

Abstract
Receptor interacting protein kinase 1 (RIP1) is a critical effector of inflammatory responses and cell death activation. Cell death pathways regulated by RIP1 include caspase‐dependent apoptosis and caspase‐independent necroptosis. The kinase activity of RIP1 has been associated with a number of inflammatory, neurodegenerative, and oncogenic diseases. In this study, we use the RIP1 kinase inhibitor GNE684 to demonstrate that RIP1 inhibition can effectively block skin inflammation and immune cell infiltrates in livers of Sharpin mutant (Cpdm; chronic proliferative dermatitis) mice in an interventional setting, after disease onset. On the other hand, genetic inactivation of RIP1 (RIP1 KD) or ablation of RIP3 (RIP3 KO) or MLKL (MLKL KO) did not affect testicular pathology of aging male mice. Likewise, infection with vaccinia virus or with mouse gammaherpesvirus MHV68 resulted in similar viral clearance in wild‐type, RIP1 KD, and RIP3 KO mice. In summary, this study highlights the benefits of inhibiting RIP1 in skin inflammation, as opposed to its lack of relevance for testicular longevity and the response to certain viral infections.
Keywords: RIP1, RIPK1, RIP3, RIPK3, MLKL, caspase
Inhibiting RIP1 has benefits to skin inflammation but not certain viral infections.
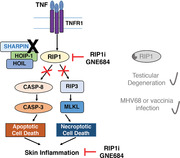
1. INTRODUCTION
Receptor interacting protein kinase 1 (RIP1; RIPK1) is critical in multiple signaling pathways downstream of the TNFR1, where it has both scaffolding and kinase activities.1, 2 Upon TNFR1 activation, RIP1 can interact with TNFR‐associated factor 2 and TNFR‐associated death domain protein and undergo ubiquitination by cellular inhibitors of apoptosis (c‐IAPs) and the linear ubiquitin assembly complex (LUBAC), allowing RIP1 to recruit Iκ B kinases (IKKs) and transforming growth factor beta‐activated kinase 1 (TAK1)/transforming growth factor beta‐activated kinase 1 (TAK1) binding protein 1 (TAB1), enabling NF‐κB signaling.3 Alternatively, RIP1 has also been shown to interact with caspase‐8 to mediate apoptosis. Notably, under conditions of caspase inhibition, RIP1 can autophosphorylate and cause RIP3 autophosphorylation resulting in mixed lineage kinase domain‐like pseudokinase (MLKL) phosphorylation and necroptosis.1, 3 Necroptosis is an inflammatory form of cell death, which is associated with the release of damage‐associated molecular patterns (DAMPs) and cytokines that perpetuate inflammatory responses.4 Although necroptosis has been proposed as an alternative means of controlling viral infections when caspase activity is inhibited,5 it has also been suggested to promote chronic inflammatory diseases by sustaining the proinflammatory environment. As such, RIP1's kinase function has been proposed as a potential therapeutic target for acute and chronic inflammatory diseases.6, 7
RIP1 kinase activity has been shown to be a critical mediator of inflammatory and cell death phenotypes in mouse models where NF‐κB signaling is disrupted.8 Conditional deletion of the NF‐κB essential modulator, NEMO, a key regulator of NF‐κB signaling, in intestinal epithelial cells results in severe necrotizing colitis, most prominently in the proximal colon, and ileal crypt degeneration and Paneth cell loss.9 NEMO‐deficient mice that express a catalytically dead RIP1(D138N) protein are protected against intestinal injury.7, 9 Similarly, treatment with a selective RIP1 kinase inhibitor GNE684 has also been shown to provide complete protection in this model, suggesting that the phenotype is driven by RIP1 kinase activity.7
SHARPIN is a component of the trimeric LUBAC complex, along with the proteins HOIP and HOIL‐1.10 The LUBAC complex linearly ubiquitinates NEMO and is necessary for NF‐κB signaling downstream of TNFR1.11, 12 Spontaneous mutations in the Sharpin gene that eliminate SHARPIN protein expression have been identified in C57BL/KaLawRij and Ocb‐3/Dem mice.13 The mutations are termed Cpdm (chronic proliferative dermatitis) and cpdmDem, respectively. The Cpdm mouse develops a progressive eosinophilic and proliferative dermatitis beginning as early as 1 wk of age, as well as immune cell infiltrates in multiple organs and abnormal development of lymphoid tissue.13, 14, 15, 16 Similar to intestinal‐specific NEMO‐deficient mice, dermatitis and immune cell infiltration in the lung, liver, and joints are absent in Cpdm mice that express a kinase dead (KD) version of RIP1.17 We previously conducted a pilot study to assess the therapeutic benefit of treating Cpdm mice with a RIP1 kinase inhibitor. Although mice were only treated for 4.5 d, a trend toward improvement of dermatitis was observed.7 Together, studies in NEMO‐ or SHARPIN‐deficient mice suggest RIP1 kinase activity is critical for the initiation and/or promotion of cell death and inflammation in the context of disrupted NF‐κB signaling, but more data are needed to gauge the benefit of RIP1 inhibition in a prolonged, chronic setting after the disease process is well underway. Therefore, in this study, we sought to determine if longer term treatment with RIP1 kinase inhibitor could provide more complete amelioration of the inflammatory phenotypes observed in SHARPIN‐deficient mice in an interventional setting, after disease onset.
In contrast to the potential proinflammatory role of RIP1 in the context of NF‐κB signaling disruption or chronic inflammation, RIP1‐dependent necroptosis has also been proposed to play a protective role in viral infections. Vaccinia virus expresses the caspase inhibitor B13R, which prevents host cells from undergoing apoptosis in response to TNF.5 Following vaccinia virus infection, RIP3 knockout (KO) mice reportedly have decreased inflammation and necrosis in the fat pads and liver, but higher viral titers and mortality rates compared to wild‐type mice.5, 18 Similarly, a subsequent study reported that both RIP3 KO and RIP1 KD mice had increased viral loads in the spleen and liver following vaccinia virus challenge compared to wild‐type mice.19 These studies suggest that RIP1‐ and RIP3‐dependent necroptosis can control vaccinia virus infection by serving as a secondary mechanism to eliminate virus‐infected cells in the event of caspase inhibition.
Given the potential for RIP1‐ and RIP3‐mediated necroptosis to mitigate viral infections, it is expected that viruses may develop strategies to inhibit necroptosis. Recently, the Epstein‐Barr virus latent membrane protein‐1 has been shown to interact with RIP1 and RIP3, where it modulates the ubiquitination of these proteins and thereby suppresses necroptosis.20 Because RIP1 kinase inhibition is a promising target for inflammatory diseases, it is important to also understand how inhibition of this pathway may affect susceptibility to viral infection.
To this end, we used a combination of our recently described mouse‐potent RIP1 kinase inhibitor GNE684 and genetically engineered mouse models, including RIP1 KD, RIP3 KO, and MLKL KO mice, to further characterize the therapeutic benefit of RIP1 inhibition in the context of ongoing inflammation, and to investigate the potential risks of RIP1 kinase deficiency in the context of viral infections and aging. The results showed that RIP1 inhibition by RIP1 kinase inhibitor GNE684 effectively blocked skin inflammation and immune cell infiltrates in livers of Sharpin mutant (Cpdm) mice, and that clearance of vaccinia virus or mouse gammaherpesvirus MHV68 were not affected in RIP1 KD or RIP3 KO mice.
2. RESULTS
2.1. RIP1 kinase inhibition protects against dermatitis in Cpdm mice, but does not prevent granulocyte infiltrates in the esophagus or intestine
Dermatitis in Sharpin mutant (Cpdm) mice reportedly begins as early as 1 wk of age with extensive dermatitis occurring by 6 wk of age.15 To determine if RIP1 kinase inhibition can mitigate dermatitis in Cpdm mice in a therapeutic setting, mice were aged to 6 wk, allowing for the development of dermatitis, and were then treated twice a day for 26 d with the mouse potent RIP1 inhibitor GNE684. This inhibitor has been reported to selectively inhibit RIP1 kinase activity without affecting NF‐κB or MAPK signaling.7 As expected, vehicle treated Cpdm mice developed alopecia, notably in the ventral cervical and thoracic regions (Fig. 1A). Histologic lesions in vehicle treated mice included mixed dermal immune cell infiltrates, fibrosis, epidermal hyperplasia, and serocellular crusting and/or ulceration (Fig. 1B, C). In contrast, RIP1 inhibitor treated mice had significantly reduced dermatitis and epidermal hyperplasia with these mice having only mild, multifocally increased dermal cellularity and minimal segmental epidermal hyperplasia (Fig. 1B, C). Improvement of dermatitis was associated with decreased serum cytokines including G‐CSF, MCP‐1, and IFNγ as well as IgM levels (Fig. S1A, B). As previously described,21, 22, 23 vehicle treated Cpdm mice had increased apoptosis as seen by cleaved caspase‐3 positive apoptotic cells distributed throughout the epidermis and hair follicles (Fig. 1C). Consistent with the improvement in dermatitis, cleaved caspase‐3 labeling was significantly reduced, but not eliminated, in Cpdm mice treated with GNE684 (Fig. 1C and S1C). pRIP3 immunohistochemistry was also performed to assess necroptotic pathway activation. Single, pRIP3 immunolabeled cells were observed in dermis of 3 of 8 vehicle treated Cpdm mice, and no pRIP3 labeling was observed in wild‐type or GNE684 treated Cpdm mice (Fig. 1C).
Figure 1.
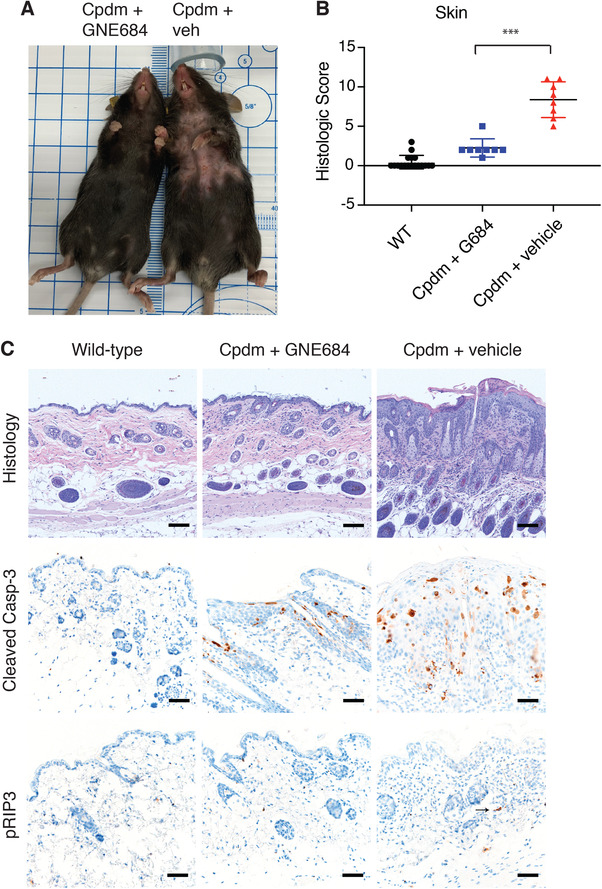
RIP1 kinase inhibition resolves dermatitis in Cpdm mice. (A) resolution of dermatitis and alopecia is observed in the Cpdm mouse treated with GNE684 (left) compared to the vehicle treated Cpdm mouse (right). (B) Histologic scores of dermatitis in wild‐type (N = 18), GNE684 treated Cpdm (N = 8), and vehicle treated Cpdm (N = 8) mice. Asterisks indicate P = 0.0003. Bar depicts mean with sd, P‐value was determined by Mann‐Whitney test. (C) Representative H&E stained, and cleaved caspase‐3 and phospho RIP3 (pRIP3) immunolabeled sections of mice from (B) demonstrating decreased dermal infiltrates, epidermal hyperplasia crusting, or ulceration, and decreased cleaved caspase‐3 and RIP3 phosphorylation in Cpdm mice following treatment with GNE684. The arrow indicates a pRIP3 positive cell
Cpdm mice also develop immune cell infiltrates in multiple organs including the liver, lung, joints, and esophagus.14, 16 In the liver, Cpdm mice develop prominent perivascular mixed immune cell infiltrates, and these infiltrates were reportedly absent in mice expressing Rip1 kinase‐dead alleles.17 Although treatment with RIP1 inhibitor did not eliminate all of the immune cell infiltrates, it significantly decreased the number and size of these infiltrates (Fig. 2). Cleaved caspase‐3 immunolabeling was observed in the livers of vehicle treated Cpdm mice (Fig. S2). Immunolabeled cells were frequently found in sinusoids and associated with perivascular immune infiltrates. This labeling was reduced to wild‐type levels in Cpdm mice treated with GNE684 (Fig. S2). In contrast, RIP1 inhibitor did not significantly protect against the eosinophilic esophagitis in the Cpdm mice, as numerous eosinophils were still present within the esophageal submucosa and mucosa of GNE684 treated mice (Fig. 2). Similarly, mildly increased proprial granulocytic infiltrates in the small and large intestine were present in both vehicle and GNE684 treated mice, with no conclusive reconstitution of Peyer's patches (Fig. 2).
Figure 2.
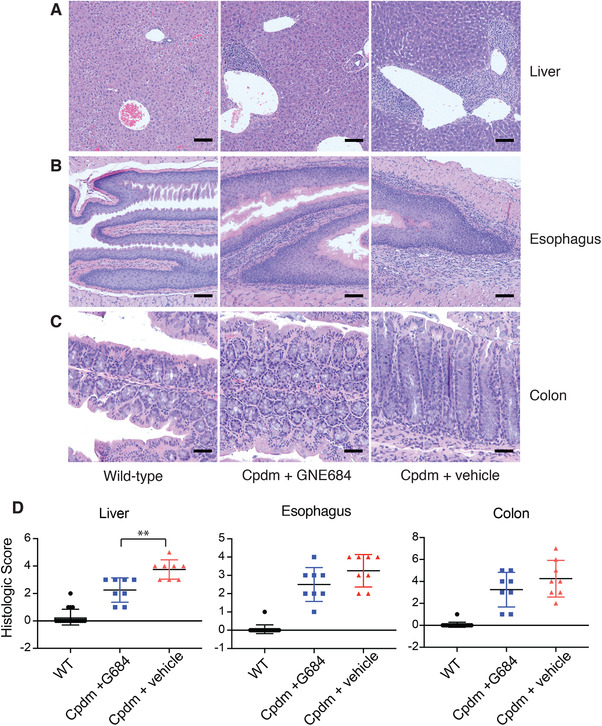
RIP1 kinase inhibition reduces liver, but not esophageal or intestinal, inflammatory infiltrates in Cpdm mice. (A–C) Histologic sections of liver (A), esophagus (B), and colon (C) demonstrated inflammatory infiltrates in Cpdm mice, although these infiltrates are relatively minimal in the colon. (D) Histologic scores of hepatic, esophageal, and colonic inflammation demonstrate abatement of inflammation in the liver, but not in the esophagus or colon, in Cpdm mice following treatment with GNE684 (G684). Asterisks indicate P = 0.0054. Bars depict mean with sd, P‐values were determined by Mann‐Whitney test. Wild‐type, N = 18; GNE684 treated Cpdm, N = 8; vehicle treated Cpdm, N = 8
2.2. Genetic inactivation or ablation of mediators of necroptosis does not affect testicular degeneration
In order to understand the long‐term consequences of deficiencies in necroptotic signaling, cohorts of Rip1D138N/D138N KD knock‐in (RIP1 KD), Rip3−/− (RIP3 KO), and Mlkl−/− (MLKL KO) male mice, as well as sibling matched wild‐type control male mice, were aged to 18 mo at which time they were necropsied and tissues were submitted for histologic examination. Mice of all genotypes were aged to 18 mo without significant increases in mortality. Terminal body weights were comparable between all genetically modified animals and their wild‐type controls (Fig. 3A). Li et al.24 previously suggested RIP1‐RIP3‐MLKL‐mediated necroptosis may play a role in age‐related testicular degeneration. To determine the consistency of these findings, we measured testis and seminal vesicle weights, and histologically assessed testes for evidence of germ cell loss. Seminal vesicle weights varied within each group, but no differences were identified between RIP1 KD, RIP3 KO, and MLKL KO mice and their wild‐type controls (Fig. 3B). Similarly, no differences in testicular weight (Fig. 3C), germ cell loss (Fig. 3D) or other histomorphologic features (Fig. 3E) were identified in this study.
Figure 3.
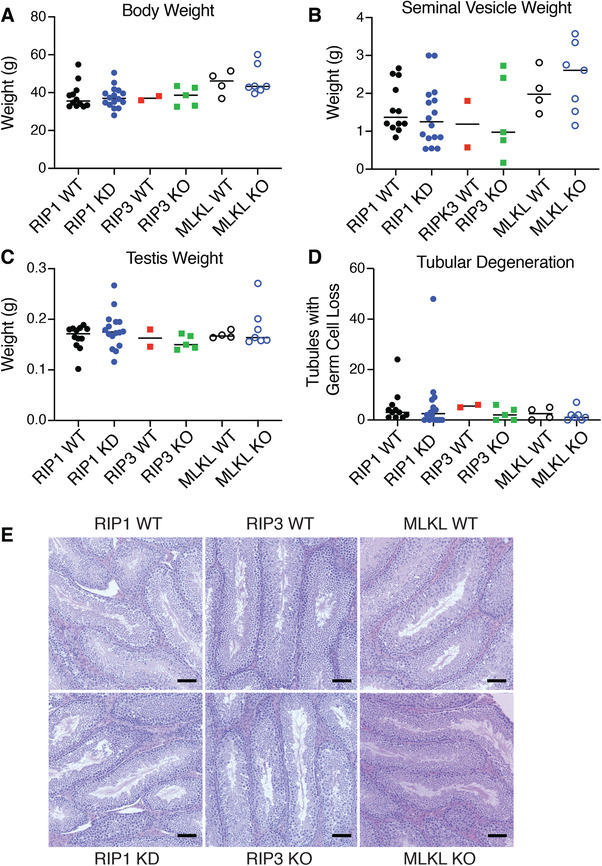
RIP1 KD, RIP3 KO, and MLKL KO mice have no evidence of differential age‐related testicular degeneration. (A–C.) Body weights (A), seminal vesicle weights (B), and testis weights (C) in RIP1 KD (N = 16), MLKL KO (N = 7), RIP3 KO (N = 5), and wild‐type mice (RIP1 WT, N = 11; MLKL WT, N = 4; RIP3 WT, N = 2) at study termination (18 mo). No group differences were noted. (D) Quantification of seminiferous tubules with loss of germ cells. Variable tubular degeneration was observed across genotypes with no clear genotype‐associated differences noted. (E) H&E stained sections of testes demonstrating similar histomorphologic features across genotypes
2.3. Loss of RIP1 kinase activity does not affect vaccinia virus or MHV68 virus clearance
Although the protective effects of RIP1 inhibitor in SHARPIN‐deficient mice demonstrates RIP1's potent role in promoting innate inflammation, we also wanted to better understand the role of RIP1 kinase activity in viral infections. First, to confirm published data, we challenged wild‐type and RIP1 KD mice with vaccinia virus intraperitoneally, and assessed viral loads in the liver, spleen, and ovary at 2 d and 14 d post‐infection. At day 2, RIP1 KD mice had slightly increased virus in the liver compared to wild‐type mice, but viral levels in the spleen and ovary were comparable (Fig. 4). At day 14 post‐infection, viral loads were below the limits of detection in both the spleen and liver, but remained comparable in ovary (Fig. 4). These data suggest that RIP1 kinase inactivation does not affect the long‐term course of vaccinia infection or viral clearance.
Figure 4.
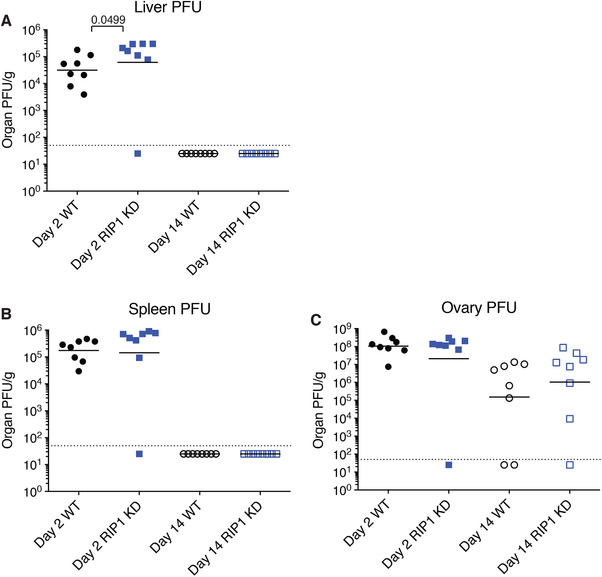
Loss of RIP1 kinase activity does not affect vaccinia virus clearance. (A‐C) Vaccinia virus plaque forming units in the liver (A), spleen (B), and ovary (C) measured 2 and 14 d post‐inoculation. Bars depict geometric mean, P‐values were determined by Mann‐Whitney test. N = 8 per group
Latent membrane proteins (LMP) are expressed by Epstein‐Barr virus, but are reportedly not present in the genome of the mouse gammaherpesvirus MHV68.25 Because LMP‐1 has been reported to inhibit necroptosis, we evaluated whether loss of RIP1 kinase activity or RIP3 gene ablation, thereby functionally blocking necroptosis, would similarly affect the course of MHV68 infection in the mouse. Wild‐type, RIP1 KD, and RIP3 KO mice were intranasally inoculated with MHV68. Viral titers were measured in the lungs on days 4 and 10–11 to assess acute infection, and in the spleen on days 21–23 and days 42–44 to assess latent infection. Similar viral titers were observed in the lungs of RIP1 KD and wild‐type mice, and RIP3 KO and wild‐type mice at 4 d (Figs. 5A and 6A). Viral titers in the lungs of all mice were significantly reduced by day 11 post‐infection (Figs. 5A and 6A). In the context of latent infection, similar viral titers were observed in the spleens of RIP1 KD and wild‐type, and RIP3 KO and wild‐type mice at days 21–23 and 42–44 (Figs. 5B and 6B). Similarly, we observed comparable levels of IFNγ, CXCL9, and CXCL10 in serum of RIP1 KD, RIP3 KO, and wild‐type mice (Figs. 5C‐D, 6C‐D, S3, S4). Together, these data suggest that inhibition of necroptosis does not potentiate viral replication in acute or latent MHV68 infection.
Figure 5.
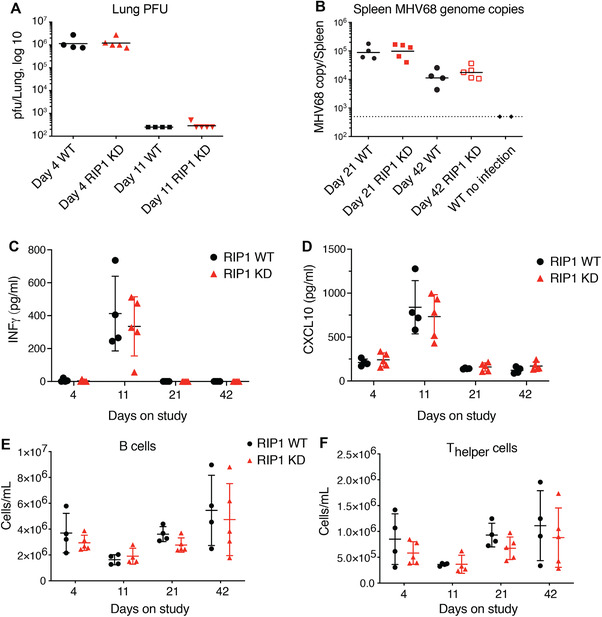
Loss of RIP1 kinase activity does not affect MHV68 virus clearance. Acute (A) and latent (B) MHV68 infections were assessed in RIP1 KD and WT mice by viral plaque assays (acute infection) or by PCR (latent infection). (C‐D) Levels of IFNγ (C) and CXCL10 (D) from animals analyzed in (A) and (B) were assessed by Luminex ELISA. (E‐F) Populations of B (E) and T‐helper (F) cells in the peripheral blood from animals analyzed in (A) and (B) were assessed by flow cytometry. WT, N = 4/ time point; RIP1 KD, N = 5/ time point; uninfected WT, N = 2. Bars depict mean with sd, P‐values were determined by multiple t tests
Figure 6.
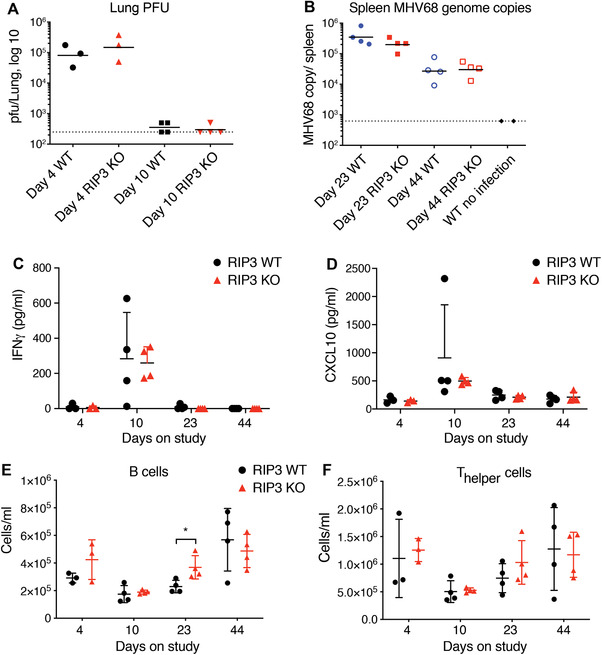
Genetic ablation of RIP3 does not affect MHV68 virus clearance. Acute (A) and latent (B) MHV68 infections were assessed in RIP3 KO and WT mice by viral plaque assays (acute infection) or by PCR (latent infection). (C‐D) Levels of IFNγ (C) and CXCL10 (D) from animals analyzed in (A) and (B) were assessed by Luminex ELISA. (E‐F) Populations of B (E) and T‐helper (F) cells in the peripheral blood from animals analyzed in (A) and (B) were assessed by flow cytometry. Day 4, N = 3/ group; other time points, N = 4/ group. Uninfected WT, N = 2. Bars depict mean with sd, P‐values were determined by multiple t tests
Given the potential for gammaherpesvirus to affect hematopoietic populations,26 we evaluated peripheral blood and splenic leukocyte populations in infected animals. Consistent differences in leukocyte populations across most time points were not observed between the wild‐type and RIP1 KD or RIP3 KO mice, but some differences were noted at individual time points (Figs. 5E‐F, 6E‐F, S3, S4). RIP1 KD mice had slightly decreased number of cytotoxic T cells in the peripheral blood at 21 d, but were comparable to wild‐type mice at all other time points (Fig. S3E). RIP3 KO mice, on the other hand, had lower number of peripheral neutrophils at 10 d, whereas peripheral B cells were increased on day 23 (Fig. 6E and S4C). At all other time points, cell counts were comparable for RIP3 KO and wild‐type mice (Fig. S4). These variations were considered to be not biologically significant, as the MHV68 viral clearance was not impacted in the RIP1 KD or RIP3 KO mice relative to wild‐type mice. Overall, these results suggest that loss of RIP1 kinase activity or RIP3‐dependent necroptosis does not have profound effects on the immune system in the context of gammaherpesvirus infection.
3. DISCUSSION
RIP1 kinase activity has been shown to drive both necroptotic and apoptotic cell death, and associated inflammatory responses. Genetically engineered mice have been critical to understand RIP1 signaling and to understand the role of RIP1 kinase activity in disease models.27 Using genetically engineered mouse models, others have shown that intestinal cell death and inflammation due to NEMO deficiency,7, 9 and dermatitis due to SHARPIN deficiency are driven by RIP1 kinase activity.17 Although these studies are critical for understanding the role of RIP1 in inflammatory cell death in the context of NF‐κB dysregulation, there was still the lingering question of whether RIP1 kinase inhibition can provide a therapeutic benefit after the inflammatory cycle has already begun. This question is critical as it provides insights as to whether RIP1 kinase activity is only involved in the initiation of the lesions or if it is involved in the continued disease propagation, giving it a larger therapeutic relevance.
To test the therapeutic benefits of RIP1 inhibition in the context of ongoing inflammation, we delayed treatment of Cpdm mice until 6 wk of age, when they are reported to have severe dermatitis.15 Delayed intervention with our RIP1 inhibitor significantly reduced the severity of dermatitis and reduced, to a lesser extent, inflammatory infiltrates in the liver. Therefore, RIP1 kinase activity is not only important in disease initiation, but is also involved in disease progression and inhibition of RIP1, even in a later stage of disease, may provide a therapeutic benefit. These data provide critical insights for the development of RIP1 inhibitors, because treatment will invariably occur after disease onset, during the progressive stage of the disease. Interestingly, GNE684 did not significantly decrease eosinophilic esophagitis in Cpdm mice. Esophageal lesions were not described in the prior genetic rescue of Cpdm mice with RIP1 kinase inactivation,17 so it is difficult to determine if the lack of rescue is due to the late stage of intervention in our study or the independence of these lesions from RIP1 kinase activity. Although the skin and esophageal lesions in SHARPIN‐deficient mice have some overlapping pathologic features and suspected drivers, differences have also been noted.16 Similarly, it has been shown that inflammation in the lungs, liver, and joints is partially dependent on lymphocytes, whereas dermatitis in Cpdm mice is not dependent on T or B cells.28 Better understanding of the mechanistic differences between lesions in the skin and other tissues may provide additional context as to when one is likely to observe the greatest benefit from RIP1 inhibition.
Apoptosis is considered a principle driver of the dermatitis in Cpdm mice, as evidenced by the increased cleaved caspase‐3 presence in the epidermis.21, 22, 23, 29 In this study, treatment with RIP1 inhibitor reduced cleaved caspase‐3 levels in the epidermis, which is consistent with the RIP1 inhibitor modulating apoptosis in these mice.29 In contrast, single pRIP3 positive cells were only observed in 3 of 8 vehicle treated Cpdm mice. Although pRIP3 is frequently difficult to detect in adult mouse tissues, the minimal pRIP3 signal is also compatible with the previous observation that loss of RIP3 delays, but does not prevent dermatitis in Cpdm mice.21, 23 Therefore, these results are consistent with the apoptosis, rather than RIP3‐mediated necroptosis, being the primary form of cell death that drives dermatitis in Cpdm mice. Similar to a previous study,21 we observed mildly increased cleaved caspase‐3 immunolabeling in livers of vehicle treated Cpdm mice. This labeling was often present in sinusoids or associated with immune cell infiltrates, and was decreased following RIP1 inhibitor treatment. Given the labeling pattern, including the lack of extensive hepatocyte labeling, decreased caspase‐3 activation in the liver following RIP1 inhibition may primarily reflect the decreased immune cell infiltration.
Abnormal lymphoid tissue development and absence of Peyer's patches have been described in Cpdm mice.14, 15 Although we didn't evaluate the spleen in this study, we did note that there was not significant reconstitution of Peyer's patches in GNE684 treated animals. The lack of Peyer's patch recovery in this study could suggest that RIP1 kinase activity does not have a role in hematopoietic defects of SHARPIN‐deficient mice or it could merely reflect the late intervention in this study. Unfortunately, the prior genetic rescue of Cpdm mice with KD RIP1 mice did not report hematopoietic phenotypes,17 so this question remains unresolved.
In this study, we found that RIP1 KD, RIP3 KO, and MLKL KO mice could survive to 18 mo without consistent disease induction. Although a previous study suggested a role for necroptosis in testicular aging,24 we did not find a similar relationship in our study. Notably, testicular degeneration or germ cell loss was not a prominent feature in our wild‐type animals either, which may have limited our ability to identify differences between genotypes. Mice used in the current study were generated by heterozygous/ heterozygous breeding, which allowed us to use sibling wild‐type controls. This approach reduces confounding strain‐dependent variability. In contrast, both KO and wild‐type animals in the previous study were generated by homozygous/homozygous mating, so wild‐type controls were not siblings of the KO animals.24 This might account for the differences in testicular degeneration identified in the previous study that were not observed in this study.
It has been proposed that necroptosis serves as a mechanism to eliminate virally infected cells when apoptosis is inhibited.5 As such, it is important to understand risks associated with inhibiting this pathway in a patient population. In this study, we evaluated the role of necroptosis in viral infections by challenging RIP1 KD with the poxvirus vaccinia and RIP1 KD and RIP3 KO mice with the gammaherpesvirus MHV68. Although RIP1 KD mice initially had slightly higher vaccinia virus titers in the liver (but not in spleen or ovaries) at day 2, viral clearance comparable to wild‐type animals was observed by day 14. Notably, RIP3 has been implicated in infection by multiple viruses including vaccinia virus, and molecularly, RIP3 can be directly engaged by different viral proteins.18, 30 Thus, our findings with RIP1 kinase‐dead mice in vaccinia infection model do not necessarily eliminate the possibility that RIP3 KO mice may exhibit a different phenotype due to RIP1‐independent RIP3 signaling.
Following MHV68 infection, RIP1 KD and RIP3 KO mice had similar responses compared to wild‐type mice. No consistent changes in peripheral leukocyte populations or IFN and cytokine/chemokine levels were identified in the context of viral infection. These data suggest that necroptosis inhibition does not predispose to increased risk or severity of viral infection, at least in the context of poxvirus and gammaherpesvirus infections. Lack of significant changes in peripheral leukocyte populations in the context of MHV68 infection further suggests that inhibition of necroptosis is not likely to affect leukocyte development or result in generalized immunosuppression. Interestingly, dermatitis in SHARPIN‐deficient mice has been shown to be T and B cell independent,28 which suggests RIP1 inhibition may have a more prominent role in prohibiting innate inflammatory responses, even in the context of chronic lesions.
In conclusion, our results suggest that RIP1 inhibition may provide therapeutic benefit in the context of ongoing active inflammation that is driven, in part, by ongoing cell death. However, we did not identify any additional risks of viral infection, immune suppression, or increased mortality associated with chronic loss of necroptotic signaling. These data suggest that RIP1 kinase inhibition is worth further evaluating as a target in inflammatory diseases.
4. MATERIALS AND METHODS
4.1. Reagents and antibodies
GNE684 was synthesized at Genentech (South San Francisco, CA, USA).7
4.2. ELISA
The levels of measured chemokines, cytokines, and IgM from animal sera were assessed using Luminex ELISA (Bio‐rad, Hercules, CA, USA or Millipore, Burlington, MA, USA) following manufacturer's specifications.
4.3. Mice for animal studies
Rip3−/−,31 Rip1D138N/D138N (KD),31 Cpdm,14 MLKL−/− 6, 32 mice were described previously. The Rip3 KO, Rip1 KD, and Mlkl KO alleles were maintained on a C57BL/6N genetic background, and wild‐type and homozygous mutants were generated by intercrossing heterozygous mice. Mlkl KO mice had been backcrossed to C57BL/6N for 9 generations. Cpdm mice and wild‐type controls were bred at Jackson Laboratories. All individuals participating in animal care and use were required to undergo training by the institution's veterinary staff. Any procedures, including handling, dosing, and sample collection mandates training and validation of proficiency under the direction of the veterinary staff prior to performing procedures in experimental in vivo studies.
4.4. Sharpin mutation (Cpdm) induced skin inflammation
Cpdm mice (Jackson Laboratories, Sacramento, CA, USA) were aged to 6 wk of age then treated with vehicle or GNE684 (50 mg/kg, BID, PO) for 26 d. All animals were euthanized at the end of treatment. Skin, liver, esophagus, small intestine, and large intestine were collected for histology. Serum was collected at the time of euthanasia for evaluation of serum cytokines, chemokines, and IgM.
4.5. Pathology
All mouse tissues submitted for histology were routinely formalin fixed, paraffin embedded, sectioned, and stained with H&E. Histologic lesions in Cpdm mice were scored according to the following criteria for inflammation, epidermal hyperplasia, and ulceration/serocellular crusts. The three individual scores were summed for a final score. Inflammation: 1) Slight, multifocal increase in dermal cellularity; 2) Mild to moderate, multifocal increase in dermal cellularity ± fibrosis; 3) Diffuse, mild to moderate increase in dermal cellularity and fibrosis; and 4) Moderate, diffuse increase in dermal cellularity and fibrosis. Epidermal hyperplasia: 1) Multifocal, 2–3 cell layer epidermal thickening; 2) Approximately 1–3 foci of 3 cell layer expansion of the epidermis; 3) >2 foci of locally extensive areas of epidermal expansion beyond 3 layers; and 4) Extensive epidermal expansion >3 layers. Ulceration/ serocellular crusts: 1) 1–2 serocellular crusts and/or increased individual pyknotic cells in the epidermis; 2) Single ulcer <2 follicles in size or >2 serocellular crusts; 3) Single ulcer >2 follicles in size or 2–5 ulcers <2 follicles in size; and 4) Multiple ulcers >2 follicles in size. Inflammatory infiltrates in the liver were scored according to the following matrix: 0) less than 3 minimal leukocyte aggregates; 1) 1–3 mild leukocyte aggregates that disrupt the hepatic architecture (approximately less than 10 hepatocytes in size); 2) 1 moderate (approximately greater than 10 hepatocytes in size) or 4–7 mild leukocyte aggregates; 3) 2–5 moderate or greater than 7 mild leukocyte aggregates; 4) 6–10 moderate leukocyte aggregates; and 5) Great than 10 moderate leukocyte aggregates. Esophageal lesions were scored as follows: 1) Minimally increased submucosal granulocytes; 2) granulocytes infiltrate the submucosa and mucosa with minimal stromal reaction; 3) Granulocytes infiltrate throughout the submucosa and mucosa with mild, often focal, stromal reaction; and 4) Granulocytes infiltrate the mucosa and submucosa with moderate to marked expansion of the stroma. The proximal, mid, and distal colon was scored individually according to the following matrix and summed for a final score: 0) Rare proprial granulocytes; 1) 1–3 minimal granulocyte aggregates that don't expand the lamina propria; 2) 1–3 mild granulocyte aggregates that expanded the lamina propria or disrupt lymphoid follicles, or locally extensive minimal granulocytes; 3) Greater than 3 mild aggregates or 1–2 moderate aggregates that expand crypts; and 4) Locally extensive or greater than 10 mild aggregates. Formalin‐fixed paraffin‐embedded tissue sections were labelled with rabbit anti472 cleaved caspase‐3 (Asp175) (cat. no. 9661, Cell Signaling Technologies, Danvers, MA, USA, 0.05 µg/ml) or rabbit anti‐phospho‐RIP3 Thr231, Ser232 (GEN135‐35‐9, Genentech, 5 µg/ml). Immunohistochemistry (IHC) for cleaved caspase‐3 and pRIP3 was performed on the Ventana Discovery XT platform with CC1 standard antigen retrieval (Ventana, Tucson, AZ, USA). Cleaved caspase‐3 was detected with the Ventana OmniMap detection system and DAB chromogen (Ventana). The pRIP3 signal was amplified with Ventana HQ Amplification and detected with HQ Discovery detection systems and DAB chromogen. Cleaved caspase‐3 labeling in the skin was labeled as follows: 0) No labeling observed in the epidermis or follicular infundibulum; 1) Less than or equal to 5 labeled cells in the epidermis or follicular infundibulum; 2) Greater than 5 individual labeled cells and/or small aggregates of approximately 5 or fewer immunolabeled cells, primarily associated with follicles; 3) Moderate, multifocal aggregates of immunolabeled cells; and 4) Locally extensive labeling in the epidermis. Cleaved caspase‐3 labeling in the liver was scored according to the following matrix: 0) No labeling observed; 1) Rare individual labeled cells, primarily in sinusoids; 2) Rare individual labeled cells and less than or equal to 3 small aggregates (less than 5 cells) of immunolabeled cells; 3) Moderate individual labeled cells or greater than 3 aggregates; and 4) extensive labeling throughout the liver. Histologic and immunohistochemical scores for Cpdm mice treated with GNE5684 versus vehicle were statistically compared using the Mann‐Whitney test.
Tissues collected for the aging study included heart, lung, liver, kidney, spleen, pancreas, thymus, small intestine, large intestine, mesenteric lymph node, skin, seminal vesicle, testis, and sternum. Whole animals, testes, and seminal vesicles were weighed at the termination of the aging study. Cross‐sections of each testis were histologically evaluated and the numbers of seminiferous tubules with loss of germ cells were counted. The Mann‐Whitney test was used to compare organ weights, body weights, and testicular degeneration between each genotype and their littermate controls.
4.6. Infection studies—vaccinia
For vaccinia viral infection study, 16 wild‐type or 16 RIP1 KD female mice were infected intraperitoneally with 2 × 106 PFU/mouse Vaccinia virus/Western Reserve strain (PAIRImmune, Laval, QC H7V 3S8, Canada). On days 2 and 14 postinfection, mice were taken down for spleen, ovary, and liver viral titers (8 mice per genotype per time point). Vaccinia viral loads were assessed by plaque forming unit (PFU) assay. Briefly, Vero cells in 24‐well plates were infected with serial dilution of tissue homogenates. Plaques were allowed to develop in the presence of methylcellulose after 3 d of incubation. Plaques were visualized by crystal violet staining after fixation. PFUs were counted at the relevant dilution and presented as PFU per gram of tissue.
4.7. Infection studies—MHV68
For infection with murine gammaherpesvirus MHV68, 16 wild‐type and 20 RIP1 KD female mice, and 15 wild‐type and 15 RIP3 KO female mice were infected intranasally with 5 × 105 PFU/mouse. On days 4 and 11 postinfection (RIP1 KD), or days 4 and 10 postinfection (RIP3 KO), mice were taken down for lung viral titers. On days 21 and 42 postinfection (RIP1 KD), or days 23 and 44 postinfection (RIP3 KO), mice were taken down for spleen viral genome copies (3–5 mice per genotype per time point).
Lung MHV68 viral load was assessed by PFU assay. Briefly, 3T12 cells in 24‐well plates were infected with serial dilution of lung tissue homogenates. Plaques were allowed to develop in the presence of methocell after 6 d of incubation. PFUs were counted at the relevant dilution and presented as PFU per lung.
Spleen MHV68 genome copies were assessed by nested quantitative PCR to detect the ORF50 gene. Briefly, spleen genomic DNA was purified using the DNeasy Kit (Qiagen, Germantown, MD, USA). The initial round of PCR was performed for 15 cycles at 94°C for 30s, 60°C for 30 s, and 72°C for 30 s with outer PCR primers, forward 5′‐AACTGGAACTCTTCTGTGGC‐3′ and reverse 5′‐GGCCGCAGACATTTAATGA‐3′. Then a second round PCR was performed with inner qPCR primers, forward 5′‐CCCCAATGGTTCATAAGTGG ‐3′, reverse 5′‐ATCAGCACGCCATCAACATC‐3′, and the probe of 5′‐CGGGCGTTGAGCCAGATTGCA‐3′.
4.8. Blood and spleen processing for flow cytometry analysis
EDTA‐anticoagulated whole peripheral blood was processed for flow cytometry analysis by reverse pipetting three technical replicates per animal to an antibody cocktail composed of PBS (‐Mg/‐Ca; Gibco) and titrated CD45‐BV785 (30F11; BioLegend, San Diego, CA, USA), CD3e‐BUV395 (145‐2C11; BD Biosciences, San Jose, CA, USA), CD4‐APCCy7 (GK1.5; BioLegend), CD8b‐PerCPCy5.5 (YTS156.7.7; BioLegend), CD44‐PE (IM7; BioLegend), CD62L‐BB515 (MEL‐14; BD Biosciences), CD19‐BV421 (6D5; BioLegend), CD335‐AF647 (29A1.4; BioLegend), NK1.1‐AF647 (PK136; BioLegend), Ly6G/C‐APCR700 (RB6‐8C5; BD Biosciences), and CD115‐PECy7 (AFS98; BioLegend). Samples were stained for 35–45 min at room temperature in 0.45 mL 96‐well round‐bottom polypropylene plates (Thermo Fisher Scientific, Waltham, MA, USA) sealed with adhesive PCR sealing foil sheets (Thermo Fisher Scientific). Samples were then diluted 1:16 in BD Pharm Lyse (BD Biosciences), centrifuged 300 × g for 5 min, gently decanted, and resuspended in an acquisition buffer composed of stain buffer (FBS) (BD Pharmingen, San Diego, CA, USA), BD TruCOUNT beads (BD Biosciences), and diamidini‐2‐phenylindole (DAPI 1 mg/mL; Thermo Fisher Scientific).
Half‐spleens were processed to single cell suspensions by manual homogenization and subsequent filtration through a 70 µM cell strainer (BD Biosciences) prior to erythroid lysis using BD Pharm Lyse. Lysed spleen samples were centrifuged and washed twice serially using stain buffer (FBS) before resuspending in a final volume of 5 mL. Processed spleen suspensions were then surface stained as described above for whole blood, except rather than using Pharm Lyse, stain buffer (FBS) was used to facilitate removal of excess antibody prior to suspension of washed cells in acquisition buffer.
4.9. Flow cytometry data analysis
A BD LSR Fortessa special order research product with high‐throughput sampler (BD Biosciences) was used to analyze peripheral blood and spleen‐derived leukocytes out of the same 96‐well plates they were stained in. In short, samples were injected at <2000 events/second and thresholded on CD45‐V785 OR PerCPCy5.5 to allow capture of beads while minimizing debris, and gated to exclude unstable sample injection, doublets, and DAPI+ dead cells before using Boolean gating for subsequent analysis using BD FACSDiva Software (v6.2; BD Biosciences). In order, B cells were defined as CD19+3‐ whereas non‐B cells were defined as CD19‐. In CD19‐ events, CD4+ T cells were defined based on co‐expression of CD3+4+ whereas CD8+ T cells were defined as CD3+4‐8+. In events that were both CD19‐ and CD3‐, a SSC‐H vs. Ly6G/C bivariate plot was used to define eosinophil‐like cells (high SSC‐H, Ly6G/C‐), neutrophils (mid SSC‐H, Ly6G/C+), and monocytes (low SSC‐H, Ly6G/C+). In remaining events that were SSC‐H low and Ly6G/C dim/‐, a bivariate plot assessing CD3e vs. NK1.1/CD335 was used to define natural killer cells as CD3e‐, NK1.1/CD335+.
Analyzed data were exported to csv, then processed on a per cell‐subset basis to cell per milliliter using the following equation:
using Excel 2016 (Microsoft). Processed data was then transferred to GraphPad Prism for visualization and statistical analysis as described below.
4.10. Data analyses
Data were analyzed and graphed using GraphPad Prism 8 Software (GraphPad Software; La Jolla, CA), Mac OS version.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We acknowledge and thank the Necropsy and Histology Laboratories at Genentech, and Catherine Sohn, Dorie Montoya, and Ryan Scott for their assistance with tissue collection, mice acquisition, and histologic sample preparation, and Kim Newton, Jessica Couch, and Michael Rothenberg for helpful suggestions and discussions.
Webster JD, Kwon YC, Park S, et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J Leukoc Biol. 2020;107:941–952. 10.1002/JLB.3MA1219-398R
Contributor Information
Joshua D. Webster, Email: webster.joshua@gene.com.
Domagoj Vucic, Email: domagoj@gene.com.
REFERENCES
- 1. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347‐353. [DOI] [PubMed] [Google Scholar]
- 2. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16:689‐697. [DOI] [PubMed] [Google Scholar]
- 3. Varfolomeev E, Vucic D. Intracellular regulation of TNF activity in health and disease. Cytokine. 2018;101:26‐32. [DOI] [PubMed] [Google Scholar]
- 4. Vanden Berghe T, Linkermann A, Jouan‐Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non‐apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135‐147. [DOI] [PubMed] [Google Scholar]
- 5. Upton JW, Kaiser WJ. DAI Another way: necroptotic control of viral infection. Cell Host Microbe. 2017;21:290‐293. [DOI] [PubMed] [Google Scholar]
- 6. Newton K, Dugger DL, Maltzman A, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel S, Webster JD, Varfolomeev E, et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF‐kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. 2017;277:113‐127. [DOI] [PubMed] [Google Scholar]
- 9. Vlantis K, Wullaert A, Polykratis A, et al. NEMO prevents RIP kinase 1‐mediated epithelial cell death and chronic intestinal inflammation by NF‐kappaB‐dependent and ‐independent functions. Immunity. 2016;44:553‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tokunaga F. Linear ubiquitination‐mediated NF‐kappaB regulation and its related disorders. J Biochem. 2013;154:313‐323. [DOI] [PubMed] [Google Scholar]
- 11. Tokunaga F, Nakagawa T, Nakahara M, et al. SHARPIN is a component of the NF‐kappaB‐activating linear ubiquitin chain assembly complex. Nature. 2011;471:633‐636. [DOI] [PubMed] [Google Scholar]
- 12. Ikeda F, Deribe YL, Skanland SS, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF‐kappaB activity and apoptosis. Nature. 2011;471:637‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seymour RE, Hasham MG, Cox GA, et al. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 2007;8:416‐421. [DOI] [PubMed] [Google Scholar]
- 14. HogenEsch H, Gijbels MJ, Offerman E, van Hooft J, van Bekkum DW, Zurcher C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am J Pathol. 1993;143:972‐982. [PMC free article] [PubMed] [Google Scholar]
- 15. Gijbels MJ, Zurcher C, Kraal G, et al. Pathogenesis of skin lesions in mice with chronic proliferative dermatitis (cpdm/cpdm). Am J Pathol. 1996;148:941‐950. [PMC free article] [PubMed] [Google Scholar]
- 16. Chien SJ, Silva KA, Kennedy VE, HogenEsch H, Sundberg JP. The pathogenesis of chronic eosinophilic esophagitis in SHARPIN‐deficient mice. Exp Mol Pathol. 2015;99:460‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berger SB, Kasparcova V, Hoffman S, et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN‐deficient mice. J Immunol. 2014;192:5476‐5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho YS, Challa S, Moquin D, et al. Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell. 2009;137:1112‐1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Polykratis A, Hermance N, Zelic M, et al. Cutting edge: rIPK1 Kinase inactive mice are viable and protected from TNF‐induced necroptosis in vivo. J Immunol. 2014;193:1539‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Li Y, Peng S, et al. Epstein‐Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018;9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumari S, Redouane Y, Lopez‐Mosqueda J, et al. Sharpin prevents skin inflammation by inhibiting TNFR1‐induced keratinocyte apoptosis. eLife. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang Y, Sundberg JP. SHARPIN regulates mitochondria‐dependent apoptosis in keratinocytes. J Dermatol Sci. 2011;63:148‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rickard JA, Anderton H, Etemadi N, et al. TNFR1‐dependent cell death drives inflammation in Sharpin‐deficient mice. eLife. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li D, Meng L, Xu T, et al. RIPK1‐RIPK3‐MLKL‐dependent necrosis promotes the aging of mouse male reproductive system. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aligo J, Brosnan K, Walker M, et al. Murine gammaherpesvirus‐68 (MHV‐68) is not horizontally transmitted amongst laboratory mice by cage contact. J Immunotoxicol. 2015;12:330‐341. [DOI] [PubMed] [Google Scholar]
- 26. Dong S, Forrest JC, Liang X. Murine gammaherpesvirus 68: a small animal model for gammaherpesvirus‐associated diseases. Adv Exp Med Biol. 2017;1018:225‐236. [DOI] [PubMed] [Google Scholar]
- 27. Webster JD, Santagostino SF, Foreman O. Applications and considerations for the use of genetically engineered mouse models in drug development. Cell Tissue Res. 2019. [DOI] [PubMed] [Google Scholar]
- 28. Potter CS, Wang Z, Silva KA, et al. Chronic proliferative dermatitis in Sharpin null mice: development of an autoinflammatory disease in the absence of B and T lymphocytes and IL4/IL13 signaling. PLoS One. 2014;9:e85666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taraborrelli L, Peltzer N, Montinaro A, et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat Commun. 2018;9:3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koehler H, Cotsmire S, Langland J, et al. Inhibition of DAI‐dependent necroptosis by the Z‐DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci U S A. 2017;114:11506‐11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Newton K, Dugger DL, Wickliffe KE, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357‐1360. [DOI] [PubMed] [Google Scholar]
- 32. Murphy JM, Czabotar PE, Hildebrand JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443‐453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information