

Abstract
Long non-coding RNAs (lncRNAs) play critical roles in plant development. However, the information of lncRNAs in Jatropha curcas remains largely unexplored. Thus, an attempt has been made in J. curcas to identify 1,850 lncRNAs based on deep sequencing of developing seeds at three typical stages. About ten percent lncRNAs (196 lncRNAs) were differentially expressed lncRNAs during seed developing process. Together with reverse transcription quantitative real-time PCR, the lncRNA expression analyses revealed the stage-specific expression patterns of some novel lncRNAs in J. curcas. The target genes of lncRNAs were annotated for their roles in various biological processes such as gene expression, metabolism, and cell growth. Besides, 10 lncRNAs were identified as the precursors of microRNAs and 26 lncRNAs were predicted to be the targets of Jatropha miRNAs. A total of 31 key lncRNAs play critical roles in the seed developing process in the context of cell growth and development, lipid metabolism, and seed maturation. Our study provides the first systematic study of lncRNAs in the developing seeds of J. curcas and facilitates the functional research of plant lncRNAs and the regulation of seed development.
Subject terms: Plant embryogenesis, Plant molecular biology, Plant physiology
Introduction
Jatropha curcas is a perennial tree belonging to Euphorbiaceae family and its seed has a high content of oil which can be used as biodiesel1,2. Gene expression profiles have been analyzed by several efforts in the developing seeds of Jatropha in order to understand the molecular processes of oil metabolism and seed development3–6. The whole genome sequence of J. curcas have been sequenced and assembled independently by Japan, China and South Korea, and these efforts lay a solid foundation for further exploration of the non-coding RNAs of J. curcas7–9.
Non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), are functional transcripts that are not translated into proteins but usually function to regulate the expression of other genes. Studies demonstrate that lncRNAs regulate gene expression in plants by DNA methylation and chromatin remodeling, and in some cases, they act as miRNA sponges to enhance the expression of mRNA targeted by miRNA10. A great body of evidence indicates that lncRNAs function in both nuclear and cytoplasmic compartments and play essential regulatory roles in plant development processes (the development of pollen, fiber and lateral root; photomorphogenesis), plant reproduction processes (vernalization, flowering time and male sterility), and plant stress responses11–15. The role of lncRNAs in plant seed development has been acknowleged in recent years, as much effort has been made to identify the lncRNAs from seeds in many plants, including maize16–18, Brassica napus19, tree peony20, castor bean21, pigeonpea22, Ginkgo biloba23, and rice24. In maize, lncRNAs might play a part in the complex regulation of genetic imprinting during maize endosperm development17, and lncRNAs probably have function in lipid metabolism regulation of Brassica napus and tree peony developing seeds19,20. Previous work also indicates that lncRNAs impact on developmental and metabolic processes as endogenous target mimics which leads to sequestering of the miRNAs18,22. In all, these reports support the fact that lncRNAs play critical roles in regulating seed development including endosperm, embryo, and fruits.
Although much effort has recently been made to identify the function of protein-coding genes and miRNAs in J. curcas25–27, the identification and role of lncRNAs have not been revealed in this biodiesel tree. Furthermore, lncRNAs are not conserved between plant species, and the potential functions of lncRNAs in plant seeds remain largely unclear, especially in endosperm of oil seeds. Therefore, it is necessary to discover novel lncRNAs and analyze their function in the developing seeds of J. curcas.
The present study aims to identify lncRNAs and compare their differential expression in the developing seeds of Jatropha. Deep sequencing was carried out in the seeds at three typical developmental stages, and functional annotation was then performed on the targets of differentially expressed lncRNAs to examine their possible roles in seed developing process.
Results and discussion
Quality assessment of sequence data and transcripts assembly
To investigate lncRNA and their expression profiles in the developing seeds of J. curcas, we sequenced nine lncRNA libraries from three stages of seed development (small, middle and large seeds representing young, intermediate and mature respectively) with three biological replicates. After trimming the adaptors, filtering out poly-N regions and low-quality reads, the reads quality was checked based on Q-score, and the percentage of Q30 base was 96.53% or more (Table 1). A total of 107.39 Gb clean reads was obtained from nine samples with at least 10.28 Gb for each sample. After mapping the clean reads to the Jatropha genome JatCur_1.0 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000696525.1/), the transcripts including protein-coding RNAs and non-coding RNAs were assembled.
Table 1.
Transcriptome sequencing results of J. curcas developing seeds.
| Samples | Read sum | Base sum | GC (%) | N (%) | Q30 (%) |
|---|---|---|---|---|---|
| L1 | 41,926,133 | 10,467,410,236 | 45.94 | 0.02 | 97.09 |
| L2 | 48,461,287 | 12,097,611,214 | 45.84 | 0.02 | 97.16 |
| L3 | 55,708,854 | 13,907,728,138 | 45.41 | 0.02 | 96.68 |
| M1 | 48,304,848 | 12,058,792,600 | 44.2 | 0.02 | 96.53 |
| M2 | 47,086,619 | 11,756,937,760 | 44.64 | 0.02 | 96.53 |
| M3 | 41,154,388 | 10,275,769,312 | 44.21 | 0.02 | 96.84 |
| S1 | 53,487,415 | 13,353,591,886 | 44.44 | 0.02 | 96.89 |
| S2 | 45,298,184 | 11,312,135,322 | 45.07 | 0.02 | 96.89 |
| S3 | 48,634,140 | 12,145,143,996 | 45.58 | 0.02 | 96.83 |
L1, L2 and L3 are larger seeds; M1, M2 and M3 are middle seeds; S1, S2 and S3 are small seeds; Read Sum: the statistical count of pair-end reads in clean data. Base Sum: total bases in clean data; GC (%): GC content in clean data; N (%): the percentage of unidentified bases in clean data; Q30 corresponds to a base calling accuracy of 99.9%.
Identification of J. curcas lncRNAs and the characteristic features
The prediction of new lncRNA includes two steps: basic screening and potential coding ability screening. Transcripts lengths, exon numbers, and FPKM were considered in the step of basic screening. Transcripts with FPKM more than 0.5 are usually considered to be convincing expression in RNA sequencing studies. In this report, transcripts with lengths > 200 nt, exons ≥ 2, and FPKM ≥ 0.5 were selected as lncRNA candidates. By basic screening, the sequence information of lncRNA candidates was ready for potential coding ability screening.
Next, all transcripts with protein-coding potential were removed. The protein-coding potential of transcripts were predicted jointly by four analyses: CPC analysis (Coding Potential Calculator)28, CNCI analysis (Coding-Non-Coding Index)29, CPAT analysis (Coding Potential Assessment Tool)30 and Pfam protein domain analysis31. CPC is a protein coding potential calculation tool based on sequence alignment with known protein databases and the biological sequence characteristics of transcripts and it is noncoding RNA when Score < 0. CNCI analysis is a method to distinguish non-coding from coding transcripts by the traits of adjacent nucleotide triplets. It does not depend on known annotation files and can effectively predict incomplete transcripts and antisense transcripts, and transcript is noncoding RNA when score < 0. CPAT analysis is a method to judge transcript encoding ability by constructing logistic regression model, calculating coding probability based on ORF length and ORF coverage. When coding probability < 0.38, it is noncoding RNA. The Pfam database is the most comprehensive classification system for protein domain annotations. The transcripts with a high similarity with known protein domain were defined as transcripts with coding ability (E-value < 0.001). Thus, four computational approaches (CPC/CNCI/Pfam/CPAT) were combined to sort lncRNA candidates from putative protein-coding RNAs in the group of unknown transcripts. The lncRNAs candidates identified using the four methods were counted statistically and plotted in a Venn diagram, and the intersection of the four sets of lncRNAs were accepted as putative lncRNAs (Fig. 1). After potential coding ability screening, lncRNA candidates with potential coding ability were removed, and a total of 1,850 lncRNAs were the newly predicted lncRNAs. Four types of lncRNAs were obtained: long intergenic lncRNAs (lincRNAs), antisense lncRNAs, intronic lncRNAs and sense lncRNAs. The results indicated that there were 893 lincRNAs (48.3%), 553 antisense-lncRNAs (29.9%), 50 intronic-lncRNAs (2.7%), and 354 sense-lncRNAs (19.1%) (Fig. 2).
Figure 1.

The Venn diagram of the noncoding transcripts identified by CPC, CNCI, CPAT, and Pfam analyses. Each circle represents an analysis method for predicting lncRNA. The number in the circle represents the number of transcripts predicted to be lncRNAs. The intersection of four circles is taken as the final result of lncRNAs.
Figure 2.
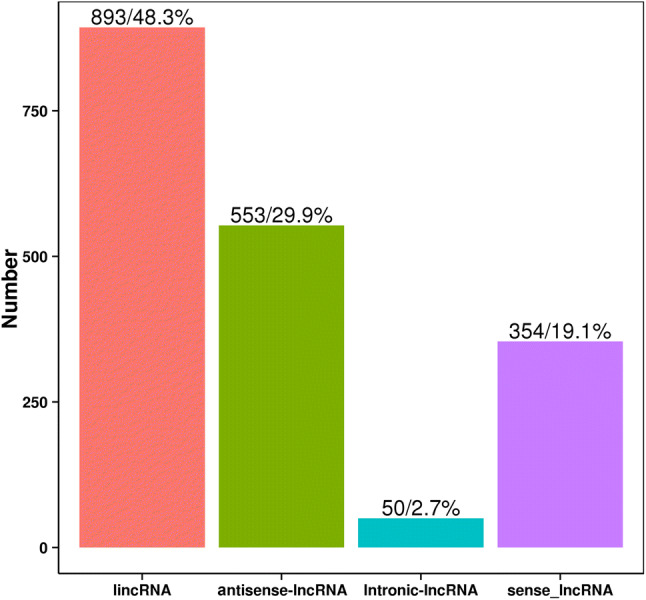
The types of lncRNA. The abscissa is the 4 types of lncRNA and the ordinate is the number of the corresponding lncRNA. LincRNA represents intergenic lncRNA.
The lengths of lncRNAs ranged from 202 to 10,587 bp, with the vast majority (84%) having lengths shorter than 2000 bp (Supplementary Table S1), which is similar to that reported for potato (90%)32. The average length of Jatropha lncRNAs was 1,238 bp, which is significantly higher than that reported for potato (895), rice (800 bp) and chickpeas (614 bp)32–34. More than half (51%) of mRNAs have lengths longer than 2000 bp, indicating that the average length of lncRNAs was lower than that of mRNAs (Supplementary Figure S1). In this study, 1,386 lncRNAs had two exons; 322, three exons; 88, four exons; and 54, between five and eleven exons. On average, the exon numbers associated with Jatropha lncRNAs were lower than those associated with mRNAs (Supplementary Figure S1). These features of lncRNAs could also be found in other plants32.
Quantitative analysis of lncRNAs
Plant lncRNAs play important functional roles in the regulation of plant growth and development. To gain insight into the roles of lncRNAs in Jatropha, the expression levels and patterns of lncRNAs in seed were determined at three different developmental stages. The lncRNA expression levels were presented as FPKM values which were comparable between different samples. Among the 1,850 lncRNAs identified in this work, about ten percent (196 lncRNAs) were differentially expressed lncRNAs (fold change > 2, and p < 0.01) (Supplementary Table S2). To compare the differential expression of lncRNAs between different development stages, the differential expression was presented as the base-2 logarithm of fold change of expression levels between small, middle and large stages (Fig. 3). In the 196 differentially expressed lncRNAs, 125 lncRNAs were up-regulated (Fig. 3a,b) whereas 69 lncRNAs were down-regulated (Fig. 3c,d), and the variation was continuous from small through large stages without fluctuation (Fig. 3). Only two lncRNAs changed without such rules. MSTRG.7207.40 lncRNA could be detected only in the middle stage of seed developing. On the contrary, MSTRG.20400.1 could NOT be detected in the middle stage of seed developing. Among the 125 up-regulated lncRNAs, about a half (61) lncRNAs were up-regulated less than five folds (Fig. 3a) while about a half (64) lncRNAs were up-regulated more than five folds (Fig. 3b); however, about 26% (18) lncRNAs were down-regulated less than five folds (Fig. 3c) while about 74% (51) lncRNAs were down-regulated more than five folds (Fig. 3d).
Figure 3.

The differentially expressed lncRNAs during seed development of J. curcas. Differential expression analyses of lncRNAs were performed based on FPKMs of lncRNAs in each sample, which were calculated using the length of lncRNAs (kilo-base) and mapped fragments (million). The variation of lncRNA levels in three different developmental stages, i.e., small (S), middle (M) and large (L) seeds were shown as the base-2 logarithm of ratios (log2 fold change) between different developmental stages (L/M, M/S and L/S). The lncRNAs shown were differentially expressed lncRNAs because their expression levels between any two of the three different developmental stages changed significantly (|log2 fold change|> 1 and p < 0.01).
Interestingly, most differentially expressed lncRNAs (135 lncRNAs) were observed during seed development from middle to large ones, and only 16 lncRNAs (Supplementary Table S2) were observed differentially expressed when small seeds developed to middle seeds. Reverse transcription (RT) quantitative real-time PCR (qPCR) of 22 differentially expressed lncRNAs was then performed for the validation of the sequencing results. The RT-qPCR results of 13 down-regulated lncRNAs, 5 up-regulated lncRNAs, and 4 lncRNAs detected only in large seeds (MSTRG.25532.1, MSTRG.18228.2, MSTRG.17363.1 and MSTRG.4651.4) agreed well with the lncRNA expression profile displayed by the high-throughput sequencing data (Supplementary Figure S2). The differentially expressed lncRNAs were outlined in a heatmap (Supplementary Figure S3). Among three biological replicates, the overviews of the 196 differentially expressed lncRNAs were similar, and three clades were obtained from the samples of three different developmental stages accordingly, suggesting that the biological replicates were reliable, and three developmental stages could be used for differentially expressed lncRNA analysis (Supplementary Figure S3). A similar profile was observed between small seeds and middle ones, whereas a huge difference was observed in large seeds when compared with the small and middle seeds. This might be caused by similar metabolism happened in the small and middle seeds, where cell division, tissue differentiation and rapid growth were in progress, and drastic changes took place during the maturation process when dry matter accumulation started27,35. These results suggested that gene regulation of by lncRNAs was more active at the late seed developmental stages, which is much similar to the miRNA profile in the developing seeds of Jatropha27.
Prediction and annotation of lncRNA targets
Based on the mode of interaction between lncRNA and its target gene, we adopted two prediction methods. First, lncRNA regulates the expression of its adjacent genes, and predicts that the adjacent genes within the range of 10 kb of lncRNA are its target genes according to the location relationship between lncRNA and gene. Second, lncRNA might play a role on RNA due to complementary base pairing, and therefore LncTar was used to predict lncRNA target gene36. The target genes were found for a total of 1563 lncRNAs based on the two prediction methods (Supplementary Table S3). The targets of differentially expressed lncRNAs were annotated on the basis of KOG/COG. According to their physiological and biochemical functions, except for those genes with unknown function, the target genes are mainly involved in three aspects of plant physiological process: gene expression regulation, metabolism, and cell growth and development. Interestingly, among those 211 target genes with explicit KOG function, nearly half of the target genes (45%) are related to gene expression regulation, including signal transduction mechanisms (T, 24), transcription (K, 24), translation (J, 13), and posttranslational modification (O, 34). The second largest group of targets are involved in metabolism (35%), including carbohydrate transport and metabolism (G, 24), lipid transport and metabolism (I, 8), amino acid transport and metabolism (E, 7), inorganic ion transport and metabolism (P, 10), secondary metabolites biosynthesis, transport and catabolism (Q, 14), and energy production and conversion (C, 11). This is consistent with previous observations in other oil crops, such as castor bean21 and Brassica napus19, in which lncRNA is an important regulator involved in carbon metabolism and lipid metabolism. The third group of target genes function in cell growth and development (11%), such as intracellular trafficking, secretion, and vesicular transport (U, 11), cytoskeleton (Z, 6), cell wall biogenesis (M, 2), and cell cycle control (D, 5) (Fig. 4). These results indicated that the differentially expressed lncRNAs might play an important role in the seed developing and substance accumulation by regulating many target genes involved in gene expression regulation, metabolism, and cell growth.
Figure 4.

The function category of KOG annotation for the targets of differentially expressed lncRNAs during seed development of J. curcas. X-axis represents KOG category. Y-axis indicates the number of lncRNA target genes. Gene function was annotated on the basis of KOG/COG (Clusters of Orthologous Groups of proteins). The genes with unknown function were not counted in. Upper panel, target genes of up-regulated lncRNAs; lower panel, target genes of down-regulated lncRNAs.
The relationship of lncRNAs and miRNAs in Jatropha
In plants, a group of endogenous small non-coding RNAs named microRNAs (or miRNAs) target mRNAs for cleavage or translational repression. miRNAs are initially transcribed as long polyadenylated transcripts called pri-miRNAs which are processed into shorter miRNA precursors (pre-miRNAs), and these pre-miRNAs are further processed into 18–24 nucleotide (nt) mature miRNAs37. A total of 10 lncRNAs was found to be miRNA precursors by mapping miRNAs which were identified from our previously sequenced small RNA libraries27 to the 1,850 lncRNAs determined in this work, suggesting that some lncRNAs encode miRNAs in Jatropha (Supplementary Table S4). These miRNAs might be important regulators for seed development. For example, a target of miR168 (coded by lncRNA MSTRG.30828.3) was AGO1, which was essential for miRNA maturation38, and the interaction between miR168 and AGO1 maintained proper embryo development39. In addition, miR168 was shown to be involved directly in lipid biosynthesis in the developing seeds of another woody oil plant (sea buckthorn)40. miR396a (from lncRNA MSTRG.24217.2) targets growth-regulating factor (GRF) which is shown to control plant seed development41.
Furthermore, 26 lncRNAs were predicted to be the targets of Jatropha miRNAs (Supplementary Table S5). These lncRNAs showing complementarity to miRNAs, might act as decoys, competing with mRNAs for binding to miRNAs to regulate genes involved in seed development. Target mimicking of miRNA is one of the most important mechanisms of lncRNAs regulating the plant development11,42. In other plant species, such as chickpea, Arabidopsis, citrus, rice, canola and maize, it was shown that lncRNAs also could act as miRNA targets, miRNA precursors or endogenous target mimics34,42–46. To sum up, the interaction between lncRNAs and miRNAs could be an important posttranscriptional regulatory mechanism for gene regulation in the developing seeds of Jatropha.
The key lncRNAs involved in seed development
To be more specific about the roles of differentially expressed lncRNAs in seeds, lncRNAs were refined in the context of possible functions in the three aspects of oil seed development, i.e., cell growth and development, lipid metabolism, and seed maturation, and eventually 31 key lncRNAs were obtained (Fig. 5 and Supplementary Table S7).
Figure 5.

Key lncRNAs in the context of cell growth and development, lipid metabolism, and seed maturation of Jatropha developing seeds. Numbers before the arrows are lncRNA ID, and the targets of lncRNA are shown after the arrows. The lncRNAs might act as endogenous target mimics of miRNA, and the miRNA targets, are shown in red (For detail, see Supplementary Table S7).
Seven lncRNAs (MSTRG.8261.1, MSTRG.8877.1, MSTRG.10126.2, MSTRG.18228.1, MSTRG.18228.2, MSTRG.25202.3, and MSTRG.30256.1) were implicated in cell division according to the annotated function of their targets in the UniProt databases. Some targets participate in several stage of cell cycle, such as G2/mitotic-specific cyclin S13-7, anaphase-promoting complex subunit, and sister chromatid cohesion protein pds5. Likewise, some targets are required for cell division, including AUGMIN subunit 5 implicated in spindle assembly, SCD2 in cytokinesis, and DUF724 domain-containing protein 3 in the polar growth of plant cells47. AP2/ERF transcription factors and squamosa promoter-binding protein are both shown to control plant seed development48–50. UVR8 (target of lncRNA MSTRG.25202.3) is required for normal progression of endocycle, which is endoreduplication or DNA replication without mitosis, leading to the formation of nuclear type endosperm. This agrees well with the fact that endosperm of Jatropha is nuclear type in the early stage of seed development51. At least 8 lncRNAs (MSTRG.1315.1, MSTRG.9561.1, MSTRG.12857.1, MSTRG.12857.2, MSTRG.13522.1, MSTRG.23348.8, MSTRG.26115.1, MSTRG.29592.1) were associated with the formation of cell wall and seed coat development, as most of their targets are related to the synthesis of cellulose and pectate which are required for cell growth. In addition, previous work showed that beta-D-xylosidase (target of MSTRG.12857.1) play important roles in seed coat development52.
Seed oil is always the focus of J, curcas, a promising biodiesel plant. Interestingly, several important proteins involved in lipid metabolism were found to be the target of 9 differentially expressed lncRNAs, including lipase, lipid-transfer protein, O-fatty acyltransferase, oleosin, and non-specific lipid-transfer protein (Fig. 5). In oil seeds, oil is stored in oil body surrounded by a half-unit phospholipid membrane containing oleosin, and oleosin is the major protein constituent of Jatropha oil body53. It agrees with previous reports that the oleosin is the target of lncRNA in the developing seed of Brassica napus and pigeonpea19,22. The non-specific lipid-transfer proteins facilitate the transfer of fatty acids and phospholipids between membranes. Mean while, phospholipase D zeta regulated by MSTRG.27126.3 through miR827 enhance diacylglycerol flux into triacylglycerol54. These results suggested that lncRNAs act pivotal part in the oil accumulation of Jatropha seeds.
Many target genes regulated by lncRNAs have critical roles in seed maturation process during which seeds suffering dehydration stress. For example, heat shock protein (including DnaJ), AP2 ERF, and heat stress transcription factor may be involved in the regulation of gene expression by stresses. Peroxidase and superoxide dismutase are known antioxidant proteins, which may protect seeds from injury of free radical. NAC domain-containing protein 67 and late embryogenesis abundant protein (LEA) may play an essential role in seed survival during seed dehydration stress by maintaining cellular stability55. This is in agreement with a previous report that LEA protein was a lncRNA target in the developing seeds of pigeonpea22. DELLA protein my contribute to the regulation of seed dormancy process, which is also an important stage of seed maturation. Collectively, these observations suggest that lncRNAs play a pivitol part in the regulation of desiccation tolerance and antioxidant system during Jatropha seed maturation.
Conclusions
In summary, we screened and identified 1,850 lncRNAs followed by examining their expression patterns at three different developmental stages of J. curcas seeds. The possible roles were investigated for lncRNAs, including target gene regulator, miRNA precursor and miRNA target. Functional analysis of the target genes of differentially expressed lncRNAs showed that lncRNAs play important parts in multiple biological processes, such as posttranslational modification, carbohydrate metabolism and signal transduction. Further analysis showed that 31 key lncRNAs could function in the seed developing process in the context of cell growth and development, lipid metabolism, and seed maturation. It indicated that the up- or down-regulation of lncRNAs is an important mode to regulate the process of seed developing. This study reveals expression profiles of lncRNAs in seed developing, providing important data for further investigation on the mechanisms of molecular regulation of seed development.
Materials and methods
Seed collection and RNA isolation
It was performed by the same method as reported previously27. Fruits from J. curcas were collected randomly from 6 plants at 5–10 (small seeds), 12–20 (middle seeds) and 25–35 (large seeds) days after flower opening (DAF). Small, middle and large seeds represent three typical stages of seed developing, i.e., young, intermediate and mature. Seeds at each stage were collected and frozen immediately in liquid nitrogen and stored at -80 °C. Total RNA was isolated from a pool of seeds from each stage with Trizol (Invitrogen, CA, USA) according to the manufacturer’s protocol. RNAs were prepared from three independent biological replicates.
Preparation for lncRNA library
RNA libraries were constructed following methods described in Zuo et al56. In brief, a total of 1.5 μg RNA per sample was used as input material for rRNA removal using the Ribo-Zero rRNA Removal Kit (Epicentre, Madison, WI, USA). Sequencing libraries were prepared by using NEBNextR Ultra™ Directional RNA Library Prep Kit for IlluminaR (NEB, USA) following manufacturer’s protocol. Fragmentation was carried out in NEB Next First Strand Synthesis Reaction Buffer (5 ×) under elevated temperature. First strand cDNA was synthesized using reverse transcriptase and random hexamer primer. Second-strand cDNA synthesis was performed subsequently using RNase H and DNA Polymerase I. NEB Next Adaptor with hairpin loop structure were ligated to prepare for hybridization after adenylation of 3′ ends of DNA fragments. The library fragments were purified with AMPure XP Beads (Beckman Coulter, Beverly, USA) to select insert fragments of preferentially 150 ~ 200 bp in length. Then PCR was performed with universal PCR primers and Phusion High-Fidelity DNA polymerase. Finally, PCR products were purified (AMPure XP system), followed by library quality assessment on the Agilent Bioanalyzer 2,100. After sequencing with Illumina HiSeq2500 platform (San Diego, CA, USA), the paired-end reads were generated. Three independent RNA libraries were constructed from each of the three stages of the developing seeds resulting in nine lncRNA libraries.
Sequence data analysis
Sequence data analysis was performed according to procedures previously reported56. In brief, from raw reads in fastq format, reads containing adapter, reads containing ploy-N and low-quality reads were removed to obtain clean data (clean reads) with high quality. Clean reads of each sample were mapped to the J. curcas genome (JatCur_1.0, https://www.ncbi.nlm.nih.gov/assembly/GCF_000696525.1/) using HISAT257, and the mapped reads was subjected to further de novo assembly and quantification by StringTie58. The gffcompare program was used to annotate the assembled transcripts. Putative lncRNAs were screened from the unknown transcripts. To overcome transcriptional noise, two or more exons, length greater than 200 bp, and abundance greater than 0.5 FPKM in at least one of the samples were selected as lncRNA candidates. These candidates were further screened using four computational approaches (CPC/CNCI/Pfam/CPAT) which can distinguish the non-coding genes from the protein-coding genes. The different types of lncRNAs include lincRNA, intronic lncRNA, anti-sense lncRNA, and sense lncRNA were selected using cuffcompare.
Quantification of gene expression levels and differential expression analysis
StringTie (1.3.1) was used to calculate FPKMs of lncRNAs in each sample. FPKM means fragments per kilo-base of transcript per million fragments mapped, calculated based on the length of the transcript length (kilo-base) and mapped fragments (million). Differential expression analysis was performed using the DESeq R package (1.10.1) following the procedures described previously27. Benjamini and Hochberg’s approach was adopted to adjust the P values to controlling the false discovery rate. Genes with absolute value of log2(Fold change) > 1 and adjusted P value < 0.01 were assigned as differentially expressed genes.
Reverse transcription quantitative real-time PCR (RT-qPCR)
RT-qPCR was performed following previously established procedures27. In brief, total RNA was treated with DNase I followed by reverse transcription using random primers and SuperScript™ III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Two-step Real-time PCR was performed in ABI StepOne (USA) following a standard SYBR Premix Ex Taq II (TaKaRa) protocol: 95 °C for 5 min, and 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The differences in gene expression were calculated using the 2−ΔΔCt analysis method59 using the actin (GenBank: HM044307.1) as internal reference gene and the young seeds as control for gene expression normalization. Each reaction was performed in triplicate. All primers used in RT-qPCR experiments were listed in Supplementary Table S6.
Prediction and annotation of lncRNA targets
The adjacent genes within the range of 10 kb of lncRNA are predicted to be its target genes according to the location relationship between lncRNAs and genes. LncTar was used to predict lncRNA target gene based on complementary base pairing36. LncTar uses complementary sequences between lncRNA and RNA to calculate the free energy and standardized free energy. When the free energy of pairing sites between RNA and lncRNA was below the threshold of standardized free energy (< − 0.1), the RNA was considered to be a target of the lncRNA. Gene function was annotated based on KOG/COG database (Clusters of Orthologous Groups of proteins; https://www.ncbi.nlm.nih.gov/KOG).
Prediction lncRNAs targeted by miRNAs
The miRNA target prediction was performed by aligning the mature miRNA sequences against J. curcas lncRNA sequences using psRNAtarget with default parameters except for a strict Expectation value 360.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31370674).
Author contributions
M.F.Y. and L.Q.M. designed the experiments, analyzed the data and wrote the main manuscript text; X.H.Y. analyzed the data and prepared all figures and tables. All authors read and approved the final version of the manuscript.
Data availability
All sequencing data were deposited in the NCBI Sequence Read Archive under ID SRR7640476 (small seeds), SRR7640477 (middle seeds) and SRR7640478 (large seeds). All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Lanqing Ma, Email: lqma@bua.edu.cn.
MingFeng Yang, Email: mfyang@bua.edu.cn.
Supplementary information
is available for this paper at 10.1038/s41598-020-67410-x.
References
- 1.Abdulla R, Chan ES, Ravindra P. Biodiesel production from Jatropha curcas: a critical review. Crit. Rev. Biotechnol. 2011;31:53–64. doi: 10.3109/07388551.2010.487185. [DOI] [PubMed] [Google Scholar]
- 2.Kandpal JB, Madan MJ. Jatropha curcus: a renewable source of energy for meeting future energy needs. Renew. Energ. 1995;6:159–160. [Google Scholar]
- 3.Natarajan P, et al. Gene discovery from Jatropha curcas by sequencing of ESTs from normalized and full-length enriched cDNA library from developing seeds. BMC Genom. 2010;11:606. doi: 10.1186/1471-2164-11-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa GGL, et al. Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genom. 2010;11:462. doi: 10.1186/1471-2164-11-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu RH, Wang RL, Liu AZ. Expression profiles of genes involved in fatty acid and triacylglycerol synthesis in developing seeds of Jatropha (Jatropha curcas L.) Biomass Bioenergy. 2011;35:1683–1692. [Google Scholar]
- 6.Jiang HW, et al. Global analysis of gene expression profiles in developing physic nut (Jatropha curcas L.) seeds. PLoS ONE. 2012;7:e36522. doi: 10.1371/journal.pone.0036522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato S, et al. Sequence analysis of the genome of an oil-bearing tree Jatrophacurcas L. DNA Res. 2011;18:65–76. doi: 10.1093/dnares/dsq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha J, et al. Genome sequence of Jatropha curcas L., a non-edible biodiesel plant, provides a resource to improve seed-related traits. Plant Biotechnol. J. 2019;17:517–530. doi: 10.1111/pbi.12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu P, et al. Integrated genome sequence and linkage map of physic nut (Jatropha curcas L.), a biodiesel plant. Plant J. 2015;81:810–821. doi: 10.1111/tpj.12761. [DOI] [PubMed] [Google Scholar]
- 10.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta R, Paul S. Long non-coding RNAs: fine-tuning the developmental responses in plants. J. Biosci. 2019;44:77. [PubMed] [Google Scholar]
- 12.Hou J, et al. Non-coding RNAs and transposable elements in plant genomes: emergence, regulatory mechanisms and roles in plant development and stress responses. Planta. 2019;250:23–40. doi: 10.1007/s00425-019-03166-7. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 2019;20:5573. doi: 10.3390/ijms20225573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Y, Zhang Y, Chen X, Chen Y. Plant noncoding RNAs: hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 2019;35:407–431. doi: 10.1146/annurev-cellbio-100818-125218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HV, Chekanova JA. Long noncoding RNAs in plants. Adv. Exp. Med. Biol. 2017;1008:133–154. doi: 10.1007/978-981-10-5203-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim ED, et al. Spatio-temporal analysis of coding and long noncoding transcripts during maize endosperm development. Sci. Rep. 2017;7:3838. doi: 10.1038/s41598-017-03878-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M, et al. Extensive, clustered parental imprinting of protein-coding and noncoding RNAs in developing maize endosperm. Proc. Natl. Acad. Sci. USA. 2011;108:20042–20047. doi: 10.1073/pnas.1112186108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu M, et al. Transcriptomic analysis of long non-coding RNAs and coding genes uncovers a complex regulatory network that is involved in maize seed development. Genes. 2017;8:274. doi: 10.3390/genes8100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen E, et al. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018;19:745. doi: 10.1186/s12864-018-5117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin DD, et al. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene. 2018;666:72–82. doi: 10.1016/j.gene.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, et al. Differential expression networks and inheritance patterns of long non-coding RNAs in castor bean seeds. Plant J. 2018;95:324–340. doi: 10.1111/tpj.13953. [DOI] [PubMed] [Google Scholar]
- 22.Das A, et al. Expressivity of the key genes associated with seed and pod development is highly regulated via lncRNAs and miRNAs in pigeonpea. Sci. Rep. 2019;9:18191. doi: 10.1038/s41598-019-54340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, et al. Identification and characterization of long non-coding RNAs involved in embryo development of Ginkgo biloba. Plant Signal. Behav. 2019;14:1674606. doi: 10.1080/15592324.2019.1674606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J, et al. Genome-wide identification of lncRNAs during rice seed development. Genes. 2020;11:243. doi: 10.3390/genes11030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galli V, et al. Identifying microRNAs and transcript targets in Jatropha seeds. PLoS ONE. 2014;9:e83727. doi: 10.1371/journal.pone.0083727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang CM, et al. Isolation and identification of miRNAs in Jatropha curcas. Int. J. Biol. Sci. 2012;8:418–429. doi: 10.7150/ijbs.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang MF, Lu HS, Xue FY, Ma LQ. Identifying high confidence microRNAs in the developing seeds of Jatropha curcas. Sci. Rep. 2019;9:1–11. doi: 10.1038/s41598-019-41189-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucl. Acids Res. 2007;35:W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucl. Acids Res. 2013;41:e166. doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, et al. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucl. Acids Res. 2013;41:e74–e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RD, et al. Pfam: the protein families database. Nucl. Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou X, et al. Genome-wide analysis of long non-coding RNAs in potato and their potential role in tuber sprouting process. Int. J. Mol. Sci. 2017;19:101. doi: 10.3390/ijms19010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang YC, et al. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014;15:512. doi: 10.1186/s13059-014-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khemka N, Singh VK, Garg R, Jain M. Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development. Sci. Rep. 2016;6:33297. doi: 10.1038/srep33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H, et al. Proteomic analysis of the seed development in Jatropha curcas: From carbon flux to the lipid accumulation. J. Proteom. 2013;91:23–40. doi: 10.1016/j.jprot.2013.06.030. [DOI] [PubMed] [Google Scholar]
- 36.Li J, et al. LncTar: a tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015;16:806–812. doi: 10.1093/bib/bbu048. [DOI] [PubMed] [Google Scholar]
- 37.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–687. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 38.Vaucheret H. AGO1 homeostasis involves differential production of 21-nt and 22-nt miR168 species by MIR168a and MIR168b. PLoS ONE. 2009;4:e6442. doi: 10.1371/journal.pone.0006442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaucheret H, Vazquez F, Crete P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding J, Ruan C, Guan Y, Krishna P. Identification of microRNAs involved in lipid biosynthesis and seed size in developing sea buckthorn seeds using high-throughput sequencing. Sci. Rep. 2018;8:4022. doi: 10.1038/s41598-018-22464-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun P, et al. OsGRF4 controls grain shape, panicle length and seed shattering in rice. J. Integr. Plant Biol. 2016;58:836–847. doi: 10.1111/jipb.12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu HJ, Wang ZM, Wang M, Wang XJ. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013;161:1875–1884. doi: 10.1104/pp.113.215962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ke L, et al. Evolutionary dynamics of lincRNA transcription in nine citrus species. Plant J. 2019;98:912–927. doi: 10.1111/tpj.14279. [DOI] [PubMed] [Google Scholar]
- 44.Liu H, Wang R, Mao B, Zhao B, Wang J. Identification of lncRNAs involved in rice ovule development and female gametophyte abortion by genome-wide screening and functional analysis. BMC Genom. 2019;20:90. doi: 10.1186/s12864-019-5442-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi RK, Megha S, Basu U, Rahman MH, Kav NN. Genome wide identification and functional prediction of long non-coding RNAs responsive to Sclerotinia sclerotiorum infection in Brassica napus. PLoS ONE. 2016;11:e0158784. doi: 10.1371/journal.pone.0158784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan C, Hao Z, Yan J, Li G. Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genom. 2015;16:793. doi: 10.1186/s12864-015-2024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao X, et al. Characterization of DUF724 gene family in Arabidopsis thaliana. Plant Mol. Biol. 2010;72:61–73. doi: 10.1007/s11103-009-9551-5. [DOI] [PubMed] [Google Scholar]
- 48.El Ouakfaoui S, et al. Control of somatic embryogenesis and embryo development by AP2 transcription factors. Plant Mol. Biol. 2010;74:313–326. doi: 10.1007/s11103-010-9674-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maes T, et al. Petunia Ap2-like genes and their role in flower and seed development. Plant Cell. 2001;13:229–244. doi: 10.1105/tpc.13.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Q, Harberd NP, Fu X. SQUAMOSA promoter binding protein-like transcription factors: targets for improving cereal grain yield. Mol. Plant. 2016;9:765–767. doi: 10.1016/j.molp.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Krishnamurthy KV. In: Embryology of Jatropha: A Review in Jatropha, Challenges for a New Energy Crop, pp 75–86. Bahadur B, Sujatha M, Carels N, editors. Berlin: Springer; 2013. [Google Scholar]
- 52.Arsovski AA, et al. AtBXL1 encodes a bifunctional beta-D-xylosidase/alpha-L-arabinofuranosidase required for pectic arabinan modification in Arabidopsis mucilage secretory cells. Plant Physiol. 2009;150:1219–1234. doi: 10.1104/pp.109.138388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang MF, et al. Proteomic analysis of oil mobilization in seed germination and postgermination development of Jatropha curcas. J. Proteome Res. 2009;8:1441–1451. doi: 10.1021/pr800799s. [DOI] [PubMed] [Google Scholar]
- 54.Yang W, et al. Phospholipase D zeta enhances diacylglycerol flux into triacylglycerol. Plant Physiol. 2017;174:110–123. doi: 10.1104/pp.17.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manfre AJ, Lanni LM, Marcotte WR., Jr The Arabidopsis group 1 LATE EMBRYOGENESIS ABUNDANT protein ATEM6 is required for normal seed development. Plant Physiol. 2006;140:140–149. doi: 10.1104/pp.105.072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zuo J, et al. Analysis of the coding and non-coding RNA transcriptomes in response to bell pepper chilling. Int. J. Mol. Sci. 2018;19:2011. doi: 10.3390/ijms19072001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016;11:1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-delta delta Ct method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 60.Dai X, Zhuang Z, Zhao PX. psRNATarget: a plant small RNA target analysis server (2017 release) Nucl. Acids Res. 2018;46:W49–w54. doi: 10.1093/nar/gky316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data were deposited in the NCBI Sequence Read Archive under ID SRR7640476 (small seeds), SRR7640477 (middle seeds) and SRR7640478 (large seeds). All data generated or analyzed during this study are included in this published article and its Supplementary Information files.