

Abstract
Objective:
Thrombocytopenia is associated with many viral infections suggesting virions interact with and affect platelets. Consistently, viral particles are seen inside platelets and platelet activation markers are detected in viremic patients. In this manuscript, we sought mechanistic insights into these virion/platelet interactions by examining how platelets endocytose, traffic, and are activated by a model virion.
Approach and Results:
Using fluorescently tagged HIV-1 pseudovirions (HIVpps), 3D-Structured Illumination Microscopy, and transgenic mouse models, we probed the interactions between platelets and virions. Mouse platelets used known endocytic machinery, e.g., Dynamin, VAMP-3, and Arf6, to take up and traffic HIVpps. Endocytosed HIVpps trafficked through early (Rab4+) and late endosomes (Rab7+), and then to a Microtubule-associated protein 1A/1B-light chain 3 decorated (LC3+) compartment. Incubation with virions induced Interleukin 1 Receptor Associated Kinase 4 (IRAK4), Akt, and IκB kinase (IKK) activation, granule secretion, and platelet-leukocyte aggregate (PLA) formation. This activation required TLRs and MyD88 but was less extensive and slower than activation with thrombin. In vivo, HIVpps injection led to virion uptake and platelet activation, as measured by IKK activation, PLA formation, and mild thrombocytopenia. All were decreased in VAMP-3−/− and, megakaryocyte/platelet-specific, Arf6−/− mice. Similar platelet activation profiles (increased PLAs, plasma platelet factor 4, and phospho-IκBα) were detected in newly diagnosed and anti-retroviral therapy (ART)-controlled HIV-1+ patients.
Conclusions:
Collectively, our data provide mechanistic insights into the “cell biology” of how platelets endocytose and process virions. We propose a mechanism by which platelets sample the circulation and respond to potential pathogens that they take up.
Keywords: Platelets, Endocytosis, Toll-like Receptor Signaling, Viremia, Cardiovascular Diseases, Infections and Inflammation, Platelet Signaling, Platelet Cell Biology
Graphical Abstract

INTRODUCTION:
A common symptom associated with viral infections is mild thrombocytopenia, suggesting an intimate relationship between circulating viral particles and platelets. Consistently, numerous studies have demonstrated evidence of platelet activation upon viral exposure, be it Dengue, HIV-1, Adenovirus, Hepatitis virus, etc. (reviewed in1). Despite this wealth of data showing different viruses activate platelets, there is less understanding of how. Viruses may bind to surface proteins and directly activate platelets via specific, signaling cascades.2 Alternatively, Ig-coated virions could activate platelets via engagement of surface Fc Receptors (i.e., FcRγIIA).3 Toll like receptors (TLRs) also play a role for some viruses (i.e. encephalomyocarditis virus; EMCV).4 An unresolved question is whether viral-mediated activation occurs at the platelet surface or whether, as in professional phagocytes, it occurs after virions have been internalized (discussed in1). This latter possibility is suggested by clear evidence that platelets take up viral particles and retain them in undefined intra-platelet compartments (i.e., “engulfosome”, or Surface Connected Canalicular System; SCCS).5–7 In this manuscript, we test the hypothesis that platelets endocytose virions and, during the particles’ transit through the platelet’s endocytic system, TLR-based signaling is activated inducing platelet activation. Similar pathways are observed in “professional” immune cells;8 however, they have not been demonstrated in platelets.
Platelet endocytosis was detected decades ago;9, 10 however, only recently have the mechanisms been examined (reviewed in11). Proteins such as: Dynamin-212, a large GTPase, important for clathrin-mediated endocytosis; the Ras-like small GTPase, ADP-ribosylation factor 6 (Arf6);13 and the v-SNARE, Vesicle-Associated Membrane Protein-3 (VAMP-3),14 both important for integrin trafficking, fibrinogen uptake and spreading; have all been shown to be active in platelets. In nucleated cells, clathrin and Dynamin are involved in endocytic trafficking of viruses such as HIV-1.15–18 Blocking Arf6 decreases HIV-1 infection;19 however, VAMP-3’s role is unclear. Given these roles of Dynamin, Arf6, and VAMP-3, we posited that they might contribute to platelet-virion interactions and thus yield insights into how platelets interact with viral particles.
Platelets contain multiple pathogens receptors e.g., complement receptors, FcγRIIa, αIIbβ3, GPIbα, CLEC2, and DC-SIGN, which could allow pathogens to directly or indirectly interact with platelets. Bacteria, bacterial products, and toxins can interact directly by binding to some of these receptors or via indirect binding to proteins that are normal ligands of these receptors, e.g., fibrinogen, fibronectin, complement C1q, vWF.20 TLRs, which recognize Pathogen-Associated Molecular Patterns (PAMPs) are also abundant in platelets.1 While many of these interactions likely occur at the surface, platelets can internalize pathogens (i.e., bacteria and viruses) where interactions with endosomal TLRs (e.g., TLR7, TLR9) could occur.4, 21–24 Endosomal TLR7 recognizes single-stranded RNA and can be activated by retroviruses.25–27 Platelet TLR7 mediates host survival in a mouse model of encephalomyocarditis virus (EMCV) infection.4 Endosomal TLR9 is activated by unmethylated 2’-deoxyribose cytidine-phosphate-guanosine (CpG) motifs, present in pathogens.28, 29 Platelet TLR9 is detected on the plasma membrane and inside the presumably endosomal “T granules”.30 TLR9 could be important for response to bacterial PAMPs since platelets do internalize some bacterial pathogens and responses are delayed, consistent with internalization being part of the activation process.7, 31, 32 Viruses, such as HIV-1, are seen in intraplatelet structures, suggesting a role for platelet endocytosis in mediating at least some of the responses to viral PAMPs.4–7 In professional immune cells, e.g., plasmacytoid dendritic cells (pDCs), responses are mediated through virus endocytosis, degradation to release TLR ligands, which bind to endosomal TLRs, and activate a Myeloid Differentiation primary response protein 88 (MyD88)- based signaling cascade.25, 33–35 Whether all or some of these steps occur in platelets is suspected but unclear.
Here we examined platelet-virus interactions using a pseudovirion model of HIV-1.36 Since several viral attachment factors are present on platelet surfaces (e.g., DC-SIGN, integrins, GPVI, CLEC-22, 37, 38), we focused on the events involved in viral entry and processing, post-binding. Using human platelets, endocytosis-deficient mice (VAMP-3−/− or lineage-specific Arf6−/−), and inhibitors of Dynamin, compartment acidification, and proteolysis, we showed that viral particles (and small-molecule, TLR7- and TLR9-agonists) required VAMP-3-, Arf6-, and Dynamin-mediated endocytosis and trafficking to activate platelets via a process involving MyD88, Akt, and IκB Kinase (IKK). Activation required an acidic compartment, active proteolysis, and led to a level of platelet secretion sufficient for PLA formation but distinct from responses to hemostatic agonists (i.e., thrombin). We demonstrate that these processes occurred in an in vivo murine model of HIV-1 pseudovirion infection and were detectable in platelets from HIV-1+ patients. Our data provides insights into the “cell biology” of platelet-virus interactions and suggest a mechanism for how platelets function as immune cells in circulation.
MATERIALS AND METHODS:
The data that support the findings of this study are available from the corresponding author upon reasonable request to Dr. Sidney W. Whiteheart.
Mice:
C57BL6 mice, global VAMP-3−/− mice14, PF4-Cre+-Arf6−/− mice (henceforth called Arf6−/−)13, 39, PF4-Cre+-IKKβ−/− mice40 and their corresponding littermates were used. Global MyD88−/− mice (strain B6. 129P2 (SJL)-MyD88<tm.1.1Defr>/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). Global TLR2, 3, 4, 7, 9 quintuple knockout (KO) mice were a generous gift from Dr. Jayakrishna Ambati, University of Virginia, VA. Homozygous GFP-LC3+ transgenic mice were a generous gift from Dr. Noboru Mizushima, University of Tokyo, Japan.41, 42 GFP-LC3+/VAMP-3−/− mice were generated by crossing VAMP-3−/− with homozygous GFP-LC3+ transgenic mice. Age-matched mice were used wherever applicable and sexes were mixed for all experiments unless otherwise mentioned. Blood was drawn via cardiac puncture and whole blood counting was done with a Hemavet (Drew Scientific, Dallas, TX) and IDEXX ProCyte Dx analyzer (IDEXX Laboratories, Westbrook, ME). For assays, platelet counts were measured using the Z2 Coulter Counter (Beckman Coulter, Inc., Miami, FL).
Preparation of HIV-1 pseudovirions:
HIV-1 pseudovirions were prepared using plasmids encoding: (i) a chimeric EcpH-TM expressing vector containing the ecliptic pHlourin, and transmembrane (TM) domain of ICAM-1 and a 3X FLAG tag, (ii) HIV-1 Gag-mCherry, (iii) HIV pR8 delta Env, and (iv) pCAGGS.HXB2 Env. Viral preps were made by the Kentucky Center for Molecular Medicine Genetic Technologies Core, by transfecting 293-LTV cell lines with all four plasmids in the following proportions (HIV pr8 delta Env (30%), HIV-Gag-mCherry (12.5%), 3X FLAG-EcpH-ICAM (12.5%), and pCAGGS.HXB2 Env (45%)). Viruses were harvested after 48–72 hr and concentrated by centrifugation onto a 20% sucrose cushion. Viral titers were quantified by measuring p24 using the ZeptoMetrix HIV-1 p24 antigen ELISA Kit (Cat No. 0801111; ZeptoMetrix, Franklin, MA). Two types of HIVpps were prepared: (i) GFP/Gag-mCherry HIVpps with both red (mCherry) and green (GFP) fluorescent tags and (ii) Gag-mCherry HIVpps with only red (mCherry) fluorescent tag. The two pseudovirions showed no differences in eliciting ex vivo or in vivo responses in platelets or mice.
Plate assay for virion endocytosis:
Washed WT and knockout (KO) platelets (100 μL, 5 × 107/mL) were added to opaque/black 96 well polystyrene plates (Corning, USA) and incubated with varying concentrations of GFP/Gag-mCherry HIV-1 pseudovirions (HIVpps) or for increasing times. Fluorescent intensities were measured using a SpectraMax plate reader after addition of 0.04% Trypan Blue to stop reactions at various time points as described in.14
Virion uptake in vitro or in vivo:
Washed platelets (1 × 109/mL) were incubated with GFP/Gag-mCherry HIV-1 pseudovirions (HIVpps) or FITC-CpG ODN2395 at 37°C for increasing times. Platelets were fixed with paraformaldehyde (2% PFA) and mixed with 0.1% Trypan Blue prior to imaging. For in vivo experiments, mice were injected with GFP/Gag-mCherry HIVpps (16 ng of p24/gm of mouse) through the retro-orbital sinus and, 24 hr post-injection, were euthanized and blood was drawn directly into fixative (0.38% sodium citrate, 2% PFA, and 0.05% glutaraldehyde). Platelets were visualized using epifluorescence and Differential Interference Contrast (DIC) microscopy with a Nikon Eclipse E600 (Nikon, Melville, NY) fitted with an AxioCam MR camera (Zeiss, Germany) and processed with Zen 2011 software (blue edition, Zeiss). The number of GFP-mCherry double positive puncta/platelet was counted.
Platelet-leukocyte or platelet-neutrophil aggregation assays:
Blood from healthy donors, or HIV-1 patients were incubated with FITC-anti-CD42b and APC-anti-CD11b/Mac-1 antibody for 30 min at RT. Mouse blood was pre-treated for 10 min with anti-mouse CD16/CD32 antibody and then incubated with FITC-anti-CD41/61 and APC-anti-Ly6G antibody for 30 min at RT. BD FACS/Lyse Solution was added to samples and incubated for 15 min at RT in dark. Samples were centrifuged at 500 x g for 7 min to remove lysed red blood cells. Samples were resuspended in 500 μL of 1x PBS pH 7.4 and analyzed on the BD LSRII flow cytometer counting 500,000 events per sample with gating around 50, 000 granulocytes. Data were analyzed using FlowJo software (v7.6.5) and plotted using SigmaPlot software (v13.0). Quantification shows platelet-neutrophil aggregates as measured by double-positive events for both CD41/61 and Ly6G for mouse, and platelet-leukocyte aggregates as double-positive events for CD42b and CD11b for human.
HIV-1 pseudovirion activation of human and mouse platelets:
Platelets (4 × 108/mL) were pooled from multiple mice (both male and female to balance potential sexual dimorphism) and humans, pre-incubated with 1 mM CaCl2 and then with GFP/Gag-mCherry HIVpps at 37°C. Where indicated, platelets were pre-treated with NH4Cl (20 mM) or Dynasore (80 μM) prior to HIVpps addition. Platelet extracts were prepared in SDS-PAGE sample buffer for western blotting.
Immunofluorescence microscopy and 3D-SIM imaging:
Washed WT, VAMP-3−/−, and Arf6−/− platelets (1 × 109/mL) were incubated in vitro with gag-mCherry HIVpps (300 ng/mL of p24) for 5 to 120 min at 37°C, and then prepared and imaged as described in14.
Measurement of platelet granule release (secretion assays):
Platelet-rich plasma (PRP) from mice (pooled from multiple male and female mice) or from informed healthy donors was prepared and labelled with 0.4 μCi/mL [3H] 5-HT (Perkin Elmer, Waltham, MA) for 30 min at 37°C. After washing, platelets were stimulated with thrombin or TLR agonists for indicated times and reactions were stopped by adding hirudin (2x) for thrombin-treated samples or centrifugation at 13,800 x g for 2 min for TLR-treated samples. The supernatant and pellet were separated post-centrifugation, and percent secretion for each marker was calculated as described in43.
Cytokine ELISA assays:
Human plasma samples were analyzed for TNF-α, IL-6, IL-1β, and PF4 using ELISA kits from R&D Systems as per manufacturer’s instructions.
Study Approval:
Animal studies were approved by the University of Kentucky IACUC, protocols 884M2005 and 2019–3384. Blood from informed human donors were obtained by a phlebotomist. All HIV-1+ patients, recruited at the Infectious Diseases Clinic at Kentucky Clinic, Lexington, KY were enrolled in our study as per the University of Kentucky IRB protocol #43971, approved in 2017 (renewed in 2020).
Statistical analyses:
Parametric tests such as paired or unpaired Student’s t-test or one-way analysis of variance (ANOVA) tests were performed where single variables where compared between two or more groups at a time. These tests were used for data that visually appeared normally distributed and thus normality and equal variance was not tested directly. Non-parametric tests such as Mann-Whitney U test, were used when data was not normally distributed, and comparisons were made between different groups to determine statistical significance.
Data sharing statement:
Supplementary data can be found in the online version of this article. For original data, analytic methods, and reagents, please contact the corresponding author’s (S.W.W.) laboratory at whitehe@uky.edu.
RESULTS
Endocytosis of HIV-1 pseudovirions: routes and requirements:
Several studies have noted internalized viral particles in platelets.4–7,44 Notably, Youssefian et al.,7 detected internalized HIV-1 particles within single-membrane-bound, intraplatelet compartments, but identified them as “engulfosomes”. Bourkour et al.,6 showed virions in two classes of compartments, one called endocytic vesicles and the other defined as part of the Surface Connected Canalicular System (SCCS), though direct connections to the surface were not demonstrated. These studies show platelets take up viral particles. To better define this process, we used fluorescently tagged HIV-1 pseudovirion particles (HIVpps36) and followed their uptake and intraplatelet trafficking using 3D Structured Illumination super-resolution fluorescence Microscopy (3D-SIM; Figure 1, Supplemental Figure I, IID–E). Wild-type (WT) platelets were incubated with Gag-mCherry HIVpps (red virus) for increasing times and virion localization was monitored. Within 5 min, HIVpps colocalized with Rab4 (early endosomes; Figure 1A–B). By 30 min, the number of Rab4+/HIVpp+ puncta had plateaued. By 60 and 120 min, HIVpps increasingly colocalized with Rab7 (late endosomes; Figure 1C–D). This suggested that the endocytosed HIVpps trafficked to a later, potentially more degradative part of the platelet’s endosomal system. HIVpps trafficking to Rab11+ recycling endosomes was minimal (Supplemental Figure 1A–B). LC3 is a marker for double-membraned, autophagosomes and single-membraned organelles known as LC3-associate phagosomes (LAPosomes45–47). Platelets contain LC3+ compartments48,49, thus, we asked whether HIVpps trafficked to such compartments. Gag-mCherry HIVpps were incubated with GFP-LC3-expressing platelets41, 42, 48 for increasing times (Figure 1E–F). There was an increased colocalization of HIVpps with GFP-LC3+ over time, with robust colocalization at 120 min. Endocytosed HIVpps also colocalized with TLR7+ compartments, in a time-dependent manner (Supplemental Figure IC–D). Collectively, these data show that platelets traffic endocytosed virions from early-to-late endosomes and to TLR7+ and LC3+ compartments.
Figure 1: HIV-1 pseudovirions (HIVpps) trafficked from early to late endosomes and into LC3+ compartments.

WT mouse platelets (1 × 109/mL) were incubated with Gag-mCherry HIVpps at 37°C for 5 to 120 min and prepared for immunofluorescence as described in the Methods. Platelets were stained with (A-B) anti-Rab4 antibody, (C-D) anti-Rab7 antibody, and then with Alexa 680-conjugated secondary antibodies, after permeabilization (see Methods). The Alexa 680-Rab signal was faux-colored blue to improve contrast. Images were taken using the 3D-SIM module of Nikon Ti-E N-STORM/N-SIM super-resolution microscope. Scale bar is 1 μm. Pearson’s correlation coefficient was calculated using the NIS-Elements v3.2 N-SIM/STORM suite software to show overlap between: (B) Alexa 680-Rab4 and Gag-mCherry HIVpps, (D) Alexa 680-Rab7 and Gag-mCherry HIVpps, at the indicated time points. Data are representative of 2 independent experiments. (E-F) Platelets (1 × 109/mL) from GFP-LC3 transgenic mice were incubated with Gag-mCherry HIVpps at 37°C for 5 to 120 min and imaged and analyzed as above. Data are representative of 3 independent experiments.
We next sought to define the molecular machinery needed for virion uptake, using endocytosis inhibitors and platelets from mice (global VAMP-3−/− and megakaryocyte/platelet-specific Arf6−/−) previously shown to have defects in endocytosis.13, 14 Initially, we used a plate-based, quenching assay to measure HIVpps uptake.14 Platelets from WT, VAMP-3−/−, and Arf6−/− mice, and WT platelets treated with Dynasore, a Dynamin-selective, endocytosis inhibitor50, were incubated with fluorescently-tagged HIVpps (Figure 2A) for increasing times or with increasing virion concentrations and the reactions were stopped with Trypan Blue (TB) to quench external GFP fluorescence.13, 14 As expected, there was a time- and dose-dependent increase in protected fluorescence, indicative of virion uptake in WT platelets (Figure 2B–C). Neither VAMP-3−/−, Arf6−/−, nor Dynasore-treated platelets showed robust increases in protected fluorescence. To further define the uptake process, we examined images of HIVpps-containing platelets, scoring platelet-associated HIVpps that contained both virion-associated fluorophores, GFP-ICAM-1 and Gag-mCherry (Figure 2D and Supplemental Figure IIA). In as early as 30 min, WT platelets internalized GFP/mCherry HIVpps. Over time, WT platelets accumulated more virions, while few, if any, were detected in VAMP-3−/− or Arf6−/− platelets, even after 6 hr (Figure 2D). To confirm our results in vivo, VAMP-3−/−, Arf6−/− and corresponding WT mice were injected, i.v., with GFP/Gag-mCherry HIVpps, and platelets were harvested 24 hr, post-injection, and imaged (Figure 2E–F and Supplemental Figure 2B–C). Pseudovirions were present in WT platelets but were rare in VAMP-3−/− and Arf6−/− platelets. Collectively, these data demonstrate that VAMP-3 and Arf6 are involved in virion uptake, in vivo. In GFP-LC3+/VAMP-3−/− platelets, HIVpps/GFP-LC3+ colocalization was severely reduced, (Supplemental Figure IID, E compared to Figure 1E, F), further confirming the need for VAMP-3 in virion endocytosis/trafficking.
Figure 2: Endocytic machinery required for HIV-1 pseudovirion (HIVpps) uptake in platelets.
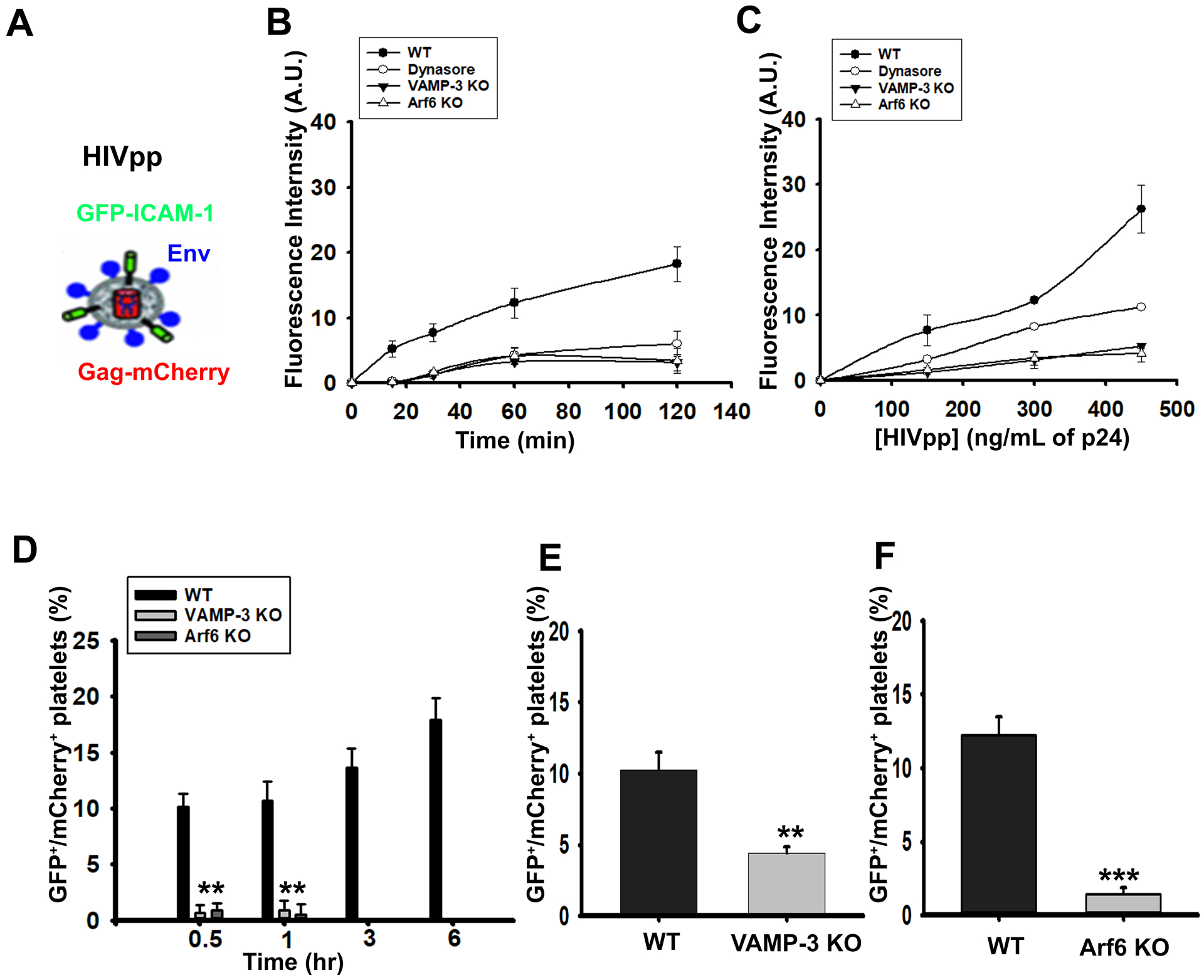
(A) Schematic representation of HIV-1 pseudovirions (HIVpps) showing envelope protein (Env) in blue, GFP-tagged-transmembrane domain (TM) of the Intercellular Adhesion Molecule-1 (ICAM-1) membrane protein in green and mCherry-tagged Gag protein in red. WT, Dynasore-pretreated (80 μM for 30 min at 37°C), VAMP-3−/− (KO) and Arf6−/− (KO) platelets (5 × 107/mL) were incubated with GFP/Gag-mCherry HIVpps (150 ng/mL of p24) for 0–120 min (B) or with 0–450 ng/mL of p24 of GFP/Gag-mCherry HIVpps for 60 min (C) at 37°C. Reactions were stopped and fluorescence intensities were measured after addition of Trypan Blue. (D) WT, VAMP-3−/− (KO) and Arf6−/− (KO) platelets (8 × 108/mL) were incubated with GFP/Gag-mCherry HIVpps (300 ng/mL of p24) at 37°C, for 0 to 6 hr, fixed with paraformaldehyde (PFA), treated with Trypan Blue, imaged, and counted. The percent of GFP/Gag-mCherry virion-containing platelets (counted as red and green puncta) was calculated. Age-matched VAMP-3−/− (KO) (n=7) and Arf6−/− (KO) mice (n=6), with their corresponding background-matched littermate (n=7 for VAMP-3 WT and n=6 for Arf6 WT) controls, were injected with HIVpps (i.v.) and platelets were harvested 24 hr post-injection and imaged. Quantification of the percent of GFP/Gag-mCherry virion-containing platelets in WT and VAMP-3−/− (KO) shown in (E) and between WT and Arf6−/− (KO) platelets shown in (F) and is shown. Statistical analyses were done by Student’s t-test where *** represents p≤0.001, ** represents p≤0.01 using the SigmaPlot software (v13.0). Data are representative of at least 2 independent experiments.
HIV-1 pseudovirion-mediated activation of platelets required endocytosis:
Given that viremic patients often have reduced platelet counts and show signs of platelet activation, we asked whether HIVpps endocytosis was important. VAMP-3−/−, Arf6−/− and their WT littermates were injected (i.v.: 16 μg of p24/mouse gm) with a single dose of GFP/Gag-mCherry HIVpps or saline and blood was harvested 24 hr post-injection. WT mice, injected with HIVpps, were mildly thrombocytopenic, with platelet counts ~50% less than the saline-injected controls (Figure 3A (i)). Platelet counts were unaffected in VAMP-3−/− or Arf6−/− mice after virion injection. The thrombocytopenia in WT mice coincided with increased PLAs in whole blood (Figure 3A (ii), Supplemental Figure XII). Conversely, infected VAMP-3−/− and Arf6−/− mice had fewer PLAs, suggesting less activation.
Figure 3: Endocytosis of HIV-1 pseudovirions (HIVpps) activated platelets in vivo and ex vivo.

Mice (20 weeks old; both male and female) WT (n=7), VAMP-3−/− (KO) (n=7) and Arf6−/− (KO) (n=6)) were either injected (i.v.) with HIVpps or saline (Sal). (A) Blood was drawn 24 hr post-injection to determine platelets counts. (Aii) PLAs in blood were measured by FACS using FITC-conjugated anti-CD41/61 and APC-anti-Ly6G antibodies and analyzed using FlowJo software (v7.6.5). (B) Platelets were isolated (ex vivo) from age-matched WT, VAMP-3−/− and Arf6−/− mice and stimulated with HIVpps for 30 min at 37°C, platelet lysates were prepared, and western blotting was performed using the indicated antibodies. (C) Volumetric quantification of (i) phospho-IRAK4, (ii) phospho-Akt, and (iii) phospho-SNAP-23 as a percent of total protein were calculated. (E) Platelets from WT, VAMP-3−/− (KO), and Arf6−/− (KO) mice were either kept resting or stimulated with HIVpps for 30 min. Samples were then incubated with FITC-anti-CD62 antibody to measure P-Selectin exposure by FACS. Data shown are representative of 3 independent experiments (mean ± SEM). Statistical analyses were done by paired Student’s t-test where ** represents p≤0.01, and * represents p≤0.05 using the SigmaPlot software (v13.0).
Previous reports indicated the importance of TLRs to a platelet’s response to viruses.4 To further probe this process, we examined the signaling steps downstream of TLRs. TLR7 activation leads to IRAK4 phosphorylation (downstream of MyD88), and IKK activation with cross-activation of PI3K-Akt pathways.4, 51 TLR9 stimulation also activates PI3K and MAPK pathways.52 Using IRAK4, Akt, and SNAP-23 (an IKK substrate40, 53) phosphorylation as metrics for TLR activation, we probed platelet lysates with phospho-peptide-specific antibodies (Figure 3B–C). Incubation of WT platelets with HIVpps, ex vivo, induced IRAK4, Akt, and SNAP-23 phosphorylation within 30 min. Consistent with defective HIVpp uptake in the mutants, neither VAMP-3−/− nor Arf6−/− platelets were responsive. WT platelets exposed more P-Selectin, post-HIVpps stimulation than either VAMP-3−/− or Arf6−/− platelets (Figure 3D). Thrombin-induced responses were unaffected in the mutant platelets (Supplemental Figure IIIA, B and13, 14).
TLR7/9-based signaling required endocytic trafficking and processing in platelets:
To determine whether endocytosis and subsequent trafficking are key to the responses of endosomal TLRs, e.g., TLR7 and TLR9, platelets were incubated with the TLR7- and TLR9-specific, small-molecule agonists. WT or VAMP-3−/− or Arf6−/− platelets were incubated with Loxoribine or CpG ODN2395 and lysates were probed for Akt and SNAP-23 phosphorylation (Supplemental Figures IV–V). In WT platelets, both TLR agonists induced Akt and SNAP-23 phosphorylation in a time-dependent manner but the responses to either agonist in VAMP-3−/− platelets were attenuated and delayed (Supplemental Figure 4). Time-dependent Loxoribine and CpG ODN2395 stimulation were also affected in Arf6−/− platelets (Supplemental Figure V). These data show that VAMP-3- and Arf6-dependent processes are important not only for responses to virions but also to these small-molecule, TLR-specific agonists. Time-dependent and dose-dependent uptake of FITC-CpG ODN2395 was significantly affected in platelets from either mutant, confirming the roles of VAMP-3 and Arf6 in CpG ODN2395 uptake (data not shown).
To further define whether the response to these TLR agonists and HIVpps required other aspects of the endocytic system, we examined the roles of Dynamin and compartment acidification using Dynasore and NH4Cl, respectively. Platelets were pre-incubated with Dynasore (a dynamin-selective inhibitor50) and stimulated with Loxoribine, CpG ODN2395, or HIVpps, and Akt and SNAP-23 phosphorylation were measured. Consistent with Dynasore-inhibited uptake of HIVpps (Figure 2B–C), pretreatment with Dynasore reduced Akt and SNAP-23 phosphorylation in response to all three agonists (Supplemental Figure VI). Similarly, pretreatment with NH4Cl reduced responses to both TLR agonists and HIVpps (Supplemental Figure VII). The concentration of Dynasore used (80 μM) did not affect thrombin-induced platelet granule secretion (data not shown). We next asked whether proteolysis was needed for the response to HIVpps. Platelets, pre-treated with a cocktail of protease enzyme inhibitors (leupeptin, aprotinin, and pepstatin, all 8 μM final) and stimulated with HIVpps, showed reduced IRAK4, Akt, and SNAP-23 phosphorylation (Supplemental Figure VIII). Together these data show that agonists for two endosomal TLRs required Dynamin, Arf6, VAMP-3, and compartmental acidification to cause platelet activation. This was equally true for HIVpps, which also required active proteolysis.
The reduction in Akt and SNAP-23 phosphorylation in Arf6−/− or VAMP-3−/− platelets could be due to loss of TLRs; thus, we probed for TLR7 and TLR9 in platelet lysates (Supplemental Figure IX). Platelets contained the full-length (FL) and cleaved (CL), ligand-binding forms of both TLRs and their levels were unchanged in the mutants. To confirm that the platelet activation occurs through the actions of TLRs, we tested platelets from a TLR2, 3, 4, 7, 9 quintuple knockout (Figure 4A). Akt and SNAP-23 phosphorylation in response to Lipopolysaccharide (LPS; TLR4-agonist), Pam3CSK4 (TLR2-agonist), Loxoribine (TLR7-agonist), and CpG ODN2395 (TLR9-agonist) were all reduced. Partial reduction in these metrics was noted in response to HIVpps, indicating the potential for an additional mechanism of post-endocytosis activation. Thrombin-induced responses were unchanged. Platelets from MyD88−/− mice also failed to activate phosphorylation of IRAK4, Akt, or SNAP-23 when treated with HIVpps, Loxoribine, or CpG ODN2395 (Figure 4B). Finally, the platelet-specific deletion of IKKβ reduced SNAP-23 phosphorylation in response to all three stimuli but had no effect on Akt activation (Figure 4C). In summary, our data outline a pathway for platelet activation by HIVpps and TLR7/9 agonists that involves endocytosis, trafficking to an acidic, degradative compartment, activation of TLRs and signaling via a MyD88-, Akt-, IRAK4-, IKK-involved pathway.
Figure 4: Responses to Loxoribine, CpG ODN2395, and HIV-1 pseudovirions (HIVpps) required TLR, MyD88, and IKKβ.
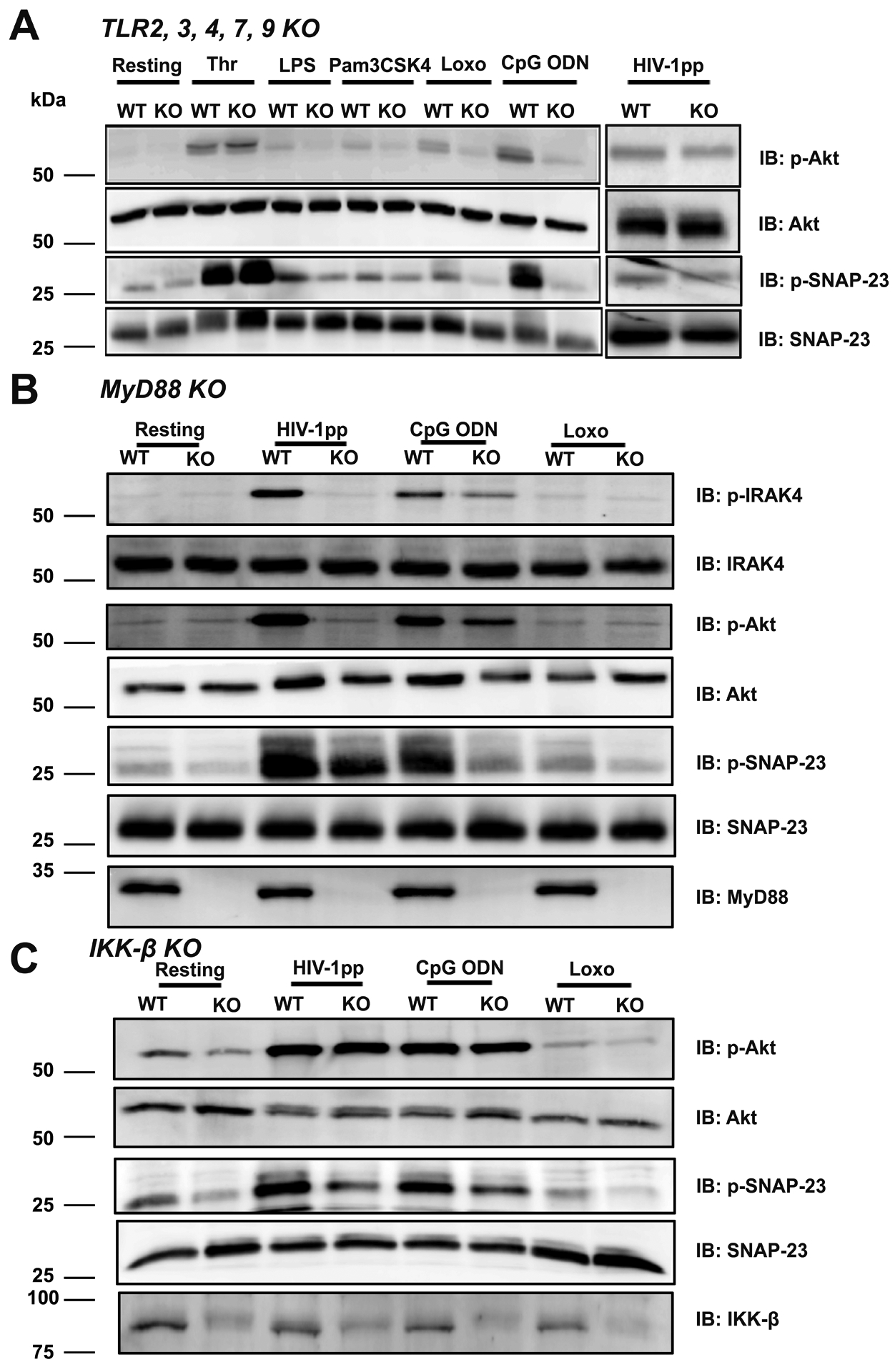
Platelets from WT and (A) TLR-2,3,4,7,9 quintuple KO mice were either kept resting or stimulated with thrombin (0.1 U/mL), LPS (100 ng/mL), Pam3CSK4 (10 μg/mL), Loxoribine (1 mM), CpG ODN2395 (5 μM), HIVpps (150 ng/mL of p24) for 60 min at 37°C. Platelets from WT or (B) MyD88−/− (KO) mice or (C) IKKβ−/− (KO) were either kept resting or stimulated with Loxoribine, CpG ODN2395, HIVpps for 60 min at 37°C and lysates were prepared. Western blotting was performed using the indicated antibodies. Data are representative of at least 2 independent experiments for (A) and (B) and 3 experiments for (C).
Endocytosed virions activated human platelets:
Given that endocytosis of HIVpps caused platelet activation in mouse platelets, we asked if this also occurred in human platelets. Human platelets were pre-incubated with GFP/Gag-mCherry-HIVpps, in the presence of Dynasore or NH4Cl. Dynasore-pretreatment inhibited virion uptake (Figure 5A–B). Pretreatment with either affected phosphorylation of IκBα (Figure 5C) and the formation of PLAs in whole blood (Figure 5D). Thus Dynamin-dependent endocytosis of HIVpps, targeting to acidic compartments, and activation of signaling cascades occurred in human platelets in a manner akin to mouse platelets.
Figure 5: Endocytosis of HIV-1 pseudovirions (HIVpps) by human platelets is Dynasore-sensitive and mediates platelet activation similar to that seen in HIV-1+ patients.
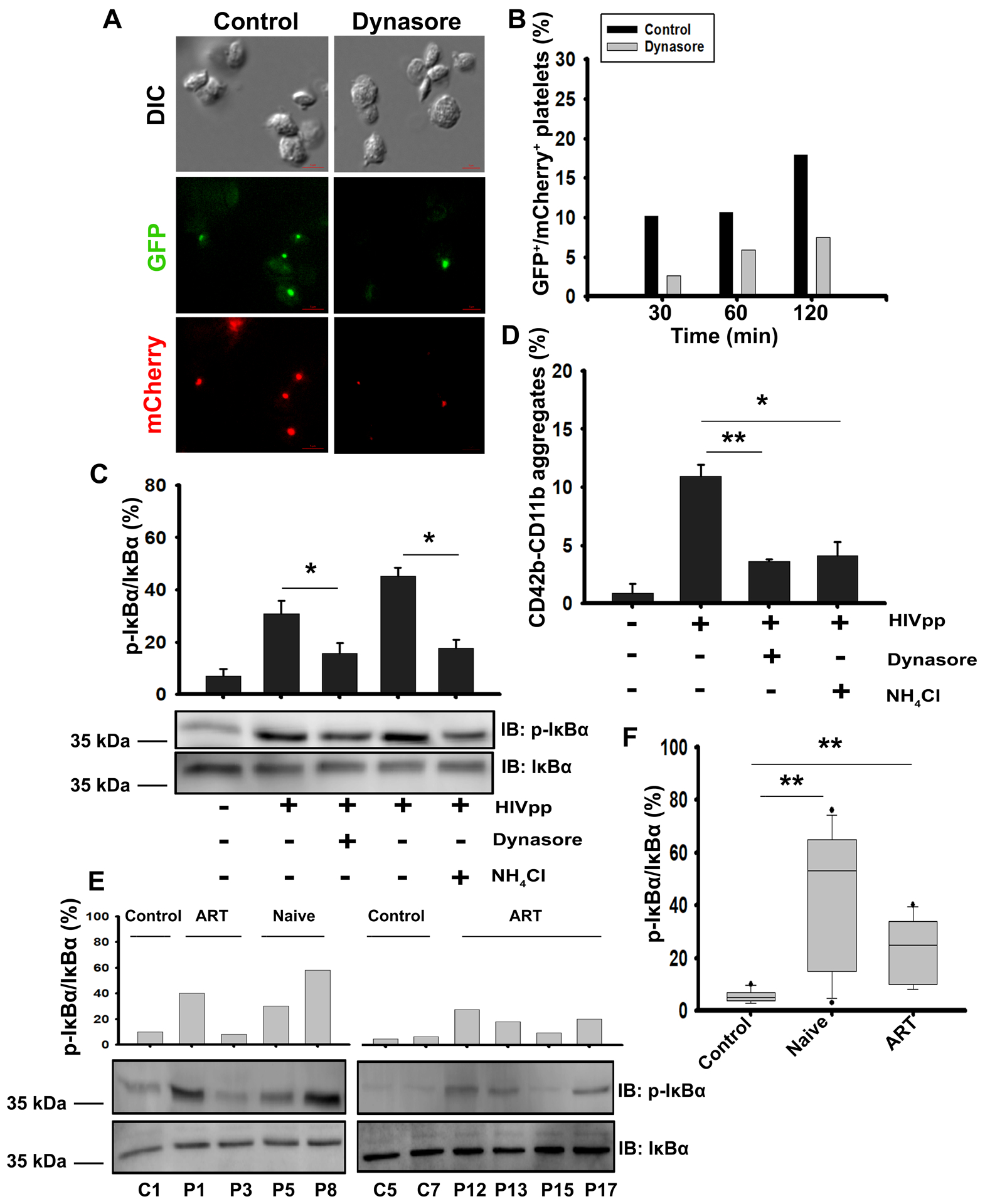
(A) Images of human platelets, either vehicle-treated (control) or Dynasore-treated (80 μM) then incubated with GFP/Gag-mCherry HIVpps, for 60 min. Scale bars are 2 μm. (B) Quantification of time-dependent uptake of GFP/Gag-mCherry HIVpps (counted as red and green puncta) into platelets. (C) Human platelets were incubated with HIVpps for 60 min either in the presence or absence of Dynasore (80 μM) or NH4Cl (20 mM) and lysates were probed by western blotting with anti-phospho-IκBα and anti-IκBα antibodies. (D) Blood was pre-incubated with either Dynasore (80 μM) or NH4Cl (20 mM), stimulated with HIVpps for 30 min, and platelet-leukocyte aggregates were detected by FACS with anti-CD42b and anti-CD11b fluorescent antibodies. Data were analyzed using FlowJo (v.7.6.5). (E) Representative western blots of platelet lysates from healthy donors (C1, C5), newly-diagnosed (HIV-1+) patients (naive, P5, P8) or from patients on anti-retroviral therapy drugs (ART-treated; P1, P3, P12, P13, P15, P17) were probed by western blot using anti-phospho-IκBα (Ser32), anti-IκBα antibodies. (F) The percent of phospho-IκBα over total IκBα were determined for platelet lysates from 10 healthy control samples, 11 newly diagnosed naive (HIV-1+) patients (naive-untreated), and 11 patients on anti-retroviral therapeutics (ART-treated). Statistical significance * represented by p ≤0.05, ** represented by p ≤0.01 was determined by Mann-Whitney U test using SigmaPlot software (v13.0). Data are representative of 3 independent experiments (mean ± SEM).
Several studies have reported signs of platelet activation in response to HIV-1 infection54–57, thus we asked whether the activation profile noted in our mouse model could be detected in HIV-1+ patients. We examined platelets from a cohort of 22 HIV-1+ patients (11 newly diagnosed and untreated (naive) and 11 on anti-retroviral therapeutics (ARTs)) and 10 healthy donors (Supplemental Table I), looking at IKK activation in platelets, PLA formation in whole blood, and plasma PF4. Representative western blots of platelet lysates show that IκBα phosphorylation was highest in naive HIV-1+ patients compared to healthy controls (Figure 5E–F and Supplemental Figure XE & G). IκBα phosphorylation was also detectable in ART-treated patients, though to lesser extents (Figure 5E–F and Supplemental Figure XE & XG). Whole blood PLAs from naive patients were significantly higher when compared to healthy donors (Supplemental Figure XF). No differences were noted between naive and ART-treated patients, consistent with the persistent platelet activation in ART-treated patients reported by Mesquita et al.57 Consistent with the course of HIV-1 infection, plasma levels of TNF-α, IL-6, and IL-1β were significantly higher in the naive patients, compared to healthy donors and ART-treated patients (Supplemental Figure XA–C). Plasma PF4 was highest in naive HIV-1+ patients and returned to normal upon ART-treatment (Supplemental Figure XD). Our data mirror previous reports of platelet activation during HIV-1 infection (e.g.57) and confirm IKK-associated platelet activation, which is consistent with the activation profiles seen in our mouse model. Further studies are underway to determine if more elements of the TLR signaling cascade are activated in platelets from HIV-1+ patients.
“Immuno-” vs. Hemostatic platelet activation:
We next explored how platelet activation induced by HIVpps or TLR7/9 agonists compared to that induced by a hemostatic agonist (i.e., thrombin). Specifically, we compared TLR-ligand-stimulated secretion to that induced by thrombin, evaluating the extents and rates of cargo release. As expected, WT platelets, stimulated with thrombin, showed rapid and robust secretion, which peaked within 3–5 min of stimulation (Figure 6A i-iii). When mouse platelets were stimulated with Loxoribine, CpG ODN2395, or HIVpps, the kinetics of release were slower, and extents of release were less. These differences in extent of release were pronounced for dense granule and lysosome release and less so for α-granule release. A similar pattern was observed when the calculated rates of release were compared (Kex; Figure 6A iv). Additionally, TLR-stimulated P-Selectin exposure and αIIbβ3 integrin activation were lower than that induced by thrombin (Figure 6B–C). Thus, hemostatic agonists, e.g., thrombin, and “immuno-agonists”, e.g., Loxoribine, CpG-ODN2395, or HIVpps elicited different secretion profiles with release from dense granules and lysosomes being more affected than α-granule release. Consistently, human platelets, when stimulated with thrombin or these “immuno-agonists” exhibited differential responses; dense granule release was the most affected, with slower kinetics and lower extent of release, in response to “immuno-agonists”. α-Granule and, interestingly, lysosomal release were less affected, showing comparable levels of secretion and similar rates of release (Kex values) (Supplemental Figure XIA, C). PAC-1 binding, a metric for αIIbβ3 activation, was lower in response to “immuno-agonists” than to thrombin (Supplemental Figure XIB). Taken together, our data clearly indicates that both human and mouse platelets demonstrate differential responses to “immuno-activation” versus “hemostatic activation”, supporting the shifting view of platelets being more diverse in their response curves to different types of stimuli, than previously thought.
Figure 6: Platelets responded differently to “hemostatic activation” vs. “immuno-activation”.
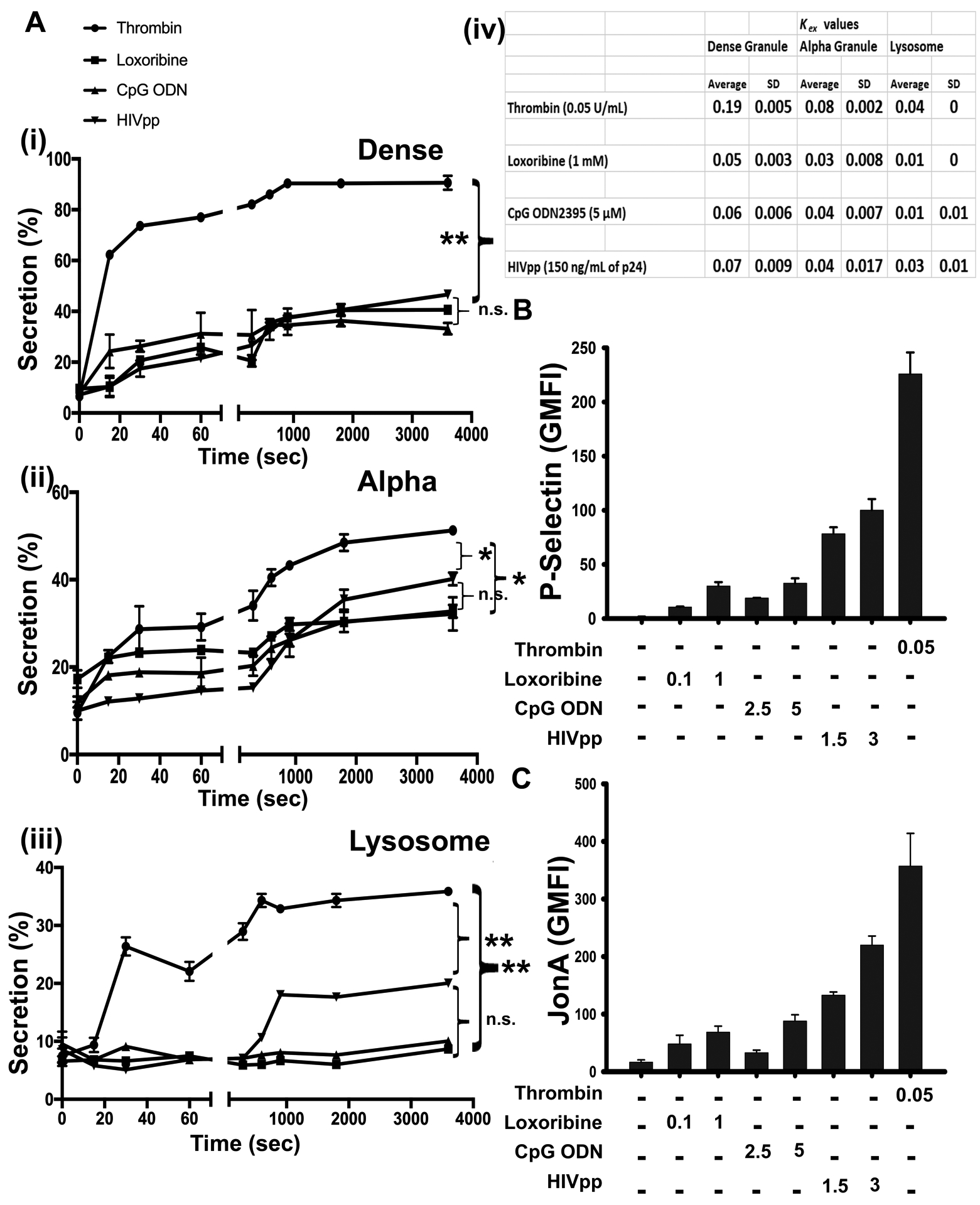
(A) WT Platelets (2.5 × 108/mL) were stimulated with Loxoribine (1 mM), CpG ODN2395 (5 μM), HIVpps (150 ng/mL of p24) or thrombin (0.05 U/mL) for the indicated times and percent secretion was measured (i) [3H]-Serotonin from dense granules, (ii) PF4 from α-granules and (iii) β-hexosaminidase from lysosomes. (iv) Release rates (Kex) were calculated for each type of secretion process and plotted as mean ± SD. WT platelets were stimulated with Loxoribine (0.1 mM and 1 mM), CpG ODN2395 (2.5 μM and 5 μM), HIVpps (1.5 × 105 pg/mL and 3 × 105 pg/mL) and thrombin (0.05 U/mL) for 30 min and incubated with (B) anti-P-Selectin or (C) anti-JonA antibody and samples were analyzed by FACS. Geometric mean fluorescence intensity (GMFI) was measured for each condition and plotted. Data shown are representative of 3 independent experiments (mean ± SEM). Statistical significance * represented by p ≤0.05, ** represented by p ≤0.01, was calculated by one-way ANOVA using GraphPad Prism 8.0 software.
DISCUSSION:
Interactions between platelets and viruses have been known for decades and much research has focused on the mild thrombocytopenia occurring during viral infections. Platelets can clearly “engulf” virions into membrane-bound compartments4–7. Our studies begin to define the molecular machinery involved and some of its outcomes. Our results demonstrate that endocytosis of virions requires VAMP-3, Arf6, and a Dynasore-sensitive step. Using endocytosis-deficient mice, we demonstrated that virion uptake leads to platelet activation, granule secretion, and formation of PLAs, both in vitro and in vivo. Using super-resolution microscopy (3D-SIM), we determined that endocytosed virions traffic through early endosomes, late endosomes and to a LC3-decorated compartment. Based on these results, we propose a general model (Figure 7) in which platelets endocytose virions, traffic/sort them into degradative compartments, to release TLR ligands (i.e., ssRNA), which activate the platelet to release granule cargo, expose P-Selectin, and form PLAs. While our data cannot quantify the impact of this pathway to systemic immunity, they demonstrate it occurs in vivo and thus constitutes part of the response to viral infections and perhaps other pathogens. Our data further suggests that the endocytic pathway in platelets is analogous to that employed by “professional” immune cells and demonstrate that circulating platelets have a specific, endocytic mechanism for pathogen recognition and response. We posit that platelet endocytosis could become a premier recognition and defense mechanism for viral pathogens more so than bacterial pathogens.
Figure 7: Endocytosis of HIVpps activates platelets.

Schematic depicting how pathogenic ligands such as HIV-1, bacterial products, etc., are endocytosed and trafficked to acidic endosomal compartments where proteolytic degradation occurs to release TLR ligands. This activates downstream signaling molecules that lead to IRAK4, Akt, and SNAP-23 phosphorylation and drives P-Selectin exposure to form platelet-leukocyte aggregates.
Based on the literature4–7, platelets take up viral particles; but, the question remains, “What happens next?” We addressed this using a fluorescently tagged virion and super resolution microscopy. Our data shows that virions are trafficked through endosomal compartments in a time-dependent manner. Strikingly, the virions end up in a compartment decorated with LC3. This is consistent with earlier electron microscopy by Cramer and colleagues6,7. In their images, virions are in two types of compartments, based on virion morphology. The first was called an endocytic compartment and the second, where virions appeared more degraded, was deemed SCCS, though no direct connections to the surface were demonstrated. Both compartments were bound by a single membrane. Our data yield more details, showing that virions first enter an early endosome (Rab4+), then colocalize with a late endosome marker (Rab7+) and ultimately with a LC3-decorated compartment. These data are consistent with platelets trafficking virions to a LC3-asscociated phagosome (LAPosome), which is a single-membrane-bound, LC3-decorated, compartment found in “professional” phagocytes, e.g., macrophages. LAPosomes are formed in a three-step process that is distinct from autophagy and begins after a pathogen is fully endocytosed.45–47 It is activated via TLR or FcR-based signaling cascades. Once the cytoplasmic leaflet of the phagosome membrane is modified, LC3 enhances fusion with lysosomes. How much of this pathway is recapitulated in platelets remains unclear; however, our results raise the exciting possibility that platelets use a TLR/LAPosome pathway to process virions, much like other professional phagocytes. Such findings may be relevant to platelet interactions with other pathogens such as bacteria, which can be internalized via several mechanisms (e.g., FcγRIIa/Ig-opsonization) and could be trafficked through different platelet compartments, much like what we have shown for HIVpps.20 Our data highlights the need to understand where (surface or endosomal compartment) pathogens activate platelets so as to better understand what platelets can do.
Multiple studies report platelet activation during viral infections with mild thrombocytopenia, PLAs, and plasma levels of platelet granule markers detected in patients with several types of viruses.57–61 However, the mechanisms and the sub-platelet locale of this activation in viremia are less clear. In our model, platelets were activated by HIVpps injection and wild-type mice were mildly thrombocytopenic, 24 hr post-injection. Thus, our model mimics early stages of a viral infection in a naive individual. This timeframe is not sufficient to mount the humoral response needed to generate opsinizing antibodies; thus, platelet activation through an Ig/FcR-based system seems unlikely. Also, mice, unlike humans, lack a relevant Fc receptor.3 Since our mutant mice, with defective virion endocytosis, failed to show thrombocytopenia, we conclude that virion uptake by platelets precipitated the loss of circulating platelets. These data also imply that, for our system, receptor signaling at the platelet surface is not sufficient for induction of thrombocytopenia.
Our in vitro experiments further define the requirement for endocytosis and confirm that activation by virions binding to and signaling through surface receptors are not sufficient. VAMP-3, Arf6, or Dynamin activity were required, as was acidification and proteolysis, thus virion-induced platelet activation required transit of the virion through an acidic, degradative compartment, like what is seen in other immune cells. None of these mutations or treatment conditions affected platelet activation induced with thrombin. Activation, expectedly, did require a TLR/MyD88/IKK-based pathway (Figure 4) consistent with previous reports for EMCV, which required TLR74. Our data further demonstrate a branchpoint in the activation pathway, since deletion of IKKβ had no effect on Akt phosphorylation. Interestingly, this mechanism was not limited to virions. Activation with TLR7- or 9-specific agonists also required endocytosis and were sensitive to NH4Cl treatment. Consistently, TLR9 was detected in an intraplatelet compartment30 and we showed TLR7 in endosomes (Supplemental Figure IC). Taken together, these data demonstrate that TLR-based signaling in platelets incubated with our model virion initiates from an intraplatelet compartment. This signaling is sufficient to activate platelet secretion, P-selectin exposure, and PLA formation in vitro. However, whether it is sufficient to induce thrombocytopenia in vivo without any input from other systems e.g., mast cells62 remains to be determined. Interestingly, the platelet granule release profiles were distinct from that seen when thrombin was the agonist. The deficit in dense granule release may be particularly relevant since other sources of serotonin (i.e., mast cells) have been noted to be important in platelet responses to Dengue virus infection.62 Further analysis of platelet activation with hemostatic agonists vs. TLR-based agonists will be required to fully understand the mechanisms and significance of these differences.
Our work expands on previous studies showing that platelets take up and can be activated by viruses. By characterizing the mechanisms of uptake and the pathways of activation, we have better defined the pallet of platelet/virus interactions. Our initial studies of human platelets and of platelets from HIV-1+ patients imply that the activation pathway, as defined in mice, is occurring in viremic humans. Future studies with the animal models described here will be needed to determine how important this pathway is during a viral infection. Is it part of the initial responses needed to alert the more classical components of an immune response? The fact that PLAs are formed, and platelets released immuno-modulator factors from their granules might suggest that this is possible. Could this also be part of some chronic response to low viral loads? Probing this possibility will require the use of a replicating virion system. Our present model likely reflects only the initial stages of an infection. It should be noted the LAPosome pathway is used by macrophages to directly control immune responses. Perhaps the same is true for platelets.45–47
A major unresolved question raised by our studies is whether this pathway is generally applicable to other viruses and pathogens. Reports of internalization of other virions (i.e., influenza44), suggest that the answer could be yes. Platelets express numerous viral “attachment factors” that could bind many different types of circulating viruses, (e.g., DC-SIGN, integrins, heparan sulfates, CLEC-2, GPVI, etc.2, 6, 37, 38), thus platelets are properly equipped to bind circulating virions. Therefore, the pathway defined by our system could be generally applicable. Consistently, severe viremia caused by SARS-CoV-2, presents with coagulation abnormalities, pulmonary microvascular thrombosis, and severe inflammatory response.63 Our preliminary data from a small pool of COVID-19 patients, shows increased platelet-leukocyte aggregates (Banerjee et al., 2020 unpublished), as reported here for HIV-1/AIDS patients. While how SARS-CoV-2 activates platelets remains to be determined, it is clear platelets sense acute viremia and become activated, which may precipitate clotting dysregulation. Future studies with other virions will be required to support this contention. Finally, given their abundance (1.5 – 4.5 × 105/μL), as opposed to leukocytes (4.0 – 11.0 × 103/μL) and the capabilities described here, platelets are well-positioned to play a significant role in the circulating, immune surveillance system.
Supplementary Material
HIGHLIGHTS:
Platelets endocytosed virions in a Dynasore-sensitive, VAMP-3-, and Arf6-dependent manner.
Virion endocytosis activated platelets and required signaling cascades involving TLRs, MyD88, and IKKβ.
Virion-mediated platelet activation was kinetically distinct from that induced with hemostatic agonists.
ACKNOWLEDGMENTS
The authors thank Dr. Jeremy P. Wood and the members of the Whiteheart Laboratory for their comments. The authors thank the University of Kentucky Flow Cytometry Core Facility, the Imaging Core Facility, and the Center for Molecular Medicine Genetic Technologies Core for their technical assistance with experiments.
SOURCES OF FUNDING
This work is supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL56652 and HL138179), from the American Heart Association Grant-in-Aid AHA16GRNT27620001, and a Department of Veterans Affairs Merit Award to S.W.W., and American Heart Association Grant-in-Aid AHA16GRNT31310020 to Q.J.W. The Center for Molecular Medicine Genetic Technologies Core is supported in part by NIH P30GM110787.
ABBREVIATIONS:
- Akt
Protein Kinase B
- Arf6
ADP-ribosylation factor
- ART
Anti-Retroviral Therapeutics
- CLEC-2
C-type lectin-like receptor-2
- DC-SIGN
Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin
- EMCV
Encephalomyocarditis virus
- GPVI
Glycoprotein VI
- HIV-1
Human Immunodeficiency Virus-1
- HIVpps
HIV-1 pseudovirions
- IKK
IκB kinase
- IRAK4
Interleukin 1 Receptor Associated Kinase 4
- LAP
LC3 Associated Phagocytosis
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- MyD88
Myeloid Differentiation Primary Response Protein 88
- PF4
Platelet Factor 4
- PLAs
Platelet-Leukocyte Aggregates
- VAMP-3
Vesicle-Associated Membrane Protein-3
- TLRs
Toll-like Receptors
- SCCS
Surface Connected Canalicular System
Footnotes
DISCLOSURES
The authors declare no competing financial interests.
REFERENCES:
- 1.Flaujac C, Boukour S, Cramer-Borde E. Platelets and viruses: An ambivalent relationship. Cell Mol Life Sci. 2010;67:545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaipan C, Soilleux EJ, Simpson P, et al. Dc-sign and clec-2 mediate human immunodeficiency virus type 1 capture by platelets. Journal of virology. 2006;80:8951–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E1550–E1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-tlr7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124:791–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zucker-Franklin D, Seremetis S, Zheng ZY. Internalization of human immunodeficiency virus type i and other retroviruses by megakaryocytes and platelets. Blood. 1990;75:1920–1923 [PubMed] [Google Scholar]
- 6.Boukour S, Masse JM, Benit L, Dubart-Kupperschmitt A, Cramer EM. Lentivirus degradation and dc-sign expression by human platelets and megakaryocytes. Journal of thrombosis and haemostasis : JTH. 2006;4:426–435 [DOI] [PubMed] [Google Scholar]
- 7.Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: Engulfment of hiv and staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. 2002;99:4021–4029 [DOI] [PubMed] [Google Scholar]
- 8.Lee BL, Barton GM. Trafficking of endosomal toll-like receptors. Trends in cell biology. 2014;24:360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Handagama P, Rappolee DA, Werb Z, Levin J, Bainton DF. Platelet alpha-granule fibrinogen, albumin, and immunoglobulin g are not synthesized by rat and mouse megakaryocytes. The Journal of clinical investigation. 1990;86:1364–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handagama PJ, Shuman MA, Bainton DF. Incorporation of intravenously injected albumin, immunoglobulin g, and fibrinogen in guinea pig megakaryocyte granules. The Journal of clinical investigation. 1989;84:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banerjee M, Whiteheart SW. The ins and outs of endocytic trafficking in platelet functions. Current opinion in hematology. 2017;24:467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bender M, Giannini S, Grozovsky R, Jonsson T, Christensen H, Pluthero FG, Ko A, Mullally A, Kahr WH, Hoffmeister KM, Falet H. Dynamin 2-dependent endocytosis is required for normal megakaryocyte development in mice. Blood. 2015;125:1014–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Joshi S, Xiang B, Kanaho Y, Li Z, Bouchard BA, Moncman CL, Whiteheart SW. Arf6 controls platelet spreading and clot retraction via integrin alphaiibbeta3 trafficking. Blood. 2016;127:1459–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee M, Joshi S, Zhang J, Moncman CL, Yadav S, Bouchard BA, Storrie B, Whiteheart SW. Cellubrevin/vesicle-associated membrane protein-3-mediated endocytosis and trafficking regulate platelet functions. Blood. 2017;130:2872–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daecke J, Fackler OT, Dittmar MT, Krausslich HG. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. Journal of virology. 2005;79:1581–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. Hiv enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pizzato M, Helander A, Popova E, Calistri A, Zamborlini A, Palu G, Gottlinger HG. Dynamin 2 is required for the enhancement of hiv-1 infectivity by nef. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6812–6817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritschet K, Donhauser N, Schuster P, Ries M, Haupt S, Kittan NA, Korn K, Pohlmann S, Holland G, Bannert N, Bogner E, Schmidt B. Cd4- and dynamin-dependent endocytosis of hiv-1 into plasmacytoid dendritic cells. Virology. 2012;423:152–164 [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Exposito L, Barroso-Gonzalez J, Puigdomenech I, Machado JD, Blanco J, Valenzuela-Fernandez A. Hiv-1 requires arf6-mediated membrane dynamics to efficiently enter and infect t lymphocytes. Molecular biology of the cell. 2011;22:1148–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamzeh-Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. Platelets and infections - complex interactions with bacteria. Frontiers in immunology. 2015;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional toll-like receptor-4. Blood. 2005;106:2417–2423 [DOI] [PubMed] [Google Scholar]
- 22.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–641 [DOI] [PubMed] [Google Scholar]
- 23.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll-like receptor molecules on human platelets. Immunology and cell biology. 2005;83:196–198 [DOI] [PubMed] [Google Scholar]
- 24.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of toll-like receptors on human platelets. Thrombosis research. 2004;113:379–385 [DOI] [PubMed] [Google Scholar]
- 25.Barton GM. Viral recognition by toll-like receptors. Seminars in immunology. 2007;19:33–40 [DOI] [PubMed] [Google Scholar]
- 26.Crozat K, Beutler B. Tlr7: A new sensor of viral infection. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6835–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded rna viruses by toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5598–5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745 [DOI] [PubMed] [Google Scholar]
- 29.Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. Tlr9 is localized in the endoplasmic reticulum prior to stimulation. Journal of immunology. 2004;173:1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE Jr. T granules in human platelets function in tlr9 organization and signaling. The Journal of cell biology. 2012;198:561–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kerrigan SW, Cox D. Platelet-bacterial interactions. Cellular and molecular life sciences : CMLS. 2010;67:513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Iwai T, Nakamura H, Inoue Y, Chen Y, Umeda M, Suzuki H. An ultrastructural study of porphyromonas gingivalis-induced platelet aggregation. Thrombosis research. 2008;122:810–819 [DOI] [PubMed] [Google Scholar]
- 33.Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, Larsson M, Gorelick RJ, Lifson JD, Bhardwaj N. Endocytosis of hiv-1 activates plasmacytoid dendritic cells via toll-like receptor-viral rna interactions. The Journal of clinical investigation. 2005;115:3265–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, Chaperot L, Plumas J, Randall RE, Si-Tahar M, Mammano F, Albert ML, Schwartz O. Innate sensing of hiv-infected cells. PLoS pathogens. 2011;7:e1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwasaki A Innate immune recognition of hiv-1. Immunity. 2012;37:389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyauchi K, Marin M, Melikyan GB. Visualization of retrovirus uptake and delivery into acidic endosomes. The Biochemical journal. 2011;434:559–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chabert A, Hamzeh-Cognasse H, Pozzetto B, Cognasse F, Schattner M, Gomez RM, Garraud O. Human platelets and their capacity of binding viruses: Meaning and challenges? BMC immunology. 2015;16:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN. Functional expression of ccr1, ccr3, ccr4, and cxcr4 chemokine receptors on human platelets. Blood. 2000;96:4046–4054 [PubMed] [Google Scholar]
- 39.Choi W, Karim ZA, Whiteheart SW. Arf6 plays an early role in platelet activation by collagen and convulxin. Blood. 2006;107:3145–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karim ZA, Zhang J, Banerjee M, Chicka MC, Al Hawas R, Hamilton TR, Roche PA, Whiteheart SW. Ikappab kinase phosphorylation of snap-23 controls platelet secretion. Blood. 2013;121:4567–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular biology of the cell. 2004;15:1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonnalagadda D, Izu LT, Whiteheart SW. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood. 2012;120:5209–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, Finberg RW, Kurt-Jones EA, Freedman JE. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. Lc3-associated phagocytosis and inflammation. J Mol Biol. 2017;429:3561–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heckmann BL, Green DR. Lc3-associated phagocytosis at a glance. J Cell Sci. 2019;132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez J Lap it up, fuzz ball: A short history of lc3-associated phagocytosis. Curr Opin Immunol. 2018;55:54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banerjee M, Huang Y, Ouseph MM, Joshi S, Pokrovskaya I, Storrie B, Zhang J, Whiteheart SW, Wang QJ. Autophagy in platelets. Methods in molecular biology. 2019;1880:511–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ouseph MM, Huang Y, Banerjee M, et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood. 2015;126:1224–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Developmental cell. 2006;10:839–850 [DOI] [PubMed] [Google Scholar]
- 51.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nature immunology. 2010;11:373–384 [DOI] [PubMed] [Google Scholar]
- 52.Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circulation research. 2013;112:103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hepp R, Puri N, Hohenstein AC, Crawford GL, Whiteheart SW, Roche PA. Phosphorylation of snap-23 regulates exocytosis from mast cells. The Journal of biological chemistry. 2005;280:6610–6620 [DOI] [PubMed] [Google Scholar]
- 54.Davidson DC, Hirschman MP, Sun A, Singh MV, Kasischke K, Maggirwar SB. Excess soluble cd40l contributes to blood brain barrier permeability in vivo: Implications for hiv-associated neurocognitive disorders. PloS one. 2012;7:e51793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang H, Duan Z, Li D, Li D, Wang Z, Ren L, Shen T, Shao Y. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased scd163 levels in hiv-1 infection. Cellular & molecular immunology. 2015;12:435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh MV, Davidson DC, Kiebala M, Maggirwar SB. Detection of circulating platelet-monocyte complexes in persons infected with human immunodeficiency virus type-1. Journal of virological methods. 2012;181:170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mesquita EC, Hottz ED, Amancio RT, Carneiro AB, Palhinha L, Coelho LE, Grinsztejn B, Zimmerman GA, Rondina MT, Weyrich AS, Bozza PT, Bozza FA. Persistent platelet activation and apoptosis in virologically suppressed hiv-infected individuals. Scientific reports. 2018;8:14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin YY, Yu XN, Qu ZY, Zhang AA, Xing YL, Jiang LX, Shang L, Wang YC. Adenovirus type 3 induces platelet activation in vitro. Mol Med Rep. 2014;9:370–374 [DOI] [PubMed] [Google Scholar]
- 59.Ojha A, Nandi D, Batra H, Singhal R, Annarapu GK, Bhattacharyya S, Seth T, Dar L, Medigeshi GR, Vrati S, Vikram NK, Guchhait P. Platelet activation determines the severity of thrombocytopenia in dengue infection. Scientific reports. 2017;7:41697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russell KE, Perkins PC, Hoffman MR, Miller RT, Walker KM, Fuller FJ, Sellon DC. Platelets from thrombocytopenic ponies acutely infected with equine infectious anemia virus are activated in vivo and hypofunctional. Virology. 1999;259:7–19 [DOI] [PubMed] [Google Scholar]
- 61.Scaradavou A Hiv-related thrombocytopenia. Blood reviews. 2002;16:73–76 [DOI] [PubMed] [Google Scholar]
- 62.Masri MFB, Mantri CK, Rathore APS, John ALS. Peripheral serotonin causes dengue virus-induced thrombocytopenia through 5ht2 receptors. Blood. 2019;133:2325–2337 [DOI] [PubMed] [Google Scholar]
- 63.Connors JM, Levy JH. Thromboinflammation and the hypercoagulability of covid-19. Journal of thrombosis and haemostasis : JTH. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.