

Abstract
Evidence for effectiveness of newborn screening (NBS) for propionic acidemia (PA) and isolated methylmalonic acidemia (MMA) is scarce. Prior to implementation in the Netherlands, we aim to estimate the expected health gain of NBS for PA and MMA. In this national retrospective cohort study, the clinical course of 76/83 Dutch PA and MMA patients, diagnosed between January 1979 and July 2019, was evaluated. Five clinical outcome parameters were defined: adverse outcome of the first symptomatic phase, frequency of acute metabolic decompensations (AMD), cognitive function, mitochondrial complications, and treatment‐related complications. Outcomes of patients identified by family testing were compared with the outcomes of their index siblings. An adverse outcome due to the first symptomatic phase was recorded in 46% of the clinically diagnosed patients. Outcome of the first symptomatic phase was similar in 5/9 sibling pairs and better in 4/9 pairs. Based on the day of diagnosis of the clinically diagnosed patients and sibling pair analysis, a preliminary estimated reduction of adverse outcome due to the first symptomatic phase from 46% to 36%‐38% was calculated. Among the sibling pairs, AMD frequency, cognitive function, mitochondrial, and treatment‐related complications were comparable. These results suggest that the health gain of NBS for PA and MMA in overall outcome may be limited, as only a modest decrease of adverse outcomes due to the first symptomatic phase is expected. With current clinical practice, no reduced AMD frequency, improved cognitive function, or reduced frequency of mitochondrial or treatment‐related complications can be expected.
Keywords: methylmalonic acidemia, MMA, NBS, newborn screening, PA, propionic acidemia
Abbreviations
- AMD
acute metabolic decompensation
- AO
adverse outcome
- BMD
bone mineral density
- EO
early onset
- LO
late onset
- MMA
methylmalonic acidemia
- NBS
newborn screening
- PA
propionic acidemia
- PY
patient year
- WHO
World Health Organisation
1. INTRODUCTION
Propionic acidemia (PA, OMIM #6060054) and isolated methylmalonic acidemia (MMA, OMIM #251000, #251100, #251110, #277400, and #277410) are caused by enzyme and cofactor deficiencies involved in propionyl‐CoA breakdown. The clinical course usually starts with an acute metabolic decompensation (AMD) in the neonatal period (early onset, EO) or thereafter (late onset, LO).1 The first symptomatic phase frequently causes irreversible neurological damage, manifesting as movement disorders in varying severity and/or psychomotor retardation. This neurological damage is often reflected by white matter lesions and/or basal ganglia hyperintensities on brain imaging. In addition, the clinical course is characterised by frequent AMD, cognitive deficits, and long‐term complications.2
A diagnosis before the first symptomatic phase, feasible through newborn screening (NBS), could theoretically prevent the neurological damage.3 However, the majority of patients is symptomatic well before NBS results can be available, limiting the potential gain.4, 5 Four studies described the potential additional value of NBS, but each with limited sample sizes and follow‐up time,6, 7, 8, 9 resulting in controversy about the potential effectiveness of NBS for PA and MMA.
Many efforts have been made to achieve consensus on which diseases are suitable for screening, both on a global level by the World Health Organisation (WHO)10, 11 (Supplementary Notes S1) and on European level.12, 13, 14 Despite these efforts, it is still debatable which diseases to screen for and it is not clear on what criteria decisions should be based, resulting in divergent conclusions. Regarding PA and MMA, NBS implementation has been recommended in the United States of America,15 but PA and MMA were not included in the European list of 26 disorders to be considered for NBS expansion.13 Nevertheless, several European countries implemented NBS for PA and MMA.5
In the Netherlands, the Health Council decided based on the expected health gain for LO patients that NBS for PA and MMA met the screening criteria and will start in October 2019.16 NBS will be performed based on increased C3‐carnitine and increased C3/C2 carnitine ratio, complemented with a second‐tier test on increased 2‐methylcitric acid and methylmalonic acid. As the heel prick is performed within 3‐7 days after birth, the aim is to report a positive second‐tier test within 11‐21 days after birth.
For the addition of new diseases to NBS, three WHO criteria seem of particular importance: clearly defined objectives (WHO#2), evidence for effectiveness (WHO#4) and careful evaluation (WHO#9)11 (Supplementary Notes S1). To date, for PA and MMA, these three criteria are not sufficiently met. Also, despite statements that good long‐term outcome is the ultimate goal and that monitoring is necessary to evaluate NBS programmes, long‐term outcome is rarely monitored in a standardised manner.13
We performed a national retrospective cohort study including 76/83 Dutch PA and MMA patients. We aim to define which objectives could be attained with NBS implementation. Hereto, we estimate the expected health gain in the Netherlands by comparing siblings diagnosed via family testing with their older index sibling. In addition, we explore how evaluation of NBS implementation can be performed by assessing newly defined clinical outcome parameters. Lastly, we use insights obtained from studying this pre‐NBS cohort to guide counselling and surveillance of NBS cohorts.
2. METHODS
2.1. Patient inclusion and compliance with ethics guidelines
In this nationwide retrospective cohort study, patients' clinical records from all six Dutch metabolic centres were analysed. Patients were eligible for inclusion if PA or MMA was confirmed by enzymatic activity analysis or mutation analysis of PCCA, PCCB, MUT, MMAA, MMAB, MCEE, or MMADHC. Patients or their legal guardians signed an informed consent form, approving analysis of their clinical records, and publication of the anonymous data, in agreement with institutional and national legislation. The study was approved by the Ethical Committees of the University Medical Centers Utrecht (17‐490/C) and Rotterdam (MEC‐2018‐1312), on behalf of all Ethical Committees of involved University Medical Centers. All procedures followed were in accordance with the ethical standards of these committees and with the Helsinki Declaration of 1975, as revised in 2000.
2.2. Data collection
Data collection was performed by two investigators (H.A.H. and F.M.). Data were extracted from both paper and electronic patient files. Data were collected from the first symptomatic phase until the last follow‐up visit. Data collection was focused on patient and family characteristics, outcome of the first symptomatic phase, AMD, hospital admissions, cognitive and psychomotor development, and long‐term complications. All data were entered in the OpenClinica open source software, version 3.12.2.
EO was defined as an episode of severely reduced clinical condition starting in the first 28 days of life, and LO as symptoms starting after 28 days of life.1 Patients identified through family testing based on an affected older (index) sibling were regarded a separate category.
2.3. Outcome parameters
To evaluate clinical outcomes, five grouped outcome parameters were defined (Supplementary Notes S2). “Adverse outcome (AO) of the first symptomatic phase” was categorised based on the presence of movement disorders that developed during or right after the first symptomatic phase, objectified during follow‐up examinations by paediatric neurologists and physiotherapists shortly after the first symptomatic phase, and/or the presence of basal ganglia hyperintensities or white matter abnormalities detected at the first time of brain imaging after the first symptomatic phase (Supplementary Notes S2). “AMD frequency” was defined based on the frequency of (impending) AMD requiring hospital admission per patient year (PY) in the first 4 years of life, other than the first symptomatic phase (Supplementary Notes S2). “Cognitive function” was distinguished in three categories based on neuropsychological test results, or in the absence of neuropsychological test results on educational level or professional employment17 (Supplementary Notes S2). Long‐term complications were divided into mitochondrial, treatment‐related, and miscellaneous complications. Patients were categorised into six categories based on the number of mitochondrial complications present at a certain age. These mitochondrial complications included 21 complications likely to be caused by mitochondrial failure2 (Supplementary Notes S2). Complications with potential treatment‐related aetiology included decreased bone mineral density (BMD), growth retardation, and obesity2 (Supplementary Notes S2). Growth retardation was defined present when height to weight was less than −2 SD at any moment during follow‐up. Other complications, with no evidence for a sole mitochondrial or treatment‐related aetiology, were regarded miscellaneous complications. These complications included pes planovalgus, port‐a‐cath infections, teeth enamel defects, urolithiasis, and gout.2
2.4. Data‐analysis
Each outcome parameter was evaluated in the complete cohort, separate for both PA and MMA patients, and separate for both EO and LO patients. In addition, for all AMDs age at decompensation, admission duration, need for intensive care unit admission, and AMD triggers were evaluated. For each complication assessed, prevalence was calculated.
As a proxy for the potential health gain of NBS, all five clinical outcome parameters were compared between patients identified by family testing and their index siblings. The expected health gain was calculated by extrapolating findings of the sibling comparison on AO of the first symptomatic phase to the entire cohort, if NBS would have been implemented. Calculations were performed for six possible days that definitive NBS results could become available: day 0, 3, 7, 11, 15, and 21 after birth.
Assessment of risk factors for each outcome parameter was performed by comparing disease types, for both PA and MMA presentation types and, for MMA vitamin B12 responsiveness.
All statistical analyses were performed in R programming language. Results are expressed as either means or medians with standard deviations and ranges for quantitative data and as number and percentage for qualitative data. Student's t tests were performed for quantitative data and Fisher's exact tests for qualitative data. P‐values were adjusted according to the Bonferroni method when multiple testing was performed, adjusted P‐values <.05 were considered statistically significant. P‐values solely reported in the text for the assessment of risk factors of clinical outcome parameters were calculated using Fisher's exact tests and were not adjusted, as no multiple testing was performed.
3. RESULTS
3.1. Patient characteristics
Of all 83 PA and MMA patients in the Netherlands diagnosed between January 1979 and July 2019, 76 patients were included. Five patients refused to participate and clinical records of two deceased MMA patients could not be retrieved. For nearly all patients, data were available from the first symptomatic phase to the last moment of follow‐up. The cohort included 31 PA patients from 24 families and 45 MMA patients from 40 families. Between 1 January 1998 and 31 December 2017, 46/76 patients (PA: n = 19, MMA: n = 27) were diagnosed, with a median of 2 patients per birth year (range: 1‐5). In this period, 3 729 128 live births were registered, providing an estimated incidence in the Netherlands of 1:81 000 for PA and MMA combined, 1:196 000 for PA and 1:138 000 for MMA. Presentation type was EO in 29, LO in 34, family testing in 12, and unknown in 1 patient (Table 1). Clinical data of five PA sibling pairs and one trio, and three MMA sibling pairs (n = 9) were available for sibling comparison (Table S1). Two MMA patients identified through family testing could not be compared with their index siblings, because medical records of the index sibling were not available.
Table 1.
Baseline characteristics of the Dutch cohort of PA and MMA patients
| Propionic acidemia (n = 31) | Methylmalonic acidemia (n = 45) | |||
|---|---|---|---|---|
| Presentation type | Early onset | n = 15; 48% | Early onset | n = 14; 31% |
| Late onset | n = 8; 26% | Late onset | n = 26; 56% | |
| Family testing | n = 7; 23% | Family testing | n = 5; 11% | |
| Unknown | n = 1; 3% | |||
| Presentation age | Early onset | 6.05 ± 6.12 days | Early onset | 2.07 ± 1.77 days |
| Late onset | 14.1 ± 22.9 years | Late onset | 1.41 ± 2.74 years | |
| Family testing | 0.07 ± 0.19 years | Family testing | 0.32 ± 0.72 years | |
| Genotype | PCCA | n = 8; 26% | MUT | n = 26; 58% |
| PCCB | n = 7; 23% | MMAA | n = 8; 18% | |
| MMAB | n = 7; 16% | |||
| No mutation analysis | n = 16; 52% | No mutation analysis | n = 4; 9% | |
| Enzyme activity | 0.0‐0.4 nmol/h/mg | n = 10; 32% | ||
| 0.5‐2.0 nmol/h/mg | n = 5; 16% | |||
| Not measured | n = 16; 52% | |||
| Vitamin B12 responsiveness | Responsive | n = 21; 47% | ||
| Not responsive | n = 24; 53% | |||
| Age at last follow‐up | 19.2 ± 15.1 years | 17.3 ± 12.1 years | ||
| Mortality | n = 7; 23% (4 EO, 1 LO, 2 family) | n = 2; 4% (1 LO, 1 family) | ||
Notes: Early onset: presentation ≤28 days of life. Late onset: presentation >28 days of life. Qualitative data are expressed as number and percentage, quantitative data as mean ± SD.
Abbreviations: EO, early onset; family, family testing; LO, late onset.
Mutation analysis was available in 52/76 patients and revealed 24 previously unreported mutations, including 7 mutations in PCCA, 5 in PCCB, 8 in MUT, 2 in MMAA, and 2 in MMAB (Table S2). Pregnancy and delivery were uncomplicated in the majority of the patients' mothers. Birth weight of MMA patients was significantly lower compared to PA patients, in line with previous reports1 (Table S3). In most patients, the first symptomatic phase was characterised by lethargy and anorexia. Significantly more LO patients presented with vomiting. Plasma ammonia at presentation was significantly higher in EO patients, resulting in significantly more EO patients requiring dialysis (Table S4).
3.2. Adverse outcome of the first symptomatic phase
An AO of the first symptomatic phase was recorded in 36/76 (47%) of all patients and in 30/64 (47%) of the clinically diagnosed patients (Figure 1). Movement disorders were recorded in 24/36 patients. Basal ganglia hyperintensities were observed in 18, white matter abnormalities in 14, and both in 6 patients. In 19/26 patients in whom white matter abnormalities and/or basal ganglia hyperintensities were observed, brain imaging was performed within 3 days to 6 months after the first symptomatic phase and in 7/26 patients later in life (range: 9 months to 23 years). In three of these patients also a movement disorder was recorded, and in all seven patients the observed abnormalities were considered to be long‐existent based on unchanged clinical symptoms and imaging characteristics.
Figure 1.
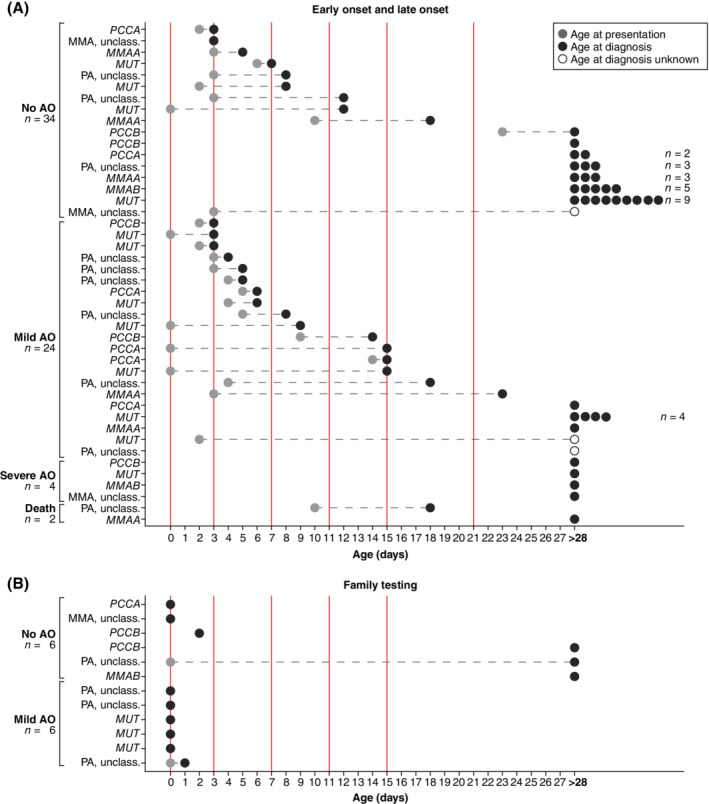
Age at onset and age at diagnosis for propionic acidemia (PA) and methylmalonic acidemia (MMA) patients. A, Patients with early or late onset presentation. B, Patients identified through family testing. The y‐axis depicts the patients, ordered on type of adverse outcome, and age at diagnosis. The x‐axis depicts the patient age in days, with a maximum of 28 days (early onset). Light grey circles depict age at presentation, dark grey circles depict age at diagnosis, open circles depict in which patients the exact age at diagnosis is unknown. For all ages at presentation and ages at diagnosis ≥28 days of age (late onset), the circles have been placed at 28 days. The vertical red solid lines depict the potential moments NBS results could become available, in line with Table 3. On the left the severity of adverse outcomes (AO) due to the first symptomatic phase are listed: no adverse outcome, mild, severe, or death due to the first symptomatic phase
In EO patients, an AO due to the first symptomatic phase was either absent or, more often, mild, whereas in LO patients most often no AO due to the first symptomatic phase was recorded. If an AO was recorded in LO patients, it was either mild or severe, which is represented by a significant difference in mild AO due to the first symptomatic phase in EO vs LO patients (Table 2 and Figure S1). AO due to the first symptomatic phase tended to be less frequent in patients identified through family testing than in their siblings (better outcome of the first symptomatic phase in 4/9 sibling pairs, similar in 5/9) (Table S1). Symptoms or signs present during the first symptomatic phase, nor presentation type in PA or MMA, nor vitamin B12 responsiveness in MMA was associated with an increased risk for an AO (Table S5).
Table 2.
Grouped outcome parameters according to presentation type
| Early onset (n = 29) | Late onset (n = 34) | P value | Bonferroni correction | |
|---|---|---|---|---|
| Adverse outcome of first symptomatic phase | ||||
| No adverse outcome | n = 11; 38% | n = 23; 64% | .024 | NS |
| Mild adverse outcome | n = 17; 59% | n = 6; 21% | .001 | .006 |
| Severe adverse outcome | n = 0; 0% | n = 4; 12% | .118 | NS |
| Death due to first presentation | n = 1; 3% | n = 1; 3% | 1.000 | NS |
| AMD frequency | ||||
| None: no AMD in first 4 years | n = 3; 10% | n = 13; 38% | .019 | NS |
| Mild: >0.0 ≤ 0.5 AMD/PY | n = 1; 3% | n = 3; 9% | .618 | NS |
| Moderate: >0.5 ≤ 1.0 AMD/PY | n = 2; 7% | n = 2; 6% | 1.000 | NS |
| Severe: >1.0 ≤ 2.0 AMD/PY | n = 6; 21% | n = 3; 9% | .280 | NS |
| Very severe: >2.0 AMD/PY | n = 11; 38% | n = 5; 15% | .045 | NS |
| Death: during or due to AMD | n = 3; 10% | n = 1; 3% | .326 | NS |
| NA | n = 3; 10% | n = 7; 21% | .319 | NS |
| Cognitive function | ||||
| Group 1: IQ >90 or regular education | n = 4; 14% | n = 15; 44% | .013 | NS |
| Group 2: IQ 60‐90 or special education | n = 10; 34% | n = 8; 24% | .407 | NS |
| Group 3: IQ <60 or no education | n = 10; 34% | n = 5; 15% | .081 | NS |
| NA | n = 5; 17% | n = 6; 18% | 1.000 | NS |
| Mitochondrial complications | ||||
| None | n = 1; 3% | n = 5; 15% | .205 | NS |
| Mild | n = 6; 21% | n = 19; 56% | .005 | .034 |
| Moderate | n = 5; 17% | n = 5; 15% | 1.000 | NS |
| Severe | n = 9; 31% | n = 2; 6% | .017 | NS |
| Very severe | n = 8; 28% | n = 3; 9% | .093 | NS |
| Death | n = 0; 0% | n = 0; 0% | 1.000 | NS |
| NA | n = 0; 0% | n = 0; 0% | 1.000 | NS |
| Treatment‐related complications | ||||
| None | n = 9; 31% | n = 17; 50% | .199 | NS |
| Mild | n = 10; 34% | n = 13; 38% | .798 | NS |
| Severe | n = 6; 21% | n = 3; 9% | .280 | NS |
| Very severe | n = 4; 14% | n = 1; 3% | .171 | NS |
| NA | n = 0; 0% | n = 0; 0% | 1.000 | NS |
Notes: Adverse outcome of first symptomatic phase: Adverse outcome of the first symptomatic phase. Early onset: presentation ≤28 days of life; Late onset: presentation >28 days of life. Statistical significance was determined by Fisher's exact tests. All P‐values were adjusted according to the Bonferroni method. Adjusted P‐values <.05 were considered statistically significant and are depicted in bold.
Abbreviations: AMD, acute metabolic decompensation; NA, not assessed; NS, not significant; PY, patient year.
The expected health gain of NBS for PA and MMA in the Netherlands was estimated based on the moment of presentation of all EO and LO patients in this cohort and based on analysis of sibling pairs (Figure 1, Table S1, and Table 3). Theoretically, the earlier NBS results are available, the more patients could be protected from an AO due to the first symptomatic phase (Figure 1 and Table 3). In this cohort, NBS results available at day 0 of life could have prevented an AO in 14 patients, reducing the percentage of an AO during the first symptomatic phase from 46% to 23%. NBS results available between day 11 and 21 could have prevented an AO in 6 or 5 patients, reducing the percentage from 46% to 36%‐38% (Table 3).
Table 3.
Expected effect of NBS implementation on occurrence of adverse outcomes due to the first symptomatic phase
| Patients | Group | ID | Formula | Day 0, % (n) | Day 3, % (n) | Day 7, % (n) | Day 11, % (n) | Day 15, % (n) | Day 21, % (n) |
|---|---|---|---|---|---|---|---|---|---|
| Potentially diagnosed through NBS (after NBS implementation) | Total | A,a | 100% (61) | 84% (51) | 61% (37) | 56% (34) | 54% (33) | 54% (33) | |
| Without AO | B,b | A‐C, a‐c | 54% (33) | 59% (30) | 65% (24) | 68% (23) | 70% (23) | 70% (23) | |
| With AO | C,c | A‐B, a‐b | 46% (28) | 41% (21) | 35% (13) | 32% (11) | 30% (10) | 30% (10) | |
| Potentially diagnosed after first symptomatic phase (after NBS implementation) | Total | D,d | 0% (0) | 16% (10) | 39% (24) | 44% (27) | 46% (28) | 46% (28) | |
| Without AO | E,e | D‐F, d‐f | 0% (0) | 30% (3) | 38% (9) | 37% (10) | 36% (10) | 36% (10) | |
| With AO | F,f | D‐E, d‐e | 0% (0) | 70% (7) | 62% (15) | 63% (17) | 64% (18) | 64% (18) | |
| Index siblings of patients identified through family testing | Total | G,g | 100% (9) | 100% (9) | 100% (9) | 100% (9) | 100% (9) | 100% (9) | |
| Without AO | H.h | 11% (1) | 11% (1) | 11% (1) | 11% (1) | 11% (1) | 11% (1) | ||
| With AO | I, i | 89% (8) | 89% (8) | 89% (8) | 89% (8) | 89% (8) | 89% (8) | ||
| Siblings identified through family testing | Without AO | J,j | 56% (5) | 56% (5) | 56% (5) | 56% (5) | 56% (5) | 56% (5) | |
| With AO | K,k | 44% (4) | 44% (4) | 44% (4) | 44% (4) | 44% (4) | 44% (4) | ||
| Estimated effectivity | L | k / i | 50% | 50% | 50% | 50% | 50% | 50% | |
| Protected from AO (after NBS implementation) | m | L * c | n = 14 | n = 11 | n = 7 | n = 6 | n = 5 | n = 5 | |
| Percentage AO first symptomatic phase (after NBS implementation) | N,n | n / (a + d) c + f − m | 23% n = 14 | 28% n = 17 | 34% n = 21 | 36% n = 22 | 38% n = 23 | 38% n = 23 | |
Notes: Total number of patients included is 61; excluding patients identified through family testing n = 12, excluding patients for whom the exact day of diagnosis is unknown, n = 3 (Figure 1). The column listing the identifiers indicates the identifier of the percentage in capitals, or the number of patients in lowercases, for the interpretation of the formula. The formula indicates how the percentages and numbers of patients were calculated. The five time points indicate the days that NBS results could become available when NBS would be implemented. All percentages and patient numbers were rounded.
Abbreviations: AO, adverse outcome; ID, identifier; NBS, newborn screening.
3.3. AMD frequency
In total, 962 AMD requiring hospital admission were reported. Hospitalisation duration was significantly shorter in MMA than in PA patients. AMD triggers were similar, with the exception of significantly more frequent feeding problems in MMA patients and constipation in PA patients (Table S6).
Among the sibling pairs, AMD frequency was similar in 4/9 pairs. The index patients of 4/9 sibling pairs died following an AMD, whereas all but one of the siblings identified through family testing are still alive, although currently only P6.3 survived his sibling in age (Table S1). AMD frequency tended to be shifted towards a more severe frequency in EO patients compared to LO patients (Table 2 and Figure S1). Between PA and MMA patients, no difference in AMD frequency was recorded. (Table S6). Among PA patients, EO patients experienced significantly more AMD/PY (P = .002) and among MMA patients, vitamin B12 unresponsive patients experienced significantly more AMD/PY (Table S6).
3.4. Cognitive function
A total of 139 neuropsychological test results were recorded in 46/76 patients, with a median of three tests per patient (range: 1‐10). In 17 patients, cognitive function was based on the patients' educational level and in 13 patients, cognitive function was not assessed (yet). An IQ >90 was noted in 24/63 patients, an IQ between 60 and 90 in 20/63 patients, and an IQ <60 in 19/63 patients (Table S7).
Cognitive function was comparable among the sibling pairs (similar in 3, better in 2, worse in 1, not assessed in 3) (Table S1), as well as between EO and LO patients (Table 2 and Figure S1). An IQ <60 was significantly more prevalent in PA patients (Table S7). Among PA patients, EO was significantly associated with cognitive function (IQ >90: EO 0%, LO 63%, P = .002). Among MMA patients, presentation type nor vitamin B12 responsiveness were associated with cognitive function.
3.5. Complications with potential mitochondrial aetiology
Mitochondrial complications were present in 70/76 patients, and were mild in 30, moderate in 13, severe in 12, and very severe in 14 patients. One PA patient with very severe mitochondrial complications died due to cardiomyopathy. Prevalence of mitochondrial complications was compared with the literature and was fairly comparable for most complications2 (Table S7). For quite some complications, especially for MMA, prevalence was here assessed for the first time in a cohort study (Table S7). Renal failure was significantly more prevalent among MMA patients than among PA patients (Table S7).
For mitochondrial complications with acute onset, age at onset was assessed (Table S7 and Figure 2). The earliest manifestation of renal failure was already reported at the age of 1.2 years in an MMAB patient and in 5/20 patients age of onset was <6 years (2 MUT, 2 MMAB, and 1 unclassified). The first manifestation of prolonged QTc interval was shortly after birth in PA, and at 3.6 years in MMA. Cardiomyopathy in PA was noted at 7.5 years, optic atrophy at 11.8 years in PA, and at 12.6 years in MMA (Table S7). Age at onset of hepatomegaly, epilepsy, and sensorineural hearing loss ranged from birth to 10 years, whereas most other complications typically arose from late infancy to adolescence (Table S7 and Figure 2).
Figure 2.
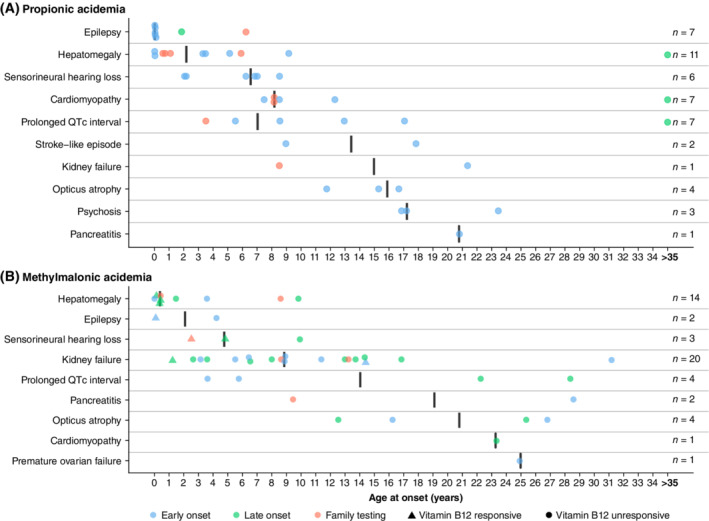
Age at onset of mitochondrial complications with acute onset. A, Depicts propionic acidemia; B, depicts methylmalonic acidemia. The y‐axis depicts the mitochondrial complications with acute onset, ordered on median age at onset. The x‐axis depicts patient age in years, with a maximum of 35 years of age. For all ages >35 years, the circles have been placed at 35 years. The black vertical lines depict the median age at onset. The filled circles and triangles depict the individual patient age at onset of a complication. Circles depict no vitamin B12 responsiveness, triangles depict vitamin B12 responsiveness of methylmalonic acidemia patients. Blue circles and triangles depict early onset, green circles and triangles depict late onset, orange circles and triangles depict diagnosis through family testing. Early onset: presentation ≤28 days of life; late onset: presentation >28 days of life
Mitochondrial complications were comparable among sibling pairs (similar in 1, better in 4, and worse in 4) (Table S1). LO patients were significantly more frequently recorded to have only mild mitochondrial complications than EO patients (Table 2 and Figure S1). PA patients had more very severe mitochondrial complications than MMA patients (Table S7), and all of these PA patients were EO patients (EO 53%, LO 0%, P = .019). Among MMA patients, mitochondrial complications were comparable between EO and LO patients but the prevalence of moderate to very severe mitochondrial complications was significantly higher in vitamin B12 unresponsive patients (unresponsive 63%, responsive 24%, P = .016).
EO for PA and vitamin B12 unresponsiveness for MMA were also found to be significantly associated with a higher risk for mitochondrial complications with acute onset (Figure 3). In addition, EO in PA tended to be associated with the occurrence of hepatomegaly, prolonged QTc interval, psychoses, and sensorineural hearing loss (Figure S2). Vitamin B12 unresponsiveness tended to be associated with the occurrence of optic atrophy, pancreatitis and prolonged QTc interval in MMA, and was significantly associated with the occurrence of renal failure (Figure S3). One of the first occurring mitochondrial complications with acute onset was hepatomegaly (Figure 2). Intriguingly, hepatomegaly was significantly associated with the onset of other mitochondrial complications with acute onset later in life, for PA and MMA as a group and for both PA and MMA separately (Figure 3).
Figure 3.
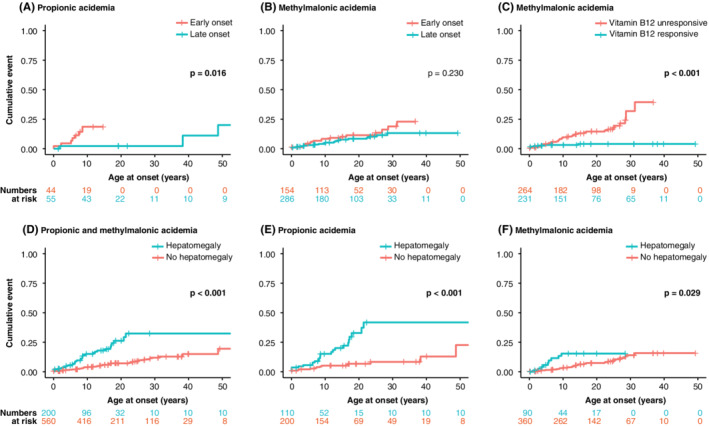
Factors associated with increased risks for mitochondrial complications with acute onset. Kaplan‐Meier plots wherein the y‐axis depicts the cumulative percentage and the x‐axis depicts patient age in years. Numbers at risk for the subgroups, indicating for how many complications the patients are at risk, are depicted below the panels, in corresponding colours. Panels A and B demonstrate late onset in blue vs early onset in orange for propionic acidemia (PA) in (A) and for methylmalonic acidemia (MMA) in (B). Early onset: presentation ≤28 days of life; late onset: presentation >28 days of life. Panel C demonstrates vitamin B12 responsiveness in blue vs vitamin B12 unresponsiveness in orange for MMA. Panels D‐F depicts the association of the presence of hepatomegaly to other mitochondrial complications with acute onset in blue, vs no hepatomegaly in orange, in (D) for both PA and MMA, in (E) for PA, and in (F) for MMA
3.6. Complications with potential treatment‐related aetiology
Reduced BMD was noted in 27, growth retardation in 21, and obesity in 16 patients. Treatment‐related complications were absent in 33, mild in 28, severe in 9, and very severe in 6 patients (Table S8).
Treatment‐related complications were comparable among sibling pairs (similar in 2, better in 5, and worse in 2) (Table S1), as well as among EO and LO patients (Table 2 and Figure S1), nor were there any differences between PA and MMA, or among PA patients between EO and LO patients (Table S8). Among MMA patients, the rate of mild, severe, and very severe treatment‐related complications was significantly higher in vitamin B12 unresponsive patients (unresponsive 71%, unresponsive 33%, P = .017).
3.7. Complications with miscellaneous aetiology
A prevalence of 29% pes planovalgus, a feature so far only described in MMA,18 was noted in PA. In one MMA and one PA patient, urolithiasis was recorded. In both patients, calculi developed due to hypercalciuria, although dehydration, chronic acidotic state, and a low‐protein diet might have contributed. Two MMA patients (MUT and MMAA) were found to have juvenile gout (Table S8). The MMAA patient had chronic renal failure when exhibiting gout, whereas the MUT patient did not.
4. DISCUSSION
4.1. Expected health gain of NBS for PA and MMA in the Netherlands (WHO#4)
Here, we present an extensive, nationwide cohort study, describing the clinical course of 76/83 Dutch PA and MMA patients. We demonstrated that the expected health gain of introducing NBS for PA and MMA in the Netherlands in overall outcome may be limited. Patients can already present shortly after birth and even if patients are diagnosed pre‐symptomatically, they can still develop an adverse outcome. We calculated an expected reduction from 46% to 36%‐38% AO due to the first symptomatic phase and we consider it unlikely that NBS could result in reduced AMD frequency, improved cognitive function, or a reduced frequency of mitochondrial and therapy‐related complications.
These findings are in line with previous reports. Although time to diagnosis is reduced in patients identified by NBS,9 NBS is unlikely to identify more than 59%‐72% of PA and MMA patients before the first symptomatic phase.8, 9 One study described no decreased AMD frequency, no better neurological outcome and no reduced frequency of long‐term complications in PA patients,8 although another study found that PA patients identified by NBS had lower cardiac manifestations,9 but follow‐up time in these patients was limited. In MMA patients, besides improved attainment of motor milestones and reduced manifestations of movement disorders in vitamin B12 unresponsive patients, NBS implementation did not result in other improved outcome parameters.9
Thus, as evidence supporting improved outcomes if treatment can be initiated in the asymptomatic phase is limited, despite reasonable consensus on therapeutic strategies for PA and MMA,19, 20 it is questionable whether the criterion regarding an accepted (effective) treatment for patients with recognised disease is met (W&J#2). As a consequence, it remains debatable whether the criteria requesting effectiveness (WHO#4) and benefits outweigh harm (WHO#10) are met (Supplementary Notes S1). We advocate that the (now limited) health gain of NBS can only be improved if treatment strategies are identified that can effectively prevent AMD, cognitive impairment, and mitochondrial complications, such as innovative therapies aiming to increase enzyme activity.20
4.2. Evaluation of NBS implementation for PA and MMA (WHO#9)
Insight in NBS efficacy is still limited, due to factors complicating proper evaluation, such as timing of evaluation. Follow‐up time of the four studies describing the potential additional value of NBS for PA and MMA was relatively short, resulting in uncertainty when drawing conclusions.6, 7, 8, 9 We propose that these countries reassess their NBS cohorts when a longer follow‐up time is available. For the Netherlands, assuming a similar incidence in the NBS cohort as in the here presented cohort, but taking into account the slightly decreasing birth rate, it will take at least till 2050 before an NBS cohort similar in size and follow‐up time as the pre‐NBS cohort is available for evaluation. More timely evaluation can only be achieved by composing larger NBS cohorts through data sharing initiatives, within Europe or worldwide.
Even if timing of evaluation is adequate, proper evaluation of the presumed effect of NBS remains challenging given the many factors that can influence the clinical course of PA and MMA. This includes genotypic factors as mutation type, residual enzyme activity, and vitamin B12 responsiveness (MMA). Also, a high variability in clinical management existed between the different metabolic centres and metabolic physicians (data not shown). Next, compliance of parents and patients to clinical management and disease insight could influence clinical outcomes. Moreover, pre‐NBS cohorts are diagnosed and treated in the era before NBS cohorts, with NBS cohorts taking advantage of general advances in clinical care, and possibly increased insights and improved therapies for PA and MMA patients.
To aid evaluation of NBS efficacy, we suggest the five clinical outcome parameters here presented and we advocate the introduction of continuation and stop criteria. We propose that NBS for PA and MMA should be continued when it results in a statistically significant and clinically relevant reduction of AO due to the first symptomatic phase, and that it should be stopped otherwise. While based on our results, NBS may be unlikely to result in reduced AMD frequency, improved cognitive function, and reduced mitochondrial complications, these outcome parameters should be assessed to gain insight in the effects of improved treatment strategies. If NBS would unexpectedly result in improvement of these parameters, this would be a supportive argument to continue NBS. NBS evaluation should also include assessment of treatment‐related complications, to gain knowledge regarding risks and harm of screening.
In NBS cohorts, patients in whom the disease is mild and who would not become symptomatic during infancy, could be detected as has been reported in Japan, where a 10 times higher incidence of PA was found during preparatory studies for NBS implementation.21 These mildly affected patients will probably be treated according to the current treatment guidelines,19 inducing a risk for developing treatment‐related complications. One could advocate that these patients should not be treated in order to prevent treatment‐related complications. However, in our cohort, two PA patients presented at 48 and 56 years of age with cardiomyopathy as first presenting symptom,22 indicating that even in very mild, untreated patients, mitochondrial complications can still occur. It is presently unknown whether earlier initiation of adequate treatment could have prevented this complication. Even if this would alter the clinical course, it is questionable whether these patients should be exposed to life‐long protein restriction, to prevent complications occurring well into adulthood. We consider overtreatment of mildly affected patients, which might or might not develop late complications,21, 22 a serious risk of NBS.
4.3. Lessons learned from the pre‐NBS cohort, to guide follow‐up of NBS cohorts
We demonstrated that mitochondrial complications with acute onset can already develop before monitoring should be initiated according to recent guidelines19 and we thus propose to adapt these guidelines. Specifically, we suggest to assess renal function in patients with MUT and MMAB type MMA from 1 year of age, to regularly check QTc intervals in PA and MMA patients starting at birth, and to maintain yearly cardiac ultrasounds and ophthalmologic assessments from 6 years onwards. Importantly, as we demonstrate that hepatomegaly is associated with the occurrence of other acute onset mitochondrial complications later in life, we advise to perform physical examination to check for hepatomegaly during every outpatient visit starting at birth, to be able to study the relevance of this finding. Lastly, we advocate awareness of all complications assessed in this cohort, including rarely described complications as exercise intolerance, acute psychosis, premature ovarian failure, pes planovalgus, gout, and urolithiasis.
For surveillance and counselling of patients in NBS cohorts, we stress the importance to take into account disease severity. For MMA patients, we demonstrated that vitamin B12 unresponsiveness increases the risk of a higher AMD frequency, more mitochondrial complications, and more treatment‐related complications. This is in line with previous reports that demonstrate that vitamin B12 unresponsiveness is associated with a decreased survival rate in EO patients, a higher frequency of AMD, an increased rate of developmental delay in EO patients, and an increased rate of disability.23 In addition, a tendency towards an increased rate of chronic renal failure has been reported.24 For PA patients, EO is associated with a higher risk for an AO of the first symptomatic phase, a higher AMD frequency, more severe cognitive impairment, and more mitochondrial complications. While this still holds true for patients that are symptomatic before NBS results are available, presentation type will be unknown for PA patients diagnosed via NBS, complicating surveillance, and counselling of these patients. This augments the need for systematic determination of propionyl‐CoA carboxylase activity in PA patients and illustrates the imminent need for a metabolic marker that can predict the risks for AMD frequency, cognitive function, and mitochondrial complications.25, 26
We could not identify specific genotype‐phenotype correlations due to the variation of the identified mutations. However, we did note that although patients with MMAB type MMA are generally reported to be vitamin B12 unresponsive,19 6/7 of our MMAB patients were responsive. In five of these, we identified the MMAB c.556C>T missense mutation in either homozygous or heterozygous fashion (Table S2). We speculate that this mutation might be related to the observed vitamin B12 responsiveness of MMAB patients, but we cannot prove this hypothesis.
4.4. Limitations and strengths
We note a few limitations. First, due to the retrospective nature of this study and variable follow‐up schedules, missing data could have affected the reliability of the analyses. We expect missing data to be random and consider it unlikely to have resulted in confounding. Nevertheless, to facilitate reliable analyses and conclusions on NBS effectivity, we advocate precise and structured prospective follow‐up of NBS cohorts. Minimum requirements for evaluation are listed in Table S9. Centres participating in data‐sharing initiatives should agree on the items to be recorded and on the timing of diagnostics studies (Table S9). Second, sample size of the sibling comparison is limited, hampering solid conclusions on the expected health gain of NBS for PA and MMA, but consolidating the urge for thorough assessment of NBS cohorts. The potential health gain might have been overestimated (a) due to the unknown number of EO patients that died undiagnosed, (b) since families with an index child are prepared for the diagnosis, and can minimise the risk for AMD more efficiently than families from individuals identified by NBS, and (c) since calculations were performed with a potential overestimation of AO due to the first symptomatic phase, as we assumed that all observed brain imaging abnormalities were caused by the first symptomatic phase instead of being caused by AMD occurring later in life (and thus under treatment). Conversely, the potential health gain of NBS might have been underestimated as the clinical course between siblings may vary and it is not known whether the sibling diagnosed through family testing would have stayed in the same condition if diagnosed later in life.
Important strengths of our study are that we provide a comprehensive overview of a nationwide cohort spanning over four decades, by systematically assessing nearly all PA and MMA patients in the Netherlands in great detail. We designed five clinical outcome parameters to guide evaluation of implementation NBS and we provide an extensive description of a pre‐NBS cohort to compare NBS cohorts with. Moreover, due to the systematic assessment of AMD, complications, and risk factor analysis, we were able to establish recommendations regarding surveillance and counselling of NBS cohorts. Lastly, continuation and stopping criteria for NBS were provided, as well as minimum requirements for follow‐up of NBS cohorts to facilitate data sharing initiatives.
5. CONCLUSIONS
The objective of NBS for PA and MMA in the Netherlands is to protect LO patients from irreversible neurological damage due to the first symptomatic phase. Implementation in the Netherlands may result in a reduction of AO due to the first symptomatic phase from 46% to 36%‐38%, if NBS results become available between day 11 and 21. We consider reduced AMD frequency, improved cognitive function, and reduced mitochondrial and treatment‐related complications unlikely given the currently available therapies.
CONFLICT OF INTEREST
All authors state that they have no competing interests to declare. None of the authors accepted any reimbursements, fees, or funds from any organisation that may in any way gain or lose financially from the results of this study. The authors have not been employed by such an organisation. The authors do not have any other competing interest.
AUTHOR CONTRIBUTIONS
H.A.H., M.W., and P.M.v.H. conceived the research. H.A.H., F.M., M.W., and P.M.v.H. designed the study protocol and gained approval of the responsible ethics committees. J.J.M.J., N.M.V.‐D., M.W., and P.M.v.H supervised the research. M.W. and J.G.L. gave advice as experts of the Dutch expertise centre on organic acidemias. M.L., M.C.J., A.M.B., F.J.v.S., M.F.M., A.T.v.d.P., M.A.W., M.E.R.G., M.C.G.J., M.C.d.V., and M.W. coordinated patient inclusion and data collection in the six Dutch metabolic centres. H.A.H. and F.M. obtained written informed consent of all patients or their legal guardians. H.A.H. and F.M. designed the electronic clinical record forms in OpenClinica, collected the data, and performed the data analyses. H.A.H. wrote the first draft of the manuscript. All authors critically reviewed the manuscript and approved the final version.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGEMENTS
We would like to thank the patients and their families for participation in this study. This work was supported by the personal Alexandre Suerman Stipend of the University Medical Centre Utrecht (H.A.H.) and by Stofwisselkracht (F.M. and M.W.).
Guarantor for the article: P. M. van Hasselt declares that he will accept full responsibility for the work and the conduct of the study. He had access to the data and controlled the decision to publish.
Haijes HA, Molema F, Langeveld M, et al. Retrospective evaluation of the Dutch pre‐newborn screening cohort for propionic acidemia and isolated methylmalonic acidemia: What to aim, expect, and evaluate from newborn screening? J Inherit Metab Dis. 2020;43:424–437. 10.1002/jimd.12193
Hanneke A. Haijes and Femke Molema contributed equally to this study; Monique Williams and Peter M. van Hasselt contributed equally to this study.
Communicating Editor: Georg Hoffmann
Funding information Alexandre Suerman Stipend, Grant/Award Number: Not applicable; Stofwisselkracht
Contributor Information
Hanneke A. Haijes, Email: h.a.siepel-3@umcutrecht.nl.
Peter M. van Hasselt, Email: p.vanhasselt@umcutrecht.nl.
REFERENCES
- 1. Kölker S, Garcia Cazorla A, Valayannopoulos V, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015;38:1041‐1057. [DOI] [PubMed] [Google Scholar]
- 2. Haijes HA, Jans JJM, Tas SY, Verhoeven‐Duif NM, van Hasselt PM (2019) Pathophysiology of propionic and methylmalonic acidemias. Part 1: complications. J Inherit Metab Dis. 42 730‐744. [DOI] [PubMed] [Google Scholar]
- 3. Sass JO, Hofmann M, Skladal D, Mayatepek E, Schwahn B, Sperl W. Propionic acidemia revisited: a workshop report. Clin Pediatr. 2004;43(9):837‐843. [DOI] [PubMed] [Google Scholar]
- 4. Leonard JV, Vijayaraghavan S, Walter JH. The impact of screening for propionic and methylmalonic acidemia. Eur J Pediatr. 2003;612:S21‐S24. [DOI] [PubMed] [Google Scholar]
- 5. Hörster F, Kölker S, Loeber JG, Cornel MC, Hoffmann GF, Burgard P. Newborn screening programmes in Europe, arguments and efforts regarding harmonisation: focus on organic acidurias. JIMD Rep. 2016;32:105‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dionisi‐Vici C, Deodato F, Röschinger W, Rhead W, Wilcken B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: long‐term outcome and effects of expanded newborn screening using tandem mass spectrometry. J Inherit Metab Dis. 2006;29(2–3):383‐389. [DOI] [PubMed] [Google Scholar]
- 7. Couce ML, Castiñeiras DE, Bóveda MD, et al. Evaluation and long‐term follow‐up of infants with inborn errors of metabolism identified in an expanded newborn screening programme. Mol Genet Metab. 2011;104(4):470‐475. [DOI] [PubMed] [Google Scholar]
- 8. Grünert SC, Müllerleile S, de Silva L, et al. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis. 2012;35:41‐49. [DOI] [PubMed] [Google Scholar]
- 9. Heringer J, Valayannopoulos V, Lund AM, et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis. 2016;39(3):341‐353. [DOI] [PubMed] [Google Scholar]
- 10. Wilson JMG, Jungner G. Principles and practice of screening for disease. Bol Oficina Sanit Panam. 1968;65(4):281‐393. [PubMed] [Google Scholar]
- 11. Andermann A, Blancquaert I, Beauchamps S, Déry V. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ. 2008;86(4):317‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cornel MC, Rigter T, Weinreich SS, et al. Newborn screening in Europe: expert opinion document. Brussels: European Union; 2011.
- 13. Burgard P, Rupp K, Lindner M, et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 2. From screening laboratory results to treatment, follow‐up and quality assurance. J Inherit Metab Dis. 2012;35(4):613‐625. [DOI] [PubMed] [Google Scholar]
- 14. Cornel MC, Rigter T, Weinreich SS. A framework to start the debate on neonatal screening policies in the EU: an expert opinion document. Eur J Hum Gen. 2014;22:12‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. American College of Medical Genetics Newborn Screening Expert Group . Newborn screening: toward a uniform screening panel and system, executive summary. Pediatrics. 2006;117:S296‐S307. [DOI] [PubMed] [Google Scholar]
- 16. Health Council of the Netherlands . Neonatal screening: new recommendations. The Hague: Health Council of the Netherlands, 2015; publication no 2015/08; 2015.
- 17. Hörster F, Baumgartner MR, Viardot C, et al. Long‐term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut‐, cblA, cblB). Pediatr Res. 2007;62(2):225‐230. [DOI] [PubMed] [Google Scholar]
- 18. Ktena YP, Paul SM, Hauser NS, et al. Delineating the spectrum of impairments, disabilities and rehabilitation needs in methylmalonic acidemia (MMA). Am J Med Genet. 2015;167A(9):2075‐2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumgartner MR, Hörster F, Dionisi‐Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haijes HA, van Hasselt PM, Jans JJM, Verhoeven‐Duif NM (2019) Pathophysiology of propionic and methylmalonic acidemias. Part 2: treatment strategies. J Inherit Metab Dis. 42:745‐761 [DOI] [PubMed] [Google Scholar]
- 21. Yorifuji T, Kawai M, Muroi J, et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: presence of a common mutation and possible clinical implications. Hum Genet. 2002;111(2):161‐165. [DOI] [PubMed] [Google Scholar]
- 22. Riemersma M, Hazebroek MR, Helderman‐van den Enden ATJM, et al. Propionic acidemia as a cause of adult‐onset dilated cardiomyopathy. Eur J Hum Genet. 2017;25(11):1195‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hörster F, Garbade SF, Zwickler T, et al. Prediction of outcome in isolated methylmalonic acidurias: combined use of clinical and biochemical parameters. J Inherit Metab Dis. 2009;32:630‐639. [DOI] [PubMed] [Google Scholar]
- 24. Kölker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015;38:1059‐1074. [DOI] [PubMed] [Google Scholar]
- 25. Manoli I, Sysol JR, Epping MW, et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight. 2018;3(23):124351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Molema F, Jacobs EH, Onkenhout W, Schoonderwoerd GC, Langendonk JG, Williams M. Fibroblast growth factor 21 as a biomarker for long‐term complications in organic acidemias. J Inherit Metab Dis. 2018;41(6):1179‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information