

Abstract
Psychotropic Psilocybe mushrooms biosynthesize their principal natural product psilocybin in five steps, among them a phosphotransfer and two methyltransfer reactions, which consume one equivalent of 5′‐adenosine triphosphate (ATP) and two equivalents of S‐adenosyl‐l‐methionine (SAM). This short but co‐substrate‐intensive pathway requires nucleoside cofactor salvage to maintain high psilocybin production rates. We characterized the adenosine kinase (AdoK) and S‐adenosyl‐l‐homocysteine (SAH) hydrolase (SahH) of Psilocybe cubensis. Both enzymes are directly or indirectly involved in regenerating SAM. qRT‐PCR expression analysis revealed an induced expression of the genes in the fungal primordia and carpophores. A one‐pot in vitro reaction with the N‐methyltransferase PsiM of the psilocybin pathway demonstrates a concerted action with SahH to facilitate biosynthesis by removal of accumulating SAH.
Keywords: adenosine kinase, enzymes, methylation, psilocybin, S-adenosylhomocysteine hydrolase
Maximum methylation: Psilocybin biosynthesis is completed by two successive methylations, which release S‐adenosyl‐l‐homocysteine (SAH) as second product. We characterized P. cubensis adenosine kinase and SAH hydrolase in vitro and show that efficient removal of SAH increases methylation and, thus, psilocybin production.
Introduction
The principal natural product of psychotropic “magic” mushrooms is psilocybin (Scheme 1), first isolated by Albert Hofmann and colleagues.1 It has prodrug character as it represents the stable precursor of the dephospho analogue psilocin (Scheme 1), the actual neuroactive compound. Psilocybin is under consideration in advanced clinical trials as a pharmaceutical to treat therapy‐resistant depression.2 In pioneering work, a biosynthetic cascade was suggested.3 Recently, the corresponding genetic locus and the enzymes were identified.4 Psilocybin biosynthesis requires four enzymes (Scheme 1): l‐tryptophan decarboxylation and hydroxylation at C‐4 are catalyzed by the decarboxylase PsiD and the monooxygenase PsiH, respectively, to form 4‐hydroxytryptamine. Subsequently, the 5′‐adenosine triphosphate (ATP)‐dependent 5‐methylthioribokinase‐like enzyme PsiK phosphorylates 4‐hydroxytryptamine.4a The biosynthesis is completed by the methyltransferase PsiM which installs the tertiary amine and requires two equivalents of S‐adenosyl‐l‐methionine (SAM). Psilocybin titers can reach up to 2 % of the fungal dry biomass.5 Consequently, SAM must be efficiently regenerated to keep up the high psilocybin production rates. Hence, we expected 1) enzymes of the SAM salvage cycle and 2) adenosine kinase (AdoK) that supports this cycle to be critical for psilocybin biosynthesis (Scheme 1). Adenosine kinases catalyze the phosphorylation of adenosine (Ado) to adenosyl‐5′‐monophosphat (AMP) using ATP as phosphate donor6 and they play an intrinsic role in purine salvage metabolism, primarily to regenerate Ado to AMP, eventually to provide new ATP in a three‐step reaction (Scheme 1).7 Most importantly, they are indirectly involved in the SAM regeneration system as well.8
Scheme 1.
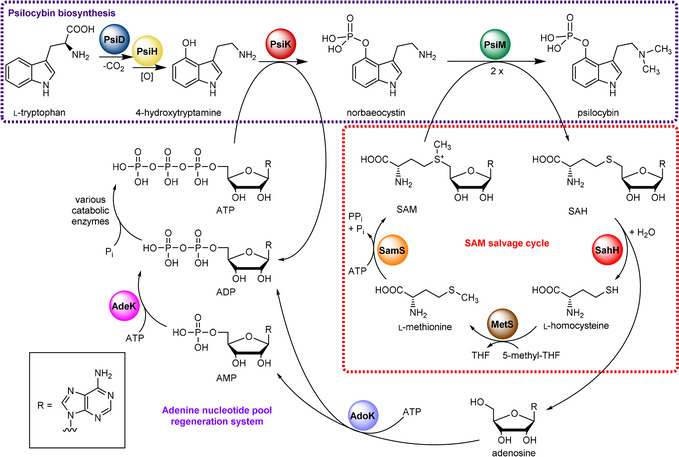
Nucleoside cofactor metabolism related to psilocybin biosynthesis by the action of PsiD, PsiH, PsiK, and PsiM (violet box). During this process, PsiM demethylates two equivalents of SAM to SAH, which is regenerated by the SAM salvage cycle comprising enzymes SahH, MetS and SamS (red box). Adenosine liberated by SahH is phosphorylated by AdoK to yield AMP. Together with ADP, AMP is regenerated to ATP by adenylate kinase (AdeK) and various enzymes of the primary metabolism (e.g., cytosolic glycolysis, mitochondrial respiratory chain and ATP synthase complex), or by use of polyphosphates (not shown).
The methyl donor SAM is regenerated by the concerted action of three enzymes: S‐adenosyl‐l‐homocysteine hydrolase (SahH) cleaves SAH to release Ado (the AdoK substrate) and l‐homocysteine (HCy), which is subsequently methylated to l‐methionine (l‐Met) by a 5′‐methyltetrahydrofolate‐dependent l‐methionine synthase (MetS). Lastly, l‐Met is adenosylated by SAM synthetase (SamS) to complete the regeneration cycle. Typically, SahH enzymes are homotetramers that noncovalently bind nicotinamide adenine dinucleotide (NAD+) via an N‐terminal domain.9 The enzymatic cleavage of SAH requires NAD+‐dependent oxidation/re‐reduction steps.10 In vitro, the equilibrium of the SahH reaction favors the synthetic direction, yet the hydrolytic direction is supported by continuous enzymatic removal of Ado under physiological conditions.11 Ado, 2′‐deoxy‐Ado and adenine (Ade) derivatives bind tightly to SahH, and accumulation of these compounds inhibits its hydrolytic activity.12 Hence, removal of Ado by AdoK is likely to maintain SahH′s activity. Collectively, both AdoK and SahH activity is required to provide sufficient methyl donor to dimethylate norbaeocystin to psilocybin (Scheme 1).
Herein we report the characterization of AdoK and SahH from the psilocybin producer Psilocybe cubensis. Kinetic parameters and substrate specificities were investigated. In addition, the impact on psilocybin production due to the metabolic interplay of both enzymes is highlighted. Information on basidiomycete primary metabolism is scanty, and neither enzyme has genetically or biochemically been characterized for any basidiomycete, that is, a division of 30 000 species. Given the poorly studied interface of primary and secondary metabolism in basidiomycetes, this study has pilot character beyond psilocybin biosynthesis.
Results and Discussion
Gene expression analysis
The genes encoding adenosine kinase (adoK) and the SAM salvage pathway (sahH, metS, samS) were identified in the P. cubensis genome4a using the genes of experimentally proven enzymes from Saccharomyces cerevisiae or Homo sapiens as query (Table 1). First, an expression analysis was carried out from fungal mycelium derived from submerse (shake flasks) and emerse cultures (agar plates) as well as from fruiting body primordia and mature carpophores (Figure 1 A).
Table 1.
P. cubensis genes involved in the SAM salvage pathway.
|
Gene |
Length [bp]/(number of introns) |
CDS [bp][b] |
Gene product[c] |
Gene locus4a |
|---|---|---|---|---|
|
adoK [a] |
1426/5 |
1041 |
adenosine kinase |
NODE_5658: 141,215–142,640 |
|
sahH [a] |
1919/10 |
1290 |
S‐adenosyl‐l‐homocysteine hydrolase |
NODE_599: 19,244–21,162 |
|
metS |
2709/7 |
2295 |
l‐methionine synthetase |
NODE_7121: 231,799–234,507 |
|
samS |
1449/5 |
1182 |
S‐adenosyl‐l‐methionine synthetase |
NODE_6392: 312,966–314,414 |
[a] Sequences were deposited in the NCBI GenBank under accession numbers MN380460 (adoK) and MN380461 (sahH). [b] CDS: coding sequence excluding introns, which were verified for adoK and sahH, and bioinformatically predicted for metS and samS. [c] Experimentally verified for AdoK and SahH, predicted for MetS and SamS.
Figure 1.
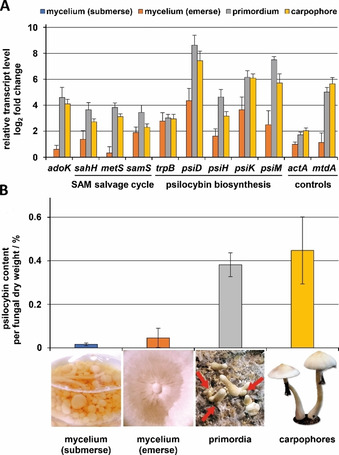
Expression analysis by qRT‐PCR and psilocybin amount from emerse or submerse mycelium, primordia and carpophores from P. cubensis. A) Expression analysis of genes of the psilocybin biosynthetic pathway (psiD, psiH, psiK, psiM), the SAM salvage pathway (sahH, metS, samS) and AdoK (adoK). actA (encoding α‐actin) and mtdA (peptide transporter) served as internal non‐regulated or carpophore‐specific gene controls, respectively. Transcript levels are expressed in ratios relative to the non‐induced calibrator cDNA derived from submerse mycelium (set to 1). The housekeeping gene gpdA (for glycerinaldehyde‐3‐phosphate dehydrogenase) served as internal reference. B) Psilocybin content and photographs from P. cubensis of various differential stages. Red arrows indicate nascent primordia.
The genes psiD, psiH, psiK, psiM as well as genes for tryptophan synthase (trpB),13 adenosine kinase (adoK) and the SAM salvage cycle (sahH, samS, metS) were tested.
A differential stage specific gene, mtdA, was included to verify the switch from asexual to sexual lifestyle. Its homolog mtd1, encoding a putative peptide transporter in the mushroom Schizophyllum commune, was previously characterized as carpophore‐specific marker gene.14 As expected, mtdA was induced in primordia and in fruiting bodies of P. cubensis, but not in undifferentiated mycelium (Figure 1 A). The trpB gene is sixfold upregulated in emerse mycelium and during mating which is consistent with an increased l‐tryptophan demand during psilocybin biosynthesis. In contrast, the psilocybin biosynthetic gene cluster (psiD, psiH, psiK, psiM) was only poorly expressed in any tested mycelial stage, but its transcription was induced during fruiting body formation which is consistent with previous observations.15 The gene for the gateway enzyme PsiD is most upregulated (395‐fold in primordia relative to submerse mycelium). However, except for psiK, expression of the psi genes is threefold downregulated in mature carpophores relative to primordia suggesting that the main biosynthetic performance occurs during the primordial stage. Moreover, all genes involved in the SAM salvage pathway including adoK and sahH were upregulated five‐ to 16‐fold in primordia and mature fruiting bodies. These results pointed to a correlation of the SAM cycle activity and psilocybin production and matched the high psilocybin content found in primordia and fruiting bodies (0.4 g g−1 dry biomass), but not in mycelia (Figure 1 B). Similar developmental stage‐specific production rates were observed for α‐ and β‐amanitin in Amanita exitialis. In this species, the toxin concentrations in the mycelium were only about 10 % of those in fruiting bodies.16 Co‐expression of adoK and sahH with the psi gene cluster under psilocybin‐ producing conditions suggested that both enzymes might be beneficial for psilocybin production. Hence, we recombinantly produced and biochemically characterized P. cubensis AdoK and SahH (Figure S1 in the Supporting Information).
Biochemical characterization of AdoK
Heterologous gene expression yielded a 36.9 kDa monomeric protein, according to size exclusion chromatography (Figure S2) which is a typical feature of eukaryotic AdoKs.17 Enzymatic activity was measured with a coupled fluorometric assay. Optimal turnover was detected at 42 °C and between pH 7.5–8.5 (Figure S3). These values are similar to wheat AdoK and to that of the parasite Toxoplasma gondii, which were optimally active at 37 °C.18 The observed slightly alkaline pH optimum correlates with previously described activities of AdoK from plants, but contrasts mammalian enzymes which prefer acidic reaction conditions.19 Mg2+ is essential for AdoK′s activity with a broad optimal concentration ranging from 0.5 to 5 mm (Figure S4). With the exception of Zn2+, various divalent cations can replace Mg2+ at <5 mm to a different extent (Mn2+>Mg2+>Co2+≫Ca2+>Zn2+; Figure 2 A). A supportive effect of Mn2+ has been described for AdoKs isolated from human placenta, Lupinus luteus seeds or the protist Leishmania donovani.19b, 20 However, Mn2+ inhibits P. cubensis AdoK activity at concentrations >1 mm.
Figure 2.
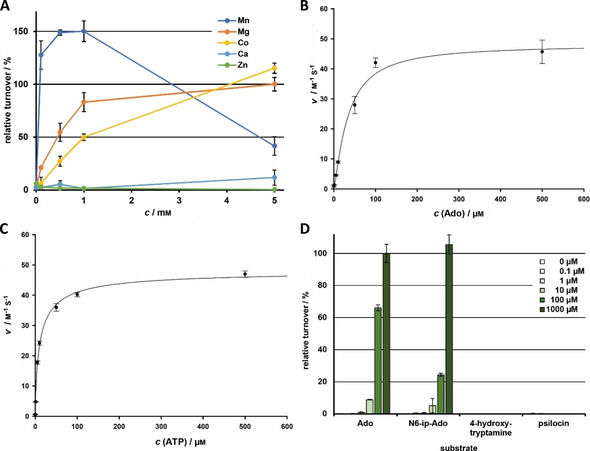
Characterization of P. cubensis AdoK. A) Impact of divalent metal cations on AdoK activity. Mn2+, Mg2+, Co2+, Ca2+, and Zn2+ were tested between 0.1 and 5 mm. Values are given in ratios relative to activity at 5 mm MgCl2. B) Michaelis–Menten kinetics for Ado. C) Michaelis–Menten kinetics for ATP. D) Substrate specificity of AdoK at 0.1–1000 μm. Ado, adenosine; N 6‐ip‐Ado: N 6‐(Δ2‐isopentenyl)adenosine.
In hamster AdoK, Mg2+ interacts with asparagine (N239) and glutamate (E242) within a highly conserved NXXE motif,21 which is present in all as yet characterized AdoK enzymes including Psilocybe AdoK. We generated a mutant gene for an AdoK variant in which this particular glutamate residue (E222) was replaced by alanine (AdoKE222A). This amino acid exchange resulted in an inactive enzyme, regardless of the cation concentration (Figure S4).
Monovalent cations had minor effects on enzyme activity. A cooperative effect of K+ in the binding ATP binding pocket has been described for mammalian AdoKs.20b, 22 In the case of P. cubensis AdoK, K+—but not Na+ or Li+—slightly stimulated activity (up to 140 %) at 10 mm, but were not essential for activity (Figure S5). In the crystal structure of murine AdoK, K+ is retained by amino acid residues D310, N312, I346, G351, and R349, which form an anion hole.22 An according mutant of Psilocybe AdoK lacking the potential K+ binding aspartate (AdoKD294A) was generated. It did not show turnover, independent of the K+ or Mg2+ concentration (Figures S4 and S6), suggesting a similar involvement of K+ in AdoK activity.
Sequence alignment with experimentally characterized representatives from various species revealed that P. cubensis AdoK clusters with the sole characterized fungal AdoK from baker's yeast S. cerevisiae and, more distantly, with those of the protists L. donovani and T. gondii (Figure S7). While most, if not all, mammalian and plant AdoKs are inhibited by Ado (at five‐ to 20‐fold K M) by a negative feedback mechanism,19b, 19d, 23 the above‐mentioned evolutionary branch comprises AdoKs, the activity of which is not or only slightly impaired by excess Ado.19b, 24 We next investigated the kinetic properties of AdoK. It followed a Michaelis–Menten type response, with a K M=33.6 μm for Ado, the k cat was 1.4×103 s−1 (Figure 2 B, Table 2). The K M value for AdoK′s second substrate ATP was 11.6 μm (Figure 2 C), which is lower than values obtained for human, plant, and other fungal representatives (200–2000 μm),19b, 19d, 24b although, enzymes with K M values of <0.05 μm (human erythrocytes, wheat germs) have been reported.18a, 19b
Table 2.
Kinetic parameters of P. cubensis AdoK and SahH.
|
Enzyme |
Substrate |
K M [μm] |
k cat [s−1] |
k cat/K M [m −1 s−1] |
|---|---|---|---|---|
|
AdoK |
Ado |
33.6 |
1.4×103 |
4.2×107 |
|
|
ATP |
11.6 |
1.4×103 |
1.2×108 |
|
|
|
|
|
|
|
SahH |
SAH |
57.2 |
0.2 |
4.1×103 |
|
|
Ado |
56.7 |
1.8 |
3.1×104 |
|
|
HCy |
19.8 |
1.8 |
9.1×104 |
We then interrogated P. cubensis AdoK′s substrate specificity. The enzyme was active with Ado but not with other naturally occurring nucleosides, such as inosine, uridine, guanosine, and thymidine (Figure S8), which reflects the substrate profile of other plant, fungal, and mammalian AdoKs.20a, 24b, 25 None of the tested nucleosides severely repressed enzymatic activity, even when present in 100‐fold excess over Ado (10 mm vs. 100 μm, Figure S8). In contrast to various bacterial, plant, or other eukaryotic AdoKs,19b, 19d, 26 the Psilocybe enzyme is not inhibited by Ado at 500 μm (Figure 2 B). The enzyme also phosphorylated the cytokinin precursor N 6‐(Δ2‐isopentenyl)adenosine, a zeatin derivative, as demonstrated both in our fluorometric assay and by activity testing by UHPLC–MS (Figures 2 D and S9). Phosphorylation of plant hormones by AdoKs has been demonstrated for plant AdoKs from Physcomitrella and Arabidopsis.19b The psilocybin congeners 4‐hydroxytryptamine and psilocin did not serve as substrates (Figure 2 D).
Biochemical characterization of SahH
Heterologous production of P. cubensis SahH yielded a 46.8 kDa protein. Size‐exclusion chromatography indicated a homotetramer (Figure S2) which was previously found as well for other SAH proteins.10 SAH hydrolysis was assayed by detecting the free thiol group of the product l‐homocysteine, using Ellman′s reagent in a colorimetric assay.9b SahH catalyzed the hydrolysis of SAH in HCy and Ado (Figure 3 A). Similar to our findings for AdoK, SahH is thermostable up to 50 °C, with an optimum turnover at 34 °C (Figure S10). Thermal stability is a common feature of SahH enzymes [27] and is most likely caused by the cofactor‐binding C‐terminal helix‐18.10 This helix is critical for NAD+ association via K426 and Y430 in the structures of human and Trypanosoma SahH10 and is present in all characterized eukaryotic SahHs including P. cubensis SahH. Activity of >90 % was detected in a range between pH 5.5 and 7.5, yet rapidly dropped above pH 8.0 (Figure S10). Optimum turnover was at pH 7.0–7.5. We further determined catalytic parameters of SahH in hydrolytic and synthetic direction (Figure 3 A–C). All substrates are accepted with similar affinity, according to their K M values (Table 2). K M values for the hydrolytic direction are slightly above the values obtained from other characterized enzymes (1–21 μm).28 The k cat values are in a similar range as previously published enzymes (k cat=0.2 s−1 (for SAH, hydrolytic direction), k cat=1.8 s−1 (Ado, HCy, synthetic direction).29 Most importantly, catalytic efficiency (k cat/K M) for Ado or HCy is 8‐ or 22‐fold higher than for SAH, respectively, suggesting that 1) SAH synthesis might be the kinetically favored reaction, at least in vitro, and, conversely, 2) SAH hydrolysis requires the activity of Ado removing enzymes, such as AdoK.
Figure 3.
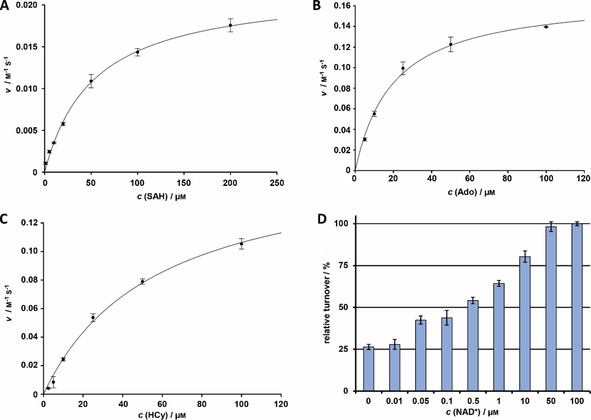
Characterization of P. cubensis SahH. Michaelis–Menten kinetics of SahH A) in hydrolytic direction (SAH as substrate), and synthetic direction with B) Ado or C) HCy as substrates. D) Impact of free NAD+ on SahH activity. Catalytic activity in samples not supplemented with NAD+ is probably due to carryover of enzyme‐bound co‐substrate from the heterologous protein production host.
The crystal structures of rat and Plasmodium SahH identified a histidine (H54) as essential for SAH binding at the ribose moiety and to abstract a proton from ribose at C‐4′ prior to elimination of HCy.9a, 30 We mutated the codon for H51, that is, the equivalent residue in P. cubensis SahH, into an alanine codon (SahHH51A). The in vitro activity assay showed a severely impaired catalytic capacity (<5 % of native SahH activity, Figure S11). The co‐substrate NAD+ binds noncovalently to the enzyme and is essential for its activity (Figure 3 D). Mechanistically, NAD+ first oxidizes the 3′‐OH group of SAH to facilitate efficient hydrolysis of SAH and the release of HCy. After addition of a water molecule to the remaining 3′‐keto‐4′,5′‐dihydroadenosine, the final reduction of the 3′‐keto group requires NADH+H+ to give Ado.31 Previous works suggested the C‐terminal lysine K425 (from the adjacent subunit) to properly position the nucleoside moiety of NAD+ via 2′‐OH and 3′‐OH.9a, 11 We verified the presumed NAD+ binding site9a in Psilocybe AdoK. A variant carrying an alanine at the C‐terminal NAD+ binding site, rather than the canonical NAD+‐binding lysine (K424 in P. cubensis SahH), was created, heterologously produced, and tested for activity in vitro. Independent of the NAD+ concentration, SahHK424A is virtually catalytically inactive (Figure S12).
SAH hydrolysis is strongly inhibited by elevated Ado concentrations and—to a lower extent—by tenfold excess of Ade over the SAH substrate (Figure S13). Feedback‐inhibition by adenosine derivatives has been described for several SahH enzymes.12, 32 However, the enzyme's activity is not product‐inhibited by AMP (Figure S13). As Ado is released during SAH hydrolysis, we reasoned that turnover by SahH may be increased when its product Ado is removed by AdoK‐mediated phosphorylation of Ado to AMP. A one‐pot enzymatic assay with SahH and AdoK showed a 2.5‐fold increased HCy release by SahH in an ATP‐concentration dependent manner (Figure S14). AdoK itself is not inhibited by high Ado concentrations (Figure 2 A), which allows for efficient conversion from SAH via Ado to AMP.
Adenosine metabolite processing increases in vitro psilocybin production
A biocatalytic in vitro route for psilocybin production from 4‐hydroxy‐l‐tryptophan was proposed and includes a three‐enzyme, one‐pot reaction, using PsiD, PsiK, and PsiM.4a The iterative methylation of norbaeocystin to psilocybin (Figure 4 A) by PsiM is thought to be a rate‐limiting critical step since N‐methyltransferases are comparably slow enzymes with k cat values ranging between 0.12 h−1 to 0.58 s−1.33 Previous studies demonstrated increased psilocybin yields, when Escherichia coli SAH nucleosidase and adenine deaminase were added to the PsiM reaction to irreversibly remove SAH from the chemical equilibrium.33d Hence, we hypothesized SAH could inhibit PsiM and ran assays in the presence of SAH, ranging between 1–16 mm. SAH competitively inhibited PsiM′s activity in a concentration‐dependent manner (Figures 4 A and B).
Figure 4.
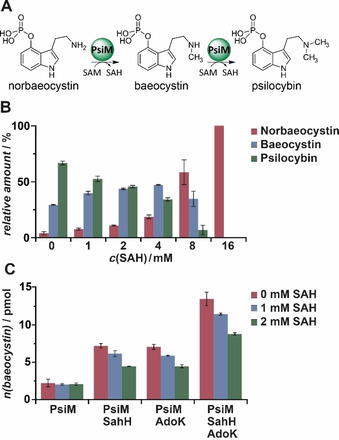
Impact of SAH, SahH and AdoK on PsiM‐mediated methylation reactions. A) Methylation reaction of norbaeocystin during psilocybin biosynthesis. B) Inhibited methylation in the presence of SAH. Relative metabolite amounts are calculated as fractions relative to the total amount of indole alkaloids. C) Impact of SahH and AdoK on baeocystin production. Reactions were carried out in absence and presence of inhibiting SAH at concentrations of 1 and 2 mm.
We further calculated the PsiM‐mediated production rates of the monomethylated product baeocystin in presence of SahH and AdoK. When solely SahH was added to the PsiM reaction, a threefold increase of enzymatic norbaeocystin methylation was observed (Figure 4 C). However, when both AdoK and SahH were present, a fivefold increase of product formation was detected indicating that the chemical equilibrium of SAH hydrolysis is on the side of SAH.11 Yet, the hydrolytic reaction is preferred in the presence of the SAH‐degrading activity of AdoK. The robust supporting effect of AdoK/SahH was likewise evident in the presence of inhibitory amounts of SAH (Figure 4 C). Interestingly, a simple two‐enzyme reaction with AdoK and PsiM elevated the norbaeocystin levels 2.5‐fold. We correlated this to a spontaneous hydrolysis of released SAH,8 which may be enhanced by downstream AdoK activity. In sum, this report provides evidence that 1) the genes of the SAM salvage cycle are co‐expressed with the psilocybin biosynthetic gene cluster and 2) SahH and AdoK can support the final step of psilocybin biosynthesis in vitro.
Conclusion
Psilocybin has entered clinical trials as low side effect medication against depression. Its simple four‐step enzymatic biosynthetic route requires continuous recycling of ATP and SAM to reach high metabolite titers. AdoK and SahH are critical players in the psilocybin pathway and their genes are expressed in Psilocybe fruiting bodies. SahH is required to efficiently remove SAH, a competitive inhibitor of the PsiM‐mediated methylation reaction. The strong product inhibition of SahH by Ado is in turn bypassed by the activity of AdoK. This close enzymatic interplay of PsiM, SahH, and AdoK allows for the efficient methyl transfer to norbaeocystin which completes psilocybin biosynthesis. This report provides insight into the primary/secondary metabolism interface in P. cubensis but also suggests novel strategies to improve yields for biocatalytic psilocybin production.
Experimental Section
Microbiological methods: P. cubensis FSU124074a was maintained on MEP (15 g L−1 malt extract, 3 g L−1 peptone, 18 g L−1 agar) plates. To collect biomass from emerse cultures, P. cubensis was cultivated for 7 days on MEP agar plates at 25 °C. For submerse cultures, the fungus was incubated in MEP liquid medium for 4 days at 25 °C and 140 rpm. Carpophore formation was induced as described.34 Mycelia were collected, shock‐frozen in liquid nitrogen and lyophilized prior to RNA isolation or metabolite quantification.
RNA isolation, cDNA synthesis and expression analysis: Fungal mycelium was lysed with 0.2 mm glass beads using a Fast Prep cell disruptor. RNA was isolated with the SV Total RNA Isolation System (Promega) using the manufacturer′s protocol. RNA (1 mg) was reverse transcribed to cDNA by using the RevertAid RT kit (Thermo) using an anchored oligo‐(dT)18 primer. Expression analysis was carried out with an AnalytikJena qTower3 using the qPCR Mix EvaGreen (Bio&SELL) and primers with a minimum primer efficiency of 91 % (Table S1, Figure S15). Amplification protocol: initial denaturation at 95 °C, 15 min followed by 40 cycles of amplification (95 °C, 15 s; 60 °C, 20 s; 72 °C, 20 s). A melting curve was obtained by heating from 60 to 95 °C. Genes encoding α‐actin (actA), glycerinaldehyde‐3‐phosphate dehydrogenase (gpdA) and enolase (enoA) served as internal housekeeping reference genes. Gene expression levels were determined by a described method.35
Metabolite quantitation: Lyophilized fungal mass (0.2–1 g) was ground to a fine powder and extracted twice with 50 mL methanol (MeOH). The pooled fractions were evaporated to dryness and the residue was dissolved in 10 mL water. After defatting in 10 mL cyclohexane, 9 mL of the aqueous phase were lyophilized. The residue was dissolved in 4 mL water/acetonitrile (ACN, 99:1), filtered and subjected to UHPLC–MS. Analyses were carried out on an Agilent Infinity II 1290 instrument, equipped with a diode array and a mass detector (6130 quadrupole) and a Phenomenex Luna Omega polar C18 column (100×2.1 mm, 1.6 μm). Eluent A was 0.1 % formic acid in water, eluent B was ACN. The following gradient was applied: 0 min, 1 % B; within 3 min to 10 % B; within another min to 100 % B; held at 100 % B for 2 min. The flow rate was 0.5 mL min−1 and the chromatogram was extracted at λ=280 nm. Psilocybin, baeocystin and norbaeocystin were quantified against a calibration curve with respective authentic reference standards.
Heterologous protein production: The construction of plasmids encoding AdoK, SahH and their respective mutants, and oligonucleotide primers, are described in the Supporting Information, and Tables S1–S2. Production of the N‐terminally His6‐tagged proteins was carried out in E. coli BL21 in 400 mL LB medium, supplemented with 50 μg mL−1 kanamycin, at 37 °C and 180 rpm. After reaching an OD600 of 0.6–0.8, the temperature was shifted to 16 °C, and gene expression was induced by adding 1 mm isopropyl‐β‐d‐1‐thiogalactopyranoside (IPTG). After 16 h, cells were harvested by centrifugation and the pellet was resuspended in binding buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole, pH 8.0). After cell lysis by a sonifier, the protein crude extract was centrifuged, and the cell‐free supernatant was transferred to an equilibrated gravity affinity column with 1 mL Protino Ni2+‐NTA resin. After incubation on ice for 30 min, the resin was washed with binding buffer containing increasing concentrations of imidazole (20 and 40 mm) and the proteins were eluted with binding buffer with 500 mm imidazole. The enzymes were re‐buffered in reaction buffer (50 mm TRIS‐HCl, 20 mm NaH2PO4, Triton X‐100 0.01 %, pH 7.8) by size exclusion chromatography (PD10, GE Healthcare). After supplementation with 10 % glycerol, enzymes were stored at −80 °C without loss of activity for up to 2 months. Protein production of PsiM was carried out as described.4a For size‐exclusion chromatography (SEC), the native and denatured (95 °C, 10 min) enzymes were re‐buffered in phosphate buffer (10 mm sodium dihydrogen phosphate, 140 mm NaCl, pH 7.5). SEC was performed by FPLC on an Äkta Pure 25 instrument with a Superdex 200 increase 10/300 GL column (GE Healthcare).
Enzymatic assays: AdoK activity was determined in kinase buffer (=reaction buffer+10 mm MgCl2) as described.22 To determine kinetic parameters, alternative substrates and activity of mutant proteins, a continuous assay was performed: 70 μL master mix (2 U mL−1 horseradish peroxidase, 1 U mL−1 pyruvate oxidase, 20 U mL−1 pyruvate kinase, 300 μm phosphoenolpyruvate, 100 μm thiamine pyrophosphate, 10 μm flavin adenine dinucleotide, 35 μm AmplifluRed in kinase buffer) was mixed with 10 μL adenosine or alternative phosphate acceptors (0–1000 μm final concentration) and 10 μL ATP (0–100 μm final). The reaction was started by adding 10 μL (34 nm final) AdoK or AdoK mutants. Fluorescence detection was carried out in black 96‐well plates at λ ex=545 nm and λ em=600 nm at 30 °C for 60 min in a ClarioStar plate reader (BMG Labtech). To determine temperature and pH optima, and metal cation dependence, a discontinuous assay was performed: ATP (200 μm), Ado (200 μm) and AdoK (34 nm) were mixed in kinase buffer, adjusted to pH 4–9 (in 50 mm acetate or TRIS buffer), or amended with divalent cations (0.01–5 mm CaCl2, CoCl2, MgSO4, MnCl2 or ZnAc2) or monovalent cations (0.1–100 mm NaCl, KCl, LiCl). The reaction was incubated for 1, 5, or 30 min at 42 °C (for temperature optimum varied between 4–60 °C). The reactions were then placed on ice and the enzyme was removed by filtering through Sartorius VIVAspin 100 spin columns (10 kDa cutoff). The ADP concentration in a 20 μL aliquot of the enzyme‐free flow‐through was determined by addition of 80 μL master mix as described above.
SahH activity was determined in hydrolase buffer (reaction buffer+100 μm NAD+). SAH hydrolysis was recorded by detecting free thiol groups of HCy, using Ellman′s reagent (5,5′‐dithiobis‐(2‐nitrobenzoic acid), DTNB) as described.9b A continuous assay was used to determine activity, kinetic parameters, pH optimum, NAD+‐dependence, and the activity of mutant proteins. The reactions contained 64 nm SahH, 200 μm DTNB and 0–100 μm SAH in hydrolase buffer and were recorded at λ abs=412 nm in transparent 384‐well plates at 34 °C for 30 min. The temperature optimum was determined discontinuously: the reaction was incubated for 30 min at 4–60 °C, briefly chilled on ice and filtered through a VIVAspin 100 column (10 kDa cutoff). The HCy concentration in a 100 μL aliquot of the enzyme‐free flow‐through was determined at λ abs=412 nm as described above.
PsiM reactions were performed in TRIS⋅HCl buffer (50 mm, pH 8.0) by quantitative detection of the products baeocystin and psilocybin by UHPLC–MS against calibrated authentic standards. The assay for SAH inhibition comprised 3 mm SAM, 1 mm norbaeocystin, 1 μm PsiM, and SAH concentrations varied between 1 and 16 mm. The coupled enzymatic assay with PsiM, SahH and AdoK was carried out in the presence of inhibiting SAH concentrations (0–2 mm) to determine if the PsiM reaction increases. The assay was composed of 3 mm SAM, 2 mm SAH, 1 mm norbaeocystin, 6 mm ATP, 6 mm MgCl2 and 100 μm NAD+. In additional reactions, SahH (64 nm) and AdoK (34 nm) were added. Reactions were started by addition of 1 μm PsiM. After incubation for 2 and 24 h at 25 °C, reaction mixtures were frozen and lyophilized. The residue was dissolved in 50 μL MeOH and subjected to UHPLC/MS as described.4a
Chemical synthesis and analysis of N 6 ‐(Δ2‐isopentenyl)adenosine: The alternative AdoK substrate, N 6‐(Δ2‐isopentenyl)adenosine was synthesized according to a published protocol in modified form.36 Details are given in the Supporting Information. The identity of the compound was confirmed by NMR spectroscopy data (Table S3) that is compatible with previously published values.37
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Jonas Wick and Julia Gressler (FSU, Jena) for providing the adoK expression vector pJG14 and Heike Heinecke (HKI, Jena) for recording NMR spectra. This work was supported by the Deutsche Forschungsgemeinschaft (DFG grant HO2515/7‐1) and by the Usona Institute (Madison, WI, USA). D.H. is also supported by the DFG Collaborative Research Center ChemBioSys 1127 (project B05).
R. Demmler, J. Fricke, S. Dörner, M. Gressler, D. Hoffmeister, ChemBioChem 2020, 21, 1364.
Contributor Information
Dr. Markus Gressler, Email: markus.gressler@leibniz-hki.de.
Prof. Dr. Dirk Hoffmeister, Email: dirk.hoffmeister@leibniz-hki.de.
References
- 1.
- 1a. Hofmann A., Heim R., Brack A., Kobel H., Experientia 1958, 14, 107–109; [DOI] [PubMed] [Google Scholar]
- 1b. Hofmann A., Heim R., Brack A., Kobel H., Frey A., Ott H., Petrzilka T., Troxler F., Helv. Chim. Acta 1959, 42, 1557–1572. [Google Scholar]
- 2. Carhart-Harris R. L., Roseman L., Bolstridge M., Demetriou L., Pannekoek J. N., Wall M. B., Tanner M., Kaelen M., McGonigle J., Murphy K., Leech R., Curran H. V., Nutt D. J., Sci. Rep. 2017, 7, 13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Agurell S., Nilsson J. L., Acta Chem. Scand. 1968, 22, 1210–1218; [DOI] [PubMed] [Google Scholar]
- 3b. Agurell S., Nilsson J. L., Tetrahedron Lett. 1968, 9, 1063–1064. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Fricke J., Blei F., Hoffmeister D., Angew. Chem. Int. Ed. 2017, 56, 12352–12355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12524–12527; [Google Scholar]
- 4b. Reynolds H. T., Vijayakumar V., Gluck-Thaler E., Korotkin H. B., Matheny P. B., Slot J. C., Evol. Lett. 2018, 2, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bigwood J., Beug M. W., J. Ethnopharmacol. 1982, 5, 287–291. [DOI] [PubMed] [Google Scholar]
- 6. Kornberg A., W. E. Pricer, Jr. , J. Biol. Chem. 1951, 193, 481–495. [PubMed] [Google Scholar]
- 7.
- 7a. Fabbro D., Cowan-Jacob S. W., Moebitz H., Br. J. Pharmacol. 2015, 172, 2675–2700; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Camici M., Garcia-Gil M., Tozzi M. G., Int. J. Mol. Sci. 2018, 19, E784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liao C., Seebeck F. P., Nat. Catal. 2019, 2, 696–701. [Google Scholar]
- 9.
- 9a. Hu Y., Komoto J., Huang Y., Gomi T., Ogawa H., Takata Y., Fujioka M., Takusagawa F., Biochemistry 1999, 38, 8323–8333; [DOI] [PubMed] [Google Scholar]
- 9b. Lozada-Ramírez J. D., Martínez-Martínez I., Sánchez-Ferrer A., García-Carmona F., J. Biochem. Biophys. Methods 2006, 67, 131–140. [DOI] [PubMed] [Google Scholar]
- 10. Cai S., Li Q. S., Fang J., Borchardt R. T., Kuczera K., Middaugh C. R., Schowen R. L., Nucleosides Nucleotides Nucleic Acids 2009, 28, 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turner M. A., Yang X., Yin D., Kuczera K., Borchardt R. T., Howell P. L., Cell Biochem. Biophys. 2000, 33, 101–125. [DOI] [PubMed] [Google Scholar]
- 12. Hershfield S., J. Biol. Chem. 1979, 254, 22–25. [PubMed] [Google Scholar]
- 13. Blei F., Baldeweg F., Fricke J., Hoffmeister D., Chem. Eur. J. 2018, 24, 10028–10031. [DOI] [PubMed] [Google Scholar]
- 14. Lengeler K. B., Kothe E., Curr. Genet. 1999, 36, 159–164. [DOI] [PubMed] [Google Scholar]
- 15. Torrens-Spence M. P., Liu C. T., Pluskal T., Chung Y. K., Weng J. K., ACS Chem. Biol. 2018, 13, 3343–3353. [DOI] [PubMed] [Google Scholar]
- 16. Zhang P., Chen Z., Hu J., Wei B., Zhang Z., Hu W., FEMS Microbiol. Lett. 2005, 252, 223–228. [DOI] [PubMed] [Google Scholar]
- 17. Song K., Li Y., He H., Liu L., Zhao P., Xia Q., Wang Y., Int. J. Mol. Sci. 2019, 20, e3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Chen C. M., Eckert R. L., Plant Physiol. 1977, 59, 443–447; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Darling J. A., W. J. Sullivan, Jr. , Carter D., Ullman B., Roos D. S., Mol. Biochem. Parasitol. 1999, 103, 15–23. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Miller R. L., Adamczyk D. L., Miller W. H., Koszalka G. W., Rideout J. L., Beacham L. M., Chao E. Y., Haggerty J. J., Krenitsky T. A., Elion G. B., J. Biol. Chem. 1979, 254, 2346–2352; [PubMed] [Google Scholar]
- 19b. Datta A. K., Bhaumik D., Chatterjee R., J. Biol. Chem. 1987, 262, 5515–5521; [PubMed] [Google Scholar]
- 19c. von Schwartzenberg K., Kuse S., Reski R., Moffatt B., Laloue M., Plant J. 1998, 13, 249–257; [DOI] [PubMed] [Google Scholar]
- 19d. Moffatt B. A., Wang L., Allen M. S., Stevens Y. Y., Qin W., Snider J., von Schwartzenberg K., Plant Physiol. 2000, 124, 1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Guranowski A., Jakubowski H., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1983, 742, 250–256; [DOI] [PubMed] [Google Scholar]
- 20b. Hurley M. C., Lin B., Fox I. H., J. Biol. Chem. 1985, 260, 15675–15681. [PubMed] [Google Scholar]
- 21. Maj M. C., Singh B., Gupta R. S., Biochemistry 2002, 41, 4059–4069. [DOI] [PubMed] [Google Scholar]
- 22. de Oliveira R. R., Morales-Neto R., Rocco S. A., Sforça M. L., Polo C. C., Tonoli C. C. C., Mercaldi G. F., Cordeiro A. T., Murakami M. T., Franchini K. G., Sci. Rep. 2018, 8, 11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Yamada Y., Goto H., Ogasawara N., Biochim. Biophys. Acta 1980, 616, 199–207; [DOI] [PubMed] [Google Scholar]
- 23b. Faye F., Le Floc′h F., Plant Physiol. Biochem. 1997, 35, 15–22. [Google Scholar]
- 24.
- 24a. Bhaumik D., Datta A. K., Mol. Biochem. Parasitol. 1988, 28, 181–188; [DOI] [PubMed] [Google Scholar]
- 24b. Barrado P., Rodriguez M. J., Jimenez A., Lobato M. F., Yeast 2003, 20, 1145–1150; [DOI] [PubMed] [Google Scholar]
- 24c. Naguib F. N. M., Rais R. H., Al Safarjalani O. N., el Kouni M. H., Comp. Biochem. Physiol. Part B 2015, 188, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palella T. D., Andres C. M., Fox I. H., J. Biol. Chem. 1980, 255, 5264–5269. [PubMed] [Google Scholar]
- 26. Long M. C., Escuyer V., Parker W. B., J. Bacteriol. 2003, 185, 6548–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakanishi M., Iwata A., Yatome C., Kitade Y., J. Biochem. 2001, 129, 101–105. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Yang X., Borchardt R. T., Arch. Biochem. Biophys. 2000, 383, 272–280; [DOI] [PubMed] [Google Scholar]
- 28b. Singhal A., Arora G., Sajid A., Maji A., Bhat A., Virmani R., Upadhyay S., Nandicoori V. K., Sengupta S., Singh Y., Sci. Rep. 2013, 3, 2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Čtrnáctá V., Stejskal F., Keithly J. S., Hrdý I., FEMS Microbiol. Lett. 2007, 273, 87–95. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka N., Nakanishi M., Kusakabe Y., Shiraiwa K., Yabe S., Ito Y., Kitade Y., Nakamura K. T., J. Mol. Biol. 2004, 343, 1007–1017. [DOI] [PubMed] [Google Scholar]
- 31. Lozada-Ramírez J. D., Sánchez-Ferrer A., García-Carmona F., Appl. Microbiol. Biotechnol. 2012, 93, 2317–2325. [DOI] [PubMed] [Google Scholar]
- 32. Knudsen R. C., Yall I., J. Bacteriol. 1972, 112, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Vit A., Misson L., Blankenfeldt W., Seebeck F. P., ChemBioChem 2015, 16, 119–125; [DOI] [PubMed] [Google Scholar]
- 33b. Misson L., Burn R., Vit A., Hildesheim J., Beliaeva M. A., Blankenfeldt W., Seebeck F. P., ACS Chem. Biol. 2018, 13, 1333–1342; [DOI] [PubMed] [Google Scholar]
- 33c. Song H., van der Velden N. S., Shiran S. L., Bleiziffer P., Zach C., Sieber R., Imani A. S., Krausbeck F., Aebi M., Freeman M. F., Riniker S., Künzler M., Naismith J. H., Sci. Adv. 2018, 4, eaat2720; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33d. Fricke J., Sherwood A., Kargbo R., Orry A., Blei F., Naschberger A., Rupp B., Hoffmeister D., ChemBioChem 2019, 20, 2824–2829. [DOI] [PubMed] [Google Scholar]
- 34. Lenz C., Wick J., Hoffmeister D., J. Nat. Prod. 2017, 80, 2835–2838. [DOI] [PubMed] [Google Scholar]
- 35. Pfaffl M. W., Nucleic Acids Res. 2001, 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pačes V., Werstiuk E., Hall R. H., Plant Physiol. 1971, 48, 775–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Casati S., Manzocchi A., Ottria R., Ciuffreda P., Magn. Reson. Chem. 2010, 48, 745–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary