

Abstract
Noble‐metal aerogels (NMAs) have drawn increasing attention because of their self‐supported conductive networks, high surface areas, and numerous optically/catalytically active sites, enabling their impressive performance in diverse fields. However, the fabrication methods suffer from tedious procedures, long preparation times, unavoidable impurities, and uncontrolled multiscale structures, discouraging their developments. By utilizing the self‐healing properties of noble‐metal aggregates, the freezing‐promoted salting‐out behavior, and the ice‐templating effect, a freeze–thaw method is crafted that is capable of preparing various hierarchically structured noble‐metal gels within one day without extra additives. In light of their cleanliness, the multi‐scale structures, and combined catalytic/optical properties, the electrocatalytic and photoelectrocatalytic performance of NMAs are demonstrated, which surpasses that of commercial noble‐metal catalysts.
Keywords: electrochemistry, gels, nanostructures, photocatalysis, sol–gel process
Coming in from the cold: By utilizing the freezing‐promoted salting‐out and the ice‐templating effect, a freeze–thaw method is developed to fabricate various noble‐metal hydrogels within one day. In light of their catalytically/optically active sites and multi‐scale structures, the impressive performance for electrocatalysis and photoelectrocatalysis is demonstrated for electro‐oxidation of ethanol.

Introduction
Aerogels are a large class of self‐supported porous materials with virtually unlimited compositions ranging from silica, polymers, nanocarbons, and semiconductors to metals.1, 2, 3, 4, 5, 6, 7 Especially for the recently emerging noble‐metal aerogels (NMAs), their combined features, including self‐supported architectures, high specific surface areas, 3D electron/mass transfer pathways, and numerous optically/catalytically active sites, have endowed them with broad application prospects such as in electrocatalysis, surface‐enhanced Raman scattering, and sensing.8, 9, 10 However, since their discovery in 2009,11 the development has been quite slow, which is presumably due to the great challenges encountered by the material fabrication.8
The aerogel is created following the route: solution → wet gel → aerogel, where the core lies in the sol–gel step. The majority of NMAs are created following this approach, producing widespread materials with tailored ligament sizes (that is, the diameter of the primary nanowires in the gel network), compositions, and microstructures. However, preparing wet gels usually requires extra additives (such as dopamine, salts, ligands, or oxidants12, 13, 14, 15, 16, 17, 18) or elevated temperatures,19, 20 thus introducing various impurities or forming less‐controlled microstructures. Additionally, long gelation times (up to a few weeks) and tedious procedures (such as ultracentrifugation) are also frequently encountered, thus impeding the material preparation.11, 21 Moreover, an efficient structure control over multiple scales, for example, from nanometers to micrometers, is still in absence.
In the field of polymer science, the freeze–thaw technique has been used to create the physical crosslinks in concentrated solutions by re‐organizing the arrangement of polymer chains, thus yielding the corresponding hydrogels (for example, poly(vinyl alcohol) (PVA) and starch gels).22, 23 A variant of this method, which is known as freeze casting, has been adopted to fabricate diverse aerogels made up of carbons and ceramics, where the precursor aqueous solution is directly freeze‐dried to form the corresponding aerogels.24, 25, 26 With this method, macro‐ and mesoscale structures of the resulting aerogels can be tailored to some extent, because of the templating effect from the ice crystals formed in situ. Recently, this method has been extended to NMA synthesis, where aerogels were produced by direct freeze‐drying of silver nanowires, gold nanowires, or various metal nanoparticle (NP) precursor solutions.27, 28, 29, 30 Nevertheless, this method does not fit well with noble‐metal systems, because highly concentrated precursor solutions (typically >100 mm) are required to support a monolithic aerogel, thus often rendering large ligament sizes (200–500 nm for gold aerogels). Additionally, because the gel formation and drying process are coupled, substantial ligands which have been introduced to stabilize the precursor solutions can inevitably contaminate the resulting aerogels. In this regard, a general method capable of synthesizing clean and structurally controllable NMAs is urgently desired, for both intrinsic properties investigations and application performance optimization.
Previously, we found that the as‐formed noble‐metal aggregates can maintain a long‐time reaction activity during the gelation process, that is, a self‐healing behavior.15, 31 Inspired by this fact, a freeze–thaw method was developed here as an additive‐free approach to directly destabilize various metal NP solutions (Au, Pd, Rh, Au‐Ag, Au‐Pd, Au‐Pt, and Au‐Rh, with metal salt concentrations c M=0.2–0.5 mm), thus creating the corresponding NMAs. Upon freezing, large aggregates are generated due to the intensified salting‐out effects incurred by the dramatically raised local solute concentration, and they are shaped concurrently at macroscale by in situ formed ice crystals. After thawing, they settle down and assemble to monolithic hydrogels in virtue of the self‐healing properties. Purified and dried, clean hydrogels and the corresponding aerogels are obtained, respectively. Ascribed to the hierarchically porous structures generated from a combined ice templating (macroscale) and aggregate stacking (meso‐ and microscale), the cleanliness, and the combined catalytic/optical properties, the resulting Au‐Pd and Au‐Pt aerogels display enhanced electrocatalytic and photoelectrocatalytic performance, which delivers a current density of up to 6.5 times higher than that of commercial Pd/C for the ethanol oxidation reaction (EOR). Therefore, the current work not only provides a new method to create clean and hierarchically structured NMAs directly from dilute precursor solutions, but also opens an avenue to probe their intrinsic electrocatalytic and photoelectrocatalytic performance. Moreover, the as‐developed strategy may also shed light on the fabrication of widespread clean hydrogels and aerogels with multi‐level structures for specific applications.
Results and Discussion
Fabrication of the Gold Hydrogels and Aerogels
As an example, the fabrication of gold aerogels is illustrated. In brief, a stable gold NP solution (0.5 mm) was prepared by reducing HAuCl4 solution with NaBH4 in absence of ligands. As seen from Figure 1 a, by directly freezing in liquid nitrogen (−196 °C), the original red solution turned to a dark‐gray solid. After sequentially thawing in air (10–20 °C) and water bath (25–30 °C), a black solution was obtained, which was further converted to a monolithic hydrogel after sedimentation. The freeze–thaw process can also be achieved in a few hours by using a fridge (−20 °C), while grounding above the freezing point (≥5 °C) will not trigger the reaction (Supporting Information, Figure S1). It is worth mentioning that the hydrogel presented in Figure 1 a is rather small because of the low precursor concentration and small volume, while an upscale production is possible which will be shown later. Subsequent purification, solvent‐exchange with tert‐butanol, and freeze‐drying eventually led to the corresponding gold aerogel. It is worth mentioning that the network structure is fixed at the hydrogel stage. The following freeze‐drying process with tert‐butanol as medium, which is regarded to accelerate the drying process and to suppress the crystal growth upon freezing32 is adopted to minimize the structure change and facilitate drying. Because organic ligands or extra initiators were not introduced, and a purification step was allowed to be inserted to remove highly water‐soluble small‐size inorganic ions (such as Na+, BH4 −, and Cl−) before freeze‐drying, clean aerogels were obtained by this freeze–thaw‐assisted process. The morphology of the resulting gold gel was revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), disclosing a distinct hierarchical structure as illustrated in Figure 1 b,c. Differing from various previously reported NMAs featuring a single‐level structure,11, 12, 15, 18, 20 the combined SEM and TEM images clearly reflected an eminent hierarchical morphology shaped by ice crystals, where extended 3D‐branched networks were observed at the microscale while compact aggregates were observed at the macroscale.
Figure 1.
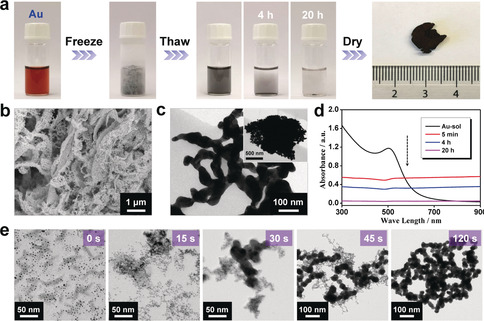
The freeze–thaw‐directed fabrication of gold gels. a) Photographs of various stages of the freeze–thaw‐directed gelation process of the Au NP solutions (c M=0.5 mm) and the corresponding aerogels. Liquid nitrogen is used for freezing. b) SEM image and c) TEM images of the obtained gold aerogel. d) Time‐lapse UV/Vis absorption spectra of the original Au NP solution, and the solution after freezing for 5 min and thawing for different periods of time. e) TEM images of original Au NP solution (freezing for 0 s), and the solution after freezing for 15 s, 30 s, 45 s or 120 s followed by thawing for about 5 min.
The aforementioned process was characterized in situ by time‐lapse UV/Vis absorption spectroscopy (Figure 1 d). The characteristic absorption peak of gold NPs centering at 502 nm disappeared after thawing. Instead, a featureless absorption spanning over the entire wavelength range was observed, suggesting the formation of large nanostructured aggregates.33 The gelation process was further unveiled by comparing the morphologies of as‐prepared gold NPs, and the formed aggregates after freezing for different periods of time. As displayed in Figure 1 e, upon freezing, two processes took place: 1) original small and separate Au NPs (size 2–5 nm) partially grew to form large NPs (size 20–40 nm) and 2) large NPs fused to form branched‐nanowire‐like gel networks. This is different from the phenomena observed in salt‐induced gelation process,15 where the gel network instantly formed upon initiation, and the ligament size of the as‐formed network gradually increased with elongated reaction time. Notably, since the multimetallic gels were prepared from multimetallic NPs (Supporting Information, Figure S2), the corresponding gel formation mechanism should be similar. After the freeze–thaw process, as‐formed large aggregates were driven to sediment by gravity owing to the large bulk density of gold (19.3 g cm−3),15 corresponding to the decrease of the absorption intensity with prolonged time. Taking advantage of their long‐term reaction activity, after automatically concentrating at the bottom along with sedimentation, these highly concentrated aggregates assembled together to a monolithic gold hydrogel with a volume much smaller than the initial NP solution.15
Mechanisms of Freeze–Thaw‐Promoted Gelation
For all of the previously reported freeze–thaw and freeze‐casting methods, a sufficiently high precursor concentration is required. Upon freezing, solutes are expelled to the boundaries between adjacent ice crystals, thus being forced to form a continuous 3D network templating ice crystals. During drying in vacuum, the ice crystals sublimate and the corresponding monolithic aerogels are obtained. However, for noble‐metal systems, organic ligands are inevitably introduced to acquire highly concentrated and stable precursor solutions (c M>100 mm), thus remaining in the final aerogel. Additionally, a high metal concentration can substantially increase the ligament size such as to 200–500 nm for the gold NP system28), considerably reducing the number of exposed reaction sites.
Previously we found that a high concentration of salt can destabilize noble‐metal NP solutions to form the corresponding hydrogels, during which process the metal aggregates exhibit a long‐term reactivity, that is, a self‐healing behavior.15, 31 On this basis, we propose that the freeze–thaw method can directly work on the dilute metal NP solutions (c M≤0.5 mm). As shown in Figure 2 a, i) starting from a gold NP solution, ii) upon freezing, the considerably reduced liquid water dramatically increases the local concentration of solutes (gold NPs, certain ions (Na+, Cl−, BH4 −, and BO2 −) resulting from HAuCl4 and NaBH4 for the NP synthesis), thus inducing a salting‐out destabilization process and generating small aggregates shaped by adjacent ice crystals; iii) after thawing, the as‐formed aggregates sediment driven by gravity,15 iv) eventually concentrating at the bottom of the vessel, where the gelation threshold is reached and a monolithic gel is acquired accordingly. It is worth mentioning that upon freezing (step II), several factors including the temperature‐dependent Brownian motion, the increasing concentration of NPs, and the increasing concentration of ions can play roles. First, previously, we found that a low temperature only retards the reaction kinetics while not affecting the network structure and ligament sizes.34 Additionally, freezing either in liquid nitrogen (−196 °C) or in a fridge (−20 °C) can produce gels (Figure 1 a; Supporting Information, Figure S1). Hence, given that the temperature is below the freezing point, it should have a minimal effect on the gelation mechanism. Second, the raising NP concentration can shorten the distance between the NPs, which will increase both the frequency of NP collisions and the van der Waals (vdW) attraction. The potential of short‐range vdW attraction (in proportion to r −6) will overwhelm the long‐range electrostatic repulsion between charged NPs (in proportion to r −1) with decreasing distance, thus overcoming the kinetically unfavorable aggregation process in the original colloidal solution. Third, the increase of the ion concentration upon freezing will activate the salting‐out effect, thereby triggering the gelation process as reported previously.15 As a proof, strong stabilizers can suppress the freezing‐induced destabilization (Supporting Information, Figure S3), which behaves similar to the salting‐out method.15 When the effects from increasing NP and ion concentrations overwhelm the low‐temperature‐induced sluggish kinetics, the gelation will occur.
Figure 2.
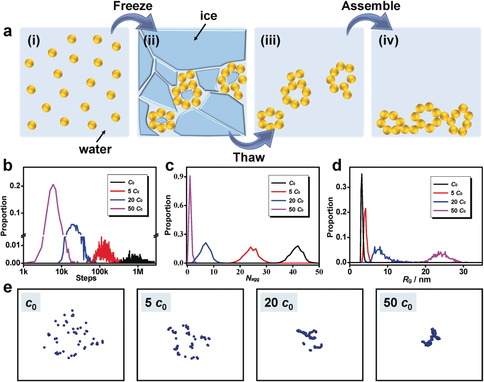
Proposed mechanisms of the freeze–thaw‐directed gelation process. a) Model for the gelation process, following (i) the original NP solution, (ii) the freeze‐induced assembly, (iii) the thaw‐induced unlocking of aggregates, and (iv) the gravity‐ and self‐healing‐driven gel formation. b)–d) MC simulations for systems with various particle concentrations (c 0, 5 c 0, 20 c 0, and 100 c 0): The distributions of b) the required steps to form one single aggregate, c) the number of remaining aggregates (N agg) after 10 000 MC steps, and d) the average radius of gyration (R g) of the resulting aggregates after 10 000 MC steps. e) Typical MC snapshots of the systems with indicated particle concentrations after 10 000 MC steps.
In this way, noble‐metal hydrogels were created by the freeze–thaw process at an initial metal concentration down to 0.2 mm, which is 2–3 orders of magnitude lower than that for the previously reported freeze‐casting method.26, 28, 29, 30 Without introduction of any ligand or additive, the gold gel was obtained, featuring a ligament size of 35.0±7.2 nm, which is about one order of magnitude smaller than the previously reported value as fabricated by freeze‐casting.28 Additionally, the ice‐templating further shapes the macrostructures of the gel, thus giving rise to a distinct hierarchical structure as evidenced from the SEM and TEM images (Figure 1 c,d). A full understanding of this gelation process in combination with precise theoretical modelling would indeed be beneficial, while it is beyond the scope of the current work.
To intuitively demonstrate the effect of freezing in the gelation process, Monto Carlo simulations can serve as a useful tool.35 Considering the calculation costs and limited available parameters, here only one of the most pronounced features, that is, the increasing concentration of the NPs, was simulated to give an intuitive impression on the freeze‐promoted gelation process without involving other interactions. Briefly, the MC algorithm lets non‐interacting particles undergo a random‐walk procedure in a spherical space. Particles that meet during movement are considered as rigid aggregates afterwards. The algorithm is terminated when all particles are aggregated to one single aggregate, and statistical analysis was performed over 1000 simulations per parameter set. To simulate the freezing effect, four systems with the same number of particles while confined in different spatial volumes (corresponding to different particle concentrations, that is, c 0, 5 c 0, 20 c 0, and 50 c 0) were adopted for comparison, where the freezing degree is positively correlated to the particle concentrations. As seen from Figure 2 b, to form one single aggregate, the number of required MC steps decreases significantly with increasing particle concentrations, implying that a freezing state can considerably promote the aggregate formation. This is also supported by analyzing the number of remaining aggregates (N agg) and the average radius of gyration (R g) of the aggregates after 10 000 MC steps for different particle concentrations. As illustrated in Figures 2 c,d, a smaller N agg and a larger R g are observed for the high‐particle‐concentration scenario, indicating that the gelation speed is substantially accelerated and the large aggregates quickly form in the freezing system. More intuitively, typical populations of the four systems after 10 000 MC steps are presented in Figure 2 e, where the particles in the low‐concentration‐system (c 0) are almost stay in the original state, while the particles in the high‐concentration system (50 c 0) have fully reacted and formed a single aggregate. These simulations can qualitatively demonstrate the underlying mechanisms for the freeze–thaw triggered gelation process.
Generality of the Freeze–Thaw‐Promoted Gelation Method
To modulate the ligament size, the specific ion effects, which had been reported previously,15 were combined with the current freeze–thaw method. As shown in Figure 1 a, without any additives, the NP solution cannot fully react as evidenced from the light red color of the solution. In contrast, a very small amount of inorganic salts (concentration of 0.2–5.0 mm) can considerably promote the gelation (Supporting Information, Figures S4–S6), in which the concentration is much below the gelation threshold (10–60 mm, without freeze–thaw treatment) and substantially smaller than that of the report elsewhere (30–100 mm).15 In agreement with the previous findings, the ligament size of the aerogel is tunable from 16.8 to 32.3 nm depending on the salts used (Figure 3 a), which indicates the good compatibility of the freeze–thaw process with other methods. This salt‐directed manipulation of the ligament size can be attributed to the different interactions between various ions and gold NPs.15
Figure 3.
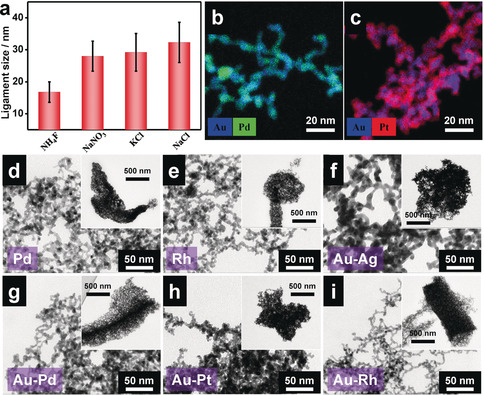
Manipulation of the noble‐metal aerogels. a) The ligament size of gold aerogels with respect to the presence of specific salts during the freeze–thaw process. b),c) EDX mappings of b) Au‐Pd and c) Au‐Pt aerogels. d)–i) TEM images of NMAs with different compositions, d) Pd, e) Rh, f) Au‐Ag, g) Au‐Pd, h) Au‐Pt, and i) Au‐Rh. Insets are low‐magnification TEM images, reflecting the closely packed morphologies.
Apart from the structure control, the freeze–thaw‐promoted gelation method is valid for preparing diverse NMAs with compositions ranging from Pd, Rh, Au‐Ag, Au‐Pd, Au‐Pt, to Au‐Rh (Supporting Information, Figure S7). In contrast, without applying the freeze–thaw treatment, all NP solutions remained stable or formed unsupported powders after long‐term standing (Supporting Information, Figure S8). The resulting NMAs were examined by various techniques (Figure 3 b–i; Supporting Information, Figure S9, Table S1). TEM images revealed that all aerogels manifested branched‐network structures at the micro‐/mesoscale (<50 nm) and compact structures at a larger scale, in agreement with the SEM observations (Figure 3 d–i; Supporting Information, Figure S9). These hierarchical structures are similar to the gold system as mentioned in the previous section, which are dictated by the ice‐templating. The ligament sizes ranges from 3.3 to 7.2 nm, depending on the specific compositions (Supporting Information, Table S1). It needs to be mentioned that, as stated before, the hydrogel matrix was fixed during the freeze–thaw process, and the following drying method (freeze drying or critical‐point drying) does not affect the morphology (Supporting Information, Figure S10).
Among these aerogels, bimetallic Au‐Pd and Au‐Pt aerogels were particularly studied for their potential electrocatalytic and photoelectrocatalytic activities, which will be elaborated in the next section (Supporting Information, Figures S11–S13). The freeze–thaw method can produce monolithic hydrogels on a large scale, allowing to obtain the corresponding aerogels at a reasonable yield (75.4±4.2 %) and to estimate their densities (35.9±4.0 and 60.6±5.5 mg cm−3 for the Au‐Pd and the Au‐Pt aerogel, respectively). Powder X‐ray diffraction (XRD) confirms the presence and the alloying of the two metals (Supporting Information, Figures S12). High‐resolution TEM images show that Au‐Pd and Au‐Pt aerogels feature a high crystallinity with randomly distributed grain orientations (Supporting Information, Figure S13). From energy‐dispersive X‐ray spectroscopy (EDX) mapping (Figure 3 b,c; Supporting Information, Figures S14, S15), as observed previously,15 various types of spatial element distributions were obtained such as relatively homogeneous distributions for the Au‐Pd and Au‐Rh aerogels, a rather inhomogeneous element distribution for the Au‐Pt aerogel, and a core–shell structure for the Au‐Ag aerogel. The reason of the core–shell architecture of the Au‐Ag aerogel can be attributed to the segregation of relatively low‐surface‐energy silver on the surface of relatively high‐surface‐energy gold, as reported elsewhere.36 Furthermore, hierarchically structured pores, which are mainly made up of mesopores (2–50 nm, seen from the pore size distribution) and macropores (>50 nm, seen from SEM and TEM observations), were found to afford substantially large Brunauer–Emmett–Teller (BET) surface areas (83.6 and 74.5 m2 g−1) and pore volumes (0.526 and 0.475 cm3 g−1) for the Au‐Pd and the Au‐Pt aerogels, respectively (Supporting Information, Figure S16, Table S1). These SSAs are larger than those of most reported NMAs (10–50 m2 g−1)11, 13, 14, 20, 28, 37, 38 and only smaller than a few, such as the Pd aerogels prepared by Ca2+ crosslinking (40–108 m2 g−1),39 Au‐Pd‐Pt aerogels prepared by salting‐out method (122.7 m2 g−1),15 and the hierarchical silver aerogels prepared by (C(NO2)4‐mediated oxidation (43–160 m2 g−1) (Supporting Information, Table S2).16 The synergy of a large specific surface area and a large pore volume can concurrently provide abundant active sites and fast mass‐transfer channels, which will be beneficial for catalytic applications as will be detailed in the next section.
Electrocatalytic and Photoelectrocatalytic Performance
Featuring hierarchically porous structures, stable and conductive 3D networks, as well as unique catalytic/optic properties, the as‐prepared Au‐Pd and Au‐Pt aerogels were examined for electrocatalytic and photoelectrocatalytic applications by using the ethanol oxidation reaction (EOR) as a model reaction. Here the gold, as the second most conductive metal only inferior to silver, can provide high stability, outstanding electrical conductivity as well as pronounced plasmonic effects, while the Pd or Pt can provide excellent catalytic properties. Notably, although the aerogel catalyst ink was prepared by sonication (in a similar way to prepare TEM samples), the networked structure was preserved at the micrometer‐scale (Supporting Information, Figure S17), which facilitates inheriting the structural advantages of the aerogels.
As illustrated in Figure 4 a, the peaks during the forward scan and the backward scan denote the oxidation of the ethanol and the oxidation of the intermediates generated during the forward scan, respectively.18, 20, 40 The Au‐Pt aerogel and the Au‐Pd aerogel exhibit forward current densities (I f) of 3.5 and 8.4 A mg−1, respectively, much larger than those of commercial Pt/C (0.9 A mg−1) and Pd/C (1.8 A mg−1). This can be attributed to the hierarchical structures of aerogels, which ensures both the smooth mass transfer and the abundant catalytic sites.
Figure 4.
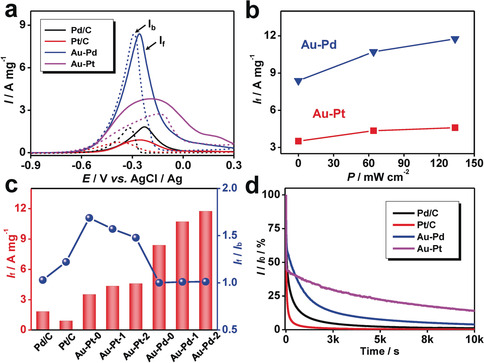
Application of NMAs in electrocatalysis and photoelectrocatalysis. a) Electrocatalytic performance of the EOR with commercial and various aerogel catalysts in dark condition. The solid and dashed lines represent the forward and backward scans, respectively. b) The dependence of I f for the Au‐Pd and Au‐Pt aerogels on the power density of the light. c) Summarized I f and I f/I b of various catalysts. The suffix 0, 1, and 2 denotes that the test was conducted under a light illumination with the power density of 0, 64.1, and 133.6 mW cm−2, respectively. d) The current retention of commercial and various aerogel catalysts under −0.23 V vs. AgCl/Ag in dark condition. All tests were performed in nitrogen‐saturated 1.0 m KOH+1.0 m ethanol aqueous solution.
Noble‐metal nanostructures, especially for Au and Ag, feature unique plasmonic properties due to the remarkable electromagnetic field enhancement confined at the nanoscale. Although photoelectrocatalysis using noble metals has been extensively studied in the metal‐semiconductor hybrid systems, the plasmon‐driven photoelectrocatalysis on sole noble metals is only considered very recently.33 Additionally, multiple absorption and scattering of incident light in the nanostructured NMAs can induce heating effect, which may also enhance catalysis. Based on these two mechanisms, here the EOR is taken as a model reaction to investigate the photoelectrocatalytic performance of NMAs. As seen from Figure 4 b and the Supporting Information, Figure S18, light irradiation promoted the EOR electrocatalytic current for both the Au‐Pt and the Au‐Pd aerogel, where 20.7 % and 40.2 % enhancement were observed (at a light power density of 133.6 mW cm−2), respectively. Since most NMAs show considerable broad‐band plasmonic absorption in the visible‐light region, the light‐directed promotion revealed here can serves as a general approach to considerably boost the electrocatalytic performance of various NMA systems. Because of the broad‐band absorption of the NMAs and the application of a white light source (wavelength 350–800 nm), the enhancing mechanisms can be attributed to either hot‐carrier transfer, the plasmonic thermal effects, or the thermal effect resulting from multiple light absorption/scattering inside the nanostructured gel networks.30, 31, 41
For an intuitive comparison, the forward current density (I f, normalized by the mass of the active species (Pd or Pt)) and the ratio of the forward/backward current density (I f/I b) are summarized in Figure 4 c. All aerogel‐based catalysts display a considerably higher I f than that of the commercial noble‐metal catalysts (Pd/C and Pt/C). Especially, as further enhanced by light irradiation, the I f of the Au‐Pd aerogel is 6.5 times higher than that of Pd/C, and the one for Au‐Pt aerogel is 5.1 times higher than that of Pt/C, which outperforms previously reported Au‐Pd aerogels (2.9 times better than Pd/C15) and various other NMAs (e.g. Pd, Pd‐Cu, Pd‐Ni, Au‐Pd‐Pt aerogels, ca. 2.0 to 6.1 times better than Pd/C15, 17, 18, 19, 20). The high performance delivered by the current materials can be attributed to the synergy of the catalytic activity of Pd or Pt, the high electrical conductivities as well as the strong plasmonic absorption of Au, and the smooth mass transfer channels enabled by hierarchical structures of aerogels. Considering I f/I b, despite a lower I f, Pt‐based catalysts (commercial Pt/C (1.22) and the Au‐Pt aerogel (1.69)) are superior than Pd‐based catalysts (commercial Pd/C (1.03) and the Au‐Pd aerogel (1.0)), which is in line with previous reports.15 This might be attributed to that Pt can promote a more complete oxidation of ethanol during the forward scan. However, for the Au‐Pt aerogel, I f/I b decreases with increasing light power density (from 1.69 to 1.48). Finally, various catalysts were evaluated at a constant potential (−0.23 V vs. AgCl/Ag; Figure 4 d), where both Au‐Pt and Au‐Pd aerogels feature a substantially better stability than that of the commercial Pd/C or Pt/C (11–28 % vs. 0–4 % at 3000 s). This can be attributed to the stable 3D networks of the aerogels, avoiding the detachment of catalysts.
Conclusion
Various synthesis challenges in the field of noble‐metal aerogels have discouraged the evaluation of the intrinsic properties as well as the optimization of the application performance. Taking advantage of the self‐healing behavior of noble‐metal aggregates, freezing‐induced phase separation, and the ice‐templating effect, here a freeze–thaw method has been proposed capable of directly preparing various noble‐metal hydrogels within only one day from the corresponding dilute solutions (Au, Pd, Rh, Au‐Ag, Au‐Pd, Au‐Pt, and Au‐Rh, with metal salt concentration c M≤0.5 mm) without introducing extra additives. The gelation mechanism is interpreted by the salting‐out effect induced by a dramatically increased solute concentration upon freezing, as partially reflected by corresponding MC simulations. In light of their multi‐scale structures, the cleanliness, and the combined catalytic/optic properties, the intrinsic electrocatalytic and light‐driven photoelectrocatalytic performance of the Au‐Pd and Au‐Pt aerogels were investigated for the electro‐oxidation of ethanol, delivering a current density of up to 6.5 times higher than that of commercial Pd/C catalysts. This can be attributed to a combination of the catalytic activity of Pd or Pt, the high conductivity as well as the plasmonic absorption of Au, and the hierarchical porous structure of the aerogels. Therefore, the present work not only overcomes the challenges for creating clean and hierarchically structured NMAs, but also demonstrates their impressive intrinsic electrocatalytic and photoelectrocatalytic performance, which may open up new space for both the fundamental and the application oriented studies of NMAs. Moreover, the as‐developed strategy may shed light on the fabrication of widespread clean and multi‐level‐structured hydrogels, aerogels, and foams for specific applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.D. acknowledges the support from the Alexander von Humboldt Foundation. We thank Shishu Zhang for assisting in the XPS measurements, and the use of HZDR Ion Beam Center TEM facilities is acknowledged. The computations were performed on an HPC system at the Center for Information Services and High Performance Computing (ZIH) at TU Dresden within the project QDSIM. This work was supported by the research funding of the Humboldt Fellowship, ERC AdG AEROCAT, German Federal Ministry of Education of Research (BMBF, Grant No. 03SF0451), the National Natural Science Foundation of China (51602226), and the Natural Science Foundation of Zhejiang Province (LY19E020008).
R. Du, J.-O. Joswig, R. Hübner, L. Zhou, W. Wei, Y. Hu, A. Eychmüller, Angew. Chem. Int. Ed. 2020, 59, 8293.
Contributor Information
Dr. Ran Du, Email: dr1581@foxmail.com.
Prof. Dr. Alexander Eychmüller, Email: alexander.eychmueller@chemie.tu-dresden.de.
References
- 1. Kistler S. S., Nature 1931, 127, 741–741. [Google Scholar]
- 2. Hüsing N., Schubert U., Angew. Chem. Int. Ed. 1998, 37, 22–45; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 22–47. [Google Scholar]
- 3. Du R., Zhao Q., Zhang N., Zhang J., Small 2015, 11, 3263–3289. [DOI] [PubMed] [Google Scholar]
- 4. Ziegler C., Wolf A., Liu W., Herrmann A.-K., Gaponik N., Eychmüller A., Angew. Chem. Int. Ed. 2017, 56, 13200–13221; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13380–13403. [Google Scholar]
- 5. Du R., Zhao Q., Zheng Z., Hu W., Zhang J., Adv. Energy Mater. 2016, 6, 1600473. [Google Scholar]
- 6. Cai B., Eychmüller A., Adv. Mater. 2019, 31, 1804881. [DOI] [PubMed] [Google Scholar]
- 7. Du R., Zheng Z., Mao N., Zhang N., Hu W., Zhang J., Adv. Sci. 2015, 2, 1400006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du R., Fan X., Jin X., Hübner R., Hu Y., Eychmüller A., Matter 2019, 1, 39–56. [Google Scholar]
- 9. Cai B., Sayevich V., Gaponik N., Eychmüller A., Adv. Mater. 2018, 30, 1707518. [DOI] [PubMed] [Google Scholar]
- 10. Du R., Jin X., Hübner R., Fan X., Hu Y., Eychmüller A., Adv. Energy Mater. 2020, 10, 1901945. [Google Scholar]
- 11. Bigall N. C., Herrmann A. K., Vogel M., Rose M., Simon P., Carrillo-Cabrera W., Dorfs D., Kaskel S., Gaponik N., Eychmüller A., Angew. Chem. Int. Ed. 2009, 48, 9731–9734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9911–9915. [Google Scholar]
- 12. Wen D., Liu W., Haubold D., Zhu C., Oschatz M., Holzschuh M., Wolf A., Simon F., Kaskel S., Eychmüller A., ACS Nano 2016, 10, 2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ranmohotti K. G., Gao X., Arachchige I. U., Chem. Mater. 2013, 25, 3528–3534. [Google Scholar]
- 14. Naskar S., Freytag A., Deutsch J., Wendt N., Behrens P., Köckritz A., Bigall N. C., Chem. Mater. 2017, 29, 9208–9217. [Google Scholar]
- 15. Du R., Hu Y., Hübner R., Joswig J.-O., Fan X., Eychmüller A., Sci. Adv. 2019, 5, eaaw4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao X., Esteves R. J., Luong T. T. H., Jaini R., Arachchige I. U., J. Am. Chem. Soc. 2014, 136, 7993–8002. [DOI] [PubMed] [Google Scholar]
- 17. Nahar L., Farghaly A. A., Esteves R. J. A., Arachchige I. U., Chem. Mater. 2017, 29, 7704–7715. [Google Scholar]
- 18. Liu W., Herrmann A. K., Geiger D., Borchardt L., Simon F., Kaskel S., Gaponik N., Eychmüller A., Angew. Chem. Int. Ed. 2012, 51, 5743–5747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5841–5846. [Google Scholar]
- 19. Cai B., Wen D., Liu W., Herrmann A. K., Benad A., Eychmüller A., Angew. Chem. Int. Ed. 2015, 54, 13101–13105; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13293–13297. [Google Scholar]
- 20. Zhu C., Shi Q., Fu S., Song J., Xia H., Du D., Lin Y., Adv. Mater. 2016, 28, 8779–8783. [DOI] [PubMed] [Google Scholar]
- 21. Liu W., Rodriguez P., Borchardt L., Foelske A., Yuan J., Herrmann A. K., Geiger D., Zheng Z., Kaskel S., Gaponik N., Angew. Chem. Int. Ed. 2013, 52, 9849–9852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10033–10037. [Google Scholar]
- 22. Stauffer S. R., Peppast N. A., Polymer 1992, 33, 3932–3936. [Google Scholar]
- 23. Baker L. A., Rayas-Duarte P., Cereal Chem. 1998, 75, 301–307. [Google Scholar]
- 24. Gutiérrez M. C., Ferrer M. L., del Monte F., Chem. Mater. 2008, 20, 634–648. [Google Scholar]
- 25. Qiu L., Liu J. Z., Chang S. L., Wu Y., Li D., Nat. Commun. 2012, 3, 1241. [DOI] [PubMed] [Google Scholar]
- 26. Gao H. L., Xu L., Long F., Pan Z., Du Y. X., Lu Y., Ge J., Yu S. H., Angew. Chem. Int. Ed. 2014, 53, 4561–4566; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4649–4654. [Google Scholar]
- 27. Sun H. Y., Xu Z., Gao C., Adv. Mater. 2013, 25, 2554–2560. [DOI] [PubMed] [Google Scholar]
- 28. Freytag A., Sánchez-Paradinas S., Naskar S., Wendt N., Colombo M., Pugliese G., Poppe J., Demirci C., Kretschmer I., Bahnemann D. W., Behrens P., Bigall N. C., Angew. Chem. Int. Ed. 2016, 55, 1200–1203; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1217–1221. [Google Scholar]
- 29. Qian F., Lan P. C., Freyman M. C., Chen W., Kou T., Olson T. Y., Zhu C., Worsley M. A., Duoss E. B., Spadaccini C. M., Baumann T., Han T. Y.-J., Nano Lett. 2017, 17, 7171–7176. [DOI] [PubMed] [Google Scholar]
- 30. Qian F., Troksa A., Fears T. M., Nielsen M. H., Nelson A. J., Baumann T. F., Kucheyev S. O., Han T. J.-J., Bagge-Hansen M., Nano Lett. 2019, 10.1021/acs.nanolett.9b03445. [DOI] [PubMed] [Google Scholar]
- 31. Du R., Joswig J.-O., Fan X., Hübner R., Spittel D., Hu Y., Eychmüller A., Matter 2020, 10.1016/j.matt.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inoué T., Osatake H., Arch. Histol. Cytol. 1988, 51, 53–59. [DOI] [PubMed] [Google Scholar]
- 33. Søndergaard T., Novikov S. M., Holmgaard T., Eriksen R. L., Beermann J., Han Z., Pedersen K., Bozhevolnyi S. I., Nat. Commun. 2012, 3, 969. [DOI] [PubMed] [Google Scholar]
- 34. Georgi M., Klemmed B., Benad A., Eychmüller A., Mater. Chem. Front. 2019, 3, 1586–1592. [Google Scholar]
- 35. Bieri M., Nguyen M.-T., Gröning O., Cai J., Treier M., Aït-Mansour K., Ruffieux P., Pignedoli C. A., Passerone D., Kastler M., J. Am. Chem. Soc. 2010, 132, 16669–16676. [DOI] [PubMed] [Google Scholar]
- 36. Feng J., Yin Y., Adv. Mater. 2019, 31, 1802349. [DOI] [PubMed] [Google Scholar]
- 37. Burpo F. J., Nagelli E. A., Morris L. A., McClure J. P., Ryu M. Y., Palmer J. L., J. Mater. Res. 2017, 32, 4153–4165. [Google Scholar]
- 38. Shi Q., Zhu C., Zhong H., Su D., Li N., Engelhard M. H., Xia H., Zhang Q., Feng S., Beckman S. P., Du D., Lin Y., ACS Energy Lett. 2018, 3, 2038–2044. [Google Scholar]
- 39. Wen D., Herrmann A.-K., Borchardt L., Simon F., Liu W., Kaskel S., Eychmüller A., J. Am. Chem. Soc. 2014, 136, 2727–2730. [DOI] [PubMed] [Google Scholar]
- 40. Du R., Wang J., Wang Y., Hübner R., Fan X., Senkovska I., Hu Y., Kaskel S., Eychmüller A., Nat. Commun. 2020, 11, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Z., Zhang C., Zheng H., Xu H., Acc. Chem. Res. 2019, 52, 2506–2515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary