

Abstract
The hydrophosphorylation of phenylacetylene with di(aryl)phosphane oxides Ar2P(O)H (Pudovik reaction) yields E/Z‐isomer mixtures of phenylethenyl‐di(aryl)phosphane oxides (1). Alkali and alkaline‐earth metal di(aryl)phosphinites have been studied as catalysts for this reaction with increasing activity for the heavier s‐block metals. The Pudovik reaction can only be mediated for di(aryl)phosphane oxides whereas P‐bound alkyl and alcoholate substituents impede the P−H addition across alkynes. The demanding mesityl group favors the single‐hydrophosphorylated products 1‐Ar whereas smaller aryl substituents lead to the double‐hydrophosphorylated products 2‐Ar. Polar solvents are beneficial for an effective addition. Increasing concentration of the reactants and the catalyst accelerates the Pudovik reaction. Whereas Mes2P(O)H does not form the bis‐phosphorylated product 2‐Mes, activation of an ortho‐methyl group and cyclization occurs yielding 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3).
Keywords: catalysis, hydrophosphorylation, phosphane oxide addition, Pudovik reaction, s-block metals
The Pudovik reaction is an atom‐economic addition of phosphane oxides across alkynes, strongly depending on the nature of the s‐block metal, the solvent, the bulkiness of P‐bound groups, and the concentration. Bulky mesityl groups can interact with the newly formed C=C bond of the alkenylphosphane oxides.
Introduction
s‐Block metal‐catalyzed processes are experiencing a vastly growing interest due to their beneficial properties including low toxicity (with highly toxic beryllium being an exception), global abundance and availability. Furthermore, tuning of reactivity succeeds by the choice of the metals (hardness, size, Lewis acidity), co‐ligands (denticity, Lewis basicity, donor strength) and solvents (polarity, denticity). Therefore, research with respect to hydrofunctionalization1 of alkenes and alkynes focusses especially on the essential metals sodium, potassium, magnesium, and calcium. However, the redox‐inert s‐block metals show a different catalytic mechanism than redox‐active transition metal‐based catalysts which also allow oxidative addition and reductive elimination steps contrary to the s‐block metals.
The addition of P−H bonds of phosphane oxides across unsaturated hydrocarbons (Pudovik reaction) like alkynes yielding alkenylated and alkylated phosphane oxides (Scheme 1) represents a versatile procedure for the formation of phosphorus–carbon bonds. Depending on the precatalyst, radical and polar reaction different mechanisms have been discussed. Thus, diverse catalyst systems have been investigated such as for example, Al2O3/KOH,2 palladium(0),3 ytterbium(II),4 rhodium(I),5, 6 nickel,7 potassium,8, 9 and bases in general.10 In the presence of air the product range deviates and radical mechanisms account for the observed products and by‐products.11, 12
Scheme 1.
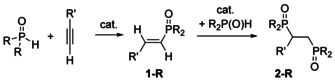
Single‐ and double‐hydrophosphorylated alkynes, yielding alkenylphosphane oxides 1‐R and 1,2‐bis(phosphorylated) alkanes 2‐R.
Potassium‐mediated addition of dimesitylphosphane oxide (R=Mes) across alkynes yielded varying mixtures of Z‐ and E‐isomeric alkenyl‐dimesitylphosphane oxides 1‐R with the isomer ratio depending on the solvent.9 Furthermore, this study revealed that these alkenes that formed during the Pudovik reaction were inert under the applied reaction conditions. Suitable substituents R′ included aryl, ester and bulky trialkylsilyl groups.
Here we elucidated the dependency of the Pudovik reaction on nature and size of the s‐block metal, the P‐bound substituent R and the solvent used. Furthermore, we studied a unique cyclization of the alkenylphosphane oxide to 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide if dimesitylphosphane oxide was applied in the Pudovik reaction. As precatalysts, we chose the bis(trimethylsilyl)amides (hexamethyldisilazanides, hmds) of the alkali and alkaline‐earth metals because these complexes are easily accessible on large scale and highly soluble in common organic reagents guaranteeing homogeneous reaction conditions. In addition, the s‐block metals sodium, potassium, magnesium and calcium are essential non‐toxic metals which are globally abundant and very inexpensive.
Results and Discussion
Influence of the metal on the Pudovik reaction
Due to the fact that alkali metal diarylphosphinites (M−OPR2) degrade and/or dismutate to the soluble phosphanides M−PR2 and to the sparingly soluble phosphinates MO2PR2,13, 14 the activity of stock solutions of M−OPR2 slowly decreases. Hill and co‐workers15 already observed under reductive conditions the formation of diphenylphosphanide in solutions containing calcium diphenylphosphinite species. Nevertheless, [(thf)4Ca(OPPh2)2] is stable in the crystalline state and hence, the single crystal structure could be determined.15 Degradation and disproportionation again yield phosphanide and phosphinate species the latter being very poorly soluble due to the formation of strand‐like polymeric structures with bridging phosphinate ligands.16
Therefore freshly prepared s‐block metal diarylphosphinites have to be used for reliable studies and the following unified procedure was applied: The s‐block metal phosphinites M−OPR2 were prepared in situ by metalation of HP(O)R2 with the hexamethyldisilazanides (hmds) of the alkali and alkaline‐earth metals yielding the corresponding metal diarylphosphinites of the type M−OPAr2 (Scheme 2). Quantitative conversion was verified by 31P NMR spectroscopy. At room temperature, 5 mol % of the amide were combined with dimesitylphosphane oxide in THF and then a slight excess amount of phenylacetylene was added. After one hour, an aliquot was protolyzed with ethanol and the product composition was investigated by 31P NMR spectroscopy.
Scheme 2.
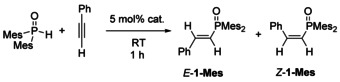
s‐Block metal‐mediated hydrophosphorylation of phenylacetylene with dimesitylphosphane oxide, using M(hmds) and M(hmds)2 of the alkali and alkaline‐earth metals, respectively, as precatalysts; c(HPOMes2) ≈0.05 mol/l.
The outcome of this study is summarized in Table 1. The precatalysts with the small metals such as Li(hmds) (entry 1), Mg(hmds)2 and Ca(hmds)2 (entries 6 and 7) could not mediate this addition reaction, whereas fast conversion was observed for the complexes of the heavier metals (entries 3–5 and 9). The formation of double‐hydrophosphorylated products is disadvantageous due to intramolecular steric strain between the phenyl group and the P‐bound mesityl substituents. The E/Z‐ isomer ratio that was observed after one hour remained unchanged and very comparable values were determined after 27 hours. A distinctive exception pertains to the hydrophosphorylation products through catalysis by Na(hmds) (entry 2). After about one day the resonance of the Z‐isomer vanished almost completely, leading to an E/Z‐ratio of 31/1. Contrary to the other metal catalysts, double‐hydrophosphorylated 2‐Mes was observed (7.5 % of 2‐Mes and 92.5 % of E/Z‐1‐Mes) and unequivocally identified by the characteristic 31P NMR parameters of two doublets at δ(31P)=37 and 44 ppm with a 3 J(P,P) coupling constant of 47.7 Hz (see below “Mechanism of phosphindole 1‐oxide formation”). No other products were formed under these reaction conditions. Steric strain is much more pronounced in the Z‐isomer of 1‐Mes than in E‐1‐Mes. Therefore, the second hydrophosphorylation step occurred preferentially at Z‐1‐Mes to significantly lower steric strain through the formation of less strained 2‐Mes decreasing the concentration of Z‐1‐Mes. It can be excluded in either case that the isomeric ratio changed after complete conversion regardless of the fact if catalyst was present or not.
Table 1.
Dependency of the Pudovik reaction (addition of dimesitylphosphane oxide across phenylacetylene) on the s‐block metal of the applied bis(trimethylsilyl)amide precatalyst [Scheme 2, THF, c(HPOMes2)∼0,05 mol/l].
|
Entry |
Metal |
After 1 h |
After 27 h |
||
|---|---|---|---|---|---|
|
|
|
Conv. [%] |
E/Z |
Conv. [%] |
E/Z |
|
1 |
Li |
0 |
– |
8 |
5/1 |
|
2 |
Na |
31 |
3/1 |
>99 |
31/1[a] |
|
3 |
K |
93 |
5/1 |
>99 |
5/1 |
|
4 |
Rb |
>99 |
7/1 |
>99 |
7/1 |
|
5 |
Cs |
>99 |
11/1 |
>99 |
13/1 |
|
6 |
Mg |
0 |
– |
0 |
– |
|
7 |
Ca |
0 |
– |
0 |
– |
|
8 |
Sr |
3 |
3/1 |
0 |
– |
|
9 |
Ba |
85 |
6/1 |
95 |
22/1 |
[a] The amount of Z‐1‐Mes is lowered due to a preferred second Na‐mediated hydrophosphorylation yielding 2‐Mes (see text).
Substituent effects on the Pudovik reaction
For the following experiments, K(hmds) was applied as precatalyst in the Pudovik reaction as depicted in Scheme 3. In a typical experiment 5 mol % of K(hmds) were combined with di(organyl)phosphane oxide in THF and then phenylacetylene was added to this solution at room temperature to elucidate the influence of the substitution pattern on the formation of single‐ and double‐hydrophosphorylated phenylacetylene. The conversion (decline of the R2P(O)H concentration) and the amount of single‐ (1‐R) and double‐hydrophosphorylation products (2‐R) were determined by 31P NMR spectroscopy.
Scheme 3.
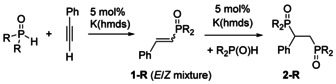
Potassium‐mediated hydrophosphorylation of phenylacetylene with di(organyl)phosphane oxide, using K(hmds) as precatalyst; c(HPOMes2) ≈0.05 mol/l.
The outcome of this study is summarized in Table 2. The conversion was determined by the decline of starting phosphane oxide (conv.). The product ratio of 1‐R and 2‐R was estimated by integration of the 31P NMR spectra. As discussed above, dimesitylphosphane oxide was only added once yielding phenylethenyl‐dimesitylphosphane oxide (styryl‐di(mesityl)phosphane oxide, 1‐Mes (entry 1) with an E/Z‐ratio of 5/1. Removal of one ortho‐methyl substituent and use of ortho‐tolyl substituents decelerates the conversion and after 1 h also double‐hydrophosphorylated phenylacetylene (2‐oTol) was observed (entry 2). After 22 h, the mono‐hydrophosphorylation product 1‐oTol was completely converted to the double‐phosphorylated ethane derivative 2‐oTol. Similar steric strain can be assumed if one mesityl group is replaced by a phenyl substituent (entry 3). Removal of all methyl groups and application of diphenylphosphane oxide accelerates the second hydrophosphorylation step and only product 2‐Ph was detected despite the fact that the conversion was incomplete after one hour (entry 4). The benzyl‐substituted phosphane oxide (entry 5) proved to be more reactive than the phenyl congener. Bis(4‐dimethylaminophenyl)phosphane oxide (entry 6) added significantly faster across the alkyne unit and only the double‐hydrophosphorylated product 2‐R was found. Alkyl‐ (entries 7 and 8), alkoxide (entry 9) and phenolato groups (entry 10) suppressed the potassium‐mediated hydrophosphorylation of phenylacetylene under these reaction conditions.
Table 2.
Dependency of the Pudovik reaction on the substituents of the applied secondary phosphane oxides R2P(O)H [Scheme 3, THF, c(HPOR2)∼0,05 mol/l].
|
Entry |
R |
After 1 h |
After 22 h |
||||
|---|---|---|---|---|---|---|---|
|
|
|
Conv. [%] |
A (E/Z) |
B |
Conv. [%] |
A (E/Z) |
B |
|
1 |
Mes |
93 |
100 (83/17) |
0 |
>99 |
100 (83/17) |
0 |
|
2 |
oTol |
55 |
66 (100/0) |
34 |
>99 |
<1 |
>99 |
|
3 |
Mes/Ph |
76 |
36 (100/0) |
64 |
>99 |
16 (100/0) |
84 |
|
4 |
Ph |
14 |
<1 |
>99 |
36 |
<1 |
>99 |
|
5 |
Bz |
64 |
<1 |
>99 |
>99 |
<1 |
>99 |
|
6 |
C6H4‐4‐NMe2 |
>99 |
<1 |
>99 |
>99 |
<1 |
>99 |
|
7 |
Cy |
0 |
– |
– |
0 |
– |
– |
|
8 |
tBu |
0 |
– |
– |
0 |
– |
– |
|
9 |
OEt |
0 |
– |
– |
0 |
– |
– |
|
10 |
OPh |
0 |
– |
– |
0 |
– |
– |
Proposed mechanism for the Pudovik reaction
The proposed mechanism of the Pudovik reaction is depicted in Scheme 4. The s‐block metal di(aryl)phosphinite adds across the C≡C functionality. Protolysis of this intermediate with Ar2P(O)H regains the catalyst MOPAr2 and yields E/Z‐mixtures of mono‐phosphorylated 1‐Ar. The newly formed alkene moiety is reactive enough to add another phosphinite leading to bis‐phosphorylated compounds. Now protolysis yields very sparingly soluble bis‐phosphorylated 2‐Ar which precipitates from the reaction mixture and hence, is removed impeding a possible equilibrium interconverting 2‐Ar back into 1‐Ar. Due to intramolecular shielding and steric strain only the mono‐hydrophosphorylation product was isolated as an isomeric mixture of 1‐Mes.
Scheme 4.
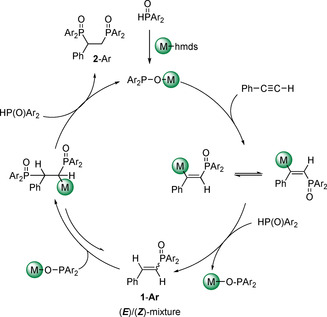
Simplified proposed mechanism for the formation of the bis‐phosphorylated products 2‐Ar. The equilibrium between 1‐Ar and the bis‐phosphorylated compounds was deduced from NMR spectroscopic observation of intermediate 2‐Mes during formation of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide 3 (see below).
Solvent effects on the Pudovik reaction
The initial study of the potassium‐mediated hydrophosphorylation of phenylacetylene with dimesitylphosphane oxide showed a strong dependency of the E/Z‐ratio on the nature of the solvent.9 Therefore we performed this catalytic addition reaction (Scheme 2) in various solvents, including aliphatic and aromatic hydrocarbons, ethers, amines, and acetonitrile. In some of these solvents (pentane, 1,4‐dioxane, NEt3, methyl tert‐butyl ether, and diethyl ether), the solubility of the hydrophosphorylation product is rather poor, leading to suspensions toward the end of the conversions. Nevertheless, in all studied solvents a complete conversion was observed within one day.
The dependency of the potassium‐mediated hydrophosphorylation of phenylacetylene with di(mesityl)phosphane oxide on the polarity of the solvent is summarized in Table 3. Several scales have been investigated to characterize polarity of solvents with larger values for more polar molecules.17 The E T values were determined with a pyridinium‐N‐phenolate‐derived dye with the color depending on the polarity of the medium. The dielectric constant ϵ r and the dipole moment μ of the solvent molecules are given as well in Table 3 and correspond satisfactory to the E T values.17
Table 3.
Dependency of the Pudovik reaction on the solvent [Scheme 2, reaction time of 1 h, c(HPOR2)∼0.05 mol/l].
|
Entry |
Solvent |
ET [a] |
ϵr [b] |
μ[c] |
Conv. [%] |
E/Z |
|---|---|---|---|---|---|---|
|
1 |
pentane |
31 |
1.84 |
0 |
70 |
70/30 |
|
2 |
dioxane |
36 |
2.27 |
0.45 |
63 |
88/12 |
|
3 |
benzene |
34.3 |
2.4 |
0 |
67 |
67/33 |
|
4 |
toluene |
32.1 |
2.43 |
0.31 |
52 |
74/26 |
|
5 |
NEt3 |
32.1 |
2.45 |
0.07 |
82 |
67/33 |
|
6 |
MeOtBu |
34.7 |
|
|
>99 |
94/6 |
|
7 |
2‐MeTHF |
36.5 |
3.05 |
|
>99 |
75/25 |
|
8 |
Et2O |
34.5 |
4.42 |
1.15 |
91 |
93/7 |
|
9 |
Me4THF |
|
5.03 |
|
>99 |
82/18 |
|
10 |
THP |
36.2 |
5.68 |
1.63 |
>99 |
93/7 |
|
11 |
DME |
35.8 |
7.2 |
1.71 |
>99 |
60/40 |
|
12 |
THF |
37.4 |
7.47 |
1.75 |
>99 |
83/17 |
|
13 |
MeCN |
45.6 |
36.0 |
3.53 |
>99 |
24/76 |
Increasing polarity and donor strength of the solvent increases the rate with quantitative conversions already after one hour for ethereal solvents (entries 6–12 with diethyl ether, entry 8, being an exception) and acetonitrile (entry 13). The E/Z ratio varies strongly generally in favor of the E‐isomer. It is noteworthy that in acetonitrile the Z‐isomer is the major product. In this table acetonitrile is not only the most polar molecule but also the strongest Brønsted acid and hence, interferences with the catalytic cycle cannot reliably be excluded.
Concentration effects on the Pudovik reaction
In another experiment, we investigated the influence of the concentration on the potassium‐mediated Pudovik reaction according to Scheme 2. Thus, the molar ratio remained unchanged (THF solution, 5 mol % of catalyst, equimolar ratio of di(mesityl)phosphane oxide and phenylacetylene), but the concentration of di(mesityl)phosphane oxide varied from 0.035 to 0.06 m and finally to 0.175 m. The E/Z ratio of the mono‐phosphorylation product styryl‐di(mesityl)phosphane oxide 1‐Mes was determined after 30 minutes by protolysis of the reaction mixture (to inactivate the catalyst) and the E/Z ratio of styryl‐di(mesityl)phosphane oxide 1‐Mes was elucidated by 31P NMR spectroscopy. The E/Z ratio decreased from 4.2/1 over 3.3/1 to 1.6/1 for the 0.035 m, 0.06 m and 0.175 m solutions, respectively, with increasing amounts of the Z‐isomer with increasing concentration. In addition, only the diluted solution still contained small amounts of starting Mes2P(O)H whereas all di(mesityl)phosphane oxide had already been consumed after 30 minutes in more concentrated solutions. The E/Z ratio of product 1‐Mes did not change for the diluted solutions if prolonged reaction times were applied. However, the concentrated reaction mixture behaved differently. After one hour, the E/Z ratio changed to 1.8/1 in favor of the E‐isomer and after 3 days, only the E‐isomer was observed. However, two new resonances with δ(31P) values of about 53 and 59 ppm with an intensity ratio of 14/1 were observed. These resonances were only observed in Lewis basic solvents like ethers and amines whereas in non‐donor solvents these resonances were absent. We showed earlier that E‐ and Z‐isomers interconvert into each other rather slowly suggesting that the Z‐1‐Mes preferably reacted to these new products.
After isolation of this new unexpected compound we determined the molar weight with 1H DOSY NMR experiments (Stalke method).18 As an internal standard adamantane with a molar weight of 136.24 g mol−1 was added to a [D8]toluene solution as depicted in Figure S10 (see Supporting Information). Comparison of the diffusion coefficients as described earlier18 gave a molar weight of 407 g mol−1 supporting a structure of a similar mass than 1‐Mes.
To elucidate the exact product composition and the reaction mechanism HRMS and X‐ray diffraction studies as well as NMR experiments were performed. The HRMS of the M+ species verified a formula of C26H29OP which is identical to the mass of the mono‐phosphorylated product (verifying a rearrangement product of 1‐Mes). Spectroscopic data and X‐ray diffraction studies verified the formation of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3) as shown in Scheme 5; the product formed formally by addition of a C−H bond of an ortho‐methyl group of the mesityl substituent onto the newly formed alkene moiety in 1‐Mes.
Scheme 5.
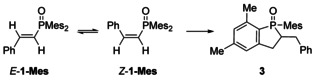
Equilibrium between E‐ and Z‐isomeric styryl‐di(mesityl)phosphane oxide and subsequent cyclization yielding 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3).
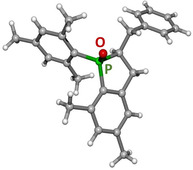
The X‐ray diffraction studies at a single crystal verified the formation of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3) as depicted in Scheme 5. The molecular structure and atom labeling are depicted in Figure 1. This molecule contains the two stereogenic centers P1 and C1 but due to the centrosymmetric monoclinic space group C2/c the crystalline state consists of a racemate of (R,S)‐ and (S,R)‐isomeric molecules. The P1=O1 bond length of 148.92(11) pm is very similar to values of simple unstrained diarylphosphane oxides (Ph2P(O)H: 148.81(11)19 Mes2P(O)H: 148.54(13) pm20) and of alkenyl‐di(mesityl)phosphane oxides like E‐1‐Mes (148.61(11) pm).9 These short P=O bonds are commonly explained by hyperconjugation from the negatively charged oxygen atom into antibonding σ*(P−C) bonds leading to slight lengthening of the P−C bonds.21 Deprotonation and formation of potassium diarylphosphinites of the type [(L)KOPAr2]n with three‐coordinate phosphorus atoms leads to an elongated P−O bond.14, 22 Contrary to the very similar P=O distances in various phosphane oxides the P−Caryl bond lengths to the aryl substituents depend on the steric strain induced by bulky groups. Thus, the P−Caryl distances increase in the row Ph2P(O)H (180.0 pm),19 Mes2P(O)H (181.6 pm)20 and E‐1‐Mes (182.7 pm).9 The latter value is comparable to the P1−C18 distance in 3 (182.65(14) pm) verifying a similar intramolecular steric strain. The endocyclic P1−C4 bond length of 179.53(15) in compound 3 is significantly smaller than the P−Caryl distances but similar to the P−C bond to the styryl group in E‐1‐Mes (179.68(15) pm)9 suggesting a slight delocalization within the P1‐C4‐C3 fragment.
Figure 1.
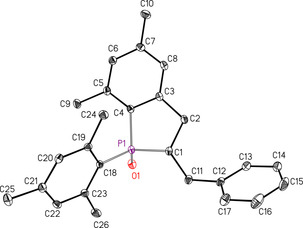
Molecular structure and atom labeling scheme of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3). The ellipsoids represent a probability of 30 %, H atoms are neglected for clarity reasons. Selected bond lengths [pm]: P1−O1 148.92(11), P1−C1 184.05(14), P1−C4 179.53(15), P1−C18 182.65(14), C1−C2 154.7(2), C2−C3 151.2(2), C3−C4 140.17(19), C1−C11 154.1(2), C11−C12 151.3(2); bond angles [°]: O1‐P1‐C1 114.66(6), O1‐P1‐C4 113.25(7), O1‐P1‐C18 110.42(6), C1‐P1‐C4 94.44(7), C1‐P1‐C18 108.98(6), C4‐P1‐C18 110.42(6).
For the investigation of the fragmentation pattern ESI‐MS experiments were performed. The fragmentation experiments revealed that the compound decayed predominantly into fragments with masses of 119, 269, and 283 as depicted in Scheme S1 (see Supporting Information). Stable isotope labeling and assignment of the observed masses was achieved with either deuterium oxide (D2O) as cosolvent during the MS study or with the use of ‐D−C≡C−Ph during the Pudovik reaction. Interestingly both independently executed experiments showed the same fragments. The most plausible explanation therefore is that the protons are exchangeable under conditions used for ESI‐MS investigations. This finding can be explained by a reversible 1,3‐hydrogen shift from C1 to O1 of 2‐benzyl‐5,7‐dimethyl‐1‐mesityl‐2,3‐dihydrophosphindole 1‐oxide (3, see Figure 1) leading to a P1=C1 double bond and a P‐bound hydroxyl functionality.
Selected NMR parameters of the racemate of the major (R,S)‐ and (S,R)‐isomers of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3) are summarized in Table 4, the numbering scheme is identical to the atom labeling of the molecular structure (Figure 2). The chemical shift of the phosphorus atom δ(31P)=53 ppm lies in the region of 1‐phenyl‐2‐phospholene 1‐oxide (CHCl3, 58.5 ppm; benzene, 57.2 ppm).23 The resonance of the minor racemic (R,R)‐ and (S,S)‐components shows a chemical shift of δ(31P)=59 ppm.
Table 4.
NMR data (chemical shifts δ [ppm] and coupling constants J [Hz]) of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide.
|
Fragment[a] |
δ(1H) |
J(P,H)[b] |
δ(13C{1H}) |
J(P,C)[c] |
|---|---|---|---|---|
|
1 |
2.81 |
n.d. |
41.2 |
1 J=69.3 |
|
2 |
3.00 |
3 J=12.6 |
34.4 |
2 J=6.8 |
|
3 |
– |
– |
146.1 |
2 J=28.7 |
|
4 |
– |
– |
131.8 |
1 J=102.3 |
|
5 |
– |
– |
140.9 |
2 J=8.7 |
|
6 |
6.90 |
4 J=3.1 |
130.0 |
3 J=9.5 |
|
7 |
– |
– |
143.1 |
4 J=2.3 |
|
8 |
6.90 |
4 J=3.1 |
124.9 |
3 J=11.3 |
|
9 |
2.37 |
– |
19.6 |
3 J=4.2 |
|
10 |
2.34 |
– |
21.7 |
– |
|
11 |
3.42/2.81 |
3 J=6.9 |
34.2 |
2 J=1.7 |
|
12 |
– |
– |
140.3 |
3 J=12.9 |
|
13,17 |
7.23 |
– |
128.9 |
– |
|
14,16 |
7.23 |
– |
128.5 |
– |
|
15 |
7.23 |
– |
128.9 |
– |
|
18 |
– |
– |
126.6 |
1 J=91.0 |
|
19,23 |
– |
– |
142.6 |
– |
|
20,22 |
6.87 |
– |
131.2 |
3 J=11.0 |
|
21 |
– |
– |
141.3 |
4 J=2.8 |
|
24,26 |
1.58/2.81[d] |
– |
23.1 |
– |
|
25 |
2.28 |
– |
21.0 |
– |
[a] The numbering of the CH fragments is identical with the atom labeling scheme of the molecular structure as depicted in Figure 1. [b] Coupling constants between hydrogen atoms and P1. [c] Coupling constants between carbon atoms and P1. [d] Chemical shifts of these methyl groups were determined at 243 K due to coalescence at RT caused by hindered rotation of the mesityl group around the P1−C18 bond.
Figure 2.
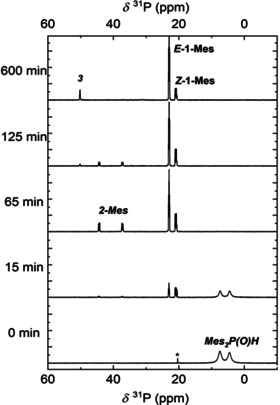
31P NMR spectroscopic monitoring at 161.98 MHz of the potassium‐mediated hydrophosphorylation of phenylacetylene with di(mesityl)phosphane oxide in [D8]THF at room temperature.
Temperature‐dependent NMR experiments allowed the elucidation of the barriers of hindered rotation of the mesityl group for an exchange between two equally occupied positions using the Gutowsky–Holm equation.24 At room temperature, the resonances of the ortho‐methyl (C24 and C26) and of the meta‐CH groups (C20 and C22) of 3 are broad due to hindered rotation around the P1−C18 bond. At low temperature a splitting of these signals is observed whereas at high temperature a rotation of the mesityl group that is fast on the NMR time scale leads to single resonances (Figure S11, Supporting Information). Determination of the coalescence temperature (Tc=253 K for the ortho‐methyl groups between 22 and 24 ppm in the 13C{1H} NMR spectrum) allowed the elucidation of a rotational barrier of 46 kJ mol−1.
Mechanism of phosphindole 1‐oxide formation
To gain mechanistic insight we performed this catalytic addition of Mes2P(O)H across phenylacetylene in THF. Thus we added 5 mol % of K(hmds) to di(mesityl)phosphane oxide producing a mixture of KOPMes2 and Mes2P(O)H with a molar ratio of 0.05:0.95. To this solution we added an equimolar amount of Ph−C≡CH, leading to a final mixture of 0.05:0.95:1 for the molar ratio of KOPMes2, Mes2P(O)H and Ph−C≡CH. Initially, the formation of 1‐Mes was observed as described above and after 15 min, 1‐Mes (E/Z‐ratio of 0.9:1) and Mes2P(O)H coexist in solution (Figure 2). After about 50 min also the resonances of bis‐phosphorylated 2‐Mes appeared with a rather low intensity which can unequivocally be recognized at the two doublets at δ(31P)=37 and 44 ppm with a 3 J(P,P) coupling constant of 47.7 Hz. After one hour di(mesityl)phosphane oxide was completely consumed and a very small singlet of KOPMes2 could be detected at δ(31P)=95 ppm. The E/Z‐ratio of 1‐Mes changed in favor of the E‐isomer to 2.1:1 (probably via an intermediate formation of 2‐Mes). Thereafter, the intensity of the resonances of KOPMes2 and 2‐Mes decreased and the resonances at δ(31P)=53 and 59 ppm, indicative of the formation of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3), appeared. After 125 min, an E/Z‐isomeric ratio of 2.8:1 was observed for 1‐Mes. While the intensity of the singlets of the isomers of 3 increased, the resonances of Z‐1‐Mes vanished (E/Z‐ratio of 4.5:1 after 10 hours) and finally, only the singlets of 3 at δ(31P)=53 and 59 ppm and of E‐1‐Mes at 23 ppm were visible. This time dependency is depicted in Figure 2.
These findings shed light on the mechanism of the potassium‐mediated Pudovik reaction and the subsequent cyclization process yielding 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3) if di(mesityl)phosphane oxide has been applied. The reaction of Mes2P(O)H with phenylacetylene or [D]phenylacetylene yields the E‐ and Z‐isomers of 1‐Mes as depicted in Scheme 6. Despite the fact that the acidity of di(aryl)phosphane oxide (pK a of Ph2P(O)H: 20.7) and phenylacetylene (pK a: 21–23.2 depending on solvent) are quite similar the deuterium is bound at the carbon atom of the styryl group adjacent to the P atom. Subsequent protonation occurs with Mes2P(O)H and the amount of double‐deuterated congeners due to protolysis with D‐C≡C−Ph is negligible.
Scheme 6.
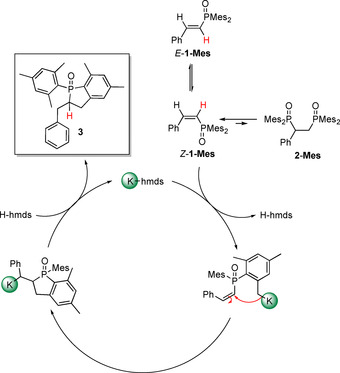
Proposed mechanism for the potassium‐mediated cyclization process of 1‐Mes yielding quantitatively 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3). Formation of very minor amounts of 2‐Mes has intermediately been observed.
A second (reversible) addition of Mes2P(O)H onto the newly formed alkenyl moiety, yielding 2‐Mes, is highly disadvantageous and only very low concentrations have been observed. The steric strain in Z‐1‐Mes is more pronounced than in the E‐isomer. Therefore, cyclization occurs much faster for Z‐isomeric 1‐Mes. Due to the fact that both Mes2P(O)H and H−C≡C−Ph are consumed yielding quantitatively 1‐Mes, the catalyst KN(SiMe3)2 is reformed. This base can deprotonate an ortho‐methyl group of the mesityl substituent in 1‐Mes. This carbanion undergoes an intramolecular nucleophilic attack at the alkenyl substituent leading to cyclization. Protonation with HN(SiMe3)2 finally leads to the formation of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3) regaining the catalytic species KN(SiMe3)2 as depicted in Scheme 6.
Conclusions
The addition of phosphane oxides across alkynes (Pudovik reaction) requires a catalyst to overcome the disadvantageous entropic influence and the approach of two electron‐rich molecules. Catalysis with s‐block metal reagents is one established procedure to activate the phosphane oxide partner by metalation and formation of the more reactive phosphinite. As precatalyst in the addition of dimesitylphosphane oxide onto phenylacetylene, we applied the bis(trimethylsilyl)amides (hmds) of the alkali and alkaline‐earth metals. The lighter s‐block metal congeners Li(hmds) as well as the M(hmds)2 derivatives of magnesium, calcium and strontium show no activity. The most reactive species are Rb(hmds) and Cs(hmds), but the reactivity of K(hmds) and Ba(hmds)2 is only slightly lower. The E/Z‐ratio of the resulting diaryl‐alkenylphosphane oxides also depends on the radius and softness of the metal ions. In summary, the alkali metal hmds complexes are more reactive than the alkaline‐earth metal congeners with increasing catalytic reactivity with increasing size of the metal ions.
These s‐block metal hmds precatalysts are able to promote and accelerate the addition of aryl‐substituted phosphane oxides across phenylacetylene whereas alkyl‐, alkoxy‐ and aryloxy‐substituted phosphane oxides cannot by added onto alkynes under these reaction conditions. Phosphane oxides Ar2P(O)H with smaller aryl groups show a two‐step addition yielding firstly the mono‐addition products, diaryl‐styrylphosphane oxides 1‐R, and then the bis‐phosphorylated derivatives, 1‐phenyl‐1,2‐bis(diarylphosphoryl)ethane 2‐R. Thereafter, we used the bulky dimesitylphosphane oxide to decelerate the addition reaction for time‐dependent studies and to avoid the second addition step, ensuring quantitative formation of the mono‐phosphorylated congener, dimesityl‐styrylphosphane oxide 1‐Mes.
The nature of the solvent plays a minor role in the s‐block metal‐mediated Pudovik reaction. In ethereal solvents the reaction is faster than in aliphatic and aromatic hydrocarbons with diethyl ether and triethylamine showing a mediocre support of this reaction. Ethereal solvents stabilize the intermediate metal phosphinites much more effectively by coordination to the metal ions and deaggregation of the catalytic species whereas in hydrocarbons larger aggregates can easily be realized as previously shown for crystalline complexes.
Without changing the molar ratios of dimesitylphosphane oxide, phenylacetylene and precatalyst, the amount of solvent has been reduced. In increasingly concentrated solutions the E/Z‐ratio of targeted dimesityl‐styrylphosphane oxide decreases. Furthermore, a second unique reaction sequence has been observed. After complete consumption of the starting substrates, the reformed precatalyst K(hmds) now slowly deprotonates an ortho‐methyl group of the P‐bound mesityl substituent. Thereafter, cyclization occurs yielding 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3). Due to two chiral atoms four isomers (two enantiomeric pairs) are observed.
The mechanism of the catalytic cycle starts with the metal diarylphosphinite, Ar2P‐OM, formed by metalation of diarylphosphane oxide with M(hmds). This phosphinite adds onto the alkyne functionality of Ar′−C≡C−H yielding diaryl‐alkenylphosphane oxides 1‐Ar after protonation with Ar2P(O)H. Labeling experiments with Ph−C≡C−D verify that the phosphane oxide is the proton source in this reaction step. A second addition occurs yielding the insoluble 1‐phenyl‐1,2‐bis(diarylphosphoryl)ethane 2‐Ar which precipitates from the reaction mixture. If bulky dimesitylphosphane oxide is applied, the second addition step remains irrelevant. Instead of that the ortho‐methyl group of the mesityl substituent is deprotonated by M(hmds), followed by the above‐mentioned cyclization to the phosphindole oxide derivative 3.
In summary, the Pudovik reaction can effectively be promoted by the heavy alkali metal bis(trimethylsilyl)amides M(hmds) with M=K, Rb, and Cs, leading to mono‐ and bis‐phosphorylated compounds. Bulky P‐bound mesityl groups hinder the second addition reaction. Mono‐phosphorylated 1‐Mes is cyclized after deprotonation of an ortho‐methyl group of the P‐bound mesityl substituent by catalytic amounts of K(hmds) finally giving phosphindole oxide 3.
Experimental Section
General remarks: All manipulations were carried out under an inert nitrogen atmosphere using standard Schlenk techniques, if not otherwise noted. The solvents were dried over KOH and subsequently distilled over sodium/benzophenone under a nitrogen atmosphere prior to use. Deuterated solvents were dried over sodium, distilled, degassed, and stored under nitrogen over sodium. 1H, 31P and 13C{1H} NMR spectra were recorded on Bruker Avance III 400 and Fourier 300 spectrometers. Chemical shifts are reported in parts per million relatively to SiMe4 (1H, 13C) or H3PO4 (31P) as an external standard referenced to the solvents residual proton signal. ASAP‐HSQC‐DEPT,25 1H{13P, 1H}26 und 1H{1H}26 were recorded using the published pulse sequences. All substrates were purchased from Alfa Aesar, abcr, Sigma Aldrich or TCI and used without further purification. [Na(hmds)],27 [Rb(hmds)],28 [Cs(hmds)],28 [Ca(hmds)2(thf)2],29 and secondary phosphane oxides (o‐tolyl)2POH,30 (C6H4‐4‐NMe2)2POH,31 (PhCH2)2POH,32 Cy2POH,33 tBu2POH33 were prepared according to literature protocols. [Li(hmds)]3 and [Mg(hmds)2] were prepared by reaction of H(hmds) with nBuLi or Mg(nBu)2 respectively. [Sr(hmds)2] and [Ba(hmds)2] were prepared starting from the metal by reaction with liquid ammonia and metalation of H(hmds) afterwards. The yields given are not optimized. Purity of the compounds was verified by NMR spectroscopy.
General protocol for catalytic studies
Secondary phosphane oxide (200 mg, 1.0 equiv) was placed in a Schlenk flask and dissolved in THF (5 mL). A solution of K(hmds) in THF (c=0.18 mol L−1, 0.19 mL, 0.05 equiv) was added through syringe and stirred for five minutes at room temperature. Phenylacetylene (0.08 mL, 0.7 mmol, 1.1 equiv.) was injected into the reaction vessel in one portion. The reaction mixture was stirred for one hour. The conversion of secondary phosphine oxide was tracked via 31P NMR spectroscopy of a hydrolyzed aliquot.
Synthesis of 2‐benzyl‐1‐mesityl‐5,7‐dimethyl‐2,3‐dihydrophosphindole 1‐oxide (3)
Dimesitylphosphane oxide (3.0 g, 10.5 mmol, 1.0 equiv) was placed in a Schlenk‐flask and dissolved in THF (30 mL). A solution of [K(hmds)] in THF (c=0.27 mol L−1, 3.9 mL, 1.1 mmol, 0.1 equiv) was added. To this yellow solution was added phenylacetylene (1.2 g, 11.6 mmol, 1.1 equiv). After a reaction time of two hours the mixture was quenched with ethanol. The solvents were evaporated under reduced pressure and the residue was purified by flash column chromatography (SiO2, PE:EtOAc 1:1). The phospholane oxide 3 (3.5 g, 9.0 mmol, 86 %) was isolated as light yellow pure product. Suitible crystals for the single crystal x‐ray diffraction were obtained by recrystallisation in n‐heptane/acetonitrile (2:0.1) at room temperature. m.p.: 102–104 °C; R f (SiO2, PE:EtOAc 1:1): 0.4; 1H NMR (400.13 MHz, CDCl3, 297 K): δ=7.33–7.15 (m, 5 H), 6.90 (d, 4 J PH=3.2 Hz, 2 H), 6.89–6.84 (m, 2 H), 3.46–3.35 (m, 1 H), 3.02–2.92 (m, 2 H), 2.88–2.72 (m, 2 H), 2.54–2.07 (m, 6 H), 2.37 (s, 3 H), 2.34 (s, 3 H), 2.28 ppm (s, 3 H); 13C{1H} NMR (100.61 MHz, CDCl3, 297 K): δ=146.1 (d, J=28.7 Hz), 143.1 (d, J=2.3 Hz), 141.3 (d, J=2.8 Hz), 140.9 (d, J=8.7 Hz), 140.3 (d, J=12.9 Hz), 131.8 (d, J=102.3 Hz), 130.0 (d, J=9.5 Hz), 128.9, 128.5, 126.6 (d, J=91.0 Hz), 124.9 (d, J=11.3 Hz), 77.4 (d, J=11.6 Hz), 77.2, 76.8, 41.2 (d, J=69.3 Hz), 34.4 (d, J=6.8 Hz), 34.2 (d, J=1.7 Hz), 23.1, 21.7, 21.0, 19.6 ppm (d, J=4.2 Hz); 31P NMR (161.99 MHz, CDCl3, 297 K): δ=53.6 (s); IR (ATR): myEqn.2 (cm−1)=3024 (w), 2917 (w), 2843 (w), 1604 (m), 1405 (m), 1188 (s), 1179 (vs.), 1088 (m), 877 (s), 756 (s), 662 (vs.), 637 ppm (s); MS: m/z (%)=777 [2 M]+ (10), 388 [M]+ (50), 373 [M−CH3]+ (60), 297 [M−Bn]+ (100), 270 [M−Mes]+ (20), 119 [Mes]+ (8), 91 [Bn]+ (10); HRMS (ESI) ([M+H]+, C26H30OP): calcd: 389.2034; found: 389.2029; Elemental analysis: C26H29OP (388 g mol−1) calc. C 80.38 % H 7.52 %; found C 80.11 % H 7.51 %
Mass spectrometric experiments
High‐resolution mass spectrometry was carried out using a THERMO (Bremen, Germany) QExactive plus Orbitrap mass spectrometer coupled to a heated electrospray source (HESI). Flow was set to 15 μL min−1 using a syringe pump. For monitoring a full scan mode was selected with the following parameters. Polarity: positive; scan range: 100 to 1500 m/z; resolution: 280 000; AGC target: 3 × 106; maximum IT: 512 ms. General settings: sheath gas flow rate: 15; auxiliary gas flow rate 5; sweep gas flow rate: 0; spray voltage: 3.0 kV; capillary temperature: 250 °C; S‐lens RF level: 90; vaporizer temperature: auxiliary gas heater temperature: 105 °C. For MS2 experiments normalized collision energy was set to 60 and the mass selection window was set to ±0.2 m/z of the protonated molecule to prevent isotope fragmentation using a mass range from 100 to 500 m/z. Compound 3 was picked with a 200 μL Eppendorf tip and dissolved in 0.5 mL of HPLC‐MS grade acetonitrile and diluted with HPLC‐MS grade water 1:1, (v/v).
X‐ray structure determination of 3
The intensity data were collected on a Nonius KappaCCD diffractometer, using graphite‐monochromated MoKα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi‐empirical basis using multiple‐scans.34, 35, 36 The structure was solved by direct methods (SHELXS)37 and refined by full‐matrix least squares techniques against Fo 2 (SHELXL‐97).37 The hydrogen atoms of compound 3 were included at calculated positions with fixed thermal parameters. All non‐hydrogen atoms were refined anisotropically.37 XP38 and POV‐Ray39 program packages were used for structure representations. Crystal Data and refinement details for 3: C26H29OP, M=388.46 g mol−1, colorless prism, size 0.124×0.112×0.088 mm3, monoclinic, space group C2/c, a=22.4180(5), b=8.5492(2), c=22.5499(5) Å, β=99.657(1)°, V=4260.58(17) Å3, T=−140 °C, Z=8, ρ calcd=1.211 g cm−3, μ(MoKα)=1.43 cm−1, multi‐scan, transmin: 0.7190, transmax: 0.7456, F(000)=1664, 15088 reflections in h(‐28/28), k(‐11/11), l(‐28/29), measured in the range 1.84°≤Θ≤27.48°, completeness Θ max=99.3 %, 4854 independent reflections, R int=0.0334, 4326 reflections with Fo >4σ(Fo), 258 parameters, 0 restraints, R1obs=0.0421, wR2obs=0.0994, R1all=0.0482, wR2all=0.1037, GOOF=1.053, largest difference peak and hole: 0.399/ −0.335 e Å−3.
CCDC 1961255 (3) contains the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge the valuable support of the NMR (www.nmr.uni‐jena.de/) and mass spectrometry service platforms (www.ms.uni‐jena.de/) of the Faculty of Chemistry and Earth Sciences of the Friedrich Schiller University Jena, Germany. The mass spectrometer used for high resolution mass spectrometric investigations was financed by the German Research Foundation (DFG, Bonn, Germany) in the frame of the Collaborative Research Center SFB 1127 (ChemBioSys). P.S. is very grateful to the German Environment Foundation (Deutsche Bundesstiftung Umwelt, DBU, grant no. 20018/578) for a generous Ph.D. grant.
B. E. Fener, P. Schüler, N. Ueberschaar, P. Bellstedt, H. Görls, S. Krieck, M. Westerhausen, Chem. Eur. J. 2020, 26, 7235.
References
- 1.
- 1a. Harder S., Chem. Rev. 2010, 110, 3852–3876; [DOI] [PubMed] [Google Scholar]
- 1b. Kobayashi S., Yamashita Y., Acc. Chem. Res. 2011, 44, 58–71; [DOI] [PubMed] [Google Scholar]
- 1c. Reznichenko A. L., Hultzsch K. C., Top. Organomet. Chem. 2011, 43, 51–114; [Google Scholar]
- 1d. Crimmin M. R., Hill M. S., Top. Organomet. Chem. 2013, 45, 191–242; [Google Scholar]
- 1e. Koshti V., Gaikwad S., Chikkali S. H., Coord. Chem. Rev. 2014, 265, 52–73; [Google Scholar]
- 1f. Coman S. M., Parvulescu V. I., Org. Process Res. Dev. 2015, 19, 1327–1355; [Google Scholar]
- 1g. Wilkins L. C., Melen R. L., Coord. Chem. Rev. 2016, 324, 123–139; [Google Scholar]
- 1h. Hill M. S., Liptrot D. J., Weetman C., Chem. Soc. Rev. 2016, 45, 972–988; [DOI] [PubMed] [Google Scholar]
- 1i. Bange C. A., Waterman R., Chem. Eur. J. 2016, 22, 12598–12605; [DOI] [PubMed] [Google Scholar]
- 1j. Krieck S., Westerhausen M. in Early Main Group Metal Catalysis: Concepts and Reactions, (Ed.: S. Harder), Wiley-VCH, Weinheim, 2020, ch. 5, pp. 123–149. [Google Scholar]
- 2. Semenzin D., Etemad-Moghadam G., Albouy D., Diallo O., Koenig M., J. Org. Chem. 1997, 62, 2414–2422. [DOI] [PubMed] [Google Scholar]
- 3. Allen A., Ma L., Lin W., Tetrahedron Lett. 2002, 43, 3707–3710. [Google Scholar]
- 4. Takaki K., Koshoji G., Komeyama K., Takeda M., Shishido T., Kitani A., Takehira K., J. Org. Chem. 2003, 68, 6554–6565. [DOI] [PubMed] [Google Scholar]
- 5. Huang Y., Hao W., Ding G., Cai D.-Z., J. Organomet. Chem. 2012, 715, 141–146. [Google Scholar]
- 6. Stone J. J., Stockland R. A., Reyes J. M., Kovach J., Goodman C. C., Tillman E. S., J. Mol. Catal. A 2005, 226, 11–21. [Google Scholar]
- 7. Liu L., Wang Y., Zeng Z., Xu P., Gao Y., Yin Y., Zhao Y., Adv. Synth. Catal. 2013, 355, 659–666. [Google Scholar]
- 8. Zhang J.-S., Zhang J.-Q., Chen T., Han L.-B., Org. Biomol. Chem. 2017, 15, 5462–5467. [DOI] [PubMed] [Google Scholar]
- 9. Härling S. M., Fener B. E., Krieck S., Görls H., Westerhausen M., Organometallics 2018, 37, 4380–4386. [Google Scholar]
- 10.
- 10a. Saga Y., Han D., Kawaguchi S.-i., Ogawa A., Han L.-B., Tetrahedron Lett. 2015, 56, 5303–5305; [Google Scholar]
- 10b. Yoshimura A., Saga Y., Sato Y., Ogawa A., Chen T., Han L.-B., Tetrahedron Lett. 2016, 57, 3382–3384. [Google Scholar]
- 11. Guo H., Yoshimura A., Chen T., Saga Y., Han L.-B., Green Chem. 2017, 19, 1502–1506. [Google Scholar]
- 12. Zhong W., Tan T., Shi L., Zeng X., Synlett 2018, 29, 1379–1384. [Google Scholar]
- 13. Beswick M. A., Cromhout N. L., Harmer C. N., Palmer J. S., Raithby P. R., Steiner A., Verhorevoort K. L., Wright D. S., Chem. Commun. 1997, 583–584. [Google Scholar]
- 14. Härling S. M., Krieck S., Görls H., Westerhausen M., Inorg. Chem. 2017, 56, 9255–9263. [DOI] [PubMed] [Google Scholar]
- 15. Hill M. S., Mahon M. F., Robinson T. P., Chem. Commun. 2010, 46, 2498–2500. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Al-Shboul T. M. A., Volland G., Görls H., Krieck S., Westerhausen M., Inorg. Chem. 2012, 51, 7903–7912; [DOI] [PubMed] [Google Scholar]
- 16b. Naktode K., Bhattacharjee J., Gupta S. D., Nayek H. P., Mallik B. S., Panda T. K., Z. Anorg. Allg. Chem. 2014, 640, 994–999; [Google Scholar]
- 16c. Rood J. A., Huttenstine A. L., Schmidt Z. A., White M. R., Oliver A. G., Acta Crystallogr. Sect. B 2014, 70, 602–607. [DOI] [PubMed] [Google Scholar]
- 16d.For a review on calcium phosphanides, phosphinites, and phosphinates see: Westerhausen M., Krieck S., Langer J., Al-Shboul T. M. A., Görls H., Coord. Chem. Rev. 2013, 257, 1049–1066. [Google Scholar]
- 17.
- 17a. Reichardt C., Chem. Rev. 1994, 94, 2319–2358; [Google Scholar]
- 17b. Reichardt C., Green Chem. 2005, 7, 339–351; [Google Scholar]
- 17c. Reichardt C., Pure Appl. Chem. 2008, 80, 1415–1432; [Google Scholar]
- 17d. Reichardt C., Welton T., Solvents and Solvent Effects in Organic Chemistry, 4th Ed; Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 18.
- 18a. Neufeld R., Stalke D., Chem. Sci. 2015, 6, 3354–3364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Bachmann S., Gernert B., Stalke D., Chem. Commun. 2016, 52, 12861–12864. [DOI] [PubMed] [Google Scholar]
- 19. Härling S., Greiser J., Al-Shboul T. M. A., Görls H., Krieck S., Westerhausen M., Aust. J. Chem. 2013, 66, 1264–1273. [Google Scholar]
- 20. Veinot A. J., Ramgoolam K., Giffin N. A., Masuda J. D., Molbank 2017, M957. [Google Scholar]
- 21. Gilheany D. G., Chem. Rev. 1994, 94, 1339–1374. [DOI] [PubMed] [Google Scholar]
- 22. Härling S. M., Görls H., Krieck S., Westerhausen M., Inorg. Chem. 2016, 55, 10741–10750. [DOI] [PubMed] [Google Scholar]
- 23. Moedritzer K., Miller R. E., Syn. React. Inorg. Met.-Org. Chem. 1977, 7, 311–332. [Google Scholar]
- 24.
- 24a. Kessler H., Angew. Chem. Int. Ed. Engl. 1970, 9, 219–235; [Google Scholar]; Angew. Chem. 1970, 82, 237–253; [Google Scholar]
- 24b. Binsch G., Kessler H., Angew. Chem. Int. Ed. Engl. 1980, 19, 411–428; [Google Scholar]; Angew. Chem. 1980, 92, 445–463; [Google Scholar]
- 24c. Bryant R. G., J. Chem. Educ. 1983, 60, 933–935; [Google Scholar]
- 24d. Günther H., NMR-Spektroskopie, 3rd Ed, Georg Thieme, New York, 1992, ch. 9, pp. 303–350. [Google Scholar]
- 25. Schulze-Sünninghausen D., Becker J., Koos M. R. M., Luy B., J. Magn. Reson. 2017, 281, 151–161. [DOI] [PubMed] [Google Scholar]
- 26. Aguilar J. A., Faulkner S., Nilsson M., Morris G. A., Angew. Chem. Int. Ed. 2010, 49, 3901–3903; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3993–3995. [Google Scholar]
- 27. Wannagat U., Niederprüm H., Chem. Ber. 1961, 94, 1540–1547. [Google Scholar]
- 28. Krieck S., Schüler P., Görls H., Westerhausen M., Dalton Trans. 2018, 47, 12562–12569. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Krieck S., Schüler P., Peschel J., Görls H., Westerhausen M., Synthesis 2019, 51, 1115–1122; [Google Scholar]
- 29b.see also: Westerhausen M., Inorg. Chem. 1991, 30, 96–101; [Google Scholar]
- 29c. Westerhausen M., Hartmann M., Makropoulos N., Wieneke B., Wieneke M., Schwarz W., Stalke D., Z. Naturforsch. B 1998, 53, 117–125. [Google Scholar]
- 30. Ke J., Tang Y., Yi H., Li Y., Cheng Y., Liu C., Lei A., Angew. Chem. Int. Ed. 2015, 54, 6604–6607; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6704–6707. [Google Scholar]
- 31. Busacca C. A., Lorenz J. C., Grinberg N., Haddad N., Hrapchak M., Latli B., Lee H., Sabila P., Saha A., Sarvestani M., Org. Lett. 2005, 7, 4277–4280. [DOI] [PubMed] [Google Scholar]
- 32. Bálint E., Tripolszky A., Jablonkai E., Karaghiosoff K., Czugler M., Murcsi Z., Kollàr L., Pongràz P., Keglevich G., J. Organomet. Chem. 2016, 801, 111–121. [Google Scholar]
- 33. Smoll K. A., Kaminsky W., Goldberg K. I., Organometallics 2017, 36, 1213–1216. [Google Scholar]
- 34.R. Hooft: COLLECT, Data Collection Software; Nonius B. V., Netherlands, 1998.
- 35. Otwinowski Z., Minor W. in Methods in Enzymology, Vol. 276, Macromolecular Crystallography (Eds.: C. W. Carter, R. M. Sweet), Academic Press, New York, 1997, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 36.SADABS 2016/2: Krause L., Herbst-Irmer R., Sheldrick G. M., Stalke D., J. Appl. Crystallogr. 2015, 48, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.XP, Siemens Analytical X-Ray Instruments Inc., Karlsruhe, Germany, 1990; Madison, WI, USA, 1994.
- 39.POV-Ray, Persistence of Vision Raytracer: Victoria, Australia, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary