

Abstract
Cytosolic sensing of pathogens and damage by myeloid and barrier epithelial cells assembles large complexes called inflammasomes, which activate inflammatory caspases to process cytokines (IL-1β) and gasdermin D (GSDMD). Cleaved GSDMD forms membrane pores, leading to cytokine release and inflammatory cell death (pyroptosis). Inhibiting GSDMD is an attractive strategy to curb inflammation. Here we identify disulfiram, a drug for treating alcohol addiction, as an inhibitor of pore formation by GSDMD, but not other members of the GSDM family. Disulfiram blocks pyroptosis and cytokine release in cells and lipopolysaccharide (LPS)-induced septic death in mice. At nanomolar concentration, disulfiram covalently modifies human/mouse Cys191/Cys192 in GSDMD to block pore formation. Disulfiram still allows IL-1β and GSDMD processing, but abrogates pore formation, thereby preventing IL-1β release and pyroptosis. The role of disulfiram in inhibiting GSDMD provides new therapeutic indications for repurposing this safe drug to counteract inflammation, which contributes to many human diseases.
Canonical inflammasomes, such as the NLRP3 inflammasome, activate caspase-1, whereas LPS and oxidized lipids form non-canonical inflammasomes to activate mouse caspase-11 or human caspase-4 and −5 1, 2, 3. As the final common effector downstream of inflammasome activation, gasdermin D (GSDMD) is cleaved by inflammatory caspases at the junction between the N-terminal domain (GSDMD-NT) and the autoinhibitory C-terminal domain (GSDMD-CT)4, 5, 6. GSDMD-NT binds to acidic phospholipids in the inner leaflet of the plasma membrane and oligomerizes to form pores that disrupt plasma membrane integrity7, 8, 9, 10, 11, 12, both enabling the release of pro-inflammatory cytokines, IL-1β and IL-18, which are processed by caspase-1, and inducing pyroptotic cell death.
GSDMD is a member of the GSDM family that also includes GSDMA, GSDMB, GSDMC, GSDME (also known as DFNA5), and GSDMF (also known as DFNB59) in humans. These GSDM family members, which are associated with genetic diseases in which inflammation, autoimmunity and/or cell death play a critical role, are a class of pore-forming proteins9. The crystal structure of full-length mouse GSDMA3 and cryo-electron microscopy structure of the GSDMA3-NT pore provide a model for pore formation by the GSDM family9, 13. Approximately 27 monomers assemble into an 18 nm inner diameter pore, through which many soluble cytosolic molecules, including caspase-1-processed pro-inflammatory cytokines, are released into the extracellular environment.
Dysregulation of inflammasome activation often contributes to human diseases, including inflammatory bowel disease, gout, type II diabetes, cardiovascular disease, Alzheimer’s disease, and sepsis, the often fatal response to systemic infection4, 6, 14, 15, as well as rare genetic diseases caused by mutations of NLRP3 and Pyrin inflammasomes. Gain-of-function mutations in the NLRP3 gene lead to cryopyrin-associated periodic syndromes including familial cold urticaria syndrome (FCAS)16, Muckle–Wells syndrome (MWS)17, and chronic infantile neurological cutaneous and articular (CINCA) syndrome that is also known as neonatal onset multisystemic inflammatory disease (NOMID)18, 19, 20. Mutations in the MEFV gene that encodes for Pyrin are associated with two clinically different autoinflammatory syndromes: familial Mediterranean fever (FMF) and Pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND); in both diseases, mutated Pyrin leads to high serum IL-1β levels during febrile episodes21.
The discovery of GSDMD as the final common step in pyroptosis and inflammatory cytokine release raises new hope for targeted therapy of these serious human diseases, which are often not adequately treated by IL-1β inhibitors or anti-IL-1β antibodies or by standard of care with methotrexate and antibodies against other inflammatory cytokines, such as anti-TNF. In particular, sepsis is fatal in about a third of patients, is the leading cause of death of newborns and small children in the world and is an important contributor to fatality of hospitalized adult patients22, 23. Currently sepsis treatment is limited to antibiotics and supportive care. Hundreds of clinical trials have failed to improve the survival of septic patients, including those designed to inhibit inflammatory cytokines and coagulation factors, which are downstream sepsis effectors. Drugs that efficiently and specifically regulate inflammasome activation, cytokine maturation and pyroptosis would be valuable therapeutic agents for treating sepsis and inflammation but are currently unavailable. Although many sensors and mediators trigger pyroptosis, pore formation by GSDMD-NT and other GSDM family members presents an attractive drug target because pore formation is a common final step required for pyroptosis and inflammatory cytokine release elicited by pathogenic or danger signals. In fact, Gsdmd genetic deficiency protects mice from LPS-induced septic death4, 6.
In this study, we performed high-throughput biochemical screening and identified tetraethylthiuram disulfide (disulfiram) as an effective inhibitor of GSDMD pore formation. Although disulfiram is a Cys-modifying drug that has other activities, examination of the steps of inflammasome activation showed that disulfiram did not substantially inhibit IL-1β and GSDMD processing in cells and in mice, but abrogated pore formation. Disulfiram is a drug used for decades to treat chronic alcohol addiction by acting on aldehyde dehydrogenase (ALDH)24. Because of its demonstrated safety, disulfiram is actively being repurposed, most notably to treat cancer due to its identified role in regulatory and stress-response pathways by directly targeting NPL4, an adaptor of the p97 segregase25. We propose that our newly identified activity of disulfiram on GSDMD may provide novel therapeutic indications for repurposing disulfiram to counteract the many human diseases that are caused or exacerbated by excessive inflammation.
Results
High-throughput screening identified disulfiram as a potent inhibitor of GSDMD-induced liposome leakage.
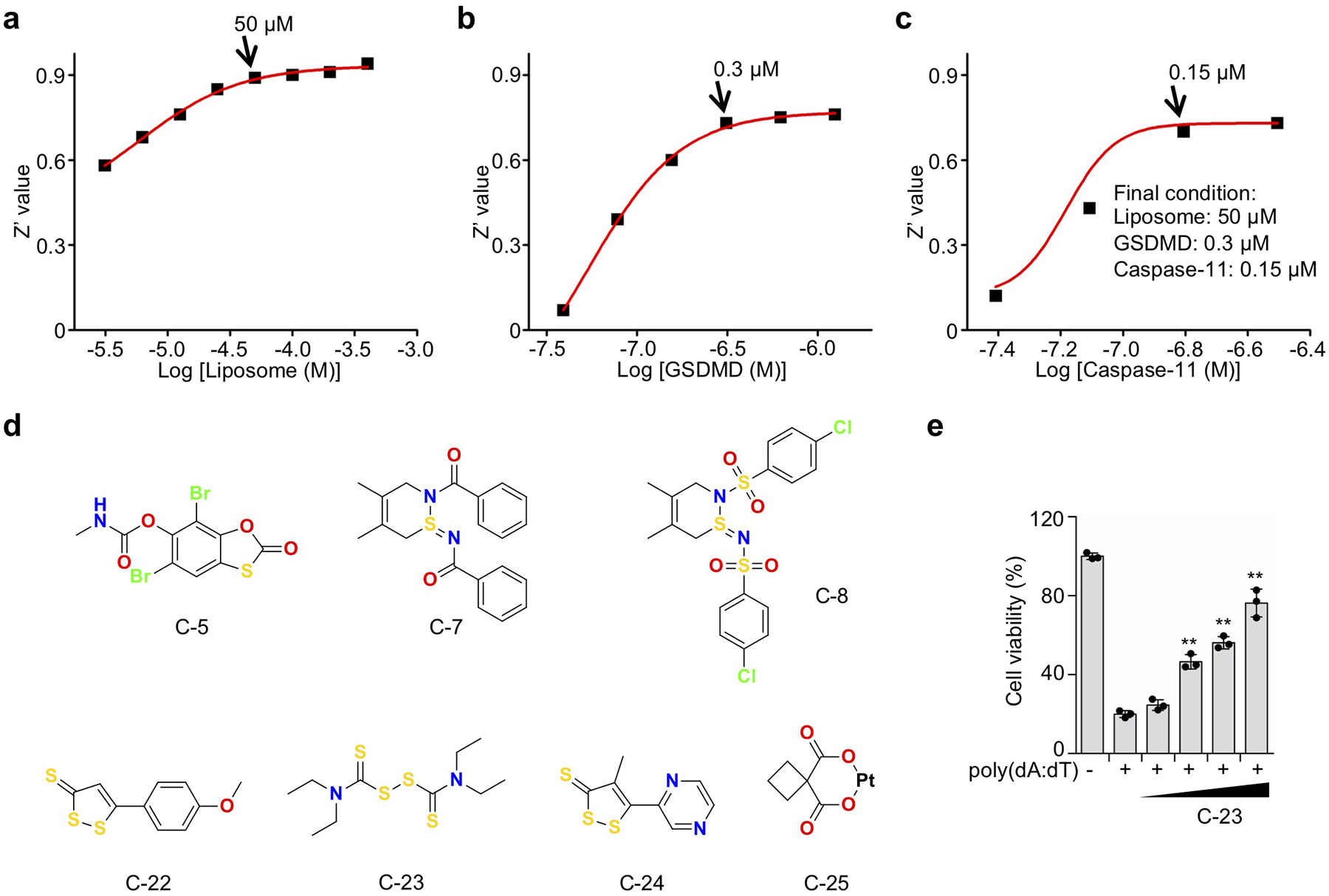
We performed high-throughput screening to discover inhibitors of GSDMD using a fluorogenic liposome leakage assay, which detects leakage of Terbium (Tb3+) from Tb3+-loaded liposomes incubated with GSDMD and caspase-11 9, 10, 26 (Fig. 1a). Concentrations of liposomes, caspase-11 and GSDMD were optimized to achieve a Z’ value of ~0.7, a cutoff that provides reproducible separation of hits from controls27 (Extended Data Fig. 1a–c). We initially screened 3,752 small molecules from a Harvard ICCB-Longwood collection to look for compounds that inhibited liposome leakage by at least 50% (Fig. 1b). After excluding pan-assay-interference compounds that non-specifically react with many biological targets and GSDMD-independent quenchers of fluorescence, we identified 22 active compounds (hit rate: 0.6%) and measured their IC50 values. The low hit rate suggests that the screen was selective. The most potent inhibitor was C-23, which had an IC50 of 0.30 ± 0.01 μM (Fig. 1c–e, Extended Data Fig. 1d). C-23 is a symmetrical molecule known as disulfiram or DSF, the active ingredient in Antabuse® that targets ALDH to deter alcohol consumption because of disulfiram-induced aversive flushing24. C-22, −23 and −24 all had low IC50 values and bound to GSDMD directly as assessed by microscale thermophoresis (MST) (Fig. 1c,f), a technique that quantifies binding by detecting a fluorescence change in a sample when its temperature is changed. Therefore, we identified three promising leads that directly inhibit GSDMD in vitro.
Figure 1.
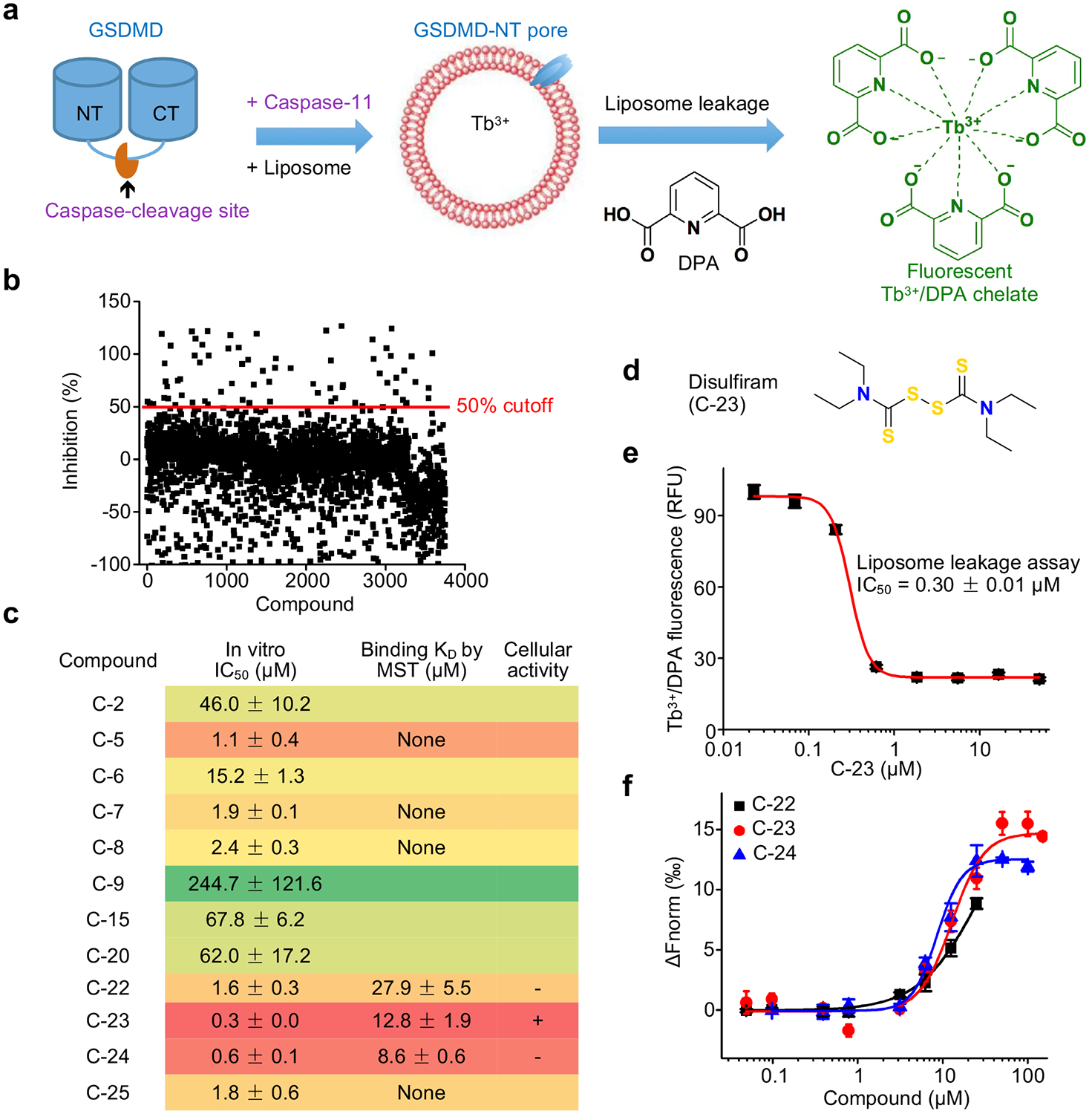
High throughput screen identifies disulfiram as an inhibitor of GSDMD pore formation. (a) Terbium (Tb3+)/dipicolinic acid (DPA) fluorescence liposome leakage assay. (b) Percentage inhibition of liposome leakage by each compound, assayed at 25 μg/mL (~50 μM for most compounds). Cutoff was 50% inhibition. (c) IC50s of the 12 screening hits after excluding Tb3+/DPA assay quenchers and hits without saturatable IC50 curve. n = 3 independent experiments. The top 7 hits were assessed for GSDMD binding by microscale thermophoresis (MST). n = 3 independent experiments. (d) Chemical structure of compound C-23 (disulfiram). (e) Dose response curve of disulfiram in liposome leakage assay. n = 3 independent experiments. The mean ± s.e.m. is shown. (f) MST measurement of the binding of Alexa 488-labeled His-MBP-GSDMD (80 nM) with C-22, C-23 or C-24. n = 3 independent experiments. The mean ± s.e.m. is shown.
Disulfiram inhibits pyroptosis in cells.
To evaluate whether C-22, −23, and −24 inhibit pyroptosis, we added these compounds to PMA-differentiated and LPS-primed human THP-1 cells before activating the canonical NLRP3 inflammasome with nigericin (Fig. 2a–d) or to mouse immortalized bone marrow-derived macrophages (iBMDMs) before activating the non-canonical inflammasome by LPS electroporation (Fig. 2e–g). Only C-23 blocked pyroptosis in both canonical human and non-canonical mouse inflammasome pathways (Fig. 2a, e). It also impaired cell death in a dose-dependent manner triggered by the AIM2 inflammasome in mouse iBMDMs transfected with poly(dA:dT) (Extended Data Fig. 1e), supporting its inhibition of a common event downstream of inflammasome action. Inhibition was shown by cell survival, assessed by CellTiter-Glo ATP luminescence (Fig. 2a,b,e,f), and membrane permeabilization, assessed by uptake of the membrane-impermeable dye SYTOX Green (Fig. 2c). In addition, disulfiram inhibited nigericin-induced IL-1β secretion in THP-1 and LPS transfection-induced IL-1β secretion in iBMDM with potency comparable to the pan-caspase inhibitor z-VAD-fmk (Fig. 2d,g). In contrast, disulfiram had no effect on necroptosis induced in HT-29 cells by treatment with TNF, second mitochondria-derived activator of caspases (SMAC) mimetic, and z-VAD-fmk, which was blocked by either necrosulfonamide (NSA) or necrostatin-1 (Nec)28, 29 (Fig. 2h). These data suggest that disulfiram inhibits pyroptosis in both human and mouse cells triggered by canonical and non-canonical inflammasomes, but not necroptosis.
Figure 2.
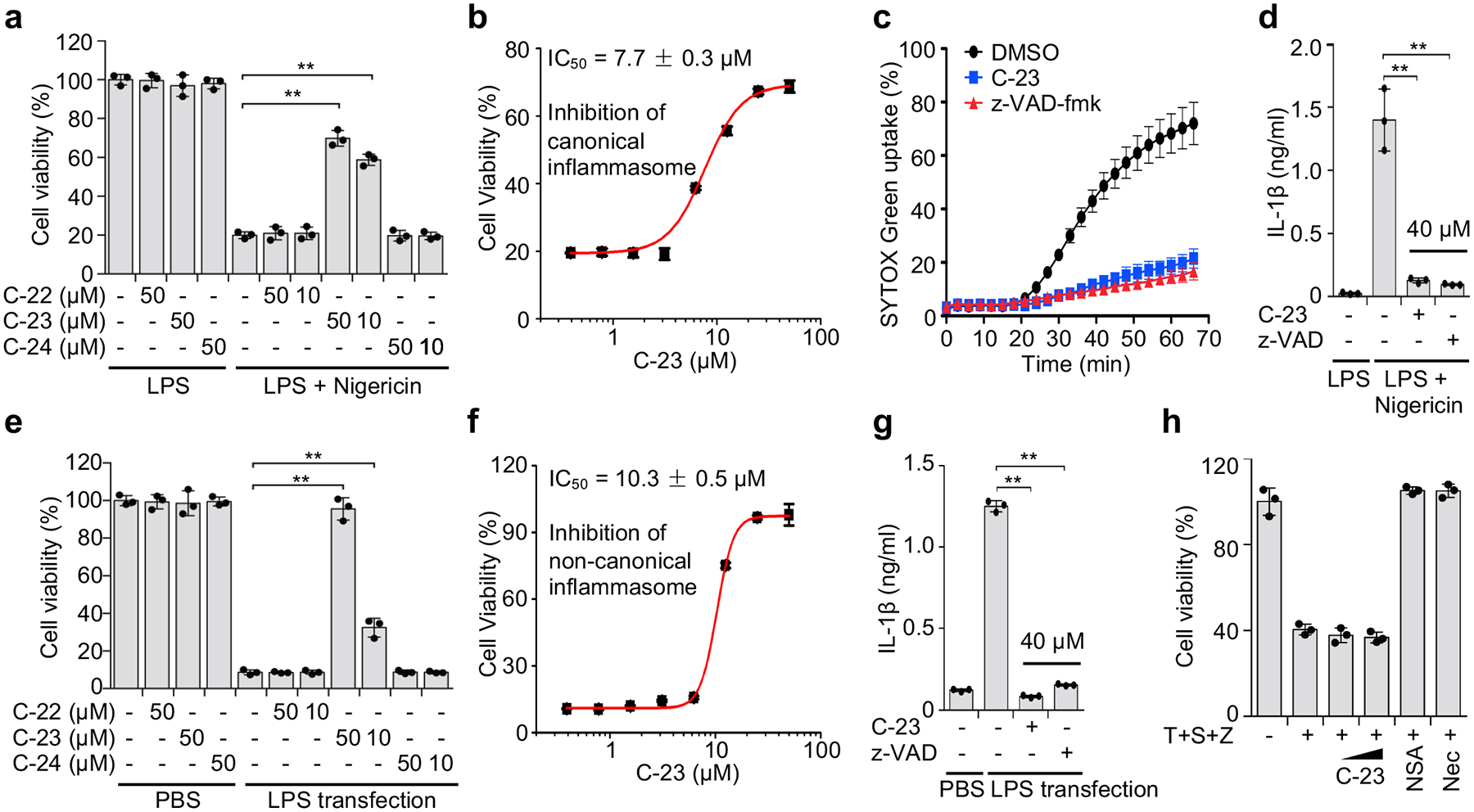
Disulfiram inhibits pyroptosis and IL-1β secretion. (a-d) PMA-differentiated LPS-primed human THP-1 were pretreated with indicated concentrations of each compound for 1 hr before adding nigericin or medium. The number of surviving cells was determined by CellTiter-Glo assay (a, b), pyroptosis was measured by SYTOX Green uptake in the presence of no inhibitor or 30 μM C-23 or z-VAD-fmk (c), and IL-1β in culture supernatants was assessed by ELISA (d) 2 hrs later. (e-g) Mouse iBMDMs were pretreated with each compound for 1 hr before electroporation with PBS or LPS. The number of surviving cells was determined by CellTiter-Glo assay (e, f); and IL-1β in culture supernatants was assessed by ELISA 2.5 h later (g). (h) HT-29 cells were pretreated (10 and 50 μM) or not with disulfiram (C-23) or 2 μM necrosulfonamide (NSA) or 10 μM Necrostatin-1 (Nec) for 1 hr before adding 20 ng/ml TNF (T), 100 nM SMAC mimetic (S), and 20 μM z-VAD-fmk (Z) and analyzed for cell viability by CellTiter-Glo assay 24 hrs later. Graphs in (a, d, e, h, g) show mean ± s.d; data are representative of three independent experiments with replicates (n= 3) and similar results. Data were analyzed using two-tailed Student’s t-test. **P < 0.01.
The measured cellular IC50 values of 7.7 ± 0.3 μM and 10.3 ± 0.5 μM for canonical human and non-canonical mouse inflammasome-dependent pyroptosis, respectively (Fig. 2b, f) are significantly higher than the submicromolar IC50 of disulfiram in the liposome leakage assay (Fig. 1e). Because previous studies have shown that disulfiram is rapidly metabolized to diethyldithiocarbomate (DTC)30, 31, and that the activity of DTC in vivo is greatly enhanced by complexation with copper gluconate [Cu(II)]25 (Extended Data Fig. 2a), we tested if Cu(II) could improve disulfiram’s cellular efficacy. Although Cu(II) weakly increased inhibition of liposome leakage by disulfiram or DTC (Extended Data Fig. 2b), it strongly enhanced protection against pyroptosis by either drugs (Extended Data Fig. 2c). With Cu(II), the IC50 of disulfiram for inhibiting pyroptosis decreased 24-fold to 0.41 ± 0.02 μM, which was similar to its potency for preventing liposome leakage (Extended Data Fig. 2b). The similar potency of disulfiram, when its principal cellular metabolite is stabilized, at inhibiting GSDMD pore formation in liposomes and pyroptosis in cells supports inhibition of GSDMD as the main mechanism of action of disulfiram.
Liposome leakage inhibition is mediated primarily by direct GSDMD pore formation.
Disulfiram has been reported to inhibit caspases in vitro by binding to the catalytic Cys responsible for proteolysis32. It is therefore likely that disulfiram inhibits both caspases and GSDMD. In a fluorogenic caspase activity assay that measures the release of 7-amino-4-methylcoumarin (AMC) from the inflammatory caspase substrate Ac-YVAD-AMC, disulfiram indeed inhibited recombinant caspase-1 and caspase-11 with IC50 of 0.15 ± 0.04 μM and 0.73 ± 0.07 μM, respectively (Extended Data Fig. 3a–d). Adding Cu(II), which stabilizes the DTC disulfiram metabolite, did not strongly change disulfiram caspase inhibition in vitro (Extended Data Fig. 3e–h).
To separate the relative contribution of caspase-11 versus GSDMD inhibition on pore formation, we first examined liposome leakage by pre-cleaved GSDMD. Disulfiram inhibited liposome leakage by pre-cleaved GSDMD with an IC50 of 0.26 ± 0.01 μM (Fig. 3a) that was comparable to its IC50 of 0.30 ± 0.01 μM for inhibiting full-length GSDMD plus caspase-11 (Fig. 1e). Disulfiram did not inhibit pore formation of pre-cleaved GSDMA3, suggesting that disulfiram does not inhibit the entire GSDM family (Fig. 3b). We then performed a time course of liposome leakage (Fig. 3c) and GSDMD cleavage by caspase-11 (Fig. 3d) in the presence or absence of disulfiram (50 μM). GSDMD processing by caspase-11 was unaffected by disulfiram, but processed GSDMD-NT did not cause liposome leakage if disulfiram was present. These data demonstrate that the inhibitory effect of disulfiram in the liposome leakage assay is mainly mediated by direct inhibition of GSDMD pore formation, rather than inhibition of caspase-11.
Figure 3.
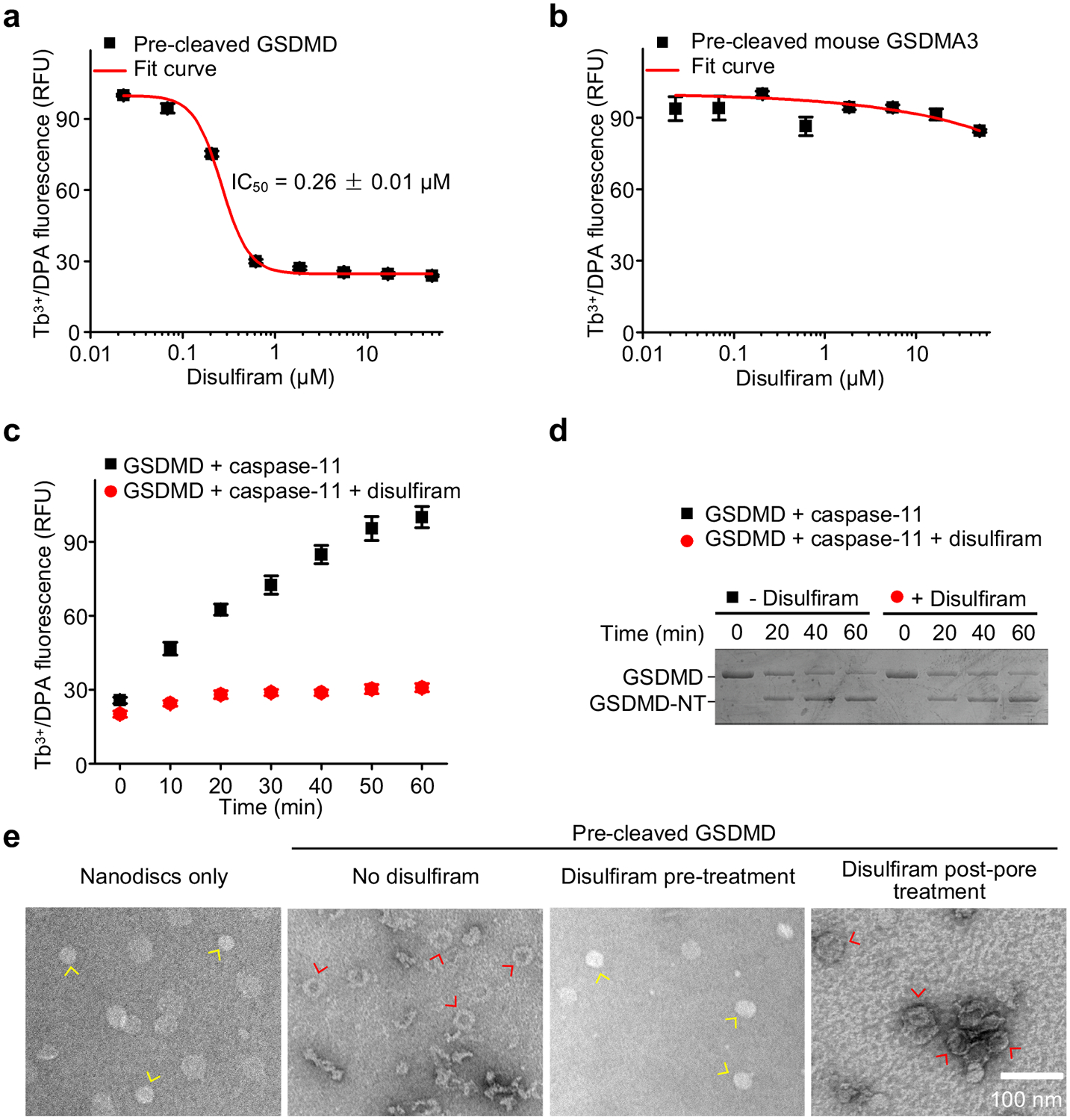
Disulfiram inhibition of liposome leakage is mediated primarily by direct inhibition of GSDMD pore formation. (a,b) Dose response of effect of disulfiram on liposome leakage induced by pre-cleaved human GSDMD (0.3 μM) (a) or pre-cleaved mouse GSDMA3 (0.3 μM) (b). n = 3 independent experiments. The mean ± s.e.m. is shown. (c) Time course of liposome leakage in the presence or absence of disulfiram. n = 3 independent experiments. The mean ± s.e.m. is shown. (d) Processing of GSDMD by caspase-11 in the presence or absence of disulfiram. Data are representative of three independent experiments. (e) Negative stain EM images of PS-containing nanodiscs that were incubated or not with pre-cleaved GSDMD. In the 3rd image from the left, disulfiram was added to pre-cleaved GSDMD before it was added to the nanodiscs; in the 4th image, disulfiram was added after the pre-cleaved GSDMD was incubated with nanodiscs when pores had formed. Data are representative of three independent experiments. Scale bar, 100 nm. Yellow arrows point to empty nanodiscs; red arrows point to pores.
To confirm that disulfiram inhibits pore formation, we used GSDMD and covalently circularized lipid nanodiscs33 constructed with phosphatidyl serine (PS), an acidic lipid, and phosphatidyl choline. Adding pre-cleaved GSDMD to assembled nanodiscs reconstituted pores that were visible by negative staining electron microscopy (EM) (Fig. 3e). When pre-cleaved GSDMD was treated with disulfiram before being added to the nanodiscs, pore formation was completely blocked (Fig. 3e). However, disulfiram addition after pore formation did not disrupt already assembled pores (Fig. 3e). Thus, disulfiram inhibits pore formation, but does not disassemble already formed pores.
Disulfiram targets Cys191 on human GSDMD.
Disulfiram has been shown to inactivate reactive Cys residues by covalent modification34. Indeed, when disulfiram was preincubated with N-acetylcysteine (NAC), which contains a reactive Cys that can inactivate Cys-reactive drugs, the ability of disulfiram to protect THP-1 cells from nigericin-mediated pyroptosis was eliminated (Extended Data Fig. 4a), consistent with disulfiram acting by modification of a reactive Cys in GSDMD. To probe the mechanism of GSDMD inhibition by disulfiram, we used nano-liquid chromatography-tandem mass spectrometry (nano-LC-MS/MS) to analyse disulfiram-treated human GSDMD. Tryptic fragments indicated a dithiodiethylcarbamoyl adduct of Cys191, in which half of the symmetrical disulfiram molecule is attached to the thiol (Fig. 4a,b, Extended Data Fig. 4b,c). Indeed, Cys191 is important for GSDMD pore formation in cells, since oligomerization was compromised by Ala mutation of the corresponding Cys192 in mouse GSDMD10. This Cys residue, conserved in GSDMD, but not in other GSDM family members, is accessible in both the full-length autoinhibited structure and the N-terminal pore form models, generated based on mouse GSDMA3 structures9, 13 (Fig. 4c, Extended Data Fig. 5a). Corresponding to Leu183 of GSDMA3, Cys191 sits at the distal tip of the membrane spanning region at the beginning of the β8 strand within the β7-β8 hairpin, which is a key element in the β-barrel that forms the pore13. Analysis of Cys reactivity using PROPKA35 suggests that Cys191 is the most reactive among all Cys residues in GSDMD. Consistent with its high reactivity, a time course analysis showed that disulfiram inhibited liposome leakage as soon as it could be measured, within 2 min of incubation (Extended Data Fig. 5b).
Figure 4.
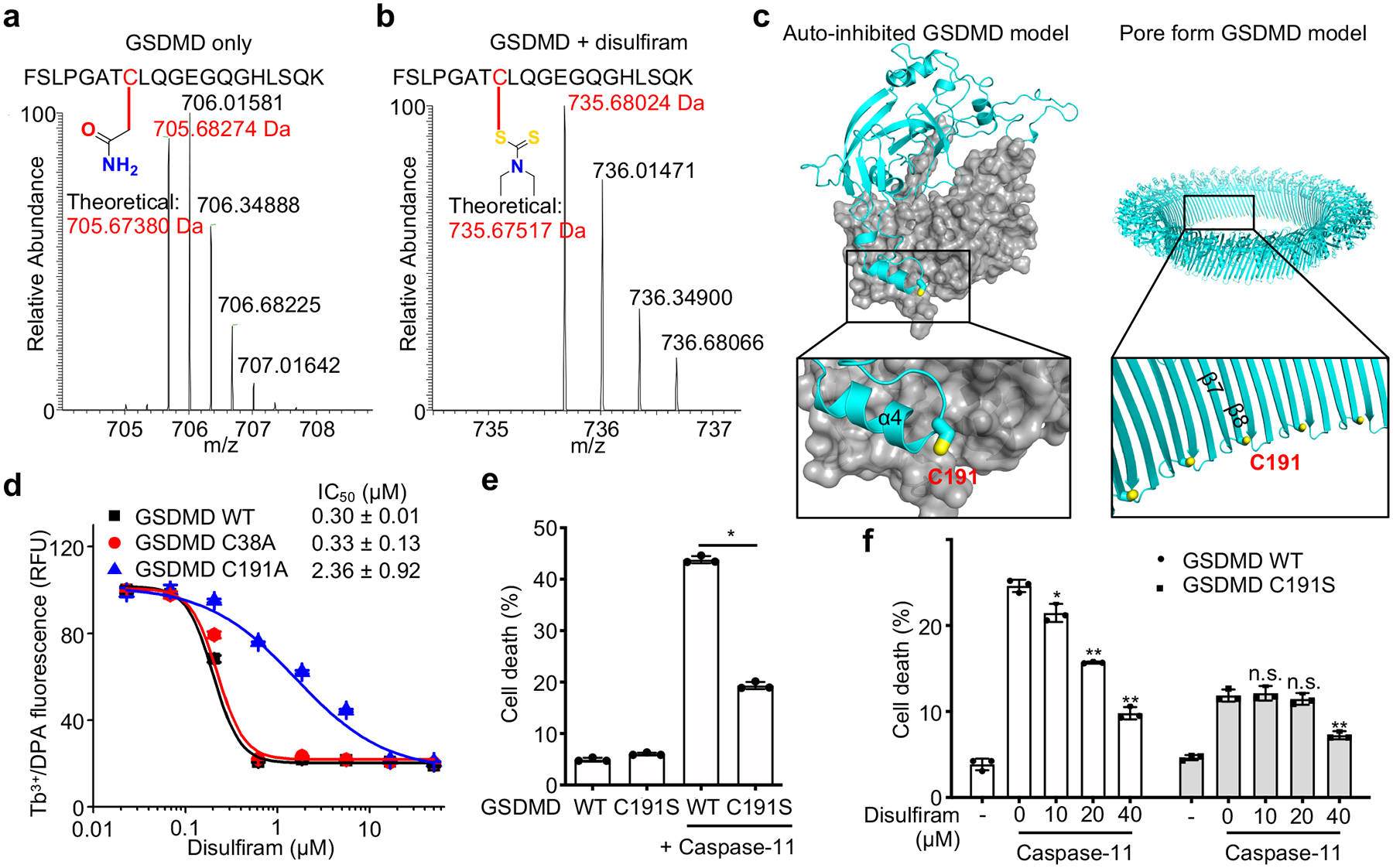
Disulfiram covalently modifies GSDMD Cys191. (a,b) MS/MS spectra of the Cys191-containing human GSDMD peptide FSLPGATCLQGEGQGHLSQK (aa 184–103; 2057.00 Da) modified on Cys191 (red) by carbamidomethyl (an increase of 57.0214 Da) [LC retention time, 22.85 min; a triplet charged precursor ion m/z 705.6827 (mass: 2114.0481 Da; delta M 2.27 ppm) was observed] (a) or of the corresponding GSDMD peptide after GSDMD incubation with disulfiram, which was modified on Cys191 (red) by the diethyldithiocarbamate moiety of disulfiram (an increase of 147.0255 Da). [LC retention time. 28.93 min; a triplet charged precursor ion m/z 735.6802 (mass: 2204.0406 Da; delta M 0.53 ppm) was observed.] (b). Data are representative of three independent experiments. (c) Models of full-length human GSDMD in its auto-inhibited form and of the pore form of GSDMD-NT based on the corresponding structures of GSDMA3 9, 13 showing the location in yellow of Cys191, modified by compound disulfiram. GSDMD-NT in cyan; GSDMD-CT in gray. d, Dose response curve of disulfiram inhibition of liposome leakage induced by wildtype (WT), C38A or C191A human GSDMD (0.3 μM) plus caspase-11 (0.15 μM). n = 3 independent experiments. The mean ± s.e.m. is shown. e, Full-length (FL) human GSDMD and GSDMD C191S were co-expressed with caspase-11 in HEK293T cells. Cell death was determined by CytoTox96 cytotoxicity assay 20 hrs after transfection. f, FL human wildtype or C191S GSDMD were co-expressed with caspase-11 in HEK293T cells. Eight h post transfection, the indicated amount of disulfiram was added and cell death was determined by LDH release 12 hrs later. (e,f) show the mean ± s.d. of 1 representative experiment of three independent experiments performed. Comparisons in (e,f) were calculated by two-tailed Student’s t-test. *P < 0.05, **P < 0.01, n.s., not significant.
To confirm that disulfiram acts on Cys191, we first compared the disulfiram IC50 values for pore formation in liposomes treated with wildtype, C38A control or C191A human GSDMD plus caspase-11. The IC50 for disulfiram acting on C191A GSDMD was ~8-fold higher than on wildtype GSDMD, while the activity on C38A was similar to wildtype GSDMD (Fig. 4d), confirming the importance of Cys191 for disulfiram activity. The residual inhibition of the Cys191 mutant may be due to disulfiram modifications of other Cys residues in the mutant GSDMD. To confirm the importance of Cys191 in pore formation, cell death was measured by LDH release in HEK293T cells ectopically expressing full-length human wildtype or C191S mutant GSDMD with or without caspase-11. Although wildtype or C191S GSDMD alone did not compromise cell survival, wildtype GSDMD and caspase-11 together caused substantial cell death, which was reduced for C191S GSDMD and caspase-11 (Fig. 4e). Similarly, cell death caused by ectopic expression of mouse GSDMD-NT (mGSDMD-NT) was significantly reduced in HEK293T cells expressing the analogous C192S mutant, but only modestly in cells expressing C39A mGSDMD-NT (Extended Data Fig. 5c). These results confirm the role of Cys191 and Cys192 in GSDMD-NT pore formation in humans and mice, respectively, consistent with previous results10, 36.
To further confirm that disulfiram acts on Cys191, we next assessed disulfiram inhibition of LDH release in HEK293T cells expressing caspase-11 and wildtype or C191S GSDMD. As expected, wildtype GSDMD-induced cell death was strongly inhibited by disulfiram in a dose-dependent manner beginning at the lowest concentration tested (10 μM), but the reduced cell death caused by expression of caspase-11 and C191S GSDMD was only inhibited when 4 times as much disulfiram was added (Fig. 4f). These data indicate that disulfiram inhibits GSDMD pore formation mainly by covalently modifying Cys191. In addition, they suggest that disulfiram inhibits cell death mainly through its effect on GSDMD-NT pore formation because if disulfiram strongly inhibited caspase-11, it would have provided better protection from death of cells expressing caspase-11 and C191S GSDMD. The residual weak inhibition of cell death by caspase-activation of C191S GSDMD could have been caused by disulfiram modification of other Cys in GSDMD when Cys191 is not present. Collectively, Cys191 in human GSDMD is the main target of covalent modification by disulfiram.
GSDMD pore formation, but not its cleavage or other earlier events in the inflammasome pathway, is the main target of disulfiram.
To further elucidate the cellular mechanism of pyroptosis inhibition by disulfiram, we analysed its effects on the entire inflammasome activation pathway. Nigericin activates the assembly of the NLRP3 canonical inflammasome using an adaptor called apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which can be visualised in immunofluorescence microscopy as specks. When LPS-primed THP-1 cells were treated with nigericin in the absence of inhibitors, ASC specks were detected in ~30% of cells (Fig. 5a,b). As expected, speck formation was not inhibited by z-VAD-fmk, since caspase activation occurs downstream of inflammasome assembly. Disulfiram, added at 1 hr before nigericin, only modestly inhibited ASC speck formation (Fig. 5a,b).
Figure 5.
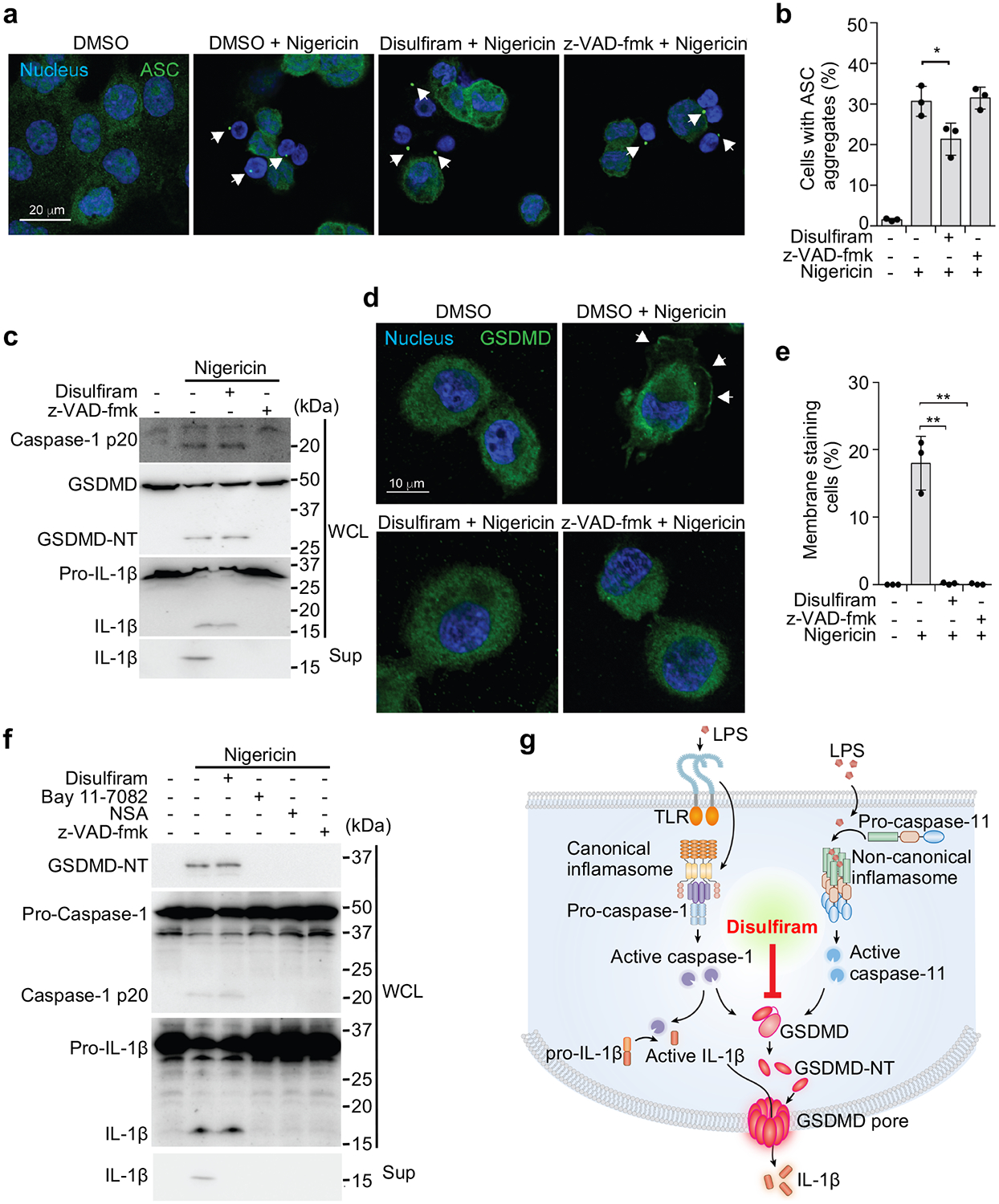
GSDMD pore formation is the main target of disulfiram. (a-e) LPS-primed THP-1 cells, pretreated or not with 30 μM disulfiram or z-VAD-fmk for 1 hr and stimulated with nigericin or medium, were analyzed for ASC specks (a, b), caspase-1, GSDMD and pro-IL-1β cleavage and IL-1 release by immunoblot of whole cell lysate (WCL) or culture supernatants (Sup) (c), and redistribution of GSDMD to the plasma membrane (d, e). (a) shows representative images of ASC specks (arrowheads) and (b) shows mean ± s.d. percent of cells with ASC specks analyzed 20 min after adding nigericin. The ratios of cells with ASC specks were calculated by counting 30 high-power fields for each sample in 5 independent experiments and analyzed using two-tailed Student’s t-test. In (c) WCL and Sup, harvested 1 hr after adding nigericin, were immunoblotted with the indicated antibodies. The GSDMD antibody used was generated in house (Extended Data Fig. 6). In (d, e) cells were fixed 30 min after adding nigericin and stained for GSDMD using a previously unreported monoclonal antibody generated in house (Extended Data Fig. 6). Shown are representative confocal microscopy images (d) and quantification (e) of the proportion of cells with GSDMD membrane staining and pyroptotic bubbles. Arrows indicate GSDMD staining of pyroptotic bubbles. The ratios of cells with GSDMD membrane staining were calculated by counting 40 high-power fields for each sample in 5 independent experiments and analyzed using two-tailed Student’s t-test. (f) Mouse iBMDMs were pretreated with disulfiram, Bay 11–7082, necrosulfonamide (NSA) or z-VAD-fmk for 1 hr before adding Nigericin. Whole cell lysates and culture supernatants, harvested 1 hr after adding nigericin, were immunoblotted with the indicated antibodies. Graphs show the mean ± s.d; data are representative of three independent experiments. Data in (c, f) are representative of three independent experiments. *P < 0.05, **P < 0.01. (g) Model of inflammasome pathway steps and their inhibition by disulfiram, with a main effect on GSDMD.
Canonical inflammasome assembly activates caspase-1, which cleaves pro-IL-1β and GSDMD, and the latter is needed to release processed IL-1β and to induce pyroptosis. To assess which steps in NLRP3-mediated inflammation were inhibited post ASC speck formation, LPS-primed THP-1 cells were treated with vehicle, 30 μM z-VAD-fmk or disulfiram 1 hr before adding nigericin, and cleavage and activation of caspase-1, GSDMD, and pro-IL-1β were analysed by immunoblot of whole cell lysates 1 hr later (Fig. 5c, Extended Data Fig. 6). Secretion of processed IL-1β was also assessed by immunoblot of culture supernatants (Fig. 5c). Caspase-1, GSDMD and pro-IL-1β cleavage to their active forms was clearly detected in the absence of inhibitors, and disulfiram treatment had no apparent effect on their proteolytic processing. In contrast and as expected, z-VAD-fmk treatment inhibited the processing of all 3 proteins. Despite lack of inhibition of pro-IL-1β processing, no IL-1β was detected in the culture supernatant in cells treated with disulfiram, suggesting blockage of GSDMD pore formation.
Next, we assessed the effect of z-VAD-fmk and disulfiram on LPS plus nigericin-induced GSDMD pore formation by immunofluorescence microscopy using a monoclonal antibody we generated that recognizes both uncleaved GSDMD and its pore form (Fig. 5d,e, Extended Data Fig. 6). In the absence of any inhibitor, the GSDMD antibody stained both the cytosol and the plasma membrane of LPS plus nigericin treated cells, which formed characteristic pyroptotic bubbles11. Both inhibitors completely blocked GSDMD membrane staining and the appearance of pyroptotic bubbles.
To determine whether disulfiram also primarily acts by blocking GSDMD pores in mouse macrophages, we treated LPS-primed iBMDMs with vehicle, z-VAD-fmk, disulfiram or two Cys-reactive compounds, NSA, which has been found to also inhibit pyroptosis36 and BAY 11–7082, an NLRP3 inhibitor 37, for 1 hr before adding nigericin, and assessed by immunoblot cleavage of caspase-1, GSDMD, and pro-IL-1β in cell lysates, and IL-1β release into culture supernatants 1 hr later (Fig. 5f). Caspase-1, GSDMD and pro-IL-1β cleavage to their active forms was clearly detected in the absence of inhibitors. Disulfiram had no apparent effect on their proteolytic processing, but completely blocked IL-1β release. In contrast the other inhibitors all acted upstream to completely suppress caspase-1 processing and hence processing of the caspase-1 substrates, GSDMD and IL-1β. Thus, disulfiram’s effect on caspase-1-mediated pyroptosis in human and mouse cells is mainly due to inhibition of GSDMD pore formation rather than inhibition of inflammatory caspase cleavage of GSDMD or other upstream events (Fig. 5g). In contrast, the other inhibitors inhibited pyroptosis and IL-1β release by blocking caspase-1 activity.
Disulfiram suppressed LPS-induced sepsis in mice.
To investigate the in vivo effect of disulfiram, we examined LPS-induced sepsis in C57BL/6J mice. Mice were treated with vehicle or disulfiram intraperitoneally before challenge with LPS using a drug dose (50 mg/kg) that was equivalent, after allometric scaling to account for body surface area, to 284 mg/day in humans, which is within the 125–500 mg/day dose range clinically approved to treat alcohol dependence38. Whereas the lowest concentration of LPS (15 mg/kg) killed 3 of 8 control mice after 96 hrs, all the disulfiram-treated mice survived (p = 0.045) (Fig. 6a). Serum IL-1β, TNF and IL-6 concentrations were strongly reduced 12 hrs after LPS challenge when all mice were alive (p ≤ 0.0003) (Fig. 6b–d). To check whether the reduction in IL-6 and TNF was secondary to GSDMD inhibition, we compared the serum levels of IL-1β, TNF and IL-6 in Gsdmd–/– and wildtype mice in the presence or absence of disulfiram (Extended Data Fig. 6a–c). Gsdmd–/– mice, as predicted, produced little IL-1β. Although Gsdmd–/– mice secreted TNF and IL-6, the serum levels of these cytokines in Gsdmd–/– mice was lower than in wildtype mice and was not significantly reduced by disulfiram. Collectively, these data indicate that disulfiram’s suppression of TNF and IL-6 secretion in wildtype mice was largely dependent on its inhibition of GSDMD pore formation. Following LPS challenge at the intermediate concentration (25 mg/kg), all the control mice died within 72 hrs, but 5 of 8 of the disulfiram-treated mice survived (p = 0.008) (Fig. 6e). At the highest LPS challenge (50 mg/kg), while all the control mice died within a day, death was significantly delayed by disulfiram treatment and 1 of 8 mice survived (p = 0.007) (Fig. 6f).
Figure 6.
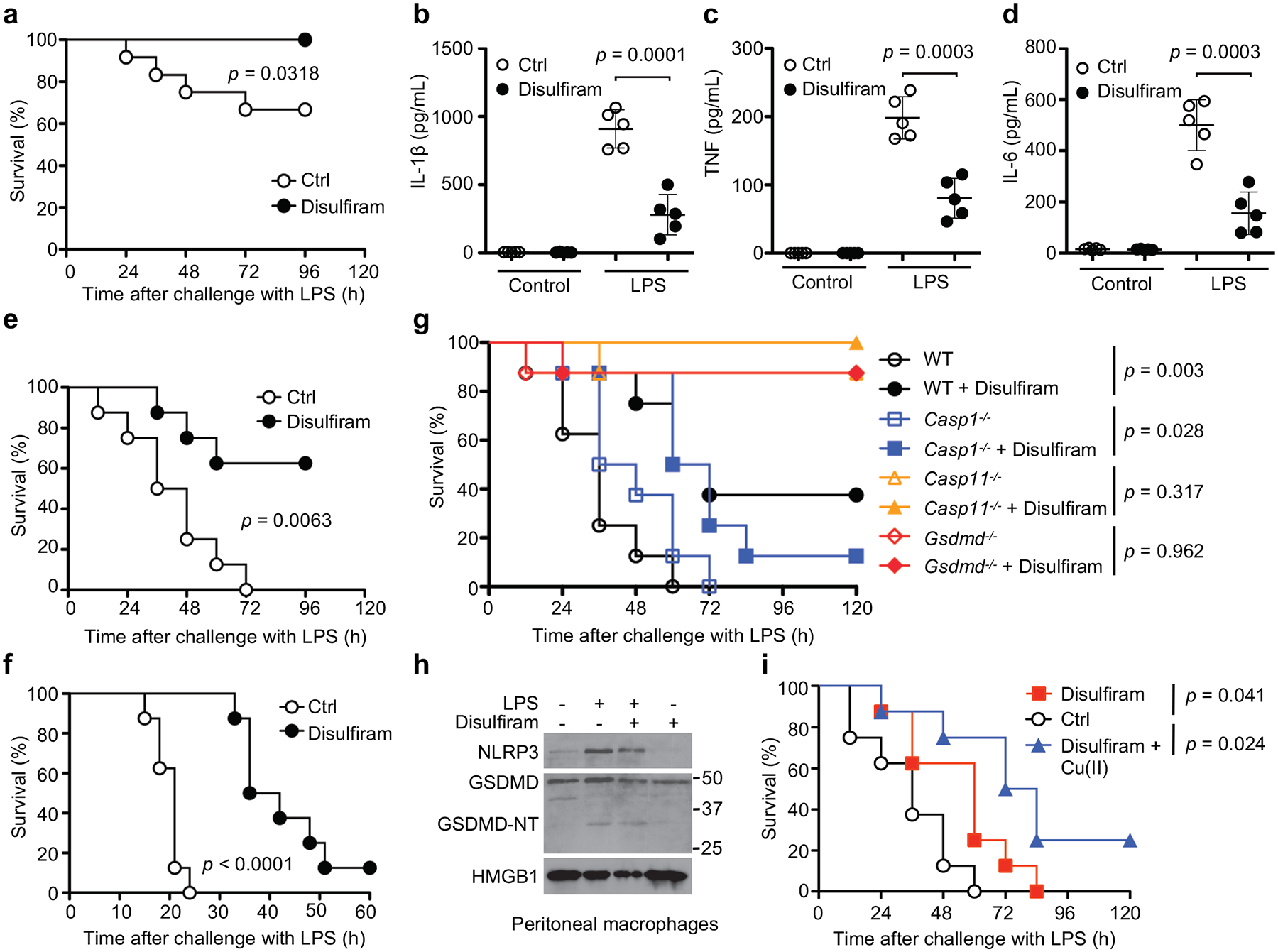
Disulfiram protects against LPS-induced sepsis. (a-f) Mice were pretreated with disulfiram (50 mg/kg) or vehicle (Ctrl) by intraperitoneal injection 24 and 4 hrs before intraperitoneal challenge with 15 (a-d), 25 (e), or 50 (f) mg/kg LPS and followed for survival (a,e,f). Statistical analysis was performed using the log-rank (Mantel-Cox) test (a, n = 12 mice/group, e,f, n = 8 mice/group). Serum IL-1β, TNF and IL-6 were measured by ELISA (n= 5 mice/group) 12 hrs post LPS challenge (b-d). Data were analyzed using two-tailed Student’s t-test. Shown are mean ± s.d. (g) Mice were pretreated with disulfiram (50 mg/kg) or vehicle (Ctrl) by intraperitoneal injection 4 hrs before and daily after intraperitoneal LPS challenge (25 mg/kg) and followed for survival. Statistical analysis was performed using the log-rank (Mantel-Cox) test (n = 8 mice/group). (h) Peritoneal macrophages from four indicated groups of mice were analyzed for NLRP3, GSDMD and HMGB1 by immunoblot. (i) Mice were challenged with 25 mg/kg LPS intraperitoneally and then treated with vehicle (Ctrl) or 50 mg/kg disulfiram given 0 and 12 hrs later. Indicated mice also received copper gluconate (0.15 mg/kg). Statistical analysis was performed using the log-rank (Mantel-Cox) test (n = 8 mice/group). Experiments were repeated three independent times.
LPS not only causes non-canonical inflammasome activation intracellularly, which does not need priming, but also primes NLRP3 inflammasome activation, which may amplify septic shock. However, genetic deficiency of NLRP3, ASC, caspase-1, or the IL-1 receptor did not offer substantial survival advantages in mice challenged with LPS in previous studies; in contrast, caspase-11 or GSDMD deficiency protected mice from septic death4, 39, suggesting that the non-canonical inflammasome pathway dominates LPS-induced sepsis. Consistent with these previous studies, Casp11–/– and Gsdmd–/– mice, but not Casp1–/– mice, were resistant to death from LPS (Fig. 6g). Further, disulfiram protected Casp1–/– mice from lethal LPS challenge but did not significantly affect the survival of Casp11–/– and Gsdmd–/– mice since all but 1 mouse in each undrugged control group survived. Peritoneal macrophages harvested 6 hrs post LPS challenge of wildtype mice also showed similar levels of NLRP3 and full length and cleaved GSDMD with or without disulfiram treatment (Fig. 6h), suggesting that disulfiram did not inhibit priming or GSDMD cleavage.
To better mimic the clinical situation in which sepsis is usually diagnosed only after the inflammatory cascade has begun, disulfiram administration was deferred until just after LPS (25 mg/kg) challenge and 12 hrs later (Fig. 6i). Post-LPS disulfiram treatment significantly delayed death. To determine if Cu(II) could improve protection from sepsis, we also examined whether Cu(II) supplementation enhanced the effectiveness of disulfiram administered post-LPS (Fig. 6i). Mice pretreated with Cu gluconate and given disulfiram after LPS challenge had improved survival compared to mock-treated control mice. Although all the control mice and mice treated with disulfiram alone died, 2 of 8 mice given both Cu(II) and disulfiram survived (Fig. 6i). Thus, disulfiram given after LPS partially protected mice and administration of Cu(II) may have improved its activity.
Other reported GSDMD inhibitors did not directly inhibit GSDMD pore formation.
The Cys-reactive necroptotic inhibitor NSA and Bay 11–7082 were shown to inhibit GSDMD-mediated pyroptosis36 and the NLRP3 inflammasome37, respectively. To elucidate whether disulfiram is different from these and other Cys-reactive compounds such as dimethyl fumarate (DMF, a drug used in the treatment for psoriasis and multiple sclerosis), afatinib (a drug that inhibits EGFR family kinases) and ibrutinib (a drug that inhibits Bruton’s tyrosine kinase), we investigated their effects on GSDMD and caspase-11-mediated liposome leakage. NSA and Bay 11–7082 moderately inhibited liposome leakage but were much less potent than disulfiram (Extended Data Fig. 8a–b), while DMF, afatinib and ibrutinib had hardly any activity (Extended Data Fig. 8c–e). In addition, the activity of NSA and Bay 11–7082 in liposome leakage was attributed to caspase inhibition because both compounds inhibited the processing of caspase-1, IL-1β and GSDMD in cells, similar to z-VAD-fmk (Fig. 5f). We also tested LDC7559, a small molecule identified from a cellular screen for inhibitors of NETosis that was also found to inhibit pyroptosis40. However, LDC7559 had no activity when assessed at up to 50 μM in the liposome leakage assay using recombinant GSDMD and caspase-11 (Extended Data Fig. 8f). These data suggest that not all Cys-reactive compounds have activity against GSDMD and that disulfiram is so far the only direct, and potent inhibitor of GSDMD.
DISCUSSION
Inflammasomes have been implicated in many human diseases. The discovery of GSDMD as the final common step in pyroptosis and inflammatory cytokine release after inflammasome activation raises a new approach for targeted therapy of inflammatory disease. A GSDMD inhibitor is an especially attractive therapeutic because it will potentially inhibit inflammation induced by activation of all inflammasome pathways triggered by any stimulus, unlike NLRP3 inhibitors that inhibit just one specific inflammasome. Moreover, blocking pore formation will inhibit the exit and inflammatory effects of all molecules whose release is increased by pyroptosis, unlike anti-IL-1β or anti-IL-18 biologics that act on just one downstream mediator. However, despite intense interest36, 40, no bona fide GSDMD inhibitors have been reported.
In this study, we found that disulfiram is a potent inhibitor of GSDMD pore formation in vitro, in cells and in vivo. In vitro, it suppresses liposome leakage mediated by caspase-11-cleaved GSDMD. In cells, it inhibits pyroptosis and inflammatory cytokine release in both canonical and non-canonical inflammasome pathways. When supplemented with Cu(II) to stabilize its metabolic intermediate, disulfiram exhibits a cellular potency similar to its in vitro potency. In vivo, it protects mice from lethal LPS-induced septic shock. These activities of GSDMD are mediated primarily by blocking GSDMD pore formation rather than its proteolytic processing. Mechanistically, disulfiram modifies Cys191 of human GSDMD, which is conserved as Cys192 of mouse GSDMD, but is not conserved in other members of the GSDM family such as mouse GSDMA3. By interfering with GSDMD pore formation, disulfiram provides an effective means to stop the cascade of inflammation that underlies sepsis.
Our data suggest that disulfiram is selective for GSDMD in several ways. First, within the steps in inflammasome pathways, disulfiram mainly blocks GSDMD pore formation, but has only a modest effect on earlier steps. Second, a number of FDA-approved Cys-modifying drugs, including the highly reactive DMF used to treat multiple sclerosis, did not show activity against GSDMD, suggesting that the presence of Cys-reactive groups per se is not sufficient to modify GSDMD and to hinder pore formation. Third, while disulfiram may also modify other proteins in a cell due to its Cys-reactive nature, the consequence of these other modifications is not important for the inflammasome pathway and is not cytotoxic. The current literature is consistent with the notion that different cellular targets become important for different cellular challenges. For example, in patients who excessively consume alcohol, disulfiram’s inhibition of ALDH makes it a useful alcohol deterrent due to its effects on acetaldehyde accumulation, which causes flushing and other aversive reactions24, 30. In contrast, disulfiram’s modification of NPL4, an adaptor of the p97 segregase, modulates multiple regulatory and stress-response pathways in cancer cells to promote their death25. The well-established safety profile of disulfiram in humans41 in over six decades of clinical use indicates that disulfiram’s modifications of other cellular targets do not lead to appreciable clinical toxicity. Hence, given the inadequacy of current treatments of inflammation, it is worth exploring repurposing this old drug for treating inflammatory diseases.
Methods
Mice.
8-week-old female C57BL/6J wildtype and Casp11–/– mice were purchased from The Jackson Laboratory. Casp1–/– mice were kindly licensed by Thirumala-Devi Kanneganti (St. Jude Children’s Research Hospital, USA) and provided by Ricardo T. Gazzinelli (University of Massachusetts Medical School, USA). Gsdmd–/– mice were a gift from Toshihiko Shiroishi (National Institute of Genetics, Japan). Mice were maintained at the SPF facility at Harvard Medical School. All mouse experiments were conducted using protocols approved by the Animal Care and Use Committees of Boston Children’s Hospital and Harvard Medical School.
Drug administration and LPS-induced sepsis in mice.
Mice were treated with disulfiram (DSF, 50 mg/kg) formulated in sesame oil (12.5 mg/mL) or vehicle (Ctrl) by intraperitoneal injection at indicated times. In the indicated group of mice in Fig. 6h, copper gluconate (0.15 mg/kg) was administered intraperitoneally 6 hr prior to the first injection of DSF. Sepsis was induced in C57BL/6 mice (8–10 weeks old) by intraperitoneal injection of LPS (E. coli O111:B4) at indicated concentrations. For Fig. 6i, mice were treated with copper gluconate (0.15 mg/kg) or vehicle by intraperitoneal injection 5 hr before LPS challenge and then given DSF (50 mg/kg) intraperitoneally dissolved in sesame oil or vehicle 4 hr before and just before LPS challenge (15 mg/kg intraperitoneally). Peritoneal cells were collected by rinsing the peritoneal cavity with ice cold PBS containing 3% FBS 6 hr after LPS challenge. To measure cytokines, blood samples were collected by tail vein bleed 12 hr post LPS challenge and allowed to clot at room temperature. Sera obtained after centrifugation at 2,000 x g for 10 min were analysed for inflammatory cytokines by ELISA.
Reagents.
β-mercaptoethanol (2ME), dithiothreitol (DTT), terbium(III) chloride (TbCl3), dipicolinic acid (DPA), disulfiram, copper gluconate, phorbol 12-myristate 13-acetate (PMA), DMSO, Necrosulfonamide, Necrostatin-1, BAY 11–7082, dimethyl fumarate, ibrutinib and afatinib were from Sigma-Aldrich. Ultra LPS and nigericin were from InvivoGen. The pan-caspase inhibitor z-VAD-fmk was from BD Bioscience. The complete protease inhibitor cocktail and the PhosSTOP phosphatase inhibitor cocktail were from Roche. LDC7559 was synthesized by Intonation Research Labs.
The monoclonal antibody against GSDMD was generated in house by immunizing 6 week-old BALB/c mice with recombinant human GSDMD and boosting with recombinant human GSDMD-NT according to standard protocols. Serum samples were collected to assess titers of reactive antibodies and spleen cells were fused with SP2/0 myeloma cells. Hybridomas were selected and supernatants from the resulting clones were screened by enzyme linked immunosorbent assay (ELISA), immunoblot and immunofluorescence microscopy. Tubulin antibody was from Sigma-Aldrich. Phospho-IκBα antibody, IκBα antibody, Phospho-NF-κB p65 antibody, cleaved human caspase-1 (Asp297) antibody and NLRP3 antibody were from Cell Signaling Technology. ASC antibody (AL177) and mouse caspase-1 p20 antibody were from AdipoGen. Human and mouse IL-1β antibodies were from R&D Systems. HMGB1 and mouse GSDMD antibodies were from Abcam.
Protein expression and purification.
Full-length human GSDMD sequence was cloned into the pDB.His.MBP vector with a tobacco etch virus (TEV)-cleavable N-terminal His6-MBP tag using NdeI and XhoI restriction sites. Human GSDMD-3C and mouse GSDMA3–3C mutants were constructed by QuikChange Mutagenesis (Agilent Technologies). For expression of full-length GSDMD, GSDMD-3C, GSDMA3, and GSDMA3–3C, E. coli BL21 (DE3) cells harbouring the indicated plasmids were grown at 18 °C overnight in LB medium supplemented with 50 μg ml−1 kanamycin after induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) when OD600 reached 0.8. Cells were ultrasonicated in lysis buffer containing 25 mM Tris-HCl at pH 8.0, 150 mM NaCl, 20 mM imidazole and 5 mM 2ME. The lysate was clarified by centrifugation at 40,000g at 4 °C for 1 hr. The supernatant containing the target protein was incubated with Ni-NTA resin (Qiagen) for 30 min at 4 °C. After incubation, the resin–supernatant mixture was poured into a column and the resin was washed with lysis buffer. The protein was eluted using the lysis buffer supplemented with 100 mM imidazole. The His6-MBP tag was removed by overnight TEV protease digestion at 16 °C. The cleaved protein was purified using HiTrap Q ion-exchange and Superdex 200 gel-filtration columns (GE Healthcare Life Sciences).
Caspase-11 sequence was cloned into the pFastBac-HTa vector with a TEV cleavable N-terminal His6-tag using EcoRI and XhoI restriction sites. The baculoviruses were prepared using the Bac-to-Bac system (Invitrogen), and the protein was expressed in Sf9 cells following the manufacturer’s instructions. His–caspase-11 baculovirus (10 ml) was used to infect 1 L of Sf9 cells. Cells were collected 48 hrs after infection and His6–caspase-11 was purified following the same protocol as for His6-MBP–GSDMD. Eluate from Ni-NTA resin was collected for subsequent assays.
Liposome preparation.
PC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 25 mg/mL in chloroform; 80 μL), PE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, 25 mg/mL in chloroform; 128 μL) and CL (1’,3’-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (sodium salt), 25 mg/mL in chloroform; 64 μL) were mixed and the solvent was evaporated under a stream of N2 gas. The lipid mixture was suspended in 1 mL Buffer A (20 mM HEPES at pH 7.4, 150 mM NaCl, 50 mM sodium citrate, and 15 mM TbCl3) for 3 min. The suspension was pushed through 100 nm Whatman® Nuclepore™ Track-Etched Membrane 30 times to obtain homogeneous liposomes. The filtered suspension was purified by size exclusion column (Superose 6, 10/300 GL) in Buffer B (20 mM HEPES, 150 mM NaCl) to remove TbCl3 outside liposomes. Void fractions were pooled to produce a stock of PC/PE/CL liposomes (1.6 mM). The liposomes are diluted to 50 μM with Buffer C (20 mM HEPES, 150 mM NaCl and 50 μM DPA) for use in high-throughput screening.
High-throughput screen for GSDMD inhibitors.
Liposome leakage was detected by an increase in fluorescence when Tb3+ bound to DPA in Buffer C. Human GSDMD (0.3 μM), dispensed into 384-well plates (Corning 3820) containing PC/PE/CL liposomes (50 μM liposome lipids), was incubated with compounds from the ICCB-Longwood Screening Facility collection for 1 hr before addition of caspase-11 (0.15 μM) to each well. The fluorescence intensity of each well was measured at 545 nm with an excitation of 276 nm 1 hr after addition of caspase-11 using a Perkin Elmer EnVision plate reader. The final percent inhibition was calculated as [(fluorescencetest compound − fluorescencenegative control)/(fluorescencepositive control − fluorescencenegative control)] × 100, where wells with GSDMD without inhibitors was used as positive control and with caspase-11 as negative control. 50% inhibition was arbitrarily chosen as a threshold. The hits were evaluated in concentration-response experiments in a dose range of 0.008–50 μM to determine IC50. For liposome leakage assay using GSDMD-NT, human GSDMD (0.3 μM), incubated with caspase-11 (0.03 μM) at 4 °C for 24 hrs, was incubated with disulfiram at indicated concentrations for 1 hr before addition of liposomes (50 μM) and fluorescence measurements.
Fluorescent protein labelling and microscale thermophoresis binding assay.
His6-MBP-GSDMD was labelled with AlexaFluor-488 using the Molecular Probes protein labelling kit. Binding of inhibitors to GSDMD was evaluated using microscale thermophoresis (MST). Ligands (49 nM - 150 μM) were incubated with purified AlexaFluor-488-labeled protein (80 nM) for 30 min in assay buffer (20 mM HEPES at pH 7.4, 150 mM NaCl, 0.05% Tween 20). The sample was loaded into NanoTemper Monolith NT.115 glass capillaries and MST carried out using 20% LED power and 40% MST power. KD values were calculated using the mass action equation and NanoTemper software.
Caspase-1 and caspase-11 inhibition assays.
The fluorogenic assay for caspase-1 and caspase-11 activity is based on release of 7-amino-4-methylcoumarin (AMC) from the caspase substrate Ac-YVAD-AMC. Compounds (8 nM - 50 μM) were incubated with 0.5 U of caspase-1 or caspase-11 for 30 min in assay buffer (20 mM HEPES, 150 mM NaCl) in 384-well plates (Corning 3820) before addition of Ac-YVAD-AMC (40 μM) to initiate the reactions. Reactions were monitored in a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, California USA) with excitation/emission wavelengths at 350/460 nm. The fluorescence intensity of each reaction was recorded every 2 min for 2 hrs.
Mass spectrometry and sample preparation.
Gel bands were cut into 1 mm size pieces and placed into separate 1.5 mL polypropylene tubes. 100 μl of 50% acetonitrile in 50 mM ammonium bicarbonate buffer were added to each tube and the samples were then incubated at room temperature for 20 min. This step was repeated if necessary to destain gel. Then, the gel slice was incubated with 55 mM iodoacetamide (in 50 mM ammonium bicarbonate) for 45 min in the dark at room temperature, before the gel was washed sequentially with 50 mM ammonium bicarbonate, water and acetonitrile. Samples were then dried in a Speedvac for 20 min. Trypsin (Promega Corp.) (10 ng/μL in 25 mM ammonium bicarbonate, pH 8.0) was added to each sample tube to just cover the gel, and samples were then incubated at 37 °C for 6 hrs or overnight.
After digestion, samples were acidified with 0.1% formic acid (FA) and 3 μl of tryptic peptide solution was injected. Nano-LC/MS/MS was performed on a Thermo Scientific Orbitrap Fusion system, coupled with a Dionex Ultimat 3000 nano HPLC and auto sampler with 40 well standard trays. Samples were injected onto a trap column (300 μm i.d. × 5mm, C18 PepMap 100) and then onto a C18 reversed-phase nano LC column (Acclaim PepMap 100 75 μm × 25 cm), heated to 50 °C. Flow rate was set to 400 nL/min with 60 min LC gradient, using mobile phases A (99.9% water, 0.1% FA) and B (99.9% acetonitrile, 0.1% FA). Eluted peptides were sprayed through a charged emitter tip (PicoTip Emitter, New Objective, 10 +/− 1 μm) into the mass spectrometer. Parameters were: tip voltage, +2.2 kV; Fourier Transform Mass Spectrometry (FTMS) mode for MS acquisition of precursor ions (resolution 120,000); Ion Trap Mass Spectrometry (ITMS) mode for subsequent MS/MS via higher-energy collisional dissociation (HCD) on top speed in 3 s.
Proteome Discoverer 1.4 was used for protein identification and modification analysis. UniPort human database was used to analyse raw data. Other parameters include the following: selecting the enzyme as trypsin; maximum missed cleavages = 2; dynamic modifications are carbamidomethyl (control), diethyldithiocarbamate (from disulfiram) on cysteine; oxidized methionine, deaminated asparagine and glutamine; precursor tolerance set at 10 ppm; MS/MS fragment tolerance set at 0.6 Da; and +2 to +4 charged peptides are considered. Peptide false discovery rate (FDR) was set to be smaller than 1% for significant match.
Cell lines and treatments.
THP-1 (obtained from ATCC) were grown in RPMI with 10% heat-inactivated fetal bovine serum, supplemented with 100 U/ml penicillin G, 100 μg/ml streptomycin sulfate, 6 mM HEPES, 1.6 mM L-glutamine, and 50 μM 2ME. HEK293T and HCT116 cells (obtained from ATCC) were cultured in DMEM with the same supplements. C57BL/6 mouse iBMDM cells were kindly provided by Jonathan Kagan (Boston Children’s Hospital) and cultured in DMEM with the same supplements. HT-29 cells were kindly provided by Junying Yuan (Harvard Medical School) and cultured in McCoy’s 5A medium, supplemented with 10% FBS and 1% penicillin and streptomycin. Cells were verified to be free of mycoplasma contamination. Transient transfection of HEK293T cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. iBMDM cells were transfected by nucleofection using the Amaxa Nucleofector kit (VPA-1009). Generally, THP-1 cells were first differentiated by incubation with 50 nM PMA for 36 hrs and then primed with LPS (1 μg/ml) for 4 hrs before treatment with nigericin (20 μM). To examine IκBα phosphorylation and degradation as well as IL-1β induction, PMA-differentiated THP-1 cells were stimulated with LPS (1 μg/ml) for 0.5, 1 and 4 hrs, respectively. For non-canonical inflammasome activation, 1 million iBMDM cells were electroporated with 1μg ultra LPS. To measure IL-1β release, iBMDMs were primed with LPS (1 mg/ml) for 2 hrs (and then removed) before disulfiram treatment and subsequent LPS transfection for 2.5 hrs. iBMDMs were not primed for other experiments. To activate the AIM2 inflammasome and subsequent pyroptotic cell death, iBMDMs were transfected with 2 μg poly(dA:dT) using Lipofectamine 2000 for 4 hrs. Necroptosis was induced in HT-29 cells by adding 20 ng/ml TNF, 100 nM SMAC mimetic, and 20 μM z-VAD-fmk for 24 hrs.
Cytotoxicity and cell viability assays.
Cell death and cell viability were determined by the lactate dehydrogenase release assay using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega) and by measuring ATP levels using the CellTiter-Glo Luminescent Cell Viability Assay (Promega), respectively, according to the manufacturer’s instructions. Luminescence and absorbance were measured on a BioTek Synergy 2 plate reader. For the cell death assay in HEK293 T cells, 0.5 μg plasmids encoding indicated proteins were transfected into HEK293T cells by calcium-phosphate transfection. Medium was changed and disulfiram was added 8 hrs post-transfection; cell death was determined by CytoTox 96 assay 12 hrs later. To assess cell permeability, cells were cultured in TBS plus 25 mM HEPES, 5 mM glucose, and 0.1% BSA containing 1 μM SYTOX Green (Molecular Probes) and fluorescence at 528 nm after excitation at 485 nm was continuously recorded for 66 min after nigericin treatment at 3 min intervals using a Biotek Synergy plate reader.
Pore reconstitution on nanodiscs and negative staining electron microscopy.
The coding sequence of the membrane scaffold protein NW50 was cloned into a pET-28a vector, and the protein was expressed in E. coli BL21(DE3), purified via a refolding procedure, and covalently circularized with sortase according to a previously described protocol42. A lipid mixture containing phosphatidylserine (PS) and phosphatidylcholine (PC) (molar ratio 3:7) was solubilized in 60 mM sodium cholate and incubated with circularized NW50 on ice for 1 h to assemble nanodiscs. Sodium cholate was then removed by incubation overnight at 4 °C with Bio-beads SM-2 (Bio-Rad). The Bio-beads were then removed using a 0.22 μm filter, and the assembled nanodiscs were further purified using a Superose 6 10/300 gel-filtration column (GE Healthcare Life Sciences) equilibrated with Buffer D (50 mM Tris-HCl at pH 8.0, 150 mM NaCl) to remove excess lipids. To form GSDMD pores on the nanodiscs, purified human GSDMD-3C was incubated with 3C protease in the presence of nanodiscs for 6 hrs on ice. The pores were further purified over a Superose 6 column equilibrated with Buffer D. To assess the effect of disulfiram, human GSDMD-3C plus 3C protease was either incubated with disulfiram (molar ratio 1:1) for 30 min on ice before adding to nanodiscs (pretreatment), or disulfiram was added for 30 min on ice to already assembled pores (post-treatment). For negative staining electron microscopy, a 5-μl sample was placed onto a glow-discharged carbon-coated copper grid (Electron Microscopy Sciences), washed twice with Buffer A, stained with 1% uranyl formate for 1 min, and air-dried. The grids were imaged on the Tecnai G2 Spirit BioTWIN electron microscope and recorded with an AMT 2k CCD camera (Harvard Medical School Electron Microscopy Facility).
Immunoblot analysis.
Cell extracts were prepared using RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with a complete protease inhibitor cocktail (Roche) and a PhosSTOP phosphatase inhibitor cocktail (Roche). Samples were subjected to SDS-PAGE and the resolved proteins were then transferred to a PVDF membrane (Millipore). Immunoblots were probed with indicated antibodies and visualized using a SuperSignal West Pico chemiluminescence ECL kit (Pierce).
Caspase-1 activity assay in cells.
To measure caspase-1 activation, THP-1 cells were seeded into 96-well plates and differentiated with PMA. After the indicated treatments, cells were incubated with a fluorescent active caspase-1 substrate FAM-YVAD-FMK (Immunochemistry Technologies). Samples were read on a BioTek Synergy 2 plate reader.
Measurement of cytokines.
Concentrations of IL-1β, TNF and IL-6 in culture supernatants or mouse serum were measured by ELISA kit (R&D Systems) according to the manufacturer’s instructions.
Immunostaining and confocal microscopy.
Cells grown on coverslips were fixed for 15 min with 4% paraformaldehyde in PBS, permeabilized for 5 min in 0.1% Triton X-100 in PBS and blocked using 5% BSA for 1 hr. Then, cells were stained with the indicated primary antibodies followed by incubation with fluorescent-conjugated secondary antibodies (Jackson ImmunoResearch). Nuclei were counterstained with DAPI (4,6-diamidino-2-phenylindole) (Sigma-Aldrich). Slides were mounted using Aqua-Poly/Mount (Dako). Images were captured using a laser scanning confocal microscope (Olympus Fluoview FV1000 Confocal System) with a 63× water immersion objective and Olympus Fluoview software (Olympus). All confocal images are representative of three independent experiments.
Statistics.
Student’s t-test was used for the statistical analysis of two independent treatments. Mouse survival curves and statistics were analysed using the Mantel-Cox Log-rank test. Multiple comparisons between two groups were performed by multiple t-test with type I error correction. Type I error is corrected by the Holm-Sidak method.
Data Availability Statement.
All relevant data are available in the Source Data or Extended Data Information of the manuscript.
Extended Data
Extended Data Fig. 1. Optimization and hits from the liposome leakage assay screen.
(a-c) Optimization of the Tb3+/DPA assay. (a) GSDMD (2.5 μM) and caspase-11 (2.5 μM) were incubated in liposome solutions at various concentrations in 20 mM HEPES buffer (150 mM NaCl) for 1 hr. The concentration of liposome lipids for the screen was set at 50 μM. n = 3 independent experiments. The mean ± s.e.m. is shown. (b) Different concentrations of GSDMD and caspase-11 (1:1 ratio) were incubated in liposome (50 μM) solutions for 1 hr. The concentration of GSDMD used in the screen was set at 0.3 μM. n = 3 independent experiments. The mean ± s.e.m. is shown. (c) Different concentrations of caspase-11 and GSDMD (0.3 μM) were incubated in liposome (50 μM) solutions for 1 hr. The concentration of caspase-11 used in the screen was set at 0.15 μM. n = 3 independent experiments. The mean ± s.e.m. is shown.The fluorescence intensity at 545 nm was measured after excitation at 276 nm. (d) Hit compounds evaluated in binding and/or cell-based assays. (e) Mouse iBMDMs were pretreated or not with disulfiram (C-23) ranging from 5–40 μM for 1 hr before transfection with PBS or poly(dA:dT) and analyzed for cell viability by CellTiter-Glo assay 4 hrs later. Graphs show mean ± s.d; data are representative of three independent experiments with replicates (n= 3) and similar results. Data were analyzed using two-tailed Student’s t-test. **P < 0.01.
Extended Data Fig. 2. The activity of disulfiram in cells is greatly increased by Cu(II).
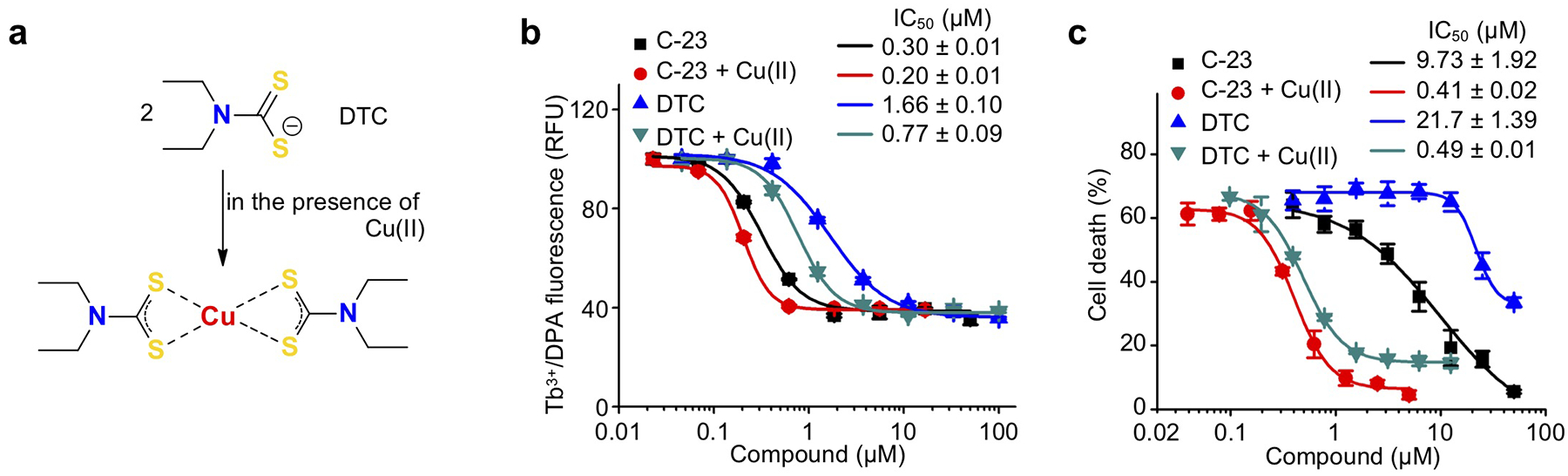
(a) DTC–copper complex formation of disulfiram metabolite diethyldithiocarbamate (DTC) with Cu(II). (b) Dose response curves of inhibition of liposome leakage by disulfiram (C-23) or DTC in the presence or absence of Cu(II). n = 3 independent experiments. The mean ± s.e.m. is shown. (c) LPS-primed THP-1 were pretreated with C-23 or DTC in the presence or absence of Cu(II) for 1 hr before adding nigericin or medium for 2 hrs. Cell death was determined by CytoTox96 assay. n = 3 independent experiments. The mean ± s.e.m. is shown.
Extended Data Fig. 3. Effect of disulfiram on caspase-1 and caspase-11.
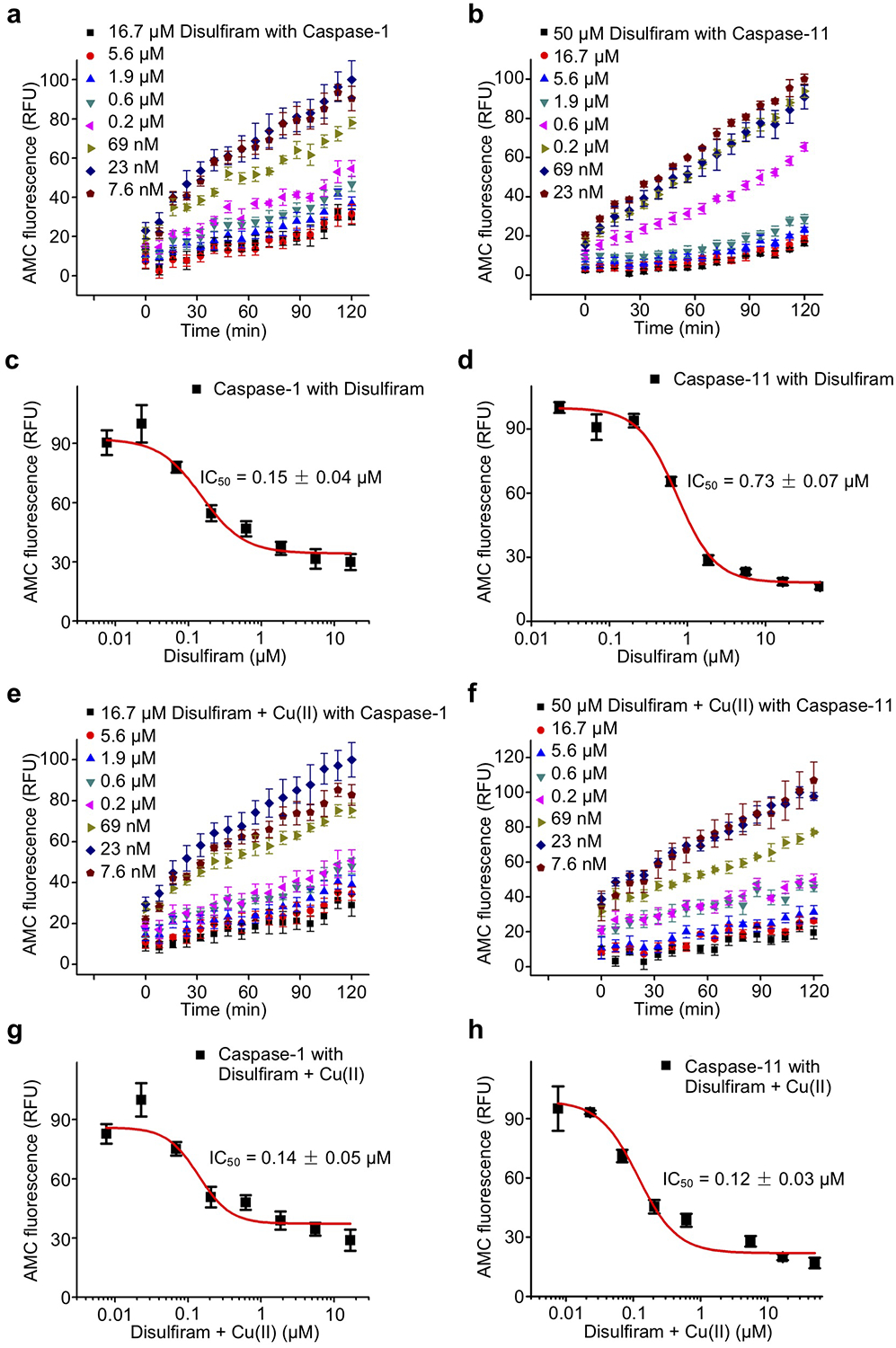
(a,b) Time course of caspase-1 (a) and caspase-11 (b) activity in the presence of indicated concentrations of disulfiram. Caspases (0.5 U) were incubated with disulfiram (at indicated concentrations for 1 hr before adding Ac-YVAD-AMC (40 μM)). (c,d) Dose response curve of disulfiram in the caspase-1 (a) and caspase-11 (b) activity assay. (e,f) Time course of caspase-1 (e) and caspase-11 (f) activity in the presence of indicated concentrations of disulfiram + Cu(II). Caspases (0.5 U) were incubated with disulfiram + Cu(II) (at indicated concentrations for 1 hr before adding Ac-YVAD-AMC (40 μM)). (g,h) Dose response curve of disulfiram + Cu(II) in the caspase-1 (e) and caspase-11 (f) activity assay. (a-h) n = 3 independent experiments. The mean ± s.e.m. is shown. Fluorescence intensity at 460 nm was measured after excitation at 350 nm.
Extended Data Fig. 4. Disulfiram covalently modifies human GSDMD on Cys 191.
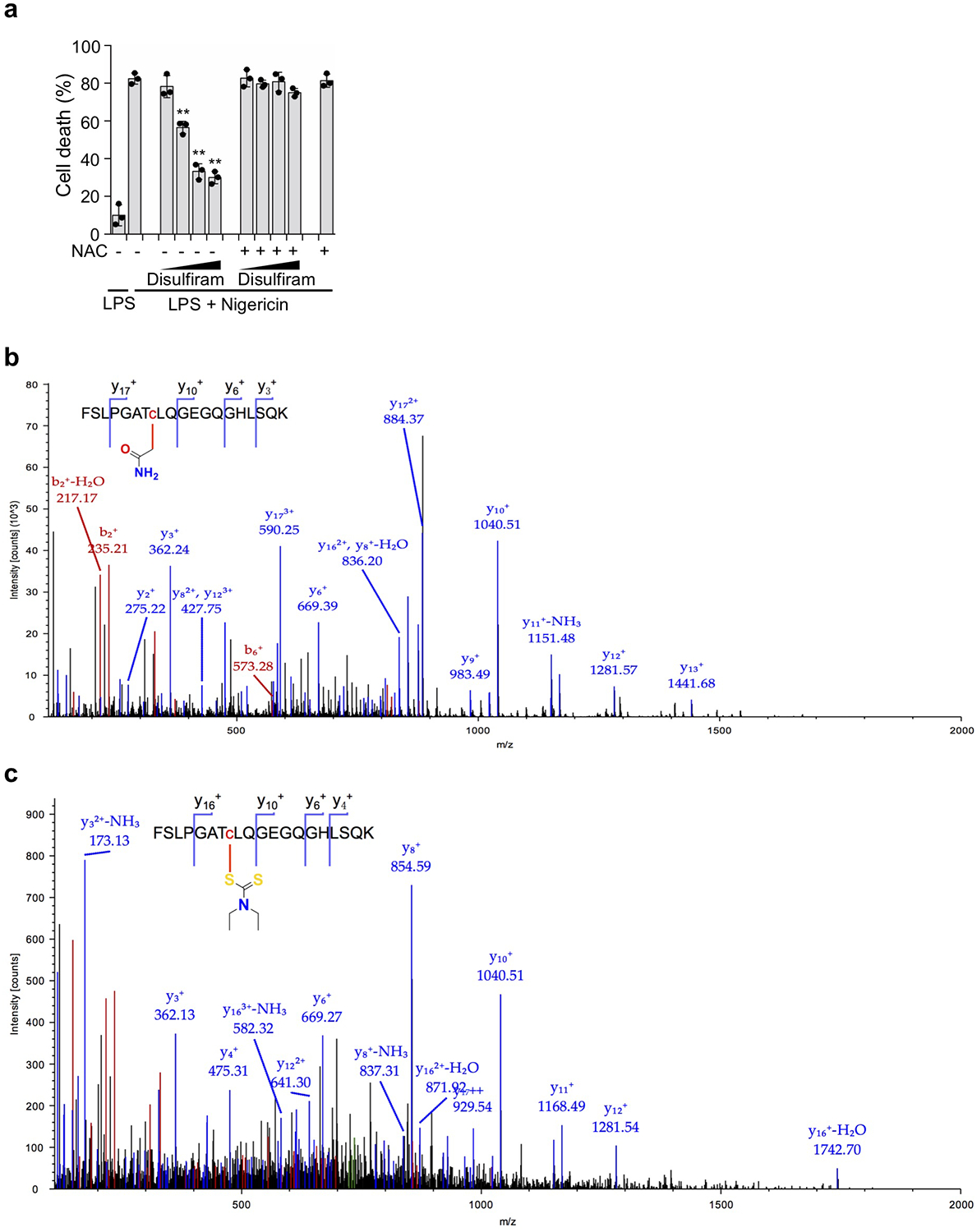
(a) Disulfiram was preincubated for 1 hr with N-acetylcysteine (NAC, 500 μM) or medium before evaluating whether it inhibited pyroptosis of LPS + nigericin treated THP-1 cells. Disulfiram 2-fold dilutions ranged from 5–40 μM. Graphs show mean ± s.d; data are representative of three independent experiments with replicates (n= 3) and similar results. Data were analyzed using two-tailed Student’s t-test. Graphs show the mean ± s.d. and data shown are representative of three independent experiments. **P < 0.01. (b,c) nano-LC-MS/MS spectrum for the peptide containing C191 in human GSDMD. Data are representative of three independent experiments. (b) MS/MS spectrum for peptide FSLPGATCLQGEGQGHLSQK modified on cysteine (red) by carbamidomethyl. Protein coverage was 73%. (c) MS/MS spectrum for peptide FSLPGATCLQGEGQGHLSQK modified on cysteine (red) by disulfiram. Protein coverage was 72%.
Extended Data Fig. 5. Disulfiram covalently modifies GSDMD Cys191.
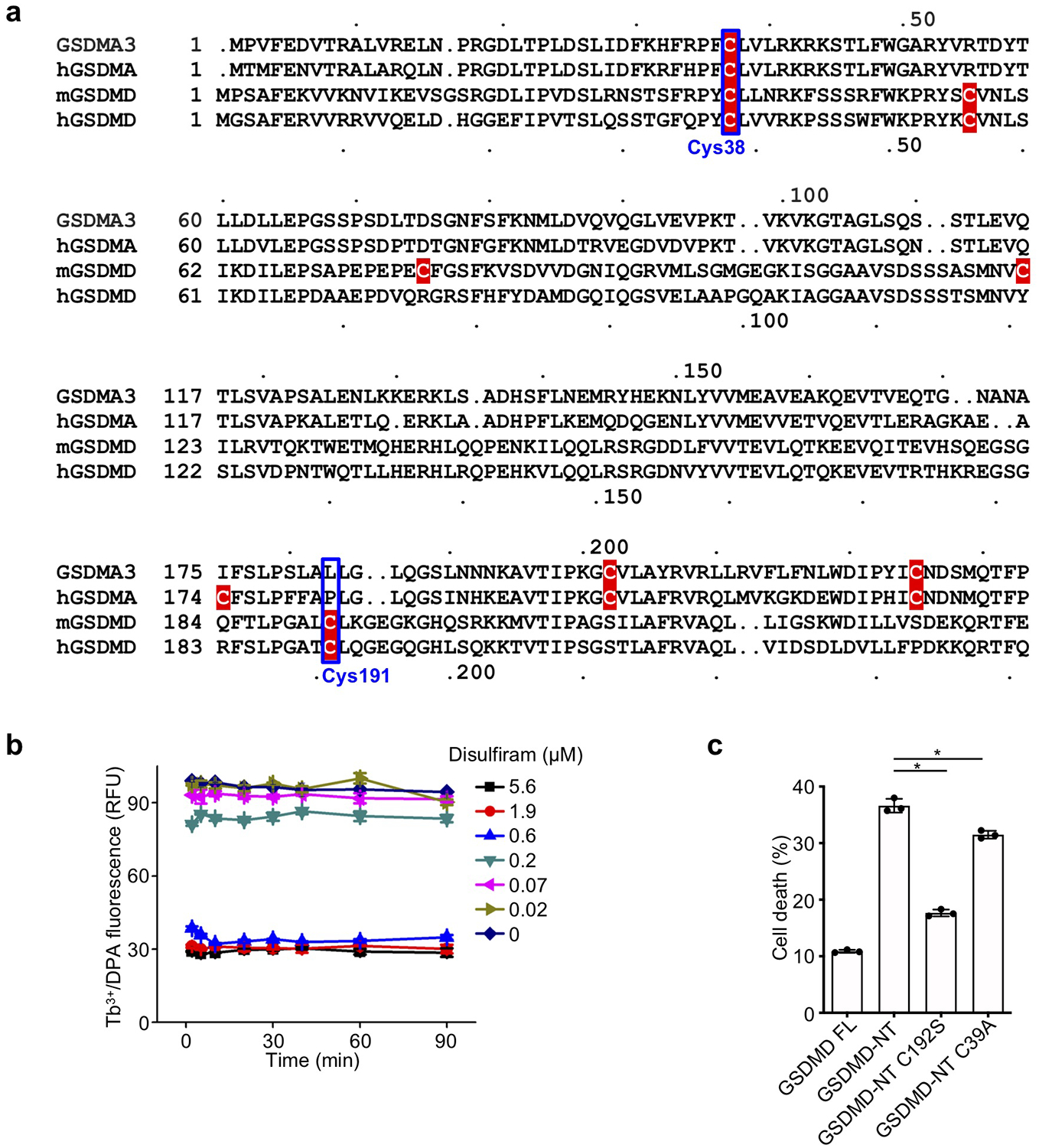
(a) Sequence alignment of GSDMA3, hGSDMA, mGSDMD and hGSDMD showing Cys residues (highlighted in red). (b) GSDMD (0.3 μM) was preincubated with the indicated concentrations of disulfiram (0–5.6 μM) for indicated times (2–90 min) before caspase-11 (0.15 μM) and liposomes (50 μM) were added. n = 3 independent experiments. The mean ± s.e.m. is shown. (c) FL mouse GSDMD or wildtype, C192S or C39A GSDMD-NT were transiently expressed in HEK293T cells. Cell death was determined by CytoTox96 cytotoxicity assay 20 hrs after transfection. (c) shows the mean ± s.d. of 1 representative experiment of three independent experiments performed. Comparison in (c) was calculated by two-tailed Student’s t-test. *P < 0.05.
Extended Data Fig. 6. Mouse monoclonal antibody recognizes full-length human GSDMD and the GSDMD-NT pore form on immunoblots and by immunofluorescence microscopy.
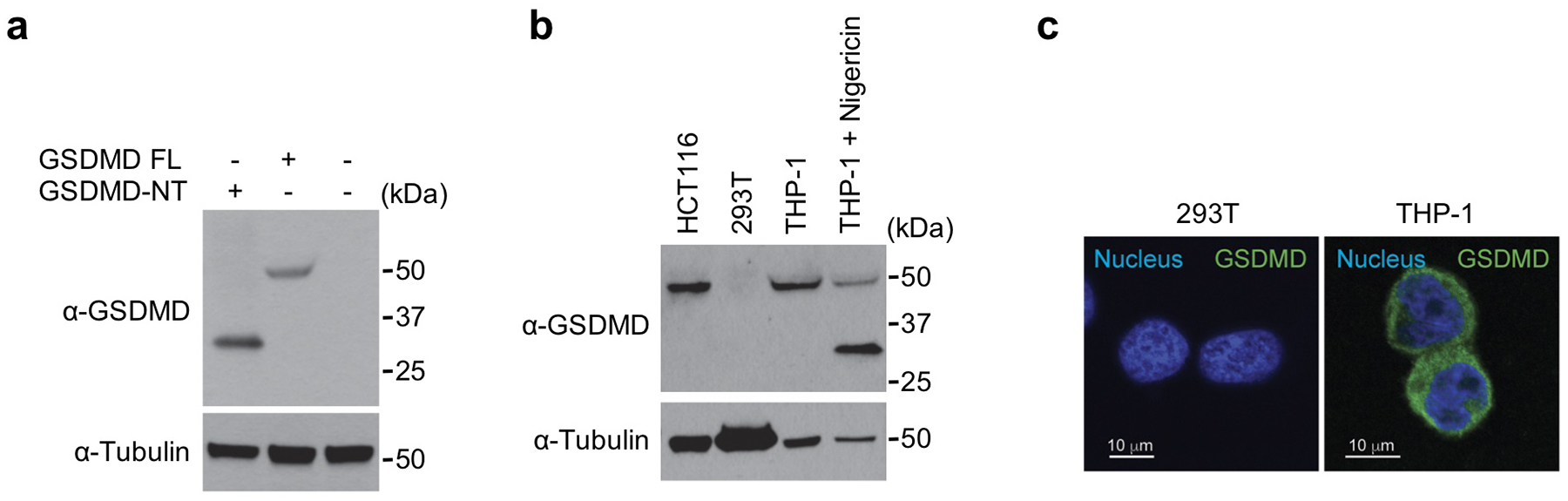
The monoclonal antibody against GSDMD was generated by immunizing mice with recombinant human GSDMD and boosting with recombinant human GSDMD-NT as described in Methods. (a) HEK293T cells were transfected with the indicated plasmids and cell lysates were analysed by immunoblot of reducing gels probed with the indicated antibodies. (b) Cell lysates of HCT116, 293T and THP-1 cells, treated or not with nigericin, were immunoblotted with the indicated antibodies. 293T cells do not express endogenous GSDMD. (c) 293T and THP-1 cells were stained with the anti-GSDMD monoclonal antibody and co-stained with DAPI (blue). 293T cells show no background staining. Data are representative of at least three independent experiments.
Extended Data Fig. 7. Disulfiram protects against LPS-induced sepsis.
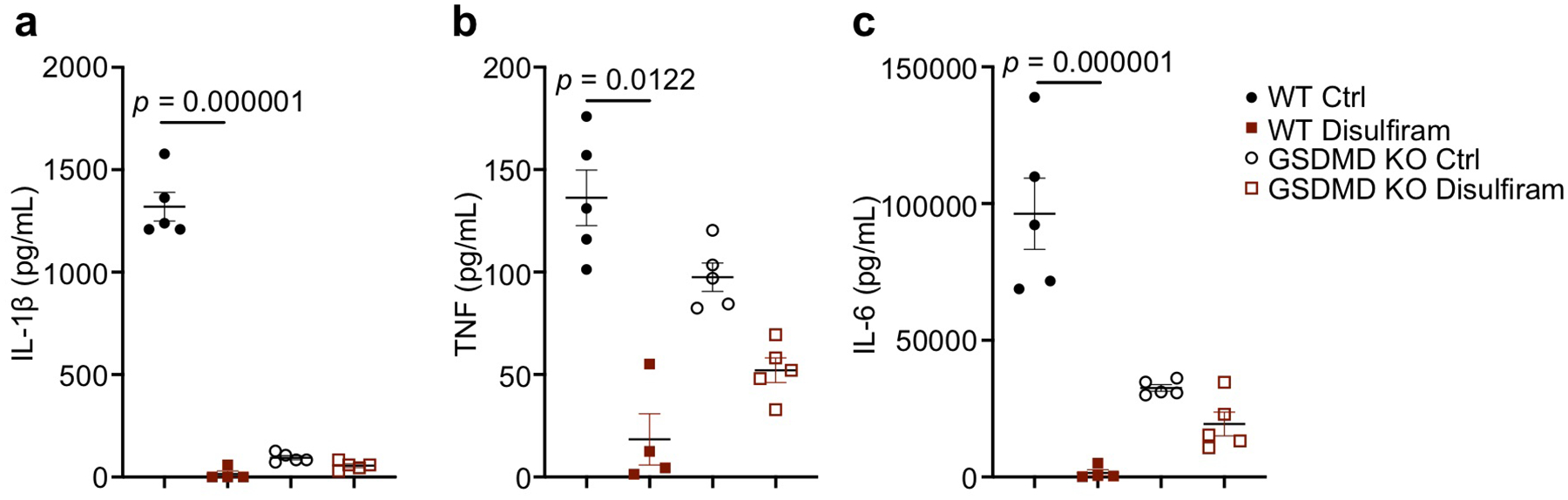
(a-c) Mice were pretreated with disulfiram (50 mg/kg) or vehicle (Ctrl) by intraperitoneal injection 24 and 4 hrs before intraperitoneal challenge with 15 mg/kg LPS and followed for survival. Serum IL-1β (a), TNF (b) and IL-6 (c) were measured by multiplex luminex assay (n= 5/group) 12 hrs post LPS challenge. Shown are mean ± s.e.m. Statistical differences between the groups were calculated by multiple t-test. Type I error was corrected by the Holm-Sidak method.
Extended Data Fig. 8. Dose response curve of other compounds in GSDMD-mediated liposome leakage assay.
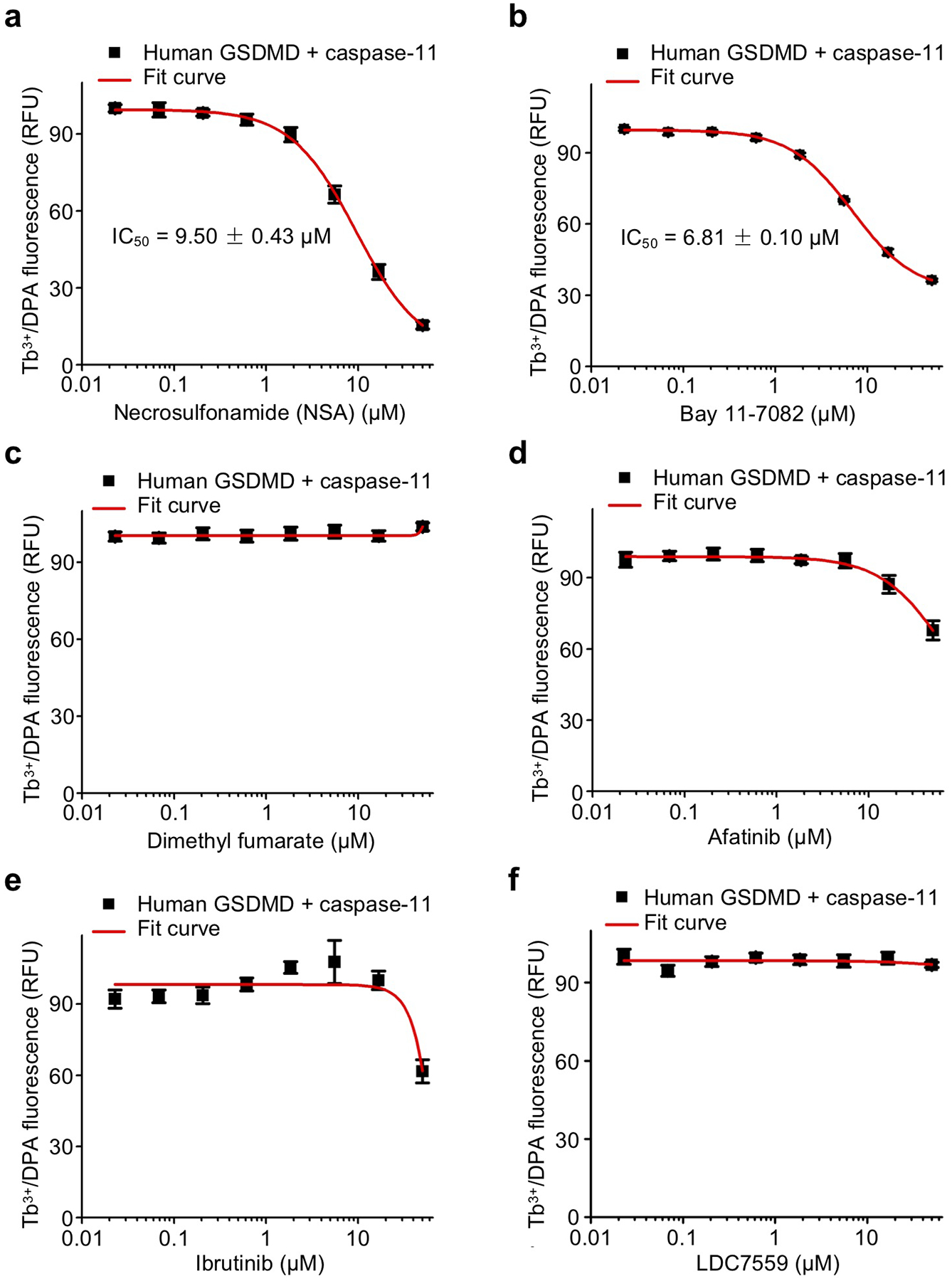
Dose response curve of necrosulfonamide (a), Bay 11–7082 (b), dimethyl fumarate (DMF) (c), afatinib (d), ibrutinib (e), and LDC7559 (f) in liposome leakage induced by 0.3 μM GSDMD plus 0.15 μM caspase-11. (a-f) n = 3 independent experiments. The mean ± s.e.m. is shown.
Acknowledgements.
This work was supported by the US National Institutes of Health (DP1HD087988 to H.W., R01Al139914 to H.W. and J.L., R01AI123265 to J.L., R01 AI142642, R01 AI145274, R01 AI141386, R01HL092020, and P01HL095489 to H.R.L), National Natural Science Foundation of China (31972897), Key Research Program of the Chinese Academy of Sciences (ZDBS-LY-SM008), Shanghai Municipal Science and Technology Major Project (2019SHZDZX02), Rising-Star Program of Shanghai Science and Technology Committee (19QA1409800) (X.L.), a grant from FAMRI (CIA 123008 to H.R.L.), Cancer Research Institute Irvington Postdoctoral Fellowship Program (J.J.H.), Charles A. King Trust Postdoctoral Fellowship Program (J.R., X.L., Z.Z.) and US DOD Breast Cancer Research Program Breakthrough Fellowship Award (Y.Z.). We thank J. Smith, G. Frey, J. Nale, D. Wrobel, and the entire staff of the ICCB-L for their outstanding technical support.
Footnotes
Competing Interests Statement. J. L. and H.W. are co-founders of Ventus Therapeutics. The other authors declare no competing interests.
References
- 1.Rathinam VA, Vanaja SK & Fitzgerald KA Regulation of inflammasome signaling. Nat Immunol 13, 333–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamkanfi M & Dixit VM Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28, 137–161 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Broz P & Dixit VM Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 6.He WT et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aglietti RA et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–7863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sborgi L et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Liu X et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26, 1007–1020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russo HM et al. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol 197, 1353–1367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruan J, Xia S, Liu X, Lieberman J & Wu H Cryo-EM structure of the gasdermin A3 membrane pore. Nature (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jo EK, Kim JK, Shin DM & Sasakawa C Molecular mechanisms regulating NLRP3 inflammasome activation. Cellular & molecular immunology 13, 148–159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frangogiannis NG, Smith CW & Entman ML The inflammatory response in myocardial infarction. Cardiovasc Res 53, 31–47 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Hoffman HM, Wanderer AA & Broide DH Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 108, 615–620 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muckle TJ & Wellsm. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med 31, 235–248 (1962). [PubMed] [Google Scholar]
- 18.Prieur AM et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 66, 57–68 (1987). [DOI] [PubMed] [Google Scholar]
- 19.Hoffman HM, Mueller JL, Broide DH, Wanderer AA & Kolodner RD Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29, 301–305 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aksentijevich I et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 46, 3340–3348 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masters SL et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Science translational medicine 8, 332ra345 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Rello J, Valenzuela-Sanchez F, Ruiz-Rodriguez M & Moyano S Sepsis: A Review of Advances in Management. Adv Ther 34, 2393–2411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dellinger RP et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 39, 165–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright C & Moore RD Disulfiram treatment of alcoholism. Am J Med 88, 647–655 (1990). [DOI] [PubMed] [Google Scholar]
- 25.Skrott Z et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 552, 194–199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sborgi L et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U S A 112, 13237–13242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD & Oldenburg KR A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Sun L et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Degterev A et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen ML, Johnson KL, Mays DC, Lipsky JJ & Naylor S Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem Pharmacol 61, 537–545 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Petersen EN The pharmacology and toxicology of disulfiram and its metabolites. Acta Psychiatr Scand Suppl 369, 7–13 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Nobel CS, Kimland M, Nicholson DW, Orrenius S & Slater AF Disulfiram is a potent inhibitor of proteases of the caspase family. Chem Res Toxicol 10, 1319–1324 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Hagn F, Nasr ML & Wagner G Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat Protoc 13, 79–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castillo-Villanueva A et al. Disulfiram as a novel inactivator of Giardia lamblia triosephosphate isomerase with antigiardial potential. Int J Parasitol Drugs Drug Resist 7, 425–432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez R, Riddle M, Woo J & Momand J Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci 17, 473–481 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rathkey JK et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Juliana C et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem 285, 9792–9802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nair AB & Jacob S A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7, 27–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Sollberger G et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Cvek B Nonprofit drugs as the salvation of the world’s healthcare systems: the case of Antabuse (disulfiram). Drug Discov Today 17, 409–412 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Nasr ML et al. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nat Methods 14, 49–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are available in the Source Data or Extended Data Information of the manuscript.