

Abstract
GABAergic interneurons comprise a small but a diverse subset of neurons in the mammalian brain, tightly regulating neuronal circuit maturation and information flow, and ultimately, animal behavior. Because of their centrality in the etiology of numerous neurological disorders, examining their molecular architecture under different physiological scenarios has piqued the interest of the broader neuroscience community. The last few years have seen an explosion in next-generation sequencing (NGS) approaches aimed at identifying genetic and state dependent subtypes in neuronal diversity. While several approaches are employed to address neuronal molecular diversity, ribosomal tagging has emerged at the forefront of identifying the translatome of the neuronal subtypes. This approach primarily relies on Cre-recombinase-driven expression of hemagglutinin A (HA)–tagged Ribotag mice, exclusively in the neuronal subtype of interest. This allows for immunoprecipitation of cell-type-specific, ribosome-engaged mRNA, expressed both in the soma and the neuronal processes, for targeted quantitative real-time PCR (qRT-PCR) or high-throughput RNAseq analyses. The current protocol will examine the typical technical caveats associated while successfully applying the Ribotag technique for the analysis of GABAergic interneurons, and in theory, for other sparse cell-types in the central nervous system.
Keywords: GABAergic, interneuron, immunoprecipitation, Ribotag, Translation affinity purification TRAP RNAseq
INTRODUCTION:
In cortico-hippocampal circuits, it is estimated that inhibitory, GABA-releasing-interneurons account for roughly 10–15% of all neurons. Despite being a minority, they exert a robust control over neural circuits; hence, information processing and animal behavior (Pelkey et al., 2017; Fishell and Rudy, 2011). While the primary output of a majority of cortico-hippocampal circuits is glutamate-releasing excitatory neurons, the diverse cohorts of the GABAergic neurons target different subcellular regions of the excitatory neurons, differentially sculpting their ability to transmit the information forward. Such domain-specific inhibition is mediated by the diverse subtypes that exist within the GABAergic interneurons. In addition to the pyramidal neuron targeting domain through their distinct axonal and dendritic morphologies, interneurons are also distinctly diverse based on their embryonic origins, molecular, and electrophysiological properties (DeFelipe et al., 2013). Decades of research has now established that GABAergic interneurons can be broadly classified based on their embryonic origins from the medial ganglionic or the caudal-ganglionic eminence (MGE/CGE) in the developing telencephalon (Wamsley and Fishell, 2017). In the adult cortex and hippocampus, MGE-derived interneurons comprise a significant proportion in comparison to the CGE-derived interneurons, roughly at a ratio of 70%:30% (Fishell and Rudy, 2011). While MGE-derived interneurons comprise interneuron classes namely, the parvalbumin (PV), somatostatin (SST), neurogliaform (NGFC) and ivy cells, the CGE-derived interneurons include cholecystokinin (CCK), vaso-intestinal peptide (VIP), calretinin (CR) and a distinct subset of NGFCs (Pelkey et al., 2017). In recent times, multiple genetic lines expressing GABAergic-subtype specific Cre-recombinase are available, targeting an even-minuscule subset of all neurons (Taniguchi, 2014; Taniguchi et al., 2011). Because of their essential and unique roles during regulation of circuit function and animal behavior, significant interest has aligned towards understanding the molecular makeup of these smaller subsets of interneurons; particularly during naïve and behaviorally active animal states (Caroni, 2015), or in different animal models of neurological disorders (Marín, 2012).
It is also clear that the neurons exploit compartmentalized mRNA translation that is permissive to nucleus-independent machinery to regulate the cellular transcriptome/proteome, thereby regulating their axonal and dendritic function (Biever et al., 2019a; Rangaraju et al., 2017; Spaulding and Burgess, 2017). While numerous next-generation sequencing (NGS) approaches are applied towards understanding the molecular diversity of these GABAergic interneurons (Zeisel et al., 2018; Tasic et al., 2016; Poulin et al., 2016; Cembrowski and Menon, 2018; Mayer et al., 2018; Close et al., 2017; Hodge et al., 2019; Harris et al., 2018; Bakken et al., 2018; Mi et al., 2018), the most popular single-cell RNAseq approaches utilize the somatic and nuclear-expressed mRNAs for sequencing. Therefore, a parallel NGS platform to examine the cellular translation machinery that is inclusive of the mRNAs from the axonal and dendritic processes are crucial in the NGS toolbox to examine the neuronal translatome. This is particularly important, considering several crucial neuronal functions during development and plasticity involve a robust mRNA translation (Sossin and Costa-Mattioli, 2019; Buffington et al., 2014), and many neurological disorders exhibit aberrant neuronal translational machinery (Kapur and Ackerman, 2018; Tahmasebi et al., 2018; Farley-Barnes et al., 2019). The Ribotag mice are ideally suited for studying the translatome through the expression of HA-tagged ribosomal subunit protein Rpl22, in a cell-type-specific, Cre recombinase-dependent manner (Sanz et al., 2009). While the existing Ribotag protocols detail the caveats of this technique (Sanz et al., 2019; Lesiak et al., 2015), it is still not ideally suited for examining sparse cell populations such as the subtypes of cortico-hippocampal GABAergic interneurons, which typically account for roughly 1–5% of all neurons. The current protocol streamlines the existing protocol to be amenable for Ribotag/translatome analysis of these sparser populations using the emerging GABAergic-subtype specific Cre-recombinase expressing mouse lines. In theory, these guidelines will also benefit applications of Ribotag during the analysis of other sparser populations.
In the current protocol, we will systematically establish the application of Ribotag technique in the Nkx2.1 Cre mouse line, which fate-maps to MGE-derived interneurons (Xu et al., 2008), ultimately revealing the translatome of the PV, SST and NGFC subsets of GABAergic interneurons. Because these neurons collectively amount to ~5–7% of all cortico-hippocampal neurons, the protocol described here is a reasonable representation of a sparse cell population. Basic Protocol 1 describes the establishment of a successful assay design specific for generating the MGE-translatome. Without accounting for every parameter described, it is futile to directly attempt the Ribotag technique, particularly for sparse cell populations. This involves in silico analyses of Rpl22 gene expression in the cell-type of interest, based on existing scRNAseq public datasets. Based on this, one can proceed to determine whether the Ribotag technique can be applied or not to the cell-type(s) of interest and whether additional tweaks are necessary to optimize the availability of Rpl22 transgene levels. Basic Protocol 2 describes the changes in the existing protocol amenable for its applicability in sparse cell populations and the ones that express low levels of Rpl22.
STRATEGIC PLANNING
Ribosomal tagging using ribotag mice has been in use for a decade now, and there are several excellent descriptions of this method available (Sanz et al., 2009, 2019; Lesiak et al., 2015). However, because the technique relies on immunoprecipitation (IP) of transgene Rpl22-HA expressed in a sparse cell population from a pool of the entire tissue lysate, every attempt should be made to enhance the enrichment level of Rpl22-HA. Based on our experience, there are a few critical parameters that need to be ascertained, unique for every rare cell-type of interest. As an example, we will establish the critical steps that will sequentially convince oneself about the success of the strategy for MGE-derived GABAergic interneurons, by utilizing the Nkx2.1 Cre recombinase mouse line readily available from Jax Laboratories. With the detailed guidelines indicated below, it will be straightforward to apply this pipeline to any cell-type-specific promotor-driven Cre recombinase transgenic lines targeting less other abundant cell-types.
Establishing appropriate cell-type-specific assay design
1. In Silico Analysis of Rpl22 Expression Levels
Because this technique relies on immunoprecipitation of Rpl22-HA, it is essential first to ascertain whether the cell-type of interest expresses any Rpl22, preferably at the animal age when the assay is performed. One assumption that is ubiquitous in the Ribotag literature and one which is rarely discussed is that, different cell-types may express different levels of Rpl22. Because of the availability of several high-throughput single-cell RNA sequencing (scRNAseq) datasets examining the mouse brain in the public domain, it is relatively straightforward to ascertain Rpl22 levels in the cell-type of interest.
Steps:
Open the Dropviz portal at http://dropviz.org/ and select the “Search by gene” tab
-
Include the Gene name “Rpl22” in the query tab and limit the “Brain Region” intended “Cell Class” “Cell Cluster” names, and hit the “Update” tab.
The term “Cell Classes” will include the broad cell-types (neurons, non-neurons, etc.) and, the term “Cell Cluster” will include the brain subregion specific Cell Class subtypes. In our present example, we test the expression of Rpl22 in Hippocampus in all cell classes and the “Interneuron” subcluster.
The results can be viewed in multiple formats indicated in the server. However, in this example, we check the “Table” format and download the results for annotation and preparing heatmaps.
Heatmaps for the downloaded cell-type-specific expression matrix for Rpl22 can be generated using the Morpheus web-resource at https://software.broadinstitute.org/morpheus/.
Copy and paste the cell-type-specific expression matrix for Rpl22 obtained from Dropviz portal into the Morpheus portal, and assign the rows and colums that need to be plotted. Heat-map visualization features can be altered by toggling with the the “Options” tab. In addition, clustering of the rows or columns can be performed with the “Tools” tab, and the heat-map can be downloaded in appropriate formats with the “Save image” tab.
In the example below, we utilize the publicly available scRNAseq dataset from the mouse brain (dropviz.org), and we observe a relatively lesser expression of Rpl22 in neurons in comparison to non-neurons and new-born neurons in the adult mouse hippocampus (Fig.1A). Additionally, it is clear that the MGE derived PV, SST, and NGFC subtypes of GABAergic interneurons express the lowest levels of Rpl22 among hippocampal neurons (Fig.1B, C). This low Rpl22 expression can negatively impact the integrity of the results unless the experimental design is tweaked.
Figure 1. In silico analysis of Rpl22 expression in adult hippocampal different celltypes (obtained from Saunders et al., 2018).
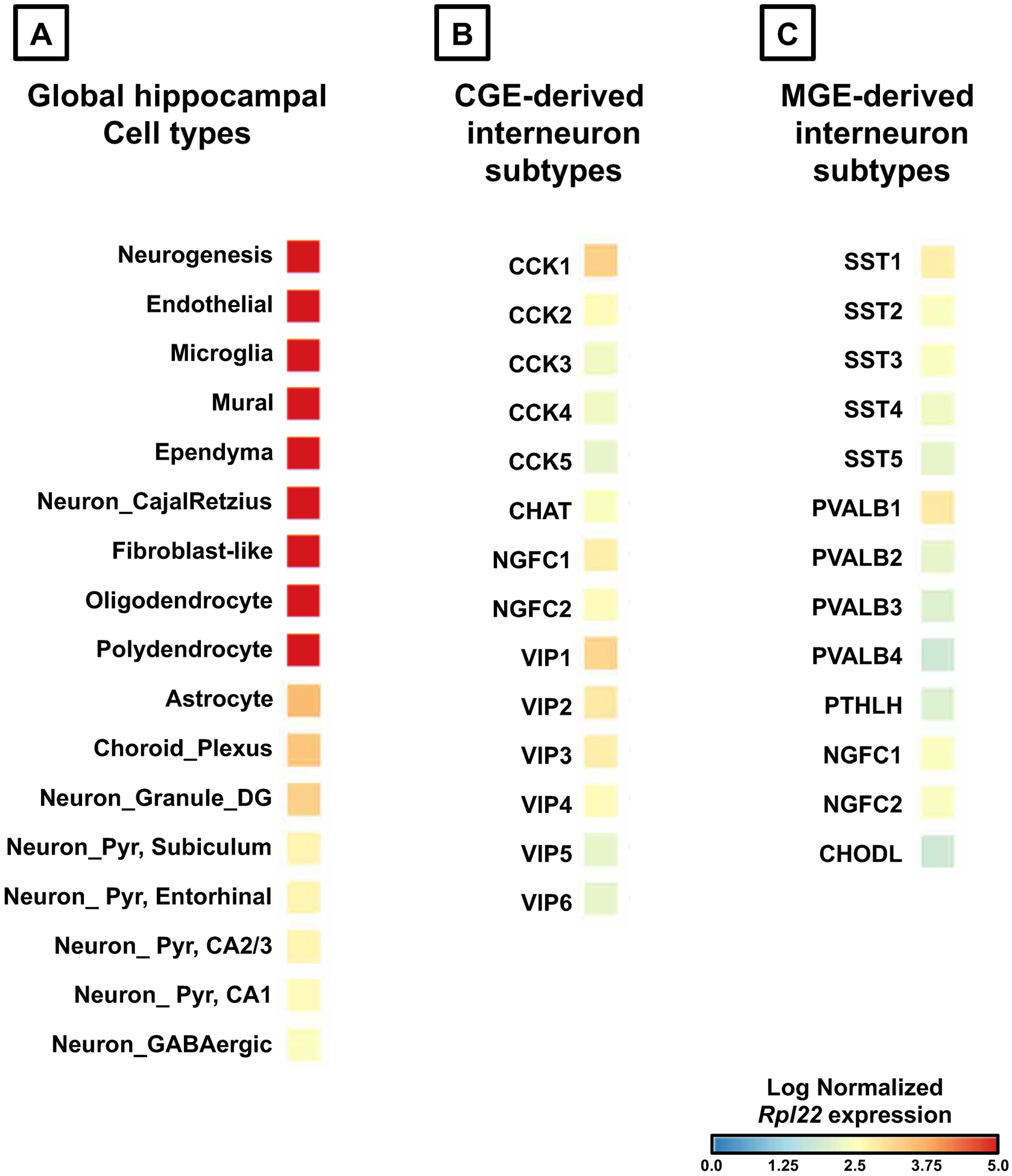
(A) Indicates the expression of Rpl22 in global cell classes, namely the neurons, and non-neurons. (B) Indicates Rpl22 expression in GABAergic interneuron subtypes derived from the CGE. (C) Indicates the expression in GABAergic interneuron subtypes derived from the MGE. The heatmaps indicate the log normalized expression levels of Rpl22 ranging from minimum of no expression (indicated in blue), to maximum expression (indicated in red). Abbreviations: CA1/2/3, Cornu Ammonis subfields in the hippocampus; DG, Dentate Gyrus; Pyr, pyramidal neurons; CGE, caudal ganglionic eminence; MGE, medial ganglionic eminence; Interneuron subtype markers: CCK, cholecystokinin; CHAT, choline acetyltransferase; NGFC, neurogliaform; VIP, vasoactive intestinal polypeptide; SST, somatostatin; PVALB, parvalbumin; PTHLH, parathyroid hormonelike hormone; CHODL, chondrolectin.
BASIC PROTOCOL 1
In this section, we demonstrate that for certain Cre recombinase-expressing mouse lines, the Ribotag allele need to be homozygously expressed to make-up for the low expression of endogenous Rpl22 gene. We describe the mouse breeding and PCR validation steps to accommodate this change.
Mouse Breeding To Obtain Ribotag Homozygosity
The published literature on the Ribotag application primarily utilizes experimental mice that are bred between Cre recombinase-expressing mice and homozygous floxed RiboTag mice (Rpl22HA/HA). Therefore, the experimental mice are heterozygous for the RiboTag allele (Rpl22HA/WT) (Sanz et al., 2019; Spiegel et al., 2014). This means that only half of the available Rpl22-assembled ribosomes and their bound mRNAs are directly available for immunoprecipitation and subsequent mRNA analyses. This is a perfectly normal strategy provided the cell-type under examination are abundant in the nervous system (for example, the ones indicated in Red, in Fig.1A. However, because of low Rpl22 levels, this may seriously impair the application of Ribotag strategy in the MGE subtypes. Therefore, for the MGE subtypes that are sparse populations, and express low levels of Rpl22, the heterozygous Ribotag mouse Rpl22HA/WT that are Nkx2.1 Cre recombinase positive, needs to be bred further to generate an Nkx2.1 Cre recombinase positive: (Rpl22HA/HA). This would ensure that all Rpl22 alleles are express the HA-tag and the entire repertoire of Rpl22-assembled ribosomes, and their associated mRNAs are directly available for immunoprecipitation, and subsequent mRNA analyses.
Materials
Nkx2.1 Cre driver line, https://www.jax.org/strain/008661
Ribotag mouse line, https://www.jax.org/strain/011029
Mouse tail-snip samples for PCR
DNeasy Blood & Tissue Kit, Qiagen (Cat. No. 69504)
Proteinase K, Qiagen (Cat. No. 19131)
PCR qualified water (DNase, RNase Protease free), Quality Biologicals (Cat. No. 351161671)
Platinum Taq DNA Polymerase, ThermoFisher Scientific (Cat. No. 10966026)
Taq DNA Polymerase PCR Buffer (10X), ThermoFisher Scientific (Cat. No. 18067017)
25mM MgCl2, ThermoFisher Scientific (Cat. No. R0971)
2.5mM dNTP mix, ThermoFisher Scientific (Cat. No. R72501)
FastStart PCR Master Mix, Roche (Cat. No. 4710436001)
- Primers for Nkx2.1 Cre recombinase genotyping
- Nkx2.1-Cre recombinase cDNA Forward: 5′- CTCTGGTGGCTGCCTAAAAC-3′
- Nkx2.1-Cre recombinase cDNA Reverse: 5′- CGGTTATTCAACTTGCACCA-3′
- Primers for Ribotag genotyping
- Ribotag loxP cDNA Forward: 5′- GGGAGGCTTGCTGGATATG -3′
- Ribotag loxP cDNA Reverse: 5′- TTTCCAGACACAGGCTAAGTACAC -3′
Eppendorf Fast PCR Tube Strips, 0.1 mL, with cap strips (Cat. No. 0030124928)
DNA Gel Loading Solution 5X, Quality Biologicals (Cat. No. 351028661)
UltraPure 10X TBE Buffer, ThermoFisher Scientific (Cat. No. 15581044)
SYBR Safe DNA Gel Stain, ThermoFisher Scientific (Cat. No. S33102)
GeneRuler 100 bp DNA Ladder, ThermoFisher Scientific (Cat. No. SM0243)
UltraPure Agarose, ThermoFisher Scientific (Cat. No. 16500100)
CoolRack XT PCR96 PCR cooler plate, Biosicion (Cat. No. BCS-529)
Milli-Q water
C1000 Touch Thermal Cycler, Biorad
Agarose gel casting and running setup
Method
-
Set up the breeding of Nkx2.1 Cre recombinase (homozygous) male mice with Ribotag (Rpl22HA/HA) female mice. The first progeny mice are going to be heterozygous for both Nkx2.1 and Ribotag alleles (Fig.2A). Set up a second generation breeding to identify the mice that are Nkx2.1 Cre recombinase positive and Ribotag homozygous Rpl22HA/HA. For the second-generation litter, perform tail snips according to institutional and ethical guidelines. Extract the genomic DNA from the tail snips using the DNeasy Blood & Tissue Kit, according to manufacturer’s guidelines.
All subsequent PCR reactions must include tail snips from the Nkx2.1 Cre recombinase homozygous parent and Ribotag homozygous parent to serve as positive controls for the genotyping. A wild-type mouse without any of the above alleles must be used as a negative control for the genotyping. Only based on the appearance / non-appearance of corresponding bands in positive and negative controls respectively, can we confidently determine whether or not the unknown tail sample is homozygous or heterozygous for the allele of interest (Fig.2B, C).
- For the Nkx2.1 Cre genotyping, prepare the following master mix per reactions on a PCR cooler plate:
- 1.8μl of the 10X Taq DNA Polymerase PCR Buffer
- 1.74μl of 25mM MgCl2
- 2.5μl of 2.5mM dNTP mix
- 0.55μl of 20μM Stock Ribotag Forward Primer
- 0.55μl of 20μM Stock Ribotag Reverse Primer
- 0.2μl of Platinum Taq DNA Polymerase
- 11.43μl of PCR qualified water
- 4μl of genomic tail DNA
- For the Ribotag genotyping, prepare the following master mix per reactions on a PCR cooler plate:
- 2μl of the 10X Taq DNA Polymerase PCR Buffer
- 2μl of 25mM MgCl2
- 2μl of 2.5mM dNTP mix
- 0.5μl of 20μM Stock Ribotag Forward Primer
- 0.5μl of 20μM Stock Ribotag Reverse Primer
- 0.1μl of Platinum Taq DNA Polymerase
- 10.9μl of PCR qualified water
- 2μl of genomic tail DNA
For both PCR reactions, calculate the master mix for 3–5 reactions more than what is required and using a clean workbench set up the reactions in Eppendorf Fast PCR Tube Strips, on a PCR cooler plate. Gently tap to mix the contents and spin the tubes to prevent any air bubble at the bottom of the tube. Also, include a sample that contains no genomic tail DNA sample, supplemented only with the same volume of PCR qualified water, to serve as a “water-only” negative control.
- Set up the following thermal cycling profile for Nkx2.1-Cre reaction:
1 cycle 94°C 2 min 10 cycles 94°C 20sec 65°C (−1.5°C per cycle) 15sec 68°C 10sec 28 cycles 94°C 15sec 50°C 15sec 72°C 10sec 1 cycle 72°C 2min 10°C hold - Set up the following thermal cycling profile for Ribotag reaction:
1 cycle 95°C 3 min 40 cycles 95°C 20sec 55°C 20sec 72°C 30sec 1 cycle 72°C 5min 10°C hold During the PCR reaction, prepare 100ml of 2% agarose gel in 0.5X TBE buffer, supplemented with 10μl of SYBR Safe DNA Gel Stain (at 1:10,000), made up with Milli-Q water.
5μl of the PCR amplicon is mixed with 1–2μl of the DNA gel loading buffer and tap to mix the contents.
Once the gel is solidified, load 5μl of GeneRuler 100 bp DNA Ladder, along with the unknown test samples, positive and negative control PCR amplicons.
Run PCR products on 2% agarose gel
- Visualize the 2% agarose gel and take a picture of results (Fig.2C).
- The Nkx2-1 Cre recombinase PCR genotyping will not distinguish between homozygosity and heterozygosity. The appearance of an amplicon band at 410 bp will indicate the presence of Nkx2.1 Cre recombinase.
- The Ribotag genotyping is capable of distinguishing between homozygosity and heterozygosity. The appearance of an amplicon band at 243bp will indicate the presence of wild-type Rpl22 (Rpl22WT/WT); a band at 290bp will indicate the presence of homozygous Ribotag (Rpl22HA/HA), and appearance of two amplicon bands at 243bp and 290bp will indicate the presence of heterozygous Ribotag (Rpl22HA/WT) (Fig.2C).
- For subsequent experiments we will require the following genotypes (Fig.2B):
- Nkx2.1 Cre positive, homozygous Ribotag (Rpl22HA/HA) subsequently termed as MGE-Ribohomo
- Nkx2.1 Cre positive, heterozygous Ribotag (Rpl22WT/HA) subsequently termed as MGE-Ribohet;
- Nkx2.1 Cre negative, homozygous Ribotag (Rpl22HA/HA) subsequently termed as MGE-Cre−/− (as a negative control).
Maintain 4–6 animals per experimental group (as indicated above), with an equal mix of males and female mice for subsequent experiments.
Figure 2. Schematic overview of the breeding strategy and Ribotag assay in Nkx2.1 cre driver line.
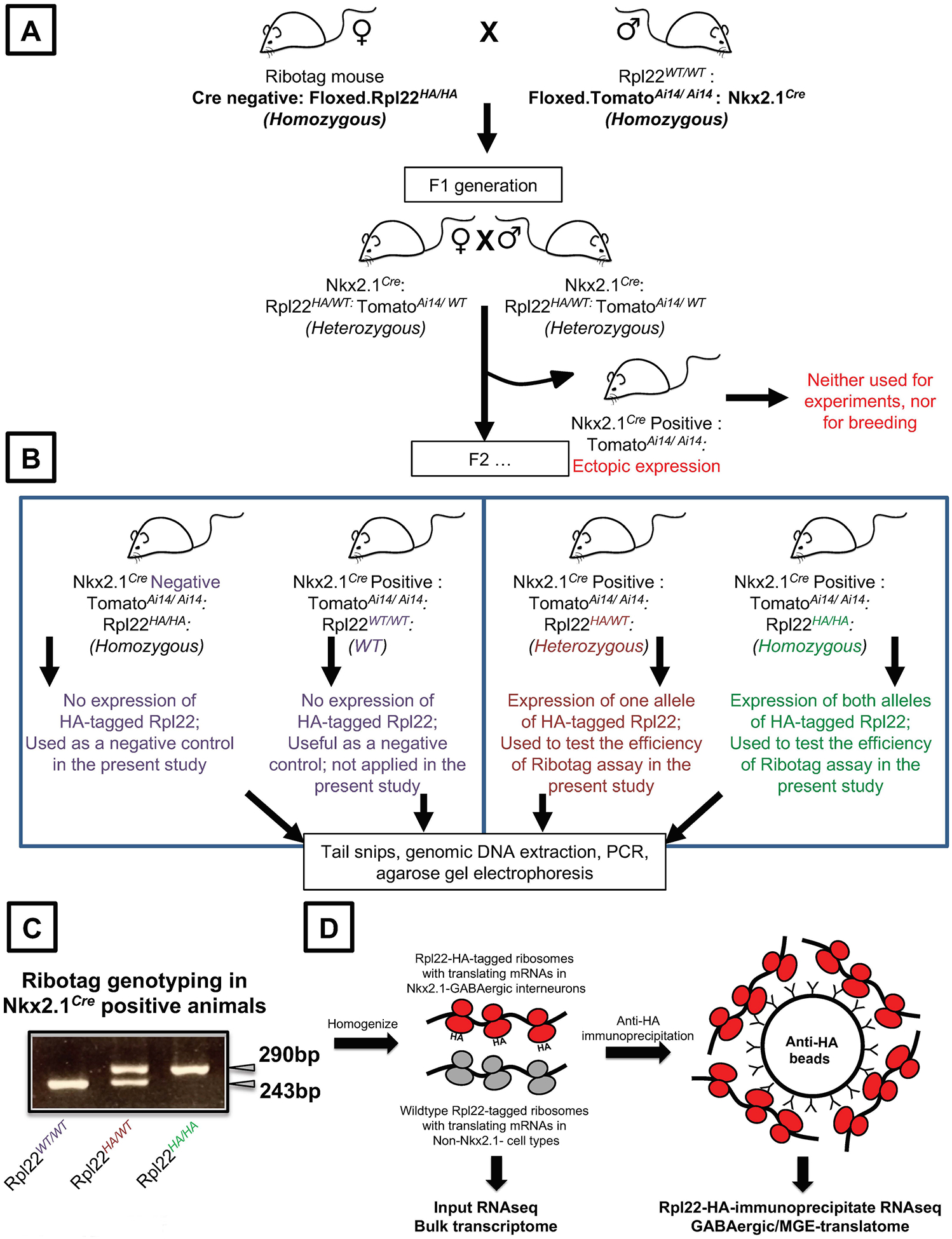
(A) Cre-negative Floxed Ribotag homozygous male mouse are bred with Nkx2.1 Cre positive, mouse containing Ai14 reporter female mouse to obtain a F1 progeny that are heterozygous for Rpl22-HA allele. All litters with ectopic Ai14 expression indicating a faulty Nkx2.1 expression, are to be discarded for further breeding or experiments (B) F1 progeny that are heterozygous for the alleles are bred to generate the F2 progeny. Only the Cre-genotyping positive, Ai14 positive, Ribotag homozygotes or heterozygotes are used for subsequent experiments and for future breeding (right). Cre negative and animals containing WT Rpl22 are used as negative control, for the Ribotag assays (left). (C) An example agarose gel electrophoresis indicating the PCR amplification of Rpl22WT/WT, Rpl22HA/WT, Rpl22HA/HA, alleles. (D) An outline of the Ribotag assay. Tissues obtained from the animals indicates in (B, C) are homogenized and the MGE-expressed, Rpl22-HA-associated mRNA are immunoprecipitated and sequenced in parallel with the bulk mRNA.
Alternate Protocol to Detect Ectopic Cre Recombinase Expression
Many Cre-driver lines are notorious in their ‘ectopic’ expression of Cre recombination, outside of the intended cell-type (Song and Palmiter, 2018; Harris et al., 2014; Luo et al., 2020). This can be detected by standard approaches described elsewhere (Sanz et al., 2019; Luo et al., 2020). Alternatively, a Cre recombinase sensitive fluorescent reporter line can be included in the genetic crosses that can readily indicate ectopic expression in the Nkx2.1 Cre driver line. In our lab, once we obtained MGE-Ribohomo, we have crossed these mice with the Ai14 mice (https://www.jax.org/strain/007908) expressing tdTomato, to visualize the MGE interneurons expressing Cre recombinase. However, when ectopic Cre recombinase expression occurs, tdTomato expression is evident in all ectopic cells, and in many cases, the entire pup will ‘glow red’ when viewed under appropriate light source (such as FS/ULS-02G2, available at http://bls-ltd.com/lightsource.html). Pups expressing ectopic Cre recombinase expression, evident by global tdTomato expression, must be identified in every litter, and the animals must be excluded from subsequent analysis (Fig.2A).
In the Nkx2.1 Cre driver line, we observe ~5–10% of litters to have ectopic Ai14 expression, similar to what is reported (Luo et al., 2020). These litters and their corresponding breeders will not be used for subsequent experiments or future breeding (Fig.2B).
In addition to ectopic expression, it is also essential to verify the expression of the Ribotag allele (homo/het) in only the cell-type of interest by using additional techniques such as immunostaining with anti-HA antibody along with appropriate control antibodies. Using standard techniques elaborated elsewhere (Sanz et al., 2009), we observe robust coexpression of anti-HA immunostaining along with tdTomato expression in mouse hippocampal layers in the MGE-Ribohomo mouse (Fig. 3A, B, C).
Figure 3. Validation of Rpl22HA/HA expression in the hippocampus of Nkx2.1 Cre mice.
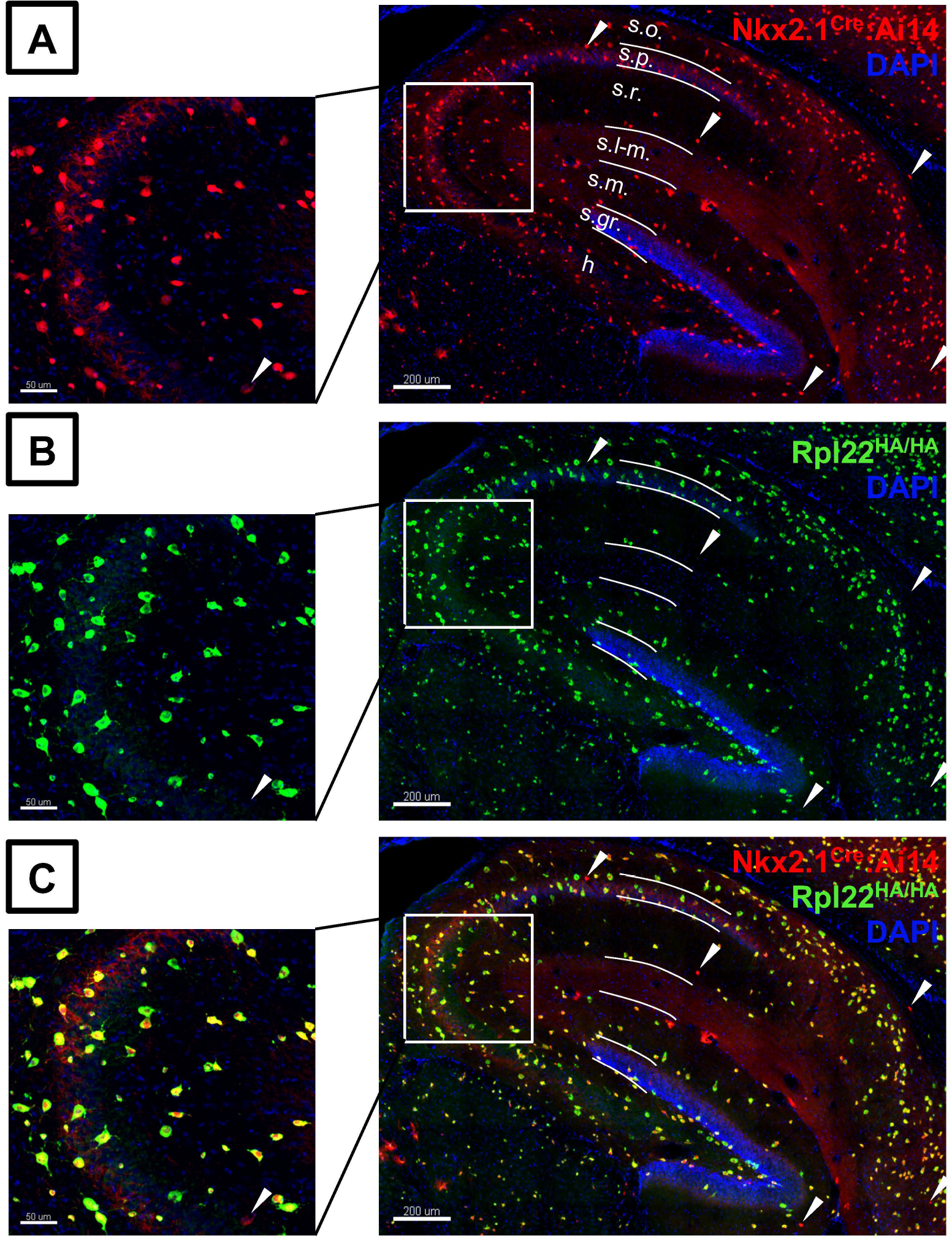
(A) tdTomato Ai14 and (B) HA expression in Nkx2.1 Cre positive, homozygous Ribotag (Rpl22HA/HA) / MGERibohomo mouse line. (C) High fidelity of HA-immunostain co-expressed with tdTomato signal across all fields of hippocampus (Scale bars 200μM. Higher magnification images in CA3 subfield indicated by the white squares (Scale bars 50μM). White arrow heads indicate minimal tdTomato signal that does not contain HA-immunostaining. Abbreviations: s.o., Stratum oriens; s.p., Stratum pyramidale; s.r., Stratum radiatum; s.l-m., Stratum lacunosum-moleculare; s.m., Stratum moleculare; S.gr., Stratum granulosum; h., Hilus.
BASIC PROTOCOL 2
The Ribotag Assay
The Ribotag assay in different cell-types along with subtle variations are described (Sanz et al., 2009, 2019; Doyle et al., 2008; Lesiak et al., 2015; Heiman et al., 2014; Srinivasan et al., 2016; Furlanis et al., 2019). In a sparse cell-type that expresses quite low activity of the endogenous Rpl22 gene locus (such as the MGE interneurons), expressing the Ribotag allele as heterozygote (Rpl22HA/WT), and after freezing and thawing the tissue, it is not surprising that one may not see any significant cell-type-specific gene enrichments, indicating an unsuccessful Ribotag experiment. Because of these experimental considerations, applying the Ribotag strategy in the same way as described in the literature for abundant cell-types is bound to fail when applied to rare cell populations. The aim of this protocol is to make the community cognizant of these critical parameters while using Ribotag for analyses of sparse cell-types, in this case, GABAergic interneurons.
Materials
DNA LoBind Tubes, 1.5 mL, 2.0 mL, Eppendorf (Cat. No. 022431021, 022431048)
ART Barrier Pipette Tips, 1000, 200, 20 & 10μl, ThermoFisher Scientific (Cat. No. 2380, 2770, 2149, 2140)
96-well Plates, Corning (Cat. No. 3596)
BCA Protein Assay Kit, ThermoFisher Scientific (Cat. No. 23227)
RNA 6000 Pico Kit, Agilent (Cat. No. 5067-1513)
RNeasy Plus Micro Kit (50), Qiagen (Cat. No. 74034)
RNase-Free DNase Set (50), Qiagen (Cat. No. 79254)
Anti-hemagglutinin (HA) tag antibody, Abcam (Cat. No. ab9110, Lot GR31995533), RRID AB_307019
(Alternate product - Anti-HA magnetic beads conjugate, ThermoFisher Scientific, Cat. No. 88836)
Dynabeads Protein G, ThermoFisher Scientific (Cat. No. 10004D)
NP-40 Surfact-Amps™ Detergent Solution, ThermoFisher Scientific (Cat. No. 28324)
RNaseZap RNase Decontamination Solution, ThermoFisher Scientific (Cat. No. AM9780)
Complete Protease Inhibitor Cocktail (50X and 10X), Millipore Sigma (Cat. No. 11697498001, 4693159001)
Refrigerated centrifuge
Standard spectrophotometer
2100 Bioanalyzer System, Agilent
DynaMag-2 Magnet, ThermoFisher Scientific (Cat. No. 12321D)
Benchtop tube revolver (such as Crystal Industries, Cat. No. TR-02UA)
Isoflurane, (also labelled as FORANE, USP) Liquid For Inhalation, Baxter (Cat. No. NDC 10019-360-40)
Decapitator, Stoelting (Cat. 51330)
Polystyrene petri dishes, Sigma-Aldrich (Cat. No. P5856)
Mouse brain dissection set, World Precision Instruments (Cat. No. MOUSEKIT)
Feather Scalpel Handle with Blade, Disposable, Electron Microscopy Sciences, VWR (Cat. No. 100499-586)
KIMBLE Dounce Homogenizer sets, Sigma (Cat. No. D8938, D9063)
Liquid nitrogen-cooled Mortar and pestle set, Thomas Scientific (Cat H37260-0100)
CoolRack M30 cooler plate, Biosicion (Cat. No. BCS-108)
Sufficient ice and dry ice
Stock Solutions:
1M Tris-Hydrochloride Buffers, pH7.5, Corning (Cat. No. 46-030-CM), Stored at 4°C
1X Phosphate-Buffered Saline, without calcium and magnesium, PH 7.4, Corning (Cat. No. 21-040-CV)
1M MgCl2, Quality Biologicals (Cat. No. 351-033-721), Stored at 4°C
Potassium chloride, 3M stock solution from prepared from, Sigma (Cat. No. P9541), Stored at 4°C
Cycloheximide (CHX), 10 mg/ml stock solution from prepared Sigma (Cat. No. C7698), Stored at −20°C
Heparin, 100 mg/ml stock solution prepared from, Sigma (Cat. No. H3393), Stored at −20°C
DL-Dithiothreitol (DTT), 2M and 100 mM stocks prepared from, Sigma (Cat. No. D9779), Stored at −20°C
RNasin Plus RNase Inhibitor, Promega (Cat. No. N2615), Stored at −20°C
UltraPure DNase/RNase-Free Distilled Water, ThermoFisher Scientific (Cat. No. 10977023)
Tissue homogenization buffer (see recipe below)
High-salt immunoprecipitation wash buffer (see recipe below)
Method
- It is essential to perform the following tasks immediately prior, or in some cases, the day before starting the Ribtoag assay:
- label and pre-chill sufficient tubes at 4°C
- Assign a separate space for the RNA-work by spraying with RNaseZap and placing bench-sheets
- clean and autoclave the dissection tools, spray equipments with copious RNaseZap before use
- clean the Dounce tissue grinder with copious RNaseZap, and UltraPure distilled water
- check the availability of MGE-Cre−/−, MGE-Ribohet, MGE-Ribohomo animals, and mark them the day prior
- On the day of the experiment, add fresh CHX, DTT, RNasein, and protease inhibitor cocktail to the homogenization buffer and add appropriate volumes of homogenization buffer into a clean Dounce Homogenizer set.
- All the steps need to be performed at 4°C, either on ice / on pre-chilled CoolRacks or inside a refrigerator (in case of the immunoprecipitation step), and the centrifuge must be pre-chilled to 4°C. Use LoBind tubes to reduce non-specific binding of RNA to the polypropylene tubes and use barrier pipette tips to prevent cross-contamination of RNA across samples.
- We use 10% (w/v) of homogenization buffer to perform the Ribotag assay in the Ribohomo genotype, and 3% (w/v) of homogenization buffer to perform the Ribotag assay in the Ribohet genotype. According to the volume used, we utilize either 2ml or 5ml Dounce homogenizer.
- Anesthetize postnatal day 60 adult animal from each genotype, MGE-CreNegative, MGE-CrePositiveRibohet, and MGE-CrePositiveRibohomo with isoflurane. Decapitate the animals using a decapitator according to protocols approved by institutional guidelines, the National Institutes of Health, in this case. Place the decapitated head on a petridish containing icecold 1XPBS and extract the hippocampus according to standard procedures using tools that are thoroughly treated with RNaseZap.
- Alternative to hippocampus, any other brain region can be extracted according to standard procedures. Subsequent to brain-regions isolation, it can be either snap-frozen in liquid-nitrogen or proceeded with fresh tissue for the Ribotag assay.
- For processing the brain-isolate from frozen tissue, pre-chill the mortar and pestle in liquid nitrogen and pulverize the tissue while kept cold. After pulverization, proceed similar to processing the assay from fresh tissue.
- Pool the hippocampus from 3 animals for assays utilizing the Ribohet genotype, and use the hippocampus from 1 animal for assays utilizing the Ribohomo genotype.
- Homogenize the hippocampus tissue using 20 slow strokes, careful not to generate air bubbles during the process. Transfer the lysates to labeled, prechilled 2.0ml LoBind tubes.
- Utilize separate sets of Dounce homogenizers to homogenize the tissues while comparing the effects of different genotypes. If using the same equipment, thoroughly rinse the Dounce homogenizer with RNaseZap and Ultrapure water between genotypes.
- After collecting lysates from all conditions being compared, centrifuge the tubes containing lysates at 20,000 × g for 10 min at 4°C.
- During the centrifuge, prepare the stock BSA and load standards for the protein estimation in 96-well plate in duplicates. Also prepare the working BCA assay according to manufacturer instructions (also see table1).
- Post-centrifugation, aliquot the clear supernatant to fresh 2.0ml LoBind tubes on ice. Add appropriate volumes of the lysates for protein estimation as triplicates/quadruplicates and perform BCA assay according to manufacturer instructions (also see table1).
- Prepare the standards according to table1; take 2μl of test sample for unknown protein estimation; makeup with 23μl of the Ultrapure Water
- Add 20μl of Reagent B to each ml of Reagent A that is needed for the entire run, called the BCA working reagent, and add 200μl of this 50:1 BCA working reagent into each well (standards and test samples)
- Incubate the plates for 5 minutes at 37˚C and measure the absorbance in a microplate reader at 562nm.
- Based on the estimated protein concentration, determine the volume of hippocampal lysate required for 5000μg of the Ribotag immunoprecipitation assay and 100μg for the Bulk / Input RNA extraction, and aliquot appropriate volumes in labeled, pre-chilled 1.5ml LoBind tubes.
- For instance, for a lysate estimated to contain 10μg/μl based on the BCA assay, we would require 500μl for 5000μg (5000/10) of the Ribotag immunoprecipitation assay, and 10μl for 100μg (100/10) for the Bulk / Input RNA extraction.
Prepare the RNA extraction buffer by adding 100μl of 2M DTT, to 5ml of RLT Plus Buffer from the RNeasy Plus Micro Kit, and thoroughly mix the buffer.
- Add 350μl of this RNA extraction buffer into the tubes containing 100μg of hippocampal lysate for Bulk / Input RNA extraction. Thoroughly mix the tube and freeze all the tubes in powdered dry ice and store it at −80°C for Bulk / Input RNA extraction.
- Remove an additional 50μg of hippocampal lysate and snap-freeze on dry ice, for subsequent Western blot analyses to compare with the Unbound fraction after the Ribotag assay (See additional comments on Step 15).
- Add 10μl of anti-HA antibody to 5000μg of hippocampal lysate and makeup all the samples to 1.5ml using the homogenization buffer. Place all the tubes on a benchtop tube revolver and perform end-over-end rotation at 15 rotations per minute, for 3 hours at 4°C
- We purchased a large quantity of the same lot of ab9110 anti-HA antibody to prevent any batch-to-batch variability while determining the antibody-beads ratio, and for reliable assay.
During the 3-hour antibody: lysate incubation, resuspend the stock Dynabeads Protein G by gentle vortexing and thorough mixing. Aliquot into a fresh 2.0ml LoBind tube, 500μl beads for every 10μl of Abcam ab9110 anti-HA antibody used.
Place the tube on a DynaMag magnetic separator for 1 minute, and remove all the supernatant. Wash the beads twice, each in 500μl homogenization buffer, and rotate end-over-end for 5 min.
At the end of 3-hour incubation of the lysate with antibody, remove all the homogenization wash buffer from the Dynabeads.
- resuspend the antibody-lysate conjugate to the washed Dynabead Protein G. Place all the tubes on a benchtop tube revolver and perform end-over-end rotation at 15 rotations per minute, for 12 hours at 4°C.
- For all subsequent steps involving washing the Dynabeads Protein G, leave tubes in the DynaMag magnetic separator for 1 min to collect all the beads, before removing the supernatant.
- We have also alternatively used Pierce monoclonal anti-HA magnetic beads conjugate (Cat. No. 88836) monoclonal antibody and performed direct immunoprecipitation for a relatively shorter duration (6-hours, as opposed to 3+12-hours using the indirect IP with ab9110 and Dynabeads Protein G), with no distinguishable difference in the RNA quality and concentrations.
- Next day, add fresh CHX, DTT, RNasein, and protease inhibitor cocktail to the high-salt immunoprecipitation wash buffer. Stop the overnight Ribotag indirect immunoprecipitation assay by removing the tubes from the rotator and place the tubes on DynaMag magnetic separator for 1 min to collect all the beads and remove all supernatant.
- Save ~50μg of this Unbound supernatant after the overnight Ribotag immunoprecipitation and snap-freeze on dry ice. Compare the levels of Rpl22-HA between the Input (collected in Step 9) and Unbound (collected in Step 15) by Western analysis, as a means to determine the efficiency of immunoprecipitation.
- Add 1ml of high-salt wash buffer to all the beads, and gently vortex each sample for 30 seconds to evenly mix the beads. Transfer all solutions to fresh 2.0ml LoBind tubes and place the tubes on DynaMag magnetic separator for 1 min to collect all the beads and remove all supernatant. Repeat this washing step for a total of 5 times.
- Previous protocols recommended 3X washes and an end-over-end rotation for 5–10 min per wash. However, we have found 5X efficient washes with shorter duration (30 seconds of gentle and thorough vortexing), as indicated by cleaner signals from negative control samples (see subsequent results).
After the last wash, remove all wash buffer using magnetic separation and place the tubes on ice.
Prepare the RNA extraction buffer by adding 100μl of 2M DTT, to 5ml of RLT Plus Buffer from the RNeasy Plus Micro Kit, and thoroughly mix the buffer.
- Add 350μl of this RNA extraction buffer into the tubes containing the washed beads containing the antibody: Rpl22-HA-bound mRNAs. Thoroughly mix the tubes and directly proceed to column purification of RNA using the RNeasy Plus Micro Kit, according to manufacturer instructions.
- The 100μg of input material stored at −80°C (from Step 8) can be thawed at this point, and RNA extraction can be performed for both Input and Ribotag immunoprecipitates simultaneously.
- Incorporate in-column digestion of DNA using the RNase-Free DNase (50Units / sample) during the RNeasy Plus Micro Kit.
After the final washes during the RNeasy Plus Micro Kit protocol, high-quality, pure RNA was eluted from the column in 25μl of Ultrapure DNase/RNase-free distilled water.
After thoroughly mixing the eluted RNA, aliquot 2.5μl for Bioanalyzer measurements. Aliquot the remaining RNA in Eppendorf Fast PCR Tube Strips in appropriate volumes and store at −80°C, so that repeated freeze-thaw cycles are reduced during subsequent assays.
- Dilute the Input / Bulk mRNA at 1:3 with Ultrapure DNase/RNase-free distilled water and dilute the MGE-specific Rpl22-HA immunoprecipitated mRNA with 1:1 with Ultrapure DNase/RNase-free distilled water. Proceed to analyze the RNA integrity using the RNA 6000 Pico Kit via Agilent 2100 Bioanalyzer, according to kit protocol and manufacturer instructions.
- The RNA 6000 Pico Kit is suited to measuring RNA quality, in samples containing 50picogm – 5nanogm of RNA. Therefore, diluting the Input and immunoprecipitated RNA to the above-recommended ratios is essential for accurately determining the sample quality and concentration using this kit. Alternatively, RNA 6000 Nano Kit can be used for samples containing 5–500 nanogm of RNA.
- All samples with RIN values >7 were used for subsequent RNA sequencing analyses.
Table 1.
Standards for Protein Estimation by Bicinchoninic Acid (BCA) Assay;
| Concentration μg/ml | Ultrapure water (in μl) | Standard BSA 1mg/ml (in μl) |
|---|---|---|
| Blank | 25 | 0 |
| 100 | 22.5 | 2.5 |
| 200 | 20 | 5 |
| 300 | 17.5 | 7.5 |
| 400 | 15 | 10 |
| 500 | 12.5 | 12.5 |
| 600 | 10 | 15 |
| 700 | 7.5 | 17.5 |
| 800 | 5 | 20 |
| 900 | 2.5 | 22.5 |
| 1000 | 0 | 25 |
SUPPORT PROTOCOL 1
Real-time Quantitative PCR (qRT-PCR)
The cell-type-specific RNA material obtained after the Rpl22-HA immunoprecipitation is the starting material for RNA sequencing. Before embarking on the sequencing approaches that constitute significant financial resources, it is prudent to verify the success of Ribotag assay using relatively cost-effective qRT-PCR assays using an array of positive and negative control genes. The design of the positive and negative control gene sets depends on the existing knowledge of the established genes that must be enriched in a cell-type under investigation and the genes that must never be enriched in the same cell-type. For instance, Nkx2.1 positive MGE interneurons must be enriched in the broad GABAergic neuron marker genes Gad1 and Gad2, and the MGE-subtype markers Nkx2-1, Lhx6, Pvalb, Sst, Hapln1 but not be enriched in the CGE-subtype marker Vip, the pyramidal neuron enriched marker Bdnf, or the Microglia enriched marker C1qa. Therefore, for the current assay, Gad1, Gad2, Nkx2-1, Lhx6, Pvalb, Sst, Hapln1 would constitute good positive control genes, while Vip, Bdnf, C1qa would constitute good negative control genes.
The enrichment of a gene can be expressed as a ratio of its expression in MGE-RNA: Bulk RNA
(IP/INPUT). Positive control genes mentioned above are typically expressed more in the MGE-RNA, resulting in an IP/INPUT ratio > 2. However, the negative control genes are typically expressed in non-MGE cell-types and hence abundant in the Bulk RNA fraction, resulting in an IP/INPUT ratio of < 1.
Materials
Input / Bulk RNA or the MGE-RNA obtained from the Ribotag assay
High-Capacity cDNA Reverse Transcription Kit, Thermofisher Scientific (Cat. No.4368814)
Power SYBR Green PCR Master Mix, Thermofisher Scientific (Cat. No. 4368577)
Applied Biosystems QuantStudio QS3 Real-Time PCR System (Cat. No. A28567)
See Table 2 for a list of suggested primers for the qRT-PCR assay, prepare the primers as 20μM stocks using DNase/RNase-Free Distilled Water, and store at −20°C
Table 2.
List of Suggested Primers for the qRT-PCR Assays for Use in Nkx2.1-Cre Mice
| Marker | GenBank no. | Sense primers | Antisense primers | Size (bp) |
|---|---|---|---|---|
| Gad1 | NM_008077.4 | ATGATACTTGGTGTGGCGTAGC | GTTTGCTCCTCCCCGTTCTTAG | 253 |
| Gad2 | NM_008078.2 | CCAAAAGTTCACGGGCGG | TCCTCCAGATTTTGCGGTTG | 375 |
| Lhx6 | NM_008500.2 | TGCCATGGCTCAGTCAGACGCA | CCTTCTCCTCAACGAGGCCGAATT | 473 |
| Pvalb | NM_013645.3 | GCCTGAAGAAAAAGAACCCG | AATCTTGCCGTCCCCATCCT | 275 |
| Sst | NM_009215.1 | ATGCTGTCCTGCCGTCTCCA | GCCTCATCTCGTCCTGCTCA | 250 |
| Vip | NM_011702.2 | TTATGATGTGTCAAGAAATGCCAG | TTTTATTTGGTTTTGCTATGGAAG | 424 |
| Slc17a7 | NM_182993.2 | CCCTTAGAACGGAGTCGGCT | TATCCGACCACCAGCAGCAG | 593 |
| GFAP | NM_010277.3 | AAGCCACCCTGGCTCGTGTG | CTGTTCGCGCATTTGCCGCT | 407 |
| Tubb3, b3tuj1 | NM_023279.2 | CTTTTCGTCTCTAGCCGCGT | CCACCCAGTGAGTGTGTCAG | 451 |
| Actb | NM_007393.5 | CCTGACCGAGCGTGGCTACAGCTTC | CTGCTGGAAGGTGGACAGTGAGGCC | 487 |
Method
While a variety of kits can be used to perform qRT-PCR, we found reliably consistent results while using 2-step RT-PCR using the High-Capacity cDNA Reverse Transcription Kit.
Generating high-quality cDNA from 10–25 nanogm of the extracted RNA, according to the manufacturer instructions.
Dilute 1:10 of the cDNA obtained in the previous step, and this will be the starting material for all qRT-PCR reactions.
Set up the qRT-PCR reactions using positive and negative control genes and a set of housekeeping genes indicated in Table 2, using SYBR Green Assay, according to manufacturer instructions.
Prepare the SYBR reaction mix containing 0.5μl each of the primer pairs, 10μl of 2X Master mix (provided with the kit), and 7μl of DNase/RNase-Free Distilled Water, per reaction.
Calculate the total reactions to be performed and prepare the SYBR reaction mix accordingly. 2μl of the 1:10 diluted cDNA is mixed with 18μl of the SYBR reaction mix per reaction, and amplified on a QS3 system.
The bulk RNA and the Rpl22-HA immunoprecipitated RNA were both subject to the 2-step qRT-PCR protocol, and the calculations were performed by normalizing the values to the housekeeping genes using the 2−DDCT method (Livak and Schmittgen, 2001). Finally, the enrichment of gene is expressed as a ratio of its expression in the Rpl22-HA immunoprecipitated RNA (MGE-translatome) over the expression in bulk RNA (bulk transcriptome).
SUPPORT PROTOCOL 2a
RNAseq Library Construction
RNAseq library construction is a multi-step process that converts the mRNA into sequencing-compatible genetic material. Briefly, the mRNA obtained from the previous steps are first converted to cDNA. Subsequently, adapters for Illumina sequencing with appropriately compatible barcodes are added to the cDNA amplicons via PCR. Next, the cDNAs generated from the ribosomal RNAs are depleted and the remaining cDNA fragments are once again amplified with universal primers. The final PCR products are purified for salt contaminations that mght obscure the sequencing reactions, and this constitutes the sequencing-compatible cDNA library. A variety of library construction kits are commercially available. However, we have observed consistent results from small starting material (250–500picogm RNA) using the SMARTer Stranded Total RNA-Seq Kit v2.
Materials
SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian, TaKaRa (Cat. No. 634413)
DNA 7500 Kit, Agilent (Cat. No. 5067-1506)
Method
250 picogm of RNA, based on the Bioanalyzer quantitation, will serve as the starting material for library construction for all bulk RNA and Rpl22-HA immunoprecipitated RNA samples.
Construct libraries according to the manufacturer protocol and the quality of the double-stranded DNA must be assessed using the DNA 7500 Kit via Agilent 2100 Bioanalyzer.
Calculate equal quantities of the samples and multiplex for subsequent sequencing reaction (Sanz et al., 2019). At this step we routinely utilize the services of Sequencing Core-facility at our institute for subsequent sequencing and primary data analyses.
SUPPORT PROTOCOL 2b
RNAseq Secondary Analyses
Previous protocols have dealt extensively with library preparation, including the basics of RNA sequencing and primary analysis (Sanz et al., 2019). In the present section, we will discuss the secondary analyses on the RNAseq dataset. Particularly, we use integrated Differential Expression and Pathway analysis (iDEP) (Ge et al., 2018), a publicly available RNAseq data secondary data analysis workflow to analyse the efficiency of Ribotag assay between Nkx2.1Cre Negative, Ribohet and Ribohomo tissue. After performing the read-alignment, normalization, and primary analyses, we will obtain a matrix with gene lists with corresponding condition-specific reads. This will serve as the input for the secondary analyses.
STEPS:
Open the iDEP.9 portal http://bioinformatics.sdstate.edu/idep/ and select the “matching species” tab
Prepare the read counts data matrix organized as the gene names in the column and a clearly defined row names representing each experimental condition and upload the read counts data matrix.
Subsequent workflow is intuitive, and the processes are described well in the resource. Briefly, after determining the quality of the sequencing, iDEP will take through quality check for the RNAsequencing data (also described well elsewhere (Ge et al., 2018).
- After qualtity assessments, it is important to perform Principal Component Analyses (PCA) on the RNAsequencing datasets to determine the relatedness between the Input and the Ribotag assay datasts. Ideally, if the Ribotag immunoprecipitations have worked well, this should ideally result in the datasets separated wide-apart in the PCA space.
- The PCA plot for our present example indicates that the Ribotag-immunoprecipitation has worked well in all three conditions, as shown by clear separation of the samples in the PCA space (Fig. 5A).
- Next, iDEP uses DESeq2 (Love et al., 2014), a widely used method to perform differential analysis of RNAsequencing counts data, to identify the genes that are differentially expressed in the MGE-derived Ribotag immunoprecipitates in comparison to the bulk RNA.
- In the present example, we identify 2020, 2455 and 1277 genes that are significantly enriched in the Ribotag-translatome, in comparison to the bulk / input RNA, using the Nkx2.1Cre Negative, Ribohet and Ribohomo tissue respectively (Fig. 5B); also see Supplemental Table. 1 for the complete list of differentially enriched genes in the Ribohomo Ribotag assay obtained through iDEP.
- Analyzing the MGE-translatome from the 3 genotypes, we determine that only the Ribohomo MGE-translatome expresses the positive control genes abundantly enriched in the top 25 enriched genes (Fig. 5C, Supplemental Table.1). In particular, we observe, a robust enrichment of the top positive controls Lhx6, Gad1, Gad2, Pvalb, Sst, Hapln1 only in the Ribohomo, but not in the Nkx2.1Cre Negative, Ribohet (Fig. 6A, B). In addition, we do not observe any enrichment in the negative control genes Vip, Slc17a7 and C1qa in the Ribohomo MGE-translatome (Fig. 6C).
- In addition, iDEP also assists in generating a variety of data representations including healmaps, volcano plots, scatter plots and gene ontology analyses of the RNAsequencing datasets.
iDEP also assists in generating a variety of data representations including heatmaps, volcano plots, scatter plots and gene ontology analyses of the RNAsequencing datasets, easy-to-use, even for first-time users, and allowing for downloading the analyses datasets. Lastly, iDEP also enables users to download the entire analysis workflow as R-scripts, and users can apply the scripts using another publicly available R-Studio software.
Figure 5. Validation of the hippocampal MGE translatome between Ribohet and Ribohomo alleles using iDEP9.0.
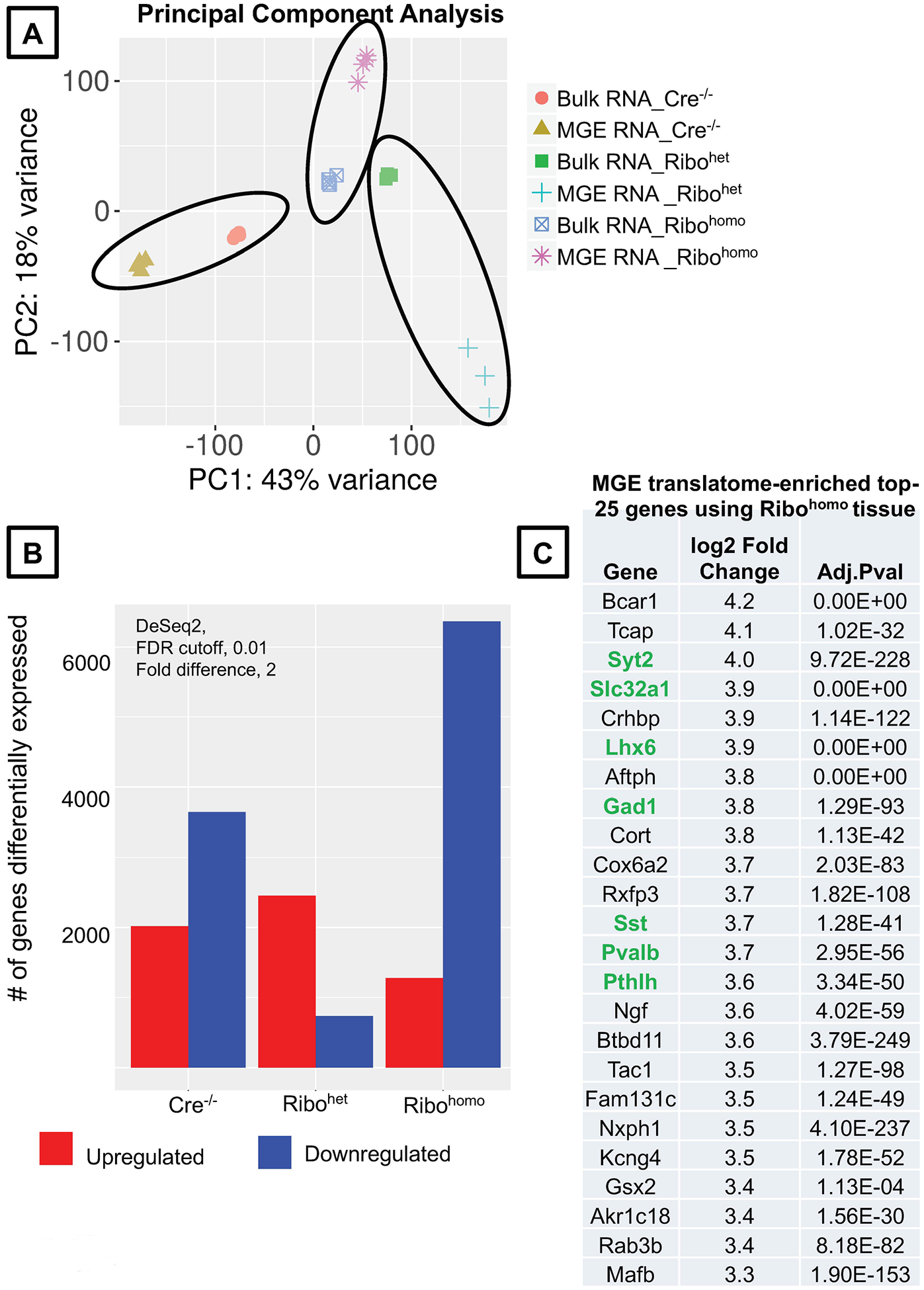
(A) Principal component analysis plot comparing the Bulk RNA vs MGE-specific Ribotag-associated RNA across different genotypes. (B) Differentially up and downregulated genes according to DeSEq2 at a stringent FDR <0.01 and fold difference >2, in the MGE-specific Ribotag-associated translatome. (C) MGE-translatome-enriched, top 25 genes obtained from Rpl22HA/HA RNAsequencing. Genes highlighted in green are established MGE-enriched positive controls (see Supplemental Table. 1 for the complete list of differentially enriched genes identified in the present Ribohomo Ribotag assay).
Figure 6. Expression of positive and negative control genes in the hippocampus of control and Nkx2.1-Ribotag mouse.
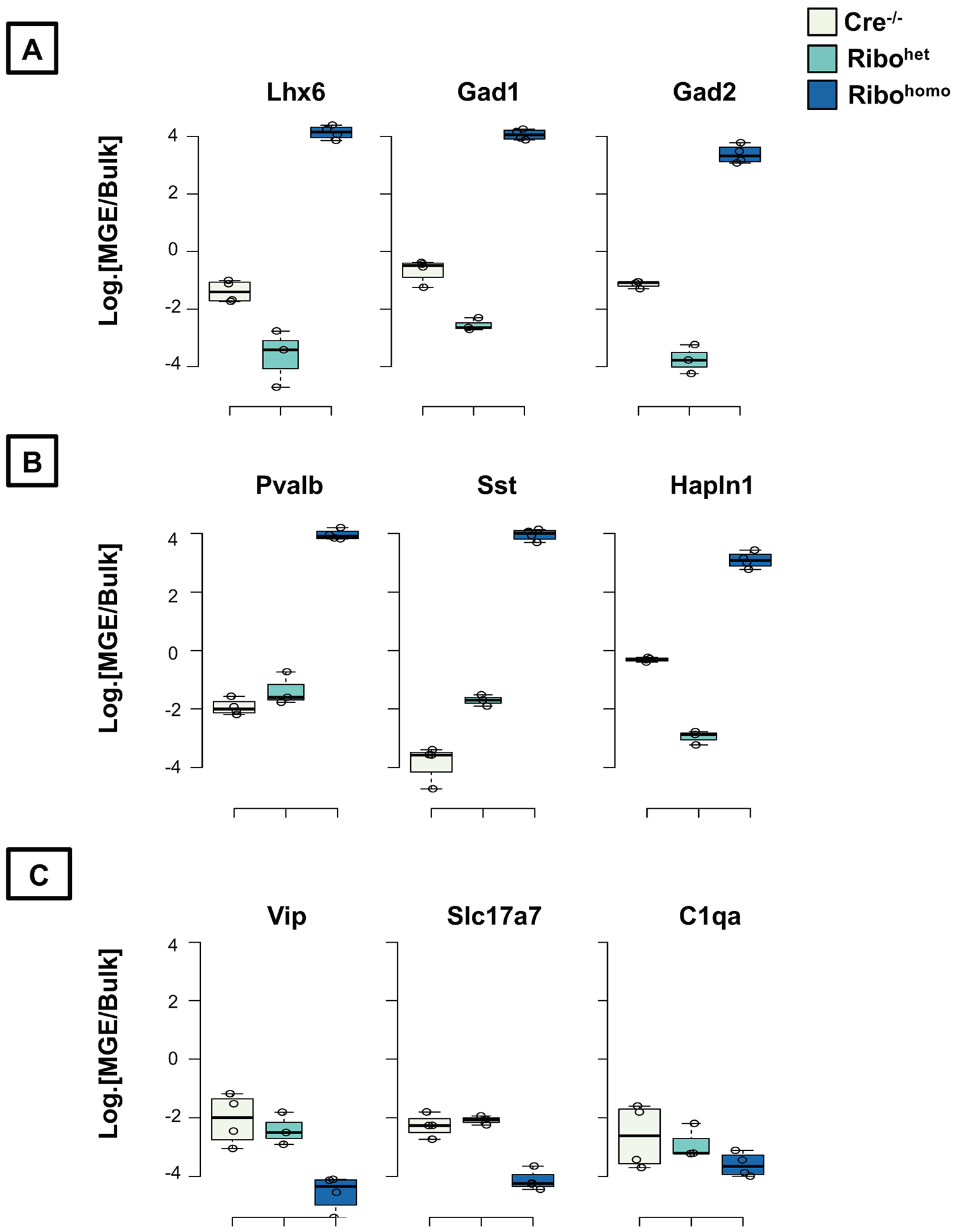
Ratio of normalized expression reads between the MGE-enriched Rpl22-HA immunoprecipitated and bulk tissue in logarithmic scale (A) established genes known to be enriched in all MGE-derived interneurons (B) established genes known to be enriched different subtypes of MGE-derived interneurons (PV, SST and NGFC), (C) established genes that are not enriched in MGE-derived interneurons, but in CGE-derived interneurons or pyramidal neurons or microglia.
REAGENTS AND SOLUTIONS:
- Tissue homogenization buffer
Stock Working For 10ml 1M Tris HCl (pH7.5) 50mM 500μl 3M KCl 100mM 333.33μl 1M MgCl2 12mM 120μl 20% (v/v) NP-40 SurfactAmps 1% 500μl 100mM DTT 1mM 100μl 100mg/ml Cyclohexamide 100μg/ml 100μl 40U/μl RNAseIN 400U/ml 100μl 10mg/ml Heparin 1mg/ml 20mg Complete mini Protease inhibitor - 1 tablet - High-salt immunoprecipitation wash buffer
Stock Working For 10ml 1M Tris HCl (pH7.5) 50mM 500μl 3M KCl 300mM 1ml 1M MgCl2 12mM 120μl 20% (v/v) NP-40 SurfactAmps 1% 500μl 100mM DTT 1mM 100μl 100mg/ml Cyclohexamide 100μg/ml 100μl 40U/μl RNAseIN 400U/ml 100μl 10mg/ml Heparin 1mg/ml 20mg Complete mini Protease inhibitor - 1 tablet
The working buffers containing Tris HCl, KCl, MgCl2, and NP-40 can be prepared the day before. However, it is best if DTT, CHX, Heparin, RNaseIn, and the protease inhibitor cocktails are added immediately prior to the Ribotag assay.
COMMENTARY
BACKGROUND INFORMATION:
The Ribotag is a robust tool to study the translatome of defined cell-types by the use of appropriate Cre recombinase expressing driver mouse lines. However, the applicability of this approach has been most successful for abundant cell populations or cell populations that express the Ribotag gene Rpl22 at relatively high levels. In the present protocol, we highlight that the expression of Rpl22 in the cell-type under investigation is a critical factor that determines the success of the assay, mainly when used for the study of sparse cell populations that are less than ~5% of all cell-types in the mouse brain. Under these circumstances, we particularly establish that the Rpl22-HA transgene must be expressed homozygously, not heterozygously, to enable this technique to fruition.
CRITICAL PARAMETERS:
This protocol emerged while studying MGE interneurons using the Nkx2.1 Cre mouse line as a result of trouble-shooting the initial Ribotag procedures that described the use of Ribohet for its wide applicability. We were surprised at the sensitivity of this assay to the abundance of Rpl22 gene expression in the cell-type. While most publications that have used this assay have successfully applied it in abundant cell-types, it was only during conference proceedings and discussions with other users concerning negative data, that the user community became aware of numerous intricacies of using this approach. So, we consider it a vital first step to examine via publicly available datasets, the levels of Rpl22 gene expression in the cell-type of interest, before attempting this strategy (Fig. 1).
The primary requirement for a successful Ribotag assay is the availability of a Cre mouse line where Cre recombinase is exclusively expressed in the cell-type of interest. Therefore, every attempt must be made to validate the specificity of the Cre mouse line. This involves determining the propensity of ectopic expression of Cre recombinase or the reporter gene outside of the cell types of interest (Song and Palmiter, 2018; Luo et al., 2020; Harris et al., 2014) (Fig. 2). In particular, one must exclude the breeders and litters having such ectopic expression for subsequent experiments or future breeding. An additional level of validation for such mouse lines must be performed by verifying whether Rpl-HA transgene expression occurs in a manner faithful of Cre recombinase expression (Fig. 3).
To determine the efficiency of tissue collection and the requirement for tissue pooling
While it is well documented that the process of freezing and thawing complex tissue such as the brain would result in a significant loss of polysome integrity (Heiman et al., 2014), similar analyses at a cell-type-specific level are not commonly observed in the literature. This is a significant assumption while attempting a multi-stage experiment such as Ribotag, particularly for rare cell-types. To circumvent the caveats associated with polysome integrity loss resulting from freezing the tissue, some studies have pooled the tissue from multiple animals to gain enough ‘mRNA material’ for downstream analysis (Spiegel et al., 2014; Mardinly et al., 2016). Therefore, it is prudent to examine these scenarios at an earlier stage of the project than as a mere afterthought. In our hands, we routinely observed a dramatic loss in RNA integrity, particularly in the Rpl22-HA immunoprecipitated RNA fractions, while performing the Ribotag assay after snap-freezing (Fig. 4Ai, Aii), in comparison to freshly prepared samples (Fig. 4Bi, Bii, Ci, Cii). In addition, we also determined that while using the MGE-Ribohet tissue, we had to pool at least three pairs of hippocampi from 3 animals to obtain detectable levels and qualities of RNA in the Rpl22-HA immunoprecipitated fraction (Fig. 4Bi, Bii). However, we could obtain good quality and quantities of RNA from a pair of hippocampi from a single mouse while using the MGE-Ribohomo tissue (Fig. 4Ci, Cii). As a negative control, we could not observe any detectable RNA from the Rpl22-HA immunoprecipitated fraction while using the MGE-Cre−/− tissue.
Figure 4. Optimizing the conditions for Ribotag assay based on ribosomal integrity.
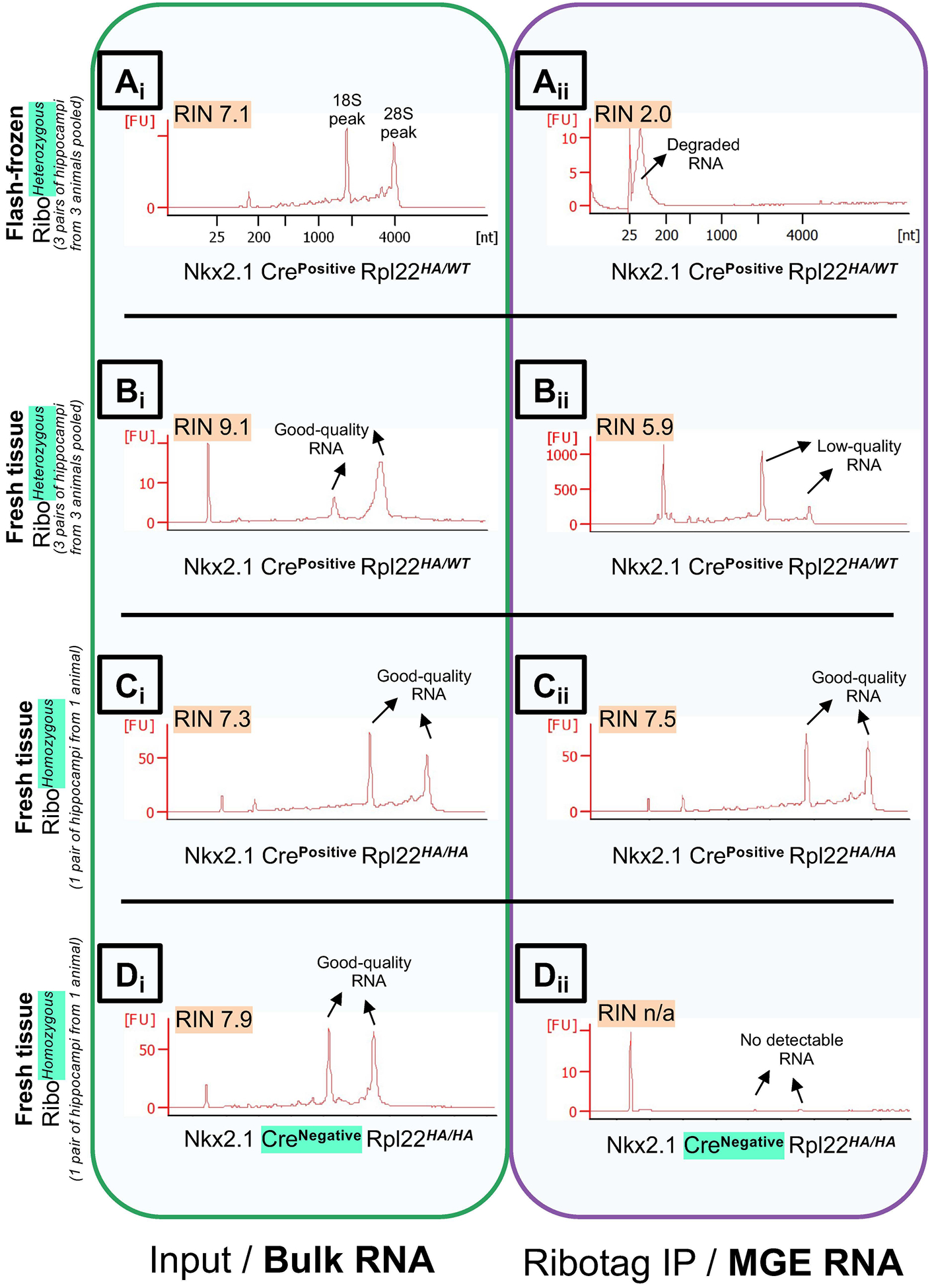
Bioanalyzer traces, measuring the RNA Integrity (RIN) Values of RNA obtained from the bulk hippocampal tissue (Input/total RNA fraction), in Green box, left; vs. hippocampal MGE interneuron-specific RNA (Rpl22-HA immunoprecipitation fraction), in Purple box, right. Comparing quality of RNA in Nkx2.1 CrePositive RibotagHeterozygous animals between (A) tissue processed after flash-freezing in liquid nitrogen Vs, (B) tissue processed from fresh animals. Comparing quality of RNA between tissue processed fresh in Nkx2.1 CrePositive (B) RibotagHeterozygous animals Vs, (C) RibotagHomozygouss animals. (D) Negative control for the Ribotag assay using Nkx2.1 CreNegative RibotagHomozygouss animals. Abbreviations: RIN, RNA Integrity; IP, immunoprecipitation; Ribo, Ribotag; MGE, Medial ganglionic eminence.
TROUBLESHOOTING:
Despite avoiding freeze-thaw of tissue, and utilizing fresh tissue for the Ribotag assay, another standard troubleshooting issue in using this technique involves poor RIN values after HA-immunoprecipitation. The likely reason for this could be unclean workspaces and the reagents not prepared fresh, or the experiment steps not performed in the cold. Apart from these common reasons, several Ribotag studies involving abundant cell-types may not have dealt with the severity of poor sample qualities, due to tissue freeze-thaw. Perhaps, the combination of low Rpl22 expression, and degrading polysomes during sample thawing / improper tissue handling while sample thawing, could collectively result in degraded RNA unviable for RNA sequencing. We have demonstrated that we could eliminate the problem resulting from tissue freeze-thaw by performing the assay with freshly prepared lysates in clean workspaces.
If the long duration of the indirect and overnight immunoprecipitation is a concern in the cell-type of interest, the duration could be reduced along with the use of direct immunoprecipitation performed with Pierce monoclonal anti-HA magnetic beads conjugate (Cat. No. 88836). However, in the present study, we did not observe any drastic RNA degradation during overnight immunoprecipitation.
UNDERSTANDING RESULTS:
The neuronal soma contains separate populations of ribosomes. When the mRNAs are bound to two or more ribosomes, they represent the polysomes and when the mRNAs are associated with single ribosomes, they are termed as monosomes. It is widely thought that the polysomes contain the mRNAs that have a higher rate of translation than the mRNAs associated with the monosomes (Noll, 2008; Warner and Knopf, 2002). However, recent studies have emerged that the monosomes can also mediate robust translation and protein synthesis (Heyer and Moore, 2016; Biever et al., 2019b). In the present study, while the translatome obtained will indicate the mRNAs associated with the Rpl22-HA-bound ribosomes, it is incorrect to assume that all Rpl22-HA are part of the machinery involved in robust translation. At the moment, it is unclear whether Rpl22-HA is part of the polysomes vs. monosomes; or whether they represent actively-translated mRNAs in the cell-type under study. The Ribotag dataset at best represents the Rpl22-HA associated mRNAs and it may be incorrect to assume that they may represent the complete repertoire of mRNAs that are ‘actively’ translated in the cell-type of interest. In this scenario, Ribotagging is a robust approach, that can easily be coupled with polysome fractionation, and during the studies that examine the rates of mRNA translation. This is especially important in neurons that have distinct functions of compartmentalized (dendrite/axon-specific) local protein synthesis, separate from the distant, nuclear-encoded translation machinery (Biever et al., 2019a).
A common approach that is currently used to verify the validity of a successful Ribotag assay is to use appropriate positive and negative controls and to express the ratio of enrichment of a gene × in the translatome vs. bulk transcriptome. Any gene that is expressed higher than 2-fold in the translatome is considered to be a bonafide marker of the cell-type under study. In the present example, we have demonstrated that in MGE-Ribohomo translatome, we could observe robust enrichment (>3 Log2-fold) of all positive control genes that are commonly expressed in all MGE interneurons such as Lhx6, Gad1, Gad2 (Fig. 6A). Additionally, the MGE-subtype marker genes, Pvalb (for the PV-cells), Sst (for the Sst cells), and Hapln1 (for the neurogliaform cells) also exhibit a high fold difference (>3 Log2-fold) only in MGE-Ribohomo translatome (Fig. 6B). Importantly, none of the non-MGE marker genes, namely Vip (a marker for CGE subtype interneurons), or Slc17a7 (vGluT1, for glutamatergic neurons), or C1qa (established microglial marker) are enriched in the MGE translatome (Fig. 6C).
In the present study, the MGE-Ribohet translatome was not enriched for any of the above established positive control genes. While the MGE-Ribohet was distinct from the Cre−/− samples, the PCA plots still showed a robust separation of the transcriptome vs. translatome in the MGE-Ribohet genotype. This clearly establishes that while the “Rpl22-HA immunoprecipitation” itself may have worked, but the existing half-copy of Rpl22-HA transgene is not sufficient to capture the full repertoire of cell-type-specific translatome. This drives the central point of the current protocols – whereby a rare cell-type with low expression of Rpl22 gene activity, is not sufficient to capture the cell-type-specific translatome by merely using the Rpl22HA/WT heterozygous allele.
Another important consideration for the data interpretation is to misinterpret a “2-fold enrichment in the translatome” as an absolute measure of expression in the cell-type of interest. While a gene may be 2-fold enriched in the translatome, it may still be a weakly expressed gene in that cell-type. Alternatively, a gene that may not exhibit 2-fold enrichment may be a highly expressed gene that is equally highly expressed across all cell-types. To further explain this, we present examples for key positive control genes Ai14/tdTomato, Lhx6, Nkx2.1 and Dlx6 from the Ribohomo dataset. All these genes show a >3 Log2-fold, MGE-translatome: Bulk transcriptome ratio (Fig. 7A). But, looking at the raw, normalized expressions of these genes, we clearly identify that only Ai14/tdTomato and Lhx6 have >10000 read counts indicating that these genes are truly highly expressed, and highly enriched in the MGE, in comparison to the bulk RNA (Fig. 7B). Meanwhile, Nkx2.1 and Dlx6 reveal a relatively much lower expression <1000 read counts indicating that these genes are in fact weakly expressed, but still highly enrichd in the MGE in comparison to the bulk RNA (Fig. 7B). This mirrors the well established literature indicating that the expression of Nkx2.1, while high in the MGE progenitors, decreases during neurodevelopment, but the expression of Lhx6 is high in the postmitotic MGE interneurons (Wamsley and Fishell, 2017; Pelkey et al., 2017).
Figure 7. Understanding the results.
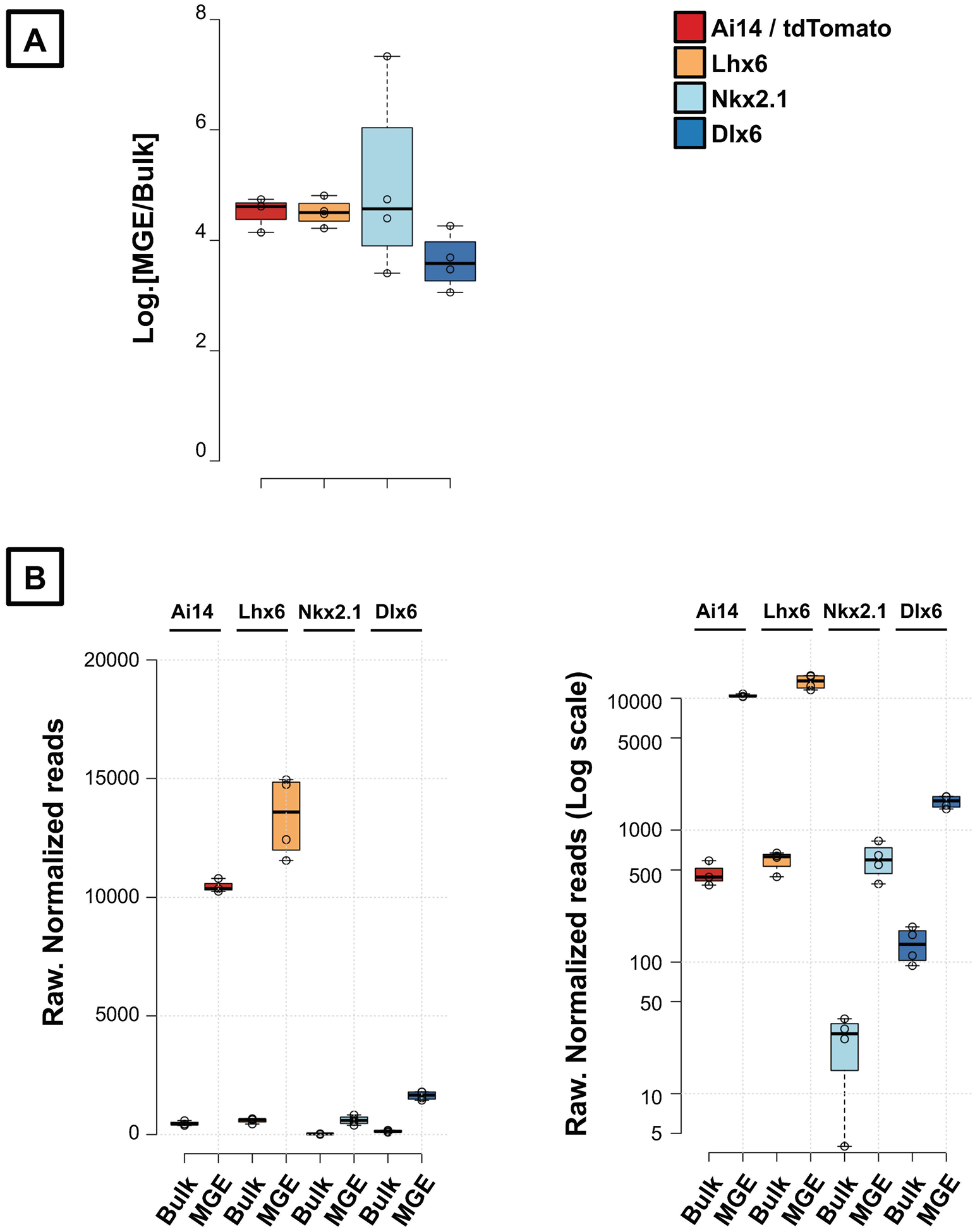
(A) MGE-translatome: bulk transcriptome Log2. enrichment ratio of hippocampal-expressed control genes, Ai14, Lhx6, Nkx2.1 and Dlx6. (B) Raw normalized read counts of the same control genes, shown in normal scale (left) and log-scale (right), to demonstrate the high MGE-expression of Ai14 reporter and Lhx6, but a low MGE-expression of Nkx2.1 and Dlx6 in the adult mouse hippocampus.
Lastly, we encourage the use of high stringency to establish the efficiency of the assay (translatome: transcriptome ratio), such as FDR <0.01. However, when applying the same stringent FDRs while studying the effect of genotype, or pharmacological treatments or, other biological considerations, the rate of false negatives could increase, and we may not obtain any significantly differentially expressed genes. Like any other unbiased screening, these results obtained are not absolute and require to be validated by alternative approaches (such as qRT-PCR, protein level, and functional assays). Therefore, it may be prudent to reduce the stringency of FDR cutoff, and observing whether pathways of related genes are commonly differentially expressed in the biological context, to make most of this technique.
TIME CONSIDERATIONS:
One drawback of utilizing this strategy to study rare cell populations in the mouse brain is the time taken to obtain the mouse that is homozygous for the Rpl22-HA allele in the background of other genetic crosses. This may typically take about six months to a year or two years, depending on how many transgenes are retained in the background. In the present study, we retained Nkx2.1 Cre recombinase and Ai14 transgenes additional to homozygous, Rpl22-HA, which took us a year to generate. Nevertheless, once the mouse is generated, this assay is ideal for studying long term effects on transcription/translation in the cell-type upon other genetic insults, or pharmacological treatments.
Performing the assay itself takes about two days once all the animals are available, and likely 2 days each for library preparation and RNA sequencing. However, when applying the translatome analyses to understand the effect of different biological contexts, it usually takes a long time to understand the implication of the differential expression in the specific context. Some examples of specific contexts may include, to study the differential pharmacological treatments or disease-causing mutations on cell-types. This will require extended pathway analyses.
Supplementary Material
Supplemental Table 1. (attached excel spreadsheet) DEG_Ribohomo_P60Hippocampus_MGEtranslatomeVsbulktranscriptome_iDEP
ACKNOWLEDGEMENTS:
CJM is funded by an NICHD intramural research award. The authors acknowledge Drs. Steve Coon, James R. Iben, Tianwei Li at the Molecular Genomics Core at the NICHD for RNA sequencing and primary data analyses. The authors also acknowledge Dr. Mayumi Miller at Dr. Brant Weinstein lab at the NICHD, and the broader Ribotag user’s community for advice on refining the technique. The authors also acknowledge Daniel Abebe, Steven Hunt for assistance with mouse colony maintenance; Drs. Ramesh Chittajallu and Ken Pelkey for critical reading of this manuscript.
LITERATURE CITED:
- Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, Barkan E, Bertagnolli D, Casper T, Dee N, et al. 2018. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLOS ONE 13:e0209648 Available at: http://dx.plos.org/10.1371/journal.pone.0209648 [Accessed December 12, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biever A, Donlin-Asp PG, and Schuman EM 2019a. Local translation in neuronal processes. Current Opinion in Neurobiology 57:141–148. [DOI] [PubMed] [Google Scholar]
- Biever A, Glock C, Tushev G, Ciirdaeva E, Langer JD, and Schuman EM 2019b. Monosomes actively translate synaptic mRNAs in neuronal processes. bioRxiv:687475. Available at: [DOI] [PubMed] [Google Scholar]
- Buffington SA, Huang W, and Costa-Mattioli M 2014. Translational Control in Synaptic Plasticity and Cognitive Dysfunction. Annual Review of Neuroscience 37:17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P 2015. Inhibitory microcircuit modules in hippocampal learning. Current Opinion in Neurobiology 35:66–73. Available at: http://www.sciencedirect.com/science/article/pii/S095943881500104X [Accessed May 25, 2016]. [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, and Menon V 2018. Continuous Variation within Cell Types of the Nervous System. Trends in Neurosciences 0 Available at: http://linkinghub.elsevier.com/retrieve/pii/S0166223618300596 [Accessed April 5, 2018]. [DOI] [PubMed] [Google Scholar]
- Close JL, Yao Z, Levi BP, Miller JA, Bakken TE, Menon V, Ting JT, Wall A, Krostag AR, Thomsen ER, et al. 2017. Single-Cell Profiling of an In Vitro Model of Human Interneuron Development Reveals Temporal Dynamics of Cell Type Production and Maturation. Neuron 93:1035–1048.e5. Available at: http://www.cell.com/neuron/fulltext/S0896-6273(17)30098-3 [Accessed March 8, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, Burkhalter A, Cauli B, Fairén A, Feldmeyer D, et al. 2013. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nature reviews. Neuroscience 14:202–16. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3619199&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. 2008. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell 135:749–762. Available at: http://www.sciencedirect.com/science/article/pii/S0092867408013664 [Accessed June 30, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley-Barnes KI, Ogawa LM, and Baserga SJ 2019. Ribosomopathies: Old Concepts, New Controversies. Trends in Genetics 35:754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishell G, and Rudy B 2011. Mechanisms of inhibition within the telencephalon: “where the wild things are”. [DOI] [PMC free article] [PubMed]
- Furlanis E, Traunmüller L, Fucile G, and Scheiffele P 2019. Landscape of ribosome-engaged transcript isoforms reveals extensive neuronal-cell-class-specific alternative splicing programs. Nature Neuroscience 22:1709–1717. Available at: http://www.nature.com/articles/s41593-019-0465-5 [Accessed October 3, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Son EW, and Yao R 2018. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. 2014. Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Frontiers in neural circuits 8:76 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25071457 [Accessed October 28, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Hochgerner H, Skene NG, Magno L, Katona L, Bengtsson Gonzales C, Somogyi P, Kessaris N, Linnarsson S, and Hjerling-Leffler J 2018. Classes and continua of hippocampal CA1 inhibitory neurons revealed by single-cell transcriptomics. PLoS Biology 16:e2006387 Available at: http://dx.plos.org/10.1371/journal.pbio.2006387 [Accessed June 20, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, and Heintz N 2014. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nature protocols 9:1282–91. Available at: http://www.scopus.com/inward/record.url?eid=2-s2.0-84901819558&partnerID=tZOtx3y1 [Accessed November 17, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer EE, and Moore MJ 2016. Redefining the Translational Status of 80S Monosomes. Cell 164:757–769. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. 2019. Conserved cell types with divergent features in human versus mouse cortex. Nature. Available at: http://www.nature.com/articles/s41586-019-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, and Ackerman SL 2018. mRNA Translation Gone Awry: Translation Fidelity and Neurological Disease. Trends in Genetics 34:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesiak AJ, Brodsky M, and Neumaier JF 2015. RiboTag is a flexible tool for measuring the translational state of targeted cells in heterogeneous cell cultures. BioTechniques 58:308–17. Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4467021&tool=pmcentrez&rendertype=abstract [Accessed May 17, 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15:550 Available at: http://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 [Accessed February 3, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Ambrozkiewicz MC, Benseler F, Chen C, Dumontier E, Falkner S, Furlanis E, Gomez AM, Hoshina N, Huang W-H, et al. 2020. Optimizing Nervous System-Specific Gene Targeting with Cre Driver Lines: Prevalence of Germline Recombination and Influencing Factors. Neuron (In press) NEURON-D-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR, Spiegel I, Patrizi A, Centofante E, Bazinet JE, Tzeng CP, Mandel-Brehm C, Harmin DA, Adesnik H, Fagiolini M, et al. 2016. Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature 531:371–375. Available at: http://www.nature.com/doifinder/10.1038/nature17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O 2012. Interneuron dysfunction in psychiatric disorders. Nature reviews. Neuroscience 13:107–20. Available at: 10.1038/nrn3155 [Accessed April 12, 2016]. [DOI] [PubMed] [Google Scholar]
- Mayer C, Hafemeister C, Bandler RC, Machold R, Brito RB, Jaglin X, Allaway K, Butler A, Fishell G, and Satija R 2018. Developmental diversification of cortical inhibitory interneurons. Nature 555:457–462. Available at: http://www.nature.com/doifinder/10.1038/nature25999 [Accessed March 5, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, et al. 2018. Early emergence of cortical interneuron diversity in the mouse embryo. Science:1–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29472441 [Accessed February 23, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll H 2008. The discovery of polyribosomes. BioEssays 30:1220–1234. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ 2017. Hippocampal GABAergic Inhibitory Interneurons. Physiological Reviews 97:1619–1747. Available at: https://www.physiology.org/doi/10.1152/physrev.00007.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin J, Tasic B, Hjerling-Leffler J, Trimarchi JM, and Awatramani R 2016. Disentangling neural cell diversity using single-cell transcriptomics. Nature neuroscience 19:1131–41. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27571192 [Accessed August 26, 2016]. [DOI] [PubMed] [Google Scholar]
- Rangaraju V, tom Dieck S, and Schuman EM 2017. Local translation in neuronal compartments: how local is local? EMBO reports:e201744045 Available at: http://embor.embopress.org/lookup/doi/10.15252/embr.201744045 [Accessed April 20, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Bean JC, Carey DP, Quintana A, and McKnight GS 2019. RiboTag: Ribosomal Tagging Strategy to Analyze Cell-Type-Specific mRNA Expression In Vivo. Current Protocols in Neuroscience 88:e77 Available at: http://doi.wiley.com/10.1002/cpns.77 [Accessed July 25, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS 2009. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proceedings of the National Academy of Sciences of the United States of America 106:13939–44. Available at: http://www.pnas.org/content/106/33/13939.full [Accessed September 3, 2015]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. 2018. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174:1015–1030.e16. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0092867418309553 [Accessed August 10, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AJ, and Palmiter RD 2018. Detecting and Avoiding Problems When Using the Cre-lox System. Trends in genetics : TIG 34:333–340. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29336844 [Accessed February 3, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS, and Costa-Mattioli M 2019. Translational Control in the Brain in Health and Disease. Cold Spring Harbor Perspectives in Biology 11:a032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding EL, and Burgess RW 2017. Accumulating Evidence for Axonal Translation in Neuronal Homeostasis. Frontiers in Neuroscience 11:312 Available at: http://journal.frontiersin.org/article/10.3389/fnins.2017.00312/full [Accessed December 12, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, and Greenberg ME 2014. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157:1216–1229. Available at: 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M, Wildenhain J, Rappsilber J, and Tyers M 2014. BoxPlotR: a web tool for generation of box plots. Nature Methods 11:121–122. Available at: http://www.nature.com/articles/nmeth.2811 [Accessed February 3, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS 2016. New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 92:1181–1195. Available at: http://www.sciencedirect.com/science/article/pii/S0896627316308984 [Accessed July 19, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi S, Khoutorsky A, Mathews MB, and Sonenberg N 2018. Translation deregulation in human disease. Nature Reviews Molecular Cell Biology 19:791–807. [DOI] [PubMed] [Google Scholar]
- Taniguchi H 2014. Genetic dissection of GABAergic neural circuits in mouse neocortex. Frontiers in cellular neuroscience 8:8 Available at: http://www.ncbi.nlm.nih.gov/pubmed/24478631%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3902216 [Accessed January 6, 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsani D, Fu Y, Lu J, Lin Y, et al. 2011. A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron 71:995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TNT, Kim TTK, Jarsky T, Yao Z, Levi BB, Gray LT, Sorensen SA, Dolbeare T, et al. 2016. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience advance on:1–37. Available at: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B, and Fishell G 2017. Genetic and activity-dependent mechanisms underlying interneuron diversity. Nature Reviews Neuroscience. [DOI] [PubMed] [Google Scholar]
- Warner JR, and Knopf PM 2002. The discovery of polyribosomes. Trends in Biochemical Sciences 27:376–380. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0968000402021266. [DOI] [PubMed] [Google Scholar]
- Xu Q, Tam M, and Anderson SA 2008. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. Journal of Comparative Neurology 506:16–29. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17990269 [Accessed April 20, 2017]. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan, J., Haring M, Braun E, Borm L, La Manno, G., et al. 2018. Molecular architecture of the mouse nervous system. Elsevier; Available at: https://www.biorxiv.org/content/early/2018/04/05/294918 [Accessed April 5, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
INTERNET RESOURCES:
- I.Dropviz - http://dropviz.org/; Dropviz is a publicly available scRNAseq dataset compiling the transcriptomes of 690,000 individual cells sampled from 9 regions of the adult mouse brain (Saunders et al., 2018).
- II.Morpheus - https://software.broadinstitute.org/morpheus/; Morpheus is a versatile, easy-to-use web-resource to produce heatmaps from gene expression matrices.
- III.iDEP - http://bioinformatics.sdstate.edu/idep/; iDEP is a publicly available RNAseq data secondary data analysis workflow that is reproducible, easy to use and intuitive for even naïve users (Ge et al., 2018).
- IV.BoxPlotR - http://shiny.chemgrid.org/boxplotr/; BoxPlotR is a publicly available, easy-to-use web-resource to generate box plots from gene expression matrices (Spitzer et al., 2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. (attached excel spreadsheet) DEG_Ribohomo_P60Hippocampus_MGEtranslatomeVsbulktranscriptome_iDEP