

Abstract
Atypical low-oxidation-state iron phases in Alzheimer’s disease (AD) pathology are implicated in disease pathogenesis, as they may promote elevated redox activity and convey toxicity. However, the origin of low-oxidation-state iron and the pathways responsible for its formation and evolution remain unresolved. Here we investigate the interaction of the AD peptide β-amyloid (Aβ) with the iron storage protein ferritin, to establish whether interactions between these two species are a potential source of low-oxidation-state iron in AD. Using X-ray spectromicroscopy and electron microscopy we found that the co-aggregation of Aβ and ferritin resulted in the conversion of ferritin’s inert ferric core into more reactive low-oxidation-states. Such findings strongly implicate Aβ in the altered iron handling and increased oxidative stress observed in AD pathogenesis. These amyloid-associated iron phases have biomarker potential to assist with disease diagnosis and staging, and may act as targets for therapies designed to lower oxidative stress in AD tissue.
Subject terms: Metals, Peptides, Dementia, Neurodegenerative diseases, Alzheimer's disease
Introduction
In the human brain, multiple functions are reliant upon iron as a co-factor, including, but not limited to, neurotransmitter synthesis, ATP production and myelin sheath formation1,2. As a result, a basal level of brain iron is required to maintain healthy tissues and brain iron homeostasis is strictly regulated1–4. Despite playing a vital role in the brain, iron can convey toxic effects when stored incorrectly or when it adopts abnormal chemical states2,4–7. The ability of iron to readily change oxidation state is an essential property required for its biological functionality. However, this ability also leads to its potential toxicity as it allows iron participation in Fenton chemistry, resulting in the production of reactive oxygen species (ROS) which are capable of inducing oxidative stress and neuronal injury8–11.
As a fundamental component of iron homeostasis, mammals store brain iron within the ferritin protein complex, in the form of a ferrihydrite-like mineral, (Fe3+)2O3•0.5H2O, a poorly crystalline ferric oxyhydroxide, typically accompanied by phosphate1,12–14. This form of storage prevents iron-associated ROS production, thereby protecting brain tissues from detrimental free radical burdens. Archetypal mammalian ferritin is comprised from two subunits: H-chain and L-chain chain ferritin, which assemble into a 24-subunit protein complex, ~450 kDa in molecular weight1,13,15,16. The complex is 12–13 nm in diameter, containing an 8 nm hollow core capable of storing up to 4500 Fe atoms13. Both H-ferritin and L-ferritin subunits have distinct functions that provide ferritin with an efficient means by which to store large numbers of iron atoms safely in vivo. H-ferritin (21 kDa) contains a di-iron binding site and possesses ferroxidase activity, catalysing the conversion of redox-active ferrous (Fe2+) iron into a redox-inactive ferric (Fe3+) state as it passes into the core. L-ferritin (19 kDa) does not possess any known enzymatic activity, but does contribute to long term iron storage, by accelerating the transfer of iron from the ferroxidase site to the ferritin core, whilst also stabilising ferritin’s complex structure1,12,13.
Evidence of disrupted iron homeostasis has been observed in the tissues of Alzheimer’s disease (AD) subjects, suggesting a link between altered iron handling in AD brain tissues and the development of the disorder2,4,6,7,17–22. AD is a progressive neurodegenerative disease that is the most common cause of dementia amongst the elderly and currently affects 5.4 million individuals in the USA alone19,23–25. The development of AD is heavily influenced by age, and due to increasing average life-expectancy, the number of AD cases is projected to nearly triple by 205025. The underlying cause of AD is poorly understood and currently no cure exists.
Pathologically, AD is characterised by the presence of two hallmark lesions: intracellular neurofibrillary tangles comprised of hyperphosphorylated tau26,27, and extracellular plaques (also referred to as senile plaques) comprised primarily of the peptide β-amyloid (Aβ)28–30. Aβ is formed through cleavage of the amyloid precursor protein by secretase enzymes, resulting in the formation of multiple peptide isoforms varying in amino acid length28,31–37. The 42-amino acid isoform of the peptide is the most fibrillogenic, and is consequently most associated with AD traits, such as senile plaques and neurotoxicity38. Although the extent to which Aβ contributes to the pathogenesis of AD is a topic of much debate, even concerning whether deposition of Aβ is a physiological response as opposed to an inherently pathological occurrence39, it is commonly accepted that Aβ accumulation in the brain parenchyma is a fundamental event in the development of the disorder28,31,40–42.
Mounting evidence shows increased levels of iron, including iron minerals, to be present within tissue areas displaying AD pathology when compared to disease-free controls20,21,43–51. Iron minerals containing iron cations in a low oxidation state, such as magnetite (Fe3O4) and pure ferrous (Fe2+) minerals (e.g. wustite), as well as zero-oxidation-state iron (Fe0), are capable of catalysing Fenton redox chemistry, driving the production of ROS and potentially inducing oxidative stress. Such low-oxidation-state iron phases are inherently unstable and thus not expected to be preserved in biological systems. However, it has been suggested that ferritin malfunction and/or the interaction of Aβ with naturally occurring iron stores may result in chemically-reduced iron phases43,45,47,50–54. Indeed, ferritin has been shown to accumulate in Aβ plaque structures in vivo55, and increased levels of ferrous iron containing minerals have been recorded in pathological ferritin isolated from AD tissues compared to disease-free ferritin controls50.
Further evidence of iron accumulation within Aβ plaque structures has been demonstrated by HR-TEM where ferritin core-sized iron deposits with a crystal structure consistent with magnetite/maghemite were characterised embedded within extracted human amyloid plaque core material47; a result supported by more recent observations in other cases of Alzheimer’s disease56. Moreover, our recent synchrotron-based X-ray absorption studies have revealed the presence of nanoscale (ca. 200 nm) deposits of chemically-reduced iron within plaque structures located in the cortical tissue of APP/PS1 transgenic mice57, and amyloid plaque core material extracted from the grey matter in donated human brains from confirmed AD cases, including phases consistent with zero-oxidation-state iron in the latter58. These studies revealed dramatic nanoscale variations in iron oxidation state within the same amyloid plaque structures, highlighting the need for nanoscale resolution chemically-sensitive imaging in the investigation of metal biochemistry in living systems.
In addition, we previously demonstrated that the Aβ(1-42) peptide fragment is capable of chemically reducing unbound iron(III) oxyhydroxide and ferrihydrite to a pure ferrous phase in vitro59,60, whilst a recent in vitro spectrophotometric-based study has suggested that Aβ(1-40) can influence ferritin iron chemistry61. This evidence strongly implicates Aβ in the formation of chemically-reduced iron phases in AD.
As increased levels of oxidative stress are characteristic of AD pathogenesis62–67, chemically reduced, redox-active iron may represent a target for therapies intended to lower oxidative burdens, thereby inhibiting disease progression68,69. Furthermore, as iron redox chemistry has a profound effect upon its physical (particularly magnetic) properties, identifying iron phases specifically associated with Aβ pathology could provide a clinical biomarker for non-invasive disease diagnosis via magnetic resonance imaging (MRI)70.
Despite this growing body of evidence, the manner in which Aβ influences iron chemistry, even within the protein encapsulated core, and the chemical by-products formed through Aβ/ferritin interaction, remain unclear. Acknowledging both that ferritin is the primary form of iron storage in the brain1, and that Aβ/ferritin co-localises within AD tissues55, studying the chemistry of Aβ/ferritin interaction is vital to understand how altered iron homeostasis may contribute to the development of AD.
In this study we employed scanning transmission X-ray microscopy (STXM), a synchrotron-based X-ray spectromicroscopy technique, to examine the interaction of Aβ(1-42) with ferritin, and establish whether this interaction could result in the formation of the nanoscale, chemically-reduced iron phases observed within amyloid structures in the brain. STXM is a powerful technique that allows the chemical speciation of a sample to be determined to a spatial resolution of ca. 20 nm. The capacity for combined nanoscale spatial and chemical sensitivity, offered by STXM, is extremely important for investigations of amyloid/iron structures as the properties of inorganic inclusions have been shown to vary at the nanoscale.
Here, STXM was used to determine the distribution and chemical composition of protein and iron within in vitro-formed Aβ/ferritin aggregate structures at a nanoscale spatial resolution. We report a detailed characterisation of iron oxidation state formed through the interaction of Aβ and ferritin.
Results
As multiple different laboratory-based and synchrotron experiments were performed for this study, it was necessary to use several different batches of the Aβ(1-42) peptide. For clarity, the different batches and associated investigations are explicitly listed in the Supplementary Information Table S1. The synchrotron work was performed using X-ray microscopes on two different synchrotron beamlines: PolLux at the Swiss Light Source; and I08 at the Diamond Light Source. As these instruments have slightly different performance characteristics (for example energy resolution), we have also stated in Table S1 which beamlines were used to obtain each dataset reported. All Aβ and ferritin incubations were performed under sterile conditions at 37 °C in a modified Krebs-Henseleit (KH) buffer medium (pH 7.4) containing physiologically relevant concentrations of Ca2+ and Mg2+ 59,60. Please refer to methods for further information regarding sample preparation/incubation.
Aβ(1-42) incorporates ferritin into amyloid aggregate structures
Time-lapse images of ferritin incubated for 18 hours in the presence and absence of Aβ are displayed in Fig. 1. When incubated in the absence of Aβ, 1.3 µM ferritin formed a stable and uniform orange/brown colloidal dispersion that remained unchanged throughout the period of examination. Conversely when ferritin was incubated in the presence of 35 µM Aβ, dense orange precipitates formed after 15–20 minutes of interaction, suggesting the co-aggregation of Aβ and ferritin. Once formed, these precipitates remained unchanged over the 18 hours of imaging.
Figure 1.
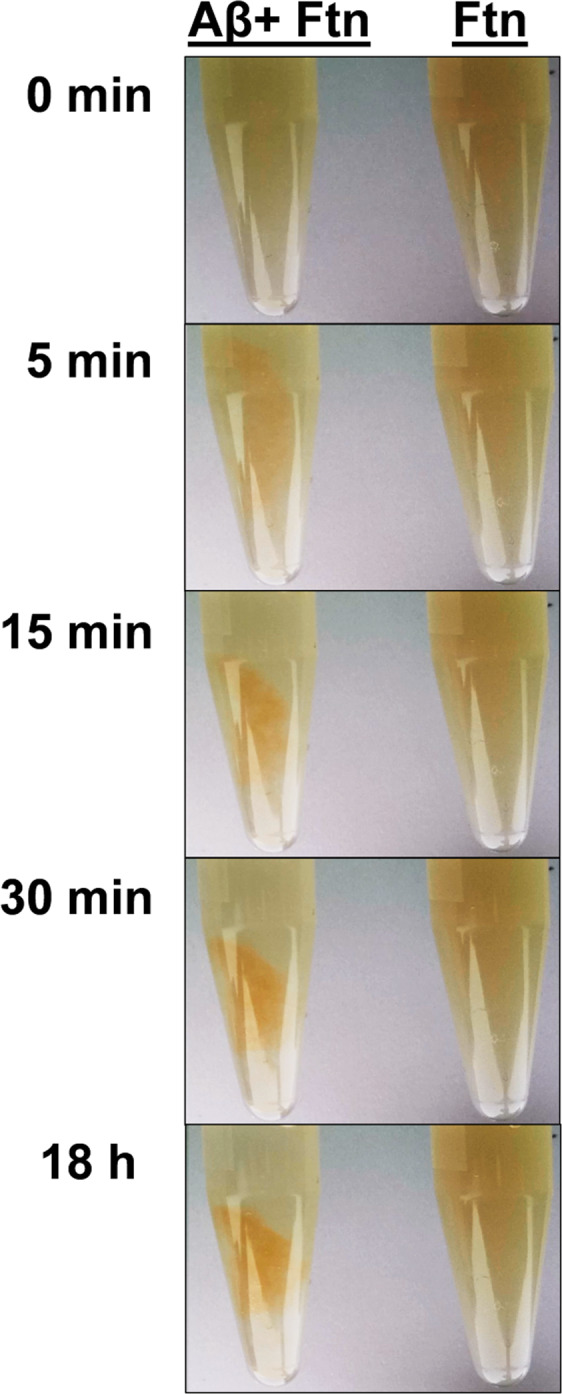
Time lapse images during incubation of ferritin and ferritin + Aβ. Images display ferritin (Ftn) incubated in the presence (left) and absence (right) of Aβ over an 18-hour period. Incubation times are shown to the left of each image.
TEM images of Aβ aggregate structures formed in the absence and presence of ferritin are displayed in Fig. 2. Where Aβ was incubated in the absence of ferritin, amyloid aggregates composed of well-defined fibrillar structures were observed (Fig. 2a), consistent in morphology with fibrillar amyloid structures reported previously71. However, when Aβ was incubated with ferritin, amyloid aggregates appeared to be composed of poorly defined structures containing a high concentration of electron dense fine particles (Fig. 2b). The average diameter (6.1 [±1.3] nm, n = 434) and morphology of these electron dense particles was similar to that of unstained ferritin standards imaged under TEM (6.8 nm [±1.1], n = 237); see Supplementary Fig. S1), where the electron dense regions correspond to ferritin’s iron oxide core. These TEM images suggest that Aβ had incorporated ferritin into its aggregate structures, complementing time lapse images of Aβ/ferritin suspensions where Aβ appeared to co-aggregate with ferritin (Fig. 1).
Figure 2.

TEM images of Aβ/ferritin aggregate structures. Images display aggregates formed in (a) the absence and (b) the presence of ferritin (minimum 48 hours of co-incubation). Inset (bottom right) shows zoomed area highlighted in the yellow box.
X-ray spectromicroscopy (STXM) of Aβ structures formed in the absence of ferritin across the carbon K-absorption edge (280–320 eV) provided an X-ray absorption spectrum (Fig. 3; red) closely resembling an albumin protein reference (Fig. 3; blue). STXM examination of ferritin structures across the carbon K-edge generated an X-ray absorption spectrum (Fig. 3; green) similar to both albumin and Aβ. Whilst sufficiently different amino acids/peptides provide distinct carbon K-edge spectra (see for example Kaznacheyev et al., and Boese et al.72,73), the subtle differences between Aβ and ferritin spectra meant that measurements at the carbon K-edge alone could not be used to distinguish between the two species.
Figure 3.
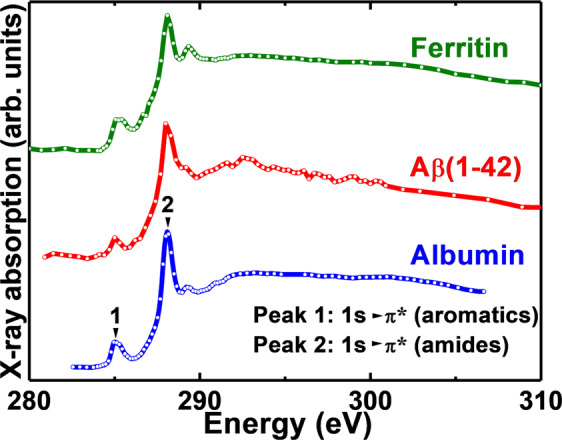
Carbon K-edge X-ray absorption spectra of albumin, Aβ(1-42) and ferritin.
Aβ/ferritin interaction leads to the chemical reduction of ferritin-stored iron
To confirm that the electron dense particles shown in Fig. 2b contained iron and to monitor the protein morphology and oxidation state of iron within Aβ/ferritin aggregates over an extended incubation period (144 hours), Aβ/ferritin aggregates were studied by STXM at the Swiss Light Source on the PolLux beamline.
Following 0.5 and 48 hours of Aβ/ferritin co-incubation, multiple protein dense aggregates were observed using STXM. A representative example of an aggregate structure formed following 48 hours of incubation is shown in Fig. 4. TEM imaging of the aggregate revealed a poorly defined fibrillar structure containing electron dense fine particles widely spread throughout the aggregate (Fig. 4a–c), indicating the iron oxide core of ferritin was preserved.
Figure 4.

TEM and STXM analysis of an Aβ/ferritin aggregate formed following 48 hours of co-incubation. (a-c) TEM images of the Aβ/ferritin aggregate. Uniformly distributed electron dense fine particles can be seen in the highest magnification image in (c). STXM speciation dependent maps (d-f) and an iron L2,3-edge X-ray absorption spectrum (g) from the aggregate. (d) Carbon K-edge protein map. (e) Iron L3-edge map. (f) Composite image displaying protein (green) and iron (red) content of the aggregate. (g) Iron L2,3-edge X-ray absorption spectrum from the iron content displayed in (e). The solid line for the spectrum corresponds to the best fit curve created by superposition of suitably scaled iron reference X-ray absorption spectra. (h) Reference iron L2,3 edge X-ray absorption spectra for ferric (ferritin standard; blue), ferrous (FeCl2; red), magnetite (Fe3O4; green) and elemental (Fe0; magenta) iron phases. Grey dashed and dotted lines indicate the energy positions of the principal Fe2+ and Fe3+ absorption peaks respectively. See also Supplementary Fig. S3 for additional ferritin iron L2,3-edge X-ray absorption spectra.
Speciation-dependent contrast maps of the aggregate collected at the carbon K-absorption edge (Fig. 4d) in order to observe the protein distribution, and the iron L3-absorption edge (Fig. 4e), showed iron to be distributed throughout the entire aggregate structure, demonstrating the co-aggregation of Aβ and ferritin. A similar distribution of iron was observed within an Aβ/ferritin aggregate formed following 0.5 hours of incubation, as displayed in Supplementary Fig. S2.
To establish the precise chemical nature of the iron within the aggregate shown in Fig. 4, STXM examination was performed across the entire iron L2,3-absorption edge (700–740 eV). A non-linear least-squares fitting procedure was employed for the iron L2,3-edge X-ray absorption spectra collected in this study to provide the relative proportion of the iron phases contributing to each spectrum. Reference iron L2,3-edge absorption spectra for iron in various oxidation states are provided in Fig. 4h. As differing ferric phases can provide subtly distinct L2,3-edge X-ray absorption spectra, thereby affecting fitting, a ferritin standard was used to fit the ferric component of the experimental iron L2,3-edge spectra to best represent the starting iron material.
Iron references were scaled for fitting as shown in our previous work (Everett et al.58), to enable accurate characterisation of the iron X-ray absorption spectra recorded here. The fit of the spectrum shown in Fig. 4g showed the iron content of the aggregate to be in an entirely ferric state, indistinguishable from ferritin standards (see Supplementary Fig. S3), indicating the preservation of ferritin’s ferric iron oxide core following 48 hours of incubation with Aβ.
Ferric materials provide an iron L2,3-edge absorption spectrum comprised of a dominant peak feature at 709.5 eV and a low intensity shoulder at 708 eV at the L3-absorption edge, and two further low intensity L2-absorption edge peaks at 721 and 723 eV, as shown in the reference spectrum in Fig. 4h (blue spectrum). The low-energy Fe3+ X-ray absorption feature at 708 eV occurs at the same energy as the principal iron L3-edge absorption feature for ferrous (Fe2+) and zero-oxidation-state (Fe0) materials. Examples of FeCl2 (red spectrum) and Fe0 (magenta spectrum) are shown in Fig. 4h. Therefore, increases in the Fe2+ and Fe0 content of an initially purely ferric iron material will manifest as an increase in the intensity of this 708 eV feature in relation to the principal L3-edge Fe3+ absorption peak at 709.5 eV. Likewise, at the iron L2-absorption edge, increases in Fe2+ and Fe0 content result in an increase in the intensity of the X-ray absorption feature at 721 eV with respect to the feature at 723 eV.
TEM and STXM images of an Aβ/ferritin aggregate formed following the full incubation period (144 hours) are displayed in Fig. 5. TEM images of this aggregate revealed a largely amorphous structure lacking any mature amyloid fibril structure (Fig. 5a,b), yet containing regions of short spiked fibrils. In contrast to the earlier time points, no electron dense fine particles were observed. STXM speciation mapping at the carbon K-absorption edge revealed this aggregate to be protein dense, in keeping with Aβ/ferritin structures formed following 0.5 and 48 hours of Aβ/ferritin co-incubation. However, speciation mapping at the iron L3-absorption edge showed the iron content of this aggregate to be confined to a few discrete regions (Fig. 5d), the largest of which being approximately 200 nm in diameter, rather than spatially correlated to the protein distribution of the aggregate as found in the earlier time points.
Figure 5.

TEM and STXM analysis of an Aβ/ferritin aggregate formed following 144 hours of co-incubation. (a,b) TEM images of the Aβ/ferritin aggregate. STXM speciation dependent maps (c-e) and iron L2.3-edge X-ray absorption spectrum (f) from the aggregate. (c) Carbon K-edge protein map. (d) Iron L3-edge map. (e) Composite image displaying protein (green) and iron (red) content of the aggregate. (f) Iron L2.3-edge X-ray absorption spectra from the iron content displayed in (d) [red] and from a time-matched ferritin-only control [blue]. The solid lines for the spectrum corresponds to the best fit curve created by superposition of suitably scaled iron reference X-ray absorption spectra. See also Supplementary Fig. S4.
Iron L2,3-edge X-ray absorption spectra from an iron region and a time-matched ferritin control are shown in Fig. 5f. Examination of the ferritin control (Fig. 5f; blue) provided an X-ray absorption spectrum characteristic of a pure ferric phase, demonstrating that the oxidation state of iron in ferritin was maintained over the incubation period. In contrast, examination of an iron deposit within the Aβ/ferritin aggregate provided an X-ray absorption spectrum characteristic of a chemically-reduced iron phase (Fig. 5f; red). As can be seen by comparing with the Fe3O4 and Fe3+ standards shown in Fig. 4h (magnetite and ferritin respectively), enhanced magnetite features and diminished Fe3+ features were recorded across the iron L2,3-absorption edges. Fitting of the spectrum showed this area to be principally composed of magnetite (ca. 84%) with a minor contribution from Fe2+ (16%).
As no evidence of iron reduction was observed in either the ferrihydrite-like iron cores within ferritin alone, or in exposed nanoparticulate ferrihydrite time-matched controls incubated in identical buffer medium (blue trace in Fig. 5f and Supplementary Fig. S3C respectively), these alterations in iron chemistry appear to be due to the interaction of Aβ with ferritin.
Further TEM examination of sample material surrounding that shown in Fig. 5 provided evidence of a poorly fibrillar aggregate structure, similar to that shown in Fig. 4, demonstrating a heterogeneity in Aβ/ferritin aggregate morphology at this incubation point (Supplementary Fig. S4).
Aβ/ferritin aggregates act as a site of carbonate deposition and calcium sequestration
Following our recent related observations of both calcium and carbonate within ex vivo human AD amyloid plaque cores using X-ray spectromicroscopy58, the protein, carbonate, calcium and iron contents of a further series of Aβ/ferritin incubations were investigated using the I08 beamline at Diamond Light Source, operating in the STXM mode. In these experiments, measurements were performed at the calcium L-edge (340–360 eV) in addition to the carbon K-edge and iron L2,3-edge.
STXM speciation maps and X-ray absorption spectra from Aβ/ferritin aggregates formed following a longer period of 240 hours of co-incubation are shown in Fig. 6. Protein speciation maps taken at 288.3 eV showed this region to contain both fibrillar-like and amorphous protein aggregates (Fig. 6a). The carbonate content of these aggregates was determined by performing speciation mapping at 290.5 eV; the energy corresponding to the absorption feature for carbonate at the carbon K-edge (see e.g. Figure 6i). Carbonate speciation mapping revealed that both fibrillar and amorphous aggregates contained carbonates, although the density of carbonate material was much higher in the amorphous structure (Fig. 6b).
Figure 6.

STXM analysis of an Aβ/ferritin aggregate formed following 240 hours of co-incubation. (a) Carbon K-edge protein map showing fibrillar (Fib) and amorphous (Am) protein structures. (b) Carbon K-edge carbonate map. (c) Calcium L-edge map. (d) Iron L3-edge map. (e) Iron oxidation state difference map showing regions containing predominately ferric (Fe3+) iron as the bright regions, and those containing predominately ferrous and/or zero-oxidation-state (Fe2+/Fe0) iron as the dark regions. (f) Composite image displaying protein (green), carbonate (light blue), calcium (blue) and iron (red) content of the aggregate. High-resolution STXM speciation (g) carbon K-edge protein map and (h) carbon K-edge carbonate map. (i) Carbon K-edge X-ray absorption spectra from the areas identified in the protein map (g). (j) High-resolution iron L3-edge map. (k,l). Iron L3-edge maps of the areas highlighted in (j). (m) Iron L2.3-edge X-ray absorption spectra from the areas labelled in (k,l). The solid lines for the spectra correspond to the best fit curves created by superposition of suitably scaled iron reference X-ray absorption spectra. See Supplementary Fig. S2 Panel 2 for additional Aβ/ferritin structures from this time point.
To further investigate the origin of this carbonate material, speciation maps were performed at the calcium L2-absorption edge (352.6 eV). Calcium was found to be present in both fibrillar and amorphous aggregates with a higher density again being present in the amorphous structure (Fig. 6c). Furthermore, co-incident calcium and carbonate deposition was observed, suggesting at least some of the calcium present to be composed of calcium carbonate. However, the calcium distribution also extended beyond the carbonated areas, indicating additional calcium phases to be present.
Iron L3-absorption edge mapping (Fig. 6d) revealed the iron content of the fibrillar aggregate to be spread throughout the aggregate structure, closely following the protein morphology as was seen earlier in Fig. 4. Conversely, the iron distribution within the amorphous aggregate was found to be concentrated into dense sub-micron foci, similar to that observed in the aggregate displayed in Fig. 5, where iron was found to be in a chemically-reduced state.
To further assess the oxidation state of the iron found within these aggregate structures, an iron oxidation state contrast map was created by subtracting the STXM image taken at 708 eV (the energy of the principal Fe2+ and Fe0 iron L2,3-edge absorption peak) from the image taken at 709.5 eV (the principal Fe3+ peak). The resulting contrast map shows Fe3+ as regions of light contrast, and Fe2+ or Fe0 as areas of dark contrast (Fig. 6e). From this oxidation difference map, it is apparent that the iron associated with the fibrillar aggregate structure was in a predominantly ferric state, whereas the dense particulate iron deposits within the amorphous protein structure were predominantly ferrous or zero-oxidation-state.
To investigate this amorphous aggregate in more detail, high resolution images of the region highlighted by the yellow box in Fig. 6e were taken across the carbon K-edge and the iron L2,3-edge (Fig. 6g–h,j–l). The resulting carbon K-edge X-ray absorption spectra from region I1 (light blue trace, Fig. 6i) highlighted in Fig. 6g,h, showed the characteristic aromatic absorption peak at 285 eV and 1s-to-π* amide peak at 288.3 eV, confirming this amorphous aggregate to be composed of protein. Additionally, a carbonate absorption peak at 290.5 eV was observed in the X-ray absorption spectrum from region I2 (red trace, Fig. 6i), identified in the carbonate speciation map shown in Fig. 6h.
High resolution iron L3-edge speciation maps of this amorphous protein region revealed multiple small iron spots 200–500 nm in size (Fig. 6j–l). The iron L2,3-edge X-ray absorption spectra for these iron deposits are shown in Fig. 6m. From these spectra it appeared that the most dense iron region (labelled M1 in Fig. 6k) was composed of a strongly reduced form of iron. Fitting for this region indicated zero-oxidation-state iron (Fe0; see Fig. 4h, magenta for reference spectrum) to be the predominant phase (ca. 90%) with minor contributions from Fe3+ and Fe3O4. Although 708 eV corresponds to the principal iron L3-edge absorption feature for both Fe2+ and Fe0, the two phases are distinguishable by the broader line-shape for the Fe0 spectrum which lacks the splitting seen for oxide spectra, and the more prominent L2 peak and post-L2 edge absorption intensity for Fe0 (see Fig. 4h).
Interestingly the high resolution iron L3-edge speciation maps displayed in Fig. 6k,l showed further regions of iron deposition beyond the region identified in the lower resolution iron difference maps (Fig. 6d,e). Iron L2,3-edge absorption spectra from these further regions (labelled M2, M3 and M4) were also consistent with a heavily reduced iron phase. Fitting of these spectra showed all of these regions to be primarily composed of Fe0, with minor contributions from Fe3O4, Fe3+ and Fe2+. This confirmed that low-oxidation-state iron material was diffusely spread throughout the aggregate, as opposed to punctate sources which could conceivably be artefacts arising from external nanoparticulate sources. Further examples of Aβ/ferritin aggregates formed from this Aβ/ferritin series following 240 hours of incubation are displayed in the Supplementary Fig. S2 (Panel 2). As was the case in Fig. 6, diffuse iron associated with these aggregates was found to be entirely ferric, whereas dense sub-micron iron deposits were found to be chemically reduced.
Discussion
From the data presented here, it is apparent that Aβ(1-42) interacts with ferritin in a manner that leads to the conversion of ferritin-encapsulated ferric iron into nanoscale deposits of chemically reduced iron. These findings implicate an Aβ/ferritin interaction in the formation of the nanoscale ferrous-rich and zero-oxidation-state iron minerals previously observed in tissue from AD cases48–50, transgenic AD mouse tissues57, and isolated AD pathological structures47,58. In addition, Aβ/ferritin aggregates were shown to sequester calcium and act as a site of carbonate deposition, resembling such mineralised deposits found in AD amyloid plaque core material58.
Electron microscopy, time-lapse imaging and X-ray spectromicroscopy (STXM) demonstrated that co-incubation of Aβ(1-42) with ferritin, results in the accumulation of ferritin within Aβ-aggregate structures. Ferritin was found to be widely spread throughout amyloid aggregates over the first 48 hours of co-incubation, with X-ray spectromicroscopy speciation mapping showing that the iron content closely follows peptide morphology, a finding in keeping with our previous in vitro examination of Aβ/ferrihydrite interaction (see Everett et al.59). The inclusion of ferritin within amyloid aggregates resulted in poorly defined peptide structures when compared to Aβ incubated in isolation. This dependence of Aβ fibril morphology on the presence/absence of ferritin may arise through interactions between the two species (potentially influencing the formation of secondary and tertiary Aβ structures), and/or through localized variations in peptide concentration arising from the rapid co-aggregation of Aβ and ferritin as shown in Fig. 1. The ability of Aβ to accumulate and incorporate ferritin into its structure as found here, also provides an explanation for observations made by Grundke-Iqbal et al. where ferritin was observed to be co-localised with senile plaque material in AD tissues55.
Prolonged incubation of Aβ(1-42) with ferritin under sterile conditions over longer time periods of 144–240 hours led to the observation of both poorly defined fibril structures (as observed over the first 48 hours of incubation) and amorphous aggregate structures. TEM imaging of these amorphous structures showed a lack of amyloid fibril morphology, coupled with an absence of the electron-dense particulate material that would indicate intact ferritin. This is consistent with either disruption of the ferritin structure and/or the removal of the iron oxide core from the ferritin cage (the ferritin iron oxide core provides the strong TEM contrast in unstained images). As mature amyloid fibril structures were observed when Aβ was incubated in the absence of ferritin, and electron-dense cores indicating intact ferritin were maintained when ferritin was incubated in isolation, these alterations to amyloid structure appear to be a direct result of the Aβ-ferritin interaction.
STXM speciation mapping showed that the iron distribution within aggregate structures formed following 144–240 hours of incubation varied dramatically, and depended on amyloid morphology. Where aggregates were composed of poorly defined fibrils, iron was diffusely spread throughout the aggregate, closely following protein distribution. Conversely, where aggregates were present in an entirely amorphous state, iron was no-longer diffusely spread throughout the aggregate, instead being localised into dense foci. Examination of aggregate structures across the entire iron L2,3-absorption edge demonstrated the oxidation state of ferritin-derived iron to also be dependent upon both amyloid and iron morphology. Where the amyloid and iron content was “fibrillar” in nature, iron was found to remain in a ferric state. However, in amorphous amyloid aggregates where iron was localised in dense deposits hundreds of nanometers in diameter, iron was in a chemically-reduced state. Thus, through the use of STXM we provide the first descriptions of intra-sample variations in amyloid morphology dependent upon iron oxidation state, heterogeneity which is undetectable with standard bulk sample measurements of fibril formation.
This association between amyloid aggregate morphology and iron oxidation state was reproducibly observed across multiple, independent, Aβ/ferritin incubation series, examined at different X-ray spectromicroscopy beamlines, and using different starting batches of Aβ. Fitting of the iron L2,3-edge absorption spectra from these chemically-reduced iron regions showed the presence of a magnetite-like ferrous-rich material and an additional further-reduced iron phase with absorption features consistent with zero-oxidation-state iron. These Fe0 phases are analogous to those we reported in our ex vivo examination of amyloid plaque material extracted from AD grey matter, suggesting a similar phase to be present58. As no evidence of reduced iron was observed where ferritin or ferrihydrite was incubated in the absence of Aβ, the creation of a reducing environment and changes in iron chemistry appear to be driven by the co-aggregation of Aβ and ferritin. The absence of detectable low-oxidation-state iron in the ferritin controls also demonstrates that the chemically-reduced iron observed within the Aβ/ferritin aggregates is unlikely to be from iron bound to the external surface of ferritin, where surface iron can arise as an artefact of ferritin purification.
The identification of nanoscale deposits of chemically-reduced iron further demonstrates the necessity for chemically-sensitive nanoscale resolution microscopy when examining the chemistry of Aβ/iron interactions. These deposits would not have been detected using bulk measurements or microfocus microscopy, where the signal from the reduced iron phases would have been lost in the prevailing signal arising from oxidized iron.
The ability of Aβ to influence the chemical composition of ferritin’s ferrihydrite core, resulting in the formation of a chemically-reduced iron phase, is entirely consistent with our previous X-ray based in vitro experiments where Aβ(1-42) was shown to induce the chemical reduction of ferric oxyhydroxide and ferrihydrite into a pure ferrous phase59,60. Our previous experiments were conducted using iron oxide phases directly exposed to Aβ. It was not known if ferritin-encapsulated ferric iron oxide cores could be affected, although our original study of iron oxide nanoparticles in extracted amyloid plaque cores pointed to ferritin as a potential source of the iron due to the size distribution of the measured particles47. In the present work, for the first time, Aβ(1-42) was shown to indeed be capable of influencing the redox chemistry of iron originating within the ferritin protein. Our previous and present findings parallel a recent in vitro study by Balejcikova et al. who utilised spectrophotometry to record a moderate increase in Fe2+ iron content when a different peptide fragment, Aβ(1-40) was incubated with ferritin, compared to where ferritin was incubated in isolation61.
What remains unclear is whether these changes to ferritin iron oxidation state occur whilst iron remains within the ferritin cage (i.e. electron transfer across the apoferritin protein as described in Watt et al.)74, or whether iron must first be extracted from the ferritin cage before reduction can take place. In either scenario, these changes would appear to be dependent on Aβ amyloid interaction with ferritin. Time-matched control experiments conducted in the absence of Aβ showed no evidence of iron leaching from the ferritin-cage, or of chemically-reduced iron (i.e. only ferric iron was detected).
In vitro evidence shows the reduction of ferritin-encapsulated iron to a ferrous state to be an effective method for the removal of iron from the ferritin cage16. If the Aβ/ferritin interaction creates an environment in which iron is chemically-reduced via electron transfer across the ferritin cage, iron may be exuded from ferritin, leading to an increased labile iron pool. Iron located in channels near the surface of the ferritin protein cage, critically involved in the transfer of iron between transferrin and ferritin, may represent a physiologically relevant iron source, susceptible to Aβ-mediated reduction75,76. Indeed, this iron has been shown to be rapidly extracted from ferritin by the iron chelator desferrioxamine75,76. This highlights the clinical importance of determining if ferritin protein quantified as a measure of iron status has retained its iron or whether it is effectively apoferritin77. In this context, quantifying apoferritin:ferritin levels (for example in the CSF) might have potential as a marker for amyloid/ferritin interaction in vivo. Additionally, the low level of free-iron we recorded in the starting ferritin suspensions may have acted for a seed for Aβ-mediated iron reduction, in a manner similar to our previous studies59,60. This scenario is certainly possible in vivo, should the ferritin protein cage be compromised, thereby exposing the ferritin iron core.
Under normal conditions in vivo, where iron homeostasis is well-managed, intracellular ferritin expression depends on levels of intracellular labile iron. If Aβ has the capacity to disrupt ferritin storage resulting in the chemical reduction and release of iron, it is conceivable that this could compromise intracellular iron metabolism to the extent that antioxidant defences are overwhelmed. The observation of chemically-reduced iron as dense iron foci is also consistent with the iron nucleation processes observed during the chemical synthesis of mixed valence78 and zero-oxidation-state iron nanoparticulates from unbound ferric iron precursors79.
Whilst these findings clearly show Aβ interaction with ferritin results in the chemical reduction of ferritin-derived iron, the reductant itself is still to be confirmed. Aβ may act as a reducing agent for iron in a manner akin to that originally proposed by Huang et al.10, with this possibility currently under further investigation. Alternatively, the co-aggregation of Aβ with ferritin could result in the formation of highly-localised compartments in which reactants capable of influencing ferritin-iron chemistry can accumulate. A possible example for this scenario is the blocking of ferritin’s hydrophilic channels by Aβ, preventing the escape of reactants such as superoxide radicals (formed at the ferritin ferroxidase centres), resulting in the chemical reduction of ferritin-iron. Indeed, the chemical reduction and subsequent release of ferritin iron via a superoxide-dependent mechanism has been demonstrated in vitro80,81.
These results suggest that not only labile iron pools but also ferritin-encapsulated iron may act as a source of chemically-reduced forms of iron in AD tissue. This may account for the increased levels of low-oxidation-state iron derived from ferritin isolated from AD tissues50, the ferritin-core sized magnetite-like deposits previously identified within amyloid plaque material using HR-TEM47, and the magnetite particles of proposed biogenic origin recorded in human brain tissues through isothermal resonance magnetisation experiments82. This mechanism is far from implausible, as shown through the artificial production of chemically-reduced iron nanoparticles within the apoferritin cage, by first removing the natural ferritin core by iron reduction, with the subsequent use of iron Fenton chemistry to biomineralise the new magnetite nanocrystals83.
Ferritin is abundant throughout the human brain1,12, and has been observed to accumulate in localised regions of Aβ deposition55,84, whilst microglia, known to secrete the ferritin protein85, have also been associated with senile plaques in AD tissues55 providing potential routes for ferritin to interact with Aβ structures in vivo. Further evidence of ferritin accumulation in regions of Aβ pathology has recently been shown in 5XFAD transgenic mouse hippocampal tissue, where antibodies specific for ferritin were found to accumulate in brain regions with both increased ferritin expression and a high loading of Aβ plaques86.
Interactions between aggregating Aβ and ferritin may represent significant sources of localised oxidative stress in AD tissue, potentially contributing to the neurodegeneration associated with AD8,9,62–64. It is noteworthy that others have previously observed associations between ferritin and aberrant tau filament structures as observed in AD and progressive supranuclear palsy patients, and that disruption to the function of ferritin can be detrimental to the many iron-dependent functions in the brain, in extreme cases (such as neuroferritinopathy) proving fatal84,87–92.
The association of largely amorphous amyloid structures with the occurrence of chemically-reduced iron deposits several hundred nanometers in diameter as observed here, was also reported in our X-ray spectromicroscopy examination of cortical tissue from APP/PS1 mice57, and isolated amyloid plaque material from AD subjects58; suggesting that Aβ/ferritin interaction may have acted as source for the formation of chemically-reduced iron in these instances. The identification of differing amyloid/ferritin aggregate subtypes ([i] fibrillar amyloid aggregates containing diffuse ferric iron, [ii] amorphous amyloid aggregates containing dense chemically-reduced iron), indicates that the chemical state of amyloid-associated iron may reflect the type of amyloid aggregate that has formed (Table 1).
Table 1.
Summary of the differing aggregate types observed in this study. Identified iron phases and the presence of calcium and carbonate loading are shown for the cases where such measurements were obtained. “—” denotes not measured.
| Sample Type | Incubation Time (hrs) | Aβ morphology | Iron Distribution | Iron oxidation state | Calcium Loading | Carbonate Loading |
|---|---|---|---|---|---|---|
| Aβ(1-42) | All time points | Fibrillar | N/A | N/A | — | None |
| Ferritin | All time points | N/A | N/A | Ferric (Fe3+) | — | None |
| Aβ(1-42) +Ferritin | 0.5 | Fibrillar | Uniform throughout aggregate | Ferric (Fe3+) | — | — |
| 48 | Fibrillar | Uniform throughout aggregate | Ferric (Fe3+) | — | — | |
| 144 | Fibrillar | Uniform throughout aggregate | — | — | — | |
| Amorphous | Localised deposits (sub micron) | Mixed Valence (Fe2+/Fe3+) | — | — | ||
| 240 | Fibrillar | Uniform throughout aggregate | Ferric (Fe3+) | Low | Low | |
| Amorphous | Localised deposits (sub micron) | Mixed Valence (Fe2+/Fe3+)/Zero Valence (Fe0) | High | High |
In addition to alterations in native iron chemistry, STXM examination of Aβ/ferritin aggregates formed following 144–240 hours of interaction at the carbon K-edge revealed the presence of additional carbon containing species. Regions of carbonate were located in Aβ/ferritin structures demonstrating carbonate to have been deposited during the co-aggregation of Aβ and ferritin, with protein N-terminal carbonates or dissolved CO2 as a potential source. As calcium was present in the buffer, examination of the amorphous Aβ/aggregate structure formed following 240 hours of incubation (shown to contain low-oxidation-state iron) was also performed at the calcium L-edge. Calcium L-edge speciation mapping revealed co-localised carbonate and calcium deposition, suggesting carbonate material to be comprised of calcium carbonate. Calcium carbonate formation therefore appears to be linked to Aβ/ferritin interaction.
Remarkably, this pattern of calcium mineralisation, carbonate loading, and nanoscale regions of low-oxidation-state iron formation within Aβ/ferritin aggregates structures is entirely consistent with that observed in amyloid plaque core material extracted from individuals with confirmed AD, as recently reported by our group58. These striking similarities are displayed in the comparison Fig. S5 of the Supplementary Information. This suggests that amyloid plaques may be formed through a similar process of Aβ/ferritin interaction in the presence of calcium in vivo. In particular, the characteristic features of dense amorphous peptide aggregates containing deposits of Fe2+, magnetite or Fe0, were replicated in the ex-vivo plaque core material.
In conclusion, the data presented here demonstrate that the interaction of Aβ with ferritin in a physiologically relevant buffer medium results in the formation of dense amorphous amyloid structures harbouring chemically-reduced iron. This points towards Aβ/ferritin interaction as a probable source of the increased levels of mixed oxidation state Fe2+ and Fe0 phases previously observed within brain tissue from the APP/PS1 model of AD, in human AD tissue, and in isolated amyloid plaque cores47–49,57,58,93. Furthermore, the chemical composition of the in vitro aggregates formed in this study was shown to be analogous to primary components of the amyloid plaque core material extracted from the grey matter of AD subjects58, consistent with Aβ/ferritin interaction playing a prominent role in the development of iron-bearing amyloid deposits in vivo.
Given the abundance of ferritin throughout the human brain1, the capacity for Aβ to create an environment in which ferritin iron is chemically reduced, could lead to the sustained production of excess ROS, and therefore to oxidative stress in AD tissues. With oxidative stress being a key characteristic of AD pathology62,63,65,94, this interaction could represent a target for therapies intended to lower oxidative burdens and delay disease progression, either through targeting Aβ-induced low-oxidation-state iron phases, or by preventing Aβ from interacting with ferritin and so averting the formation of chemically-reduced iron. Moreover, as specific iron and calcium phases provide marked and quantifiable impact on MRI contrast, it is possible that forms of iron and calcium preferentially associated with amyloid deposits could be used as endogenous markers to facilitate screening for amyloid deposition in at-risk populations, before clinical manifestations arise.
Methods
The Interaction of Aβ and Ferritin
Preparation of Aβ/ferritin Suspensions
Horse spleen ferritin (Type I; 125 mg/mL; 1% saline solution) was purchased from Sigma Aldrich and stored at 4 °C until time of use. Characterisation of ferritin size, crystal structure, oxidation state and iron content are provided in the Supplementary Information (Figs. S1, S3 and S6). Ferritin was diluted in a modified Krebs-Henseleit (KH) buffer (pH 7.4; 100 mM PIPES (piperazine-N,N′-bis(2-ethanesulfonic acid), 118.5 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.4 mM CaCl2, 11 mM glucose) to achieve a 0.7 mg/mL (440 µM iron) ferritin suspension. KH buffer was made on the day of use, and was filtered (0.2 µm pore size) in sterile conditions prior to use, to inhibit bacterial growth. This buffer medium, modelled on the cerebral spinal fluid which bathes the brain, contains physiologically relevant concentrations of Ca2+ and Mg2+, both of which have been shown to influence amyloid peptide aggregation dynamics95,96. The buffer PIPES was chosen as it does not strongly interact with metal ions97.
Synthetic monomeric Aβ(1-42) (Bachem) was thawed and dissolved in 1 mM NaOH (Sigma Aldrich) to create a 1 mg/mL (220 μM) stock. Aβ stock was allowed to sit at room temperature for 30 minutes to ensure complete peptide dissolution before being added to 0.7 mg/mL ferritin suspensions in KH buffer under sterile conditions. Final ferritin and Aβ concentrations were 0.6 mg/mL (1.3 µM, 370 μM iron content) and 0.16 mg/mL (35 μM) respectively. A ferritin concentration of 0.6 mg/mL was used as this value corresponded to the iron concentration used in our previous in vitro experiments examining Aβ interaction with ferric iron59,60. This ferritin concentration ensures sufficient iron is present to enable detection of iron using X-ray spectromicroscopy when employing the sampling technique described below. Aβ-free ferritin controls were created as above, with the substitution of 1 mM NaOH for Aβ. 35 μM Aβ solutions were prepared in KH buffer as an amyloid reference. 370 µM 6-line ferrihydrite suspensions were prepared in KH buffer as an unencapsulated iron reference. Ferrihydrite was synthesised as described in Schwertmann et al.98.
Incubation of suspensions
All suspensions/solutions were incubated under aerobic conditions at 37 °C within sealed microcentrifuge tubes until the time of sampling. For time lapse imaging a camera was mounted inside the incubator, allowing images of Aβ/ferritin suspensions to be taken in situ without physical manipulation of the sample tubes. Time lapse images demonstrating the precipitation of ferritin by Aβ were created using Lapse it™ time lapse software. Images of microcentrifuge tubes containing Aβ/ferritin and a ferritin only control were taken over a period of 18 hours.
The incubation of ferritin suspensions in KH buffer did not result in the leaching of ferritin iron, as demonstrated through iron quantification measurements performed over a 240 hour incubation period (see Supplementary Information).
Scanning Transmission X-ray Microscopy Sample Preparation
15 μL of Aβ/ferritin suspensions were deposited onto either silicon oxide membranes (DuneSciences; 75 nm thickness) or silicon nitride membranes (Agar Scientific; 75 nm thickness) and excess liquid was removed using filter paper. For the Aβ/ferritin series examined on the PolLux beamline at the Swiss Light Source sampling was performed following 0.5, 48 and 144 hours of incubation. An additional Aβ/ferritin series was examined using the I08 beamline at Diamond Light Source, with sampling being performed following 240 hours of incubation. Additional Aβ and ferritin control samples were examined to provide reference carbon K-edge and iron L-edge reference X-ray absorption spectra.
As STXM was the primary technique employed to assess iron oxidation state following Aβ/ferritin incubation, conscientious effort was taken to maintain anoxic conditions during sampling, transfer and examination of Aβ and ferritin materials. All STXM samples were prepared within a nitrogen filled glove bag (Glas-col), and membranes containing Aβ/ferritin structures were stored within a nitrogen filled O-ring sealed jar (Oxoid) to prevent changes in iron oxidation states. Membranes were mounted onto STXM sample holders for X-ray spectromicroscopy. Sample mounting was conducted within a nitrogen filled glove bag. Anoxic conditions were maintained during sample transportation by using a nitrogen filled vessel for sample transfer and purging of the STXM endstation with nitrogen prior to sample loading.
Electron microscopy sample preparation
Small volumes of Aβ/ferritin suspensions were deposited onto carbon/formvar coated copper TEM grids and excess liquid removed. Samples were taken following 0.5, 48, 96 and 144 hours of incubation.
X-ray spectromicroscopy: scanning transmission X-ray microscopy
X-ray spectromicroscopy was performed at the Swiss Light Source on the PolLux beamline using the STXM endstation, and at the Diamond Light Source on beamline I08. Focussed X-ray spot size was approximately 20 nm on the PolLux beamline and 50 nm on I08. Energy-specific images of sample material were created by raster scanning the sample across a focussed X-ray beam and recording the intensity of the transmitted X-rays. To minimise potential photoreduction effects caused by the incident X-ray beam thereby preserving native sample chemistry, scanning (exposure) times were kept to a minimum (typically 1–2 ms per point).
To create contrast maps displaying the chemical speciation of a given sample material, paired images were taken at the energy of a feature of interest (e.g. the main ferric (Fe3+) iron peak in the Fe L3-edge absorption region) and a few eV below this feature. The “off-peak” images were then subtracted from the “on-peak” images revealing the chemical speciation for the examined sample region.
X-ray absorption spectra were collected by taking a series of images (called a stack) of a region of interest at multiple energies across a desired energy range (e.g. the iron L2,3-absorption edge [700–740 eV], or the carbon K-absorption edge [280–310 eV]). Transmitted X-ray intensities for the stack images were converted to optical density using background regions that did not contain any sample material, thereby removing background X-ray absorption features attributable to the beamline. This method of spectromicroscopy allows an X-ray absorption spectrum to be created from every pixel of a stack image, thus allowing spectral information to be realised from highly localised regions of interest.
Carbon K-edge X-ray spectromicroscopy was performed prior to higher energy iron L2,3-edge examination as to minimize X-ray beam induced damage to carbon structures. Only a sub-set of Aβ/ferritin structures were examined using STXM due to experimental time constraints.
X-ray spectromicroscopy data were processed using the aXis 2000 software package (http://unicorn.mcmaster.ca/aXis2000. html). The brightness and contrast of X-ray microscopy images were adjusted using ImageJ software. Grey scale X-ray microscopy images were converted to false colour before being recombined as overlays to create coloured composite images.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed using a JEOL 1230 microscope operating at 100 kV. A low operating energy was used to minimise electron beam damage of sample materials. No positive or negative stains were used during TEM microscopy. Where both techniques were employed, TEM was performed following STXM examination to prevent electron beam induced changes to sample chemistry.
Quantification and statistical analysis
Analysis of X-ray Absorption Spectra
To estimate the relative proportion of iron phases contributing to the iron L2,3-edge X-ray absorption spectra measured in these experiments, iron L2,3-edge X-ray absorption spectra were fitted to reference X-ray absorption spectra from Fe3+, Fe2+, Fe3O4 and Fe0 standards using non-linear least squares fitting procedures. Accurate scaling of these standards was required to provide precise quantitative determination of the phase proportions contributing to the experimental data. The scaling factors were determined by normalising the X-ray absorption intensity for each reference iron phase to the integrated intensity over the iron L2,3 absorption edges, as described in our previous work (see Everett et al.58). An iron L2,3-edge X-ray absorption spectrum from a ferritin standard was used for fitting the Fe3+ contribution to the experimental iron L2,3-edge X-ray absorption spectra. The absorption features from this ferritin spectrum are consistent with a pure ferric phase, and it was therefore chosen as a suitable ferric (Fe3+) standard for fitting. The Fe0 reference spectrum used for fitting was obtained from Fe0 film standards prepared and measured under vacuum to prevent oxidation99.
Supplementary information
Acknowledgements
This work was supported by the Diamond Light Source, EPSRC doctoral training grant EP/P503981, EPSRC project grants EP/N033191/1 and EP/N033140/1, and University of Warwick alumni donations (JE). Measurements were performed on the PolLux beamline at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland, and we thank Dr. Joerg Raabe and Dr. Benjamn Watts for their support. Measurements were also performed on beamlines I08 and I10 at Diamond Light Source, and we are grateful for the support provided by Dr. Burkhard Kaulich, Dr. Tohru Araki, Dr. Majid Abyaneh and Dr. Paul Steadman. We thank Karen Walker (Keele University) for TEM support.
Author contributions
J.E. wrote the first draft and J.B., F.L., P.O.C., P.J.S., J.D., J.F.C. and N.D.T. read and commented on the manuscript prior to submission. J.E. and N.D.T. contributed equally to the experimental design, sample preparation and measurement, J.E., N.D.T. and J.F.C. contributed equally to interpretation of the data. J.B. and F.L. contributed to sample measurement.
Data availability
The datasets generated during and/or analysed during the current study are available through the Keele Research Repository at https://eprints.keele.ac.uk.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-67117-z.
References
- 1.Connor JR, Menzies SL, Burdo JR, Boyer PJ. Iron and iron management proteins in neurobiology. Pediatric Neurology. 2001;25:118–129. doi: 10.1016/s0887-8994(01)00303-4. [DOI] [PubMed] [Google Scholar]
- 2.Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nature Reviews Neuroscience. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 3.Munoz P, Humeres A. Iron deficiency on neuronal function. Biometals. 2012;25:825–835. doi: 10.1007/s10534-012-9550-x. [DOI] [PubMed] [Google Scholar]
- 4.Ke Y, Qian ZM. Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurology. 2003;2:246–253. doi: 10.1016/s1474-4422(03)00353-3. [DOI] [PubMed] [Google Scholar]
- 5.Bolt HM, Marchan R. Iron dysregulation: an important aspect in toxicology. Archives of Toxicology. 2010;84:823–824. doi: 10.1007/s00204-010-0610-0. [DOI] [PubMed] [Google Scholar]
- 6.Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. Bmc Medical Genomics. 2009;2:2. doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kell DB. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Archives of Toxicology. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honda K, Casadesus G, Petersen RB, Perry G, Smith MA. Oxidative stress and redox-active iron in Alzheimer’s disease. Redox-Active Metals in Neurological Disorders. 2004;1012:179–182. doi: 10.1196/annals.1306.015. [DOI] [PubMed] [Google Scholar]
- 9.Honda K, et al. Redox active iron at the center of oxidative stress in Alzheimer disease. Letters in Drug Design & Discovery. 2005;2:479–482. [Google Scholar]
- 10.Huang XD, Moir RD, Tanzi RE, Bush AI, Rogers JT. Redox-active metals, oxidative stress, and Alzheimer’s disease pathology. Redox-Active Metals in Neurological Disorders. 2004;1012:153–163. doi: 10.1196/annals.1306.012. [DOI] [PubMed] [Google Scholar]
- 11.Smith DG, Cappai R, Barnham KJ. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochimica Et Biophysica Acta-Biomembranes. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Chasteen ND, Harrison PM. Mineralization in ferritin: An efficient means of iron storage. Journal of Structural Biology. 1999;126:182–194. doi: 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- 13.Cowley JM, Janney DE, Gerkin RC, Buseck PR. The structure of ferritin cores determined by electron nanodiffraction. Journal of Structural Biology. 2000;131:210–216. doi: 10.1006/jsbi.2000.4292. [DOI] [PubMed] [Google Scholar]
- 14.Treffry A, Harrison PM, Cleton MI, de Bruijn WC, Mann S. A note on the composition and properties of ferritin iron cores. Journal of Inorganic Biochemistry. 1987;31:1–6. doi: 10.1016/0162-0134(87)85001-8. [DOI] [PubMed] [Google Scholar]
- 15.Yoon JH, et al. Oxidative modification of ferritin induced by hydrogen peroxide. Bmb Reports. 2011;44:165–169. doi: 10.5483/BMBRep.2011.44.3.165. [DOI] [PubMed] [Google Scholar]
- 16.Carmona F, et al. Ferritin iron uptake and release in the presence of metals and metalloproteins: Chemical implications in the brain. Coordination Chemistry Reviews. 2013;257:2752–2764. [Google Scholar]
- 17.Becerril-Ortega J, Bordji K, Fréret T, Rush T, Buisson A. Iron overload accelerates neuronal amyloid-β production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiology of Aging. 2014;35:2288–2301. doi: 10.1016/j.neurobiolaging.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 18.Goodman L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. The Journal of nervous and mental disease. 1953;118:97–130. [PubMed] [Google Scholar]
- 19.Ong WY, Farooqui AA. Iron, neuroinflammation, and Alzheimer’s disease. Journal of Alzheimers Disease. 2005;8:183–200. doi: 10.3233/jad-2005-8211. [DOI] [PubMed] [Google Scholar]
- 20.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. Journal of the Neurological Sciences. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 21.Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deibel MA, Ehmann WD, Markesbery WR. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. Journal of the Neurological Sciences. 1996;143:137–142. doi: 10.1016/s0022-510x(96)00203-1. [DOI] [PubMed] [Google Scholar]
- 23.Thompson PM, et al. Dynamics of gray matter loss in Alzheimer’s disease. Journal of Neuroscience. 2003;23:994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fox NC, et al. Imaging of onset and progression of Alzheimer’s disease with voxel-compression mapping of serial magnetic resonance images. Lancet. 2001;358:201–205. doi: 10.1016/S0140-6736(01)05408-3. [DOI] [PubMed] [Google Scholar]
- 25.Alzheimer’s disease facts and figures. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 12, 459-509, 10.1016/j.jalz.2016.03.001 (2016). [DOI] [PubMed]
- 26.Goedert M, Sisodia SS, Price DL. Neurofibrillary tangles and β-amyloid deposits in Alzheimer’s disease. Current Opinion in Neurobiology. 1991;1:441–447. doi: 10.1016/0959-4388(91)90067-h. [DOI] [PubMed] [Google Scholar]
- 27.Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proceedings of the National Academy of Sciences. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardy JA, Higgins GA. Alzheimer’s Disease - The Amyloid Cascade Hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 29.Rogers J, Morrison JH. Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer’s-disease. Journal of Neuroscience. 1985;5:2801–2808. doi: 10.1523/JNEUROSCI.05-10-02801.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiala JC. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathologica. 2007;114:551–571. doi: 10.1007/s00401-007-0284-8. [DOI] [PubMed] [Google Scholar]
- 31.Mucke, L. & Selkoe, D. J. Neurotoxicity of Amyloid beta-Protein: Synaptic and Network Dysfunction. Cold Spring Harbor Perspectives in Medicine. 2, 10.1101/cshperspect.a006338 (2012). [DOI] [PMC free article] [PubMed]
- 32.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends in Cell Biology. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 33.Hensley K, et al. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide – relevance to Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann T, et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nature Medicine. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 35.Sisodia SS, Price DL. Role of the beta-amyloid protein in Alzheimer’s disease. The FASEB Journal. 1995;9:366–370. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 36.Tay WM, Huang D, Rosenberry TL, Paravastu AK. The Alzheimer’s Amyloid-β(1–42) Peptide Forms Off-Pathway Oligomers and Fibrils That Are Distinguished Structurally by Intermolecular Organization. Journal of Molecular Biology. 2013;425:2494–2508. doi: 10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wildburger NC, et al. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Scientific Reports. 2017;7:9520. doi: 10.1038/s41598-017-10422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid-beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Castellani RJ, Perry G. The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol. 2014;88:671–676. doi: 10.1016/j.bcp.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 40.Butterfield DA, Swomley AM, Sultana R. Amyloid β-Peptide (1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxidants & Redox Signaling. 2013;19:823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardy J, Selkoe DJ. Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 42.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 43.Dobson J. Nanoscale biogenic iron oxides and neurodegenerative disease. Febs Letters. 2001;496:1–5. doi: 10.1016/s0014-5793(01)02386-9. [DOI] [PubMed] [Google Scholar]
- 44.Dobson J. Investigation of age-related variations in biogenic magnetite levels in the human hippocampus. Experimental Brain Research. 2002;144:122–126. doi: 10.1007/s00221-002-1066-0. [DOI] [PubMed] [Google Scholar]
- 45.Dobson J. Magnetic iron compounds in neurological disorders. Redox-Active Metals in Neurological Disorders. 2004;1012:183–192. doi: 10.1196/annals.1306.016. [DOI] [PubMed] [Google Scholar]
- 46.Collingwood J, Dobson J. Mapping and characterization of iron compounds in Alzheimer’s tissue. Journal of Alzheimers Disease. 2006;10:215–222. doi: 10.3233/jad-2006-102-308. [DOI] [PubMed] [Google Scholar]
- 47.Collingwood JF, et al. Three-dimensional tomographic Imaging and characterization of iron compounds within Alzheimer’s plaque core material. Journal of Alzheimers Disease. 2008;14:235–245. doi: 10.3233/jad-2008-14211. [DOI] [PubMed] [Google Scholar]
- 48.Hautot D, Pankhurst QA, Khan N, Dobson J. Preliminary evaluation of nanoscale biogenic magnetite in Alzheimer’s disease brain tissue. Proceedings of the Royal Society B-Biological Sciences. 2003;270:S62–S64. doi: 10.1098/rsbl.2003.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pankhurst Q, Hautot D, Khan N, Dobson J. Increased levels of magnetic iron compounds in Alzheimer’s Disease. Journal of Alzheimers Disease. 2008;13:49–52. doi: 10.3233/jad-2008-13105. [DOI] [PubMed] [Google Scholar]
- 50.Quintana C, Cowley JM, Marhic C. Electron nanodiffraction and high-resolution electron microscopy studies of the structure and composition of physiological and pathological ferritin. Journal of Structural Biology. 2004;147:166–178. doi: 10.1016/j.jsb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Quintana C, Gutierrez L. Could a dysfunction of ferritin be a determinant factor in the aetiology of some neurodegenerative diseases? Biochimica Et Biophysica Acta-General Subjects. 2010;1800:770–782. doi: 10.1016/j.bbagen.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 52.Khan A, Dobson JP, Exley C. Redox cycling of iron by A beta(42) Free Radical Biology and Medicine. 2006;40:557–569. doi: 10.1016/j.freeradbiomed.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Quintana C, et al. Morphological and chemical studies of pathological human and mice brain at the subcellular level: Correlation between light, electron, and nanosims microscopies. Microscopy Research and Technique. 2007;70:281–295. doi: 10.1002/jemt.20403. [DOI] [PubMed] [Google Scholar]
- 54.Gallagher JJ, et al. Modest Amyloid Deposition is Associated with Iron Dysregulation, Microglial Activation, and Oxidative Stress. Journal of Alzheimers Disease. 2012;28:147–161. doi: 10.3233/JAD-2011-110614. [DOI] [PubMed] [Google Scholar]
- 55.Grundkeiqbal I, et al. Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathologica. 1990;81:105–110. doi: 10.1007/BF00334497. [DOI] [PubMed] [Google Scholar]
- 56.Plascencia-Villa G, et al. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Scientific Reports. 2016;6:24873. doi: 10.1038/srep24873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Telling ND, et al. Iron Biochemistry is Correlated with Amyloid Plaque Morphology in an Established Mouse Model of Alzheimer’s Disease. Cell Chemical Biolog.y. 2017;24:1205–1215. doi: 10.1016/j.chembiol.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 58.Everett J, et al. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale. 2018;10:11782–11796. doi: 10.1039/c7nr06794a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Everett J, et al. Evidence of Redox-Active Iron Formation Following Aggregation of Ferrihydrite and the Alzheimer’s Disease Peptide beta-Amyloid. Inorganic Chemistry. 2014;53:2803–2809. doi: 10.1021/ic402406g. [DOI] [PubMed] [Google Scholar]
- 60.Everett J, et al. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1-42) Journal of the Royal Society Interface. 2014;11:20140165. doi: 10.1098/rsif.2014.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balejcikova, L., Siposova, K., Kopcansky, P. & Safarik, I. Fe(II) formation after interaction of the amyloid β-peptide with iron-storage protein ferritin. Journal of Biological Physics, 10.1007/s10867-018-9498-3 (2018). [DOI] [PMC free article] [PubMed]
- 62.Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer’s disease. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2000;1502:139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 63.Nunomura A, et al. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 64.Pratico D. Oxidative imbalance and lipid peroxidation in Alzheimer’s disease. Drug Development Research. 2002;56:446–451. [Google Scholar]
- 65.Cai ZY, Zhao B, Ratka A. Oxidative Stress and beta-Amyloid Protein in Alzheimer’s Disease. Neuromolecular Medicine. 2011;13:223–250. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- 66.Castellani RJ, et al. Contribution of redox-active iron and copper to oxidative damage in Alzheimer disease. Ageing Research Reviews. 2004;3:319–326. doi: 10.1016/j.arr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Castellani RJ, et al. Iron: The redox-active center of oxidative stress in Alzheimer disease. Neurochemical Research. 2007;32:1640–1645. doi: 10.1007/s11064-007-9360-7. [DOI] [PubMed] [Google Scholar]
- 68.Devos D, et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxidants & Redox Signaling. 2014;21:195–210. doi: 10.1089/ars.2013.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li X, Jankovic J, Le W. Iron chelation and neuroprotection in neurodegenerative diseases. J Neural Transm. 2011;118:473–477. doi: 10.1007/s00702-010-0518-0. [DOI] [PubMed] [Google Scholar]
- 70.Meadowcroft MD, Connor JR, Smith MB, Yang QX. MRI and Histological Analysis of Beta-Amyloid Plaques in Both Human Alzheimer’s Disease and APP/PS1 Transgenic Mice. Journal of Magnetic Resonance Imaging. 2009;29:997–1007. doi: 10.1002/jmri.21731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Serpell LC. Alzheimer’s amyloid fibrils: structure and assembly. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 72.Kaznacheyev K, et al. Innershell Absorption Spectroscopy of Amino Acids. The Journal of Physical Chemistry A. 2002;106:3153–3168. [Google Scholar]
- 73.Boese J, Osanna A, Jacobsen C, Kirz J. Carbon edge XANES spectroscopy of amino acids and peptides. Journal of Electron Spectroscopy and Related Phenomena. 1997;85:9–15. [Google Scholar]
- 74.Watt GD, Jacobs D, Frankel RB. Redox reactivity of bacterial and mammalian ferritin – is reductant entry into the ferritin interior a necessary step for iron release. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:7457–7461. doi: 10.1073/pnas.85.20.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harris DC. Iron exchange between ferritin and transferrin in vitro. Biochemistry. 1978;17:3071–3078. doi: 10.1021/bi00608a020. [DOI] [PubMed] [Google Scholar]
- 76.Harrison PM. The structure and function of ferritin. Biochemical Education. 1986;14:154–162. [Google Scholar]
- 77.Ayton S, et al. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nature Communications. 2015;6:6760. doi: 10.1038/ncomms7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baumgartner J, et al. Nucleation and growth of magnetite from solution. Nature Materials. 2013;12:310–314. doi: 10.1038/nmat3558. [DOI] [PubMed] [Google Scholar]
- 79.Jia H, Wang C. Comparative studies on montmorillonite-supported zero-valent iron nanoparticles produced by different methods: reactivity and stability. Environmental Technology. 2013;34:25–33. doi: 10.1080/09593330.2012.679698. [DOI] [PubMed] [Google Scholar]
- 80.Aliaga ME, Carrasco-Pozo C, López-Alarcón C, Olea-Azar C, Speisky H. Superoxide-dependent reduction of free Fe3+ and release of Fe2+ from ferritin by the physiologically-occurring Cu(I)–glutathione complex. Bioorganic & Medicinal Chemistry. 2011;19:534–541. doi: 10.1016/j.bmc.2010.10.064. [DOI] [PubMed] [Google Scholar]
- 81.Paul T. Effect of a Prolonged Superoxide Flux on Transferrin and Ferritin. Archives of Biochemistry and Biophysics. 2000;382:253–261. doi: 10.1006/abbi.2000.2027. [DOI] [PubMed] [Google Scholar]
- 82.Gilder SA, et al. Distribution of magnetic remanence carriers in the human brain. Scientific Reports. 2018;8:11363. doi: 10.1038/s41598-018-29766-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meldrum F, Heywood B, Mann S. Magnetoferritin: in vitro synthesis of a novel magnetic protein. Science. 1992;257:522–523. doi: 10.1126/science.1636086. [DOI] [PubMed] [Google Scholar]
- 84.Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MBH. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Gen Sect. 1990;2:327–340. doi: 10.1007/BF02252926. [DOI] [PubMed] [Google Scholar]
- 85.Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. 2006;54:795–804. doi: 10.1002/glia.20416. [DOI] [PubMed] [Google Scholar]
- 86.Fernández T, Martínez-Serrano A, Cussó L, Desco M, Ramos-Gómez M. Functionalization and Characterization of Magnetic Nanoparticles for the Detection of Ferritin Accumulation in Alzheimer’s Disease. ACS Chemical Neuroscience. 2018;9:912–924. doi: 10.1021/acschemneuro.7b00260. [DOI] [PubMed] [Google Scholar]
- 87.Curtis ARJ, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nature Genetics. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 88.Perez M, et al. Ferritin is associated with the aberrant tau filaments present in progressive supranuclear palsy. American Journal of Pathology. 1998;152:1531–1539. [PMC free article] [PubMed] [Google Scholar]
- 89.Connor JR, Snyder BS, Arosio P, Loeffler DA, Lewitt P. A Quantitative analysis of isoferritins in select regions of aged, Parkinsonian, and, Alzheimers diseased brains. Journal of Neurochemistry. 1995;65:717–724. doi: 10.1046/j.1471-4159.1995.65020717.x. [DOI] [PubMed] [Google Scholar]
- 90.Dixon SJ, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baraibar MA, Barbeito AG, Muhoberac BB, Vidal R. A mutant light-chain ferritin that causes neurodegeneration has enhanced propensity toward oxidative damage. Free Radical Biology and Medicine. 2012;52:1692–1697. doi: 10.1016/j.freeradbiomed.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Friedman A, Arosio P, Finazzi D, Koziorowski D, Galazka-Friedman J. Ferritin as an important player in neurodegeneration. Parkinsonism & Related Disorders. 2011;17:423–430. doi: 10.1016/j.parkreldis.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 93.Collingwood JF, et al. In situ characterization and mapping of iron compounds in Alzheimer’s disease tissue. Journal of Alzheimers Disease. 2005;7:267–272. doi: 10.3233/jad-2005-7401. [DOI] [PubMed] [Google Scholar]
- 94.Conrad CC, et al. Oxidized proteins in Alzheimer’s plasma. Biochemical and Biophysical Research Communications. 2000;275:678–681. doi: 10.1006/bbrc.2000.3356. [DOI] [PubMed] [Google Scholar]
- 95.Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of Amyloid β-Peptide Aggregation by Physiological Concentrations of Calcium. Journal of Biological Chemistry. 2006;281:27916–27923. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garai K, Sengupta P, Sahoo B, Maiti S. Selective destabilization of soluble amyloid β oligomers by divalent metal ions. Biochemical and Biophysical Research Communications. 2006;345:210–215. doi: 10.1016/j.bbrc.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 97.Benn MH, Rauk A, Swaddle TW. Measurement of the interaction of aqueous copper(II) with a model amyloid-β protein fragment — Interference from buffers. Canadian Journal of Chemistry. 2011;89:1429–1444. [Google Scholar]
- 98.Schwertmann, U. & Cornell, R.M. Ferrihydrite in Iron Oxides in the Laboratory: Preparation and Characterization 103-112 (Wiley-VCH, 2000).
- 99.Telling ND, Laan GVD, Georgieva MT, Farley NRS. Facility for combined in situ magnetron sputtering and soft x-ray magnetic circular dichroism. Review of Scientific Instruments. 2006;77:073903. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available through the Keele Research Repository at https://eprints.keele.ac.uk.