotopically graded in postnatal and adult type I and type II spiral ganglion neurons. Journal of Comparative Neurology, 519(8), 1455–1475. 10.1002/cne.22576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores EN, Duggan A, Madathany T, Hogan AK, Márquez FG, Kumar G, … García-Añoveros J (2015). A non-canonical pathway from cochlea to brain signals tissue-damaging noise. Current Biology, 25(5), 606–612. 10.1016/j.cub.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Barbacid M, & Silos-Santiago I (1998). The combined effects of trkB and trkC mutations on the innervation of the inner ear. International Journal of Developmental Neuroscience, 16(6), 493–505. 10.1016/S0736-5748(98)00043-4 [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Fariñas I, & Reichardt LF (1997). Lack of Neurotrophin 3 Causes Losses of Both Classes of Spiral Ganglion Neurons in the Cochlea in a Region-Specific Fashion. The Journal of Neuroscience, 17(16), 6213–6225. 10.1523/jneurosci.17-16-06213.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, & Steller H (2011). Programmed cell death in animal development and disease. Cell, 147(4), 742–758. 10.1016/j.cell.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SH, Bailey E, Wang Q, & Davis RL (2012). The Trk A, B, C’s of Neurotrophins in the Cochlea. Anatomical Record, 295, 1877–1895. 10.1002/ar.22587 [DOI] [PubMed] [Google Scholar]
- Hafidi A (1998). Peripherin-like immunoreactivity in type II spiral ganglion cell body and projections. Brain Research, 805(1–2), 181–190. 10.1016/S0006-8993(98)00448-X [DOI] [PubMed] [Google Scholar]
- Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao YD, Yee AG, … Corey DP (1997). Unconventional myosins in inner-ear sensory epithelia. Journal of Cell Biology, 137(6), 1287–1307. 10.1083/jcb.137.6.1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T, & Mooseker MS (1994). Porcine myosin-VI: Characterization of a new mammalian unconventional myosin. Journal of Cell Biology, 127(2), 425–440. 10.1083/jcb.127.2.425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydemann A, Nguyen LC, & Crenshaw EB (2001). Regulatory regions from the Brn4 promoter direct LACZ expression to the developing forebrain and neural tube. Developmental Brain Research, 128(1), 83–90. 10.1016/S0165-3806(01)00137-7 [DOI] [PubMed] [Google Scholar]
- Ikenaga T, Urban JM, Gebhart N, Hatta K, Kawakami K, & Ono F (2011). Formation of the spinal network in zebrafish determined by domain-specific pax genes. Journal of Comparative Neurology, 519(8), 1562–1579. 10.1002/cne.22585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, & Kohsaka S (1996). A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochemical and Biophysical Research Communications, 224(3), 855–862. 10.1006/bbrc.1996.1112 [DOI] [PubMed] [Google Scholar]
- Jessen KR, & Mirsky R (2005). The origin and development of glial cells in peripheral nerves. Nature Reviews Neuroscience, 6(9), 671–682. 10.1038/nrn1746 [DOI] [PubMed] [Google Scholar]
- Ji MM, Wang L, Zhan Q, Xue W, Zhao Y, Zhao X, … Zhao WL (2015). Induction of autophagy by valproic acid enhanced lymphoma cell chemosensitivity through HDAC-independent and IP3-mediated PRKAA activation. Autophagy, 11(12), 160–2171. 10.1080/15548627.2015.1082024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wang X, Zhang J, Kachelmeier A, Lopez IA, & Shi X (2019). Microvascular networks in the area of the auditory peripheral nervous system. Hearing Research, 371, 105–116. [DOI] [PubMed] [Google Scholar]
- Kiang NYS, Rho JM, Northrop CC, Liberman MC, & Ryugo DK (1982). Hair-Call Innervation by Spiral Ganglion Cells in Adult Cats. Science, 217, 175–177. [DOI] [PubMed] [Google Scholar]
- Kidokoro Y, Karasawa K, Minowa O, Sugitani Y, Noda T, Ikeda K, & Kamiya K (2014). Deficiency of transcription factor Brn4 disrupts cochlear gap junction plaques in a model of DFN3 non-syndromic deafness. PLoS ONE, 9(9). 10.1371/journal.pone.0108216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok YJM De, Maarel SM Van Der, Bitner-glindzicz M, Malcolm S, Pembrey ME, Ropers H, … Cremers M (1995). Association Between X-Linked Mixed Deafness and Mutations in the POU Domain Gene POU3F4 Published by : American Association for the Advancement of Science Stable URL : http://www.jstor.org/stable/2885951. Science, 267(5198), 685–688. [DOI] [PubMed] [Google Scholar]
- Koundakjian EJ, Appler JL, & Goodrich LV (2007). Auditory neurons make stereotyped wiring decisions before maturation of their targets. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 27, 14078–14088. 10.1523/JNEUROSCI.3765-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen M, & Ham J (2014). Programmed cell death during neuronal development: The sympathetic neuron model. Cell Death and Differentiation, 21(7), 1025–1035. 10.1038/cdd.2014.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, & Wegner M (1998). Sox10, a novel transcriptional modulator in glial cells. Journal of Neuroscience, 18(1), 237–250. 10.1523/jneurosci.18-01-00237.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Rebhun LI, & Frankfurter A (1990). Posttranslational modification of class III β-tubulin. Proceedings of the National Academy of Sciences of the United States of America, 87(18), 7195–7199. 10.1073/pnas.87.18.7195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, & Ginty DD (2014). The structure and organization of lanceolate mechanosensory complexes at mouse hair follicles. ELife, 2014(3), 1–24. 10.7554/eLife.01901.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mo Z, Yang X, Price SM, Shen MM, & Xiang M (2004). Foxn4 controls the genesis of amacrine and horizontal cells by retinal progenitors. Neuron, 43(6), 795–807. 10.1016/j.neuron.2004.08.041 [DOI] [PubMed] [Google Scholar]
- Liberman LD, & Liberman MC (2016). Postnatal maturation of auditory-nerve heterogeneity, as seen in spatial gradients of synapse morphology in the inner hair cell area. Hearing Research, 339, 12–22. 10.1016/j.heares.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman M Charles, & Oliver ME (1984). Morphometry of intracellularly labeled neurons of the auditory nerve: Correlations with functional properties. Journal of Comparative Neurology. 10.1002/cne.902230203 [DOI] [PubMed] [Google Scholar]
- Liberman MC (1982). Single-neuron labeling in the cat auditory nerve. Science (New York, N.Y.), 216, 1239–1241. 10.1126/science.7079757 [DOI] [PubMed] [Google Scholar]
- Liu C, Glowatzki E, & Fuchs PA (2015). Unmyelinated type II afferent neurons report cochlear damage. Proceedings of the National Academy of Sciences, 112(47), 14723–14727. 10.1073/pnas.1515228112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen Z, Pompa L De, & Anderson DJ (1998). neurogenin1 Is Essential for the Determination of Neuronal Precursors for Proximal Cranial Sensory Ganglia. Neuron, 20(3), 469–482. [DOI] [PubMed] [Google Scholar]
- Malgrange B, Rogister B, Lefebvre PP, Mazy-Servais C, Welcher AA, Bonnet C, … Moonen G (1998). Expression of growth factors and their receptors in the postnatal rat cochlea. Neurochemical Research, 23(8), 1133–1138. 10.1023/A:1020724506337 [DOI] [PubMed] [Google Scholar]
- Mammano F, & Bortolozzi M (2017). Ca2+signaling, apoptosis and autophagy in the developing cochlea: Milestones to hearing acquisition. Cell Calcium. 10.1016/j.ceca.2017.05.006 [DOI] [PubMed] [Google Scholar]
- Matei V, Pauley S, Kaing S, Rowitch D, Beisel K, Morris K, … Fritzsch B (2005). Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell terminal mitosis. Developmental Dynamics, 234(3), 633–650. 10.1002/dvdy.20551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcfadden SL, Ding D, Jiang H, & Salvi RJ (2004). Time course of efferent fiber and spiral ganglion cell degeneration following complete hair cell loss in the chinchilla, 997, 40–51. 10.1016/j.brainres.2003.10.031 [DOI] [PubMed] [Google Scholar]
- Minowa O, Ikeda K, Sugitani Y, Oshima T, Nakai S, Katori Y, … Noda T (1999). Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science, 285(5432), 1408–1411. https://doi.org/7787 [pii] [DOI] [PubMed] [Google Scholar]
- Nadol J, Burgress B, & Reisser C (1990). Morphometric Analysis of Normal Human Spiral Ganglion Cells. Ann Otol Rhinol Laryngol, 99, 340–348. [DOI] [PubMed] [Google Scholar]
- Parakalan R, Jiang B, Nimmi B, Janani M, Jayapal M, Lu J, … Dheen ST (2012). Transcriptome analysis of amoeboid and ramified microglia isolated from the corpus callosum of rat brain. BMC Neuroscience, 13(1). 10.1186/1471-2202-13-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitpré C, Wu H, Sharma A, Tokarska A, Fontanet P, Wang Y, … Lallemend F (2018). Neuronal heterogeneity and stereotyped connectivity in the auditory afferent system. Nature Communications, 9(1), 3691 10.1038/s41467-018-06033-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phippard D, Lu L, Lee D, Saunders JC, & Crenshaw EB (1999). Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 19(14), 5980–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phippard Deborah, Heydemann A, Lechner M, Lu L, Lee D, Kyin T, & Crenshaw EB (1998). Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hearing Research, 120(1–2), 77–85. 10.1016/S0378-5955(98)00059-8 [DOI] [PubMed] [Google Scholar]
- Raft S, Coate TM, Kelley MW, Crenshaw EB, & Wu DK (2014). Pou3f4-mediated regulation of ephrin-B2 controls temporal bone development in the mouse. PLoS ONE, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft S, Koundakjian EJ, Quinones H, Jayasena CS, Goodrich LV, Johnson JE, … Groves AK (2007). Cross-regulation of Ngn1 and Math1 coordinates the production of neurons and sensory hair cells during inner ear development. Development, 134, 4405–4415. 10.1242/dev.009118 [DOI] [PubMed] [Google Scholar]
- Romand R, & Romand M (1985). Qualitative and quantitative observations of spiral ganglion development in the rat, 18, 111–120. [DOI] [PubMed] [Google Scholar]
- Ruben RJ, Van de Water T, & Rubel EW (1986). The biology of change in otolaryngology. In Proceedings of the symposium of the 9th ARO Mid-winter Research Meeting (pp. 1–404). [Google Scholar]
- Sadanaga M, & Morimitsu T (1995). Development of endocochlear potential and its negative component in mouse cochlea. Hearing Research, 89(1–2), 155–161. 10.1016/0378-5955(95)00133-X [DOI] [PubMed] [Google Scholar]
- Samadi DS, Saunders JC, & Crenshaw EB (2005). Mutation of the POU-domain gene Brn4/Pou3f4 affects middle-ear sound conduction in the mouse. Hearing Research, 199(1–2), 11–21. 10.1016/j.heares.2004.07.013 [DOI] [PubMed] [Google Scholar]
- Schimmang T, Tan J, Müller M, Zimmermann U, Rohbock K, Köpschall I, … Knipper M (2003). Lack of Bdnf and TrkB signalling in the postnatal cochlea leads to a spatial reshaping of innervation along the tonotopic axis and hearing loss. Development, 130(19), 4741–4750. 10.1242/dev.00676 [DOI] [PubMed] [Google Scholar]
- Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, & Goodrich LV (2018). Sensory Neuron Diversity in the Inner Ear Is Shaped by Activity. Cell, 174(5), 1229–1246.e17. 10.1016/j.cell.2018.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly DJ, Campbell L, Uschakov A, Saief ST, Lam M, & O’Leary SJ (2016). Applying neurotrophins to the round window rescues auditory function and reduces inner hair cell synaptopathy after noise-induced hearing loss. Otology and Neurotology, 37(9), 1223–1230. 10.1097/MAO.0000000000001191 [DOI] [PubMed] [Google Scholar]
- Stankovic KM, & Corfas G (2003). Real-time quantitative RT-PCR for low-abundance transcripts in the inner ear: Analysis of neurotrophic factor expression. Hearing Research, 185(1–2), 97–108. 10.1016/S0378-5955(03)00298-3 [DOI] [PubMed] [Google Scholar]
- Stöver T, Gong TWL, Cho Y, Altschuler RA, & Lomax MI (2000). Expression of the GDNF family members and their receptors in the mature rat cochlea. Molecular Brain Research, 76(1), 25–35. 10.1016/S0169-328X(99)00328-9 [DOI] [PubMed] [Google Scholar]
- Stöver T, Nam YJ, Gong TWL, Lomax MI, & Altschuler RA (2001). Glial cell line-derived neurotrophic factor (GDNF) and its receptor complex are expressed in the auditory nerve of the mature rat cochlea. Hearing Research, 155(1–2), 143–151. 10.1016/S0378-5955(01)00227-1 [DOI] [PubMed] [Google Scholar]
- SUGAWARA M, MURTIE JC, STANKOVIC KM, LIBERMAN MC, & CORFAS G (2007). Dynamic Patterns of Neurotrophin 3 Expression in the Postnatal Mouse Inner. J Comp Neurol, 501(2), 30–37. 10.1002/cne [DOI] [PubMed] [Google Scholar]
- Sun S, Babola T, Pregernig G, So KS, Nguyen M, Su S-SM, … Müller U (2018). Hair Cell Mechanotransduction Regulates Spontaneous Activity and Spiral Ganglion Subtype Specification in the Auditory System. Cell, 174(5), 1247–1263.e15. 10.1016/j.cell.2018.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Zhang YX, Ellis-Davies G, & Bergles DE (2010). ATP-induced morphological changes in supporting cells of the developing cochlea. Purinergic Signalling, 6(2), 155–166. 10.1007/s11302-010-9189-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanevski F, & Xu B (2015). HuD interacts with Bdnf mRNA and is essential for activity-induced Bdnf synthesis in dendrites. PLoS ONE, 10(2), 1–20. 10.1371/journal.pone.0117264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Thaller C, & Eichele G (2004). GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Research, 32, D552–D556. 10.1093/nar/gkh029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-z, Ibrahim LA, Kim YJ, Gibson DA, Leung HC, Yuan W, … Zhang LI (2013). Slit/Robo Signaling Mediates Spatial Positioning of Spiral Ganglion Neurons during Development of Cochlear Innervation. Journal of Neuroscience, 33(30), 12242–12254. 10.1523/jneurosci.5736-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefstaedt P, Scheper V, Rieger H, Lenarz T, & Stöver T (2006). Neurotrophe faktoren der GDNF-familie und ihre rezeptoren lassen sich in spiralganglienzellen hörender, aber auch ertaubter ratten nachweisen. Laryngo- Rhino- Otologie, 85(11), 802–808. 10.1055/s-2006-925287 [DOI] [PubMed] [Google Scholar]
- Weisz C, Glowatzki E, & Fuchs P (2009). The postsynaptic function of type II cochlear afferents. Nature, 461, 1126–1129. 10.1038/nature08487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlon DS, Grover M, Tristano J, Williams T, & Coulson MT (2007). Culture conditions determine the prevalence of bipolar and monopolar neurons in cultures of dissociated spiral ganglion. Neuroscience, 146(2), 833–840. 10.1016/j.neuroscience.2007.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlon D, Zhang X, & Kusakabe M (1999). Tenascin-C in the cochlea of the developing mouse. Journal of Comparative Neurology, 406(3), 361–374. 10.1002/(SICI)1096-9861(19990412)406:3<361::AID-CNE5>3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- Wise AK, Tu T, Atkinson PJ, Flynn BO, Sgro BE, Hume C, … Richardson RT (2011). The effect of deafness duration on neurotrophin gene therapy for spiral ganglion neuron protection. Hearing Research, 278(1–2), 69–76. 10.1016/j.heares.2011.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SHC, Eddy RL, … Nathans J (1995). The Brn-3 family of POU-domain factors: Primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. Journal of Neuroscience, 15(7 I), 4762–4785. 10.1523/jneurosci.15-07-04762.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Mengqing, Gan L, Zhou L, Klein WH, & Nathans J (1996). Targeted deletion of the mouse POU domain gene Brn-3a causes a selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proceedings of the National Academy of Sciences of the United States of America, 93(21), 11950–11955. 10.1073/pnas.93.21.11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Type I SGN subtype specification of Pou3f4 mutants at P14 and P28. (a-h) Images of P14 Pou3f4y/+ and Pou3f4y/lacZ cochleae at the middle turn, labeled with anti-Tuj1 to label all SGNs and either anti-Calb2, -Calb1, -Pou4f1 or -Prph. (i-p) Images of P28 Pou3f4y/+ and Pou3f4y/lacZ cochleae at the middle turn, labeled with anti-Tuj1 to label all SGNs and either anti-Calb2, -Calb1, -Pou4f1 or -Prph. Scale = 50 μm.