

Abstract
We here report the discovery and early characterization of Compound 3, a representative of a novel class of small molecule bradykinin (BK) B2 receptor antagonists, and its superior profile to the prior art B2 receptor antagonists Compound 1 and Compound 2. Compound 3, Compound 2, and Compound 1 are highly potent antagonists of the human recombinant B2 receptor (Kb values 0.24, 0.95, and 1.24 nM, respectively, calcium mobilization assay). Compound 3 is more potent than the prior art compounds and icatibant in this assay (Kb icatibant 2.81 nM). The compounds also potently inhibit BK-induced contraction of endogenous B2 receptors in a human isolated umbilical vein bioassay. The potencies of Compound 3, Compound 2, Compound 1, and icatibant are (pA2 values) 9.67, 9.02, 8.58, and 8.06 (i.e. 0.21, 0.95, 2.63, and 8.71 nM), respectively. Compound 3 and Compound 2 were further characterized. They inhibit BK-induced c-Fos signaling and internalization of recombinant human B2 receptors in HEK293 cells, and do not antagonize the venous effects mediated by other G protein-coupled receptors in the umbilical vein model, including the bradykinin B1 receptor. Antagonist potency of Compound 3 at cloned cynomolgus monkey, dog, rat, and mouse B2 receptors revealed species selectivity, with a high antagonist potency for human and monkey B2 receptors, but several hundred-fold lower potency for the other B2 receptors. The in vitro off-target profile of Compound 3 demonstrates a high degree of selectivity over a wide range of molecular targets, including the bradykinin B1 receptor. Compound 3 showed a lower intrinsic clearance in the microsomal stability assay than the prior art compounds. With an efflux ratio of 1.0 in the Caco-2 permeability assay Compound 3 is predicted to be not a substrate of efflux pumps. In conclusion, we discovered a novel chemical class of highly selective and very potent B2 receptor antagonists, as exemplified by Compound 3. The compound showed excellent absorption in the Caco-2 assay, predictive of good oral bioavailability, and favourable metabolic stability in liver microsomes. Compound 3 has provided a significant stepping stone towards the discovery of the orally bioavailable B2 antagonist PHA-022121, currently in phase 1 clinical development.
Keywords: bradykinin, B2 receptor, small molecule antagonists, icatibant, human umbilical vein
Introduction
Bradykinin (BK)-related peptides, the kinins, are released from kininogens by kallikreins and exert various physiological and pathological effects via 2 related G protein-coupled receptors (GPCRs) termed the bradykinin B1 and B2 receptors (Leeb-Lundberg et al., 2005). While kinins and their receptors mediate compensatory and protective vasodilator effects under various pathological conditions, they are also mediators of inflammation, producing plasma extravasation and edema and pain (Leeb-Lundberg et al., 2005). Despite considerable efforts invested in antagonist drug development (Marceau and Regoli, 2004; Whalley et al., 2012), only one bradykinin receptor ligand is currently used in clinical practice, the B2 receptor antagonist icatibant, initially described decades ago (Hock et al., 1991). This synthetic peptide is short-lived and not orally bioavailable. When given subcutaneously, icatibant (Firazyr) aborts or limits attacks of hereditary angioedema (HAE) of type I and type II and attacks in patients with normal C1 inhibitor (HAE-nC1 INH) (Cicardi et al., 2014; Wu et al., 2016; Bouillet et al., 2016).
Many small molecule B2 receptor antagonists belonging to various chemotypes have been described (Leeb-Lundberg et al., 2005; Whalley et al., 2012). Two small molecules reached the clinic, but were injectables and discontinued due to lack of efficacy: anatibant (Shakur et al., 2009), for the treatment of traumatic brain injury and fasitibant for osteoarthritis (Tenti et al., 2016). The orally bioavailable B2 receptor antagonist FK 3657 was reported to be in clinical development but little is know about the results of the clinical studies and the fate of this compound (Abe et al., 2005). The feasibility to develop potent orally bioavailable B2 receptor antagonists was reported, but no clinical development candidate has been described from this series (Gibson et al., 2009).
The objective of the present work is to describe the key properties of a B2 receptor antagonist as a representative of a novel chemical class and how it compares to two related prior art compounds. We describe the in vitro pharmacology properties including: (1) in vitro antagonist potency vs. icatibant at the cloned recombinant human B2 receptor; (2) species specificity, as several B2 antagonists exhibit large potency differences as a function of the mammalian species (e.g., the bradyzide series; Marceau et al., 2003); (3) antagonist potency at the endogenous human B2 receptor according to the pA2 scale (Neubig et al., 2003) in the isolated umbilical vein, a standard model used to characterize B2 receptor ligands (Marceau et al., 1994; Marceau et al., 2003; Bawolak et al., 2007; Bawolak et al., 2008; Bawolak et al., 2009; Gera et al., 2016); (4) the competitive (surmountable) and reversible behavior in this model; (5) activity at the human bradykinin B1 receptor, itself antagonized by high concentrations of icatibant (Bastian et al., 1997), and at other receptor types represented in the umbilical vein; (6) The selectivity profile over a battery of more than 120 molecular targets. Furthermore we determined the permeability in a bidirectional Caco-2 assay and metabolic stability in a rat liver microsomes assay as first indicators of the drugability of the corresponding class of small molecule B2 receptor antagonists.
Methods
Materials
BK was purchased from Bachem Bioscience (Torrance, CA), icatibant, from Phoenix Pharmaceuticals (Burlingame, CA) and the B1 receptor agonist Sar-[D-Phe8]des-Arg9-BK, from Tocris Bioscience (Minneapolis, MN). In selectivity experiments, serotonin creatinine sulfate (Millipore-Sigma) and U-46619 (Enzo Life Sciences, Farmingdale, NY) were also exploited.
Compound 1: N-((5-chloro-4-((2-methyl-4-(1-methyl-1H-1,2,4-triazol-5-yl)quinolin-8-yloxy)methyl)pyridin-3-yl)methyl)cyclohexanecarboxamide; Compound 2: rac-N-(1-(5-chloro-4-((2-methyl-4-(1-methyl-1H-1,2,4-triazol-5-yl)quinolin-8-yloxy)methyl)pyridin-3-yl)ethyl)-2-(difluoromethoxy)acetamide; Compound 3: (S)-N-(1-(3-chloro-5-fluoro-2-((2-methyl-4-(1-methyl-1H-1,2,4-triazol-5-yl)quinolin-8-yloxy)methyl)phenyl)ethyl)-2-(difluoromethoxy)acetamide. (Figure 1). The compounds have been synthesized as described previously (Gibson et al., 2019). For detailed experimental procedures for the synthesis of Compound 3 we refer to example 6 in the patent (preparation of Compound no. 6). Purities by HPLC (215 nm) for all three compounds was >99%, the supplier of the compounds was AnalytiCon Discovery GmbH.
Figure 1.

Structures of Compound 1, Compound 2, and Compound 3 and their selected key properties. Key structural elements of the novel series indicated in red. Caco-2 AB, permeation rate apical to basolateral direction. Caco-2 BA/AB, ratio of permeation rates basolateral to apical direction and apical to basolateral direction.
Competition Binding Assay
The ability of the test items to inhibit [3H]BK binding to the human B2 receptors was determined in human recombinant CHO cells expressing the cloned human B2 receptor. Cell membrane homogenates were incubated for 60 min at 22°C with 0.3 nM [3H]BK (Kd value 0.32 nM) in the absence or presence of the test compound in a buffer containing 50 mM Tris/HCl (pH 7.4), 0.2 g/L 1-10-phenanthroline and 0.1% BSA. Nonspecific binding is determined in the presence of 1 µM BK. Following incubation, the samples are filtered rapidly under vacuum through glass fiber filters presoaked with 0.3% PEI and rinsed several times with ice-cold 50 mM Tris-HCl using a 96-sample cell harvester (Packard). The filters are dried then counted for radioactivity in a scintillation counter (Topcount, Packard) using a scintillation cocktail. The results are expressed as percent inhibition of the control radioligand specific binding. The standard reference compound used is the B2 antagonist NPC 567 (Bachem), which was tested in each experiment at several concentrations to obtain a competition curve from which its IC50 was calculated. Test items and icatibant were evaluated at final concentrations between 10-11 and 10-6 M.
Ca2+ Mobilization Assay
Human B2 Receptor
The agonist and antagonist potency of the test items at the human B2 receptor were evaluated in transfected CHO cells by measuring their direct effect (agonism) and their inhibitory effect of BK-induced cytosolic Ca2+ mobilization using a fluorimetric detection method, according to the methods established at Eurofins (Celle l'Evescault, France). The cells were suspended in DMEM buffer, then distributed in microplates. The fluorescent probe (Fluo4 Direct, Invitrogen) was mixed with probenecid in HBSS buffer complemented with 20 mM Hepes (pH 7.4) and was added to each well and equilibrated with the cells for 60 min at 37°C, then 15 min at 22°C. First, for the measurement of agonist potency, test compound and reference agonist BK were added to the cells at several concentrations to generate a concentration-response curve, and the change in fluorescence intensity was measured. The results are expressed as a percent of the control response to 3 nM BK. Where possible, EC50 values of test items were calculated. Second, the antagonist activity of the compounds was evaluated by measuring their effect on BK-induced cytosolic Ca2+ mobilization. For this, test compound, reference antagonist icatibant or HBSS buffer were added to the cells at several concentrations to generate concentration–response curves, left for 5 min, then 0.03 nM BK was added and the changes in free cytosolic Ca2+ concentration was measured as a change in fluorescence intensity. The EC50 value of BK in this assay was 6.05 ± 2.86 pM (mean ± S.D.). The results are expressed as a percent inhibition of the control response to 0.03 nM BK. IC50 values were calculated where possible. Test items and icatibant were evaluated at final concentrations between 10-11 and 10-6 M.
Cynomolgus B2 Receptor
HEK293 cells expressing the B2 receptor of cynomolgus monkeys (Macaca fascicularis) were constructed in the laboratories of Prof. Alexander Faußner, Ludwig-Maximilians-Universität München. The B2 receptor gene was introduced via a pcDNA5/FRT derived plasmid. The cells are cultured in DMEM medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 200 units/mL of potassium penicillin and 100 µg/mL of streptomycin sulfate, 10 µg/mL blasticidin and 250 µg/mL hygromycin. Experiments were conducted at AnalytiCon (Potsdam, Germany) according to their established methods and conditions. The cells were suspended in culture medium with supplemental fetal calf serum reduced to 5% into 96 well plates coated with 0.2 mg/L PDL at an appropriate density. The cells were loaded with a Ca2+- dependent fluorescent dye (FLIPR® Calcium 6 Assay Kit). For the intrinsic agonist response, test compound was added to the cells using FlexStation 3 (Molecular Devices, LLC) and change of fluorescence was recorded. Test item was then left on the cells for 30 min, and BK was added at its EC80 concentration to measure antagonism of BK, recorded as a change in fluorescence of the calcium sensitive dye (excitation 485 nM, emission 525 nm). The EC80 concentration of BK used, was determined in the morning of each experiment and ranged between 365 and 710 pM. The average EC50 value of BK was 155 ± 43 pM, the average EC80 value was 539 ± 160 pM (mean ± S.D.). Compound 3 and icatibant were tested between 10-11 and 10-6 M.
Dog, Rat, And Mouse B2 Receptor
The determination of antagonist potencies of test items to inhibit BK activation of rat, mouse and dog B2 receptors was conducted by Multispan Inc (Hayward, California USA). HEK293T cells, stably expressing the recombinant rat, mouse, and dog bradykinin B2 receptor, were grown in DMEM, 10% FBS, 1 μg/mL puromycin. On the day of assay, cells were seeded in 384-well plate into HBSS assay buffer at an appropriate density. The calcium assay was conducted on the same day. For this, the calcium dye loading buffer (Multispan Inc., Cat# MSCA01-1), containing Fluo-8 loading dye and probenecid, was added to the cells and incubated for 30 min at 37°C and 30 min at room temperature. Cells were preincubated with Compound 3 for 30 min at room temperature prior to the addition of BK. Ca2+- flux was monitored when test compound was added to the wells, and again 30 min later, when the agonist BK is added at the EC80 concentration (4.32, 0.53 and 1.86 nM for rat, mouse and dog B2 receptor, respectively). Excitation/emission wavelengths were 490 and 525 nm respectively. Compound 3 was tested between 10-10 and 10-5 M, icatibant was tested between 10-15 and 10-6 M.
Profiling Across a Panel of Molecular Targets
The binding affinity or functional activity of Compound 3 was measured for a range of 138 molecular targets, including receptors, ion channels, enzymes and transporters, according to methods established by Eurofins (Celle l'Evescault, France). Assay conditions are summarized in Table S2 (Supplementary material).
Antibody Internalization Assay
B2 receptor-mediated transport of immune complexes constructed at the cell surface was previously reported (Bawolak et al., 2011). Intact HEK 293a cells stably expressing the N-terminally myc-tagged B2 receptor construction (human sequence, Charest-Morin et al., 2015) were treated with the monoclonal anti-myc antibody (clone 4A6) conjugated with AlexaFluor-488 (Millipore, dilution 1:500) added to the culture medium. Cells were further incubated at 37°C. Without rinsing, a selected antagonist or their DMSO vehicle was added 5 min later, and BK (100 nM) or its saline vehicle 15 min later. After an additional 30 min incubation period at 37°C, cells were rinsed 3 times with Hank's balanced salt solution (Multicell Wisent, St. Bruno, Canada) and photographed in transmission and epifluorescence using an Olympus BX51 microscope coupled to a CoolSnap HQ digital camera (filters for AlexaFluor 488: excitation 460–500 nm, emission 510–560 nm).
Human Umbilical Vein Pharmacology
The local ethics committee (CHU de Québec-Université Laval) had approved the project (File no. 2017-3720). Umbilical cords were obtained following elective caesarean sections; all problematic deliveries are excluded. Informed consent was obtained from the mothers. The cords were transported from the maternity ward to the laboratory in sterile Hank's balanced salt solution containing penicillin and streptomycin. Segments of umbilical veins were dissected carefully from the cords and a metal rod was inserted in the lumen. Excess connective tissue was excised and rings (2–3 mm wide) were cut. Rings of umbilical veins were suspended in 5-mL organ chambers containing oxygenated (95% O2, 5% CO2) and warmed (37°C) Krebs' solution between a metal hook and a thread loop. This solution had the following composition: NaCl 117.5 mM, KCl 4.7 mM, KH2PO4 1.2 mM, MgSO4 1.18 mM, CaCI2 2.5 mM, NaHCO3 25.0 mM, and D-glucose 5.5 mM. A baseline of one gram-weight was applied to these preparations. Changes of tension were recorded by using isometric transducers (model 52-9545, Harvard Bioscience, South Natick MA). Eight tissue baths are operated in parallel.
For the evaluation of drugs as B2 receptor antagonists, the tissues were equilibrated for 3 h before starting the experiments. The full cumulative concentration-effect curve of synthetic BK (1 to 10,000 ng/mL, optionally 25,000 ng/mL) was constructed with 7 or 8 concentrations of BK in the presence or absence of the tested antagonist drug or its vehicle (saline or DMSO, not exceeding 0.1% v/v final concentration for DMSO). The antagonist drugs were applied to the bathing fluid 30 min before the construction of the curve. The usual design was to test the vehicle control and 4 concentrations of the antagonist (e.g., 1, 10, 100 nM, 1 μM, and possibly 10 μM). The set of 8 tissues can be reused at time 5 h with ample washing with fresh Krebs, allowing the testing of the same or larger antagonist concentrations in each tissue bath. The maximal response to BK can be estimated from the first concentration-effect curve if a high concentration of antagonist (usually tested at time 5 h) is believed to depress the maximal response at the maximal cumulative BK concentration tested, 34.1 μM.
We evaluated the selectivity of Compound 2, Compound 3 and icatibant for B2 receptors versus other endogenously expressed receptors in the human umbilical vein. For this purpose, contractile responses to serotonin, acting on the 5-HT2A receptor, and U-46619, acting at the prostanoid TP receptor, were measured (Rogines-Velo et al., 2002; Daray et al., 2003). The experiments were conducted in the presence or absence of Compound 2, Compound 3 or icatibant, applied 30 min before the agonist.
Compound 3 and icatibant were also tested against an agonist of the kinin B1 receptor, Sar-[D-Phe8]des-Arg9-BK. This highly selective agonist of the B1 receptor is resistant to multiple peptidases and adapted to the umbilical vein preparation (Houle et al., 2000; Jean et al., 2016). In this special procedure, tissues were incubated with interleukin-1β (5 ng/mL) and tumor necrosis factor-α (10 ng/mL) during the 3-hour incubation period to upregulate the expression of B1, and then amply washed (Jean et al., 2016). The cumulative concentration-effect of Sar-[D-Phe8]des-Arg9-BK was constructed 4 h after tissue mounting and antagonists were applied 30 min before this.
Data Analysis for Umbilical Vein Pharmacology
Each curve allows the determination of a maximal effect (expressed in % maximal BK effect for competitive antagonists) and sensitivity (EC50) of each preparation to BK. Prism 5.0 (GraphPad) was used to draw concentration–effect curves (least square fitting of sigmoidal dose–response equation with variable slope) and to derive contractile EC50 values. The effect of each antagonist drug at each tested concentration was calculated as the rightward shift (dose ratio, DR) of the averaged concentration–effect curve relative to the EC50 established in control tissues from the same donor exposed only to the vehicle of the drugs. When applicable, Schild plot parameters (pA2 ± S.E.M., slope ± S.E.M.) were estimated from the Schild plot (abscissa: -log[antagonist], ordinate: log(DR-1), linear regression) using the computer program Pharm/PCS (Tallarida and Murray, 1987). For this calculation, each point of the regression was calculated using data from each separate day of experiments because day-to-day contingencies, that include the identity of tissue donor, were felt to be the most important source of variability. This computerized procedure also calculated the slope of the regression ± S.E.M. that should be close to −1 for competitive agonists. Two-way ANOVA has been applied to the data used for generating the pA2 values. In this analysis, the 2 independent categories are the antagonist drug identity (4 drugs) and concentration (only the range common to all 4 antagonists: 10, 100, and 1000 nM). The numerical variable was log(DR-1) (data sets compatible with normal distribution and homogenous variances). The Bonferroni post-hoc test was used to compare the effect of the drugs at each concentration (Prism 5.0).
Caco-2 Permeation Assay
The permeability of the test compounds was determined at 10 µM across monolayers formed by the human colon carcinoma cell line Caco-2 according to standard operating procedures at Pharmacelsus (Saarbruecken, Germany). Caco-2 cell cultures (ACC 169, Leibniz Institut DSMZ GmbH) were maintained and subcultured at 37°C, 5% CO2 and 95% humidity. For the assays, the cells were seeded at a concentration of 1.5 x 104 cells/well on permeable membrane inserts with a growth surface of 1.1 cm2 (Transwell® plates). The cells were cultured for 21 days and the medium was renewed 3 times a week. Once a week the cell monolayer was quality controlled. The bidirectional Caco-2 assay was performed as described by Hubatsch et al., 2007, with minor modifications. Prior to assay start, the cells were washed twice with HBSS and then the monolayer was equilibrated in pre-warmed apical and basolateral buffer for 20 min at 37°C. The assay was started by replacing the apical or basolateral buffer by the test or reference item solutions depending on the permeation direction. During the experiment, the Transwell® plates were incubated at 37°C at 20 rpm in a shaking water bath. Samples of 200 µl were taken from the basolateral and apical side, respectively, after 60 min and were replaced by drug-free basolateral or apical buffer. In order to assess the mass balance (recovery) of the experiments, a sample from each basolateral compartment was taken after an additional 60 min. Samples were processed for ACN precipitation and analysis by LC/MS.
The apparent permeation coefficient (Papp) was calculated using the following equation:
Equation 1: Papp [cm/sec] = ΔQ/Δt *1/A *1/Co
ΔQ/Δt = diffusion rate of compound [μg/s] (i.e. slope of concentration vs. time relation)
A = total membrane surface [cm2]
Co = initial concentration [μg/mL]
To consider the efflux by P-glycoprotein and other transporters, the ratio of both transport directions was calculated:
Equation 2: Efflux = Papp (B-A) / Papp (A-B)
Papp B-A = apparent permeation from basolateral chamber to apical chamber
Papp A-B = apparent permeation from apical chamber to basolateral chamber
Microsomal Stability
The metabolic stability of the test compounds was determined according to Obach, 1999. Assays were performed in 96-deepwell-plates using liver microsomes pooled from male Wistar rats (Corning). Rat liver microsomal incubations were conducted in duplicate and incubation mixtures consisted of liver microsomes (0.5 mg microsomal protein/mL), test compound (1 µM), MgCl2 (2 mM), and NADPH (1 mM) in a total volume of 0.7 mL sodium phosphate buffer (100 mM, pH 7.4). Reactions were commenced with the addition of NADPH and shaken on a horizontal shaker with a fitted heating block at 37°C. At t = 0 min and the time points 10 min, 30 min and 60 min, aliquots (70 µL) were removed from the incubations and added to 140 µL termination mixtures. Termination mixtures consisted of acetonitrile supplemented with diazepam, diclofenac and griseofulvin as internal analytical standards. The quenched samples were processed by mixing and centrifugation (2,200 x g, 5 min). The particle free supernatant was diluted (1:1, v/v) with deionized water and subsequently subjected to LC-MS for quantitative bioanalysis in terms of test compound depletion (pump flow rate: 600 µL/min; Kinetex Phenyl-Hexyl analytical column 2.6 µm, 50x2.1 mm (Phenomenex, Germany)). Incubations containing verapamil at a concentration of 1 µM were used as high clearance positive control (PC; n=2), and incubations without NADPH (70 µL phosphate buffer (supplemented with 2 mM MgCl2) instead of 70 µL NADPH solution), in order to verify that any apparent loss of test item in the assay incubation was due to metabolism, were used as negative control (NC; n=2). The rate of disappearance of a test compound over time is measured and used to calculate in vitro intrinsic clearance (CLint).
Data Analysis
Numerical values are reported as means ± standard deviation (S.D.) or standard error of the mean (S.E.M.), as indicated for each assay. Sets of values were compared with ANOVA followed by Dunnett's or Tukey's post-hoc test (Prism 5.0).
Results
Binding to Recombinant Human B2 Receptors
[3H]BK had previously shown saturable binding to the membrane fraction of the CHO cells stably expressing recombinant human B2 receptors with an apparent Kd of 0.32 nM, in line with the reported affinity of [3H]BK (0.45 nM) for the recombinant human B2 receptor (Pruneau et al., 1998). NPC 567, the internal reference antagonist showed a Ki value in our experiments of 7.2 nM also in line with its reported affinity (4.1 nM) for the cloned human B2 receptor (Paquet et al., 1999).
All compounds fully inhibited 0.3 nM [3H]BK binding to the recombinant human B2 receptors. IC50 and Ki values are summarized in Table 1. Compound 3 and icatibant exhibited subnanomolar affinity for the human B2 receptor (Ki values 0.50 and 0.60 nM, respectively). The affinities of Compound 1 and Compound 2 were lower, but still in the low nanomolar range (Ki values 3.8 and 3.9 nM, respectively).
Table 1.
Affinities, IC50 and Ki values at the human B2 receptor derived from the [3H]BK binding competition assay.
| IC50 (nM) | Ki (nM) | n | |||||
|---|---|---|---|---|---|---|---|
| Mean | S.D. | Geometric mean | Mean | S.D. | Geometric mean | ||
| Icatibant | 1.21 | 0.46 | 1.15 | 0.64 | 0.26 | 0.60 | 6 |
| Compound 1 | 7.4 | – | 7.4 | 3.8 | – | 3.8 | 1 |
| Compound 2 | 7.6 | – | 7.6 | 3.9 | – | 3.9 | 1 |
| Compound 3 | 0.97 | 0.03 | 0.97 | 0.50 | 0.02 | 0.50 | 3 |
Antagonism of Recombinant Human B2 Receptors
Administration of BK to CHO cells stably expressing recombinant human B2 receptors, elicited Ca2+- ion mobilization with an average EC50 value of 6 pM, in line with the reported BK agonist potency of 9 pM at the cloned human B2 receptor in CHO cells (Zhang et al., 2001).
Table 2 summarizes the potencies of Compound 1, Compound 2, Compound 3, and icatibant to inhibit BK activation of recombinant human B2 receptors expressed in CHO cells, using a calcium mobilization assay.
Table 2.
Antagonist potencies, IC50 and Kb values at the human B2 receptor in the calcium mobilization assay.
| IC50 (nM) | Kb (nM) | n | |||||
|---|---|---|---|---|---|---|---|
| Mean | S.D. | Geometric mean | Mean | S.D. | Geometric mean | ||
| Icatibant | 27.8 | 7.6 | 25 | 3.19 | 1.47 | 2.81 | 13 |
| Compound 1 | 11.3 | 4.1 | 11 | 1.30 | 0.46 | 1.24 | 3 |
| Compound 2 | 9.57 | 6.46 | 8.3 | 1.08* | 0.71 | 0.95 | 3 |
| Compound 3 | 2.13 | 0.12 | 2.13 | 0.24** | 0.02 | 0.24 | 3 |
ANOVA, P < 0.01. *P < 0.05; **P < 0.01 for comparison with the common control value, icatibant Kb (Dunnett's test).
Compound 1, Compound 2 and Compound 3 all demonstrated high antagonist potency towards the human B2 receptor. Compounds 2 and 3 were significantly more potent as compared to icatibant with Compound 3 being the most active compound. The Kb values were 1.24, 0.95 and 0.24 nM, respectively, versus 2.81 nM for icatibant.
Species Selectivity
As the bradykinin B2 receptor is well known for its species selective pharmacology (Paquet et al., 1999; Burgess et al., 2000), we investigated the antagonist potency of Compound 3 in cells expressing B2 receptors from human, cynomolgus monkey, dog, rat, and mouse (Table 3). The Kb value of Compound 3 to antagonize BK activation of the cynomolgus monkey B2 receptor was found to be in the same range as for the human B2 receptor (1.36 nM versus 0.24 nM). Compound 3 was however more than 500-fold less potent in antagonizing the rat, and mouse B2 receptor, and more than 10,000-fold less potent towards the dog B2 receptor. Icatibant in contrast, showed a more or less stable antagonist potency across species (Table 3).
Table 3.
Antagonist potencies of Compound 3 and icatibant at the B2 receptor from different species.
| Compound 3 | Icatibant | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | Kb (nM) | n | IC50 (nM) | Kb (nM) | n | |||||
| Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | |||
| Human | 2.13 | 0.12 | 0.24 | 0.02 | 3 | 27.8 | 13.4 | 3.19 | 1.55 | 16 |
| Monkey | 6.25 | 2.11 | 1.36 | 0.34 | 5 | 17.1 | 5.64 | 4.06 | 0.99 | 5 |
| Dog | ~7,600 | NA | ~2,700 | – | 1 | 87.1 | NA | 30.7 | – | 1 |
| Rat | 1,034 | 329 | 147 | 60 | 3 | 37.4 | 6.4 | 5.60 | 2.20 | 3 |
| Mouse | ~1,200 | NA | ~350 | – | 1 | 15.5 | NA | 4.4 | – | 1 |
Pharmacology in the Human Umbilical Vein
The construction of a cumulative concentration-effect curve for BK-induced contraction of the umbilical vein in the absence or presence of an antagonist or the DMSO vehicle is graphically illustrated in Figure 2 and further explained in Figure 2 legend. None of the four B2 receptor antagonists tested showed any intrinsic partial agonist effect when applied to human umbilical vein rings (illustrated for Compound 3 and Compound 2 in Figure 2). The clinically used peptide antagonist icatibant was studied in the umbilical vein contractility assay for comparison with the small molecule compounds; its vehicle was 0.9% saline. Icatibant behaved as a competitive (surmountable) antagonist of BK in the isolated human umbilical vein preparation (Figure 3A). This can be further described as a dose-dependent rightward shift of the BK concentration-effect curve with no depression of the maximal effect. Further evidence of a lack of effect on BK contractile Emax is provided in Table S1 (Supplementary material): when expressed in force units (gram-weight), the maximal BK-induced contractions recorded in the absence or presence of various icatibant concentrations were similar. Figure 4A shows a Schild plot where each experimental point in the regression represents the log(DR-1) value for each experimental day, as the tissue donor identity is felt to be a major determinant of variability. Using this method applied by the computer program Pharm/PCS, the pA2 value for icatibant is 8.06 ± 0.37, comparable to the previously reported value of 8.2 (Marceau et al., 1994), and the slope of the regression, -0.85 ± 0.16 (Table 4), compatible with competitive antagonism.
Figure 2.
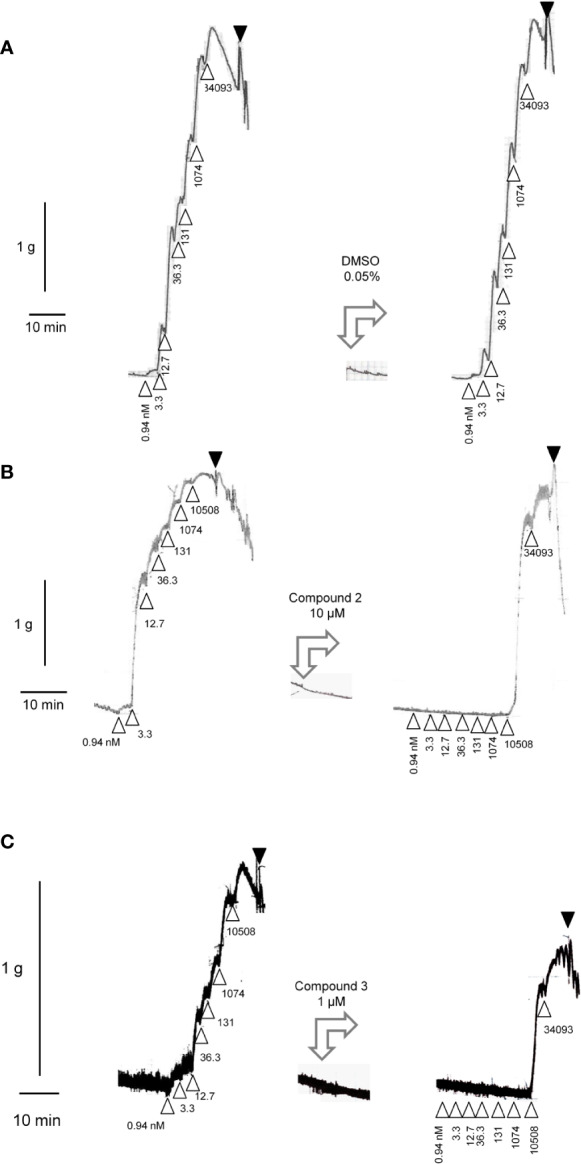
All panels, left tracings: Construction of a cumulative concentration-effect curve for BK-induced contraction by using a ring of human umbilical vein pre-equilibrated for 3 h. Abscissa scale: time; ordinate scale: isometric contraction, grams. Δ indicates the application of BK (cumulative nanomolar concentration indicated) and ▼ the first of a series of washouts. All panels, middle tracings: the tested small molecule antagonists or their DMSO vehicle exerted no contractile effects. All panels, right tracings are recorded in the same tissues at time 5 h. The DMSO vehicle has no major effect on the previously recorded response to BK (sensitivity, maximal effect) (A). Compound 2 (B) or Compound 3 (C) at a high concentration depressed both the sensitivity of the same ring to BK and, slightly, the maximal effect of the peptide (estimated from the previously recorded Emax).
Figure 3.

Effect of four B2 receptor antagonists on BK-induced contraction in the human isolated umbilical vein. The identity of the tested antagonist is icatibant (A), Compound 1 (B), Compound 2 (C) and Compound 3 (D). Values are the means ± S.E.M. of the number determinations indicated between parentheses. For the sake of clarity, several error bars below or above the average values have been omitted.
Figure 4.

Schild plot analysis of Figure 3 data where a point in the regression represents one day of experimental data for a given antagonist concentration. The identity of the tested antagonist is icatibant (A), Compound 1 (B), Compound 2 (C) and Compound 3 (D). Two-way ANOVA indicated that the antagonist drug identity and concentration determined the variation of log(DR-1) values (P < 10-4 for each). Values different from those of icatibant: *P < 0.05; **P < 0.01; values different from those of Compound 1: ‡P < 0.05 (Bonferroni post-hoc test).
Table 4.
pA2 value estimates for compounds tested as B2 receptor antagonists using the umbilical vein contractility assay.
| Compound | Parameters calculated using Pharm/PCS (Figure 4) | |||
|---|---|---|---|---|
| pA2 | pA2 S.E.M. | Slope | Slope S.E.M. | |
| icatibant | 8.06 | 0.37 | −0.85 | 0.16 |
| Compound 1 | 8.58 | 0.26 | −0.62 | 0.07 |
| Compound 2 | 9.02 | 0.31 | −0.65 | 0.07 |
| Compound 3 | 9.67 | 0.42 | −0.76 | 0.13 |
The vehicle for the three small molecule antagonists was DMSO 0.05% v/v. The compounds were more potent than icatibant in the following order: Compound 1 (pA2 8.58), Compound 2 (9.02) and Compound 3 (9.67) (Figures 3B–D and 4B–D and Table 4). Thus, Compound 3 showed a picomolar potency in this model (0.21 nM), approximately 40-fold more potent than icatibant (8.7 nM). Two-way analysis of variance of Figure 4 data showed that the significant sources of variation for log(DR-1) values were the antagonist drug identity (P < 10-4) and concentration (P < 10-4). Further, the Bonferroni post-hoc test evidenced that Compound 3 was more potent than either icatibant or Compound 1 at specific concentrations (Figure 4). The small molecule antagonists were surmountable, based on lack of effect on BK contractile Emax (Figure 3, Table S1), but the Schild plot slopes were somewhat inferior to 1 (Table 4), possibly indicating a not fully competitive behavior.
The umbilical vein contractile assay also allows to assess other GPCRs (Altura et al., 1972; Gobeil et al., 1996). We performed additional experiments to characterize the specificity of the most potent antagonists, Compound 2 and Compound 3 (Figure 5, Table 5). Compound 2, at 10 μM, failed to modify the serotonin (5-hydroxytryptamine)-mediated concentration–effect curve which is predominantly mediated by 5-HT2A receptors (Rogines-Velo et al., 2002; Figure 5A) and failed to modify the thromboxane A2 mimetic U-46619-induced contractile response, which activates the TP prostanoid receptors in the umbilical vein (Daray et al., 2003; Figure 5B). The same conclusions applied to Compound 3 tested at 1 μM against serotonin and U-46619 (Figures 5C, D). Both Compound 3 and icatibant, at 1 μM, failed to antagonize the contractile effect of the B1 receptor agonist Sar-[D-Phe8]des-Arg9-BK in the umbilical vein (Figures 5E, F). EC50 values of each agonist in the presence or absence of each antagonist are reported in Table 5 along with overlapping 95% confidence limits on the values.
Table 5.
Agonist EC50 values in specificity experiments (Figure 5).
| Antagonist | Agonist | Antagonist concentration | Agonist EC50 (nM) | 95% confidence limits on EC50 values |
|---|---|---|---|---|
| Compound 2 | Serotonin | 0 | 7.5 | 5.9 – 9.5 |
| 10 μM | 4.0 | 2.6 – 6.1 | ||
| U-46619 | 0 | 24.9 | 17.2 – 36.2 | |
| 10 μM | 24.5 | 18.3 – 32.9 | ||
| Compound 3 | Serotonin | 0 | 17.2 | 11.5 – 25.8 |
| 1 μM | 20.5 | 14.5 – 28.9 | ||
| U-46619 | 0 | 14.1 | 9.7 – 20.5 | |
| 1 μM | 11.3 | 8.8 – 15.5 | ||
| Sar-[D-Phe8]des-Arg9-BK | 0 | 92.0 | 62.9 – 134.7 | |
| 1 μM | 72.3 | 43.9 – 119.2 | ||
| icatibant | Sar-[D-Phe8]des-Arg9-BK | 0 | 112.1 | 92.2 – 139.3 |
| 1 μM | 68.2 | 42.5 – 109.4 |
Figure 5.

Specificity of the antagonist effect of Compound 2 (A, B), Compound 3 (C–E) or icatibant (F) in the human umbilical vein preparation (concentrations as indicated, replicate number indicated by n). (A, C) Lack of antagonist effect on serotonin-induced contraction. (B, D) Lack of antagonist effect on U-46619-induced contraction. (E, F) Lack of Compound 3 and icatibant on the B1 receptor agonist Sar-[D-Phe8]des-Arg9-BK-induced contraction. Values are the means ± S.E.M. of the number of determinations indicated by n. For the sake of clarity, several error bars below or above the average values have been omitted. Agonist EC50 values and overlapping confidence limits are reported in Table 5.
In other experiments, the reversibility of the most potent antagonist, Compound 3, has been tested (Figure 6). A 100 nM concentration, that strongly antagonizes BK without depressing its maximal effect, has been applied 30 min before the initial construction of the BK concentration effect curve (performed at 3 h after tissue mounting). At the end of the BK concentration curve, the antagonism was fully overcome by high BK concentrations, suggesting full displacement. Then the tissues were amply washed for about 100 min. The BK curve construction was repeated (at about 5 h after tissue mounting) to assess reversibility (Figure 6A). The potency of BK after Compound 3 antagonism and washout, was about 7.2-fold lower as compared to the potency of BK in control tissue tested at 5 h, indicating partial or slow reversibility (Table 6). In contrast, the EC50 of BK remained approximately constant from the 3 hour to the 5 hour curves following treatment with 1 μM icatibant, a concentration that produced approximately the same rightward shift intensity as that selected for 100 nM Compound 3 (Figure 6D, BK EC50 values and 95% confidence limits of these experiments reported in Tables 6 and 7). The effects of the more therapeutically relevant Compound 3 concentrations of 1 and 10 nM were fully reversible when this protocol was applied (Figures 6B, C and Table 6).
Figure 6.
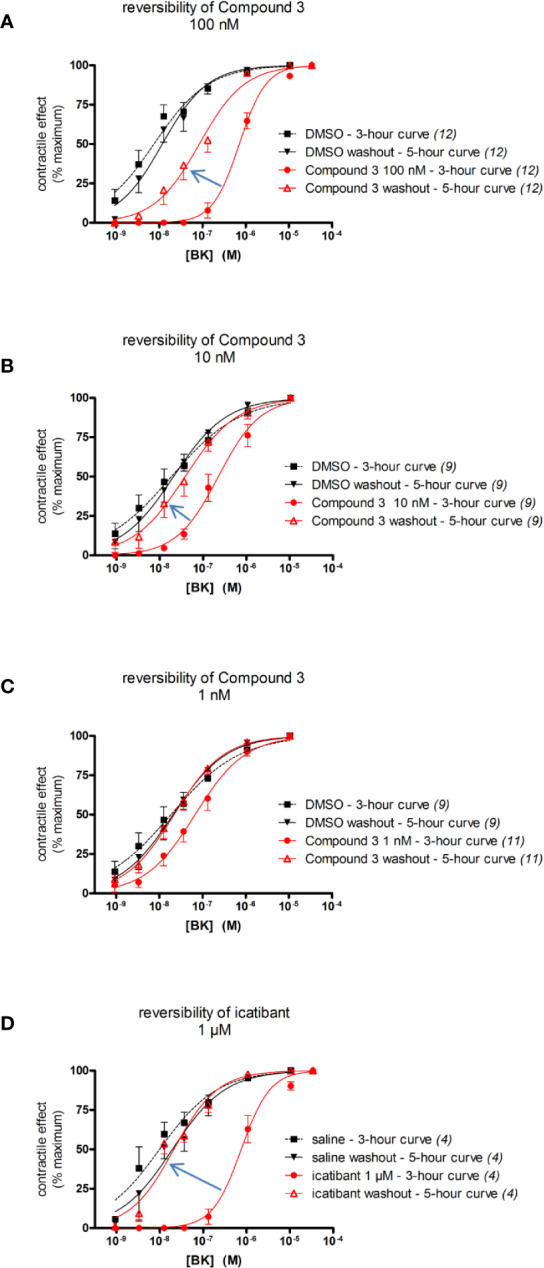
Reversibility of the antagonist effect of selected B2 receptor antagonists (concentrations as indicated, replicate number indicated between parentheses) in the human umbilical vein preparation following ample washing with fresh Kreb's solution. The apparent BK EC50 values are reported in Tables 6 and 7. (A–C) Reversibility of Compound 3 (100 nM, 10 nM, and 1 nM, respectively). (D) Reversibility of icatibant, 1 μM. The experiments reported in (B) and (C) have been conducted in parallel and share the same DMSO controls (presented twice in these panels). Values are the means ± S.E.M. of the number of determinations indicated by n. For the sake of clarity, several error bars below or above the average values have been omitted.
Table 6.
Apparent BK EC50 values in the Compound 3 reversibility experiment (Figures 6A–C).
| Compound 3 concentration | Curve, time point | BK EC50 (nM) | 95% confidence limits on EC50 values |
|---|---|---|---|
| 100 nM (Figure 6A) | Vehicle, 3 h | 7.5 | 5.1 – 11.1 |
| Vehicle washout, 5 h | 11.7 | 8.3 – 16.5 | |
| Compound 3 100 nM, 3 h | 713 | 613 – 829 | |
| Compound 3 washout, 5 h | 84.1 | 57.9 – 122.2 | |
| 10 nM (Figure 6B) | Vehicle, 3 h | 18.7 | 11.8 – 29.4 |
| Vehicle washout, 5 h | 20.8 | 15.0 – 28.7 | |
| Compound 3 10 nM, 3 h | 224.9 | 164.5 – 307.4 | |
| Compound 3 washout, 5 h | 38.5 | 25.1 – 59.1 | |
| 1 nM (Figure 6C) | Vehicle, 3 h | 18.7 | 11.8 – 29.4 |
| Vehicle washout, 5 h | 20.8 | 15.0 – 28.7 | |
| Compound 3 1 nM, 3 h | 67.4 | 47.9 – 94.9 | |
| Compound 3 washout, 5 h | 22.0 | 16.8 – 28.6 |
Table 7.
Apparent BK EC50 values in the icatibant reversibility experiment (Figure 6D).
| Curve, time point | BK EC50 (nM) | 95% confidence limits on EC50 values |
|---|---|---|
| Vehicle, 3 h | 9.9 | 6.0 – 16.2 |
| Vehicle washout, 5 h | 19.5 | 11.8 – 32.5 |
| icatibant 1 μM, 3 h | 756 | 599 – 954 |
| icatibant washout, 5 h | 21.1 | 15.9 – 27.9 |
Effect of Compound 2 and Compound 3 on BK-Induced Signaling in HEK 293a Cells
c-Fos accumulation is a distal response to the stimulation of numerous receptors, including the bradykinin B2 receptor (Bawolak et al., 2009). Over 60 min, 10 nM BK induced c-Fos accumulation in HEK 293a that stably expressed human myc-B2 receptor, but not in untransfected cells (Figure S1, Supplementary material). Co-treatment with Compound 2 or Compound 3 inhibited this response in a concentration-dependent manner (between 10 nM and 1 μM). The 10-fold excess of the former antagonist (100 nM) significantly reduced the response to BK (10 nM) whereas the latter had a significant effect at equimolar concentration (Figures S1 and S2, Supplementary material). The antagonists applied alone did not promote c-Fos accumulation.
BK and its B2 receptor constitute an excellent example of a cycling system of ligand-GPCR complex, starting with agonist-induced receptor phosphorylation by GPCR kinases (Blaukat et al., 2001), receptor mediated ligand endocytosis (Munoz and Leeb-Lundberg, 1992; Charest-Morin et al., 2013), and subsequently, essentially complete receptor recycling at the cell surface upon agonist washout (Blaukat et al., 1996; Bawolak et al., 2007). The anti-myc antibodies, when bound to the myc-B2 receptor construction in intact cells, do not functionally antagonize BK but are submitted to agonist-induced endocytosis along with the receptor (Bawolak et al., 2011). This was replicated using a fluorophore-conjugated anti-myc monoclonal antibody, which essentially located at the plasma membrane, but completely internalized in response to BK (100 nM, 30 min) in HEK 293a stably expressing myc-B2 receptor (human sequence; Figure 7). Despite certain subjectivity in assessing drug effects in this assay, it was observed that pretreating the cells with Compound 2 or Compound 3 (between 1 nM and 1 μM, administered 15 min before BK) inhibited the endocytosis induced by BK (100 nM) in a concentration-dependent manner, with clear efficacy at and above 100 nM for both compounds (Figure 7). This represents an agonist/antagonist ratio comparable to one that produces a full antagonism of BK-induced Ca2+ mobilisation or venous contraction. The antagonists did not promote significant receptor endocytosis if applied alone.
Figure 7.

Endocytosis of the fluorescent anti-myc 4A6 monoclonal antibody by intact HEK 293a cells expressing myc-tagged human B2 receptors (h myc-B2) treated as indicated at 37°C (BK 0 or 100 nM, the antagonists Compound 2 or Compound 3 at the indicated concentrations 15 min before). The intact cells had been labeled with the anti-myc tag antibodies prior to stimulation (see Methods). Each field is shown as a pair of images: epifluorescence in intact cells (left); transmission (right). Original magnification 1,000 ×; the sides of each square field are 120 µm long. Cells incubated without the antibody were photographed using the same settings to evaluate the autofluorescence.
Off-Target Profile
Compound 3 was screened at 10 μM against a selected panel of G protein-coupled receptors (GPCRs), ion channels, enzymes and transporters (see Table S2, Supplementary material). Compound 3 did not significantly interact (<50% inhibition of binding or functional activity at 10 μM) with most targets (including the bradykinin B1 and the angiotensin AT1 receptor), except for the vasopressin receptor 1a (V1a receptor), the corticotropin-releasing factor receptor 1 (CRF1 receptor), the prostanoid EP1 receptor, the TXA2/PGH2 receptor (TP receptor), and the neurokinin 1 receptor (NK1 receptor). Full concentration–response curves were established for these targets (except for the prostanoid EP1 receptor) in agonist and antagonist mode, and for NK3 receptors in addition. Compound 3 was found to show no agonism at these targets, but to be a weak antagonist with Kb values in the micromolar range (Table 8). These data indicate that Compound 3 is a very selective B2 receptor antagonist: at least thousand-fold more potent at the human B2 receptor (Kb 0.24 nM) as compared to a wide set of off-targets.
Table 8.
In vitro target profile of Compound 3.
| Radioligand competition binding | Functional antagonism | |||
|---|---|---|---|---|
| GPCR | % Inhibition at 10 µM | IC50 (µM) | Kb (µM) | Fold selectivity over the human B2 receptor* |
| V1a | 84 | 9.1 | 0.31 | 1,292 |
| CRF1 | 74 | 10 | 3.6 | 15,000 |
| TP | 62 | 3.6 | 0.51 | 2,125 |
| NK1 | 83 | 17 | 2.4 | 10,000 |
| NK2 | 21 | – | – | – |
| NK3 | 6.1 | 17 | 1.6 | 6,667 |
| EP1 | 80 | – | – | – |
*Relative to the Kb at the human B2 receptor (0.24 nM).
Caco-2 Permeability Assay
We measured the permeability of Compound 1, Compound 2, and Compound 3 across the polarized human colon carcinoma Caco-2 cell monolayer. This bidirectional assay is a standard test that allows a first prediction of absorption rate across human intestinal epithelial cells. Compound 1, Compound 2 and Compound 3 displayed an apparent permeation rate of 5.5 x 10-6 cm/s, 3.9 x 10-6 cm/s and 17.3 x 10-6 cm/s in apical to basolateral direction and a permeation rate of 20.9 x 10-6 cm/s, 29.9 x 10-6 cm/s, 17.6 x 10-6 cm/s in basolateral to apical direction. With a permeability rate >10-5 cm/s, and an efflux ratio of 1.0, Compound 3 is predicted to be highly permeable, and not a substrate of efflux pumps such as P-glycoprotein (P-gp) in contrast to Compound 1 and Compound 2 that showed an efflux ratio of 5.5 and 7.7.
Microsomal Stability
Compound 1, Compound 2, and Compound 3 have been evaluated in a rat liver microsome assay fortified with the phase I metabolism cofactor NADPH. Verapamil was used as the positive control and showed a degradation in the range of 97.00 to 99.97% within 30 min. With a CLint of 960 μL/min/mg protein and 497 μL/min/mg protein Compound 1 as well as Compound 2 revealed a very low stability in the rat microsome assay. Compound 3 showed with a CLint of 27.9 μL/min/mg protein a substantially lower conversion rate and thus potentially a largely improved metabolic stability.
Discussion
This paper describes the characterization of the highly potent B2 receptor antagonist Compound 3 as a representative of a novel chemical class. The difluoromethoxy, the 1-methyl-1H-1,2,4-triazol-5-yl, the m-chlorofluorophenyl as well as the S-configurated benzylic methyl group of compound 3 were identified as key structural elements for achieving high permeability, metabolic stability and activity towards the B2 receptor (Figure 1). In the patent literature, only six B2 receptor antagonists have been described previously that feature a 1-methyl-1H-1,2,4-triazol-5-yl group (Gibson et al., 2008; Gibson et al., 2010) with Compound 1 being the closest analog to Compound 3. In comparison to the prior art Compound 1, Compound 3 showed a 5.2-fold improved potency on the B2 receptor in the calcium mobilization assay and a 34-fold improved intrinsic clearance in the rat microsomal stability assay. With an efflux ratio of 1.0 in the Caco-2 assay Compound 3 is predicted to be not a substrate of efflux pumps such as P-gp whereas with an efflux ratio of 5.5 Compound 1 seems obviously to be a substrate of efflux pumps. In order to demonstrate the contribution of the novel structural elements (m-chlorofluorophenyl group in combination with the defined stereochemical configuration of the benzylic stereocenter) of the new B2 antagonists series, the comparator Compound 2 has been synthesized. Compound 2 is furnished with the identical structural elements as Compound 3 except for the pyridine core and the undefined stereo center, both of which are distinctive features of the previously described series (Gibson et al., 2008; Gibson et al., 2009; Gibson et al., 2010). With a 4.0-fold lower potency on the B2 receptor, a 18-fold lower intrinsic clearance in the rat microsomal stability assay and a 7.7-fold higher efflux ratio in the Caco-2 permeability assay the comparator Compound 2 clearly revealed a substantially inferior profile when compared with Compound 3.
The potencies of the compounds to antagonize BK-induced Ca2+ mobilization at recombinant human B2 receptors in CHO cells were very similar to the potencies to antagonize BK-induced contraction at endogenous human B2 receptor in the umbilical vein model (Kb values in nM and pA2 value converted into nM, respectively): Compound 1 (1.24 and 2.63 nM), Compound 2 (0.95 and 0.95 nM), Compound 3 (0.24 and 0.21 nM) and icatibant (2.81 and 8.71 nM). This alignment between these 2 very different assays supports the high potency of Compound 3 at the human B2 receptor and confirms the superior potency towards the precedent B2 antagonists.
Interestingly, in radioligand binding assays using membranes of a mammalian cell line that express recombinant human B2 receptors, the prior art Compound 1 and Compound 2 showed about 7-fold lower affinity for the human B2 receptor as compared to Compound 3 and icatibant. In contrast to the 10-40-fold higher functional B2 receptor antagonist potency of Compound 3 as compared to icatibant, in this assay, icatibant and Compound 3 showed a similar affinity. The latter is not well understood, but may be related to the low ionic strength applied in the binding assay which may (1) produce an artificially high affinity for [3H]BK for the human B2 receptor, and (2) increase artificially the affinity of icatibant for this receptor because these conditions increase the protein-peptide interactions, but not the protein-small molecule interaction.
Species selectivity has been a hurdle in the development of B2 receptor antagonists (Leeb-Lundberg et al., 2005). In some cases, rodent specific compounds were discovered, not suitable for further therapeutic development (Hess et al., 1994; Jones et al., 1999; Burgess et al., 2000; Marceau et al., 2003). We here describe that Compound 3 shows high antagonist potency at the human and monkey B2 receptors but is several hundred fold less active at rodent and dog B2 receptors. This finding is not unexpected, considering the relatively low amino acid sequence identity between B2 receptors from human and mouse (84%, Hess et al., 1994), rat (82%, Ma et al., 1994), and dog (81%, Hess et al., 2001). Indeed, several amino acids in the B2 receptor known to be engaged in the binding of peptides or small molecule ligands differ between human and other species (Leeb-Lundberg et al., 2005).
The small molecule compounds tested herein are completely devoid of agonist activity in all bioassays: Ca2+ mobilization, venous contraction and c-Fos activation. This is of relevance since small molecule partial agonists of the B2 receptor have been described in the past, notably Fujisawa's compound 47a, a very active compound able to contract the human umbilical vein (Bawolak et al., 2009). Furthermore, agonist activity at the B2 receptor could be associated with injection pain, as hypothesized for icatibant (Floccard et al., 2012).
Compound 3 presumably inhibits all types of B2 receptor-initiated signaling: in the persistent contraction of the vascular smooth muscle, protein kinase C (PKC) is involved, as well as rho kinase and myosin light chain kinase and phosphatase (Liu and Khalil, 2018); PKC, the MEK1 and ERK1/2 MAP kinases are involved in c-Fos induction (Bawolak et al., 2009) and the agonist-induced B2 receptor endocytosis is dependent on GRKs and β-arrestins (Charest-Morin et al., 2013).
Compound 1, Compound 2, and Compound 3 are competitive B2 receptor antagonists, as indicated by the surmountable behavior in the human umbilical vein assay. Indeed, very high concentrations of BK can fully overcome the antagonism of the small molecules, showing no reduction of BK Emax value in the presence of antagonist. Despite this undisputable competitive signal, the Schild plot slopes of the small molecule antagonist were somewhat inferior to unity (Table 4), possibly indicating a distortion caused by a lack of equilibrium at the B2 receptor level due to intracellular entry of the drugs at higher concentrations. Cell permeability can be expected of small molecules, this in contrast to the heavier and hydrophilic peptide icatibant.
Our reversibility experiments in the human umbilical vein contraction model (Figure 6), indicate that following Compound 3 treatment and washout, the BK potency was reduced when pretreatment was performed at a concentration of 100 nM, but not at 10 and 1 nM Compound 3. Clearly, Compound 3 bound in a reversible fashion to the B2 receptor, as is obvious by the full efficacy of high concentrations of BK (fully surmountable behavior seen in the 3 h concentration response curve). Therefore we hypothesize that cell permeability of Compound 3 may also here be a plausible explanation of the slower/partial reversibility seen at high concentration of this antagonist (100 nM, Figure 6A). Perhaps the compound enters the intracellular space, and is released slowly after washing. Similar partial/slow reversibility was described for the small molecule B2 receptor antagonist MEN16132 (Meini et al., 2009).
Compound 2 and Compound 3 inhibited endocytosis of the human B2 receptor expressed in HEK 293a cells, in a concentration-dependent manner, with clear efficacy at and above 100 nM for both compounds (Figure 7). The block of BK-induced internalization is a direct consequence of their competitive interaction with BK. BK-induced internalization of the B2 receptor population in cells is fully reversible and not associated with receptor degradation (Blaukat et al., 1996; Bawolak et al., 2007). Further, there is evidence that the naturally expressed B2 receptor is long-lived at the cell surface, relative to the rapid clearance of the highly regulated B1 receptors (Fortin et al., 2003; Enquist et al., 2007). By inhibiting B2 receptor endocytosis, B2 receptor antagonists may be expected to increase surface levels of the B2 receptor, and may theoretically be expected to elicit a BK-mediated rebound effect following washout or metabolism of the small molecules. However, there is currently no evidence of antagonist-induced upregulation of B2 receptor expression, likely indicating that the synthesis rate of the B2 receptor is slow (Marceau et al., 1999; Khosravani et al., 2015), and no indication of an increased BK potency after treatment with a B2 receptor antagonist and washout (Gobeil et al., 1996 and present Figure 6). In man, it was shown that the B2 receptor antagonist icatibant retains its efficacy in patients with hereditary angioedema, even after 5 consecutive doses (Baş et al., 2013; Malbrán et al., 2014). Together these data point to the maintenance of antagonist activity following long-term treatment.
Compound 3, was further characterized for its off-target profile. We found that out of a list of 138 molecular targets, including GPCRs, receptors, ion channels, enzymes and transporters, Compound 3 only interacted with a handful of GPCRs, and only at micromolar concentrations. Compound 3 has no affinity for the human B1 receptor. Comparing Kb values across targets, Compound 3 is shown to be a highly selective B2 receptor antagonist with over 1,000 - fold selectivity versus the following human receptors CRF1, NK1, NK3, thromboxane TP, prostanoid EP1 and vasopressin V1a (i.e. a hitrate of 4.3%). Interestingly, although Compound 3 is an antagonist of U-44069-induced Ca2+ mobilization at recombinant human TP receptors (IC50 value 3.6 μM), when tested against U-46619-induced contractile response mediated by the endogenous human prostanoid TP receptors in the umbilical vein model, no functional antagonism was observed up to 1 μM, confirming the low potency. Also of relevance is the low antagonist potency of Compound 3 for the neurokinin receptors NK1 and NK3 (IC50 values were twice 17 μM), and the low affinity for NK2 (21% inhibition of radioligand binding at 10 μM) this is striking as icatibant binds potently to NK2 receptors with an IC50 value of 420 nM (FDA Pharmacology Review Icatibant).
The discovery of Compound 3, with picomolar antagonist potency and high specificity for the human B2 receptor and predicted oral bioavailability, has provided a significant stepping stone towards the development of a widely usable potent and selective, orally bioavailable B2 receptor antagonist. PHA-022121, currently in phase 1 clinical development, is a close analogue of Compound 3.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité d'éthique de la recherche, CHU de Québec-Université Laval. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FM designed experiments involving the umbilical vein contractility assay, analyzed results and drafted the corresponding parts of the manuscript. XC-M executed experiments related to signaling in HEK 293a cells and performed preliminary analyses. WK executed and analyzed Calcium mobilization assays with HEK293 cells. AL designed the in vitro signal transduction experiments for recombinant B2 receptors from different species, the off-target screening panel, analyzed results and wrote related sections to the manuscript. BL collected and summarized data and contributed to the manuscript writing. CG, H-DA and JS designed Compound 3, supervised the synthesis of Compound 1, Compound 2, and Compound 3 and drafted the corresponding parts of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that this study received funding from Pharvaris Netherlands B.V. XC-M held a Studentship award from Fonds de recherche Santé du Québec.
Conflict of Interest
AL, BL, and JK are consultants for Pharvaris Netherlands B.V. FM served as a consultant and received research funds from Pharvaris Netherlands B.V. CG, H-DA, JS, and WK were employed by AnalytiCon Discovery GmbH.
The remaining author declares that the research was conducted in the absence of any commercialor financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Pharvaris Netherlands B.V. The funder was involved in the decision to submit the paper for publication. The funder had no role in the study design, collection, analysis, interpretation of data, or the writing of this article.
Acknowledgments
We thank Ms. Johanne Bouthillier for technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.00916/full#supplementary-material
References
- Abe Y., Sawada Y., Imai K., Hiroshi Kayakiri H. (2005). Discovery of Non peptide Bradykinin B2 Receptor Antagonists and Agonists. J. Syn. Org. Chem. Jpn. 63, 852–863. 10.5059/yukigoseikyokaishi.63.852 [DOI] [Google Scholar]
- Altura B. M., Malaviya D., Reich C. F., Orkin L. R. (1972). Effects of vasoactive agents on isolated human umbilical arteries and veins. Am. J. Physiol. 222, 345–355. 10.1152/ajplegacy.1972.222.2.345 [DOI] [PubMed] [Google Scholar]
- Baş M., Greve J., Hoffmann T. K., Reshef A., Aberer W., Maurer M., et al. (2013). Repeat treatment with icatibant for multiple hereditary angioedema attacks: FAST-2 open-label study. Allergy 68, 1452–1459. 10.1111/all.12244 [DOI] [PubMed] [Google Scholar]
- Bastian S., Loillier B., Paquet J. L., Pruneau D. (1997). Stable expression of human kinin B1 receptor in 293 cells: pharmacological and functional characterization. Br. J. Pharmacol. 122, 393–399. 10.1038/sj.bjp.0701380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawolak M. T., Gera L., Morissette G., Stewart J. M., Marceau F. (2007). B-9972 (D-Arg-[Hyp3,Igl5,Oic7,Igl8]-bradykinin) is an inactivation-resistant agonist of the bradykinin B2 receptor derived from the peptide antagonist B-9430 (D-Arg-[Hyp3,Igl5,D-Igl7,Oic8]-bradykinin): pharmacologic profile and effective induction of receptor degradation. J. Pharmacol. Exp. Ther. 323, 534–546. 10.1124/jpet.107.123422 [DOI] [PubMed] [Google Scholar]
- Bawolak M. T., Gera L., Bouthillier J., Stewart J. M., Adam A., Marceau F. (2008). A fluorescent version of the bradykinin B2 receptor antagonist B-9430: pharmacological characterization and use in live cell imaging. Peptides 29, 1626–1630. 10.1016/j.peptides.2008.05.007 [DOI] [PubMed] [Google Scholar]
- Bawolak M. T., Fortin S., Bouthillier J., Adam A., Gera L., C-Gaudreault R., et al. (2009). Effects of inactivation-resistant agonists on the signalling, desensitization and down-regulation of bradykinin B2 receptors. Br. J. Pharmacol. 158, 1375–1386. 10.1111/j.1476-5381.2009.00409.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawolak M. T., Lodge R., Morissette G., Marceau F. (2011). Bradykinin B2 receptor-mediated transport into intact cells: anti-receptor antibody-based cargoes. Eur. J. Pharmacol. 668, 107–114. 10.1016/j.ejphar.2011.06.041 [DOI] [PubMed] [Google Scholar]
- Blaukat A., Alla S. A., Lohse M. J., Müller-Esterl W. (1996). Ligand-induced phosphorylation/dephosphorylation of the endogenous bradykinin B2 receptor from human fibroblasts. J. Biol. Chem. 271, 32366–32374. 10.1074/jbc.271.50.32366 [DOI] [PubMed] [Google Scholar]
- Blaukat A., Pizard A., Breit A., Wernstedt C., Alhenc-Gelas F., Muller-Esterl W., et al. (2001). Determination of bradykinin B2 receptor in vivo phosphorylation sites and their role in receptor function. J. Biol. Chem. 276, 40431–40440. 10.1074/jbc.M107024200 [DOI] [PubMed] [Google Scholar]
- Bouillet L., Boccon-Gibod I., Launay D., Gompel A., Kanny G., Fabien V., et al. (2016). Hereditary angioedema with normal C1 inhibitor in a French cohort: Clinical characteristics and response to treatment with icatibant. Immun. Inflammation Dis. 5, 29–36. 10.1002/iid3.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess G. M., Perkins M. N., Rang H. P., Campbell E. A., Brown M. C., McIntyre P., et al. (2000). Bradyzide, a potent non-peptide B(2) bradykinin receptor antagonist with long-lasting oral activity in animal models of inflammatory hyperalgesia. Br. J. Pharmacol. 129, 77–86. 10.1038/sj.bjp.0703012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest-Morin X., Fortin S., Lodge R., Roy C., Gera L., Gaudreault R. C., et al. (2013). Inhibitory effects of cytoskeleton disrupting drugs and GDP-locked Rab mutants on bradykinin B2 receptor cycling. Pharmacol. Res. 71, 44–52. 10.1016/j.phrs.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Charest-Morin X., Raghavan A., Charles M. L., Kolodka T., Bouthillier J., Jean M., et al. (2015). Pharmacological effects of recombinant human tissue kallikrein on bradykinin B2 receptors. Pharmacol. Res. Perspect. 3, e00119. 10.1002/prp2.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicardi M., Aberer W., Banerji A., Bas M., Bernstein J. A., Bork K., et al. (2014). Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 69, 602–616. 10.1111/all.12380 [DOI] [PubMed] [Google Scholar]
- Daray F. M., Minvielle A. I., Puppo S., Rothlin R. P. (2003). Pharmacological characterization of prostanoid receptors mediating vasoconstriction in human umbilical vein. Br. J. Pharmacol. 139, 1409–1416. 10.1038/sj.bjp.0705375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist J., Skröder C., Whistler J. L., Leeb-Lundberg L. M. (2007). Kinins promote B2 receptor endocytosis and delay constitutive B1 receptor endocytosis. Mol. Pharmacol. 71, 494–507. 10.1124/mol.106.030858 [DOI] [PubMed] [Google Scholar]
- Floccard B., Hautin E., Bouillet L., Coppere B., Allaouchiche B. (2012). An evidence-based review of the potential role of icatibant in the treatment of acute attacks in hereditary angioedema type I and II. Core Evid. 7, 105–114. 10.2147/CE.S24743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J. P., Bouthillier J., Marceau F. (2003). High agonist-independent clearance of rabbit kinin B1 receptors in cultured cells. Am. J. Physiol. Heart Circ. Physiol. 284, H1647–H1654. 10.1152/ajpheart.00884.2002 [DOI] [PubMed] [Google Scholar]
- Gera L., Charest-Morin X., Jean M., Bachelard H., Marceau F. (2016). Infrared-emitting, peptidase-resistant fluorescent ligands of the bradykinin B2 receptor: application to cytofluorometry and imaging. BMC Res. Notes 9, 452. 10.1186/s13104-016-2258-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson C., Schnatbaum K., Tradler T., Pfeifer J., Scharn D., Reimer U., et al. (2008). 8-Oxy-quinoline derivatives as bradykinin B2 receptor modulators. WIPO Pantent No. WO2008116620. Geneva, Switzerland: World Intellectual Property Organization. [Google Scholar]
- Gibson C., Schnatbaum K., Pfeier J. R., Locardi E., Paschke M., Reimer U., et al. (2009). Novel small molecule bradykinin B2 receptor antagonists. J. Med. Chem. 52, 4370–4379. 10.1021/jm9002445 [DOI] [PubMed] [Google Scholar]
- Gibson C., Tradler T., Schnatbaum K., Pfeifer J., Locardi E., Scharn D., et al. (2010). Small molecule bradykinin B2 receptor modulators. WIPO Patent No. WO2010031589. Geneva, Switzerland: World Intellectual Property Organization. [Google Scholar]
- Gibson C., Saupe J., Ambrosi H.-D., Haustedt L. O. (2019). Novel bradykinin B2 receptor antagonists. WO2019101906. Geneva, Switzerland: World Intellectual Property Organization. [Google Scholar]
- Gobeil F., Pheng L. H., Badini I., Nguyen-Le X. K., Pizard A., Rizzi A., et al. (1996). Receptors for kinins in the human isolated umbilical vein. Br. J. Pharmacol. 118, 289–294. 10.1111/j.1476-5381.1996.tb15401.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J. F., Borkowski J. A., Macneil T., Stonesifer G. Y., Fraher J., Strader C. D., et al. (1994). Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol. Pharmacol. 45, 1–8. [PubMed] [Google Scholar]
- Hess J. F., Hey P. J., Chen T. B., O'Brien J., Omalley S. S., Pettibone D. J., et al. (2001). Molecular cloning and pharmacological characterization of the canine B1 and B2 bradykinin receptors. Biol. Chem. 382, 123–129. 10.1515/BC.2001.018 [DOI] [PubMed] [Google Scholar]
- Hock F. J., Wirth K., Albus U., Linz W., Gerhards H. J., Wiemer G., et al. (1991). Hoe 140 a new potent and long acting bradykinin-antagonist: in vitro studies. Br. J. Pharmacol. 102, 769–773. 10.1111/j.1476-5381.1991.tb12248.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle S., Landry M., Audet R., Bouthillier J., Bachvarov D. R., Marceau F. (2000). Effect of allelic polymorphism of the B1 and B2 receptor genes on the contractile responses of the human umbilical vein to kinins. J. Pharmacol. Exp. Ther. 294, 45–51. [PubMed] [Google Scholar]
- Hubatsch I., Ragnarsson E. G., Artursson P. (2007). Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2, 2111–2119. 10.1038/nprot.2007.303 [DOI] [PubMed] [Google Scholar]
- Jean M., Raghavan A., Charles M. L., Robbins M. S., Wagner E., Rivard GÉ, et al. (2016). The isolated human umbilical vein as a bioassay for kinin-generating proteases: An in vitro model for therapeutic angioedema agents. Life Sci. 155, 180–188. 10.1016/j.lfs.2016.05.010 [DOI] [PubMed] [Google Scholar]
- Jones C., Phillips E., Davis C., Arbuckle J., Yaqoob M., Burgess G. M., et al. (1999). Molecular characterisation of cloned bradykinin B1 receptors from rat and human. Eur. J. Pharmacol. 374, 423–433. 10.1016/s0014-2999(99)00315-5 [DOI] [PubMed] [Google Scholar]
- Khosravani F., Suvorava T., Dao V. T., Brockmann N., Kocgirli O., Herbst F. F., et al. (2015). Stability of murine bradykinin type 2 receptor despite treatment with NO, bradykinin, icatibant, or C1-INH. Allergy 70, 285–294. 10.1111/all.12556 [DOI] [PubMed] [Google Scholar]
- Leeb-Lundberg L. M., Marceau F., Müller-Esterl W., Pettibone D. J., Zuraw B. L. (2005). International Union of Pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57, 27–77. 10.1124/pr.57.1.2 [DOI] [PubMed] [Google Scholar]
- Liu Z., Khalil R. A. (2018). Evolving mechanisms of vascular smooth muscle contraction highlight key targets in vascular disease. Biochem. Pharmacol. 153, 91–122. 10.1016/j.bcp.2018.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.-X., Wang D.-Z., Ward D. C., Chen L., Dessai T., Chao J., et al. (1994). Structure and chromosomal localisation of the gene (BDKRB2) encoding human bradykinin B2 receptor. Genomics 23, 362–369. 10.1006/geno.1994.1512 [DOI] [PubMed] [Google Scholar]
- Malbrán A., Riedl M., Ritchie B., Smith W. B., Yang W., Banerji A., et al. (2014). Repeat treatment of acute hereditary angioedema attacks with open-label icatibant in the FAST-1 trial. Clin. Exp. Immunol. 177, 544–553. 10.1111/cei.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau F., Regoli D. (2004). Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug Discovery 3, 845–852. 10.1038/nrd1522 [DOI] [PubMed] [Google Scholar]
- Marceau F., Levesque L., Drapeau G., Rioux F., Salvino J. M., Wolfe H. R., et al. (1994). Effects of peptide and nonpeptide antagonists of bradykinin B2 receptors on the venoconstrictor action of bradykinin. J. Pharmacol. Exp. Ther. 269, 1136–1143. [PubMed] [Google Scholar]
- Marceau F., Larrivée J. F., Bouthillier J., Bachvarova M., Houle S., Bachvarov D. R. (1999). Effect of endogenous kinins, prostanoids, and NO on kinin B1 and B2 receptor expression in the rabbit. Am. J. Physiol. 277, R1568–R1578. 10.1152/ajpregu.1999.277.6.R1568 [DOI] [PubMed] [Google Scholar]
- Marceau F., Fortin J. P., Morissette G., Dziadulewicz E. K. (2003). A non-peptide antagonist unusually selective for the human form of the bradykinin B2 receptor. Int. Immunopharmacol. 3, 1529–1536. 10.1016/S1567-5769(03)00180-2 [DOI] [PubMed] [Google Scholar]
- Meini S., Cucchi P., Catalani C., Bellucci F., Giuliani S., Santicioli P., et al. (2009). Pharmacological characterization of the bradykinin B2 receptor antagonist MEN16132 in rat in vitro bioassays. Eur. J. Pharmacol. 615, 10–16. 10.1016/j.ejphar.2009.04.057 [DOI] [PubMed] [Google Scholar]
- Munoz C. M., Leeb-Lundberg L. M. F. (1992). Receptor-mediated internalization of bradykinin. DDT1 MF-2 smooth muscle cells process internalized bradykinin via multiple degradation pathways. J. Biol. Chem. 267, 303–309. [PubMed] [Google Scholar]
- Neubig R. R., Spedding M., Kenakin T., Christopoulos A., International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification (2003). VIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 55, 597–606. 10.1124/pr.55.4.4 [DOI] [PubMed] [Google Scholar]
- Obach R. S. (1999). Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 27, 1350–1359. [PubMed] [Google Scholar]
- Paquet J.-L., Luccarini J.-M., Fouchet C., Defrêne E., Loillier B., Robert C., et al. (1999). Pharmacological characterization of the bradykinin B2 receptor: inter-species variability and dissociation between binding and functional responses. Br. J. Pharmacol. 126, 1083–1090. 10.1038/sj.bjp.0702403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneau D., Luccarini J.-M., Fouchet C., Defrêne E., Franck R.-M., Loillier B., et al. (1998). LF 16.0335, a novel potent and selective nonpeptide antagonist of the human bradykinin B2 receptor. Br. J. Pharmacol. 125, 365–372. 10.1038/sj.bjp.0702083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogines-Velo M., Pelorosso F., Zold C., Nowak W., Pesce G., Sardi S., et al. (2002). Characterization of 5-HT receptor subtypes mediating contraction in human umbilical vein. 1. Evidence of involvement of 5-HT2A receptors using functional and radioligand binding assays. Naunyn-Schmied Arch. Pharmacol. 366, 587–595. 10.1007/s00210-002-0636-9 [DOI] [PubMed] [Google Scholar]
- Shakur H., Andrews P., Asser T., Balica L., Boeriu C., Quintero J. D., et al. (2009). The BRAIN TRIAL: a randomised, placebo controlled trial of a bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 10, 109. 10.1186/1745-6215-10-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida R. J., Murray R. B. (1987). Manual of Pharmacologic Calculations with Computer Programs (New York: Springer; ). [Google Scholar]
- Tenti S., Pascarelli N. A., Cheleschi S., Guidelli G. M., Fioravanti A. (2016). The emerging role of bradykinin in the pathogenesis of osteoarthritis and its possible clinical implications. Curr. Rheumatol. Rev. 12, 177–184. 10.2174/1573397112666160331143305 [DOI] [PubMed] [Google Scholar]
- Whalley E. T., Figueroa C. D., Gera L., Bhoola K. D. (2012). Discovery and therapeutic potential of kinin receptor antagonists. Expert Opin. Drug Discovery 7, 1129–1148. 10.1517/17460441.2012.729038 [DOI] [PubMed] [Google Scholar]
- Wu M. A., Zanichelli A., Mansi M., Cicardi M. (2016). Current treatment options for hereditary angioedema due to C1 inhibitor deficiency. Expert Opin. Pharmacother. 17, 27–40. 10.1517/14656566.2016.1104300 [DOI] [PubMed] [Google Scholar]
- Zhang S.-P., Wang H.-Y., Lovenberg T. W., Codd E. E. (2001). Functional studies of bradykinin receptors in Chinese hamster ovary cells stably expressing the human B bradykinin receptor. Int. Immunopharmacol. 1, 955–965. 10.1016/s1567-5769(01)00032-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.