

Abstract
The enzyme tyrosinase contains a reactive side‐on peroxo dicopper(II) center as catalytically active species in C−H oxygenation reactions. The tyrosinase activity of the isomeric bis(μ‐oxo) dicopper(III) form has been discussed controversially. The synthesis of bis(μ‐oxo) dicopper(III) species [Cu2(μ‐O)2(L1)2](X)2 ([O1](X)2, X=PF6 − , BF4 −, OTf−, ClO4 −), stabilized by the new hybrid guanidine ligand 2‐{2‐((dimethylamino)methyl)phenyl}‐1,1,3,3‐tetramethylguanidine (L1), and its characterization by UV/Vis, Raman, and XAS spectroscopy, as well as cryo‐UHR‐ESI mass spectrometry, is described. We highlight selective oxygenation of a plethora of phenolic substrates mediated by [O1](PF6)2, which results in mono‐ and bicyclic quinones and provides an attractive strategy for designing new phenazines. The selectivity is predicted by using the Fukui function, which is hereby introduced into tyrosinase model chemistry. Our bioinspired catalysis harnesses molecular dioxygen for organic transformations and achieves a substrate diversity reaching far beyond the scope of the enzyme.
Keywords: copper catalysis, dioxygen activation, guanidines, phenazines, tyrosinase
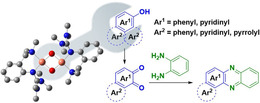
Guanidines strike back! The synthesis of bis(μ‐oxo) dicopper(III) species [Cu2(μ‐O)2(L1)2](X)2 ([O1](X)2, X=PF6, BF4, OTf, ClO4), stabilized by an aromatic hybrid guanidine ligand, and its characterization by UV/Vis, Raman and X‐ray absoption spectroscopy, as well as cryo‐UHR‐ESI mass spectrometry, are described. Selective oxidation of a plethora of phenolic substrates mediated by [O1](PF6)2, which results in mono‐ and bicyclic quinones far beyond the enzyme's scope and provides an attractive strategy to design new phenazines, is highlighted (see scheme).
The use of dioxygen as a readily available oxidizing agent is crucial for many biological and biomimetic oxidation processes, as well as industrial applications.1 Natural copper enzymes, such as tyrosinase or particulate methane monooxygenase, successfully activate molecular dioxygen.2 Tyrosinase, in particular, is essential in living organisms catalyzing phenol oxidation in melanin biosynthesis.3 It converts phenols, for instance, l‐tyrosine, via an electrophilic aromatic substitution through its catechol form to the final quinone form.4 Although structure and reactivity of tyrosinases were studied extensively, many details of the oxidation mechanism are still under debate.5 Recently, second shell residues at the active site (ca. 5.5–16 Å distance to Cu ions) were considered to direct the reactivity of the enzyme.2b, 6
For understanding the mechanism of activation and transfer of O2, synthetic model systems were developed mimicking enzymatic properties. Some well‐studied Cu/O2 species represent μ‐η2:η2‐peroxo CuII [P] and bis(μ‐oxo) CuIII [O] complexes, which exist in a dynamic equilibrium due to a small isomerization barrier.2b, 7 In many functional tyrosinase systems, [P] cores are found,8 but an increasing number of examples of functional [O] species has also been reported.9 However, only few tyrosinase model systems feature catalytic oxygenation reactivity.10 Réglier and co‐workers presented the first system using the imine–pyridine ligand BiPh(impy)2,11 followed by Casella and co‐workers by using the benzimidazole ligand L66,12 which both stabilize binuclear Cu catalysts. Later, Tuczek and co‐workers reported the mononucleating benzimidazole ligand Limpy promoting catalytic conversion of 2,4‐di‐tert‐butyl phenol.13 Over the years, Lumb and co‐workers have focused on the chemo‐ and regioselectivity of tyrosinase‐like reactions by using the amine DBED giving rise to several quinones, oxidative coupling, and cyclization products.14 More complex phenolic substrates were oxygenated by Herres‐Pawlis group by using bis(pyrazolyl)methane and guanidine ligands.15
Herein, we report the synthesis and characterization of hybrid guanidine‐stabilized bis(μ‐oxo) complex [Cu2(μ‐O)2(L1)2]2+ ([O1]2+, Scheme 1) along with its high catalytic activity in oxygenation and oxidation reactions of phenolic substrates and subsequent condensation reactions, offering a facile synthetic pathway to new phenazines.
Scheme 1.
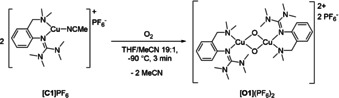
Synthesis of bis(μ‐oxo) species [O1](PF6)2.
Guanidines feature high basicity and strong N‐donor abilities, enabling stabilization of metal ions with high oxidation states.16 For instance, TMG3tren is known to stabilize highly reactive CuII superoxo complexes.17 By using [O1](PF6)2, it is possible to promote selective C−H functionalization of complex phenolic substrates. To the best of our knowledge, no activity of tyrosinases towards complex phenolic substrates has been reported before. Compound [O1](PF6)2 highlights a beneficial interplay of steric and electronic factors of the ligand regarding substrate accessibility of [Cu2O2] core leading to a remarkable extension of the substrate scope. Using a variety of phenolic substances allows the synthesis of new quinones. In a consecutive reaction, quinones can condense with 1,2‐phenylenediamine to phenazines, which feature antibacterial, antimalarial, and antitumor activities, and are used in dyes and pesticides.18 We targeted to selectively obtain bent phenazines as special benefit of the [O1](PF6)2‐mediated hydroxylation, because the calculation of the Fukui function of the hydroxylation products indicated preferential formation of the corresponding quinone.
Inspired by a propylene‐bridged hybrid guanidine ligand system that showed promising phenolase activity,9k, 9l we developed a related ligand system with a more rigid aromatic backbone. Compound 2‐{2‐((dimethylamino)methyl)phenyl}‐1,1,3,3‐tetramethylguanidine (L1) was synthesized in a three‐step reaction and isolated in high yield (see the Supporting Information). Synthesis of the colorless, air‐ and moisture‐sensitive CuI complex [C1]PF6 was achieved by mixing equimolar amounts of L1 and [Cu(MeCN)4]PF6 in acetonitrile at room temperature. Oxygenation of [C1]PF6 in THF at −90 °C led to the formation of the khaki colored species [O1](PF6)2 (Scheme 1).19 Compound [O1](PF6)2 showed ligand‐to‐metal‐charge transfer (LMCT) features at 280 nm (ϵ=40000 m −1 cm−1) and 392 nm (21 000 m −1 cm−1) in the UV/Vis spectrum20 (Figure 1 a), which are characteristic for bis(μ‐oxo) dicopper(III) species.8, 21 Similar UV/Vis features were obtained by using different weakly coordinating anions (BF4 −, ClO4 −, OTf−; Figure S17 and Table S3 in the Supporting Information). Incorporated O2 of [O1](PF6)2 was resistant to cycles of evacuation and purging with N2. Laser excitation at 420 nm led to a resonance Raman spectrum with a characteristic vibration at 620 cm−1, which was attributed to the symmetrical Cu2O2 core expansion (breathing mode), thus evidencing the formation of a bis(μ‐oxo) species (Figure 1 b). 16O2/18O2 isotope exchange measurements in THF exhibited a shift to 591 cm−1, which is in good agreement with theoretical calculations (Table S13 in the Supporting Information). The signal at 530 cm−1 is caused by the N(amine)−Cu vibration.
Figure 1.
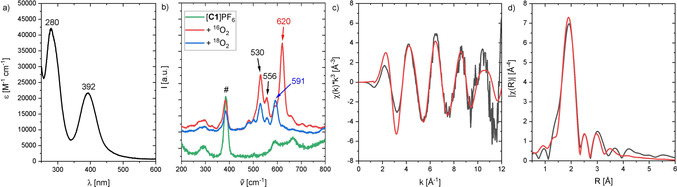
a) UV/Vis spectrum of [O1](PF6)2 (0.5 mm) in THF at −90 °C; b) resonance Raman spectra of [O1](PF6)2 in THF with excitation at 420 nm (blue: 18O2, red: 16O2, green: [C1]PF6, #: solvent); c) k3‐weighted Cu K‐edge EXAFS of [O1](PF6)2 (black: experimental, red: calculated fit); and d) phase‐corrected Cu‐K‐edge Fourier transform of EXAFS of [O1](PF6)2 (black: experimental, red: calculated fit).
Theoretical studies on [O1](PF6)2 were performed to investigate geometry and Raman features (Figure 2). The calculations showed that the O species is favored by 10 kcal mol−1 over the P species (Table S11 in the Supporting Information). Key bond lengths around the Cu atoms were determined by Cu K‐edge EXAFS (Figure 1 c–d, and section 1.3 in the Supporting Information) and agree well with the theoretical model. Moreover, the edge position is in accordance with the assignment as CuIII. For quantification of the formation of [O1](PF6)2, spectrophotometric back titration of [O1](PF6)2 was performed by using ferrocene monocarboxylic acid (FcCOOH).9k, 9l Following the decay of the LMCT band at 392 nm, titration of [O1](PF6)2 with FcCOOH revealed >90 % formation of [O1](PF6)2 (Figures S18 and 19 in the Supporting Information). To elucidate the Cu−O2 stoichiometry of [O1](PF6)2, cryo‐UHR‐ESI mass spectrometry was performed (Figure S20). The isotopic pattern and corresponding m/z values are in accordance with the calculated spectrum of the monocationic species [O1](PF6)+ with a 2:1 Cu‐O2 ratio.
Figure 2.
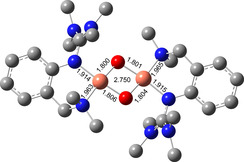
DFT model of [O1]2+ (TPSSh/def2‐TZVP, THF‐PCM), selected bond lengths [Å] and Cu⋅⋅⋅Cu vector [Å].
Thermal decomposition kinetics of [O1](PF6)2 revealed a first‐order decay at low temperatures (Figures S21 and 22 in the Supporting Information). Half‐life times of [O1](PF6)2 in THF of 1 h at −80 °C and five minutes at −74 °C were determined. Thermal decomposition products of [O1](PF6)2 were identified by crystallization as a dicationic μ‐alkoxo‐μ‐hydroxo copper(II) complex [H1](PF6)2 with a Cu⋅⋅⋅Cu distance of 2.953(1) Å (Figure 3). Each copper atom is coordinated in a distorted square‐planar fashion. The average Cu−O bond length (1.92 Å) is shorter compared to that in the mixed phenolato hydroxo‐bridged dicopper(II) species (1.96 Å) reported by Karlin and co‐workers.22 Concomitant formation of yellow blocks was observed, which contain the protonated ligand [(L1)H2](PF6)2.
Figure 3.
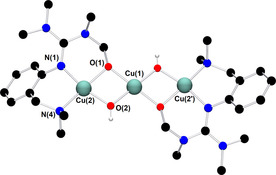
Molecular structure of [H1]2+ in crystals of [H1](PF6)2. H atoms, except for the H atom of μ‐OH groups, counterions, and solvent molecules are omitted for clarity. Selected interatomic distances [Å] and angles [°]: Cu(1)−O(1) 1.912(2), Cu(1)−O(2) 1.893(2), Cu(2)−O(1) 1.923(2), Cu(2)−O(2) 1.932(2), Cu(1)⋅⋅⋅Cu(2) 2.953(1), Cu(2)−N(1) 1.986(2), Cu(2)−N(4) 2.019(3); N(1)‐Cu(2)‐N(4) 94.9(1), O(1)‐Cu(2)‐O(2) 76.8(1), Cu(2)‐O(1)‐Cu(1) 100.7(1), Cu(2)‐Cu(1)‐Cu(2′) 180.0.
Bis(μ‐hydroxo) species are commonly observed as decay products9, 18a and often resulted from intramolecular hydroxylation of C−H bonds in α‐ or β‐position of N‐donor groups.23 Interestingly, [H1]2+ is the first example of a trinuclear μ‐alkoxo‐μ‐hydroxo copper(II) complex cation. Only few examples of alkoxo‐hydroxo copper(II) species have been reported to date.7b, 23 Mechanistic studies on intramolecular hydroxylation of the supporting ligand upon warming of the bis(μ‐oxo) species were proposed by Itoh, Tolman and co‐workers stating a mixed alkoxo‐hydroxo copper(II) complex as intermediate species to form thermodynamically stable bis(μ‐hydroxo) and bis(μ‐alkoxo) complexes.23a, 24 In case of [H1]2+, the chemoselective attack of the equatorially located methyl groups of the guanidine moiety over the axially disposed aminomethyl groups is favored due to geometric constraints, similar to observations made by Tolman and co‐workers.23c, 25
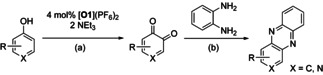
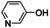
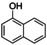
Aiming to expand the commonly used substrate scope of simple phenols, catalytic hydroxylation activity of [O1](PF6)2 was evaluated towards a variety of challenging phenolic substrates, including phenols, pyridinols, naphthols, quinolinols, and indolols (Table 1 and section 2.6 in the Supporting Information), quinones and phenazines of which are biologically relevant and difficult to synthesize. We report the hydroxylation of a multitude of these substrates by [O1](PF6)2 along with transformation of the quinone into a phenazine. Remarkably that until now, catalytic conversion of phenols was mostly reported for side‐on peroxo copper complexes as catalytically active species.11, 12, 13, 15 Oxygenation reactions were performed following a protocol established by Bulkowski and modified by Tuczek and co‐workers by using 25 equivalents of substrate and 50 equivalents of NEt3 (Table 1, Reaction (a)).13a, 26 Reaction (a) was monitored by UV/Vis spectroscopy at −90 °C for one hour as optical spectra remained constant after that time. Reactive quinones were subsequently converted in a one‐pot reaction into their corresponding phenazines at room temperature by using 1,2‐phenylenediamine (Table 1, Reaction (b)).
Table 1.
Catalytic oxygenation of phenolic substrates[a] and subsequent condensation of the quinone by using 1,2‐phenylenediamine.[b]
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
Substrate |
Conv. |
Product |
Yield[c] |
TON[d] |
|
|
|
|
[%] |
(quinone/phenazine) |
[%] |
|
|
|
1 |
|
>99 |
|
(Q1) |
[e] |
[f] |
|
2 |
|
>99 |
(Q1) |
[e] |
[f] |
|
|
3 |
|
80 |
|
(P1) |
22 |
11 |
|
4 |
|
89 |
(P1) |
31 |
16 |
|
|
5 |
|
95 |
|
(P2) |
32 |
16 |
|
6 |
|
87 |
(P2) |
21 |
11 |
|
|
7 |
|
>99 |
|
(P3) |
30 |
15 |
|
8 |
|
28 |
|
(Q2) |
– |
14[g] |
|
9 |
|
24 |
|
(Q3) |
– |
12[g] |
|
10 |
|
81 |
|
(P4) |
19 |
10 |
|
11 |
|
88 |
(P4) |
26 |
13 |
|
|
12 |
|
92 |
|
(P5) |
27 |
14 |
|
13 |
|
84 |
(P5) |
31 |
16 |
|
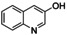
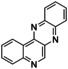
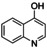
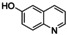
[a] Conditions: THF, −90 °C, 1 h. [b] Conditions: THF, −90 °C, then rt, overnight. [c] Isolated yield after column chromatography. [d] Based on isolated yield in correlation with conc. of [O1](PF6)2. [e] Quinone too reactive to be isolated. [f] No extinction coefficient of the quinone reported. [g] Determined after reaction (a) by UV/Vis spectra and based on conc. of [O1](PF6)2.
When simple phenols were used, [O1](PF6)2 showed, besides the expected hydroxylation activity, also C−O coupling chemistry (Table S8 in the Supporting Information, entries 1–4). UV/Vis spectra showed a low intensity absorption band at 510 and 530 nm (Figures S39 and 40 in the Supporting Information). EPR measurements revealed the hyperfine splitting pattern typical for a radical‐free, metal‐centered CuII species, ruling out the formation of a semiquinone species (Figure S41).27
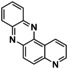
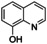
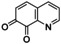
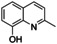
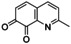
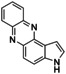
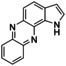
An interesting substrate for the catalytic oxygenation is 8‐quinolinol.15, 28 Although 7,8‐quinolinedione (Q2) was formed with 14 turnovers (entry 8), methyl substitution in 2‐position resulted in a TON of 12 (Q3, entry 9). However, formation of the respective phenazine was observed in neither case (Figures S50 and 51 in the Supporting Information), presumably due to the smaller Fukui function f+ at the corresponding C atoms of Q2 and Q3 (Table S15). Conversion of other quinolinols was not reported before in Cu/O2 chemistry. Compounds 3‐ and 4‐quinolinol were transformed into their quinone (absorption band at 370 nm, Figures S47 and 48 in the Supporting Information) and isolated as quinolino[3,4‐b]quinoxaline (P2; entries 5–6). 6‐Quinolinol was oxidized into 5,6‐quinolinedione (entry 7), which showed absorption bands at 325, 340, and 370 nm (Figure S49). Pyrido[3,2‐a]phenazine (P3) was isolated in 15 turnovers after column chromatography and sublimation, and crystallized from DMSO (see the Supporting Information).
In contrast to reactions with phenols and 8‐quinolinol, no conversion of pyridinols has been reported to date. When 3‐ and 4‐pyridinol (entries 1–2) were used, an intense absorption band at 375 nm, similar to that in 3‐ and 4‐quinolinol, was observed in both cases within less than one minute (Figures S42 and 43 in the Supporting Information), indicating formation of 3,4‐pyridoquinone (Q1). A fast drop in intensity illustrates high reactivity of the quinone even at low temperatures, leading to C−O coupled dimers (see the Supporting Information).29 Theoretical studies revealed lesser stability of 3,4‐pyridoquinone compared to its most stable 2,5‐isomer.30 However, no other study exists that reports on pyridoquinones.
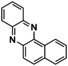
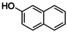
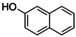
Naphthoquinones are accessible from 1‐ and 2‐naphthol in the presence of an oxidizing agent.31 Both naphthols were converted via [O1](PF6)2 into the corresponding quinone, which resulted in the formation of benzo[a]phenazine31c (P1) upon treatment with 1,2‐phenylenediamine (entries 3–4, Figures S44 and 45 in the Supporting Information). Compound P1 was purified by column chromatography and isolated with a TON of 11–16. Consistent with observations made by Krohn and co‐workers, no formation of the linear phenazine was observed.31c The control experiment by using 1‐methyl 2‐naphthol (Table S8, entry 7) showed no product formation as was expected (Figure S46), because the 1‐position is occupied by the methyl substituent inhibiting the oxygenation process.
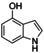
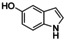
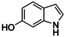
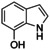
Although related bicyclic indolols possess an easily accessible pyrrole ring, C−H functionalization of the phenyl ring remains challenging.32 When [O1](PF6)2 was used, 4‐ and 5‐indolol were converted into 4,5‐indoledione, which was captured as pyrrolo[3,2‐a]phenazine (P4) in 10–13 turnovers (entries 10–11, Figures S52 and 53). Similarly, 6‐ and 7‐indolol (Figures S54 and 55) were transformed into pyrrolo[2,3‐a]phenazine (P5, TON=14–16) and crystallized from hexane (entries 12–13). The control experiment revealed no oxygenation activity on 6‐indolol in the absence of supporting ligand L1 (Figure S36), thus evidencing the necessity of the stabilizing ligand system.
The accompanying DFT calculations based on the negative Fukui function were used to determine the location of the electrophilic attack, which are in good agreement with the observed products (for substrates with two product possibilities, see Tables 1 and 2 and section 3.2 in the Supporting Information). The Fukui function describes the electron density in a frontier orbital, in this case fk − denotes the initial part for an electrophilic reaction. A large value for fk − indicates the preferred site for an electrophilic attack—in this case, the tyrosinase‐like hydroxylation. In all cased studied herein, the Fukui function points to the experimentally observed hydroxylation site finally yielding the bent phenazines.
Table 2.
Summary of calculated Fukui function [fk −] of phenolic substrates with two possible products.
|
Entry |
Substrate |
fk − |
C atom position |
|---|---|---|---|
|
1 |
|
21.14 0.74 |
1 3 |
|
2 |
|
1.38 15.17 |
2 4 |
|
3 |
|
23.93 0.91 |
5 7 |
|
4 |
|
19.93 1.05 |
4 6 |
|
5 |
|
2.03 11.99 |
5 7 |
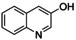
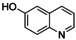
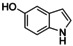
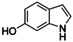
It has to be highlighted that the phenazines P3, P4, and P5 are new and fully characterized.33 Thus, the combination of tyrosinase‐like hydroxylation reactivity with the condensation of quinones with diamines allows synthetic access to a multitude of new phenazines.
Although tyrosinase (from Aspergillus oryzae) oxidizes phenol quantitatively,34 the enzyme exhibited no reactivity towards naphthols, quinolinols, and indolols (Table S10 in the Supporting Information). Thus, the bis(μ‐oxo) species [O1](PF6)2 demonstrates an exceptional broad substrate scope to which the enzyme itself gave no access.
In summary, we established the synthesis of the aromatic hybrid guanidine‐stabilized bis(μ‐oxo) species [O1]2+, which was clearly evidenced by its spectroscopic properties. Compound [O1](PF6)2 revealed a moderate stability at low temperatures with a distinguished activity towards a large variety of phenolic substrates. DFT calculations enabled a prognosis of the hydroxylation position by Fukui function, which fully agreed with experimental results. The Fukui function was introduced as new predictive tool for Cu/O2 chemistry. Moreover, our present findings clearly show efficient C−H activation of mono‐ and bicyclic phenolic substrates, stating an atom‐economic strategy to design new phenazines. A bioinspired model system, such as [O1](PF6)2, indicates tyrosinase‐like activity of O cores and exceeds evidently the enzymatically limited substrate scope. This study opens the door to future developments on tyrosinase model systems with high relevance to atom‐economic oxygen‐transfer reactions with synthetic importance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support provided by the German Research Foundation (DFG), in framework of Priority Program “Reactive Bubbly Flows” SPP 1740 (HE 5480/10‐2, http://www.dfg‐spp1740.de/), International Research Training Group SeleCa and further projects (RU 773/8‐1). We also acknowledge computing time and support provided by OCuLUS Cluster at PC2 Paderborn and support at beamline P64 by Marcel Görlitz and Dr. Wolfgang Caliebe.
M. Paul, M. Teubner, B. Grimm-Lebsanft, C. Golchert, Y. Meiners, L. Senft, K. Keisers, P. Liebhäuser, T. Rösener, F. Biebl, S. Buchenau, M. Naumova, V. Murzin, R. Krug, A. Hoffmann, J. Pietruszka, I. Ivanović-Burmazović, M. Rübhausen, S. Herres-Pawlis, Chem. Eur. J. 2020, 26, 7556.
References
- 1.
- 1a. Campbell A. N., Stahl S. S., Acc. Chem. Res. 2012, 45, 851–863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. McCann S. D., Stahl S. S., Acc. Chem. Res. 2015, 48, 1756–1766. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Solomon E. I., Heppner D. E., Johnston E. M., Ginsbach J. W., Cirera J., Qayyum M., Kieber-Emmons M. T., Kjaergaard C. H., Hadt R. G., Tian L., Chem. Rev. 2014, 114, 3659–3853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Elwell C. E., Gagnon N. L., Neisen B. D., Dhar D., Spaeth A. D., Yee G. M., Tolman W. B., Chem. Rev. 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Matoba Y., Kumagai T., Yamamoto A., Yoshitsu H., Sugiyama M., J. Biol. Chem. 2006, 281, 8981–8990; [DOI] [PubMed] [Google Scholar]
- 3b. Bijelic A., Pretzler M., Molitor C., Zekiri F., Rompel A., Angew. Chem. Int. Ed. 2015, 54, 14677–14680; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14889–14893. [Google Scholar]
- 4.
- 4a. Kahn V., Ben-Shalom N., Pigment Cell Res. 1998, 11, 24–33; [DOI] [PubMed] [Google Scholar]
- 4b. del Mar Garcia-Molina M., Muñoz-Muñoz J. L., Garcia-Molina F., García-Ruiz P. A., Garcia-Canovas F., J. Agric. Food Chem. 2012, 60, 6447–6453. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Large T. A. G., Mahadevan V., Keown W., Stack T. D. P., Inorg. Chim. Acta 2019, 486, 782–792; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Chiang L., Wasinger E. C., Shimazaki Y., Young V., Storr T., Stack T. D. P., Inorg. Chim. Acta 2018, 481, 151–158; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Quist D. A., Diaz D. E., Liu J. J., Karlin K. D., J. Biol. Inorg. Chem. 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Kanteev M., Goldfeder M., Fishman A., Protein Sci. 2015, 24, 1360–1369; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Deri B., Kanteev M., Goldfeder M., Lecina D., Guallar V., Adir N., Fishman A., Sci. Rep. 2016, 6, 34993; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Singha A., Rana A., Dey A., Inorg. Chim. Acta 2019, 487, 63–69. [Google Scholar]
- 7.
- 7a. Cramer C. J., Smith B. A., Tolman W. B., J. Am. Chem. Soc. 1996, 118, 11283–11287; [Google Scholar]
- 7b. Mahadevan V., Hou Z., Cole A. P., Root D. E., Lal T. K., Solomon E. I., Stack T. D. P., J. Am. Chem. Soc. 1997, 119, 11996–11997. [Google Scholar]
- 8.P. Liebhäuser, A. Hoffmann, S. Herres-Pawlis in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 10.1016/B978-0-12-409547-2.11554-9. [DOI]
- 9.
- 9a. Handley D. A., Hitchcock P. B., Lee T. H., Leigh G. J., Inorg. Chim. Acta 2001, 316, 59–64; [Google Scholar]
- 9b. Mukherjee J., Mukherjee R., Dalton Trans. 2006, 6, 1611; [DOI] [PubMed] [Google Scholar]
- 9c. Mirica L. M., Stack T. D. P., Inorg. Chem. 2005, 44, 2131–2133; [DOI] [PubMed] [Google Scholar]
- 9d. Kunishita A., Kubo M., Ishimaru H., Ogura T., Sugimoto H., Itoh S., Inorg. Chem. 2008, 47, 12032–12039; [DOI] [PubMed] [Google Scholar]
- 9e. Hong S., Hill L. M. R., Gupta A. K., Naab B. D., Gilroy J. B., Hicks R. G., Cramer C. J., Tolman W. B., Inorg. Chem. 2009, 48, 4514–4523; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Taki M., Teramae S., Nagatomo S., Tachi Y., Kitagawa T., Itoh S., Fukuzumi S., J. Am. Chem. Soc. 2002, 124, 6367–6377; [DOI] [PubMed] [Google Scholar]
- 9g. Cole A. P., Mahadevan V., Mirica L. M., Ottenwaelder X., Stack T. D. P., Inorg. Chem. 2005, 44, 7345–7364; [DOI] [PubMed] [Google Scholar]
- 9h. Citek C., Lyons C. T., Wasinger E. C., Stack T. D. P., Nat. Chem. 2012, 4, 317–322; [DOI] [PubMed] [Google Scholar]
- 9i. Chiang L., Keown W., Citek C., Wasinger E. C., Stack T. D. P., Angew. Chem. Int. Ed. 2016, 55, 10453–10457; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10609–10613; [Google Scholar]
- 9j. McCrory C. C. L., Devadoss A., Ottenwaelder X., Lowe R. D., Stack T. D. P., Chidsey C. E. D., J. Am. Chem. Soc. 2011, 133, 3696–3699; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9k. Herres-Pawlis S., Verma P., Haase R., Kang P., Lyons C. T., Wasinger E. C., Flörke U., Henkel G., Stack T. D. P., J. Am. Chem. Soc. 2009, 131, 1154–1169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9l. Herres-Pawlis S., Haase R., Verma P., Hoffmann A., Kang P., Stack T. D. P., Eur. J. Inorg. Chem. 2015, 5426–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamann J. N., Herzigkeit B., Jurgeleit R., Tuczek F., Coord. Chem. Rev. 2017, 334, 54–66. [Google Scholar]
- 11. Réglier M., Jorand C., Waegell B., J. Chem. Soc. Chem. Commun. 1990, 1752–1755. [Google Scholar]
- 12. Casella L., Gullotti M., Radaelli R., Di Gennaro P., J. Chem. Soc. Chem. Commun. 1991, 1611. [Google Scholar]
- 13.
- 13a. Rolff M., Schottenheim J., Peters G., Tuczek F., Angew. Chem. Int. Ed. 2010, 49, 6438–6442; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6583–6587; [Google Scholar]
- 13b. Herzigkeit B., Flöser B. M., Meißner N. E., Engesser T. A., Tuczek F., ChemCatChem 2018, 10, 5402–5405; [Google Scholar]
- 13c. Hamann J. N., Tuczek F., Chem. Commun. 2014, 50, 2298–2300; [DOI] [PubMed] [Google Scholar]
- 13d. Herzigkeit B., Flöser B. M., Engesser T. A., Näther C., Tuczek F., Eur. J. Inorg. Chem. 2018, 3058–3069; [Google Scholar]
- 13e. Herzigkeit B., Jurgeleit R., Flöser B. M., Meißner N. E., Engesser T. A., Näther C., Tuczek F., Eur. J. Inorg. Chem. 2019, 2258–2266; [Google Scholar]
- 13f. Schottenheim J., Gernert C., Herzigkeit B., Krahmer J., Tuczek F., Eur. J. Inorg. Chem. 2015, 3501–3511. [Google Scholar]
- 14.
- 14a. Esguerra K. V. N., Fall Y., Lumb J. P., Angew. Chem. Int. Ed. 2014, 53, 5877–5881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5987–5991; [Google Scholar]
- 14b. Askari M. S., Rodríguez-Solano L. A., Proppe A., McAllister B., Lumb J.-P., Ottenwaelder X., Dalton Trans. 2015, 44, 12094–12097; [DOI] [PubMed] [Google Scholar]
- 14c. Huang Z., Kwon O., Huang H., Fadli A., Marat X., Moreau M., Lumb J.-P., Angew. Chem. Int. Ed. 2018, 57, 11963–11967; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12139–12143. [Google Scholar]
- 15.
- 15a. Hoffmann A., Citek C., Binder S., Goos A., Rübhausen M., Troeppner O., Ivanović-Burmazović I., Wasinger E. C., Stack T. D. P., Herres-Pawlis S., Angew. Chem. Int. Ed. 2013, 52, 5398–5401; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5508–5512; [Google Scholar]
- 15b. Strassl F., Hoffmann A., Grimm-Lebsanft B., Rukser D., Biebl F., Tran M., Metz F., Rübhausen M., Herres-Pawlis S., Inorganics 2018, 6, 114. [Google Scholar]
- 16.
- 16a. Stanek J., Rösener T., Metz A., Mannsperger J., Hoffmann A., Herres-Pawlis S., Top. Heterocycl. Chem. 2015, 51, 95–164; [Google Scholar]
- 16b. Herres-Pawlis S., Binder S., Eich A., Haase R., Schulz B., Wellenreuther G., Henkel G., Rübhausen M., Meyer-Klaucke W., Chem. Eur. J. 2009, 15, 8678–8682; [DOI] [PubMed] [Google Scholar]
- 16c. Hoffmann A., Wern M., Hoppe T., Witte M., Haase R., Liebhäuser P., Glatthaar J., Herres-Pawlis S., Schindler S., Eur. J. Inorg. Chem. 2016, 4744–4751; [Google Scholar]
- 16d. Schurr D., Strassl F., Liebhäuser P., Rinke G., Dittmeyer R., Herres-Pawlis S., React. Chem. Eng. 2016, 1, 485–493. [Google Scholar]
- 17.
- 17a. Schatz M., Raab V., Foxon S. P., Brehm G., Schneider S., Reiher M., Holthausen M. C., Sundermeyer J., Schindler S., Angew. Chem. Int. Ed. 2004, 43, 4360–4363; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4460–4464; [Google Scholar]
- 17b. Maiti D., Lee D.-H., Gaoutchenova K., Würtele C., Holthausen M. C., Narducci Sarjeant A. A., Sundermeyer J., Schindler S., Karlin K. D., Angew. Chem. Int. Ed. 2008, 47, 82–85; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 88–91. [Google Scholar]
- 18.
- 18a. Cross B., Dunn C. L., Payne D. H., Tipton J. D., J. Sci. Food Agric. 1969, 20, 340–344; [DOI] [PubMed] [Google Scholar]
- 18b. Guttenberger N., Blankenfeldt W., Breinbauer R., Bioorg. Med. Chem. 2017, 25, 6149–6166; [DOI] [PubMed] [Google Scholar]
- 18c. de Andrade-Neto V. F., Goulart M. O. F., da Silva Filho J. F., da Silva M. J., do C. F. R. Pinto M., Pinto A. V., Zalis M. G., Carvalho L. H., Krettli A. U., Bioorg. Med. Chem. Lett. 2004, 14, 1145–1149; [DOI] [PubMed] [Google Scholar]
- 18d. Jana A. K., J. Photochem. Photobiol. A 2000, 132, 1–17; [Google Scholar]
- 18e. Gulevskaya A. V., Eur. J. Org. Chem. 2016, 4207–4214; [Google Scholar]
- 18f. Mortzfeld F. B., Pietruszka J., Baxendale I. R., Eur. J. Org. Chem. 2019, 5424–5433; [Google Scholar]
- 18g. Hamann J. N., Rolff M., Tuczek F., Dalton Trans. 2015, 44, 3251–3258; [DOI] [PubMed] [Google Scholar]
- 18h. Chaudhary A., Khurana J. M., Res. Chem. Intermed. 2018, 44, 1045–1083; [Google Scholar]
- 18i. Xiao Y., Hu W., Sun S., Yu J.-T., Cheng J., Synlett 2019, 30, 2113–2122; [Google Scholar]
- 18j. Banerjee S., Arkivoc 2016, 82–110; [Google Scholar]
- 18k. Bilal M., Guo S., Iqbal H. M. N., Hu H., Wang W., Zhang X., World J. Microbiol. Biotechnol. 2017, 33, 191; [DOI] [PubMed] [Google Scholar]
- 18l. Cimmino A., Evidente A., Mathieu V., Andolfi A., Lefranc F., Kornienko A., Kiss R., Nat. Prod. Rep. 2012, 29, 487–501. [DOI] [PubMed] [Google Scholar]
- 19.Formation of [O1](PF6)2 at −80 °C led to smaller extinction at 392 nm (20000 m −1 cm−1, −5 %) in the UV/Vis spectrum compared to the formation at −90 °C. Formation of [O1](PF6)2 at −74 °C led to a significant loss in quantity (16000 m −1 cm−1, −24 %) (see Supporting Information).
- 20.High extinction of [O1](PF6)2 at 280 nm is caused by overlapping in-plane πσ* to dxy CT transition and π to π* transition of the aromatic ligand backbone.
- 21. Mirica L. M., Ottenwaelder X., Stack T. D. P., Chem. Rev. 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]
- 22. Karlin K. D., Hayes J. C., Gultneh Y., Cruse R. W., McKown J. W., Hutchinson J. P., Zubieta J., J. Am. Chem. Soc. 1984, 106, 2121–2128. [Google Scholar]
- 23.
- 23a. Itoh S., Taki M., Nakao H., Holland P. L., Tolman W. B., L. Que, Jr. , Fukuzumi S., Angew. Chem. Int. Ed. 2000, 39, 398–400; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 409–411; [Google Scholar]
- 23b. Arii H., Saito Y., Nagatomo S., Kitagawa T., Funahashi Y., Jitsukawa K., Masuda H., Chem. Lett. 2003, 32, 156–157; [Google Scholar]
- 23c. Mahapatra S., Halfen J. A., Tolman W. B., J. Am. Chem. Soc. 1996, 118, 11575–11586; [Google Scholar]
- 23d. Enomoto M., Aida T., J. Am. Chem. Soc. 1999, 121, 874–875. [Google Scholar]
- 24.
- 24a. Lewis E. A., Tolman W. B., Chem. Rev. 2004, 104, 1047–1076; [DOI] [PubMed] [Google Scholar]
- 24b. Halfen J. A., Young V. G., Tolman W. B., Inorg. Chem. 1998, 37, 2102–2103; [DOI] [PubMed] [Google Scholar]
- 24c. Itoh S., Nakao H., Berreau L. M., Kondo T., Komatsu M., Fukuzumi S., J. Am. Chem. Soc. 1998, 120, 2890–2899. [Google Scholar]
- 25. Mahapatra S., Kaderli S., Llobet A., Neuhold Y.-M., Palanché T., Halfen J. A., Young V. G., Kaden T. A., Que L., Zuberbühler A. D., Tolman W. B., Inorg. Chem. 1997, 36, 6343–6356. [Google Scholar]
- 26. Bulkowski J. E., Binucleating Ligand–Metal Complexes as Oxidation Catalysts, US Patent 4,545,937, 1985.
- 27. Verma P., Weir J., Mirica L., Stack T. D. P., Inorg. Chem. 2011, 50, 9816–9825. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Liebhäuser P., Keisers K., Hoffmann A., Schnappinger T., Sommer I., Thoma A., Wilfer C., Schoch R., Stührenberg K., Bauer M., Dürr M., Ivanović-Burmazović I., Herres-Pawlis S., Chem. Eur. J. 2017, 23, 12171–12183; [DOI] [PubMed] [Google Scholar]
- 28b. Wilfer C., Liebhäuser P., Hoffmann A., Erdmann H., Grossmann O., Runtsch L., Paffenholz E., Schepper R., Dick R., Bauer M., Dürr M., Ivanovic-Burmazovic I., Herres-Pawlis S., Chem. Eur. J. 2015, 21, 17639–17649. [DOI] [PubMed] [Google Scholar]
- 29.Turnovers could not be determined, because no extinction coefficient of the quinone is reported elsewhere in the literature.
- 30. Yavari I., Nader Z., Dye. Pigment. 2007, 75, 474–478. [Google Scholar]
- 31.
- 31a. Mishra A. K., Moorthy J. N., J. Org. Chem. 2016, 81, 6472–6480; [DOI] [PubMed] [Google Scholar]
- 31b. Wu A., Duan Y., Xu D., Penning T. M., Harvey R. G., Tetrahedron 2010, 66, 2111–2118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31c. Hussain H., Specht S., Sarite S. R., Saeftel M., Hoerauf A., Schulz B., Krohn K., J. Med. Chem. 2011, 54, 4913–4917. [DOI] [PubMed] [Google Scholar]
- 32. Leitch J. A., Bhonoah Y., Frost C. G., ACS Catal. 2017, 7, 5618–5627. [Google Scholar]
- 33.Furthermore, we report NMR, IR, and MS analytical data for P2 Another synthetic route and elemental analysis data have been published previously: Morgan L. R., Schunior R. J., Boyer J. H., J. Org. Chem. 1963, 28, 260–261. [Google Scholar]
- 34.
- 34a. Krug R., Schröder D., Gebauer J., Suljić S., Morimoto Y., Fujieda N., Itoh S., Pietruszka J., Eur. J. Org. Chem. 2018, 1789–1796; [Google Scholar]
- 34b. Penttinen L., Rutanen C., Jänis J., Rouvinen J., Hakulinen N., ChemBioChem 2018, 19, 2348–2352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary