

Abstract
Objective
Guillain‐Barré syndrome (GBS) is a rare, life‐threatening disorder of the peripheral nervous system. Immunoglobulin G Fc‐gamma receptors (FcγRs) mediate and regulate diverse effector functions and are involved in the pathogenesis of GBS. We investigated whether the FcγR polymorphisms FcγRIIa H/R131 (rs1801274), FcγRIIIa V/F158 (rs396991), and FcγRIIIb NA1/NA2, and their haplotype patterns affect the affinity of IgG‐FcγR interactivity and influence GBS susceptibility and severity.
Methods
We determined FcγR polymorphisms in 303 patients with GBS and 302 ethnically matched healthy individuals from Bangladesh by allele‐specific polymerase chain reaction. Pairwise linkage disequilibrium and haplotype patterns were analyzed based on D ´statistics and the genotype package of R statistics, respectively. Logistic regression analysis and Fisher’s exact test with corrected P (Pc) values were employed for statistical comparisons.
Results
FcγRIIIa‐V158F was associated with the severe form of GBS compared to the mild form (P = 0.005, OR = 2.24, 95% CI = 1.28–3.91; Pc = 0.015); however, FcγR genotypes and haplotype patterns did not show any association with GBS susceptibility compared to healthy controls. FcγRIIIa‐V/V158 and FcγRIIIb‐NA2/2 were associated with recent Campylobacter jejuni infection (P ≤ 0.001, OR = 0.36, 95% CI = 0.23–0.56; Pc ≤ 0.003 and P = 0.004, OR = 1.70, 95% CI = 1.18–2.44; Pc ≤ 0.012, respectively). Haplotype 1 (FcγRIIa‐H131R‐ FcγRIIIa‐V158F‐ FcγRIIIb‐NA1/2) and the FcγRIIIb‐NA2/2 genotype were more prevalent among anti‐GM1 antibody‐positive patients (P = 0.031, OR = 9.61, 95% CI = 1.24–74.77, Pc = 0.279; P = 0.027, OR = 1.62, 95% CI = 1.06–2.5, Pc = 0.081, respectively).
Interpretation
FcγR polymorphisms and haplotypes are not associated with susceptibility to GBS, though the FcγRIIIa‐V158F genotype is associated with the severity of GBS.
Introduction
Guillain‐Barré syndrome (GBS) is a post‐infectious autoimmune disorder of the peripheral nervous system that can lead to significant morbidity, long‐term disability or death.
Cross‐reactive immune responses induced by molecular mimicry between the outer core structure of infectious agents that trigger GBS and host nerve gangliosides 1 result in a blockade of nerve conduction. 1 , 2 Campylobacter jejuni has been identified as the predominant causative microbial infectious agent in GBS. 3 , 4 , 5 In addition to multifarious microorganism‐derived factors, host immunogenic factors are likely to affect GBS susceptibility as only a subset of C. jejuni‐infected individuals (1 in 1000–5000 cases) develop GBS. 6 , 7 , 8 , 9 Natural variations in genetic host susceptibility factors have become a focus of research on the susceptibility and severity of disease pathogenesis in GBS.
Immunoglobulin G Fc‐gamma receptors (FcγRs) are important immune‐response modulating molecules that link the cellular and humoral immune system by interacting with IgG subtypes (IgG1‐4). The most common autoantibodies in GBS are produced against GM1, GD1a and GQ1b gangliosides. 5 , 10 , 11 These autoantigens may influence nerve disruption, demyelination or axonal degeneration via diverse mechanisms, 12 including induction of inflammatory immune responses, by interacting with Fc receptors. FcγR polymorphisms can determine the vigor of inflammatory responses, affect downstream functions such as phagocytosis, antibody‐dependent cellular cytotoxicity (ADCC) and the release of inflammatory mediators, and have been implicated in the development of autoimmune disease. 13 , 14 Thus, FcγRs may represent important effector molecules in the pathogenesis of GBS. 15 Three subclasses of FcγRs, namely FcγRIIa, FcγRIIIa and FcγRIIIb, exhibit allelic variation. 13 , 16 The most widely distributed receptor, FcγRIIa, is expressed on all types of white blood cells and has two allelic forms: FcγRIIa‐H131 and FcγRIIa‐R131. These alleles differ by the replacement of histidine by arginine at position 131 due to an A → G single nucleotide exchange at position 494. 17 , 18 FcγRIIa‐H131 is reported to bind human IgG2 with a higher affinity than FcγRIIa‐R131. 19 FcγRIIIa is expressed on macrophages, dendritic cells, γ/δ T‐cells and natural killer (NK) cells. 20 A functional polymorphism at nucleotide 559 results in either a valine (V) or phenylalanine (F) at amino acid position 158, which affects the receptor binding capacity of IgG1, IgG3, and IgG4. 21 FcγRIIIb is expressed on neutrophils and exhibits two allelic forms, neutrophil antigen 1 (NA1) and neutrophil antigen 2 (NA2). NA1 and NA2 differ by five base substitutions (nucleotides 141, 147, 227, 277, and 349) that lead to four amino acid changes (at positions 36, 65, 82, and 106) within exon 3. 18 , 22 However, these allelic forms of FcγR (NA1/NA2) have different affinities for IgG1 and IgG3. Thus, the various allelic forms of FcγR may possibly determine the extent of inflammatory responses and thereby influence autoimmune diseases, including GBS.
Several studies have already evaluated the relationship between FcγR polymorphisms and the pathogenesis of GBS. 23 , 24 , 25 , 26 , 27 FcγRIIa‐H/H131 was significantly associated with susceptibility to GBS and was also a potent risk factor for the development of GBS in a Dutch population. 23 These findings were consistent with a study of Indian patients with GBS, but not with a report on Norwegian Caucasian patients. 24 , 26 One meta‐analysis indicated that every FcγRIIIb‐NA2 allele cumulatively increases the GBS severity score, though none of the genotypes or alleles were associated with susceptibility to GBS. 25 However, consensus regarding the role of FcγR polymorphisms in the pathogenesis of GBS has not yet been established due to the inadequate statistical power of studies with small sample sizes and differences in the ethnicities of the populations tested. Thus, we aimed to evaluate whether candidate gene polymorphisms in FcγR are a major causative factor for GBS susceptibility or severity in Bangladeshi patients with C. jejuni‐triggered GBS, which represents the world’s largest cohort.
Materials and Methods
Research participants
The GBS cohort used in this study includes 303 patients with GBS (208 males, 95 females; median age: 30 years [interquartile range, 17–42]; Table 1) and 302 ethnically matched healthy controls (204 males, 99 females; median age: 34 years [interquartile range, 28–46]). Patients with GBS were diagnosed based on the previously established diagnostic criteria described by Asbury and Cornblath 28 and enrolled from Dhaka Medical College and Hospital (DMCH), Dhaka, Bangladesh. No preference was given to race, religion, or socioeconomic status during study subject selection. Genetically unrelated healthy individuals who did not have neurological diseases, antecedent infections, recent surgery or other illnesses were included in this study following informed consent and matched with patients. Clinical, electrophysiological, and serological data were obtained from patients with informed consent.
Table 1.
Demographic and clinical characteristics of the patients with GBS.
| Characteristic | Number of patients, n = 303 (%) |
|---|---|
| Sex | |
| Male/female | 208/95 |
| Age | |
| Median (IQR) | 30 (17–42) |
| Preceding illness, n = 303 | |
| Diarrhea | 129/303 (43) |
| Respiratory tract infections | 45/303 (15) |
| Fever | 25/303 (8) |
| Other | 28/303 (9) |
| None/unknown | 76/303 (25) |
| Electrophysiological classification, n = 247 | |
| Axonal | 146/247 (59) |
| Demyelinating | 68/247 (27) |
| Unclassified | 33/247 (13) |
| MRC sum score (at entry) | |
| Severely affected patients | 232/303 (77) |
| Mildly affected patients | 71/303 (23) |
| Serological characteristics | |
| Anti‐GM1‐Ab‐seropositive | 118/303 (39) |
| C. jejuni‐seropositive | 186/303 (61) |
| Disease prognosis at 6 months, n = 303 | |
| Good outcome | 209/303 (69) |
| Poor outcome | 94/303 (31) |
GBS, Guillain‐Barré syndrome; IQR, interquartile range; MRC, Medical Research Council; Ab, antibody; C. jejuni, Campylobacter jejuni.
Blood specimens were collected by venipuncture before patients received medication and disease outcome was evaluated by assessing clinical data at specific standard time‐points (at entry, 2 weeks, 4 weeks and 6 months). In this cohort, 75% (227/303) patients had an antecedent illness; most frequently diarrhea (43%; 129/303), followed by respiratory infection (15%, 45/303), fever (8%, 25/303) or other illnesses (9%, 28/303); 25% (76/303) of patients had history of unknown infections or no infection. Serological tests, that is, antibodies against C. jejuni or GM1, GD1a and GQ1b gangliosides were measured using enzyme‐linked immunosorbent assays (ELISAs). 5 , 29
Electrophysiological studies of 82% (247/303) of the GBS patients indicated 59% (146/247) of patients had an axonal subtype of GBS, including acute motor axonal neuropathy (AMAN) and acute motor and sensory axonal neuropathy (AMSAN), 27% (68/247) of patients had acute inflammatory demyelinating polyradiculoneuropathy (AIDP) and 13% (33/247) of cases were unclassified with inexcitable nerves or equivocal findings. 30 Severity of disease (degree of muscle weakness) was assessed using the Medical Research Council (MRC) sum score 31 , 32 ranging from 0 to 60 at nadir (maximum muscle weakness); GBS patients at nadir with MRC sumscore < 40 were defined as severely affected patients and with MRC sumscore ≥ 40 were defined as mildly affected patients. 33 The outcome of the disease was measured using the GBS disability score after 6 months of follow‐up. 34 This study was reviewed and approved by the Institutional Review Board (IRB) and ethical committees of the icddr, b, Dhaka, Bangladesh.
Genomic DNA isolation
Whole blood samples were collected from 605 study subjects into lithium heparin‐coated anti‐coagulation tubes for genomic DNA isolation. Genomic DNA was extracted using the QIAamp® DNA Blood Midi Kit (100) (Qiagen, Hilden, Germany), dissolved in 1 × TE buffer (10 mmol/L Tris‐Cl, pH 8.0, 1 mmol/L EDTA), stored at −80°C, diluted to 10 ng/µL with Milli‐Q water and then stored at −20°C until SNP detection.
FcγR polymorphism detection and genotype analysis
The FcγR polymorphisms FcγRIIa H/R131 (rs1801274), FcγRIIIa V/F158 (rs396991) and FcγRIIIb NA1/NA2 were genotyped via a previously described allele‐specific polymerase chain reaction (AS‐PCR) method using published primer sequences and reaction conditions. 18 , 21 Human growth hormone (HGH) primers (5`‐GCCTTCCCAACCATTCCCTTA‐3′ and 5′‐CTCACGGATTTCTGTTGTGTTTC‐3′) were used as an internal positive control. 18 The PCR products were visualized on 2% agarose gels using a Molecular Imager® Gel Doc™ XR + system (Bio‐Rad Laboratories Inc).
Statistical analysis
Statistical analysis was performed using logistic regression analysis and Fisher’s exact test with Yates' continuity correction to assess associations between the FcγR polymorphisms and disease susceptibility or subgroups. In the control group, all SNPs were within Hardy‐Weinberg equilibrium. P values less than 0.05 were considered statistically significant. The Bonferroni method was applied to correct the P values for multiple comparisons: each Pvalue was multiplied by the number of comparisons and denoted Pc (Pc, P corrected). Genotype/allelic frequencies were estimated by a simple counting method and the data were processed using Microsoft Excel 2010 (Microsoft, Redmond, WA, USA), GraphPad prism (version 5.01, GraphPad software, Inc., La Jolla, CA) or SPSS (version 16.0, Company, Chicago, IL). Haplotype patterns and frequencies were analyzed using the genotype package of R statistics and their associations with GBS susceptibility and subgroups were assessed using logistic regression analysis.
Results
FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphisms and haplotype in patients with GBS and healthy individuals
No significant associations were observed between the FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphisms and susceptibility to GBS compared to healthy controls (Table 2). The comparison of axonal variants of GBS versus healthy controls or demyelinating subtypes versus healthy subjects showed no relation with disease susceptibility (Table 3). The haplotype distributions of the three loci were compared between patients with GBS and healthy individuals. Haplotype analysis revealed 27 possible different patterns for the FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphic loci (Fig. 1). The nine most predominant patterns (haplotypes 1–9; frequency > 5%), representing 61.5% of total variation, were selected for further haplotype analysis (Fig. 2). No significant association was observed between any haplotype and GBS susceptibility when each haplotype was analyzed individually.
Table 2.
FcγR genotype and allelic distributions in Bangladeshi patients with GBS and healthy controls.
| FcγR genotype/allele | HC, n = 302 (%) | GBS patients, n = 303 (%) | P value | Odds ratio (95% CI) |
|---|---|---|---|---|
| FcγR‐IIa | ||||
| H/H‐131 | 116 (38.4) | 114 (37.6) | Reference | |
| H/R‐131 | 136 (45) | 124 (40.9) | 0.283 | 0.93 (0.65–1.32) |
| R/R‐131 | 50 (16.6) | 65 (21.5) | 1.32 (0.84–2.08) | |
| R‐131 | 236 (39.1) | 254 (41.9) | 0.320 | 0.89 (0.71–1.12) |
| H‐131 | 368 (60.9) | 352 (58.1) | Reference | |
| FcγR‐IIIa | ||||
| F/F‐158 | 110 (36.4) | 120 (39.6) | Reference | |
| V/F‐158 | 150 (49.7) | 143 (47.2) | 0.723 | 0.87 (0.62–1.23) |
| V/V‐158 | 42 (13.9) | 40 (13.2) | 0.87 (0.53–1.45) | |
| V‐158 | 234 (38.7) | 223 (36.8) | 1.09 (0.86–1.37) | |
| F‐158 | 370 (61.3) | 383 (63.2) | 0.514 | Reference |
| FcγR‐IIIb | ||||
| NA1/1 | 69 (22.9) | 56 (18.5) | Reference | |
| NA1/2 | 126 (41.7) | 125 (41.2) | 0.311 | 1.22 (0.79–1.88) |
| NA2/2 | 107 (35.4) | 122 (40.3) | 1.41 (0.91–2.18) | |
| NA1 | 264 (43.7) | 237 (39.1) | 0.115 | 1.21(0.96–1.52) |
| NA2 | 340 (56.3) | 369 (60.9) | Reference | |
GBS, Guillain‐Barré syndrome; HC, healthy controls; 95% CI, 95% confidence interval.
Table 3.
Distribution of FcγR genotypes and alleles among axonal and demyelinating cases of GBS compared to healthy controls.
| FcγR | Subtype | Axonal versus HC | Demyelinating versus HC | ||||
|---|---|---|---|---|---|---|---|
| Genotypes/Alleles | Axonal, n = 146 (%) | Demyelinating, n = 68 (%) | Healthy control (HC), n = 302 (%) | P value | OR (95% CI) | P value | OR (95% CI) |
| FcγR IIa | |||||||
| H/H −131 | 50 (34.2) | 28 (41.2) | 116 (38.4) | Reference | Reference | ||
| H/R‐131 | 63 (43.2) | 24 (35.3) | 136 (45) | 0.289 | 1.1 (0.69–1.68) | 0.242 | 0.7 (0.40–1.33) |
| R/R −131 | 33 (22.6) | 16 (23.5) | 50 (16.6) | 1.5 (0.88–2.66) | 1.3 (0.66–2.67) | ||
| R‐131 | 129 (44.2) | 56 (41.2) | 236 (39.1) | Reference | Reference | ||
| H‐131 | 163 (55.8) | 80 (58.8) | 368 (60.9) | 0.147 | 1.2 (0.93–1.64) | 0.698 | 1.1 (0.75–1.59) |
| FcγR IIIa | |||||||
| F/F‐158 | 57 (39) | 33 (48.5) | 110 (36.4) | Reference | Reference | ||
| V/F‐158 | 74 (50.7) | 27 (39.7) | 150 (49.7) | 0.542 | 0.9 (0.6–1.4) | 0.178 | 0.6 (0.3–1.0) |
| V/V‐158 | 15 (10.3) | 8 (11.8) | 42 (13.9) | 0.7 (0.4–1.3) | 0.6 (0.3–1.5) | ||
| V‐158 | 104 (35.6) | 43 (31.6) | 234 (38.7) | Reference | Reference | ||
| F‐158 | 188 (64.4) | 93 (68.4) | 370 (61.3) | 0.378 | 0.9 (0.65–1.17) | 0.141 | 0.7 (0.49–1.09) |
| FcγR IIIb | |||||||
| NA1/1 | 27 (18.5) | 17 (25) | 69 (22.8) | Reference | Reference | ||
| NA1/2 | 61 (41.8) | 25 (36.8) | 126 (41.7) | 0.506 | 0.8 (0.5–1.4) | 0.753 | 1.2 (0.6–2.4) |
| NA2/2 | 58 (39.7) | 26 (38.2) | 107 (35.4) | 0.7 (0.4–1.2) | 1.0 (0.5–2.0) | ||
| NA1 | 115 (39.4) | 59 (43.4) | 264 (43.7) | Reference | Reference | ||
| NA2 | 177 (60.6) | 77 (56.6) | 340 (56.3) | 0.248 | 0.8 (0.6–1.1) | 1.0 | 1.0 (0.7–1.4) |
OR, odds ratio; 95% CI, 95% confidence interval.
Figure 1.
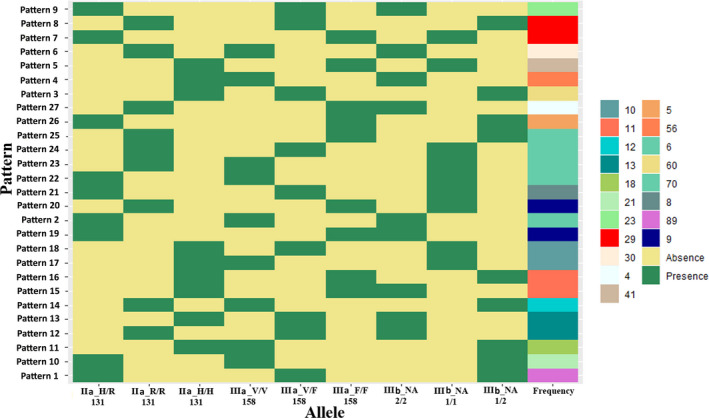
Haplotype analysis of the FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphic loci for the study subjects from Bangladesh. Twenty‐seven different haplotype patterns were observed; pattern 1 was the most common (pink). Green indicates the presence and yellow indicates the absence of specific FcγR polymorphisms for each of the three loci. The polymorphism frequencies are presented as a color gradient on the right.
Figure 2.
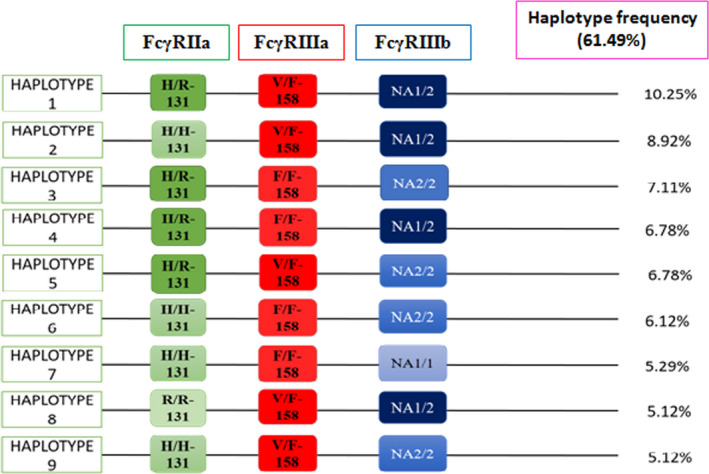
Haplotype frequencies for FcγRIIa, FcγRIIIa, and FcγRIIIb (FcγRs) polymorphisms for the study subjects from Bangladesh. The nine most predominant patterns (haplotypes 1–9; frequency >5%) represented 61.49% of total variation and were selected for haplotype analysis. The frequencies of specific haplotypes are presented on the left.
FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphisms and haplotypes in anti‐GM1 antibody‐positive GBS
The frequency of FcγRIIIb‐NA2/2 genotypes was predominant among anti‐GM1 antibody‐positive patients compared to healthy individuals but association was not significant (P = 0.051, OR = 1.93, 95% CI = 1.03–3.62; Table 4). Haplotype 1 (FcγRIIa‐H131R‐ FcγRIIIa‐V158F‐ FcγRIIIb‐NA1/2) and the FcγRIIIb‐NA2/2 genotype were significantly prevalent among anti‐GM1 antibody‐positive patients than antibody‐negative patients with GBS; however, these associations were lost after Bonferroni correction (P = 0.031, OR = 9.61, 95% CI = 1.24–74.77; Pc = 0.279 and P = 0 .027, OR = 1.62, 95% CI = 1.06–2.5; Pc = 0.081; respectively; Table 5). The homozygous FcγRIIIb NA1/1 genotype was predominant in healthy individuals compared to anti‐GM1 antibody‐positive patients (22.9% vs. 14.2%; Table 4) and significantly present in anti‐GM1 antibody‐negative patients with GBS than antibody‐positive patients (P = 0.002, OR = 0.43, 95% CI = 0.25–0.73; Pc = 0.006; Table 5). Except haplotype 1, no other haplotypes (haplotype 2–9) were associated with anti‐GM1 antibody positivity (Table 5).
Table 4.
Distribution of FcγR genotypes and alleles between healthy controls versus C. jejuni‐seropositive patients and healthy controls versus Anti‐GM1 antibody‐seropositive patients with GBS.
| FcγR genotype/allele | Healthy controls (a), n = 302 (%) | C. jejuni seropositivepatients (b), n = 186 (%) | Anti‐GM1‐Ab‐seropositive patients (c), n = 119 (%) | a versus b P value | a versus b P corrected (Pc) | Odds ratio (95% CI) | a versus c P value | a versus c P corrected (Pc) | Odds ratio (95% CI) |
|---|---|---|---|---|---|---|---|---|---|
| FcγR‐IIa | |||||||||
| H/H‐131 | 116 (38.4) | 67 (36.0) | 42 (35.3) | Reference | Reference | ||||
| H/R‐131 | 136 (45) | 81 (43.6) | 53 (44.5) | 0.917 | na | 1.03 (0.69–1.55) | 0.809 | na | 1.08 (0.67–1.73) |
| R/R‐131 | 50 (16.6) | 38 (20.4) | 24 (20.2) | 0.351 | na | 1.32 (0.78–2.21) | 0.354 | na | 1.33 (0.73–2.42) |
| R‐131 | 236 (39.1) | 157 (42.2) | 101 (42.4) | 0.88 (0.68–1.14) | 0.87 (0.64–1.18) | ||||
| H‐131 | 368 (60.9) | 215 (57.8) | 137 (57.6) | 0.347 | na | Reference | 0.391 | na | Reference |
| FcγR‐IIIa | |||||||||
| F/F‐158 | 110 (36.4) | 70 (37.6) | 44 (37.3) | Reference | Reference | ||||
| V/F‐158 | 150 (49.7) | 90 (48.4) | 55 (46.6) | 0.839 | na | 0.94 (0.63–1.40) | 0.722 | na | 0.92 (0.57–1.46) |
| V/V‐158 | 42 (13.9) | 26 (14.0) | 20 (16.1) | 1.0 | na | 0.97 (0.55–1.73) | 0.623 | na | 1.20 (0.63–2.25) |
| V‐158 | 234 (38.7) | 142 (38.2) | 95 (39.9) | 1.02 (0.78–1.34) | 0.95 (0.70–1.29) | ||||
| F‐158 | 370 (61.3) | 230 (61.8) | 143 (60.1) | 0.892 | na | Reference | 0.754 | na | Reference |
| FcγR‐IIIb | |||||||||
| NA1/1 | 69 (22.9) | 27 (14.3) | 17 (14.2) | Reference | Reference | ||||
| NA1/2 | 126 (41.7) | 86 (46.2) | 51 (42.9) | 0.041 | 0.123 | 1.74 (1.03–2.94) | 0.134 | na | 1.64 (0.88–3.06) |
| NA2/2 | 107 (35.4) | 73 (39.3) | 51 (42.9) | 0.048 | 0.144 | 1.74 (1.02–2.98) | 0.051 | na | 1.93 (1.03–3.62) |
| NA1 | 264 (43.7) | 140 (37.6) | 85 (35.7) | 1.29 (0.98–1.68) | 1.40 (1.02–1.91) | ||||
| NA2 | 340 (56.3) | 232 (62.4) | 153 (64.3) | 0.071 | na | Reference | 0.036 | 0.072 | Reference |
OR, odds ratio; 95% CI, 95% confidence interval; C. jejuni, Campylobacter jejuni; Anti‐GM1 Ab, Anti‐GM1 antibody; na, not applicable.
Table 5.
Associations between FcγR genotypes and haplotypes with severe disease, anti‐GM1 antibody‐seropositivity and C. jejuni‐seropositivity among patients with GBS.
| Variables | FcγR genotype/haplotype | P value | Odds ratio | 95% CI | P corrected (Pc) |
|---|---|---|---|---|---|
| Mildly affected (n = 71) versus severely affected (n = 232) patients | FcγRIIIa | ||||
| F/F‐158 | 0.03 | 0.55 | 0.32–0.94 | 0.09 | |
| V/F‐158 | 0.005 | 2.24 | 1.28–3.91 | 0.015 | |
| V/V‐158 | 0.25 | 0.68 | 0.32–1.41 | – | |
| FcγRIIIb | |||||
| NA1/1 | 0.007 | 0.41 | 0.22–0.77 | 0.021 | |
| NA1/2 | 0.054 | 1.75 | 0.99–3.08 | 0.162 | |
| NA2/2 | 0.891 | 1.06 | 0.62–1.82 | – | |
| Anti‐GM1 Ab‐seropositive (n = 118) versus seronegative (n = 185) | FcγRIIIb | ||||
| NA1/1 | 0.002 | 0.43 | 0.25–0.73 | 0.006 | |
| NA1/2 | 0.482 | 1.16 | 0.76–1.77 | – | |
| NA2/2 | 0.027 | 1.62 | 1.06–2.47 | 0.081 | |
| Haplotype 1 | 0.031 | 9.61 | 1.24–74.77 | 0.279 | |
| C. jejuni‐seropositive (n = 186) versus seronegative (n = 117) | FcγRIIIa | ||||
| F/F‐158 | 0.038 | 1.47 | 1.02–2.11 | 0.114 | |
| V/F‐158 | 0.025 | 1.49 | 1.05–2.10 | 0.025 | |
| V/V‐158 | ≤0.001 | 0.36 | 0.23–0.56 | ≤0.003 | |
| FcγRIIIb | |||||
| NA1/1 | ≤0.001 | 0.32 | 0.21–0.49 | ≤0.003 | |
| NA1/2 | 0.026 | 1.48 | 1.05–2.10 | 0.078 | |
| NA2/2 | 0.004 | 1.70 | 1.18–2.44 | 0.012 |
OR, odds ratio; 95% CI, 95% confidence interval; MRC sum scores < 40 at nadir were defined as severely affected; MRC sum scores ≥ 40 were defined as mildly affected 33 ; Anti‐GM1 Ab, Anti‐GM1 antibody; Pc, Bonferroni‐corrected P values.
Associations of FcγRIIa, FcγRIIIa, and FcγRIIIb polymorphisms and haplotype patterns with disease severity and outcome
FcγRIIa, FcγRIIIa, and FcγRIIIb genotypes and haplotype patterns were investigated in patients with severe and mild form of GBS (Table 5). The haplotype patterns were not associated with disease severity, though homozygous FcγRIIIa‐F158 was significantly associated with the mild form of disease before Bonferroni correction (P = 0.03, OR = 0.55, 95% CI = 0.32–0.94; Pc = 0.09; Table 5). Heterozygous FcγRIIIa‐V158F was significantly associated with the severe form of disease (compared to the mild form) after correcting the P value (P = 0.005, OR = 2.24, 95% CI = 1.28–3.91; Pc = 0.015; Table 5). FcγRIIIa‐NA1/NA1 was significantly predominant in the mild form of GBS than the severe form (P = 0.007, OR = 0.41, 95% CI = 0.22–0.77; Pc = 0.021; Table 5). FcγRIIIa‐NA1/NA2 tended to be more common in severe GBS (P = 0.054, OR = 1.75, 95% CI = 0.99–3.08; Pc = 0.162; Table 5). However, the FcγRIIa‐H131 and FcγRIIa‐R131 alleles and genotypes were not associated with the severity of GBS. Individual FcγR genotypes were not associated with disease outcome at 6‐month follow‐up.
FcγRIIa, FcγRIIIa, and FcγRIIIb genotypes in patients with recent C. jejuni infection
The homozygous FcγRIIIb‐NA2 and heterozygous FcγRIIIb‐NA1/2 genotypes were associated with recent C. jejuni infection in patients with GBS; however, the association for the heterozygous FcγRIIIb‐NA1/2 genotype lost significance after Bonferroni correction (P = 0.004, OR = 1.70, 95% CI = 1.18–2.44; Pc = 0.012 and P = 0.026, OR = 1.48, 95% CI = 1.05–2.10; Pc = 0.078; respectively; Table 5). Frequency of homozygous FcγRIIIb‐NA2 and heterozygous FcγRIIIb‐NA1/2 genotypes were significantly prevalent in C. jejuni infected patients with GBS compared to healthy controls. But P‐value lost its significance after Bonferroni correction (P = 0.041, OR = 1.74, 95% CI = 1.03–2.94; Pc = 0.123 and P = 0.048, OR = 1.74, 95% CI = 1.02–2.98; Pc = 0.144; respectively; Table 4). The FcγRIIIa‐V/V158 genotype was less frequent in C. jejuni ‐seropositive patients (P ≤ 0.001, OR = 0.36, 95% CI = 0.23–0.56; Pc ≤ 0.003; Table 5); however, the FcγRIIIa‐F/F158 and FcγRIIIa‐V/F158 genotypes were significantly prevalent among C. jejuni ‐seropositive patients than seronegative patients before correcting the P values (P = 0.038, OR = 1.47, 95% CI = 1.02–2.11; Pc = 0.114 and P = 0.025, OR = 1.49, 95% CI = 1.05–2.10; Pc = 0.075, respectively; Table 5).
Discussion
This study investigated the association of three functionally relevant polymorphisms in FcγR and the resulting haplotype patterns with the susceptibility and severity of GBS among patients compared to healthy controls in a large cohort of GBS in Bangladesh. We found no significant associations between individual FcγR alleles or genotypes and susceptibility to GBS; however, the FcγRIIIa‐V/F158 genotype influenced the severity of disease. Moreover, associations between the FcγRIIIa and FcγRIIIb genotypes and haplotype patterns were evident in patients with an antecedent C. jejuni infection and anti‐GM1 antibody‐positive patients, respectively.
Associations between FcγR polymorphisms and susceptibility to GBS have previously been studied in patients with different ethnic backgrounds (Table 6). 23 , 24 , 25 , 26 We observed no significant differences in the FcγR allele or genotype frequencies and haplotype patterns between Bangladeshi patients with GBS and healthy controls. These findings confirm a previous meta‐analysis of British, Dutch, and Norwegian GBS cases, 25 which suggested FcγR polymorphisms were not related to disease susceptibility, regardless of ethnic variation.
Table 6.
Summary of population‐association studies of Fc‐gamma receptor polymorphisms with GBS disease susceptibility and severity in various ethnicities.
| Study (Author, year) | Ethnic origin/population | Country | Participants (n) (GBS vs. controls) | Reported association |
|---|---|---|---|---|
| van der Pol WL, 2000 | Caucasian | Netherlands | 31 versus 187 | FcγRIIa‐H/H131 more frequent in patients than controls (OR, 2.45; P = 0.037). |
| FcγRIIa‐H/H131 associated with disease severity (OR, 18.57; P = 0.007). | ||||
| Vedeler, 2000 | Caucasian | Norway | 62 versus 89 | FcγRIIIb‐NA1/NA1 associated with mild GBS (P = 0.027). |
| van Sorge, 2005 | Caucasian | Netherlands | 192 versus 514 | FcγRIIIb‐NA2/2 more frequent in severe GBS (OR, 2.03; P = 0.03). |
| van Sorge, 2005 | British | United Kingdom | 91 versus 111 | FcγRIIa‐H/H131 more frequent in patients than controls (OR, 2.48; P = 0.02) |
| FcgRIIIa‐F158 allele more frequent in patients than controls (OR, 1.56; P = 0.03). | ||||
| Sinha, 2010 | Asian | India | 80 versus 80 | FcγRIIa‐H/H131 and FcγRIIa‐H131 more frequent in patients than controls (P ≤ 0.0001 and P ≤ 0.0001) |
| FcγRIIIa‐V/V158 more frequent in patients than controls (P ≤ 0.0001) | ||||
| This study | Asian | Bangladesh | 303 versus 302 | FcγRIIIa‐V/F158 associated with severe GBS (OR, 2.24; P = 0.015). FcγRIIIb NA1/NA1 associated with mild GBS (OR, 0.41; P = 0.02) |
GBS, Guillain‐Barré syndrome; OR, odds ratio.
In addition, we found the FcγRIIIa‐F/F158 genotype was associated with the mild form of GBS based on MRC sum score at nadir, while the FcγRIIIa‐V/F158 genotype was associated with the severe form of GBS. As phagocytosis, cellular cytotoxicity, cytokine production, and other immune responses depend on efficient FcγR‐IgG interactions, the higher frequency of FcγRIIIa‐F/F158 among patients with the mild form of GBS may indicate this genotype reduces the affinity of IgG binding and in turn impairs immune complex clearance and decreases subsequent inflammation. 13 , 35 , 36 Patients with FcγRIIIa‐V/F158 genotypes may have better ability to clear immune complexes (ICs) via degranulation and phagocytosis more efficiently, resulting in more severe disease. 36 We observed a higher frequency of FcγRIIIb‐NA1/NA1 genotypes in patients with the mild form of GBS, similar to a previous study of Norwegian patients with GBS. 24 The NA1/NA1 genotype has a high affinity for IgG1 and IgG3, 37 which are the most common among the anti‐GM1 and anti‐GQ1b antibodies. 38 Autoantibodies such as anti‐ganglioside antibodies are neutralized in the circulation, thus cross‐reaction of these auto‐antibodies with the peripheral nerves may be partially prevented in patients with GBS who are homozygous for FcγRIIIb‐NA1. 24
Ganglioside‐specific IgG have been reported to damage nerve tissues by activating effector functions (eg, phagocytosis and/or degranulation) via FcγR. 35 , 39 Homozygous FcγRIIIb‐NA1 was less frequent among both C. jejuni ‐seropositive patients and anti‐GM1 antibody‐positive patients with the mild form of the disease. In contrast, FcγRIIIb‐NA2/2 was associated with recent C. jejuni infection and anti‐GM1 antibody production. In addition, C. jejuni ‐seropositive patients had higher frequencies of the FcγRIIIa‐F/F158 and FcγRIIIa‐V158F genotypes. These findings indicate C. jejuni ‐seropositive patients with higher frequency of the FcγRIIIa‐V158F genotype may suffer severe muscle weakness.
One limitation of this study is that polymorphisms of FcγRIIIb receptor gene, FcγRIIIb‐SH alleles were not investigated; however, it is not yet known whether FcγRIIIb‐SH polymorphisms influence the function of FcγRIIIb or not. 16 , 40
The present study strengthens the evidence that FcγR polymorphisms and haplotypes influence the clinical and serological subgroup of GBS, as well as the strength of the immune responses that ultimately trigger the development of GBS and affect disease severity. In addition, the FcγRIIIa‐V158F genotype was more frequent among patients with recent C. jejuni infection and was found to contribute to disease severity. Variation in the FcγR gene differs greatly between populations of different ethnicities, thus it will be important and interesting to confirm our findings in a multiethnic population, such as the International GBS Outcome Study (IGOS) population. 41
Authors’ Contributions
ZI and SH conceived and designed the study. SH and MGB contributed to data acquisition. SH, MGB, and AD performed data analysis and interpreted the data. ZI and SH drafted the manuscript, which was critically reviewed by MGB, AD, ZHH, and IM for intellectual content. All authors read and approved the final manuscript before submission.
Conflict of Interest
ZI received funding from the Fogarty International Center, National Institute of Neurological Disorders and Stroke of the National Institutes of Health, USA under Award Number K43 TW011447) and Annexon Biosciences (South San Francisco, CA 94080, USA). SH, MGB, AD ZHH and IM have no conflicts of interest to declare.
Acknowledgments
This research activity was funded by icddr,b, Dhaka, Bangladesh. icddr,b acknowledges with gratitude the commitment of the Government of Bangladesh to its research efforts, and also gratefully acknowledges the unrestricted support provided by the Governments of the People’s Republic of Bangladesh, Canada, Sweden, and the UK. We also thank the neurologists who referred their patients to us.
References
- 1. Islam Z, Gilbert M, Mohammad QD, et al. Guillain‐Barré syndrome‐related Campylobacter jejuni in Bangladesh: ganglioside mimicry and cross‐reactive antibodies. PLoS One 2012;7:e43976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ang CW, Jacobs BC, Laman JD. The Guillain‐Barré syndrome: a true case of molecular mimicry. WTrends Immunol 2004;25:61–66. [DOI] [PubMed] [Google Scholar]
- 3. Jacobs BC, Rothbarth PH, van der Meché FGA, et al. The spectrum of antecedent infections in Guillain‐Barré syndrome. Neurology 1998;51:1110–1115. [DOI] [PubMed] [Google Scholar]
- 4. Perera VN, Nachamkin I, Ung H, et al. Molecular mimicry in Campylobacter jejuni : role of the lipo‐oligosaccharide core oligosaccharide in inducing anti‐ganglioside antibodies. FEMS Immunol Med Microbiol 2007;50:27–36. [DOI] [PubMed] [Google Scholar]
- 5. Islam Z, Jacobs BC, van Belkum A, et al. Axonal variant of Guillain‐Barré syndrome associated with Campylobacter infection in Bangladesh. Neurology 2010;74:581–587. [DOI] [PubMed] [Google Scholar]
- 6. Tauxe VR Epidemiology of Campylobacter jejuni infections in the United States and other industrialized nations. . Campylobacter jejuni: Curr Status Futur Trends 1992;9–19. [Google Scholar]
- 7. Geleijns K, Schreuder G, Neurology BJ, et al. HLA class II alleles are not a general susceptibility factor in Guillain‐Barré syndrome. AAN Enterp 2005;11:44–49. [DOI] [PubMed] [Google Scholar]
- 8. Nyati KK, Nyati R. Role of campylobacter jejuni infection in the pathogenesis of Guillain‐Barré syndrome: an update. Biomed Res Int 2013;2013:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jin PP, Sun LL, Ding BJ, et al. Human leukocyte antigen DQB1 (HLA‐DQB1) polymorphisms and the risk for Guillain‐Barré syndrome: a systematic review and meta‐analysis. PLoS One 2015;10:e0131374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuki N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve 2007;35:691–711. [DOI] [PubMed] [Google Scholar]
- 11. Willison HJ, Plomp JJ. Anti‐ganglioside antibodies and the presynaptic motor nerve terminal. Ann N Y Acad Sci 2008;1132:114–123. [DOI] [PubMed] [Google Scholar]
- 12. Sorge NV, van der Pol WL, Jansen M. D., van den Berg, L. H . Pathogenicity of anti‐ganglioside antibodies in the Guillain‐Barré syndrome. Autoimmunity 2004;3:61–68. [DOI] [PubMed] [Google Scholar]
- 13. Van Der Pol WL, Van De Winkel JGJ. IgG receptor polymorphisms: Risk factors for disease. Immunogenetics 1998;48:222–232. [DOI] [PubMed] [Google Scholar]
- 14. Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2002;2:580–592. [DOI] [PubMed] [Google Scholar]
- 15. Vedeler CA, Myhr KM, Nyland H. Fc receptors for immunoglobulin G ‐ A role in the pathogenesis of Guillain‐Barré syndrome and multiple sclerosis. J Neuroimmunol 2001;118:187–193. [DOI] [PubMed] [Google Scholar]
- 16. Van Sorge NM, Van Der Pol WL, Van De Winkel JGJ. FcγR polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tissue Antigens 2003;61:189–202. [DOI] [PubMed] [Google Scholar]
- 17. Flesch BK, Bauer F, Neppert J. Rapid typing of the human Fcγreceptor IIA polymorphism by polymerase chain reaction amplification with allele‐specific primers. Transfusion 1998;38:174–176. [DOI] [PubMed] [Google Scholar]
- 18. Kuwano ST, Bordin JO, Chiba AK, et al. Allelic polymorphisms of human Fcγ receptor IIa and Fcγ receptor IIIb among distinct groups in Brazil. Transfusion 2000;40:1388–1392. [DOI] [PubMed] [Google Scholar]
- 19. Warmerdam PA, van de Winkel JG, Vlug A, et al. A single amino acid in the second Ig‐like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol 1991;147. [PubMed] [Google Scholar]
- 20. Nimmerjahn F, Ravetch JV. Fc‐Receptors as regulators of immunity. Adv Immunol 2007;96:179–204. [DOI] [PubMed] [Google Scholar]
- 21. Wu J, Edberg JC, Redecha PB, et al. Novel polymorphism of Fc RIIIa (CD16) and autoimmune disease a novel polymorphism of Fc RIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest 1997;100:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hessner M, Curtis B, Endean D, Aster R. Determination of neutrophil antigen gene frequencies in five ethnic groups by polymerase chain reaction with sequence‐specific primers. Transfusion 1996;36:895–899. [DOI] [PubMed] [Google Scholar]
- 23. van der Pol WL, van den Berg LH, Scheepers RHM, et al. IgG receptor IIa alleles determine susceptibility and severity of Guillain‐Barré syndrome. Neurology 2000;54:1661–1665. [DOI] [PubMed] [Google Scholar]
- 24. Vedeler C, Raknes G, Myhr K, et al. IgG Fc‐receptor polymorphisms in Guillain‐Barré syndrome. Neurology 2000;55:705–707. [DOI] [PubMed] [Google Scholar]
- 25. van Sorge N, van der Pol W, Of MJEA. Severity of Guillain‐Barré syndrome is associated with Fcγ Receptor III polymorphisms. J Neuroimmunol 2005;162:157–164. [DOI] [PubMed] [Google Scholar]
- 26. Sinha S, Prasad KN, Jain D, et al. Immunoglobulin IgG Fc‐receptor polymorphisms and HLA class II molecules in Guillain‐Barré syndrome. Acta Neurol Scand 2010;122:21–26. [DOI] [PubMed] [Google Scholar]
- 27. Wu LY, Zhou Y, Qin C, Hu BL. The effect of TNF‐alpha, FcγR and CD1 polymorphisms on Guillain‐Barré syndrome risk: evidences from a meta‐analysis. J Neuroimmunol 2012;243:18–24. [DOI] [PubMed] [Google Scholar]
- 28. Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain‐Barré syndrome. Ann Neurol 1990;27:S21–S24. [DOI] [PubMed] [Google Scholar]
- 29. Kuijf ML, van Doorn PA, Tio‐Gillen AP, et al. Diagnostic value of anti‐GM1 ganglioside serology and validation of the INCAT‐ELISA. J Neurol Sci 2005;239:37–44. [DOI] [PubMed] [Google Scholar]
- 30. Hadden RDM, Cornblath DR, Hughes RAC, et al. Electrophysiological classification of Guillain‐Barré syndrome: clinical associations and outcome. Ann Neurol 1998;44:780–788. [DOI] [PubMed] [Google Scholar]
- 31. Kleyweg RP, Van Der Meché FGA, Schmitz PIM. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain‐Barré syndrome. Muscle Nerve 1991;14:1103–1109. [DOI] [PubMed] [Google Scholar]
- 32. Hayat S, Jahan I, Das A, et al. Human leukocyte antigen‐DQB1 polymorphisms and haplotype patterns in Guillain‐Barré syndrome. Ann Clin Transl Neurol 2019;6:1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geleijns K, Emonts M, Laman J, et al. Genetic polymorphisms of macrophage‐mediators in Guillain‐Barré syndrome. J Neuroimmunol 2007;190:127–130. [DOI] [PubMed] [Google Scholar]
- 34. Hughes RAC, Newsom‐Davis JM, Perkin GD, Pierce JM. Controlled trial of prednisolone in acute polyneuropathy. Lancet 1978;312:750–753. [DOI] [PubMed] [Google Scholar]
- 35. van Sorge NM, van den Berg LH, Geleijns K, et al. Anti‐GM1 IgG antibodies induce leukocyte effector functions via Fcγ receptors. Ann Neurol 2003;53:570–579. [DOI] [PubMed] [Google Scholar]
- 36. Binstadt BA, Geha RS, Bonilla FA. IgG Fc receptor polymorphisms in human disease: Implications for intravenous immunoglobulin therapy. J Allergy Clin Immunol 2003;111:697–703. [DOI] [PubMed] [Google Scholar]
- 37. Deo Y, Graziano R, Repp R, et al. Clinical significance of IgG Fc receptors and FcγR‐directed immunotherapies. Immunol Today 1997;18:127–135. [DOI] [PubMed] [Google Scholar]
- 38. Yuki N, Ichihashi Y, Taki T. Subclass of IgG antibody to GM1 epitope‐bearing lipopolysaccharide of Campylobacter jejuni in patients with Guillain‐Barré syndrome. J. Neuroimmunol 1995;60:161–164. [DOI] [PubMed] [Google Scholar]
- 39. van der Meché FGA, Schmitz PIM. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain‐Barré syndrome. N Engl J Med 1992;326:1123–1129. [DOI] [PubMed] [Google Scholar]
- 40. Koene HR, Kleijer M, Roos D, et al. FcγRIIIB gene duplication: evidence for presence and expression of three distinct FcγRIIIB genes in NA(1+,2+)SH(+) individuals. Blood 1998;91:673–679. [PubMed] [Google Scholar]
- 41. Doets AY, Verboon C, van den Berg B, et al. Regional variation of Guillain‐Barré syndrome. Brain 2018;141:2866–2877. [DOI] [PubMed] [Google Scholar]