

Abstract
High‐grade periodate is relatively expensive, but is required for many sensitive applications such as the synthesis of active pharmaceutical ingredients. These high costs originate from using lead dioxide anodes in contemporary electrochemical methods and from expensive starting materials. A direct and cost‐efficient electrochemical synthesis of periodate from iodide, which is less costly and relies on a readily available starting material, is reported. The oxidation is conducted at boron‐doped diamond anodes, which are durable, metal‐free, and nontoxic. The avoidance of lead dioxide ultimately lowers the cost of purification and quality assurance. The electrolytic process was optimized by statistical methods and was scaled up in an electrolysis flow cell that enhanced the space–time yields by a cyclization protocol. An LC‐PDA analytical protocol was established enabling simple quantification of iodide, iodate, and periodate simultaneously with remarkable precision.
Keywords: boron-doped diamond, electrolysis, flow chemistry, oxidation, periodate
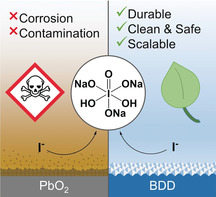
The electrochemical synthesis of periodate from iodide at boron‐doped diamond (BDD) anodes was developed, which in contrast to lead dioxide anodes are sustainable and durable. Toxic contaminations are hereby prevented, ultimately reducing the cost of purification. This opens the door for the broad application of periodate for demanding applications, such as the synthesis of active pharmaceutical ingredients.
Periodate has emerged as an important oxidizing agent in organic synthesis.1 It is used in the Malaprade oxidation for the cleavage of vicinal glycols, and as a primary oxidant in OsO4‐ or RuO4‐catalyzed transformations, such as in the Lemieux–Johnson oxidation.2 More recently, it has been used for difficult iodinations of alkenes and arenes,3 for the preparation of aryl boronic acids,4 and for the cleavage of allyl protecting groups.5 It was also used in the synthesis of N‐protected α‐ and β‐amino acids,6 as well as for vinyl sulfones.7 Therefore, periodate enables a rich chemistry in the synthesis of active pharmaceutical ingredients (APIs).8
Unfortunately, periodate is expensive and less readily available, which impedes most technical applications. Ph.Eur.‐grade periodate is not available and BioUltra‐grade periodate costs approximately 274 € per mole. Periodate in lower quality is indeed less costly and more available (109 € per mole, ACS), but might be contaminated with traces of highly toxic metals that are strongly regulated for pharmaceuticals.9 The high costs can be attributed to the purification protocols. Periodate is mostly generated electrochemically, which is the method with the lowest cost and environmental impact, and which is considered to be inherently safe.10 The regeneration of hypervalent iodine reagents can be achieved electrochemically and represents a hot topic.11 However, lead dioxide is generally used as the anode, due to electrocatalytic effects and a high overpotential for the oxygen evolution in aqueous media.12 It is known to disintegrate slowly during electrolysis which causes contaminations.13 The mass loss of the anodes is up to 2.5 g (A h)−1, and even the loss of particles was reported.14, 15 The toxicity of lead is inacceptable for regulated products, and its removal is cumbersome and economically prohibitive. Thus, the utilization of lead‐related anodes for such purposes increases the costs for purification, quality assurance, anode maintenance, and idle time.16 Alternative anode materials, such as platinum, nickel, manganese, titanium, graphite, oxides of ruthenium, and iridium have been investigated, but showed poorer performance and inferior durability in general.17, 18, 19 Further drawbacks are the use of toxic additives and expensive starting materials. In the most advanced synthetic access, periodate is directly generated from iodide, which is currently the source for commercial scale with the lowest molar cost in high quality (Scheme 1 a).20 However, the undivided cell necessitated the use of highly toxic anti‐reducing agents.21 Further electrochemical syntheses of periodate are known starting from iodine14, 15 and iodate.17, 22, 23 However, iodate is more than twice as expensive as iodide, and iodine is difficult to handle on a technical scale.
Scheme 1.

Electrochemical synthesis of periodate. (BDD=boron‐doped diamond; CE=current efficiency; conv.=conversion).
The research for innovative electrode materials in the last decades has resulted in boron‐doped diamond (BDD) anodes, which exhibit strongly improved properties.24 BDD is sustainable since it can be made from methane and it has a similar overpotential for oxygen evolution at PbO2 but a superior durability.25 The favorable overpotential of BDD anodes was confirmed by Janssen and co‐workers for the oxidation of iodate to periodate and indeed a similar current efficiency was achieved (Scheme 1 b).26, 27 For solubility reasons, however, mostly lithium iodate was studied and yields, analytical data, and scale have not been reported. Lithium salts are extremely expensive and therefore impractical. Another study on BDD electrodes was published for the oxidation of chloride to perchlorate—a process related to the iodide oxidation (Scheme 1 c). However, only a low current efficiency of 65 % was obtained as a result of the high current density of j=760 mA cm−2.28 This approach is not applicable, since a low pH was used, in which the electrochemical oxidation is less favored based on the oxidation potentials. Furthermore, iodine will precipitate, which lowers the yield and current efficiency due to evaporation and deposition on the anode.29
For this reason, we developed a clean and cost‐efficient periodate synthesis at BDD. Common alkali iodides were used as the commercial source and alkaline conditions were chosen to favor a high current efficiency and the solubility of iodine. The use of toxic anti‐reducing agents was avoided by using a Nafion membrane. Ultimately, the process was scaled‐up into a flow electrolysis.
Prior to addressing the synthesis, a method for the quantification of iodate and periodate was developed due to inaccurate iodometry protocols available. In the literature, iodate and periodate were analyzed by iodometry or comparative methods with arsenic compounds. Unfortunately, the exact protocols were not reported.18, 20, 27 We tested the standard protocol for iodometry and Belcher's approach for the sequential determination of iodate and periodate, but only found low accuracy and precision in both methods.30 For this reason, we established a protocol for liquid chromatography photodiode array (LC‐PDA), since the UV/Vis activity of periodate is known in literature.31 Periodate, iodate, and iodide were isocratically separable in less than 2 min on a stationary reversed‐phase column. The anions were detected photometrically and the concentrations of iodate and periodate were quantified by external calibration with high accuracy and precision (Figure 1).
Figure 1.

Calibration plot for the liquid chromatography photodiode array (LC‐PDA) analysis.
We initiated our research with cyclic voltammetry, which suggested a hydroxyl radical based mechanism, which is well known for BDD anodes.32 The first electrolysis was carried out in a divided batch‐type cell made of Teflon equipped with a Nafion membrane, a stainless‐steel cathode (EN1.4401; AISI/ASTM), and a commercial BDD anode. Caustic soda (C=1 m) was used as both strong electrolyte and base. NaI was added to the anodic compartment. The electrolysis was conducted at a low current density of j=3.3 mA cm−2 and an applied charge of Q=9 F. After electrolysis, the solution in the anodic compartment was observed to have a yellowish color and a white precipitate had formed, which was assumed to correspond to the iodate and/or periodate as a sodium hydroxide adduct (para‐periodate, Na3H2IO6), respectively.33 The reaction mixture was acidified with aqueous NaHSO4 to dissolve solids and convert para‐ into meta‐ and ortho‐periodate. The diluted solution was then analyzed by LC‐PDA and to our delight, we found the quantitative conversion of the iodide to iodate and periodate, which we considered to be a good starting point for further investigation (Table 1, entry 1).
Table 1.
DoE optimization of the batch electrolysis (selected results, for more details see Tables S5–S8 in the Supporting Information).

|
Entry |
j [mA cm−2] |
Q [F] |
C(NaOH) [m] |
C(NaI) [m] |
|
LC‐PDA yields [%] |
||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
IO3 − |
IO4 − |
Σ |
|
1 |
3.3 |
9 |
1 |
0.11 |
|
84 |
13 |
97 |
|
2 |
60 |
10.5 |
2 |
0.22 |
|
37 |
25 |
62 |
|
3 |
100 |
9 |
3 |
0.33 |
|
6 |
87 |
93 |
|
4 |
100 |
9 |
3 |
0.33 |
|
8 |
90 |
98 |
|
5 |
100 |
9 |
3 |
0.33 |
|
10 |
89 |
99 |
|
6 |
100 |
9 |
3 |
0.33 |
|
11 |
87 |
98 |
|
7 |
100 |
9 |
3 |
0.55 |
|
66 |
29 |
95 |
|
8 |
100 |
9 |
2 |
0.55 |
|
79 |
10 |
89 |
|
9 |
100 |
9 |
1 |
0.33 |
|
75 |
10 |
85 |
BDD=boron‐doped diamond, Q=applied charge, j=current density, LC‐PDA=liquid chromatography photodiode array.
Optimizations were done by a statistical approach also known as Design of Experiments (DoE).11d, 34 The screenings investigated the concentrations of hydroxide C(NaOH) and iodide C(NaI) in the range from 1 m to 3 m and from 0.11 m to 0.55 m, respectively.35 The current density j was screened from 20 mA cm−2 to 100 mA cm−2 and the applied charge Q from 9 F to 12 F (Table 1). After electrolysis, the anodic reaction mixtures often showed a pale yellow color, and a white precipitate had formed. The formation of iodine was observed as a brown‐violet solid precipitating upon acidification with NaHSO4 due to comproportionation. That was the case when the total yield was low (entry 2). Best results were obtained for molarities of C(NaOH)=3 m and C(NaI)=0.33 m, roughly a ratio of 10:1. Such experiments reproducibly produced periodate in around 90 % yield (entries 3–6). Higher concentrations of NaI in relation to NaOH resulted in <29 % yield (entry 7) and worst results were obtained for low hydroxide concentrations below 3 m with an modest periodate yield of 10 % (entries 8 and 9). The statistical interpretation confirmed a significant main effect for C(NaOH) on the yield, while C(NaI), j, and Q had only a low level of significance. The interaction chart indicated two‐factor interactions between C(NaOH) and C(NaI), and between j and Q. Additionally, the current density was further screened until j=300 mA cm−2, which corresponds to shorter electrolysis times. However, higher current densities led to a significant evaporation of iodine due to heat generation, while having no significant positive main effect. The optimized electrolysis conditions were tested with graphite, glassy carbon, nickel, platinum, lead, and lead dioxide anodes, but only a periodate yield of maximum 58 % was reached for the lead dioxide. All electrodes beside nickel and platinum corroded severely, confirming that BDD anodes are clearly superior. We did not observe any surface abrasion on BDD during the entire study. The process was also feasible for other iodide sources, that is, LiI, KI, CsI, RbI, CuI, and ZnI2, although lower yields were obtained.
Next, a flow process was developed for scale‐up. Based on optimization results under batch conditions, the flow electrolysis was tested and optimized using a 12‐cm2 flow cell made of Teflon in cycling mode.21, 36 BDD was used as the anode and stainless steel as the cathode. The cell was divided by Nafion and both electrolytes (V=25 mL) were pumped in two independent cycles.
The optimization investigated the hydroxide and the iodide concentrations, the current density, the applied charge, and the flow rate. During electrolysis, the anodic solution had a pale yellow color that vanished at around 4.5 F, while a white solid precipitated and formed a suspension while stirring (Figure 2 b,c). Hereby, the terminal voltage increased from ≈5.5 V to ≈6.5 V. With low molarities of NaOH, I2 precipitated as brown‐violet solid (Figure 2 a). After electrolysis, the flow system was rinsed with aqueous NaHSO4 and water, and the resulting solution was analyzed by LC‐PDA. In accordance with the batch optimization, best yields were obtained for molar base‐to‐iodide ratios ≥10:1 (Table 2, entries 1–4), whereby the highest periodate yield of 94 % was obtained for the concentration ratio of 4:0.4 m/m (entry 5). Variations, again, led to diminished yields of 19 % (entry 6) and higher applied charges were not able to drive the reaction to completion. In the statistical interpretation, large positive main effects and two‐factor interactions were observed for the concentrations of NaOH and NaI, and the flow rate, while the current density and the applied charge had only small main effects. Beside the concentration ratio, optimal conditions in flow were found at a current density of j=100 mA cm−2, an applied charge of Q=9 F, and a fast flow rate of fr=7.5 L h−1.
Figure 2.

Reaction mixtures from flow electrolysis. a) Low hydroxide concentration caused iodine precipitation; b,c) mixtures with sufficient hydroxide concentration (C(NaOH)=3 m) in chronological order.
Table 2.
DoE optimization of the flow electrolysis (selected results; for more details see Tables S10–S14 in the Supporting Information).

|
Entry |
C(NaOH) [m] |
C(NaI) [m] |
|
LC‐PDA yields [%] |
||
|---|---|---|---|---|---|---|
|
|
|
|
|
IO3 − |
IO4 − |
Σ |
|
1 |
3 |
0.1 |
|
10 |
90 |
100 |
|
2 |
5 |
0.3 |
|
8 |
91 |
99 |
|
3 |
5 |
0.3 |
|
7 |
91 |
98 |
|
4 |
5 |
0.3 |
|
7 |
93 |
100 |
|
5 |
4 |
0.4 |
|
6 |
94 |
100 |
|
6 |
1 |
0.3 |
|
69 |
19 |
88 |
BDD=boron‐doped diamond, Q=applied charge, j=current density, fr=flow rate, LC‐PDA=liquid chromatography photodiode array.
The optimal NaI/NaOH ratio of 1:10 can be rationalized by the reactions in the individual half‐cells (Scheme 2). In theory, 8 equivalents (eq.) of NaOH are consumed in the anodic compartment as part of the oxidation. In the cathodic compartment, 8 eq. of NaOH are generated as part of the reduction. Another 2 eq. of NaOH are consumed to form the double salt. For the maximum yield at 0.4:4 m/m NaI/NaOH, kinetic effects are hypothesized.
Scheme 2.

Half‐cell reactions at the anode and cathode, and the double salt formation of meta‐periodate (IO4 −) to para‐periodate (H2IO6 3−). Negative charges are balanced out by Na+ and are omitted for clarity.
The flow electrolysis was scaled up with respect to the anode surface, the current density, and the amount of substance. A commercial electrolysis cell made of stainless steel was employed first, having an anode surface of 37 cm2. The standard conditions and higher current densities were tested in the range of 100–1892 mA cm−2, but periodate was producted in only 24–65 % yield (3.7–70 A, Table 3, entries 1–3). We observed slight corrosion of the stainless steel casing at sites exposed to the anolyte and hypothesized an metal‐induced decay of periodate to O2.37 Another explanation could be the larger gap in the anodic compartment leading to higher cell resistance and energy loss due to increased heat generation. Significantly higher and more reproducible yields were obtained with the 48‐cm2 cell made of Teflon.38 Periodate was produced in up to 83 % yield in the screening of the current density (entries 4 and 5), which improved slightly to 86 % when the volume was increased 10‐fold (entries 6–8). The determined yield corresponded to a mass of 101 g para‐periodate.
Table 3.
Scale up (selected results; for more details see Tables S15–S17 in the Supporting Information).

|
Entry |
a [cm2] |
V [mL] |
j [mA cm−2] |
|
LC‐PDA yields [%] |
||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
IO3 − |
IO4 − |
Σ |
|
1 |
37 |
100 |
100 |
|
47 |
24 |
71 |
|
2 |
37 |
100 |
541 |
|
24 |
73 |
97 |
|
3 |
37 |
100 |
1892 |
|
31 |
63 |
94 |
|
4 |
48 |
100 |
100 |
|
6 |
80 |
86 |
|
5 |
48 |
100 |
500 |
|
21 |
80 |
101 |
|
6 |
48 |
250 |
500 |
|
5 |
73 |
78 |
|
7 |
48 |
500 |
500 |
|
3 |
63 |
66 |
|
8 |
48 |
1000 |
500 |
|
4 |
86 |
90 |
BDD=boron‐doped diamond, Q=applied charge, j=current density, fr=flow rate, LC‐PDA=liquid chromatography photodiode array, a=anode surface area.
After electrolysis, the para‐periodate was isolated by simple filtration and was obtained in 90 % yield with a purity of 97 % periodate by LC‐PDA. It was converted to meta‐periodate in an acidic recrystallization according to literature procedures.21, 37 The solution was acidified to pH 1–2 with concentrated nitric acid and was concentrated at elevated temperatures until crystallization started. The mixture was allowed to cool to room temperature and the formed crystals were filtered off. The meta‐periodate was obtained in 71 % yield, however, containing traces of sulfate (Figure S16 in the Supporting Information). The identity of the prepared periodate was confirmed by IR analysis. Commercial meta‐periodate was converted with 2 eq. of NaOH to yield para‐periodate and was isolated as above. Both meta‐ and para‐periodate were analyzed by IR spectroscopy, and the spectra were compared to those of the corresponding commercial materials (Figure S16 in the Supporting Information) and were in good accordance with the Bio‐Rad database.40 Furthermore, the electrochemically synthesized meta‐periodate was tested in the sulfide and the Malaprade oxidation (Scheme 3). These reactions were compared to those with commercial meta‐periodate.41 In both oxidations, full conversion was achieved with both the synthesized and the commercial meta‐periodate, confirming the high quality of the periodate generated by this electrochemical method.
Scheme 3.

Synthetic confirmation of the electrochemically synthesized meta‐periodate. Yields were determined by 1H NMR spectroscopy versus caffeine as an internal standard. All reactions showed full conversion of the starting material.
In summary, the direct electrochemical synthesis of periodate from common iodides has been established at a BDD anode. The conventional use of nondurable metal‐based electrodes, in particular lead dioxide, was hereby avoided, which prevents contamination and enables the use of periodate in sensitive applications. In contrast to previous reports, the total number of synthetic steps was reduced by starting from iodide, thereby facilitating a simple, robust and cost‐efficient synthesis. The conditions have been optimized by statistical experiments where the optimal stoichiometry of hydroxide to iodide was determined to be 10:1. The current density had only a small effect on the yield, allowing the application of low current densities below 500 mA cm−2. Under these conditions, the heat generation is moderate, which contributes to an energy‐efficient process and a yield of 94 %. The synthesis was scaled‐up to produce 100 g para‐periodate in a flow‐electrolysis cell and the space–time yield was enhanced in a cyclization protocol. This electrochemical approach will promote the use of periodate in several fields where toxic heavy metals are considered to be critical. Furthermore, a novel analysis method was developed based on high‐performance liquid chromatography, which enables the time‐ and material‐efficient determination of periodate along with iodate and iodine. This method will accelerate future research in the field of hypervalent iodic species.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation in frame of FOR 2982—UNDODE Wa1276/23‐1).
S. Arndt, D. Weis, K. Donsbach, S. R. Waldvogel, Angew. Chem. Int. Ed. 2020, 59, 8036.
Contributor Information
Dr. Sebastian Arndt, https://www.aksw.uni‐mainz.de.
Prof. Siegfried R. Waldvogel, Email: waldvogel@uni-mainz.de.
References
- 1.
- 1a. Sudalai A., Khenkin A., Neumann R., Org. Biomol. Chem. 2015, 13, 4374–4394; [DOI] [PubMed] [Google Scholar]
- 1b. Fuchs P. L., Charette A. B., Rovis T., Bode J. W., Essential Reagents for Organic Synthesis, Wiley-VCH, Weinheim, 2016. [Google Scholar]
- 2.
- 2a. Abdel-Akher M., Smith F., J. Am. Chem. Soc. 1959, 81, 1718–1721; [Google Scholar]
- 2b. Jackson E. L., Hudson C. S., J. Am. Chem. Soc. 1939, 61, 959–960; [Google Scholar]
- 2c. Sklarz B., Q. Rev. Chem. Soc. 1967, 21, 3; [Google Scholar]
- 2d. Zhou M., Crabtree R. H., Chem. Soc. Rev. 2011, 40, 1875–1884; [DOI] [PubMed] [Google Scholar]
- 2e. Plietker B., Synthesis 2005, 2453–2472. [Google Scholar]
- 3.
- 3a. Kraszkiewicz L., Sosnowski M., Skulski L., Synthesis 2006, 1195–1199; [Google Scholar]
- 3b. Chouthaiwale P., Karabal P., Suryavanshi G., Sudalai A., Synthesis 2010, 3879–3882. [Google Scholar]
- 4. Murphy J. M., Tzschucke C. C., Hartwig J. F., Org. Lett. 2007, 9, 757–760. [DOI] [PubMed] [Google Scholar]
- 5. Kitov P. I., Bundle D. R., Org. Lett. 2001, 3, 2835–2838. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Chen Y. K., Lurain A. E., Walsh P. J., J. Am. Chem. Soc. 2002, 124, 12225–12231; [DOI] [PubMed] [Google Scholar]
- 6b. Palomo C., Oiarbide M., Halder R., Kelso M., Gómez-Bengoa E., García J. M., J. Am. Chem. Soc. 2004, 126, 9188–9189; [DOI] [PubMed] [Google Scholar]
- 6c. Juhl K., Jørgensen K. A., J. Am. Chem. Soc. 2002, 124, 2420–2421. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Das B., Lingaiah M., Damodar K., Bhunia N., Synthesis 2011, 2941–2944; [Google Scholar]
- 7b. Smith J. M., Dixon J. A., deGruyter J. N., Baran P. S., J. Med. Chem. 2019, 62, 2256–2264. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Satyanarayana K., Srinivas K., Himabindu V., Reddy G. M., Org. Process Res. Dev. 2007, 11, 842–845; [Google Scholar]
- 8b. Wang X., Zeng Y., Sheng L., Larson P., Liu X., Zou X., Wang S., Guo K., Ma C., Zhang G., et al., J. Med. Chem. 2019, 62, 2305–2332; [DOI] [PubMed] [Google Scholar]
- 8c. Zhou S., Jia Y., Org. Lett. 2014, 16, 3416–3418; [DOI] [PubMed] [Google Scholar]
- 8d. Gao X., Woo S. K., Krische M. J., J. Am. Chem. Soc. 2013, 135, 4223–4226; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Bihelovic F., Saicic R. N., Angew. Chem. Int. Ed. 2012, 51, 5687–5691; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5785–5789. [Google Scholar]
- 9.Molar costs were calculated based on the best price offers from Sigma Aldrich/Merck KGaA. https://www.sigmaaldrich.com/germany.html, 2019.
- 10.
- 10a. Röckl J., Pollok D., Franke R., Waldvogel S. R., Acc. Chem. Res. 2020, 53, 45–61; [DOI] [PubMed] [Google Scholar]
- 10b. Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721; [Google Scholar]
- 10c. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 10d. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706–6765. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Francke R., Curr. Opin. Electrochem. 2019, 15, 83–88; [Google Scholar]
- 11b. Haupt J. D., Berger M., Waldvogel S. R., Org. Lett. 2019, 21, 242–245; [DOI] [PubMed] [Google Scholar]
- 11c. Herszman J. D., Berger M., Waldvogel S. R., Org. Lett. 2019, 21, 7893–7896; [DOI] [PubMed] [Google Scholar]
- 11d. Möckel R., Babaoglu E., Hilt G., Chem. Eur. J. 2018, 24, 15781–15785; [DOI] [PubMed] [Google Scholar]
- 11e. Elsherbini M., Wirth T., Chem. Eur. J. 2018, 24, 13399–13407; [DOI] [PubMed] [Google Scholar]
- 11f. Elsherbini M., Winterson B., Alharbi H., Folgueiras A. A., Génot A. C., Wirth T., Angew. Chem. Int. Ed. 2019, 58, 9811–9815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9916–9920. [Google Scholar]
- 12.
- 12a. Xia Y., Dai Q., Chen J., J. Electroanal. Chem. 2015, 744, 117–125; [Google Scholar]
- 12b. Kim J. K., Choi B. S., Nam C. W., Kongop Hwahak 1996, 7, 1105–1114. [Google Scholar]
- 13.
- 13a. Wang Y.-s., Yang F., Liu Z.-h., Yuan L., Li G., Catal. Commun. 2015, 67, 49–53; [Google Scholar]
- 13b. Mukimin A., Vistanty H., Zen N., Chem. Eng. J. 2015, 259, 430–437. [Google Scholar]
- 14. Aiya Y., Fujii S., Sugino K., Shirai K., J. Electrochem. Soc. 1962, 109, 419–424. [Google Scholar]
- 15. Mehltretter C. L., US2830941A, 1958.
- 16.
- 16a.European Medicines Agency, ICH guideline Q3D (R1) on elemental impurities can be found under https://www.ich.org/page/quality-guidelines, 2019;
- 16b. Abernethy D. R., Destefano A. J., Cecil T. L., Zaidi K., Williams R. L., Pharm. Res. 2010, 27, 750–755. [DOI] [PubMed] [Google Scholar]
- 17. Hickling A., Richards S. H., J. Chem. Soc. 1940, 256–264. [Google Scholar]
- 18. Müler E., Z. Elektrochem. 1901, 38, 509–517. [Google Scholar]
- 19.
- 19a. Müller E., Z. Elektrochem. 1904, 4, 51–68; [Google Scholar]
- 19b. Kong D., Wan P., Chen Y., Khan Z. U. l. H., Tang Y., Int. J. Electrochem. Sci. 2015, 6422–6432; [Google Scholar]
- 19c. Gan Y., Yao B., Zhang W., Huang H., Xia Y., Zhang J., Liang C., He X., CN 110158112, 2019.
- 20. Nam C. W., Kim H. J., J. Korean Chem. Soc. 1974, 18, 373–380. [Google Scholar]
- 21. Hirakata K., Mochizuki M., Kanai H., Itai R., US4687565A, 1987.
- 22. Willard H. H., Ralston R. R., Trans. Electrochem. Soc. 1932, 62, 239–254. [Google Scholar]
- 23.
- 23a. Nam C. W., Kim H. J., J. Korean Chem. Soc. 1971, 15, 324–329. [Google Scholar]
- 24.
- 24a. Waldvogel S. R., Mentizi S., Kirste A., Top. Curr. Chem. 2012, 320, 1–31; [DOI] [PubMed] [Google Scholar]
- 24b. Yang N., Yu S., Macpherson J. V., Einaga Y., Zhao H., Zhao G., Swain G. M., Jiang X., Chem. Soc. Rev. 2019, 48, 157–204; [DOI] [PubMed] [Google Scholar]
- 24c. Lips S., Waldvogel S. R., ChemElectroChem 2019, 6, 1649–1660. [Google Scholar]
- 25.
- 25a. Luong J. H. T., Male K. B., Glennon J. D., Analyst 2009, 134, 1965–1979; [DOI] [PubMed] [Google Scholar]
- 25b. Einaga Y., Bull. Chem. Soc. Jpn. 2018, 91, 1752–1762; [Google Scholar]
- 25c. Gleede B., Yamamoto T., Nakahara K., Botz A., Graßl T., Neuber R., Matthée T., Einaga Y., Schuhmann W., Waldvogel S. R., ChemElectroChem 2019, 6, 2771–2776. [Google Scholar]
- 26. Janssen L. J. J., NL1013348C2, 2001.
- 27. Janssen L. J. J., Blijlevens M. H. A., Electrochim. Acta 2003, 48, 3959–3964. [Google Scholar]
- 28. Lehmann T., Stenner P., DE10258652A1, 2004.
- 29.
- 29a. Bejerano T., Gileadi E., J. Electroanal. Chem. 1977, 82, 209–225. [Google Scholar]
- 30.
- 30a. Belcher R., Townshend A., Anal. Chim. Acta 1968, 41, 395–397; [Google Scholar]
- 30b. Hofmann H., Jander G., Qualitative Analyse, De Gruyter, München, 2011; [Google Scholar]
- 30c. Al-Zamil I. Z., Anal. Chim. Acta 1984, 158, 383–387. [Google Scholar]
- 31. Buist G. J., Tabatabai S. M., J. Chem. Soc. Faraday Trans. 1 1979, 75, 631. [Google Scholar]
- 32.
- 32a. Kasahara S., Ogose T., Ikemiya N., Yamamoto T., Natsui K., Yokota Y., Wong R. A., Iizuka S., Hoshi N., Tateyama Y., et al., Anal. Chem. 2019, 91, 4980–4986; [DOI] [PubMed] [Google Scholar]
- 32b. Azizi O., Hubler D., Schrader G., Farrell J., Chaplin B. P., Environ. Sci. Technol. 2011, 45, 10582–10590. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Hill A. E., J. Am. Chem. Soc. 1928, 50, 2678–2692; [Google Scholar]
- 33b. Valkai L., Peintler G., Horváth A. K., Inorg. Chem. 2017, 56, 11417–11425. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Santi M., Seitz J., Cicala R., Hardwick T., Ahmed N., Wirth T., Chem. Eur. J. 2019, 25, 16230–16235; [DOI] [PubMed] [Google Scholar]
- 34b. Murray P. M., Bellany F., Benhamou L., Bučar D.-K., Tabor A. B., Sheppard T. D., Org. Biomol. Chem. 2016, 14, 2373–2384; [DOI] [PubMed] [Google Scholar]
- 34c. Weissman S. A., Anderson N. G., Org. Process Res. Dev. 2015, 19, 1605–1633. [Google Scholar]
- 35. Gütz C., Klöckner B., Waldvogel S. R., Org. Process Res. Dev. 2016, 20, 26–32. [Google Scholar]
- 36.
- 36a. Pletcher D., Green R. A., Brown R. C. D., Chem. Rev. 2018, 118, 4573–4591; [DOI] [PubMed] [Google Scholar]
- 36b. Elsherbini M., Wirth T., Acc. Chem. Res. 2019, 52, 3287–3296; [DOI] [PubMed] [Google Scholar]
- 36c. Gütz C., Bänziger M., Bucher C., Galvão T. R., Waldvogel S. R., Org. Process Res. Dev. 2015, 19, 1428–1433; [Google Scholar]
- 36d. Gütz C., Stenglein A., Waldvogel S. R., Org. Process Res. Dev. 2017, 21, 771–778. [Google Scholar]
- 37. Mills A., Hazafy D., Elouali S., O'Rourke C., J. Mater. Chem. A 2016, 4, 2863–2872. [Google Scholar]
- 38. Gleede B., Selt M., Gütz C., Stenglein A., Waldvogel S. R., Org. Process Res. Dev. 2019, 10.1021/acs.oprd.9b00451. [DOI] [Google Scholar]
- 39. Mehltretter C. L., Wise C. S., US2989371A, 1961.
- 40.Infrared spectral data were obtained from the Bio-Rad/Sadtler IR Data Collection, Bio-Rad Laboratories, Philadelphia, PA (US) and can be found under https://spectrabase.com. Spectrum ID (meta-periodate): 3ZPsHGmepSu, Spectrum ID (para-periodate): GtfOwBZh0NM.
- 41.
- 41a. Ruff F., Fábián A., Farkas Ö., Kucsman Á., Eur. J. Org. Chem. 2009, 2102–2111; [Google Scholar]
- 41b. Leonard N. J., Johnson C. R., J. Org. Chem. 1962, 27, 282–284. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary