

Abstract
Retinol‐binding protein‐4 (RBP4) is elevated in serum and adipose tissue (AT) in obesity‐induced insulin resistance and correlates inversely with insulin‐stimulated glucose disposal. But its role in insulin‐mediated suppression of lipolysis, free fatty acids (FFA), and endogenous glucose production (EGP) in humans is unknown. RBP4 mRNA or protein levels were higher in liver, subcutaneous adipose tissue (SAT), and visceral adipose tissue (VAT) in morbidly obese subjects undergoing Roux‐en‐Y gastric bypass surgery compared to lean controls undergoing elective laparoscopic cholecystectomy. RBP4 mRNA expression in SAT correlated with the expression of several macrophage and other inflammation markers. Serum RBP4 levels correlated inversely with glucose disposal and insulin‐mediated suppression of lipolysis, FFA, and EGP. Mechanistically, RBP4 treatment of human adipocytes in vitro directly stimulated basal lipolysis. Treatment of adipocytes with conditioned media from RBP4‐activated macrophages markedly increased basal lipolysis and impaired insulin‐mediated lipolysis suppression. RBP4 treatment of macrophages increased TNFα production. These data suggest that elevated serum or adipose tissue RBP4 levels in morbidly obese subjects may cause hepatic and systemic insulin resistance by stimulating basal lipolysis and by activating macrophages in adipose tissue, resulting in release of pro‐inflammatory cytokines that impair lipolysis suppression. While we have demonstrated this mechanism in human adipocytes in vitro, and correlations from our flux studies in humans strongly support this, further studies are needed to determine whether this mechanism explains RBP4‐induced insulin resistance in humans.
Keywords: endogenous glucose production, inflammation, insulin resistance, lipolysis, obesity, RBP4
Abbreviations
- ADA
adenosine deaminase
- AT
adipose tissue
- EGP
endogenous glucose production
- FFA
free fatty acids
- GLUT4
glucose transporter‐4
- KRB
Krebs‐Ringer
- PEPCK
phosphoenolpyruvate carboxykinase
- PI3K
phosphoinositide 3‐kinase
- PIA
phenylisopropyl adenosine
- RBP4
retinol‐binding protein‐4
- Rd
rate of disappearance
- RYGB
Roux‐en‐Y gastric bypass
- SAT
subcutaneous adipose tissue
- SVF
stromal vascular fraction
- T2DM
type 2 diabetes mellitus
- VAT
visceral adipose tissue
1. INTRODUCTION
Insulin resistance in obesity is a complex metabolic state consisting of increased endogenous glucose production (EGP), reduced muscle glucose uptake and increased lipolysis.1 Many studies have now demonstrated low grade inflammation, systemically and in adipose tissue in the obese state1 but the causal relationship between inflammation and insulin resistance in humans is still unclear. Inflammation in adipose tissue is associated with increased lipolysis which may be driven by proinflammatory cytokines.2 This lipolysis can play a causal role in insulin resistance because the FFAs released can increase EGP and reduce muscle glucose uptake.3
Retinol‐binding protein 4 (RBP4) is an adipose and liver secreted protein which is elevated in adipose tissue and serum of humans with obesity and/or type 2 diabetes (T2D).4, 5, 6 Adipose expression and serum levels of RBP4 are associated with insulin resistance in most, but not all studies.4, 5, 6, 7 Data that support a causative role for RBP4 in insulin resistance/T2D include the fact that a gain‐of‐function polymorphism in the RBP4 promoter which increases adipose RBP4 expression8 is associated with an 80% increased risk of T2D in humans.9 Furthermore, obese, insulin‐resistant women with breast cancer that were treated with the synthetic retinoid fenretinide which lowers circulating RBP4 levels, for 2 years had a sevenfold greater probability to normalize insulin sensitivity than women that were treated with tamoxifen or placebo.10 While fenretinide has other effects in addition to lowering RBP4, these results are consistent with preclinical studies.4, 11, 12, 13
Genetic or pharmacologic elevation of RBP4 in mice induces insulin resistance4 and lowering RBP4 in obese mice improves insulin sensitivity.11, 12, 13 Mechanistically, RBP4 elevation increases the expression of the major gluconeogenic enzyme phosphoenolpyruvate carboxykinase (PEPCK) in liver and impairs insulin signaling in skeletal muscle.4 In cultured hepatocytes, RBP4 increases basal EGP and reduces insulin‐mediated EGP suppression,4 but this remains to be investigated definitively in vivo in humans or rodents. RBP4 also induces adipose tissue inflammation in mice by activating both innate and adaptive immune responses14, 15, 16 and blocking antigen presentation in RBP4 overexpressing mice reduces adipose tissue inflammation and improves insulin sensitivity.16 Adipose‐specific RBP4 overexpression in mice results in elevated plasma FFA and higher expression of lipolysis genes as well as increased adipose inflammation,17 but lipolysis itself has not been measured. Clamp studies in humans have shown an inverse correlation between serum RBP4 and glucose disposal rates5, 7 but there is limited information in humans regarding whether RBP4 reduces insulin suppression of EGP or lipolysis. Because RBP4 levels in serum correlate with systemic low‐grade inflammation18 and subcutaneous adipose tissue (SAT) RBP4 expression correlates with inflammatory markers19 in humans, it is possible that RBP4‐induced inflammation could result in increased lipolysis and impaired suppression of EGP.
Therefore, in this study, we aimed to determine the relationship between circulating and tissue RBP4 levels, lipolysis, and hepatic insulin resistance in humans in vivo. We show that adipose tissue and hepatic RBP4 mRNA expression are both increased in obesity. Serum RBP4 is inversely correlated with insulin‐mediated suppression of lipolysis and EGP as well as with peripheral insulin‐mediated glucose disposal. In addition, adipose RBP4 expression correlates with inflammatory markers. Our mechanistic studies show that RBP4 directly stimulates basal lipolysis, and media from macrophages incubated with RBP4 augments isoproterenol‐ and 8‐Br‐cAMP‐stimulated lipolysis and impairs insulin suppression of lipolysis ex vivo in adipocytes from obese subjects. The media from RBP4‐treated macrophages has a marked increase in TNFα protein. Thus, RBP4 activation of macrophages increases lipolysis which may result at least in part, from proinflammatory cytokine release and this may play an important role in RBP4‐induced hepatic and systemic insulin resistance.
2. MATERIAL AND METHODS
2.1. Subjects
Twenty morbidly obese female subjects scheduled for laparoscopic Roux‐en‐Y gastric bypass (RYGB) surgery were studied. In these subjects, we performed a two‐step hyperinsulinemic euglycemic clamp and during surgery, we collected liver, subcutaneous (SAT), and visceral AT (VAT) biopsies.20 As controls for tissue RBP4, we included six lean and age‐matched women undergoing elective laparoscopic cholecystectomy. Liver and adipose tissue biopsies were collected in the same way as in the obese subjects. To avoid confounding by impaired glucose tolerance, the control subjects needed to have a normal glucose tolerance test according to the criteria of the ADA.21 Other eligibility criteria for the controls were an age older than 18 years, understanding the objective of the study, competent to give informed consent, and a stable body mass index (BMI) between 20 and 25 kg/m2. Exclusion criteria were performance of vigorous exercise, family history of T2DM, a recent history (6 months or less) of substantial alcohol or drug abuse and the use of any medication. For in vitro lipolysis experiments stromal vascular fraction (SVF) cells from six female subjects with a BMI of 32.5 ± 2.2 kg/m2 were obtained from the Boston Nutrition and Obesity Research Center Adipocyte Core. The study was performed according to the guidelines of the Declaration of Helsinki (2008) and approved by the Medical Ethical Committees of the Academic Medical Center (AMC) of the University of Amsterdam and Boston University Medical Center. After a complete description of the study had been given, written informed consent was obtained.
2.2. Two‐step hyperinsulinemic euglycemic clamp
Glucose metabolism and lipolysis were assessed in the obese subjects only.20 The subjects were admitted to the Metabolic Clinical Research Unit of the AMC at 08:00 am after an overnight fast. A catheter was inserted into an antecubital vein for infusion of [6,6‐2H2]glucose, [1,1,2,3,3‐2H5]glycerol (>99% enriched; Cambridge Isotopes, Andover, MA, USA), glucose 20%, and insulin. Another catheter was inserted into a contralateral hand vein and kept in a thermoregulated (60°C) plexiglas box for sampling of arterialized venous blood. Saline was infused as NaCl 0.9% at a rate of 50 mL/h to sustain catheter patency. [6,6‐2H2]‐glucose and [1,1,2,3,3‐2H5]glycerol were infused as tracers to study glucose kinetics and lipolysis (total triacylglycerol hydrolysis), respectively. At 09:00 am (T = −2 hours), after drawing a blood sample for background enrichment of plasma glucose and glycerol, a primed‐continuous infusion of [6,6‐2H2]glucose and of [1,1,2,3,3‐2H5]glycerol were started at a rate of 0.11 µmol/kg/min after a priming dose equivalent to 120‐minute infusion. After 110, 115, and 120 minutes, blood samples were drawn for determination of glucose and glycerol enrichments, glucoregulatory hormones and FFA. Subsequently, at 11:05 am (T = 0), a continuous infusion of insulin (Actrapid 100 U/mL; Novo Nordisk Farma, Alphen a/d Rijn, the Netherlands) was started for 2 hours at a rate of 20 mU/m2 body surface area/min. At T = 2 hours, the infusion rate of insulin was increased to 60 mU/m2 body surface area/min. Plasma glucose was measured every 10 minutes and glucose 20% (enriched with [6,6‐2H2]glucose to approximate plasma enrichment) was infused at a variable rate to maintain plasma glucose at 5.0 mmol/L. At T = 2 hours and T = 4 hours, five blood samples with a 5‐minute interval were drawn to measure glucose and glycerol enrichments and two samples were drawn to measure glucoregulatory hormones and FFA. During the study, the participants were allowed to drink water only.
2.3. Biopsies
Roux‐en‐Y gastric bypass surgery was performed by experienced bariatric surgeons in two medical centers (Rijnstate Hospital, Arnhem and the former Slotervaart Hospital, Amsterdam, the Netherlands). The laparoscopic cholecystectomies were performed by an experienced surgeon in one medical center (Northwest Clinics, Alkmaar, the Netherlands). Before starting the RYGB procedure, tissue biopsies were taken from the liver and visceral and subcutaneous abdominal adipose tissue. Local hemostasis was checked directly after the biopsies and at the end of the surgical procedure. Tissue samples were obtained after a comparable fasting period (10‐12 hours). The samples were immediately frozen in liquid nitrogen and stored at −80°C until further analyses.
2.4. Gene expression
Total RNA was isolated using TRIzol reagent (Invitrogen, Breda, the Netherlands), followed by the NucleoSpin II extraction kit (Machterey & Nagel Gm bH, Duren, Germany) according to the manufacturer’s recommendations. Briefly, 1 mL of TRIzol and glass beads (Biospec Products Inc., Bartlesville, OK, USA) were added to the tissue. After vigorous shaking using a Fast Prep‐24 machine for 20 seconds. At 4.5 m/s (MP Biomedicals, Santa Ana, CA, USA), the homogenate was centrifuged (10 minutes at 12 000×g at 4°C). The nonlipid containing fraction was transferred and 200 μL of chloroform was added. The mixture was subsequently vortexed and centrifuged (15 minutes at 16 100×g at 4°C). The aqueous phase was transferred to a new tube and an equal volume of 70% ethanol was added. Afterward, we continued with the NucleoSpin II extraction kit according to the manufacturer’s instructions. RNA concentrations were measured using the Nanodrop Spectrophotometer 1 (Nanodrop Technologies, Wilmington, NC, USA). cDNA was synthesized using Superscript II (Invitrogen) according to the manufacturer’s instructions. After synthesis, the cDNA was diluted 20×. mRNA expression of RBP4, GLUT4, and inflammatory markers was normalized to the housekeeping gene Acidic Ribosomal Phosphoprotein P0 (RPLP0).
2.5. Protein isolation and detection of RBP4 in serum and tissue
Cell lysates were prepared in RIPA buffer (150 mM NaCl, 50 mM Tris HCl pH 7.4, 2 mM EDTA, 0.5% deoxycholaat, 1 mM Na3VO4, 20 mM NaF, 0.5% Triton X‐100), supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland), and PMSF.
Serum was diluted 30× in a standard lysis buffer. Adipose tissue was lysed in standard lysis buffer. One microliter equivalent of human serum and 50 μg of protein from human adipose tissue were separated by 18% SDS‐PAGE and transferred to nitrocellulose membranes. Human RBP4 proteins were detected using anti‐human RBP4 polyclonal antisera (Dako, Glostrup, Denmark, catalog #A0040). For quantitation of serum RBP4 levels, a standard curve of purified human RBP4 was run on each gel.22
2.6. Differentiation of human stromal vascular fraction (SVF) cells into adipocytes
Isolation, proliferation, and differentiation of human SVF cells were performed as described.23 Briefly, AT biopsies were washed, minced, treated with collagenase, and cells were filtered and centrifuged. Cells were resuspended in growth media, counted and plated. After cells (preadipocytes) reached 70%‐80% confluence, they were re‐plated from 10‐cm dish to 12‐well plates, with density of 5000‐15000 cells/cm2. Cells were grown in growth media (alpha‐MEM with 10% FBS and 1% pen/strep antibiotics) and at 2 days after confluence cells were considered ready for differentiation. Growth media was changed to differentiation media (18.5 mM glucose DMEM/F12 media, 15 mM HEPES, 25 mM NaHCO3, 1% pen/strep antibiotics, 33 µM d‐Biotin, 17 uM pantothenate, 100 nM dexamethasone, 100 nM insulin, 1 µM rosiglitazone, 0.5 mM IBMX (1‐methyl‐3‐isobutylxanthine), 2 nM T3, 10 µg/mL transferrin). After 3 days of differentiation, differentiation media was replaced by maintenance media (18.5 mM glucose DMEM/F12 media, 15 mM HEPES, 25 mM NaHCO3, 1% pen/strep antibiotics, 33 uM d‐Biotin, 17 µM pantothenate, 100 nM dexamethasone, 100 nM insulin) which was changed every second day. At day 12 after induction of differentiation, adipocytes were used for the experiment.
2.7. Activation of macrophages (RAW 264.7 cells) by RBP4 to obtain conditioned media
RAW 264.7 cells were maintained in DMEM media (4.5 g/L glucose, L‐glutamine, sodium pyruvate, 10% FBS, 1% pen/strep antibiotics). For the production of conditioned media, cells were treated with RBP4 (50 µg/mL) for 18‐24 hours. Subsequently, media were harvested and any cell debris removed by centrifugation.
Differentiated human SVF‐adipocytes at day 12 of differentiation were treated with RBP4 (50 µg/mL)/serum and insulin‐free maintenance media or DMSO/serum and insulin‐free maintenance media (control) for 3 or 18 hours. After treatment, cells were additionally serum and insulin starved for 1 hour followed by the lipolysis experiment.
Day 12 differentiated human SVF adipocytes were treated with conditioned media from RBP4‐treated macrophages or conditioned media from untreated macrophages (25% conditioned media/75% serum and insulin free maintenance media) for 3 hours. After treatment, cells were additionally serum‐ and insulin‐starved for 1 hour and the lipolysis experiment was performed.
2.8. Basal and stimulated lipolysis in human adipocytes
The lipolysis experiment was performed as described previously.23 After DMSO or RBP4 treatment and serum/insulin starvation, the lipolysis assay was performed using Krebs‐Ringer (KRB) buffer with 5 mM glucose and 4% BSA, pH 7.4 with adenosine deaminase (ADA [1 U/mL]), and phenylisopropyl adenosine (PIA [20 nM]). For stimulated lipolysis, isoproterenol (10−8‐10−6 M) or 8‐Br‐cAMP (1 mM) was also added. Inhibition of lipolysis by insulin was measured under 8‐Br‐cAMP‐stimulated conditions, with insulin at 0, 5, 10, 15, 30, and 600 pM concentrations.
Cells were incubated with KRB and reagents for basal, stimulated or insulin‐suppressed lipolysis at 37°C for 2 hours. After incubation, cell culture plates were placed on ice to stop the reaction and media was saved for glycerol measurement and cells for cell number calculations.
Glycerol concentration in culture media was measured using a standard enzymatic fluorometric assay in neutralized perchloric acid extracts.24 Cell number/double‐stranded DNA was measured using Quant‐iT PicoGreen ds DNA Assay Kit (ThermoFisher Scientific cat #P7589).
TNFα levels in media from the RAW macrophages that were treated with RBP4 (50 µg/mL) were measured by Elisa as described.14
2.9. Analytical procedures
In human plasma, glucose was measured using a Biosen C‐line plus glucose analyzer (EKF Diagnostics, Barleben/Magdeburg, Germany). Insulin was determined using an Immulite 2000 system (Diagnostic Products Corporation, Los Angeles, CA, USA), with a chemiluminiscent immunometric assay (intra‐assay variation 4%‐5%; inter‐assay variation 5%; detection limit 15 pmol/L). FFA were measured using an enzymatic method (NEFA‐Cc, Wako Chemicals, Neuss, Germany; intra‐assay variation 1%; inter‐assay variation 4%‐15%; detection limit 0.02 mmol/L). [6,6‐2H2]glucose and [1,1,2,3,3‐2H5]glycerol enrichment were measured as described previously.20
2.10. Calculations
EGP, lipolysis rate, and peripheral glucose disposal (rate of disappearance [Rd]) were calculated using modified versions of the Steele equations as described previously.25, 26 EGP is expressed as μmol/(kg fat‐free mass)/min. Lipolysis and Rd are both expressed as μmol/kg/min. Insulin‐mediated suppression of lipolysis, FFA, and EGP is expressed as the % suppression of the basal values and was assessed during the first step of the hyperinsulinemic‐euglycemic clamp.
2.11. Statistical analysis
In vivo data are presented as median and range. Due to the small sample size and nonnormal distribution of study parameters, nonparametric tests were used. Between‐group differences were tested using the Mann‐Whitney U test. Correlations were calculated using the Spearman correlation test. For in vitro experiments, data are presented as means ± SEM. Paired t test and two‐way ANOVA were used to investigate differences between the groups. Subjects with missing data were excluded from analyses. A P value < 0.1 was considered a trend and a P value <.05 was considered statistically significant. All statistical analyses were run on IBM SPSS version 24 (SPSS, Chicago, IL, USA) and GraphPad PRISM 7 software (La Jolla, CA, USA).
3. RESULTS
3.1. Demographic and metabolic characteristics
Table 1 shows the clinical and metabolic characteristics. The subjects were all female and similar in age. Due to technical issues, the clamp was not completed in two of the obese subjects. In one lean control, visceral biopsies were not performed because it was considered not safe because of the low amount of visceral adipose tissue (VAT). The obese subjects were insulin‐resistant reflected in higher fasting insulin levels.27 Since reduced expression of GLUT4 specifically in adipocytes is a central feature of whole‐body insulin resistance,28 we measured GLUT4 mRNA expression in the adipose tissue depots of both groups. GLUT4 mRNA expression was lower in the obese subjects in both the subcutaneous (P < .001) and visceral (P = .019) adipose tissue depots (Figure 1). Subcutaneous and visceral adipose GLUT4 mRNA expression were both inversely correlated with BMI (rs = −0.601, P = .001; rs = −0.502, P = .011, respectively) and in the obese subjects subcutaneous GLUT4 mRNA expression correlated with Rd (rs = 0.591, P = .013), whereas there was no correlation between visceral GLUT4 mRNA expression and Rd (rs = 0.397, P = .115).
Table 1.
Demographic and metabolic characteristics
| Lean (N = 6) | Obese (N = 20) | P | |
|---|---|---|---|
| Age (years) | 37.5 [26‐45] | 41 [26‐58] | .120 |
| BMI (kg/m2) | 22.1 [20‐24.3] | 42.9 [38.7‐61.3] | <.001 |
| Fasting glucose (mmoL/L) | 5.0 [4.5‐5.6] | 5.4 [4.4‐8.8] | .200 |
| Fasting insulin (pmoL/L) | 25 [14‐63] | 81 [20‐142] | .002 |
Data are presented as median [range].
Figure 1.
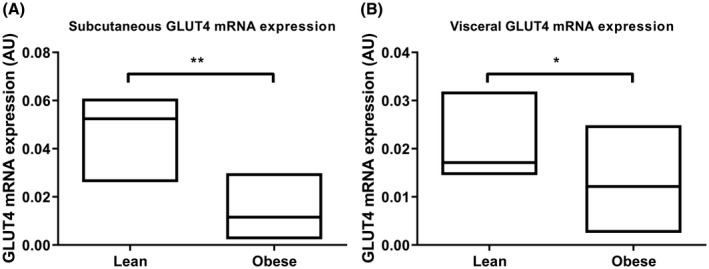
Relative adipose tissue GLUT4 mRNA expression of lean versus obese subjects. Data presented as median (range). n = 6 for panel A and n = 5 for panel B lean and 20 obese women. *P < .05, ** P < .001
3.2. Adipose and hepatic RBP4 mRNA expression is increased in obese women
Subcutaneous adipose RBP4 mRNA expression (P = .015) but not RBP4 protein expression (P = .570) was increased in the obese subjects. In VAT from obese subjects, RBP4 protein was increased (P = .009) and RBP4 mRNA expression tended to be increased (P = .062). Hepatic RBP4 mRNA expression was increased in the obese subjects compared to lean controls (P < .001) (Figure 2). Neither hepatic nor adipose mRNA expression of RBP4 correlated with serum RBP4 levels. A limitation of this study is that we were not able to measure RBP4 protein levels in liver due to insufficient sample size.
Figure 2.
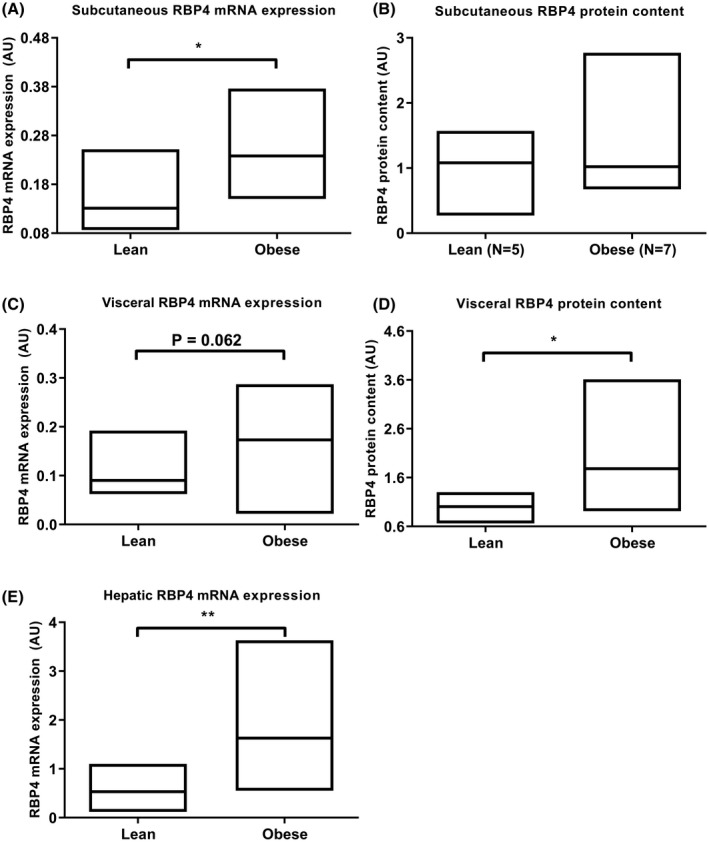
Relative mRNA expression and protein content of RBP4 in subcutaneous AT (A and B), visceral AT (C and D), and relative RBP4 mRNA expression in liver (E) of lean versus obese subjects. Data presented as median (range). *P < .05, **P < .001. n = 6 lean for panels A and E and n = 5 lean for panels B‐D. n = 7 obese in panel B, n = 12 obese in panel D, n = 20 obese in panels A, C, and E
3.3. Serum RBP4 levels correlate inversely with insulin sensitivity
Circulating RBP4 can interfere with insulin signaling in skeletal muscle and liver.4 In line, in the obese subjects, serum RBP4 correlated inversely with Rd (rs = −0.551, P = .022). There was also a trend for an inverse correlation between SAT RBP4 protein and Rd (rs = −0.771, P = .072). In contrast, there was a strong inverse correlation between serum RBP4 levels and insulin‐mediated suppression of lipolysis (rs = −0.635, P = .008), FFA (rs = −0.672, P = .003), and EGP (rs = −0.637, P = .006) (Figure 3). Serum RBP4 levels did not correlate with fasting lipolysis, FFA, or EGP.
Figure 3.
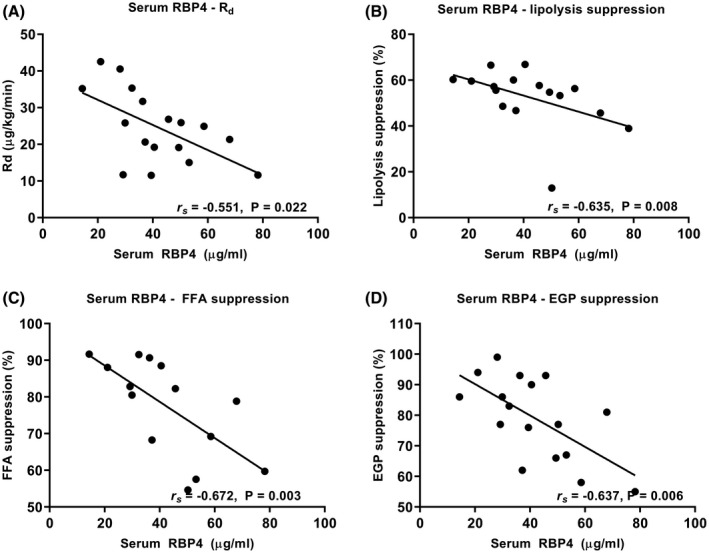
Serum RBP4 levels correlate inversely with peripheral glucose disposal (A), and insulin‐mediated suppression (% of basal) of lipolysis (B), free fatty acids (C), and endogenous glucose production (D) in the obese subjects
3.4. RBP4 mRNA expression correlates with expression of inflammatory markers in SAT, but not in VAT
Recent studies have shown that RBP4 activates pro‐inflammatory pathways in cell lines and animal models, which may lead to insulin resistance.14, 15, 16 To investigate the relationship between RBP4 and adipose inflammation, we measured the expression of several known inflammatory markers involved in adipose tissue inflammation. RBP4 mRNA expression was associated with mRNA expression of adipose macrophage (CD68, MRC1) and T cell (CD4, CD25) markers as well as with expression of members of the chemokine family (CCL18, CCR7) and TLR4 in SAT, but not VAT (Table 2 and Figure 4).
Table 2.
Correlation between RBP4 mRNA expression and inflammatory marker mRNA expression in white adipose tissue
| SAT | VAT | |
|---|---|---|
| CD68 | 0.433a | 0.168 |
| CD163 | 0.281 | 0.220 |
| MRC1 | 0.512b | 0.305 |
| CCL18 | 0.413a | 0.254 |
| CCR7 | 0.596b | 0.026 |
| CCL4 | 0.346 | −0.007 |
| CCL2 | 0.294 | −0.080 |
| TLR4 | 0.520b | 0.283 |
| IL10 | 0.520b | 0.129 |
| CD200R | 0.595b | 0.408a |
| TNFA | 0.139 | 0.065 |
| CD11C | 0.192 | 0.150 |
| CD4 | 0.475a | 0.391 |
| CD25 | 0.471a | 0.250 |
| FOXP3 | 0.069 | 0.366 |
Correlations performed using Spearman correlation test.
Abbreviations: SAT, subcutaneous adipose tissue; VAT, visceral adipose tissue.
n = 23‐26 for SAT. n = 21‐25 for VAT.
P < .05,
P < .01.
Figure 4.
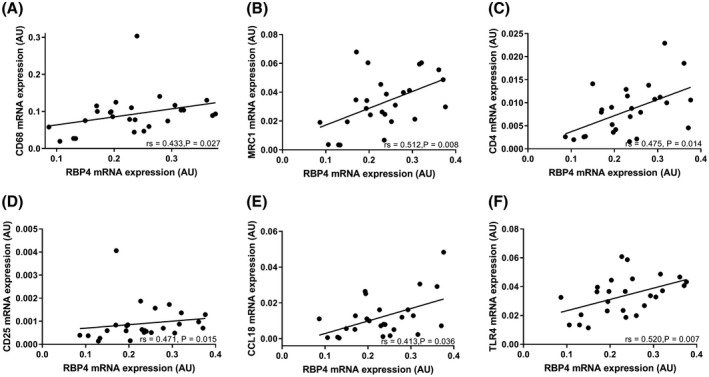
RBP4 mRNA expression correlates with the mRNA expression of CD68 (A), MRC1 (B), CD4 (C), CD25 (D), CCL18 (E), and TLR4 (F) in subcutaneous adipose tissue
3.5. Direct and indirect effects of RBP4 on lipolysis in human adipocytes
Direct treatment of human adipocytes with RBP4 increased basal lipolysis (Figure 5A) but did not impair the effect of insulin to suppress lipolysis (Figures 5B,C). However, conditioned media from RBP4‐treated macrophages markedly stimulated basal lipolysis, and augmented isoproterenol‐stimulated and 8‐Br‐cAMP‐stimulated lipolysis in human adipocytes (Figure 6A). This conditioned media also impaired insulin inhibition of lipolysis (Figure 6B). To explore the mechanisms by which RBP4 increases lipolysis, we measured TNFα protein levels in the conditioned media from macrophages treated with RBP4, as TNFα is known to increase lipolysis29 and a previous study showed that RBP4 increased TNFα production from mouse and human macrophages.14 Compared to the conditioned media of untreated macrophages, the conditioned media of macrophages treated with RBP4 had at least 40‐fold higher TNFα protein levels (Figure 6C). Thus, treatment of adipocytes with conditioned media from RBP4‐treated macrophages stimulates lipolysis and causes resistance to insulin‐mediated inhibition of lipolysis. This could result from increased RBP4‐induced secretion of pro‐inflammatory cytokines such as TNFα.
Figure 5.
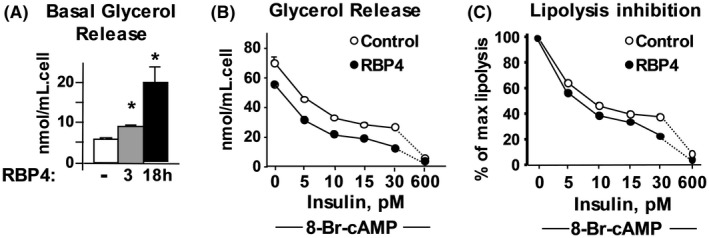
Direct effect of RBP4 on human SVF‐derived adipocytes leads to increased basal lipolysis (A) but does not impair the ability of insulin to inhibit lipolysis (B, C). A, Human SVF‐derived adipocytes were incubated with RBP4 (50 µg/mL) for 3 or 18 hours, *P < .01 versus all other conditions. B, After RBP4 incubation, a dose‐response to insulin of 8‐Bromo‐cAMP‐activated lipolysis was performed. C, Results in panel B are expressed as % of maximal 8‐Bromo‐cAMP‐activated lipolysis
Figure 6.
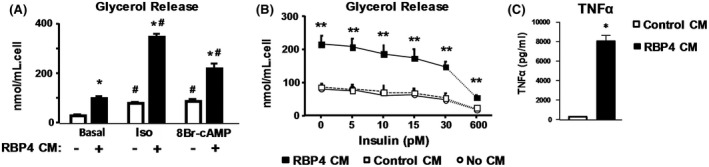
Indirect effect of RBP4 on human SVF‐derived adipocytes (A). Media from RAW macrophages incubated with RBP4 (RBP4 CM) increases basal (−) and stimulated (+) lipolysis in human adipocytes. *P < .05 versus (−) same condition, #P < .05 versus Basal same media condition. (B) Insulin inhibition of RBP4 CM‐induced lipolysis has an ED50 over 30 pM compared to 10 pM for insulin inhibition of isoproterenol‐induced lipolysis (data not shown). Control CM: media from RAW cells incubated with vehicle only without RBP4. No CM: adipocytes incubated without any conditioned media. **P < .05 versus all other conditions. (C) Concentration of TNFα in media of RAW cells incubated without (open bar) or with RBP4 (50 µg/mL) (black bar). *P < .05 versus conditioned media without RBP4
4. DISCUSSION
Obesity‐induced insulin resistance is associated with increased lipolysis in adipose tissue.3 Because increased RBP4 levels induce insulin resistance, we hypothesized that RBP4 may have a direct effect on basal lipolysis and/or the ability of insulin to suppress lipolysis and this could be an important mechanism by which elevated RBP4 levels result in whole body insulin resistance. We found a strong relationship between serum RBP4 levels and resistance to suppression of lipolysis by insulin in vivo in obese human subjects. In parallel, our mechanistic studies showed that in cultured human adipocytes, RBP4 directly stimulated lipolysis in the basal state but did not alter insulin suppression of lipolysis. Previous studies in human and rodent cells14 and animal models have shown that RBP4 activates macrophages and other antigen‐presenting cells. RBP4 activates both CD4‐positive T cells and macrophages through TLR4‐ and JNK‐dependent pathways, resulting in increased production of pro‐inflammatory cytokines such as TNF‐α, IL‐1β, and IL‐6.14, 15, 16, 17 These cytokines have been shown to stimulate lipolysis and also to impair insulin suppression of lipolysis.29, 30, 31, 32 The possibility that RBP4 increases lipolysis is strongly supported by the observation that mice overexpressing human RBP4 selectively in adipocytes have increased mRNA expression of the lipolytic enzymes ATGL, HSL, and LPL in adipose tissues and elevated plasma FFA levels.17
We tested whether media from RBP4‐activated macrophages could affect lipolysis. We found that conditioned media from RBP4‐activated macrophages augments basal‐, isoproterenol‐, and 8‐Br‐cAMP‐stimulated lipolysis in adipocytes and causes resistance to insulin suppression of lipolysis. We hypothesized that the impaired suppression of lipolysis in vivo may result from increased adipose tissue inflammation and pro‐inflammatory effects of RBP4 on macrophages,14, 15, 16 since we find in this study that RBP4 mRNA expression correlates with several macrophage markers and other pro‐inflammatory markers in SAT in obese humans.
RBP4 induces TNFα expression through a TLR4‐dependent pathway in macrophages14 and suppression of TLR4, either through a transgenic knockout or pharmacologically through a TLR4 inhibitor, attenuates RBP4‐dependent TNFα production.14, 33, 34 Because RBP4 induces TNFα production, and TNFα stimulates lipolysis,29 we measured TNFα protein levels in the media from macrophages treated with RBP4 which was used as conditioned media for the human adipocyte experiments. We found that the levels of TNFα protein were increased at least 40‐fold in media from macrophages after RBP4 treatment. These results further support the hypothesis that RBP4 affects lipolysis by activating macrophages to produce proinflammatory cytokines which can stimulate lipolysis and also impair insulin signaling.
These results are consistent with our findings that elevation of RBP4 in adipose tissue inhibits insulin‐stimulated phosphoinositide 3‐kinase (PI3K) activity in vivo4 through an indirect mechanism involving activation of antigen‐presenting macrophages and dendritic cells and release of pro‐inflammatory cytokines.14, 15 Since PI3K activity is a major pathway mediating the anti‐lipolytic effect of insulin,35 this could explain, at least in part, how RBP4 impairs insulin action on lipolysis in adipose tissue.
Surprisingly, adipose tissue RBP4 mRNA expression did not correlate with insulin‐mediated suppression of lipolysis in vivo. It has been shown that the mRNA expression of RBP4 in adipocytes may not always correlate with serum levels.36 This is partly because circulating RBP4 can accumulate in adipose tissue where it can activate macrophages as demonstrated in mice overexpressing human RBP4 driven by a muscle promoter.15 In this model, RBP4 is secreted from muscle and elevates circulating levels. Western blotting showed a greater than sixfold higher accumulation of human RBP4 in perigonadal adipose tissue compared to liver. Therefore, the RBP4 in adipose tissue may not be entirely derived from adipocytes but may originate in part from the circulation. Thus, mRNA expression may not reflect all the RBP4 present in adipose tissue.
In our study, in the obese subjects, hepatic RBP4 mRNA expression also did not correlate with serum RBP4 levels. Recently, a study in mice suggested that the main source of circulating RBP4 is from hepatocytes and that in liver‐specific RBP4 knockout mice on a high‐fat, high‐sucrose diet, adipose tissue RBP4 expression was increased, while RBP4 was undetectable in serum.36 However, another study showed that overexpression of human RBP4 selectively in adipocytes in mice resulted in increased adipocyte and serum RBP4 levels when mice were fed a high‐fat diet.17 The adipocytes were the source of the increased serum RBP4 since the human RBP4 protein was detected in the mouse serum and this could only be derived from the transgene. This suggests that under at least some obese/high‐fat diet conditions, increased adipose secretion of RBP4 contributes to elevated serum levels. These studies underscore the complicated relationship between tissue levels of RBP4 and circulating RBP4 concentrations and explain the lack of a direct linear correlation.
Another novel finding is that serum RBP4 levels inversely correlate with insulin‐mediated EGP suppression, that is, hepatic insulin sensitivity. One previous study showed that serum RBP4 levels inversely correlated with the absolute difference in hepatic glucose production between the basal state and upon insulin infusion in humans. However, in that study, after adjusting for sex, age, and body fat percentage, plasma RBP4 was not an independent predictor of EGP suppression.37 In our study, serum RBP4 remained a negative predictor of EGP suppression after correction for both age and BMI (data not shown). Another study evaluated the correlation between plasma RBP4 levels and the hepatic insulin resistance index, a surrogate marker for hepatic insulin sensitivity, in Mexican Americans, ranging from lean to obese in weight and from normal glucose tolerant to T2DM. This study did not find a correlation.38 However, a two step clamp was not performed and the insulin infusion rate seems too high to adequately assess hepatic insulin sensitivity. The results of the previous studies may differ from ours because of their inclusion of subjects of both sexes and subjects with both normal glucose tolerance and overt diabetes, which may have confounded the relationship between serum RBP4 levels and EGP suppression. The effect of RBP4 on hepatic insulin sensitivity might be through its stimulation of lipolysis, thus increasing hepatic delivery of glycerol and acetyl CoA.39 In support of this, adipose‐specific overexpression of human RBP4 results in increased expression of lipolytic genes in white adipose tissue with increased fatty acid uptake in liver and hepatic steatosis.17 It is possible that the effects of elevated RBP4 on insulin suppression of EGP may also have a direct component since liver PEPCK expression is increased in mice treated chronically with RBP4 and RBP4 impairs EGP suppression in cultured hepatocytes.4 Thus, RBP4 might act through more than one mechanism including a retinol‐dependent mechanism since retinoids induce the expression of some gluconeogenic enzymes such as PEPCK.40
Another finding in this study is that hepatic RBP4 mRNA expression was increased in obese subjects compared to lean controls. The causative factor is not known, but factors that increase RBP4 expression include retinol, glucagon, estrogen, and stimulation of cAMP.41, 42, 43, 44 There are mixed results in the literature in terms of hepatic RBP4 regulation in obesity and whether elevated liver RBP4 levels could contribute to nonalcoholic hepatic steatosis based on either mRNA or protein measurements. Studies have shown that hepatic RBP4 mRNA and protein expression is increased in obese rodents.45 In humans, one study showed no difference in hepatic RBP4 mRNA expression between 45 obese women and 4 lean controls.46 However, a recent study indicated that both mRNA and protein levels of RBP4 are increased in liver of humans with nonalcoholic fatty liver disease and RBP4 mRNA expression correlates with hepatic triglyceride content.47 Adeno‐associated viral overexpression of RBP4 in liver does not impair whole body glucose tolerance in mice but serum RBP4 levels were not elevated in that study and hepatic insulin sensitivity was not assessed.48 Finally, adipocyte‐selective overexpression of RBP4 and global overexpression of RBP4 both result in glucose intolerance and increased hepatic lipid accumulation even on a chow diet.17, 47
In our study, in addition to the inverse correlation of serum RBP4 levels with suppression of lipolysis and hepatic glucose output, serum RBP4 levels correlated inversely with peripheral glucose disposal, which is in line with the effect of elevated RBP4 to impair insulin signaling in muscle in vivo.4 We did not measure RBP4 protein levels in skeletal muscle or liver due to insufficient sample size. Therefore, we cannot exclude direct effects of tissue RBP4 protein on the measured metabolic fluxes.
Limitations of this study include the relatively small number of subjects for which we had tissue biopsies, the smaller number of lean controls compared to the obese subjects and the inclusion of female subjects only. Generalization to the general population needs to occur with caution as sexual dimorphism in the expression of RBP4 has been observed.6
We conclude that in morbid obesity, hepatic, visceral adipose and subcutaneous adipose RBP4 mRNA and/or protein levels are increased and circulating RBP4 levels correlate inversely with insulin suppression of lipolysis and EGP, and with glucose disposal. Furthermore, RBP4 mRNA levels correlate with the mRNA levels of inflammation markers in WAT. RBP4 acts directly and indirectly through macrophage activation to augment lipolysis and induce resistance to insulin suppression of lipolysis in human adipocytes. Thus, RBP4‐induced inflammation of adipose tissue increases lipolysis in vitro and our flux studies in humans in vivo show a corresponding correlation between RBP4 and resistance to insulin suppression of lipolysis. These results, combined with previous in vitro studies, suggest that elevated RBP4 levels in serum and/or adipose tissue may contribute to causing insulin resistance in humans by impairing lipolysis suppression. While we demonstrate a causative role in vitro in human adipocytes, our flux studies in vivo provide the basis for future experiments to test the potential causative role of RBP4 on increased lipolysis and the extent to which this contributes to insulin resistance in obese humans.
CONFLICT OF INTEREST
B.B.K. is an inventor on a patent on RBP4. There are no other potential conflicts of interest relevant to this article.
AUTHOR CONTRIBUTIONS
M.J. Serlie and B.B. Kahn designed the research; M. Kilicarslan, B.A. de Weijer, and K. Simonyté Sjödin performed the research; M. Kilicarslan, M.J. Serlie, and B.B. Kahn analyzed data and wrote the paper; H. Cakir, I.M. Janssen, F.J. Berends, A.W. van de Laar, A.P. Houdijk collected the data; P. Aryal, K.W. ter Horst, M.T. Ackermans provided experimental technical support; and J.A. Romijn made revisions to the manuscript.
ACKNOWLEDGMENTS
We thank Martine van Vessem at the Academic Medical Center, Amsterdam, the Netherlands, for her assistance with the clamps and Odile Peroni at Beth Israel Deaconess Medical Center, Boston, MA, USA, for assistance with data analysis and preparation of figures for the cell‐based assays. We thank S.K. Fried and MJ Lee at the BNORC NIDDK P30DK046200 Adipose Biology Core for assistance with differentiation of human stromovascular cells to adipocytes and lipolysis assays. This work is supported by National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK43051 to B.B.K. M.J.S. received funding from Mediq‐TEFA and was in part supported by EU grant FP7‐EU 305707. K.S. was supported by the Swedish Research Council.
Kilicarslan M, de Weijer BA, Simonyté Sjödin K, et al. RBP4 increases lipolysis in human adipocytes and is associated with increased lipolysis and hepatic insulin resistance in obese women. The FASEB Journal. 2020;34:6099–6110. 10.1096/fj.201901979RR
Murat Kilicarslan, Barbara A. de Weijer, Kotryna Simonyté Sjödin, Barbara B. Kahn and Mireille J. Serlie contributed equally to this study.
Contributor Information
Barbara B. Kahn, Email: bkahn@bidmc.harvard.edu.
Mireille J. Serlie, Email: m.j.serlie@amsterdamumc.nl.
REFERENCES
- 1. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grant RW, Stephens JM. Fat in flames: influence of cytokines and pattern recognition receptors on adipocyte lipolysis. Am J Physiol Endocrinol Metab. 2015;309:E205‐E213. [DOI] [PubMed] [Google Scholar]
- 3. Morigny P, Houssier M, Mouisel E, Langin D. Adipocyte lipolysis and insulin resistance. Biochimie. 2016;125:259‐266. [DOI] [PubMed] [Google Scholar]
- 4. Yang Q, Graham TE, Mody N, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356‐362. [DOI] [PubMed] [Google Scholar]
- 5. Graham TE, Yang Q, Blüher M, et al. Retinol‐binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552‐2563. [DOI] [PubMed] [Google Scholar]
- 6. Kotnik P, Fischer‐Posovszky P, Wabitsch M. RBP4: a controversial adipokine. Eur J Endocrinol. 2011;165:703‐711. [DOI] [PubMed] [Google Scholar]
- 7. Kloting N, Graham TE, Berndt J, et al. Serum retinol‐binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra‐abdominal fat mass. Cell Metab. 2007;6:79‐87. [DOI] [PubMed] [Google Scholar]
- 8. Munkhtulga L, Nagashima S, Nakayama K, et al. Regulatory SNP in the RBP4 gene modified the expression in adipocytes and associated with BMI. Obesity. 2010;18:1006‐1014. [DOI] [PubMed] [Google Scholar]
- 9. van Hoek M, Dehghan A, Zillikens MC, Hofman A, Witteman JC, Sijbrands EJ. An RBP4 promoter polymorphism increases risk of type 2 diabetes. Diabetologia. 2008;51:1423‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johansson H, Gandini S, Guerrieri‐Gonzaga A, et al. Effect of fenretinide and low‐dose tamoxifen on insulin sensitivity in premenopausal women at high risk for breast cancer. Cancer Res. 2008;68:9512‐9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Preitner F, Mody N, Graham TE, Peroni OD, Kahn BB. Long‐term fenretinide treatment prevents high‐fat diet‐induced obesity, insulin resistance, and hepatic steatosis. Am J Physiol Endocrinol Metab. 2009;297:E1420‐E1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma X, Zhou Z, Chen Y, Wu Y, Liu Y. RBP4 functions as a hepatokine in the regulation of glucose metabolism by the circadian clock in mice. Diabetologia. 2016;59:354‐362. [DOI] [PubMed] [Google Scholar]
- 13. Zemany L, Bhanot S, Peroni OD, et al. Transthyretin antisense oligonucleotides lower circulating RBP4 levels and improve insulin sensitivity in obese mice. Diabetes. 2015;64:1603‐1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Norseen J, Hosooka T, Hammarstedt A, et al. Retinol‐binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c‐Jun N‐terminal kinase‐ and toll‐like receptor 4‐dependent and retinol‐independent mechanism. Mol Cell Biol. 2012;32:2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moraes‐Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 activates antigen‐presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014;19:512‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moraes‐Vieira PM, Castoldi A, Aryal P, Wellenstein K, Peroni OD, Kahn BB. Antigen presentation and T‐cell activation are critical for RBP4‐induced insulin resistance. Diabetes. 2016;65:1317‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee SA, Yuen JJ, Jiang H, Kahn BB, Blaner WS. Adipocyte‐specific overexpression of retinol‐binding protein 4 causes hepatic steatosis in mice. Hepatology. 2016;64:1534‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balagopal P, Graham TE, Kahn BB, Altomare A, Funanage V, George D. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: association with subclinical inflammation. J Clin Endocrinol Metab. 2007;92:1971‐1974. [DOI] [PubMed] [Google Scholar]
- 19. Yao‐Borengasser A, Varma V, Bodles AM, et al. Retinol binding protein 4 expression in humans: relationship to insulin resistance, inflammation, and response to pioglitazone. J Clin Endocrinol Metab. 2007;92:2590‐2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Weijer BA, Aarts E, Janssen IM, et al. Hepatic and peripheral insulin sensitivity do not improve 2 weeks after bariatric surgery. Obesity. 2013;21:1143‐1147. [DOI] [PubMed] [Google Scholar]
- 21. Association AD. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33(suppl 1):S62‐S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graham TE, Wason CJ, Bluher M, Kahn BB. Shortcomings in methodology complicate measurements of serum retinol binding protein (RBP4) in insulin‐resistant human subjects. Diabetologia. 2007;50:814‐823. [DOI] [PubMed] [Google Scholar]
- 23. Lee MJ, Fried SK. Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods Enzymol. 2014;538:49‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laurell S, Tibbling G. An enzymatic fluorometric micromethod for the determination of glycerol. Clin Chim Acta. 1966;13:317‐322. [DOI] [PubMed] [Google Scholar]
- 25. Steele R. Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci. 1959;82:420‐430. [DOI] [PubMed] [Google Scholar]
- 26. Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose production during hyperinsulinemic‐euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes. 1987;36:914‐924. [DOI] [PubMed] [Google Scholar]
- 27. ter Horst KW, Gilijamse PW, Koopman KE, et al. Insulin resistance in obesity can be reliably identified from fasting plasma insulin. Int J Obes (Lond). 2015;39:1703‐1709. [DOI] [PubMed] [Google Scholar]
- 28. Shepherd PR, Kahn BB. Glucose transporters and insulin action‐implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248‐257. [DOI] [PubMed] [Google Scholar]
- 29. Cawthorn WP, Sethi JK. TNF‐alpha and adipocyte biology. FEBS Lett. 2008;582:117‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao D, Madi M, Ding C, et al. Interleukin‐1beta mediates macrophage‐induced impairment of insulin signaling in human primary adipocytes. Am J Physiol Endocrinol Metab. 2014;307:E289‐E304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang Y, Ju D, Zhang M, Yang G. Interleukin‐6 stimulates lipolysis in porcine adipocytes. Endocrine. 2008;33:261‐269. [DOI] [PubMed] [Google Scholar]
- 32. Lee MJ, Fried SK. Glucocorticoids antagonize tumor necrosis factor‐alpha‐stimulated lipolysis and resistance to the antilipolytic effect of insulin in human adipocytes. Am J Physiol Endocrinol Metab. 2012;303:E1126‐E1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yore MM, Aryal P, Kahn BB. Retinol‐binding protein 4 (RBP4) signals through TLR2 and TLR4/MD2 [abstract]. Diabetes. 2012;61(suppl 1):A423‐A424. [Google Scholar]
- 34. Deng ZB, Poliakov A, Hardy RW, et al. Adipose tissue exosome‐like vesicles mediate activation of macrophage‐induced insulin resistance. Diabetes. 2009;58:2498‐2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi SM, Tucker DF, Gross DN, et al. Insulin regulates adipocyte lipolysis via an Akt‐independent signaling pathway. Mol Cell Biol. 2010;30:5009‐5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson SJ, Sargsyan A, Lee SA, et al. Hepatocytes are the principal source of circulating RBP4 in mice. Diabetes. 2017;66:58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ribel‐Madsen R, Friedrichsen M, Vaag A, Poulsen P. Retinol‐binding protein 4 in twins: regulatory mechanisms and impact of circulating and tissue expression levels on insulin secretion and action. Diabetes. 2009;58:54‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chavez AO, Coletta DK, Kamath S, et al. Retinol-binding protein 4 is associated with impaired glucose tolerance but not with whole body or hepatic insulin resistance in Mexican Americans. Am J Physiol Endocrinol Metab. 2009;296:E758–764. [DOI] [PubMed] [Google Scholar]
- 39. Perry RJ, Camporez JP, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shin DJ, Joshi P, Hong SH, Mosure K, Shin DG, Osborne TF. Genome‐wide analysis of FoxO1 binding in hepatic chromatin: potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Res. 2012;40:11499‐11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dixon JL, Goodman DS. Studies on the metabolism of retinol‐binding protein by primary hepatocytes from retinol‐deficient rats. J Cell Physiol. 1987;130:14‐20. [DOI] [PubMed] [Google Scholar]
- 42. Smith JE, Borek C, Goodman DS. Regulation of retinol‐binding protein metabolism in cultured rat liver cell lines. Cell. 1978;15:865‐873. [DOI] [PubMed] [Google Scholar]
- 43. Bianconcini A, Lupo A, Capone S, et al. Transcriptional activity of the murine retinol‐binding protein gene is regulated by a multiprotein complex containing HMGA1, p54 nrb/NonO, protein‐associated splicing factor (PSF) and steroidogenic factor 1 (SF1)/liver receptor homologue 1 (LRH‐1). Int J Biochem Cell Biol. 2009;41:2189‐2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan BK, Chen J, Lehnert H, Kennedy R, Randeva HS. Raised serum, adipocyte, and adipose tissue retinol‐binding protein 4 in overweight women with polycystic ovary syndrome: effects of gonadal and adrenal steroids. J Clin Endocrinol Metab. 2007;92:2764‐2772. [DOI] [PubMed] [Google Scholar]
- 45. Tan Y, Sun LQ, Kamal MA, Wang X, Seale JP, Qu X. Suppression of retinol‐binding protein 4 with RNA oligonucleotide prevents high‐fat diet‐induced metabolic syndrome and non‐alcoholic fatty liver disease in mice. Biochem Biophys Acta. 2011;1811:1045‐1053. [DOI] [PubMed] [Google Scholar]
- 46. Terra X, Auguet T, Broch M, et al. Retinol binding protein‐4 circulating levels were higher in nonalcoholic fatty liver disease vs. histologically normal liver from morbidly obese women. Obesity. 2013;21:170‐177. [DOI] [PubMed] [Google Scholar]
- 47. Liu Y, Mu D, Chen H, et al. Retinol binding protein 4 induces hepatic mitochondrial dysfunction and promotes hepatic steatosis. J Clin Endocrinol Metab. 2016;101:4338‐4348. [DOI] [PubMed] [Google Scholar]
- 48. Fedders R, Muenzner M, Weber P, et al. Liver‐secreted RBP4 does not impair glucose homeostasis in mice. J Biol Chem. 2018;293:15269‐15276. [DOI] [PMC free article] [PubMed] [Google Scholar]