

Abstract
Objective
The presence of autoantibodies to citrullinated proteins (ACPAs) often precedes the development of rheumatoid arthritis (RA). Citrullines are arginine residues that have been modified by peptidylarginine deiminases (PADs). PAD4 is the target of autoantibodies in RA. ACPAs could arise because PAD4 is recognized by T cells, which facilitate the production of autoantibodies to proteins bound by PAD4. We previously found evidence for this hapten–carrier model in mice. This study was undertaken to investigate whether there is evidence for this model in humans.
Methods
We analyzed antibody response to PAD4 and T cell proliferation in response to PAD4 in 41 RA patients and 36 controls. We tested binding of 65 PAD4 peptides to 5 HLA–DR alleles (DRB1*04:01, *04:02, *04:04, *01:01, and *07:01) and selected 11 PAD4 peptides for proliferation studies using samples from 22 RA patients and 27 controls. Peripheral blood lymphocytes from an additional 10 RA patients and 7 healthy controls were analyzed by flow cytometry for CD3, CD4, CD154, and tumor necrosis factor expression after PAD4 stimulation.
Results
Only patients with RA had both antibodies and T cell responses to PAD4. T cell response to peptide 8, a PAD4 peptide, was associated with RA (P = 0.02), anti‐PAD4 antibodies (P = 0.057), and the shared epitope (P = 0.05).
Conclusion
ACPA immunity is associated with antibodies to PAD4 and T cell responses to PAD4 and PAD4 peptides. These findings are consistent with a hapten–carrier model in which PAD4 is the carrier and citrullinated proteins are the haptens.
INTRODUCTION
The influence of HLA–DRB1 genes on the development of rheumatoid arthritis (RA) has been known since 1969, when it was observed that lymphocytes from RA patients do not stimulate each other in mixed lymphocyte cultures 1. Since that time, HLA–DRB1 genes and their products have been characterized, and their function, peptide presentation to CD4+ T cells, has been identified. HLA–DRB1 polymorphism has been described, and it has been shown that HLA–DRB1 alleles associated with RA share a similar motif, called the shared epitope, in the third hypervariable region of their DRβ1 chains 2.
Similarly, the definition of RA has continued to improve with regular revisions of classification criteria. The American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2010 criteria include the presence of RA‐specific anti–citrullinated protein antibodies (ACPAs) 3. ACPAs recognize citrulline residues on many different proteins 4, 5, 6, 7, 8, 9. Citrulline is a modified form of arginine obtained after a posttranslational modification called deimination and carried by enzymes called peptidylarginine deiminases (PADs) 4. ACPAs are present in >75% of patients with RA. They often precede the development of RA.
However, how shared epitope–positive HLA–DRB1 genes contribute to the development of RA is still unknown. It has been suggested that shared epitope–positive HLA–DRB1 alleles might bind citrullinated peptides to present them to T helper cells specific for citrullinated proteins 10. However, extensive HLA–DR peptide binding data from our previous study and others showed no evidence of preferential binding of citrullinated peptides to shared epitope–positive HLA–DR alleles 11, 12, 13.
While screening human protein chips in order to identify new autoantibodies in RA, we found that PAD4, one of the 5 isoenzymes of PADs, was the target of autoantibodies in ACPA‐positive and ‐negative RA 14, 15. This implied the existence of T cells capable of facilitating the production of IgG anti‐PAD4. PAD4 is an enzyme that binds and citrullinates multiple proteins. Thus, B cells specific for PAD4 might internalize PAD4 and any associated substrate and present peptides of PAD4 and its substrates to T helper cells. In other words, PAD4, because it binds many proteins, could behave as a carrier and aid in the development of antibodies to the proteins it binds and citrullinates that behave as haptens. We demonstrated this point in normal mice: after immunization with PAD4 or PAD2, they developed IgG autoantibodies to citrullinated fibrinogen peptides 16.
In this study, we addressed the question of whether the same hapten–carrier mechanism triggers the production of ACPA in patients with RA. We looked for evidence of recognition of PAD4 by antibodies and T cells in patients with RA, patients with psoriatic arthritis (PsA), and healthy controls. Having both a proliferative response and an antibody response to PAD4 was characteristic of RA. We identified a peptide of PAD4, peptide 8, that triggered T cell proliferation in 40% of the patients with RA and whose recognition was associated with antibodies to PAD4, shared epitope–positive HLA–DR alleles, and RA.
PATIENTS AND METHODS
Patients
We tested 41 patients with RA and 25 patients with PsA from the rheumatology unit at Sainte Marguerite Hospital (Marseille, France) and 11 healthy controls from the staff of the laboratory and the rheumatology unit. RA patients fulfilled the ACR/EULAR 2010 criteria and had ACPA titers higher than 3 times the upper limit of normal 3. PsA patients fulfilled the Classification of Psoriatic Arthritis Study Group criteria 17. HLA–DRB1 typing of all patients and controls was performed by polymerase chain reaction/sequence‐specific oligonucleotide analysis 18. IgG anti–cyclic citrullinated peptide antibodies were detected using a second‐generation enzyme‐linked immunosorbent assay (ELISA) (Immunoscan RA Mark 2; Euro‐Diagnostica). Rheumatoid factor (RF) was detected by ELISA using an Orgentec kit. The baseline characteristics of the subjects are presented in [Link], [Link], available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41189/abstract.
Proteins
Human PAD4 was produced in a baculovirus expression system (ProteoGenix) and purified. PAD4 activity and autocitrullination status were tested, and a T cell proliferation assay was then performed. Human fibrinogen (Merck Millipore) was incubated in a buffer containing 1M Tris HCl (pH 7.4), 100 mM CaCl2, and 50 mM dithiothreitol at a concentration of 1 mg/ml with rabbit PAD2 protein (Sigma‐Aldrich). Citrullination was performed for 2 hours at 37°C. Noncitrullinated fibrinogen was treated identically, except that water was added instead of PAD.
Synthetic peptides of human PAD4
Peptides were synthesized using a solid‐phase system (Neosystems) and purified (>70%). We synthesized 65 20‐mer peptides, encompassing residues 1–663 of wild‐type PAD4 (residues S55, A82, and A112, on locus NM_012387) and overlapping on 10 amino acids. Residues S55, A82, and A112, which can be polymorphic, were detected in their native, unmutated forms on peptides 5–6, 8–9, and 11–12, respectively.
Detection of anti‐PAD4 antibodies
Plates were coated with 0.5 μg of human PAD4 and blocked with 2% bovine serum albumin (BSA). Sera were diluted 1:100 and incubated on plates. After washing, peroxidase‐conjugated anti‐human IgG or IgM was added. Optical density (OD) was read at 405 nm. Background OD was obtained by adding each serum to a well without protein. Positive sera were defined as those with OD values higher than twice the background OD for IgG and higher than 3 times the background OD for IgM.
T cell proliferation assay
Mononuclear cells were isolated from 20 ml of heparinized blood with Ficoll‐Histopaque (Sigma‐Aldrich). Cells were cultured at a density of 106/ml in RPMI 1640 with 10% self‐serum in the presence of 1 μg/ml of human PAD4, human fibrinogen, or phytohemagglutinin or 5 μg/ml of PAD4 peptide. After 6 days of culture at 37°C, the proliferative response to proteins was evaluated using a colorimetric bromodeoxyuridine kit (Roche Diagnostics) 19. Positive T cell responses were defined as OD values higher than twice the OD values for cells cultured without protein or peptide.
Purification of HLA–DRB1 molecules from lymphoblastoid cell lines
The HLA homozygous lymphoblastoid cell lines JESTHOM (HLA–DRB1*01:01), SAVC (HLA–DRB1*04:01), YAR (HLA–DRB1*04:02), PEYSSON (HLA–DRB1*04:04), and MOU (HLA–DRB1*07:01) were cultured in RPMI 1640 with 10% fetal calf serum. Cells (2 × 109) were lysed in 10 mM Tris (pH 8), 10 mM NaCl, 10 mM MgCl2, 1% Triton X‐100, 0.05 mg/ml DNase, and protease inhibitors. These homozygous cell lines were chosen because they express 3 shared epitope–positive alleles, HLA–DRB1*01:01, *04:01, and *04:04, and 2 shared epitope–negative alleles not associated with RA, DRB1*04:02 and DRB1*07:01. Total protein extracts were immunoprecipitated by anti–HLA–DR LB3.1 antibody covalently coupled on CNBr–activated Sepharose 4B (Sigma‐Aldrich). After washing, HLA–DR molecules were eluted in phosphate buffered saline (pH 2) with 0.5% octyl glucoside, neutralized in 1M Tris, and quantified 11.
HLA–DR peptide binding assay
ELISA plates were coated with 10 μg of PAD4 peptide/well and blocked in 1% BSA. One microgram of purified HLA–DR molecule was added to each well. After washing, bound HLA–DR was detected with biotinylated anti–HLA–DR antibody B8122 (Immunotech), followed by peroxidase‐conjugated avidin. Peroxidase‐conjugated anti‐mouse IgG and peroxidase‐conjugated avidin were supplied by Sigma‐Aldrich. OD was read at 405 nm. The binding of each of the purified HLA–DR alleles was assayed on ELISA plates coated with PAD4 peptides (each in duplicate wells). As a control, 2 wells were coated with a classic positive binder, influenza hemagglutinin peptide (PKYVKQNTLKLAT). Positive binding was defined as an OD value equal to the OD for the hemagglutinin peptide 11.
Flow cytometric analysis
Whole blood samples were obtained from patients and healthy donors and processed within 24 hours. Two hundred fifty–microliter aliquots of whole blood from each subject were then incubated for 5 hours at 37°C with or without 2 μg of PAD4. The aliquot without PAD4 was not treated with any stimulating agent but underwent the same incubation protocol and was used as a negative control. After incubation, an adapted PerFix‐nc kit protocol (Beckman Coulter) was implemented, with 125 μl of fixation reagent initially added to the mixture and left to react for 5 minutes at room temperature. Lysis and permeabilization reagent (1.5 ml) was then added, and the mixture obtained was incubated for 15 minutes at room temperature. After a quick centrifugation step (at 500g for 5 minutes), the supernatant was discarded by aspiration. Fixed and permeabilized leukocytes were then stained in the dark for 1 hour at room temperature with 150 μl of previously reconstituted dry and ready‐to‐use DURAClone staining reagents (PC5.5‐conjugated anti‐CD3, Alexa Fluor 750–conjugated anti‐CD4, Alexa Fluor 700–conjugated anti–tumor necrosis factor [anti‐TNF], and phycoerythrin‐conjugated anti‐CD154) (all from Beckman Coulter). Cells were subsequently washed with 3 ml of final reagent, concentrated by a quick centrifugation step (at 500g for 5 minutes), and finally reconstituted in 250 μl of final reagent. The whole sample was then analyzed on a Cytoflex flow cytometer (Beckman Coulter) with an acquisition threshold set on CD4‐positive events.
Study approval
All experimental protocols were approved by the Comité de Protection des Personnes, Sud‐Méditerranée II, Ministère de l'Enseignement Supérieur et de la Recherche. Samples were collected under collection number DC‐2008‐327. All participants gave written informed consent.
Statistical analysis
Comparisons between groups for T cell and antibody assays were performed using Fisher's exact test.
RESULTS
Association of anti‐PAD4 antibodies with ACPA‐positive RA but not PsA or healthy controls
IgG anti‐PAD4 antibodies were detected in 11 (27%) of 41 patients with RA versus 1 (4%) of 25 patients with PsA and 0 of 11 healthy controls (both P = 0.004 by Fisher's exact test) (Figure 1). IgM antibodies to human PAD4 were detected in 14 (34%) of 41 patients with RA versus 3 (12%) of 25 patients with PsA and 0 of 11 healthy controls (both P = 0.01 by Fisher's exact test) (Figure 1).
Figure 1.
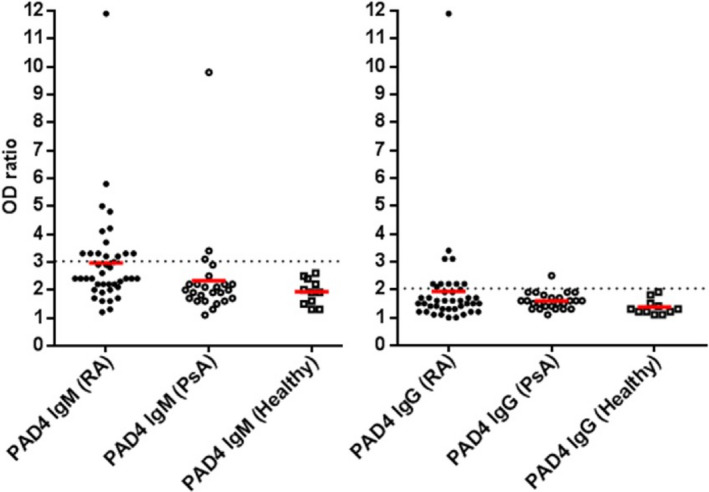
Antibody responses to human peptidylarginine deiminase 4 (PAD4) in patients with anti–citrullinated protein antibody–positive rheumatoid arthritis (RA; n = 41), patients with psoriatic arthritis (PsA; n = 25), and healthy controls (n = 11). Subjects were tested for IgM anti‐PAD4 and IgG anti‐PAD4 by enzyme‐linked immunosorbent assay. Each antibody assay was performed in duplicate. The OD ratio was the ratio of the OD for a well with serum and PAD4 protein to the OD for a well with serum but without PAD4 protein. Positivity was defined as an OD ratio of >3 for IgM and an OD ratio of >2 for IgG (dotted lines). Symbols represent individual subjects; red lines indicate the mean OD ratio.
Positive IgM anti‐PAD4 responses in RA patients are most likely caused by recognition of IgM‐RF bound to IgG anti‐PAD4 antibodies by the peroxidase‐labeled anti‐IgM antibody used in the ELISA. Indeed, 11 of the 14 patients with RA who tested positive for IgM anti‐PAD4 also tested positive for IgM‐RF. Thus, we pooled IgM and IgG anti‐PAD4 antibodies and referred to them as “anti‐PAD4 antibodies.” Anti‐PAD4 antibodies were present in 21 (51%) of 41 patients with RA versus 4 (16%) of 25 patients with PsA and 0 of 11 healthy controls (both P = 0.0002 by Fisher's exact test) (data not shown).
T cell responses to human PAD4 in patients with RA, patients with PsA, and healthy controls
Peripheral blood leukocytes from 41 patients with ACPA‐positive RA, 25 patients with PsA, and 11 healthy controls were tested for T cell proliferation in response to human PAD4 and human native and citrullinated fibrinogen by bromodeoxyuridine incorporation (Figure 2). Nineteen (46%) of 41 RA patients versus 8 (32%) of 25 PsA patients and 4 (36%) of 11 controls had a proliferative response to PAD4 (both P = 0.35 by Fisher's exact test [nonsignificant difference]). A proliferative response to citrullinated fibrinogen was observed in 1 patient with RA (2.4%) and none of the PsA patients or controls.
Figure 2.
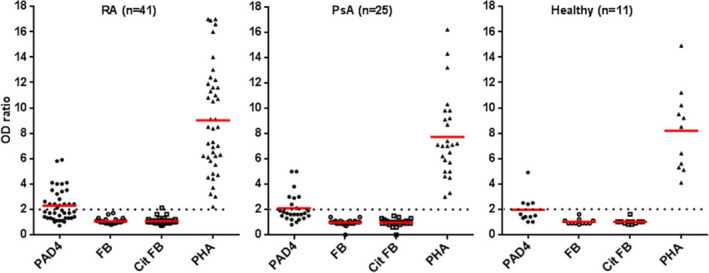
Proliferation of T cells in response to human PAD4, native fibrinogen (FB), citrullinated fibrinogen (cit FB), and phytohemagglutinin (PHA) in patients with RA, patients with PsA, and healthy controls. Proliferative response was evaluated by bromodeoxyuridine incorporation (n = 4 replicates per protein). The OD ratio was the ratio of the OD for a well with cells and protein to the OD for a well with cells but without protein. Positivity was defined as an OD ratio of >2 (dotted lines). Symbols represent individual subjects; red lines show the mean OD ratio. See Figure 1 for other definitions.
To confirm that the proliferative response observed in peripheral blood leukocytes cultured with PAD4 was due to T cells, we analyzed samples from an additional 10 RA patients and 7 healthy controls by flow cytometry. In this analysis, CD4+ T cells showed increased expression of CD154, a T cell early activation marker, and increased production of TNF, in whole blood samples stimulated with PAD4 (Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.41189/abstract).
Association of the shared epitope with anti‐PAD4 antibodies but not with T cell proliferation in response to PAD4
We studied the HLA–DR shared epitope status in the 77 subjects (including patients with RA, patients with PsA, and healthy controls) tested for antibody and proliferative response to PAD4. Twenty‐five had antibodies to PAD4, of whom 16 expressed shared epitope–positive HLA–DR alleles. Fifty‐two were negative for anti‐PAD4, of whom 20 expressed shared epitope–positive HLA–DR alleles (P = 0.051 by Fisher's exact test) (Figure 3). Thirty‐one subjects had a proliferative response to PAD4, of whom 15 expressed shared epitope‐positive HLA–DR alleles, and 46 subjects had no proliferative response to PAD4, of whom 21 expressed shared epitope–positive HLA–DR alleles (P = 0.8 by Fisher's exact test) (Figure 3). Thus, the HLA–DR shared epitope is associated with anti‐PAD4 antibodies, but not with anti‐PAD4 proliferative response.
Figure 3.
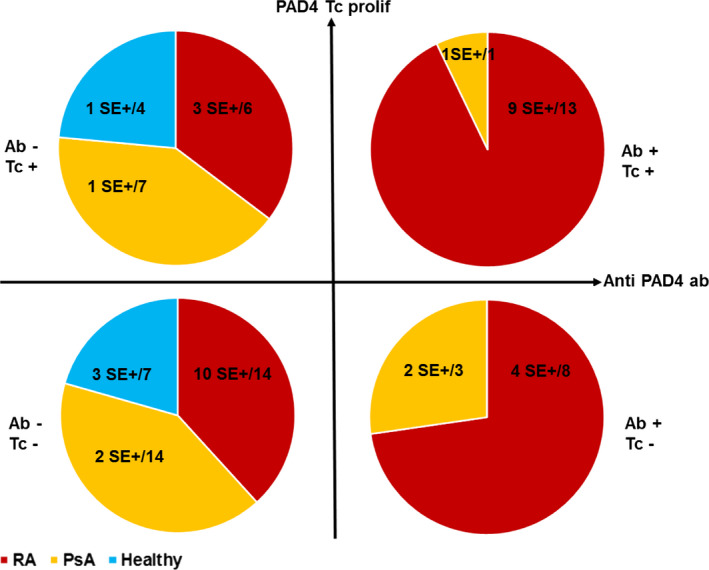
Antibody response to PAD4 and proliferation of T cells in response to PAD4 in patients with RA (n = 41), patients with PsA (n = 25), and healthy controls (n = 11). Subjects were tested for antibody response to PAD4 and proliferative response to PAD4 and classified into the following 4 groups: negative for PAD4 antibody (Ab−) and positive for T cell proliferation (Tc+), positive for both PAD4 antibody and T cell proliferation, negative for both PAD4 antibody and T cell proliferation, or positive for PAD4 antibody and negative for T cell proliferation. Values are the number of shared epitope (SE)–positive subjects/total number of subjects in the indicated group. See Figure 1 for other definitions.
Binding of PAD4 peptides to 5 different HLA–DR alleles
To help us identify potential T cell epitopes on PAD4, we tested the binding of 65 20‐mer peptides of PAD4, overlapping on 10 amino acids, to 5 purified HLA‐DR molecules (HLA‐DRB1*04:01, *04:02, *01:01, *04:04, and *07:01). Each purified HLA‐DRB1 molecule bound 2–9 of 65 PAD4 peptides (Figure 4).
Figure 4.
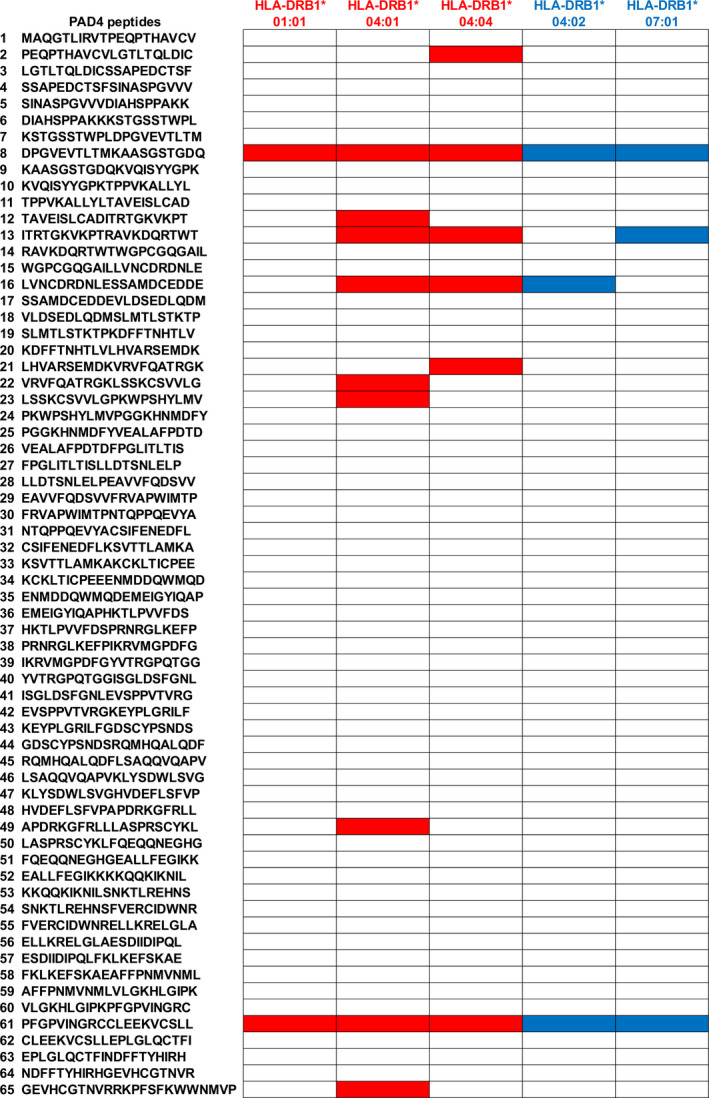
Binding of peptidylarginine deiminase 4 (PAD4) peptides to 5 different HLA–DR alleles. The binding of each purified HLA–DR allele was assayed on enzyme‐linked immunosorbent assay plates coated with 65 PAD4 peptides (each in duplicate wells). Two control wells were coated with a positive binder, hemagglutinin peptide. Positive binding was defined as an OD value equal to that for hemagglutinin peptide. Open boxes indicate no binding, red boxes indicate binding to a shared epitope–positive allele, and blue boxes indicate binding to a shared epitope–negative allele.
T cell proliferation in response to PAD4 peptides in RA patients and controls
We studied proliferative responses to the 11 20‐mer peptides of PAD4 that we had found to be good binders of HLA–DRB1*04:01, *04:04, *01:01, *04:02, or *07:01 in 22 RA patients, 16 PsA patients, and 11 healthy controls (Figure 5). Proliferative response to peptide 22 (VRVFQATRGKLSSKCSVVLG), a peptide found to bind HLA–DRB1*04:01 only, was observed in 2 (9%) of 22 RA patients versus 2 (12.5%) of 16 PsA patients and none of the 11 controls (both P not significant, by Fisher's exact test).
Figure 5.
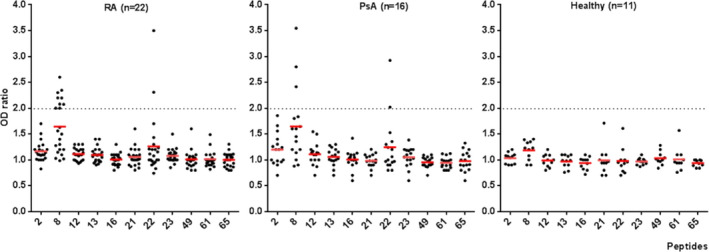
Proliferation of T cells in response to PAD4 peptides in patients with RA, patients with PsA, and healthy controls. Proliferative response was evaluated by bromodeoxyuridine incorporation (n = 4 replicates per peptide). The OD ratio was the ratio of the OD for a well with cells and peptide to the OD for a well with cells but without peptide. Positivity was defined as an OD ratio of >2 (dotted lines). Symbols represent individual subjects; red lines show the mean OD ratio. See Figure 1 for definitions.
Proliferative response to peptide 8 (DPGVEVTLTMKAASGSTGDQ), a peptide that bound all 5 HLA–DRB1 alleles tested, was associated with RA. A proliferative response to peptide 8 was observed in 9 (41%) of 22 RA patients versus 3 (19%) of 16 PsA patients and 0 of 11 controls (both P = 0.02 by Fisher's exact test) (Figure 5). A proliferative response to peptide 8 was associated with the shared epitope, which was present in 9 (75%) of 12 responders versus 15 (40%) of 37 nonresponders (P = 0.05 by Fisher's exact test) (Figure 6). Proliferative response to peptide 8 was associated with anti‐PAD4 antibodies (P = 0.057 for responders versus nonresponders, by Fisher's exact test) (Figure 6).
Figure 6.
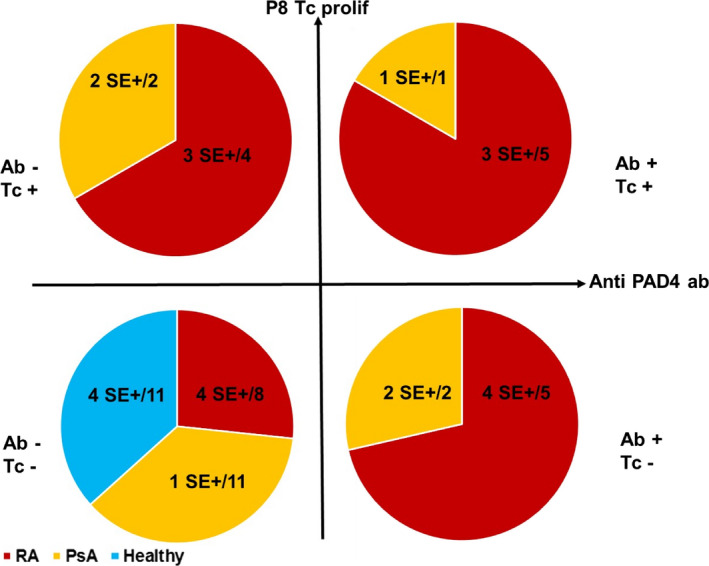
Antibody response to PAD4 and proliferation of T cells in response to peptide 8 in patients with RA (n = 22), patients with PsA (n = 16), and healthy controls (n = 11). Subjects were tested for antibody response to PAD4 and proliferative response to peptide 8 and classified into the following 4 groups: negative for PAD4 antibody (Ab−) and positive for T cell proliferation (Tc+), positive for both PAD4 antibody and T cell proliferation, negative for both PAD4 antibody and T cell proliferation, or positive for PAD4 antibody and negative for T cell proliferation. Values are the number of shared epitope (SE)–positive subjects/total number of subjects in the indicated group. See Figure 1 for other definitions.
DISCUSSION
How HLA–DRB1 molecules contribute to the development of RA is unknown, but since they are associated with ACPAs and RA, it is likely that they influence the production of ACPAs. Indeed, HLA–DRB1 molecules present peptides to T helper cells to allow IgG antibody production. The classic hypothesis that the shared epitope binds citrullinated peptide suggests that RA‐associated HLA–DRB1 molecules preferentially bind citrullinated peptides because of a P4 pocket rendered basic by the shared epitope. T helper cells recognizing citrullinated peptides presented by shared epitope–positive HLA–DRB1 alleles would then deliver specific help to B cells specific for individual citrullinated peptides, allowing the production of IgG antibodies to many citrullinated antigens. Preferential binding of citrullinated peptides to shared epitope–positive HLA–DRB1 alleles was first suggested by a binding study performed on one vimentin peptide in citrullinated or native form 10 and later by binding and structural studies performed with a few highly selected peptides 20, 21. However, thorough HLA–DRB1 binding studies involving hundreds of peptides of fibrinogen, vimentin, collagen, or Epstein‐Barr nuclear antigen 1 in their native or citrullinated forms found no evidence of preferential binding of citrullinated peptides to shared epitope–positive HLA–DRB1 alleles 11, 12, 13.
We have developed an alternative to the “shared epitope binds citrullinated peptides” hypothesis. Indeed, the development of RA is accompanied, sometimes preceded, by the emergence of IgG antibodies to PAD4 (the citrullinating enzyme) 14, 15, 22, 23, 24, 25, 26. This suggests the existence of T helper cells specific for PAD4 in patients with RA. Since PAD4 binds and citrullinates multiple proteins, any protein bound and being citrullinated by PAD4 may benefit from the help of a classic hapten–carrier mechanism. Indeed, B cells specific for citrullinated residues on proteins bound to PAD4 could internalize and process the PAD4–citrullinated protein complex and present PAD4 peptides to T helper cells, which would lead to the production of IgG antibodies to multiple citrullinated proteins. To demonstrate this point, we previously immunized normal nonautoimmune mice with human and/or murine PADs and found that 20% of the mice developed anti–citrullinated fibrinogen antibodies in the absence of any T cell response to citrullinated fibrinogen 16.
In this study, we investigated antibody and proliferative responses to PAD4 in the peripheral blood of patients with RA, patients with PsA, and healthy controls. Anti‐PAD4 antibodies were present in 21 (51%) of 41 patients with RA versus 4 (16%) of 25 patients with PsA and 0 of 11 healthy controls (both P = 0.0002 by Fisher's exact test). T cell proliferation in response to PAD4 was common in patients with RA, patients with PsA, and healthy controls. However, when associated with anti‐PAD4 antibodies it was characteristic of ACPA‐positive RA (P = 0.0009 for RA versus PsA or healthy controls, by Fisher's exact test).
To identity the PAD4 peptides recognized by the T cells that proliferate in response to PAD4, we performed proliferation studies using 11 PAD4 peptides predicted to bind shared epitope–positive HLA–DRB1 alleles. Samples from 22 patients with RA, 16 patients with PsA, and 11 healthy controls were included. We identified one peptide of PAD4, peptide 8, that was recognized by 9 of 22 RA patients, 3 of 16 PsA patients, and no healthy controls and another peptide of PAD4, peptide 22, that was recognized by 2 of 22 RA patients, 2 of 16 PsA patients, and no healthy controls. Proliferative response to peptide 8 was associated with RA (P = 0.02 by Fisher's exact test), the HLA‐DRB1 shared epitope (P = 0.05 by Fisher's exact test), and antibodies to PAD4 (P = 0.057 by Fisher's exact test).
These data suggest a model of the development of ACPAs in which T cells specific for PAD4 help, as expected, B cells specific for PAD4, leading to the production of IgG antibodies to PAD4, and, at the same time, B cells specific for citrullinated antigens that have internalized and processed the PAD4–citrullinated antigen complex and present PAD4 peptides. This PAD4 hapten–carrier model is only tentative. Indeed, flow cytometric analysis confirmed the presence of CD4+CD154+ cells in RA patient peripheral blood leukocytes stimulated with PAD4. However, we did not directly demonstrate that these CD4+ T cells help B cells specific for PAD4 and citrullinated antigens. We found immune responses to PAD4 in 27 of 41 RA patients and observed that having both T cell and antibody response to PAD4 is characteristic of RA patients. Still, we found no evidence of anti‐PAD4 immunity in 14 of 41 RA patients, a finding possibly caused by immunosuppressive drugs used to treat RA or by lack of sensitivity of our detection tests. Besides, PAD4 may not be the only target of T cells capable of providing help to ACPA‐secreting B cells. In this respect, any protein capable of binding and citrullinating peptides can be as relevant as PAD4 for activating T cells that can facilitate the production of ACPAs by the same hapten–carrier mechanism. This includes, for instance, PAD2, a PAD4 isoform also expressed in the RA synovium, or bacterial PADs that are not homologous to eukaryotic PADs but still bind and citrullinate peptides.
Our results suggest that a hapten–carrier mechanism, in which one of the candidate carriers is PAD4 and the haptens are the multiple proteins citrullinated by PAD4, may be at work in the development of ACPAs in RA. Thus, PAD4 stands at the center of the immunologic conflict leading to RA. The gene encoding PAD4 is associated with RA in Asians and in some European populations 27. Finally, if anti–citrullinated protein immunity develops under the influence of T helper cells specific for peptide(s) of PAD proteins, as opposed to citrullinated peptide(s) of multiple proteins, it improves the prospects for the prevention of RA by PAD peptide tolerization in high‐risk individuals identified by their HLA–DRB1 genotypes 18.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Roudier had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Auger, Balandraud, Roudier.
Acquisition of data
Auger, Balandraud, Massy, Hemon, Peen, Arnoux, Mariot, Martin, Lafforgue, Busnel, Roudier.
Analysis and interpretation of data
Auger, Balandraud, Massy, Hemon, Roudier.
ROLE OF THE STUDY SPONSOR
Pfizer had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Pfizer.
ADDITIONAL DISCLOSURES
Author Busnel is an employee of Beckman Coulter Life Sciences.
Supporting information
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1 Legend
ACKNOWLEDGMENT
We thank Leonor Nogueira (INSERM UMRs 1056) for providing citrullinated fibrinogen.
Supported by INSERM, Fondation ARTHRITIS, and Pfizer.
1Isabelle Auger, PhD, Marie F. Hemon, PhD, Elisa Peen, MD, Fanny Arnoux, PhD, Charlotte Mariot, Marielle Martin: INSERM UMRs 1097 and Aix‐Marseille University, Marseille, France; 2Nathalie Balandraud, MD, PhD, Emmanuel Massy, MD, Jean Roudier, MD, PhD: INSERM UMRs 1097, Aix‐Marseille University, and Assistance Publique‐Hôpitaux de Marseille, Marseille, France; 3Pierre Lafforgue, MD: Assistance Publique‐Hôpitaux de Marseille, Marseille, France; 4Jean‐Marc Busnel, PhD: Beckman Coulter Life Sciences, Marseille, France.
Drs. Auger and Balandraud contributed equally to this work.
Dr. Balandraud has received speaking fees from Bristol‐Myers Squibb and Chugai (less than $10,000 each). No other disclosures relevant to this article were reported.
References
- 1. Astorga GP, Williams RC Jr. Altered reactivity in mixed lymphocyte culture of lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum 1969;12:547–54. [DOI] [PubMed] [Google Scholar]
- 2. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205–13. [DOI] [PubMed] [Google Scholar]
- 3. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 4. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis‐specific autoantibodies. J Clin Invest 1998;101:273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res 2000;2:249–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simon M, Girbal E, Sebbag M, Gomès‐Daudrix V, Vincent C, Salama G, et al. The cytokeratin filament aggregating protein filaggrin is the target of the so‐called “antikeratin antibodies”, autoantibodies specific for rheumatoid arthritis. J Clin Invest 1993;92:1387–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Girbal‐Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, et al. The epitopes targeted by the rheumatoid arthritis‐associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol 1999;162:585–94. [PubMed] [Google Scholar]
- 8. Vossenaar E, Després N, Lapointe E, van der Heiden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti‐Sa antibodies target citrullinated vimentin. Arthritis Res Ther 2004;6:R142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masson‐Bessière C, Sebbag M, Girbal‐Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial target of the rheumatoid arthritis‐specific antifilaggrin autoantibodies are deiminated forms of the α‐ and β‐chains of fibrin. J Immunol 2001;166:4177–84. [DOI] [PubMed] [Google Scholar]
- 10. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high‐affinity peptide interaction with the rheumatoid arthritis‐associated HLA‐DRB1*0401 MHC class II molecule. J Immunol 2003;171:538–41. [DOI] [PubMed] [Google Scholar]
- 11. Auger I, Sebbag M, Vincent C, Balandraud N, Guis S, Nogueira L, et al. Influence of HLA–DR genes on the production of rheumatoid arthritis–specific autoantibodies to citrullinated fibrinogen. Arthritis Rheum 2005;52:3424–32. [DOI] [PubMed] [Google Scholar]
- 12. Pratesi F, Petit‐Teixeira E, Sidney J, Teixeira VH, Puxeddu I, Sette A, et al. Effect of rheumatoid arthritis (RA) susceptibility genes on the immune response to viral citrullinated peptides in RA. J Rheumatol 2012;39:1490–3. [DOI] [PubMed] [Google Scholar]
- 13. Sidney J, Becart S, Zhou M, Duffy K, Lindvall M, Moore EC, et al. Citrullination only infrequently impacts peptide binding to HLA class II MHC. PLoS One 2017;12:e0177140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Auger I, Balandraud N, Rak J, Lambert N, Martin M, Roudier J. New autoantigens in rheumatoid arthritis (RA): screening 8268 protein arrays with sera from patients with RA. Ann Rheum Dis 2009;68:591–4. [DOI] [PubMed] [Google Scholar]
- 15. Auger I, Martin M, Balandraud N, Roudier J. Rheumatoid arthritis–specific autoantibodies to peptidyl arginine deiminase type 4 inhibit citrullination of fibrinogen. Arthritis Rheum 2010;62:126–31. [DOI] [PubMed] [Google Scholar]
- 16. Arnoux F, Mariot C, Peen E, Lambert N, Balandraud N, Roudier J, et al. Peptidyl arginine deiminase immunization induces anticitrullinated protein antibodies in mice with particular MHC types. Proc Natl Acad Sci U S A 2017;114:E10169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chandran V. Spondyloarthritis: CASPAR criteria in early psoriatic arthritis. Nat Rev Rheumatol 2012;8:503–4. [DOI] [PubMed] [Google Scholar]
- 18. Balandraud N, Picard C, Reviron D, Landais C, Toussirot E, Lambert N, et al. HLA‐DRB1 genotypes and the risk of developing anti citrullinated protein antibody (ACPA) positive rheumatoid arthritis. PLoS One 2013;18:64108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Porstmann T, Ternynck T, Avrameas S. Quantitation of 5‐bromo‐2‐deoxyuridine incorporation into DNA: an enzyme immunoassay for the assessment of the lymphoid cell proliferative response. J Immunol Methods 1985;82:169–79. [DOI] [PubMed] [Google Scholar]
- 20. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA‐DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med 2013;210:2569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ting YT, Petersen J, Ramarathinam SH, Scally SW, Loh KL, Thomas R, et al. The interplay between citrullination and HLA‐DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J Biol Chem 2018;293:3236–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takizawa Y, Sawada T, Suzuki A, Yamada R, Inoue T, Yamamoto K. Peptidylarginine deiminase 4 (PADI4) identified as a conformation‐dependent autoantigen in rheumatoid arthritis. Scand J Rheumatol 2005;34:212–5. [DOI] [PubMed] [Google Scholar]
- 23. Roth EB, Stenberg P, Book C, Sjöberg K. Antibodies against transglutaminases, peptidylarginine deiminase and citrulline in rheumatoid arthritis—new pathways to epitope spreading. Clin Exp Rheumatol 2006;24:12–8. [PubMed] [Google Scholar]
- 24. Halvorsen EH, Pollmann S, Gilboe IM, van der Heijde D, Landewé R, Ødegård S, et al. Serum IgG antibodies to peptidylarginine deiminase 4 in rheumatoid arthritis and associations with disease severity. Ann Rheum Dis 2008;67:414–7. [DOI] [PubMed] [Google Scholar]
- 25. Zhao J, Zhao Y, He J, Jia R, Li Z. Prevalence and significance of anti‐peptidylarginine deiminase 4 antibodies in rheumatoid arthritis. J Rheumatol 2008;35:969–74. [PubMed] [Google Scholar]
- 26. Kolfenbach JR, Deane KD, Derber LA, O'Donnell CI, Gilliland WR, Edison JD, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum 2010;62:2633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 2003;34:395–402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1 Legend