

Abstract
Familial adult myoclonic epilepsy 1 (FAME1), first recognised in Japanese families, was recently shown to be caused by a TTTCA repeat insertion in intron 4 of SAMD12 on chromosome 8. We performed whole genome sequencing on two families with FAME, one of Sri Lankan origin and the other of Indian origin, and identified a TTTCA repeat insertion in SAMD12 in both families. Haplotype analysis revealed that both families shared the same core ancestral haplotype reported in Japanese and Chinese families with FAME1. Mutation dating, based on the length of shared haplotypes, estimated the age of the ancestral haplotype to be ~670 generations, or 17,000 years old. Our data extend the geographic range of this repeat expansion to Southern Asia and potentially implicate an even broader regional distribution given the age of the variant. This finding suggests patients of Asian ancestry with suspected FAME should be screened for the SAMD12 TTTCA expansion.
Subject terms: Epilepsy, DNA sequencing, Haplotypes
Introduction
Familial adult myoclonic epilepsy (FAME) is an autosomal-dominant, adolescent–adult onset epilepsy characterised by a cortical myoclonic tremor, often with myoclonic and generalised tonic-clonic seizures presenting later in life [1]. This disorder is also known as benign adult familial myoclonus epilepsy (BAFME), familial cortical myoclonic tremor with epilepsy (FCMTE) and autosomal dominant cortical myoclonus and epilepsy (ADCME). FAME has been previously linked to loci at 8q24 (FAME1 [MIM: 601068]), 2p11-2q11 (FAME2 [MIM: 607876]), 5p15 (FAME3 [MIM: 613608]) and 3q26-3q28 (FAME4 [MIM: 615127]) [2–5].
FAME1 was recently identified to be caused by a TTTCA repeat insertion in intron 4 of SAMD12 [6]. The TTTCA repeat insertion is located immediately 3′ to a TTTTA repeat expansion and was found in 85 affected individuals from 49 families from Japan, suggesting a founder effect. The TTTTA short tandem repeat (STR) is present in the human reference genome and expansions of this motif were observed in 5.9% of controls [6]. The TTTCA repeat was not observed in any controls at this locus and is not present in the human reference genome [6]. Similar repeat expansions were also identified in one family each in two new genes: TNRC6A (FAME6 [MIM: 618074]) and RAPGEF2 (FAME7 [MIM: 618075]) [6]. The FAME1 SAMD12 intronic TTTTA repeat expansion and TTTCA repeat insertion were then confirmed in 23 Chinese families [7–9]. Haplotypes for five Chinese families were compared with three of the reported Japanese families, which found that a core haplotype containing the TTTCA repeat insertion was shared by all families, again suggesting a common ancestor [7].
Recently, several methods have been developed to identify repeat expansions in whole exome and whole genome sequencing data [10–14]. We applied these methods to search for TTTCA repeat insertions in two families with FAME, one of Sri Lankan origin and one of Indian origin. We found the same FAME1 repeat insertion and shared haplotype in these families as was reported in Japanese and Chinese families and estimated the age of the ancestral haplotype to be 17,000 years old.
Materials and methods
Subjects and whole genome sequencing
We studied two families with FAME. Family A is of Sri Lankan origin and Family B is of Indian origin (Fig. 1, phenotypic data in Table 1, Supplementary Movie 1). Both families had relatively mild phenotypes dominated by tremor with rare convulsive or focal seizures. Overt myoclonic seizures were observed or reported in a minority, but the tremor was a ‘cortical tremor’ [1, 4] with polymyoclonus.
Fig. 1. Family pedigrees.
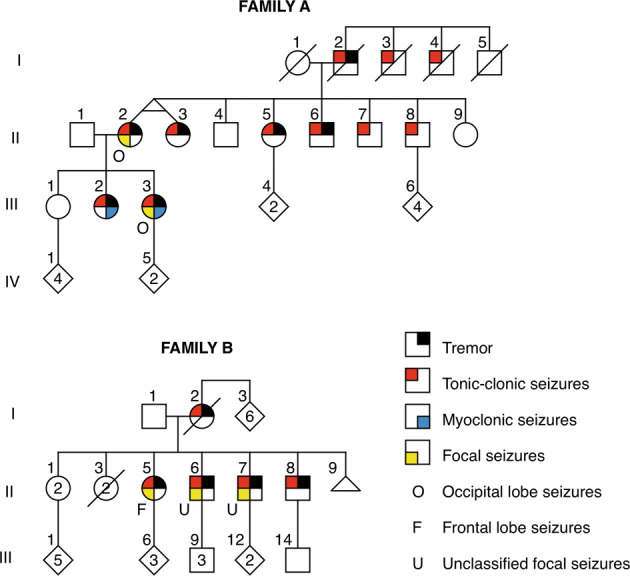
Family A of Sri Lankan origin and Family B of Indian origin.
Table 1.
Phenotypic information and repeat expansion results from WGS and repeat-primed PCR for individuals in Family A and B.
| Family | Individual | Ethnicity | Age studied (y)/gender | FAME affected status | Age of symptom onset | Cortical tremor (onset) | Myoclonic seizures (onset) | Seizure types (onset) | EEG abnormalities | WGS repeat expansion predictions | Repeat-Primed PCR | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FAME1-TTTTA | FAME1-TTTCA | FAME1-AAAAT | FAME1-TTTCA | ||||||||||
| A | II-1 | Sri Lankan | 72 M | Unaffected | – | – | – | – | – | Not done | Not done | No expansion | No expansion |
| A | II-2 | Sri Lankan | 71 F | Affected | 21 y | Y (unknown) | N |
FIAS (21 y) TCS (21 y) |
Not available | Expansion | Expansion | Expansion | Expansion |
| A | II-5 | Sri Lankan | 68 F | Affected | 25 y | Y (25 y) | N | TCS (25 y) | Not available | Not done | Not done | Expansion | Expansion |
| A | III-1 | Sri Lankan | 46 F | Unaffected | – | – | – | – | – | Expansion | No expansion | Expansion | No expansion |
| A | III-2 | Sri Lankan | 30 F | Affected | 25 y | Y (unknown) | Y (25 y) | Photic-induced TCS (27 y) | Generalized 4–6 Hz spike-wave, HV and photic induced 5-6 Hz occipital spike wave and poly-spike wave | Expansion | Expansion | Expansion | Expansion |
| A | III-3 | Sri Lankan | 41 F | Affected | 25 y | Y (27 y) | Y (25 y) |
FIAS (27 y) TCS (27 y) |
Left temporoparietal spikes | Not done | Not done | Expansion | Expansion |
| B | II-5 | Indian | 76 F | Affected | 21 y | Y (21 y) | N | Focal NCSE (60 y) | Ictal recording—electrographically in NCSE with right frontal predominance | Expansion | Expansion | Expansion | Expansion |
| B | II-6 | Indian | 73 M | Affected | 29 y | Y (29 y) | N | TCS (36 y) | Right temporal slowing | Not done | Not done | Expansion | Expansion |
| B | II-7 | Indian | 68 M | Affected | Late teens | Y (late teens) | N |
TCS (55 y) FIAS (unknown) |
Normal | Expansion | Expansion | Expansion | Expansion |
| B | II-8 | Indian | 65 M | Affected | Late 20s | Y (unknown) | Unknown | TCS (adult life) | Not available | Not done | Not done | Expansion | Expansion |
EEG electroencephalogram, F female, FIAS focal impaired awareness seizures, HV hyperventilation, M male, N no, NCSE non-convulsive status epilepticus, TCS tonic-clonic seizures, y years, Y yes
Whole genome sequencing was performed on five individuals, four affected (A-II-2, A-III-2, B-II-5, and B-II-7) and one unaffected (A-III-1), from the two families with FAME. A cohort of 69 individuals without FAME was used for comparison (see Supplementary methods).
Whole genome sequencing data are available in the European Genome-phenome Archive: accession EGAS00001004012, https://ega-archive.org/studies/EGAS00001004012.
Repeat expansion detection
We applied several bioinformatic tools for the detection of repeat expansions, exSTRa [10], ExpansionHunter [11], TREDPARSE [12], STRetch [13], and GangSTR [14] (see Bahlo et al. [15] for a review), with custom target files to search for the TTTCA and TTTTA expansions associated with FAME in SAMD12, TNRC6A and RAPGEF2 (see Supplementary methods). Repeat primed PCR (RP-PCR) was carried out as previously described [6] with minor modifications to the primer sequences (Supplementary methods, Table S1).
Shared haplotype dating
Haplotype analysis was performed for both families and compared with the reported Japanese and Chinese FAME1 haplotypes [6, 7]. We estimated the haplotype age based on the lengths of shared regions using an algorithm that makes use of recombination rates to predict the age of the most recent common ancestor [16]. This method was implemented as a web application and is publicly accessible at: https://shiny.wehi.edu.au/rafehi.h/mutation-dating/ (see Supplementary methods).
Results
Repeat expansion detection
Expansions of the TTTCA repeat in SAMD12 associated with FAME1 were identified in all four affected individuals with whole genome sequencing data (Figs. 2 and S1; ClinVar accession VCV000800660, https://www.ncbi.nlm.nih.gov/clinvar/variation/800660/; Leiden Open Variation Database variant ID 0000630767, https://databases.lovd.nl/shared/variants/0000630767). All methods predicted TTTCA repeat expansions in the four individuals with FAME and none of the controls, with the exception of GangSTR which performed poorly for the TTTCA insertions. ExpansionHunter and TREDPARSE predicted non-zero TTTCA repeat size only for the samples with FAME and no evidence for the presence of TTTCA alleles in any of the remaining samples. The inferred repeat size is not limited by the 150 bp read length and is predicted to exceed this for some samples. ExpansionHunter does not call the smaller allele size equal to zero for the individuals with the TTTCA repeat because it is agnostic to the genetic model and cannot distinguish the contributions to the expansion from the different alleles and hence sometimes attributes expanded reads to either rather than just one allele. exSTRa and STRetch identified all four samples with FAME as significant outliers for the TTTCA repeat expansion (p < 10−5) and no significant outliers in the remaining samples. No bioinformatic method predicted expansions for any individuals in Family A or B at the TNRC6A (FAME6) or RAPGEF2 (FAME7) loci (Figs. S2 and S3). Snapshots visualising the FAME1 expansions in the aligned sequencing data using Integrative Genomics Viewer [17] are presented in Fig. S4.
Fig. 2. FAME1 repeat expansion analysis with ExpansionHunter, exSTRa and STRetch.

Repeat expansion analysis output for the TTTTA repeat expansion (a–c) and the TTTCA repeat insertion (d–f) in SAMD12 associated with FAME1. Two affected individuals (A-II-2, A-III-2) and one unaffected individual (A-III-1) from Family A and two affected individuals (B-II-5, B-II-7) from Family B are shown in colour, while 69 control individuals without FAME are shown in light grey for comparison. a and d show smaller and larger allele sizes predicted by ExpansionHunter with a small amount of noise jitter to distinguish samples that share the same genotype. b and e show empirical cumulative distribution functions (CDFs) of the number of bases matching the repeat motif created from the exSTRa scores. c and f show histograms of negative log base-10 transformed false discovery rate adjusted p values returned by STRetch (padj). The red dashed vertical line represents padj = 0.05. TREDPARSE and GangSTR results are shown in Fig. S1.
Repeat-primed PCR was performed to validate the expansions and test additional family members (Table S2). Results from the repeat expansion detection methods and the repeat-primed PCR assays were concordant (Table 1). The TTTCA repeat expansion was identified in all tested individuals with FAME and not in the unaffected relative, nor in any of the controls. Individual A-III-1, who is clinically unaffected, had a positive result for the TTTTA repeat expansion in both the whole genome sequencing and repeat-primed PCR. This expansion was inherited from individual A-II-2 (affected mother), however expansions of this STR were observed in 5.9% of controls [6] and are not diagnostic for FAME1. We confirmed that individual A-III-1 did not inherit the haplotype on which the TTTCA expansion is located (Fig. S5). The repeat-primed PCR results suggest that all individuals with the TTTCA repeat insertion have the most frequently observed repeat configuration (5′-(TTTTA)n(N)n(TTTCA)n-3′) in the context of the SAMD12 gene on the reverse strand of chromosome 8.
Shared haplotype dating
Haplotype analysis using the same set of marker SNPs as Cen et al. [7] identified a core haplotype surrounding the TTTTA and TTTCA repeat expansions which is shared by all FAME1 families (Families A and B reported here along with five Chinese families and three Japanese families previously reported [7]) and is of length 84 kilobase pairs (kbp) (Table S3). Most families share this core haplotype across a larger region 210 kbp in size, however beyond this region, two distinct haplotypes emerge. One haplotype (haplotype Anc1) is shared by all reported Japanese families [6, 7] and two of the five Chinese families [7]. The second haplotype (haplotype Anc2) is shared by Families A and B, who are of Sri Lankan and Indian origin, and three Chinese families [7].
We estimate the age of the most recent common ancestor to be 672 generations old (95% confidence interval: 213–1240 generations), or ~16,800 years old (assuming 25 years per generation), based on the regions where each family shares haplotype Anc2, the more ancient haplotype (Table S4). Haplotype Anc1 extends across a wider genomic region than Anc2, suggesting that it originates from a more recent common founder than the original ancestral variant. The age of most recent common founder of the families sharing haplotype Anc1 (Table S4) is estimated to be 172 generations (95% confidence interval: 149–341 generations) or ~4300 years (assuming 25 years per generation).
Discussion
We identified the SAMD12 TTTTA repeat expansion and TTTCA repeat insertion, which causes FAME1, in families of Sri Lankan and Indian origin. Previously, the FAME1 repeat expansion was identified in Japanese and Chinese families who were found to share a common haplotype [6, 7]. We showed that the families reported here share the same haplotype and estimated the age of the most recent common ancestor to be 17,000 years old.
We searched for the TTTCA repeat insertions associated with FAME [6] by applying methods recently developed to search short-read sequencing data for repeat expansions [10–14]. Our findings further demonstrate the ability of these methods to detect repeat expansions in whole genome sequencing data. Analysis pipelines typically focus on single nucleotide variants and short insertions and deletions. Searching for repeat expansions is a valuable addition to analysis pipelines, particularly for neurological disorders, and increases the diagnostic potential of whole genome sequencing.
A previous comparison of repeat expansion methods found that a consensus approach, identifying repeat expansions called by multiple methods, gave better performance than any single method alone [10]. We recommend using exSTRa and ExpansionHunter to search for known repeat expansions as both performed strongly and are computationally efficient compared with STRetch, which also performed well but requires realignment of sequencing data. We hypothesise that GangSTR did not perform well for the FAME1 TTTCA expansion because it is a repeat insertion, not present in the reference genome. GangSTR has not been previously tested on such repeat expansions, however it has been demonstrated to work well for other pentamer expansions where the repeat is present in the reference genome [18].
Our data extend the occurrence of the FAME1 repeat expansion beyond the Japanese and Chinese populations to a wider region of Southern Asia. Several Indian families with FAME have been previously reported [19, 20], who, based on these results, should be screened for the SAMD12 expansion. We infer that this repeat expansion is an old founder event which probably spread to Japan, rather than having a Japanese origin, given the extended geographic range we have identified. Any patients with suspected FAME of Asian ethnicity should thus be tested for the SAMD12 repeat expansion.
Supplementary information
Acknowledgements
We thank the families for their participation in our research and Stephanie Leech who provided research support.
Funding
MSH was supported by a National Health and Medical Research Council (NHMRC) Project Grant (1079058) and R. D. Wright Career Development Fellowship (1063799). MAC was supported by the Cerebral Palsy Alliance Research Foundation. JG was supported by NHMRC Senior Research Fellowship (1155224) and Program Grant (1091593). IES was supported by a NHMRC Program Grant (1091593) and Senior Practitioner Fellowship (1104831) and by the Health Research Council of New Zealand. SFB was supported by a NHMRC Program Grant (1091593) and Project Grant (1129054). MB was supported by a NHMRC Senior Research Fellowship (GNT1102971). This work was supported by the Victorian Government’s Operational Infrastructure Support Program and the NHMRC Independent Research Institute Infrastructure Support Scheme (IRIISS).
Compliance with ethical standards
Conflict of interest
IES has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia and Xenon Pharmaceuticals; editorial boards of the Annals of Neurology, Neurology and Epileptic Disorders; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has received speaker honoraria from GlaxoSmithKline, Athena Diagnostics, UCB, BioMarin, Eisai and Transgenomics; has received funding for travel from Athena Diagnostics, UCB, Biocodex, GlaxoSmithKline, Biomarin and Eisai. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-020-0606-z) contains supplementary material, which is available to authorized users.
References
- 1.van den Ende T, Sharifi S, van der Salm SMA, van Rootselaar AF. Familial cortical myoclonic tremor and epilepsy, an enigmatic disorder: from phenotypes to pathophysiology and genetics. a systematic review. Tremor Other Hyperkinet Mov. 2018;8:503. doi: 10.7916/D85155WJ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mikami M, Yasuda T, Terao A, Nakamura M, Ueno S, Tanabe H, et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am J Hum Genet. 1999;65:745–51. doi: 10.1086/302535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henden L, Freytag S, Afawi Z, Baldassari S, Berkovic SF, Bisulli F, et al. Identity by descent fine mapping of familial adult myoclonus epilepsy (FAME) to 2p11.2-2q11.2. Hum Genet. 2016;135:1117–25. doi: 10.1007/s00439-016-1700-8. [DOI] [PubMed] [Google Scholar]
- 4.Depienne C, Magnin E, Bouteiller D, Stevanin G, Saint-Martin C, Vidailhet M, et al. Familial cortical myoclonic tremor with epilepsy: the third locus (FCMTE3) maps to 5p. Neurology. 2010;74:2000–3. doi: 10.1212/WNL.0b013e3181e396a8. [DOI] [PubMed] [Google Scholar]
- 5.Yeetong P, Ausavarat S, Bhidayasiri R, Piravej K, Pasutharnchat N, Desudchit T, et al. A newly identified locus for benign adult familial myoclonic epilepsy on chromosome 3q26.32-3q28. Eur J Hum Genet. 2013;21:225–8. doi: 10.1038/ejhg.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581–90. doi: 10.1038/s41588-018-0067-2. [DOI] [PubMed] [Google Scholar]
- 7.Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain. 2018;141:2280–8. doi: 10.1093/brain/awy160. [DOI] [PubMed] [Google Scholar]
- 8.Zeng S, Zhang MY, Wang XJ, Hu ZM, Li JC, Li N, et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J Med Genet. 2019;56:265–70. doi: 10.1136/jmedgenet-2018-105484. [DOI] [PubMed] [Google Scholar]
- 9.Lei XX, Liu Q, Lu Q, Huang Y, Zhou XQ, Sun HY, et al. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur J Neurol. 2019;26:513–8. doi: 10.1111/ene.13848. [DOI] [PubMed] [Google Scholar]
- 10.Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet. 2018;103:858–73. doi: 10.1016/j.ajhg.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017;27:1895–903. doi: 10.1101/gr.225672.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang H, Kirkness EF, Lippert C, Biggs WH, Fabani M, Guzman E, et al. Profiling of short-tandem-repeat disease alleles in 12,632 human whole genomes. Am J Hum Genet. 2017;101:700–15. doi: 10.1016/j.ajhg.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dashnow H, Lek M, Phipson B, Halman A, Sadedin S, Lonsdale A, et al. STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 2018;19:121. doi: 10.1186/s13059-018-1505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mousavi N, Shleizer-Burko S, Yanicky R, Gymrek M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 2019;47:e90. doi: 10.1093/nar/gkz501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahlo M, Bennett MF, Degorski P, Tankard RM, Delatycki MB, Lockhart PJ. Recent advances in the detection of repeat expansions with short-read next-generation sequencing. F1000 Res. 2018;7:736. doi: 10.12688/f1000research.13980.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandolfo LC, Bahlo M, Speed TP. Dating rare mutations from small samples with dense marker data. Genetics. 2014;197:1315–27. doi: 10.1534/genetics.114.164616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NLM, Pope K, Smith KR, et al. Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet. 2019;105:151–65. doi: 10.1016/j.ajhg.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma CM, Nath K, Kumawat BL, Khandelwal D. Autosomal dominant cortical tremor, myoclonus, and epilepsy (ADCME): probable first family from India. Ann Indian Acad Neurol. 2014;17:433–6. doi: 10.4103/0972-2327.144025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahadevan R, Viswanathan N, Shanmugam G, Sankaralingam S, Essaki B, Chelladurai RP. Autosomal dominant cortical tremor, myoclonus, and epilepsy (ADCME) in a unique south Indian community. Epilepsia. 2016;57:e56–9. doi: 10.1111/epi.13303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.