

Abstract
PF‐05221304 is a liver‐targeted inhibitor of acetyl‐CoA carboxylase, an enzyme that catalyzes the first committed step in de novo lipogenesis (DNL). This first‐in‐human study investigated safety/tolerability and pharmacokinetics of single and multiple ascending oral PF‐05221304 doses, and fructose‐stimulated DNL inhibition with repeated oral doses. Healthy subjects (n = 96) received single (1‐240 mg) or repeated (2‐200 mg daily) doses for 14 days or single 100‐mg doses with and without food. PF‐05221304 was well tolerated at all doses. Repeated PF‐05221304 doses inhibited hepatic DNL in a dose‐dependent manner, with near‐complete inhibition seen at higher doses. With doses yielding ≥90% DNL inhibition, asymptomatic increases in fasting/postprandial serum triglyceride levels (≥40 mg/day) and declines in platelet count (≥60 mg/day) occurred; these were not observed at ≤80% DNL inhibition. Steady‐state pharmacokinetics generally increased dose‐proportionally, with a half‐life of 14‐18 hours and a minimal food effect on plasma exposure. The observed safety and tolerability, pharmacokinetics, and pharmacodynamics support the continued evaluation of PF‐05221304 for the treatment of nonalcoholic steatohepatitis.
Keywords: clinical research, lipid metabolism, liver disease, pharmacodynamics, pharmacokinetics and drug metabolism
Nonalcoholic fatty liver disease (NAFLD) is characterized by excessive accumulation of triglycerides (steatosis) in the liver and is emerging as one of the predominant causes of chronic liver disease.1, 2 Nonalcoholic steatohepatitis (NASH) is an advanced subtype of NAFLD characterized by cell injury and inflammation; progression to NASH significantly increases the risk of developing cirrhosis, hepatocellular carcinoma, and liver failure.2 Globally, no approved medications for treating NAFLD/NASH exist to date.3
Elevated de novo lipogenesis (DNL) in hepatocytes due to excessive carbohydrate consumption and hyperinsulinemia is thought to contribute to hepatic fat accumulation in NAFLD.4, 5 Acetyl‐CoA carboxylase (ACC) plays a key regulatory role in DNL and lipid metabolism,6 by catalyzing the conversion of acetyl‐CoA to malonyl‐CoA, which is the first committed and rate‐limiting step in DNL.6 Malonyl‐CoA is an allosteric inhibitor of carnitine palmitoyltransferase 1, an enzyme responsible for the transport of long‐chain fatty acyl‐CoAs to the mitochondria for oxidation.7, 8, 9 Two isoforms of ACC exist: ACC1 and ACC2.6, 10, 11
ACC inhibitors have been shown to inhibit hepatic DNL12, 13, 14 and reduce hepatic steatosis.13 However, hepatic ACC inhibition can activate the transcription factor sterol regulatory element‐binding protein (SREBP), resulting in secretion of very low‐density lipoproteins (VLDLs) and hypertriglyceridemia.13 Reductions in lipoprotein lipase‐mediated clearance have also been hypothesized to contribute to elevated circulating triglyceride levels.15 Furthermore, a systemically acting ACC inhibitor, PF‐05175157, decreased platelet count in humans.16 Mechanistic studies revealed that ACC activity and DNL play critical roles in megakaryocyte maturation and platelet production in humans and primates.16
PF‐05221304 is a potent, selective, orally bioavailable, and reversible dual ACC1/2 inhibitor (Supplementary Figure 1). PF‐05221304 was designed for preferential distribution to the liver to maximize hepatic DNL inhibition while minimizing DNL inhibition in peripheral tissues (including bone marrow),17 yielding an improved therapeutic index relative to effects on platelet count.16 To achieve this, PF‐05221304 is a substrate for cellular organic anion‐transporting polypeptide transporters (OATPs) in the liver, which confer ≥100‐fold asymmetric hepatic distribution (demonstrated in rats and monkeys), with robust hepatic DNL inhibition (demonstrated in humans)16 and suppression of markers for hepatic inflammation and fibrogenesis (demonstrated in humans and rats).17 Furthermore, unpublished preclinical data indicate that PF‐05221304 is cleared via liver uptake mediated by OATPs and sodium taurocholate cotransporting polypeptide, followed by oxidative/reduction metabolism via a variety of metabolic enzymes.18
We describe a 3‐part first‐in‐human study of PF‐05221304 in healthy adults. Part 1 of the study involved single, ascending PF‐05221304 doses after an overnight fast to evaluate safety and tolerability and the plasma pharmacokinetic (PK) profile. Part 2 involved repeated ascending PF‐05221304 doses with food for 14 days to assess safety and tolerability, the plasma and urine PK profile, and pharmacodynamics (PD) ascertained through effects of PF‐05221304 on fructose‐stimulated hepatic DNL. Part 3 evaluated the effect of a high‐fat/high‐calorie meal versus a fasted state on the single‐dose plasma PK of PF‐05221304. The study was registered with ClinicalTrials.gov (NCT02871037).
Methods
Subjects
The protocol was approved by an independent institutional review board, IntegReview, in Austin, Texas. All subjects provided informed consent before screening. This phase 1 study was conducted at the sponsor's Clinical Research Unit in New Haven, Connnecticut, from August 12, 2016 (first subject first visit) to March 27, 2017 (last subject last protocol‐specified follow‐up visit). The study was conducted in compliance with ethical principles of the Declaration of Helsinki and the International Council for Harmonization Good Clinical Practice guidelines. All local regulatory requirements were followed.
Subjects were healthy adults aged 18‐55 years (women were of nonchildbearing potential), with a body mass index of 17.5‐30.5 kg/m2 and a body weight of >50 kg. Exclusion criteria included history of clinically significant hematologic, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, neurologic, or allergic disease; conditions affecting drug absorption; or known history of hereditary fructose intolerance (part 2). Each subject participated in 1 part of the study.
Study Design
Part 1. This was a randomized, double‐blind, 4‐period, crossover, interleaving, placebo‐controlled design (Figure 1). Subjects received PF‐05221304 or matching placebo with water, following a ≥10‐hour overnight fast. The interleaving design enabled both within‐ and between‐subject assessments. There were 2 cohorts of 8 subjects, with each cohort receiving 4 single ascending doses of PF‐05221304 or matching placebo with ≥10 days between doses. Cohort 1 received 1, 10, 100, and 200 mg, whereas those in cohort 2, who started 5 days after cohort 1, received 3, 30, 200, and 240 mg (Figure 1). The days between doses were used to review safety and PK data from each dose level before decisions were made on the next dose.
Figure 1.
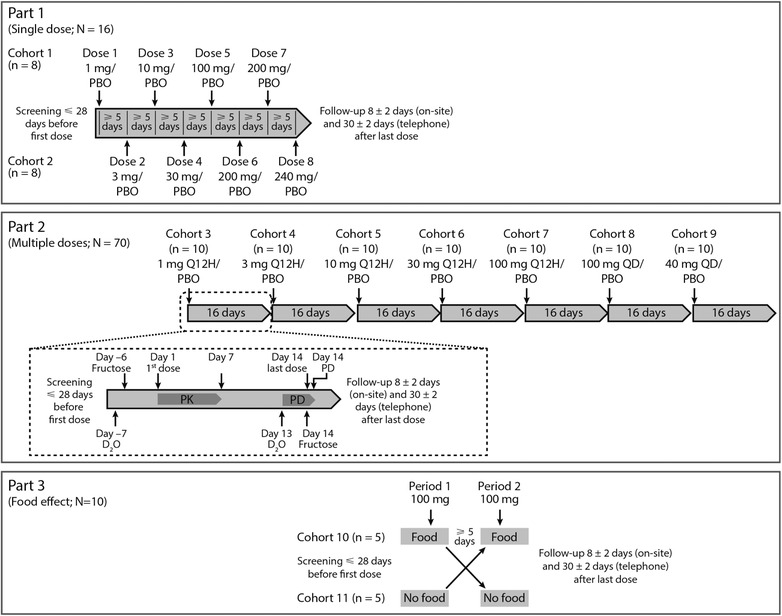
Design of the 3 parts of the study, with PF‐05221304 dose indicated. Part 2 overlapped with part 1, and part 3 overlapped with part 2 (indicated by the overlapping timelines). The first dosing cohort of part 2 is expanded to outline the procedures performed. PBO, placebo; PD, pharmacodynamics; PK, pharmacokinetics; Q12H, every 12 hours; QD, once daily.
Part 2. This was a randomized, double‐blind, placebo‐controlled, sequential, ascending multiple‐dose design (Figure 1). Subjects were admitted to the phase 1 unit from 8 days before dosing (day −8) to day 16 and were randomized 4:1 to receive PF‐05221304 or placebo on days 1‐14. The study drug was administered with food, either once daily (with breakfast) or every 12 hours (with meals), except on day 14, when the morning dose followed an oral fructose dose. On day −6 and day 14, subjects received an oral fructose solution (0.25 g/kg) every 30 minutes from 0 hours (when PF‐05221304/placebo was administered) to 9.5 hours. In the evenings/nights of day −7 and day 13 (5‐16 hours before fructose dosing was initiated), subjects received 4 oral doses of 70% deuterated water (D2O; 60 mL each). Fructose and D2O were provided as open‐label challenge agents.
Parts 1 and 2 were investigator‐ and subject‐blinded but sponsor‐open to allow members of the sponsor's study team to monitor safety, PK, and PD data in real time. Dose escalation could be stopped if the safety and tolerability limit was reached. Progression to the next dose level occurred if the previous dose was well tolerated following a review of the safety, PK, and, when available, PD data.
Part 3. This was a randomized, open‐label, crossover design with a single oral dose administered with and without a high‐fat/high‐calorie meal (Figure 1). Subjects stayed at the unit from the day before period 1 to day 3 after period 2. Dosing was in the morning, 30 minutes after subjects had eaten or following a ≥10‐hour fast.
Randomization was performed for all parts using a sponsor‐provided randomization schedule.
Selection of Doses
Starting doses in parts 1 and 2 were derived from nonclinical information on PK and metabolism. A projected efficacious dose of 7 mg twice daily or 18 mg once daily was predicted to reduce hepatic DNL by 70%, with a predicted effective terminal half‐life (t½) of ∼5 hours, and was selected using a physiologically based PK model (sponsor data on file). A starting dose of 1 mg was selected, where 1 mg every 12 hours would inhibit DNL by ∼25%, and 3, 10, and 30 mg every 12 hours would inhibit DNL by 49%, 76%, and 90%, respectively. The part 3 dose was based on observed plasma PK from parts 1 and 2.
Pharmacokinetics
Blood samples were collected at intervals ≤72 hours (part 1) and ≤48 hours (part 3) postdose. In part 2, plasma samples were collected at postdose intervals on days 1, 7, and 14, and predose on days 0‐2, 4, 7, 8, 10, and 12‐14 (plus 24 and 48 hours post–day 14 dose). Urine was collected at intervals on day 7 in part 2 (the Supporting Information to this article provides details).
PF‐05221304 and its stable isotope‐labeled internal standard, PF‐05221304‐2H, were isolated from plasma or urine modified with 5% triton:acetonitrile (50:50, v:v) using a protein precipitation extraction procedure. Analysis was performed using an Xbridge, C8, 2.1 × 50 mm, 3.5‐µm column. Mobile phase A contained 0.1% formic acid (aqueous), phase B contained 0.1% formic acid in acetonitrile, and phase C in tetrahydofuran.
The samples were analyzed by electrospray ionization liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS) using positive ionization mode. The mass spectrometer used was the SCIEX API 4000 (Covance Laboratories Inc., West Trenton, New Jersey) with m/z set to 503.2. Calibration was linear from 2.00 to 1000 ng/mL using l/concentration2 linear regression. The lower limit of quantification (LLOQ) for PF‐05221304 was 2.00 ng/mL in plasma and urine. PK parameters were calculated using sponsor‐validated electronic noncompartmental analysis software (version 2.2.4).
For plasma and urine, interday (between‐day) accuracy was expressed as percent relative error (%RE) for quality control (QC) concentrations and ranged from ‐0.3% to 8.1% and ‐3.2% to 1.3%, respectively. Assay precision was expressed as the interday percent coefficient of variation (%CV) of the mean estimated concentrations of QC samples, and was ≤5.9% and ≤4.1% for plasma and urine, respectively, for low (6.00 ng/mL), medium (50.0 and 450 ng/mL), high (800 ng/mL), and dilution (16 000 ng/mL) concentrations.
Pharmacodynamics (Part 2 Only)
Hepatic DNL in healthy subjects is an inherently variable process depending on diet, exercise, and other factors; however, repeated oral dosing of fructose over 10 hours increases the fractional contribution of hepatic DNL to VLDL palmitate, producing a consistent, measurable hepatic DNL signal.12, 13, 14, 19 This study used a fructose‐loading procedure to induce a DNL signal, using D2O as a tracer to measure the fractional contribution of hepatic DNL to triglyceride‐palmitate.20 Fractional hepatic DNL was assessed at baseline and following the administration of PF‐05221304/placebo for 14 days.
Samples were analyzed by Metabolic Solutions Inc. (Nashua, New Hampshire), except where specified. Samples were analyzed for deuterium enrichment in total body water using a validated and sensitive cavity ring‐down spectroscopy method, with percent deuterium in total body water quantified using a Los Gatos Research Liquid Water Isotope Analyzer.
The stable isotopic composition of hydrogen was reported as delta values in parts per thousand (denoted as ‰ or per mil) enrichments or depletions relative to a standard (D2O). The delta values were calculated by: delta per mil (‰) = (Rsample/Rstandard − 1) × 1000, where R is the ratio of the heavy to light isotope in the sample or standard. Stable hydrogen isotopic ratios were reported relative to Vienna Standard Mean Ocean Water standard; therefore, measurements were calibrated according to International Atomic Energy Agency guidelines for expression of delta values relative to available reference materials on normalized per mil scales. The delta per mil values were then converted to % deuterium.
Percent deuterium enrichment in body water was determined using a linear regression model. The range for this method in hexane was 0.0131% to 0.0515% with an upper limit of quantification (ULOQ) of 0.0457% and LLOQ of 0.0145%. The overall interday assay accuracy ranged from 0.62% to 0.73% for the low (0.0208%), medium (0.0293%), high (0.0420%), and diluted (0.0295%) QC concentrations. The overall interday assay precision was ≤1.04% for low, medium, high, and diluted QC concentrations.
Deuterium‐enriched triglyceride‐palmitate was extracted from 150 µL of K2EDTA plasma by thin‐layer chromatography (TLC; Pfizer Internal Medicine Research Unit, Cambridge, Massachusetts) using the classic Folch technique on day 1. On day 2, the sample was applied to a prescored 19‐lane silica TLC plate. To aid in the identification of the triglyceride band, a mixture of cholesteryl olein, trioleate, and dioleate was applied to 3 lanes (left, right, and center) reserved for this purpose. The plate was developed in hexane:diethyl ether:acetic acid (75:24:1), and the lipid bands visualized by spraying with dihydrofluoroscein in methyl alcohol. The silica containing the triglyceride band was scraped from the plate and lipids extracted by the addition of 2 × 2 mL of diethyl ether. The pooled ether extracts were dried under nitrogen gas and then sent to Metabolic Solutions Inc. for analysis. The TLC dried extracted sample was initially saponified and extracted using basified ethanol. The carboxylic acid of the free palmitate was reacted with TMS‐diazomethane to form the methyl ester derivative.
Palmitate methyl ester preparations were analyzed for deuterated palmitate using a Thermo Finnigan Delta V Isotope Ration Mass Spectrometer coupled to a Thermo Trace Ultra Gas Chromatograph (GC) with a GC combustion interface III and Conflow IV. The methyl ester of 2H‐palmitate was analyzed using a splitless injection with CTC Pal autosampler (1 µL) at an injection temperature of 250°C, and using a Zebron ZB‐5 column of 30 m × 0.25 mm × 0.50 µm film. The GC oven was programmed with an initial column temperature of 100°C with a 1‐minute hold, followed by a ramp of 10°C per minute to 150°C, and a final ramp of 30°C per minute to 340°C. Compounds eluting off the column were directed into the pyrolysis reactor, heated at 1450°C, and converted to hydrogen gas with monitoring of masses 2 and 3.
The ULOQ for % deuterium in triglyceride‐palmitate was 0.1866%, and the LLOQ was 0.0123%. The overall interday assay accuracy ranged from 1.79% to 7.63% for the low (0.0234%), medium (0.0545%), and high (0.0934%) concentrations, and ranged from 1.62% to 2.24% for the TLC QC medium (0.0298%) and TLC QC high (0.0797%) concentrations. The overall interday assay precision was ≤1.71% for low, ≤7.72% for medium, and ≤2.92% for high concentrations, and was ≤2.61% for TLC QC medium and ≤2.51% for TLC QC high concentrations.
Safety
All observed and self‐reported adverse events (AEs), any clinically significant changes in physical examination results, and abnormal test results were recorded. Serious AEs were those that resulted in death or were life‐threatening, requiring hospitalization/prolongation of hospitalization, and resulting in persistent or significant disability/incapacity, or congenital anomalies/birth defects, or that were considered an important medical event. Laboratory evaluations included hematology, chemistry, and urinalysis. Twelve‐lead electrocardiogram (ECG) and vital signs were monitored throughout.
In part 2, AEs were reported separately for days 1‐13 (PF‐05221304 alone) and day −6 and day 13 post‐D2O dose onward (PF‐05221304 plus fructose/D2O) to identify any AE profile differences.
In part 2, blood samples for lipid parameters were collected following overnight fasts over the 16‐day period. Postprandial serum triglycerides were assessed on days 0 (baseline) and 12, with 12 serial blood samples collected over a 24‐hour period, and the area under the effect curve (AUEC) calculated using the trapezoid rule.
Evaluations for total cholesterol, and direct low‐density lipoprotein (LDL) and high‐density lipoprotein (HDL) were performed using the Roche Cobas 6000 automated chemistry analyzer. Total cholesterol was analyzed using an enzymatic, colorimetric assay, in which cholesterol esters were cleaved by the action of cholesterol esterase to yield free cholesterol and fatty acids. Subsequent reactions involved cholesterol oxidase and cholesterol peroxidase to form a red quinone‐imine dye, the color intensity of which is directly proportional to cholesterol concentration and was assessed by an increase in absorbance.
Assessments for LDL used a similar enzymatic colorimetric assay as total cholesterol, but conducted in the presence of surfactants, which selectively solubilize LDL. Enzymatic reactions to lipoproteins, excluding LDL, are inhibited by surfactants and a sugar compound. As such, cholesterol in HDL, VLDL, and chylomicrons are not evaluated by this method.
The Cobas HDL‐cholesterol (HDL‐C) diagnostic test system was used for direct assessment of lipids. Detergents, cholesterol esterase, cholesterol oxidase, and peroxidase were used to form colored pigments, which were measured optically. Non‐HDL‐C lipoproteins were combined with polyanions and a detergent, creating a water‐soluble entity that blocked the enzymatic actions of cholesterol oxidase and cholesterol esterase toward non‐HDL‐C lipoproteins, thus allowing assessment of HDL alone.
Statistical Analyses
In part 1, a sample size of 8 subjects per cohort was chosen to minimize the first exposure in humans of a new chemical entity while providing safety information for each dose. In part 2, 10 subjects per cohort (8 receiving PF‐05221304 and 2 receiving placebo) were selected as per part 1. In part 3, 10 subjects were deemed to be sufficient, based on the observed PK variability in part 1, to obtain 7% precision in area under the concentration‐time curve from time 0 extrapolated to infinity (AUC0‐∞) and 12% precision in maximum plasma concentration (Cmax).
No formal inferential statistics were applied to plasma PK data in parts 1 and 2. PK parameters were summarized descriptively by dose. In part 3, the geometric means, adjusted mean ratio, and 90% confidence intervals (CIs) for the Cmax and AUC0‐∞ adjusted mean ratios were calculated.
In part 2, on DNL assessment days, the percentage of new triglyceride‐palmitate synthesized under fructose load was calculated. The AUEC10 for basal‐adjusted DNL response was calculated using the basal‐adjusted percentage of newly formed triglyceride‐palmitate at each time over the 10‐hour fructose‐loading period. The percentage of newly formed triglyceride‐palmitate was computed using an equation derived elsewhere21:
The safety analysis set comprised all subjects receiving ≥1 dose of study drug (and subjects receiving fructose/D2O in part 2); AEs, ECGs, blood pressure, pulse rate, and safety laboratory data were reviewed on an ongoing basis. Safety data were summarized descriptively.
For the derivation of postprandial serum triglycerides (part 2), average AUEC24/24 was calculated in subjects with at least the first, last, and 75% of total sample numbers available, divided by 24. Triglyceride baseline level was calculated from averaged measurements at 0 hours on days 0‐1. Effects on postprandial serum triglyceride levels were summarized descriptively.
Results
Subject Demographics and Disposition
Ninety‐six healthy subjects (93 men) were randomized and assessed for PK and safety (parts 1, 2, and 3), and PD (part 2 only). All randomized subjects completed the study, with the exception of 1 subject in part 2 who discontinued. Subject age ranged from 23 to 55 years, with mean body mass index ranging from 20.3 to 30.5 kg/m2.
In part 1, 16 subjects (15 men) were randomized, with 6 receiving PF‐05221304 and 2 receiving placebo at each dose level. In part 2, 70 subjects (68 men) were randomized, with 8 receiving PF‐05221304 and 2 receiving placebo at each dose. Ten subjects were enrolled in part 3 (Figure 1).
Pharmacokinetics
Part 1. At each dose, serial blood samples were collected to characterize the PK profile for 48‐72 hours postdose (see Supporting Information). Following single oral doses of 1‐240 mg administered after fasting, PF‐05221304 was absorbed relatively rapidly, with a median time to maximum plasma concentration (Tmax) of 1.5‐3 hours (Table 1). AUC0–∞ and Cmax increased in a dose‐proportional manner up to 100 mg and slightly greater than dose‐proportionally at doses > 100 mg (Table 1). No visually discernable trends between doses were observed for Tmax and t½. After Cmax was reached, concentrations declined in a biphasic manner (Figure 2), with a terminal elimination phase half‐life of approximately 14‐18 hours. Between‐subject variability in plasma PF‐05221304 PK parameters was low to moderate (Table 1).
Table 1.
Plasma PF‐05221304 Pharmacokinetic Parameter Values Following Single Oral Doses (Part 1 and Part 3)
| PF‐05221304 Dose | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Part 1 | Part 3 | ||||||||
| 1 mg | 3 mg | 10 mg | 30 mg | 100 mg | 200 mg | 240 mg | 100 mg | 100 mg | |
| Cohort 1 | Cohort 2 | Cohort 1 | Cohort 2 | Cohort 1 | Cohorts 1 & 2 | Cohort 2 | Fasted State | Fed State | |
| Parametera | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 12) | (n = 6) | (n = 10) | (n = 10) |
| AUC0‐∞, ng·h/mL | 670.5 (182.2) | 2508 (476.0) | 8603 (1603.8) | 26 950 (4308.0) | 81 950 (21 910.0) | 216 300 (49 526.0) | 268 800 (48 734.0) | 82 200 (23071.0) | 75 090 (20 969.0) |
| Cmax, ng/mL | 48.3 (8.2) | 177.0 (46.3) | 608.5 (134.0) | 2008 (421.7) | 7435 (1904.5) | 19 100 (3505.6) | 23 000 (5372.5) | 7671 (1699.8) | 5947 (1982.1) |
| Median Tmax, h (range) | 2.0 (1.0‐3.0) | 2.5 (1.1‐4.0) | 3.0 (1.0‐5.0) | 2.0 (0.5‐4.0) | 2.0 (1.0‐4.0) | 2.0 (0.5‐4.0) | 1.5 (1.0‐4.0) | 2.5 (1.0‐3.0) | 5.0 (2.0‐8.0) |
| t½, h | 13.6 ± 3.4 | 13.5 ± 3.5 | 18.3 ± 2.3 | 17.6 ± 2.6 | 16.0 ± 3.6 | 15.3 ± 3.4 | 15.2 ± 2.8 | 12.3 ± 2.0 | 13.2 ± 3.5 |
| CL/F, mL/min | 27.55 (12.1) | 20.48 (3.7) | 19.98 (4.0) | 18.97 (3.0) | 21.82 (7.0) | 16.63 (6.2) | 15.35 (3.3) | 21.8 (6.4) | 24.0 (7.4) |
AUC0‐∞, area under the concentration–time curve from time 0 extrapolated to infinity; CL/F, apparent clearance; Cmax, maximum plasma concentration; h, hours; min, minutes; SD, standard deviation; t½, terminal half‐life; Tmax, time to maximum plasma concentration.
Arithmetic mean (SD), except where stated.
Figure 2.
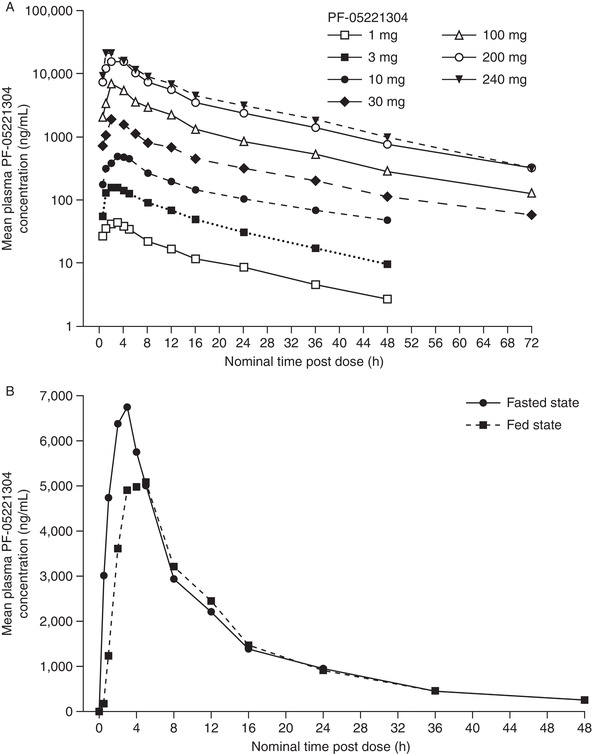
Mean plasma PF‐05221304 concentration‐time profile following (A) single oral doses (part 1), and (B) food effect (part 3). Summary statistics were calculated by setting concentration values below the lower limit of quantification to 0 (lower limit of quantification was 2.00 ng/mL). h, hours.
Part 2. Subjects were dosed for 14 days; full PK profiles were assessed on days 1 and 7 (meal with dosing) and day 14 (no meal with dosing because of fructose loading and PD measurements). Day 7 was considered the primary assessment of steady‐state PK, as PF‐05221304 absorption on day 14 was potentially affected by fructose and D2O administration. Following multiple oral doses of 1‐100 mg every 12 hours (total daily doses of 2‐200 mg) and 40‐100 mg once daily with food, PF‐05221304 was absorbed at a moderate rate (Table 2); median Tmax ranged from 3 to 5 hours with no dose‐related trend. AUC from time 0 to the dosing interval tau (AUCtau) and Cmax increased in an approximately dose‐dependent manner at steady state (Table 2). Based on the observed accumulation ratio for AUC24 (Rac) values in Table 2, accumulation for AUCtau of PF‐05221304 was approximately 2‐fold with every‐12‐hour dosing and approximately 1.5‐fold with once‐daily dosing. Less than 1% of PF‐05221304 was excreted in urine as unchanged drug over the dosing interval (Aeτ), indicating that urinary excretion is not a major clearance pathway of PF‐05221304. Based on visualization of trough concentrations, steady state was achieved by day 7. Variability in plasma PF‐05221304 exposure at steady state was 16%‐34%, based on geometric mean percentage coefficient of variation (CV%) for AUCtau and Cmax (Table 2).
Table 2.
Plasma PF‐05221304 Pharmacokinetic Parameter Values on Days 1 and 7 of Multiple‐Dose Administration (Part 2)
| PF‐05221304 Dose | |||||||
|---|---|---|---|---|---|---|---|
| Parametera | 1 mg Every 12 Hours (n = 8) | 3 mg Every 12 Hours (n = 8) | 10 mg Every 12 Hours (n = 8) | 30 mg Every 12 Hours (n = 8) | 100 mg Every 12 Hours (n = 8) | 40 mg Once Daily (n = 8) | 100 mg Once Daily (n = 8) |
| Day 1 | |||||||
| AUCtau, ng·h/mL | 384.5 (71.1) | 1038 (131.4) | 3811 (767.3) | 10 380 (1955.3) | 41 230 (6694.7) | 19 330 (2787.1) | 54 760 (9607.6) |
| Cmax, ng/mL | 54.4 (9.0) | 142.7 (21.2) | 584.6 (91.2) | 1689 (256.7) | 6831 (1388.6) | 1986 (591.8) | 5965 (1252.2) |
| Median Tmax, h (range) | 3.0 (3.0‐5.0) | 5.0 (2.0‐6.0) | 3.5 (1.0‐5.0) | 5.0 (3.0‐5.0) | 3.5 (0.5‐5.0) | 4.0 (2.0‐8.0) | 4.0 (2.0‐6.0) |
| Day 7 | |||||||
| AUCtau, ng·h/mL | 760.4 (202.4) | 2264 (484.8) | 8334 (2131.6) | 23 650 (6971.2) | 90 540 (16 282.0) | 27 590 (6449.2) | 89 510 (15 771.0) |
| Cmax, ng/mL | 91.4 (19.3) | 283.1 (49.1) | 1009 (215.9) | 3229 (741.0) | 12 100 (1926.3) | 2654 (674.6) | 7894 (1117.5) |
| Median Tmax, h (range) | 3.0 (2.0‐5.0) | 5.0 (3.0‐5.1) | 5.0 (5.0‐6.0) | 5.0 (3.0‐6.0) | 3.5 (3.0‐5.0) | 4.0 (3.0‐6.0) | 4.0 (2.0‐6.0) |
| Cmin, ng/mL | 41.6 (13.3) | 133.1 (32.5) | 493.3 (172.5) | 1240 (462.7) | 4471 (1128.6) | 432.6 (157.3) | 1475 (398.9) |
| PTR | 2.3 (0.33) | 2.2 (0.42) | 2.2 (0.45) | 2.9 (0.95) | 2.8 (0.54) | 6.7 (2.76) | 5.6 (1.49) |
| CL/F, mL/min | 23.3 (6.2) | 23.1 (5.3) | 21.5 (6.8) | 23.2 (8.4) | 19.1 (4.5) | 25.1 (4.9) | 19.3 (4.2) |
| Rac b | 2.0 (0.23) | 2.2 (0.32) | 2.2 (0.54) | 2.3 (0.39) | 2.2 (0.38) | 1.4 (0.18) | 1.6 (0.14) |
| RacCmax c | 1.7 (0.24) | 2.0 (0.22) | 1.7 (0.34) | 1.9 (0.37) | 1.8 (0.34) | 1.4 (0.55) | 1.4 (0.32) |
AUCtau, area under the plasma concentration‐time profile from time 0 to tau, the dosing interval (where tau = 12 hours for 12‐hour dosing or 24 hours for once‐daily dosing); CL/F, apparent clearance; Cmax, maximum plasma concentration; Cmin, minimum observed concentration during the dosing interval; h, hours; min, minutes; PTR, peak‐to‐trough ratio; Rac, observed accumulation ratio for AUC24; RacCmax, observed accumulation ratio for Cmax; SD, standard deviation; Tmax, time to maximum plasma concentration.
Arithmetic means (SD), except where stated.
Rac was calculated as the ratio of AUCtau at steady state/AUCtau on day 1.
RacCmax was calculated as the ratio of Cmax at steady state/Cmax on day 1.
Part 3. A single oral 100‐mg dose of PF‐05221304 was administered with a high‐fat meal or following an overnight fast in a 2‐period, crossover design. Food had a minimal impact on AUC0‐∞ (decrease of ∼9%). Geometric mean AUC0‐∞ was 72 360 and 79 320 ng·h/mL for the fed and fasted states, respectively, with an adjusted mean ratio of 91.2% (90%CI, 83.2%‐100.1%), where the 90%CI was within the bioequivalence range of 80%‐125%. However, Tmax was delayed (median, 5.0 hours [range, 2.0‐8.0 hours] versus 2.5 hours [range, 1.0‐3.0 hours]), and plasma Cmax was reduced by 25% (geometric mean, 5592 ng/mL [CV%, 41%] versus 7479 ng/mL [CV%, 25%]) with food, suggesting that food may delay the PF‐05221304 oral absorption rate.
Pharmacodynamics (Part 2 Only)
DNL inhibition was assessed by computing the AUEC for the percentage of newly made triglyceride‐palmitate (by subtracting the percentage of basal newly made triglyceride‐palmitate before fructose loading) on day 14 versus baseline for each subject.
Following 14 days of PF‐05221304 administration, the dose‐dependent inhibition of DNL (based on the AUEC of newly made triglyceride‐palmitate) from baseline (day −6) was observed, with near‐maximal inhibition (>90%) at doses ≥40 mg/day (Figure 3). Some variability between cohorts at baseline was observed (Figure 3A); this was expected, and the study design included a baseline measurement for each subject to account for this. Inhibition of fructose‐stimulated DNL on day 14 was maintained throughout the 10 hours of fructose loading at doses ≥20 mg/day (Figure 3B). Doses of 1, 3, and 10 mg every 12 hours (total daily doses of 2, 6, and 20 mg) resulted in average DNL inhibition of 47.4%, 52.6%, and 80.0%, respectively (Figure 3C). DNL inhibition, as assessed by the percentage of newly made triglyceride‐palmitate at the end of fructose loading (10 hours), provided very similar results (Figure 3D).
Figure 3.
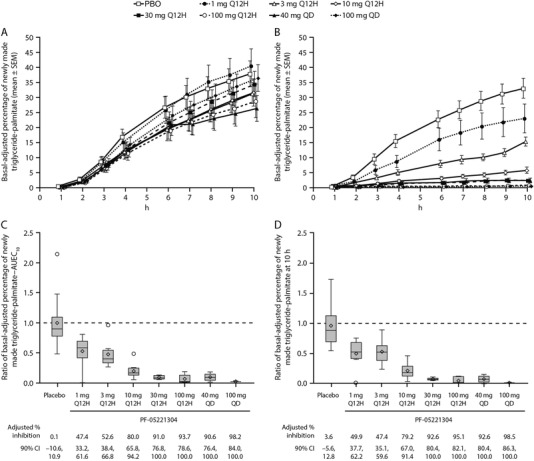
Basal‐adjusted percentage of newly made triglyceride‐palmitate following oral fructose administration. (A and B) Basal‐adjusted percentage of newly made triglyceride‐palmitate on (A) day −6 and (B) day 14. (C and D) Box‐and‐whiskers plots of individual ratios of (C) AUEC10 and (D) at 10 hours (basal‐adjusted percentage inhibition of newly made triglyceride‐palmitate is shown below graphs C and D). The box plots provide medians with 25% and 75% quartiles, with whiskers to the last point within 1.5 times the interquartile range. Diamonds represent the arithmetic means, and circles represent the outlier values. Basal‐adjusted results accounted for preoral fructose triglyceride‐palmitate, with basal results defined as the average of the measurements at −15 minutes and 0 hours on each day. AUEC10, area under the effect curve from 0 to 10 hours; CI, confidence interval; h, hours; PBO, placebo; Q12H, every 12 hours; QD, once daily; SEM, standard error of the mean.
Safety
All AEs reported upon initiation of PF‐05221304 or placebo were mild in severity, with the exception of 1 moderate AE (see below). No serious AEs, deaths, or severe AEs occurred. There was no clear evidence of a dose‐related increase in AE frequency, with increasing single (up to 240 mg) or repeated (up to 200 mg/day) PF‐05221304 doses.
In parts 1 and 3, AEs occurred in 5 and 3 subjects, respectively. In part 2, 26 subjects reported AEs during days 1‐13, of which headache was the most common (Table S1A). Following D2O and fructose administration, diarrhea was the most common AE (Table S1B); these AEs have been previously described.12
One moderate AE of decreased platelet count, with a maximum decline from day 0 of 53 000/µL in platelet count on day 13 + 24 hours, was noted in a 45‐year‐old man receiving PF‐05221304 100 mg every 12 hours in part 2. This subject was also noted to have elevated fasting (2.1‐fold) and postprandial (2.6‐fold) serum triglyceride levels, which were considered mild in intensity. The subject's platelet count had already decreased considerably before PF‐05221304 administration, from 194 000/µL at screening to 167 000/µL on day 0. Furthermore, his fasting triglyceride levels had risen between day −8 (55 mg/dL) and day 0 (142 mg/dL) and generally continued to rise until day 13 (297 mg/dL). Although these observations were not accompanied by any signs/symptoms, the subject was withdrawn after the day 13 morning dose. Ten days following discontinuation, his platelet count increased to that on day 0, and his fasting triglycerides returned to below day 0 levels.
In part 2, both fasting and postprandial serum triglyceride levels increased with repeated, daily PF‐05221304 doses ≥40 mg/day (Figure 4A, Table S2). There appeared to be a threshold effect, whereby triglyceride levels increased by a similar degree across all doses ≥40 mg/day, despite a 5‐fold increase in doses from 40 to 200 mg/day (Figure 4A). These increases were associated with ≥90% inhibition of hepatic DNL (Figure 4A). Other lipid levels, including total cholesterol, HDL‐C, and direct LDL‐cholesterol (LDL‐C), exhibited small, albeit statistically significant, changes, which were generally decreases (Table S3A).
Figure 4.
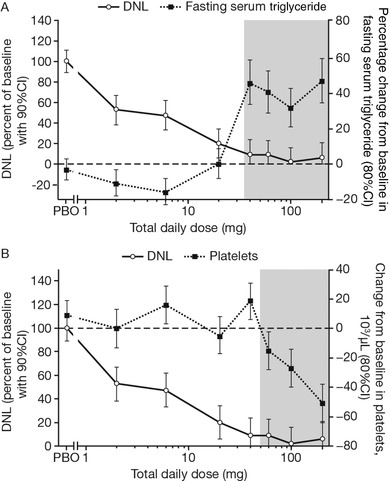
Basal‐adjusted change from baseline in DNL on day 14 in part 2, relative to the percentage change from baseline in fasting serum triglyceride on day 14 (A), and change from baseline in platelet counts on day 15 (B). Geometric mean is shown for serum triglyceride, and arithmetic mean for platelet count. Gray shading represents those doses at which elevated triglyceride levels from baseline (A) or declines in platelet count from baseline (B) were observed. CI, confidence interval; DNL, de novo lipogenesis; PBO, placebo; Q12H, every 12 hours; QD, once daily.
Platelet count did not change with single doses of PF‐05221304 across the 1‐ to 240‐mg dose range. In part 2, the arithmetic mean ± standard deviation (SD) platelet count at baseline ranged from 209 400/µL ± 35 800/µL to 255 100/µL ± 40 600/µL across the doses evaluated. Repeated dosing led to a gradual decline in platelet count from day 7 onward at PF‐05221304 doses ≥60 mg/day (Figure 5A, Table S3B); however, a platelet count below 100 000/µL was not observed. Decreases in platelet count were only observed with >90% inhibition of DNL (Figure 4B). Visual inspection did not suggest the attainment of a clear steady state for the effect of platelet count over the 14 days of dosing, although the decline reverted toward baseline by 8 ± 3 days after the last dose. Declines in platelet count were accompanied by increases in platelet volume apparent at doses ≥20 mg/day administered every 12 hours. For doses administered once daily, a decline in platelet count and increase in platelet volume were observed at 100 mg, but not 40 mg (Figure 5B). No clinical signs or symptoms due to platelet count reduction were observed; there was a lack of dose‐related trends in venipuncture bruising‐related AEs (eg, contusions) despite repeated direct venipunctures being performed for serial blood collections. There were no clinically significant changes in vital signs or cardiac conduction intervals as assessed via 12‐lead ECG (data not shown; a statement regarding the sponsor's policy on sharing clinical data appears at the end of this article).
Figure 5.
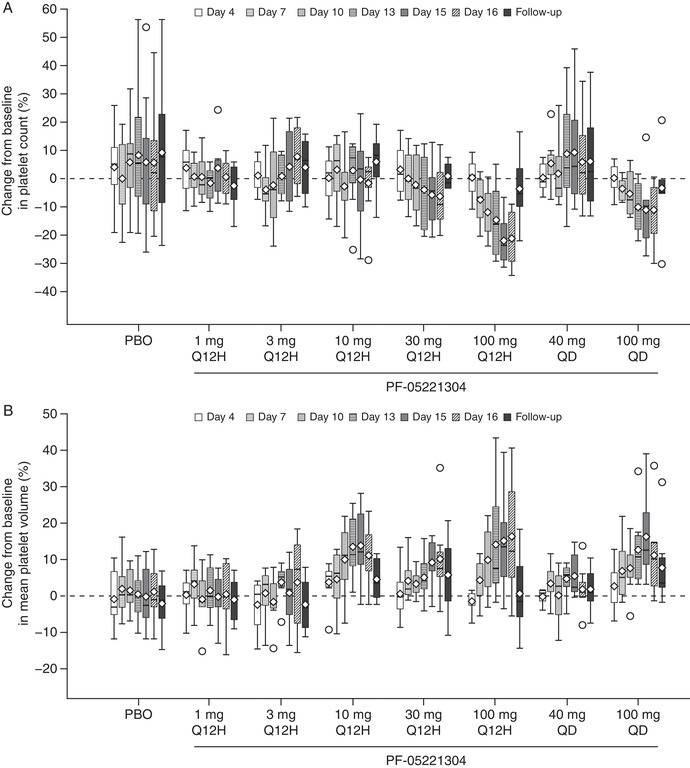
Percentage change from baseline in platelet count (A) and mean platelet volume (B) on repeated dosing. The box plots provide medians with 25% and 75% quartiles, with whiskers to the last point within 1.5 times the interquartile range. Diamonds represent the arithmetic means and circles represent the outliers. Unplanned readings were not included. Follow‐up visits were 8 ± 3 days after the last dose of study drug. PBO, placebo; Q12H, every 12 hours; QD, once daily.
Discussion
In the first clinical study to evaluate safety, tolerability, PK, and PD, PF‐05221304 was well tolerated with an acceptable safety profile, no serious or severe AEs, and no increase in AE severity associated with increasing dose, across the 240‐fold dose range studied. Althouh the maximum tolerated dose was not identified, repeated doses > 200 mg/day were not administered as near‐complete DNL was achieved with doses ≥40 mg/day. Plasma PK indicated a dose‐proportional increase in exposure up to 100 mg (single dose) and 200 mg/day (repeated doses). The observed t½ (14‐18 hours), robust inhibition of DNL with once‐daily dosing, and the modest effects of a high‐fat/high‐calorie meal on PK, indicate that PF‐05221304 can be dosed once daily, irrespective of food intake.
DNL is elevated approximately 3‐fold in patients with NAFLD relative to individuals with healthy/normal liver fat.21 Thus, DNL inhibition of approximately two‐thirds would be expected to return the elevated DNL rate observed in NAFLD to normal. PF‐05221304 achieved DNL inhibition of approximately 70% (up to ∼98%), consistent with projected efficacious doses based nonclinical PK and PD modelling. Similar efficacy data have been observed with other liver‐targeted ACC inhibitors, which have reported 70%‐104% DNL inhibition AUEC with a single‐dose ACC inhibitor in overweight healthy subjects12 and near maximal inhibition of DNL following repeated dosing in healthy subjects.13
No change in platelet count was observed with doses ≤40 mg/day, the doses at which ≤91% DNL inhibition occurred. An asymptomatic decrease in platelet count was observed with higher PF‐05221304 doses. Changes in platelet count and volume appear to be a result of ACC inhibition within the bone marrow, demonstrating the importance of DNL and ACC activity for megakaryocyte maturation and platelet production.16 Therefore, the asymptomatic decrease in platelet count observed at higher PF‐05221304 doses suggests that peripheral exposure at these doses was sufficient to at least partially inhibit DNL in tissues beyond the liver. An increase in mean platelet volume was also observed at some doses where platelet count had not declined, but the clinical relevance is unclear. Although no direct clinical consequence of decreased platelet count was observed in this study, it remains to be seen whether there is a potential adverse clinical consequence of reduced platelet count in the target patient population at efficacious doses.
Serum triglyceride levels increased with higher repeated PF‐05221304 doses. The increase in both fasting and postprandial serum triglyceride levels appeared to be a threshold effect at doses yielding ≥90% DNL inhibition in the first 10 hours of a dosing interval (ie, doses ≥40 mg/day) and suggest that near‐complete inhibition of ACC leads to other compensatory mechanisms, such as SREBP activation.13 A previous study reported an approximate 2‐fold increase in serum triglyceride levels after 4 weeks’ treatment with an ACC inhibitor in patients with hepatic steatosis.13 However, that study used a higher dose (400 mg/day) than the dose of the same ACC inhibitor used in a separate study (140 mg/day) that achieved near‐complete DNL inhibition in healthy subjects.13 Another study reported a 15%‐17% increase in triglycerides relative to placebo after 12 weeks’ treatment with an ACC inhibitor in patients with NASH, albeit at doses reported to inhibit DNL by ≤70% over a 10‐hour postdose period in healthy subjects.12, 22
Small changes in other lipid levels were noted; this has been observed with other ACC inhibitors where VLDL, LDL‐C, and HDL‐C levels increased13 or HDL‐C slightly decreased with no significant associations between lipid changes and changes in glycemic parameters or body weight.23 However, lipid monitoring is warranted to assess any atherogenic risk.
By establishing a dose response, we identified that PF‐05221304 doses <40 mg/day exhibited considerable (≤80%) hepatic DNL inhibition over a 10‐hour period postdose, but were not associated with declines in platelet count or elevation in serum triglyceride levels. To examine if a similar dose response occurs in patients, doses of 2, 10, 25, and 50 mg once daily are being assessed in the phase 2a dose‐ranging trial (ClinicalTrials.gov; NCT03248882).
Although this study adds to our published understanding of ACC inhibition, it has limitations. It was conducted in healthy subjects, so it is not known if the results translate to the target patient population. Repeated dosing was performed for 14 days, but changes in platelet count did not reach steady state over this period, warranting longer‐term studies in patients. DNL was assessed in healthy subjects under controlled conditions (10‐hour fructose dosing), and it is not known if DNL inhibition in the patient population will be similar without fructose loading. Last, additional studies are warranted to quantify the relationship between DNL inhibition and reductions in hepatic steatosis, downstream improvements in inflammation, hepatocyte ballooning, and fibrosis.
Conclusions
In conclusion, single and repeated doses of PF‐05221304 were well tolerated, with an acceptable safety profile seen in healthy subjects. Fructose‐stimulated hepatic DNL was inhibited by PF‐05221304, with near‐complete DNL inhibition at higher doses. The higher doses were accompanied by elevated serum triglyceride levels and declines in platelet count; these changes were not observed at lower doses at which DNL was inhibited by up to 80%, a level of target engagement that may be sufficient to normalize the elevated DNL seen in patients with NAFLD. These results support further clinical investigation of PF‐05221304 in the target patient population.
Conflicts of Interest
All authors are employees of Pfizer Inc, which funded this study.
Funding
This study was funded by Pfizer Inc.
Author Contributions
A.B., S.C.‐G., and N.B.A. wrote the article; A.B., S.C.‐G., W.P.E., and N.B.A. designed the research; A.B., S.C.‐G., S.T., A.R.S., and N.B.A. performed the research; and A.B., S.C.‐G., S.T., A.R.S., W.P.E., and N.B.A. analyzed the data. All authors critically reviewed the article and approved the final draft for submission.
Data Accessibility Statement
On request and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the United States and/or European Union or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions and for which an exception does not apply via a secure portal. To gain access, data requesters must enter into a data access agreement with Pfizer.
Supporting information
Figure S1. Chemical structure of PF‐05221304.
Table S1
Table S2
Table S3
Supporting Information
Acknowledgments
The authors thank the subjects who participated and the site staff who executed this study. The authors also thank Jill Sutt for contributions to the protocol and study conduct, Trenton Ross, David Beebe, and Nancy Raha for contributions to sample analysis to assess the degree of hepatic DNL inhibition (de novo lipogenesis), and Sakambari Tripathy for work on the PF‐05221304 PK assay. Medical writing support, under the direction of the authors, was provided by Kate Silverthorne, PhD, at CMC Connect, McCann Health Medical Communications, and was funded by Pfizer Inc, New York, New York, in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163[6]:461‐464).
None of the authors are members of the American College of Clinical Pharmacology.
References
- 1. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10(11):686‐690. [DOI] [PubMed] [Google Scholar]
- 2. LaBrecque DR, Abbas Z, Anania F, et al. World Gastroenterology Organisation global guidelines: nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Clin Gastroenterol. 2014;48(6):467‐473. [DOI] [PubMed] [Google Scholar]
- 3. Oseini AM, Sanyal AJ. Therapies in non‐alcoholic steatohepatitis (NASH). Liver Int. 2017;37(Suppl 1):97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asrih M, Jornayvaz FR. Diets and nonalcoholic fatty liver disease: the good and the bad. Clin Nutr. 2014;33(2):186‐190. [DOI] [PubMed] [Google Scholar]
- 5. Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(31):12587‐12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61(5):1282‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saggerson D. Malonyl‐CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253‐272. [DOI] [PubMed] [Google Scholar]
- 8. Abu‐Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl‐CoA carboxylase 2. Proc Natl Acad Sci U S A. 2000;97(4):1444‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 10. Thampy KG, Wakil SJ. Regulation of acetyl‐coenzyme A carboxylase. II. Effect of fasting and refeeding on the activity, phosphate content, and aggregation state of the enzyme. J Biol Chem. 1988;263(13):6454‐6458. [PubMed] [Google Scholar]
- 11. Thampy KG, Wakil SJ. Regulation of acetyl‐coenzyme A carboxylase. I. Purification and properties of two forms of acetyl‐coenzyme A carboxylase from rat liver. J Biol Chem. 1988;263(13):6447‐6453. [PubMed] [Google Scholar]
- 12. Stiede K, Miao W, Blanchette HS, et al. Acetyl‐coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: A randomized, double‐blind, crossover study. Hepatology. 2017;66(2):324‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim CW, Addy C, Kusunoki J, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26(2):394‐406 e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffith DA, Kung DW, Esler WP, et al. Decreasing the rate of metabolic ketone reduction in the discovery of a clinical acetyl‐CoA carboxylase inhibitor for the treatment of diabetes. J Med Chem. 2014;57(24):10512‐10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goedeke L, Bates J, Vatner DF, et al. Acetyl‐CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68(6):2197‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelly K, Reagan W, Sonnenberg G, et al. De novo lipogenesis is critical for platelet production; the liver directed ACC inhibitor PF‐05221304 for the treatment of NASH shows improved plately safety profile over systemic ACC inhibition [abstract]. Hepatology. 2019;70(S1):1272A. [Google Scholar]
- 17. Ross T, Kelly K, Rinaldi A, et al. The acetyl‐CoA carboxylase inhibitor PF‐05221304 exerts direct effects on hepatic inflammation and fibrosis independent of benefits on steatosis. J Hepatol. 2019;70(1):e86. [Google Scholar]
- 18.Pfizer. Data on file 2019. (unpublished).
- 19. Beysen C, Ruddy M, Stoch A, et al. Dose‐dependent quantitative effects of acute fructose administration on hepatic de novo lipogenesis in healthy humans. Am J Physiol Endocrinol Metab. 2018;315(1):E126‐E132. [DOI] [PubMed] [Google Scholar]
- 20. Strawford A, Antelo F, Christiansen M, Hellerstein MK. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. Am J Physiol Endocrinol Metab. 2004;286(4):E577‐588. [DOI] [PubMed] [Google Scholar]
- 21. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loomba R, Kayali Z, Noureddin M, et al. Acetyl‐CoA carboxylase inhibitor GS‐0976 leads to significant improvements in MRI‐PDFF in a phase 2, randomized, placebo‐controlled trial of patients with NASH. Hepatology. 2017;66(6):1709‐2102. [Google Scholar]
- 23. Lawitz EJ, Coste A, Poordad F, et al. Acetyl‐CoA carboxylase inhibitor GS‐0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2018;16(12):1983‐1991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemical structure of PF‐05221304.
Table S1
Table S2
Table S3
Supporting Information
Data Availability Statement
On request and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the United States and/or European Union or (2) in programs that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions and for which an exception does not apply via a secure portal. To gain access, data requesters must enter into a data access agreement with Pfizer.