

To the Editor:
1.
The hematopoietic stem and progenitor cell marker CD34 is implicated in cell‐cell interactions in the bone marrow niche. Upon hematopoietic maturation its expression is switched off. Aberrant CD34 expression on mature platelets has been associated with monogenic bleeding disorders that are caused by mutations in the transcription factors GFI1B (Growth Factor Independence 1B) and RUNX1 (RUNT related transcription factor 1).1, 2 These familial disorders are characterized by a bleeding diathesis, caused by disturbed megakaryocyte development, which results in the formation of dysfunctional platelets. In addition, these disorders are accompanied by abnormal retention of CD34 on platelets, which is not lost during differentiation. In this letter, we report a patient with CD34 expression on platelets associated with a partial deletion of the RUNX1 binding partner CBFB (Core Binding Factor B). CBFB is part of a heterodimeric transcription factor complex with either RUNX1, −2 or − 3. CBFB enables the function of RUNX1‐3 through stabilization of their interaction with DNA. Together with GFI1B and RUNX1, CBFB is now the third hematopoietic transcriptional regulator that associates with abnormal platelet CD34 expression.
The patient we report here, presented to our outpatient clinic with multiple bleeding events. A Tosetto bleeding score of 11 was established, consisting of spontaneous epistaxis, prolonged bleeding from small wounds, spontaneous bruising and menorrhagia. In addition, she experienced prolonged bleeding after wisdom teeth extraction, and severe bleeding following hysterectomy requiring relaparotomy and blood transfusion. However, she did not experience postpartum bleeding after giving birth to a son, and correction of clavicle hypoplasia as a child was uncomplicated. Both the parents and the patientʼs son were not affected by a bleeding tendency (Figure 1A). Her medical history revealed mild asthma, migraines and a clinical depression for which she used a selective serotonin reuptake inhibitor (SSRI). Platelet counts, as well as mean platelet volume, ADP levels, β‐thromboglobulin and platelet factor 4 expression, and the ADP/ATP ratio were within the normal range (Table S1). The platelets showed no abnormalities in a May‐Grünwald Giemsa stained blood film (data not shown). The platelet function analyzer measured prolonged closure time after stimulation with the collagen/epinephrine membrane (>300 seconds). Platelet light transmission aggregometry revealed an aggregation defect upon stimulation with epinephrine (10 μM), ADP (10 μM) and low concentration of collagen (1 μg/mL) (Table S1). Stimulation with high concentrations of collagen or ADP resulted in normal aggregation responses, similar to the strong agonists arachidonic acid, thrombin receptor‐activating peptide and ristocetin. The ATP release from δ‐granules was measured following stimulation with a high concentration of collagen (5 μg/mL), epinephrine (5 μM) and the thromboxane analogue U46619 (1 μM) (Table S1). The ATP release was normal after stimulation with a high concentration of collagen, as expected. No ATP was released upon stimulation with epinephrine and low levels were measured after stimulation with U46619. Cessation of the SSRI for 8 weeks did not correct platelet aggregation defects. Thin section electron microscopy (EM) showed normal platelets, with occasional platelets carrying few α‐granules (Figure 1B). Whole mount EM showed increased numbers of δ‐granules at two independent time points, compared to controls (on average 5.84 δ‐granules vs 3.66 per platelet, respectively) (Figure 1C, D).2 Together, these data suggest that the index patient suffers from a mild δ‐granule secretion defect. Remarkably, the platelets from the son showed defects in the light transmission aggregometry tests upon stimulation with epinephrine and low concentrations of collagen and ADP, similar to the index patient (Table S1). The adult son did not experience hemostatic challenges and had no clinical history of bleedings, which may suggest that the bleeding disorder observed in the index patient is not solely caused by the δ‐granule secretion defect.
Figure 1.
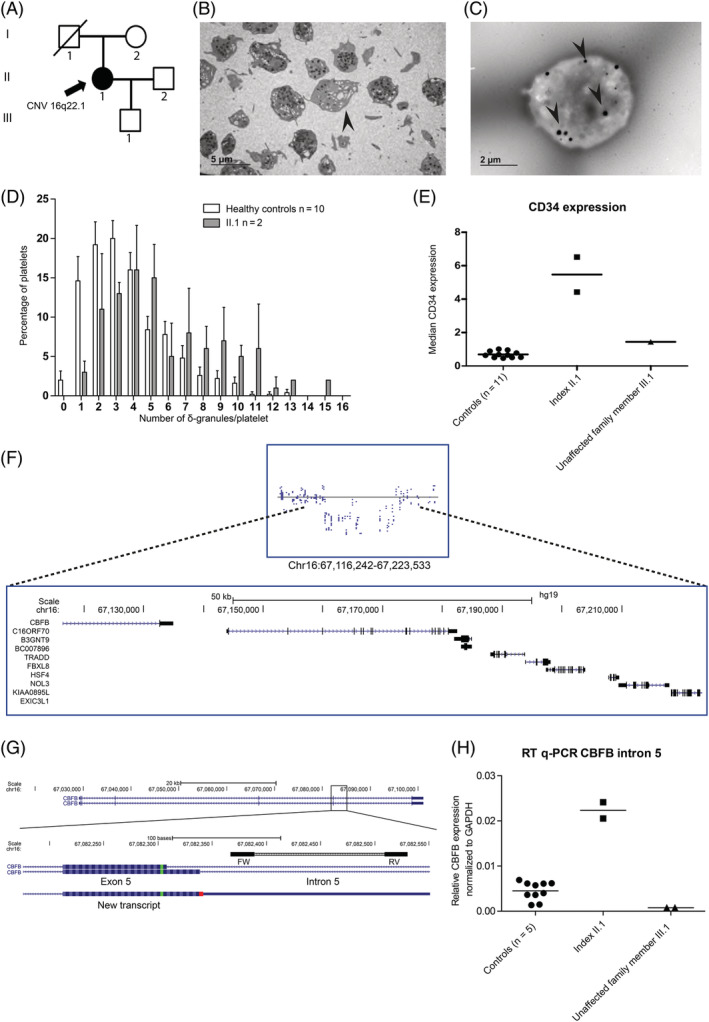
Family history, platelet phenotyping and genetic analyses in a patient with a partial deletion of CBFB. A, Family pedigree with one affected family member with a bleeding disorder as well as a heterozygous deletion of chromosome 16q22.1. Besides patient II.1 none of the other family members had a clinical presentation of a bleeding tendency. The father of the index patient (I1) was deceased and unavailable for further testing. No clinical abnormalities were reported. □ represents male, ○ represents females. B, Transmission electron microscopy images show platelets with few α‐granules (as depicted with an arrowhead) as well as platelets with normal amounts of α‐granules. (JEM 1400 Flash, Jeol, Tokyo, Japan) C, D, Whole mount electron microscopy image revealed on average increased numbers of δ‐granules (5.84 granules/platelet, black dots depicted with arrowheads) in the platelets from index II.1 compared to healthy controls (3.66 granules/platelet). (JEM 1400 Flash, Jeol, Tokyo, Japan) Whole mount EM was performed twice at different time points, showing similar results. E, CD34 expression was measured on CD42b positive platelets. CD34 was expressed on platelets derived from the index patient II.1 (n = 2), whereas the platelets from the son (III.1) as well as healthy controls (n = 10) showed no CD34 expression. F, The last exon of CBFB together with eight other genes were deleted as observed in WES (not shown) and SNP array. The proximal breakpoint is situated within intron 5 of CBFB. The father (I1) of index patient II.1 was unavailable for genotyping. The mother (I2) did not carry the deletion nor did the son (III.1). G, 3′‐RACE using an oligo‐dT primer was performed, followed by amplification of the cDNA from exon5 onwards and subsequently a semi‐nested PCR was performed. Sanger sequencing identified a transcript that predicts the coding for 11 amino acids prior to encountering a stop codon into CBFB intron 5. H, High expression of intron 5 sequences was observed in blood mononuclear cells from the index patient II.1, whereas low expression was observed for five healthy controls and the index case her son (III.1). Expression was measured with RT‐qPCR with primers indicated with the black boxes in panel G. Expression was normalized against the housekeeping gene Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Primer sequences are given in Table S2
To identify a genetic cause for the bleeding tendency, a panel of thrombosis and hemostasis associated genes was screened following whole exome sequencing.3 No disease‐causing variants were called. Exome‐wide analysis detected a ~100 Kb heterozygous chromosome 16q22.1 deletion that was confirmed by single nucleotide polymorphism (SNP) array. This deletion covers nine genes, including the last exon of CBFB (Figure 1F). No disease‐causing variants were observed in the remaining wild type CBFB allele. The mother and son of the index patient did not have the 16q deletion. The father was deceased and could therefore not be tested for the deletion, however, no clinical history of bleedings was reported. The expression of CBFB transcripts was studied by 3′‐Rapid Amplification of cDNA Ends (RACE) and Sanger sequencing. Readthrough from exon 5 into intron 5 was observed, predicting the formation of a protein that exceeds 11 amino acids from the splice junction. To quantify RNA expression of this transcript in leukocytes, a quantitative real‐time polymerase chain reaction (RT‐qPCR) was designed within intron 5 (Figure 1G). Next to the patient (II.1) and her son (III.1) five additional healthy controls were tested for expression of this transcript. This showed on average a 10‐fold increase of this transcript in the index case compared to the controls and her son (Figure 1H). Based on findings in the son we concluded that the δ‐granule defect was not caused by the 16q deletion. Yet, the 16q deletion in combination with the δ‐granule defect might contribute to the bleedings in the index patient. To date, none of the 16q‐deleted genes have been implicated in inherited bleeding or platelet disorders. However, conditional Cbfb knockout in mice resulted in disturbed megakaryopoiesis and thrombocytopenia.4 Because bleeding disorders caused by RUNX1 mutations result in platelet CD34 expression and an interaction with CBFB is required for the function of RUNX1, the platelets were tested for CD34 expression.2 Flow cytometry clearly detected platelet CD34 expression in the index patient while platelets from her son and 10 nonrelated healthy controls did not. Thus, the 16q deletion associates with platelet CD34 expression (Figure 1E).
Entire heterozygous CBFB deletions have been reported in rare cases with congenital bone abnormalities and mutations in its binding partner RUNX2 may cause cleidocranial dysplasia, associated with clavicle hypoplasia.5 The medical record of the patient indicated that she had undergone surgery at the age of 13 for a congenital hypoplastic clavicle. The mother and son of the index patient had no signs of clavicle hypoplasia and the father was reported not to have bone abnormalities either. Thus, the 16q deletion associates with platelet CD34 expression, and a congenital clavicle hypoplasia but not with disturbed platelet δ‐granule secretion.
To our knowledge, the patient reported here is the first in whom a deletion of chromosome 16q22.1 partially encompassing CBFB associates with platelet CD34 expression. The deletion is not causal to the platelet δ‐granule abnormalities, because the son who did not inherit the deletion showed similar δ‐granule defects as the mother. Recently, we and others reported CD34 expression on platelets from patients with GFI1B variants that did not necessarily cause bleedings on their own. This suggests that platelet CD34 expression may be uncoupled from a bleeding tendency for other transcription factor mutations as well.6, 7 During endothelial to hematopoietic transition GFI1B is a crucial RUNX1/CBFB target gene, which may explain the similarities between RUNX1, CBFB and GFI1B mutants.8 As the son had a δ‐granule secretion defect, similar to the index patient, but no clinical history of bleedings, the index patientʼs bleeding tendency cannot solely be explained by the δ‐granule dysfunction. This case underscores that mutations in various hematopoietic transcription factors may result in abnormal platelet development associated with increased CD34 expression, which may aggravate but are not necessarily the sole cause of the clinically manifested bleeding tendency.
AUTHOR CONTRIBUTIONS
M.G.J.M.v.B., J.L.S., B.A.P.L.‐v.G., S.E.M.S., M.C.J.J., J.H.J., and B.A.v.d.R. designed and coordinated research; M.G.J.M.v.B., J.L.S., A.S., K.M.H., Y.M.C.H., W.B., E.H., and F.W.P. collected and analyzed data; B.A.P.L.‐v.G., and M.C.J.J. arranged for obtaining patient informed consent; and M.G.J.M.v.B. and B.A.v.d.R. wrote the manuscript that was critically revised by all authors.
Supporting information
Table S1. Platelet phenotyping index case II.1 and III.1.
ACKNOWLEDGMENTS
This work was supported by the Landsteiner Foundation for Blood Transfusion Research (project 1531).
Funding information Landsteiner Foundation for Blood Transfusion Research, Grant/Award Number: 1531
REFERENCES
- 1. Monteferrario D, Bolar NA, Marneth AE, et al. A dominant‐negative GFI1B mutation in the gray platelet syndrome. N Engl J Med. 2014;370(3):245‐253. [DOI] [PubMed] [Google Scholar]
- 2. Marneth AE, Van Heerde WL, Hebeda KM, et al. Platelet CD34 expression and α/δ‐granule abnormalities in GFI1B‐and RUNX1‐related familial bleeding disorders. Blood. 2017;129(12):1733‐1736. [DOI] [PubMed] [Google Scholar]
- 3. Saes JL, Simons A, de Munnik SA, et al. Whole exome sequencing in the diagnostic workup of patients with a bleeding diathesis. Haemophilia. 2019;25(1):127‐135. [DOI] [PubMed] [Google Scholar]
- 4. Wang CQ, Chin DW, Chooi JY, et al. Cbfb deficiency results in differentiation blocks and stem/progenitor cell expansion in hematopoiesis. Leukemia. 2015;29(3):753‐757. [DOI] [PubMed] [Google Scholar]
- 5. Khan A, Hyde RK, Dutra A, Mohide P, Liu P. Core binding factor beta (CBFB) haploinsufficiency due to an interstitial deletion at 16q21q22 resulting in delayed cranial ossification, cleft palate, congenital heart anomalies, and feeding difficulties but favorable outcome. Am J Med Genet A. 2006;140(21):2349‐2354. [DOI] [PubMed] [Google Scholar]
- 6. van Oorschot R, Marneth AE, Bergevoet SM, et al. Inherited missense variants that affect GFI1B function do not necessarily cause bleeding diatheses. Haematologica. 2019;104(6):e260‐e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rabbolini DJ, Morel‐Kopp MC, Chen Q, et al. Thrombocytopenia and CD34 expression is decoupled from alpha‐granule deficiency with mutation of the first growth factor‐independent 1B zinc finger. J Thromb Haemost. 2017;15(11):2245‐2258. [DOI] [PubMed] [Google Scholar]
- 8. Thambyrajah R, Patel R, Mazan M, et al. New insights into the regulation by RUNX1 and GFI1(s) proteins of the endothelial to hematopoietic transition generating primordial hematopoietic cells. Cell Cycle. 2016;15(16):2108‐2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Platelet phenotyping index case II.1 and III.1.