

Abstract
Objectives
In the TOURMALINE‐MM1 phase 3 trial in relapsed/refractory multiple myeloma, ixazomib‐lenalidomide‐dexamethasone (IRd) showed different magnitudes of progression‐free survival (PFS) benefit vs placebo‐Rd according to number and type of prior therapies, with greater benefit seen in patients with >1 prior line of therapy or 1 prior line of therapy without stem cell transplantation (SCT).
Methods
RNA sequencing data were used to investigate the basis of these differences.
Results
The PFS benefit of IRd vs placebo‐Rd was greater in patients with tumors expressing high c‐MYC levels (median not reached vs 11.3 months; hazard ratio [HR] 0.42; 95% CI, 0.26, 0.66; P < .001) compared with in those expressing low c‐MYC levels (median 20.6 vs 16.6 months; HR 0.75; 95% CI, 0.42, 1.2). Expression of c‐MYC in tumors varied based on the number and type of prior therapy received, with the lowest levels observed in tumors of patients who had received 1 prior line of therapy including SCT. These tumors also had higher expression levels of CD19 and CD81.
Conclusions
PFS analyses suggest that lenalidomide and ixazomib target tumors with different levels of c‐MYC, CD19, and CD81 expression, thus providing a potential rationale for the differential benefits observed in the TOURMALINE‐MM1 study. This trial was registered at www.clinicaltrials.gov as: NCT01564537
Keywords: c‐myc Proto‐Oncogenes, multiple myeloma, mutation, progression‐free survival, RNA sequencing
Novelty statement.
What is the new aspect of your work? We explore the hypothesis that the progression‐free survival outcomes observed in the phase 3 TOURMALINE‐MM1 trial of ixazomib‐lenalidomide‐dexamethasone (IRd) versus placebo‐lenalidomide‐dexamethasone (placebo‐Rd) in patients with relapsed/refractory multiple myeloma are linked to molecular differences between tumors in patients whose disease has relapsed after various prior therapies.
What is the central finding of your work? Our analyses show that tumors in patients whose disease has relapsed after autologous stem cell transplantation tend to have lower levels of c‐MYC and higher levels of CD19 and CD81 expression and that IRd and placebo‐Rd target tumors with different expression levels of these markers, potentially explaining in part the differential progression‐free survival benefits with IRd versus placebo‐Rd observed in the TOURMALINE‐MM1 trial according to number and type of prior therapies.
What is (or could be) the specific clinical relevance of your work? Our results suggest that differences in cell maturity add to the molecular heterogeneity observed in multiple myeloma and that selective pressure from treatment may lead to more mature/differentiated tumors, thereby impacting the relative magnitude of anti‐myeloma effects of therapies with different mechanisms of action.
1. INTRODUCTION
The advent and widespread use of next‐generation sequencing has revealed the full complexity of tumor heterogeneity and its implication for cancer prognosis and therapeutic response.1 Multiple myeloma (MM), a clonal plasma cell disorder, is genetically complex and heterogeneous, with clonal diversity evolving over the disease course.2 Several studies have investigated the genomic heterogeneity of relapsed/refractory MM (RRMM),3, 4, 5 but few have explored the relationship between clonal heterogeneity and types of prior therapies received, and the impact of the resulting differences in tumor biology on the outcomes seen with novel therapies.
The phase 3 TOURMALINE‐MM1 study compared the oral proteasome inhibitor (PI) ixazomib plus lenalidomide‐dexamethasone (IRd) vs placebo‐Rd in 722 patients with RRMM.6 Results from this study demonstrated a significant progression‐free survival (PFS) benefit in the intent‐to‐treat (ITT) analysis. A subgroup analysis showed that while IRd was associated with clinical benefit compared with placebo‐Rd, the greatest benefit was observed in patients who had received 2 or 3 prior lines of therapy (LoT; 2/3LoT patients) compared to patients who had received 1 prior LoT (1LoT patients).7 We therefore conducted analyses of RNA sequencing (RNAseq) data from tumors collected during TOURMALINE‐MM1 and investigated the molecular differences between tumors from patients according to number and/or type of prior therapies.
In particular, we evaluated outcomes according to expression of c‐MYC. Increased expression of c‐MYC is involved in MM pathogenesis/progression,8 making c‐MYC one of the key genes deregulated in MM. Translocations of c‐MYC are present in approximately 15% of patients with newly diagnosed MM (NDMM) and RRMM.2, 9, 10 Gene expression studies indicate that the transcriptional signature of c‐MYC is present in 67% of primary MM but not in the pre‐malignant condition monoclonal gammopathy of undetermined significance.11 Recent genetic evidence also suggests a novel role for c‐MYC during the first steps of hematopoietic stem cell differentiation,12 along with the B‐cell receptor (BCR) complex, of which CD19, CD81, and CD79A make up part of the co‐receptor.13 Thus, differences in c‐MYC expression and level of cell maturity may be associated with differential activity of agents and regimens.
2. METHODS
2.1. Study design and patients
TOURMALINE‐MM1 (NCT01564537) study design and clinical results have been reported previously.6 Briefly, 722 patients with RRMM after 1‐3 LoT were randomized to receive IRd or placebo‐Rd until disease progression or unacceptable toxicity. Bone marrow aspirates were collected from patients at screening, and purified CD138‐positive cells were used for RNAseq. Data reported here are from the first prespecified statistical analysis of PFS (median follow‐up ~15 months; range 13.6‐15.4), as reported by Moreau et al.6
2.2. Myeloma cell purification and RNA isolation
Bone marrow aspirate samples were collected at screening from patients enrolled in the trial for tumor gene expression analysis. CD138‐positive cells were isolated within 24‐48 hours using the Miltenyi CD138+ isolation kit according to the manufacturer's instructions. Frozen CD138‐positive cell pellets were then shipped to a different laboratory for DNA and RNA co‐extraction using the Qiagen AllPrep DNA/RNA kit. Extracted DNA and RNA were measured for quantity, quality, and purity. Each sample was aliquoted in two vials, frozen at −80ºC and shipped to the Broad Institute (Cambridge, MA) for sequencing. All RNA samples were quantified again using a Nanodrop spectrophotometer (Thermo Scientific), and their integrity was assessed using the Agilent Bioanalyzer and normalized before starting the sequencing. The minimum Nanodrop and BioAnalyzer input requirement was ≥250 ng RNA and an RNA integrity number (RIN) ≥ 6.
2.3. Gene expression profiling and QC steps
Whole transcriptome sequencing (RNAseq) of bone marrow‐sorted CD138‐positive cells was performed as described in the Methods S1. The purity of the MM samples was assessed, and samples with at least one specific variable region isoform (either heavy or light chain) having more than 90% of the reads mapped were automatically deemed as having high purity and were used in the present analyses (“RNAseq population”).
2.4. Gene expression analyses in the RNAseq population
Details of the gene expression analyses in the RNAseq population are provided in the Methods S1. c‐MYC, CD19, CD81, and CD138 expression were evaluated by treatment arm and according to the number (1 vs 2/3) and type (stem cell transplant [SCT] vs no SCT) of prior therapies received. A multivariate analysis, using logistic regression with stepwise selection, was performed to understand the influence of different disease characteristics on c‐MYC expression level among 1LoT patients. For subsequent analyses, patients were dichotomized into two subgroups, defined as c‐MYC‐high and c‐MYC‐low, using median c‐MYC expression as the cutoff, or into four subgroups based on c‐MYC expression quartiles. Patients’ data were analyzed based on median expression levels of CD19 and/or CD81 (CD19‐high vs CD19‐low; CD81‐high vs CD81‐low). PFS was analyzed in subgroups based on prior LoTs (1LoT, 2/3LoT) and by whether patients had SCT in a prior line of therapy (1LoT‐SCT, 1LoT‐noSCT, 2/3LoT‐SCT, 2/3LoT‐noSCT). All analyses were exploratory; no correction for multiplicity of testing was done.
3. RESULTS
3.1. Baseline characteristics of patients with RNAseq data
Of 419 samples collected, 399 were deemed of high purity for the genomic analyses, and so RNAseq data were available for 399 of 722 (55.2%) patients enrolled in TOURMALINE‐MM1, including 189 and 210 randomized to IRd and placebo‐Rd, respectively (RNAseq population). Baseline characteristics were similar between patients with or without RNAseq data, including disease stage, performance status, frequency of high‐risk vs standard‐risk cytogenetic abnormalities, prior LoTs, and prior exposure to immunomodulatory drugs or PIs (Table S1).
3.2. c‐MYC expression levels differ between patients with different numbers of prior LoT
Since cereblon is essential for lenalidomide activity in MM, we initially assessed if cereblon's expression level varied across patients with different types and numbers of prior LoT and did not observe any significant differences (data not shown). We then analyzed c‐MYC expression across these subgroups of patients, as prior observations had suggested that c‐MYC activity was likely to increase benefit in patients treated with bortezomib‐based therapy.11 c‐MYC was significantly upregulated in tumors from 2/3LoT vs 1LoT patients (P = .0275) (Figure 1A). Analysis of 1LoT patients according to prior SCT showed that tumors from 1LoT‐SCT patients had significantly lower c‐MYC expression levels vs those from 1LoT‐noSCT patients (P = .013) and vs those from 2/3LoT patients (P = .0019). No differences were observed between 1LoT‐noSCT and 2/3LoT patients (P = .71) (Figure 1B). To investigate if other parameters were correlated with lower c‐MYC expression in tumors, we performed a multivariate analysis and showed that the best predictor for c‐MYC expression level (above vs below the median) in 1LoT patients was prior SCT (no vs yes; odds ratio 2.296, P = .0018). Using c‐MYC expression level as a continuous dependent variable, linear regression analyses with stepwise selection showed a strong correlation between c‐MYC expression level and prior SCT (no vs yes; coefficient .696; P = .0015), disease stage (I vs II or III; coefficient .510; P = .0282), and baseline serum M‐protein (coefficient .0270; P = .0001).
FIGURE 1.
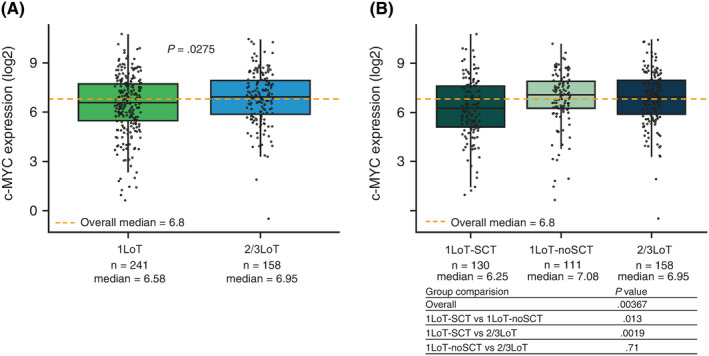
c‐MYC expression by prior treatment exposure. A, Median c‐MYC expression was significantly lower in 1LoT vs 2/3LoT patients. B, Median c‐MYC expression was significantly lower in 1LoT‐SCT vs 1LoT‐noSCT or 2/3LoT patients. No significant difference was observed between 1LoT‐noSCT and 2/3LoT patients. Overall P‐values are analysis of variance F‐test P‐values, and pairwise comparison P‐value are two sample t test P‐values). Solid black horizontal lines show the medians, dotted orange lines show the overall medians, boxes show interquartile range, lines show range excluding outliers. LoT, line of therapy; SCT, stem cell transplantation [Colour figure can be viewed at wileyonlinelibrary.com]
3.3. Prognostic impact of c‐MYC expression based on treatment received
Since c‐MYC is a known negative prognostic factor in MM,9, 11, 14, 15, 16, 17 we assessed the impact of its expression on PFS in the RNAseq population, dichotomized per median c‐MYC expression level and regardless of therapy (IRd and placebo‐Rd patients pooled). Median PFS was similar—18.5 vs 17.6 months—in the c‐MYC‐high vs c‐MYC‐low groups (HR 0.99; 95% CI, 0.73, 1.4; P > .05), suggesting that in this population c‐MYC expression did not per se have a negative impact on outcome. Similar results were obtained in an analysis of patients in the top vs bottom quartiles of c‐MYC expression (median PFS 18.4 vs 15.7 months; HR 0.92; 95% CI, 0.6, 1.4; P > .05) (Figure 2A). PFS was then assessed based on c‐MYC expression level and study treatment. A statistically significant improvement in PFS was seen in the c‐MYC‐high group with IRd vs placebo‐Rd (median PFS not reached vs 11.3 months; HR 0.42; 95% CI, 0.26, 0.66; P < .001). In the c‐MYC‐low subgroup, median PFS was 20.6 vs 16.6 months for IRd vs placebo‐Rd (HR 0.75; 95% CI, 0.47, 1.2; P > .05). While a PFS benefit with IRd vs placebo‐Rd was observed in both the c‐MYC‐high and c‐MYC‐low subgroups, the magnitude of benefit was larger in the c‐MYC‐high subgroup (Figure 2A).
FIGURE 2.
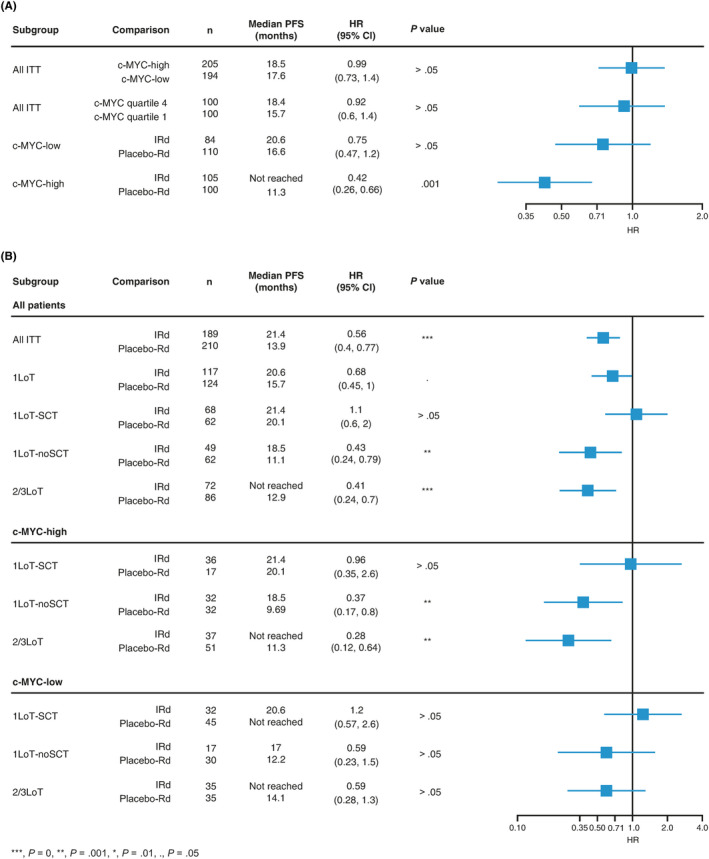
c‐MYC as prognostic factor in RNAseq population. A, c‐MYC level did not appear to be a negative prognostic factor in the population analyzed. A significant difference in outcome was observed when treatment arm was included in the analysis. A significant difference in PFS with IRd vs placebo‐Rd was observed only in c‐MYC‐high patients. B, PFS with IRd vs Rd according to prior therapy and by high or low c‐MYC expression level. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; ITT, intent‐to‐treat; LoT, line of therapy; PFS, progression‐free survival; Rd, lenalidomide‐dexamethasone; RNAseq, RNA sequencing; SCT, stem cell transplantation [Colour figure can be viewed at wileyonlinelibrary.com]
Baseline characteristics were similar between the c‐MYC‐high and c‐MYC‐low subgroups (Table 1), except for a higher frequency of IgA MM, higher rates of refractory and relapsed and refractory disease, and a higher percentage of 2/3LoT patients in the c‐MYC‐high subgroup. The overall rate of high‐risk cytogenetics was similar between subgroups; however, del(17p) appeared more common in the c‐MYC‐high vs c‐MYC‐low subgroup (15% vs 9%), and t(4;14) appeared less common (7% vs 14%), acknowledging the small patient numbers.
TABLE 1.
Baseline patient characteristics and TOURMALINE‐MM1 treatment information in patients with RNAseq data (N = 399) by c‐MYC expression subgroup
| c‐MYC‐high (n = 199) | c‐MYC‐low (n = 200) | |
|---|---|---|
| Age, years, n (%) | ||
| ≤65 | 89 (45) | 95 (48) |
| >65, ≤75 | 75 (38) | 81 (41) |
| >75 | 35 (18) | 24 (12) |
| ECOG PS, n (%) | ||
| 0 | 97 (49) | 94 (47) |
| 1 | 89 (45) | 86 (43) |
| 2 | 10 (5) | 16 (8) |
| Missing | 3 | 4 |
| Measurable disease, n (%) | ||
| SPEP only | 109 (55) | 96 (48) |
| UPEP only | 22 (11) | 34 (17) |
| SPEP and UPEP | 47 (24) | 42 (21) |
| FLC only | 17 (9) | 25 (13) |
| Not measurable | 4 (2) | 3 (2) |
| MM subtype, n (%) | ||
| IgG | 115 (58) | 116 (58) |
| IgA | 46 (23) | 24 (12) |
| Other | 12 (6) | 13 (7) |
| Missing | 26 | 47 |
| ISS stage a , n (%) | ||
| I or II | 166 (83) | 174 (87) |
| III | 33 (17) | 26 (13) |
| Cytogenetics, n (%) | ||
| Standard risk | 138 (69) | 135 (68) |
| High risk | 47 (24) | 45 (23) |
| del(17) | 30 (15) | 17 (9) |
| t(4;14) | 13 (7) | 27 (14) |
| t(14;16) | 4 (2) | 1 (1) |
| Not available | 14 (7) | 20 (10) |
| Prior lines a , n (%) | ||
| 1 | 106 (53) | 128 (64) |
| 2 or 3 | 93 (47) | 72 (36) |
| Prior LoT b , n (%) | ||
| 1 | 112 (56) | 129 (65) |
| SCT | 51 (26) | 79 (40) |
| No SCT | 61 (31) | 50 (25) |
| 2 | 64 (32) | 48 (24) |
| SCT | 41 (21) | 27 (14) |
| No SCT | 23 (12) | 21 (11) |
| 3 | 23 (12) | 23 (12) |
| SCT | 10 (5) | 13 (7) |
| No SCT | 13 (7) | 10 (5) |
| Time from SCT, months, n (%) | ||
| <12 | 8 (4) | 6 (3) |
| ≥12, <24 | 19 (10) | 20 (10) |
| ≥24, <36 | 19 (10) | 35 (18) |
| ≥36, <48 | 23 (12) | 20 (10) |
| ≥48 | 33 (17) | 38 (19) |
| Missing | 97 | 81 |
| MM status, n (%) c | ||
| Relapsed | 142 (71) | 165 (83) |
| Refractory | 22 (11) | 17 (9) |
| Relapsed and refractory | 35 (18) | 18 (9) |
| Prior PI, n (%) | ||
| Exposed | 136 (68) | 139 (70) |
| Naïve | 63 (32) | 61 (31) |
| Refractory | 15 (8) | 5 (3) |
| V‐refractory | 5 (3) | 1 (1) |
| Prior IMiD, n (%) | ||
| Exposed | 120 (60) | 115 (58) |
| T‐exposed | 97 (49) | 96 (48) |
| R‐exposed | 26 (13) | 27 (14) |
| Naïve | 79 (40) | 85 (43) |
| T‐naïve | 102 (51) | 104 (52) |
| R‐naïve | 173 (87) | 173 (87) |
| Refractory | 33 (17) | 14 (7) |
| T‐refractory | 32 (16) | 14 (7) |
| R‐refractory | 0 | 0 |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; FLC, free light chain; IgG/A, immunoglobulin G/A; IMiD, immunomodulatory drug; IRd, ixazomib‐lenalidomide‐dexamethasone; ISS, International Staging System; LoT, lines of therapy; MM, multiple myeloma; PI, proteasome inhibitor; R, lenalidomide; Rd, lenalidomide, dexamethasone; RNAseq, RNA sequencing; SCT, stem cell transplantation; SPEP, serum protein electrophoresis; T, thalidomide; UPEP, urine protein electrophoresis; V, bortezomib.
Per stratification.
Per Sponsor review.
’Relapsed’ includes patients who had relapsed from at least 1 previous treatment but were not refractory to any previous treatment; ‘Refractory’ includes patients who were refractory (disease progressed on or within 60 d of last study drug) to at least 1 prior line of therapy; ‘Relapsed and refractory’ includes patients who relapsed from at least 1 previous treatment and additionally were refractory to at least 1 previous treatment; ‘Primary refractory’ is a subgroup of ‘refractory’ patients who never responded to prior therapy.
3.4. PFS by c‐MYC expression and prior treatment exposure
Similar to what was reported previously for the ITT population,6 analyses of PFS in the RNAseq population according to prior therapy showed that the PFS benefit with IRd vs placebo‐Rd was greater in 2/3LoT patients (median not reached vs 12.9 months; HR 0.41; 95% CI, 0.24, 0.7; P < .001) compared to 1LoT patients (median 20.6 vs 15.7 months; HR 0.68; 95% CI, 0.45, 1.0; P > .05). When 1LoT patients were divided by prior SCT, a difference in PFS benefit was observed only for 1LoT‐noSCT patients. In this group, the median PFS with IRd vs placebo‐Rd was 18.5 vs 11.1 months (HR 0.43; 95% CI, 0.24, 0.79; P = .01; Figure 2B). When each prior therapy subgroup was stratified as c‐MYC‐high or c‐MYC‐low (based on median c‐MYC expression level), the magnitude of PFS benefit with IRd vs placebo‐Rd appeared greater among the c‐MYC‐high vs c‐MYC‐low patient subgroups (based on the respective HRs). While PFS with IRd and placebo‐Rd was similar among the c‐MYC‐high 1LoT‐SCT patients, the PFS HR favored placebo‐Rd in the c‐MYC‐low 1LoT‐SCT subgroup (Figure 2B). Notably, the longest median PFS with placebo‐Rd was observed in 1LoT‐SCT patients.
3.5. Characteristics of 1LoT‐SCT patients
To understand if any baseline characteristics of 1LoT‐SCT patients might have influenced the differential PFS benefit with IRd vs placebo‐Rd, we compared the baseline characteristics of 1LoT‐SCT patients to those of 1LoT‐noSCT and 2/3LoT patients combined (Table S2). We found that 1LoT‐SCT patients had better prognostic features compared with the rest of the RNAseq population: more of them had an Eastern Cooperative Oncology Group performance status of 0 (65% vs 39%); they were younger (65% vs 37% aged ≤ 65 years); fewer had renal involvement (5% vs 17%); fewer had tumors with high‐risk cytogenetics features (del(17), t(4;14), and t(14;16); 18% vs 25%); slightly more of them had stage I or II disease (89% vs 83%); and the frequency of patients with relapsed‐only (non‐refractory) MM was higher (93% vs 70%). Notably, the majority of 1LoT‐SCT patients (71%) had received SCT ≥ 24 months prior to TOURMALINE‐MM1 study entry, indicating that they had not relapsed shortly after transplantation.
3.6. Gene expression analyses identified markers of early differentiation in 1LoT‐SCT patients
We compared RNAseq data from 1LoT‐SCT patients to pooled data from 1LoT‐noSCT and 2/3LoT patients to evaluate differences at the molecular level. As shown in Table 2, the 10 most differentially expressed genes were the immunoglobulin heavy constant delta and epsilon genes, IGHD and IGHE (3.57 and 2.08 fold higher), components of the BCR complex (CD19, CD81, CD79A) (1.97, 1.51, and 1.54 fold higher), and genes involved in different steps of cell maturation (PAX5, VPREB1, IGLL1) (1.54, 1.81, and 1.68 fold higher). All these genes were significantly upregulated in 1LoT‐SCT patients, while c‐MYC expression was significantly lower (0.65 fold lower).
TABLE 2.
Top 10 differentially expressed genes that are associated with cell maturation. Gene expression profile of 1LoT‐SCT patients vs 1LoT‐noSCT and 2/3LoT patients with RNAseq data, pooled across TOURMALINE‐MM1 treatment arms
| Gene |
1LoT‐SCT patients (n = 130) Mean expression |
1LoT‐noSCT and 2/3LoT patients (n = 269) Mean expression |
Fold change |
t test P‐value |
|---|---|---|---|---|
| IGHD | 29.92 | 8.38 | 3.57 | .0000 |
| CD19 | 2.51 | 1.27 | 1.97 | .0001 |
| CD22 | 0.95 | 0.55 | 1.74 | .0006 |
| MYC | 74.39 | 114.45 | 0.65 | .0014 |
| IGHE | 1.79 | 0.86 | 2.08 | .0015 |
| CD81 | 27.57 | 18.32 | 1.51 | .0034 |
| PAX5 | 1.36 | 0.89 | 1.54 | .0108 |
| VPREB1 | 0.39 | 0.21 | 1.81 | .0110 |
| CD79A | 138.61 | 89.96 | 1.54 | .0123 |
| IGLL1 | 1.31 | 0.78 | 1.68 | .0145 |
Abbreviations: LoT, line of therapy; RNAseq, RNA sequencing; SCT, stem cell transplantation.
3.7. 1LoT‐SCT patients demonstrated higher levels of CD19 and CD81 expression
Pairwise comparison of CD19 or CD81 expression showed statistically significantly higher expression of both markers in 1LoT‐SCT patients. CD19 expression was significantly higher in 1LoT‐SCT vs 1LoT‐noSCT (P < .001) or 2/3LoT patients (P = .0079); no difference was observed between 1LoT‐noSCT and 2/3LoT (P = .11) patients (Figure 3A). Similarly, CD81 expression was higher in 1LoT‐SCT vs 1LoT‐noSCT (P = .006) or 2/3LoT (P = .02) patients, with no difference between 1LoT‐noSCT and 2/3LoT patients (P = .55) (Figure 3B). We also looked at CD138 expression levels and, while we did not see any significant difference between the three groups of patients (Figure S1), we observed lower CD138 expression among patients with tumors with high CD19 or CD81 expression (P = .00155) (Figure S2). Analyses of CD19 and CD81 expression in patients in the top and bottom quartiles of c‐MYC expression, independent of line of therapy, confirmed the inverse correlation of these markers. CD19, CD81, and CD79A expression was significantly greater in tumors from patients in the lowest vs highest c‐MYC expression quartile (P = .0164, P = .00114, and P = .0223, respectively), independent of the number or type of prior therapies (Figure 3C). These data suggest that tumors displaying these molecular characteristics are present throughout the RNAseq population but are more frequent in 1LoT‐SCT patients.
FIGURE 3.
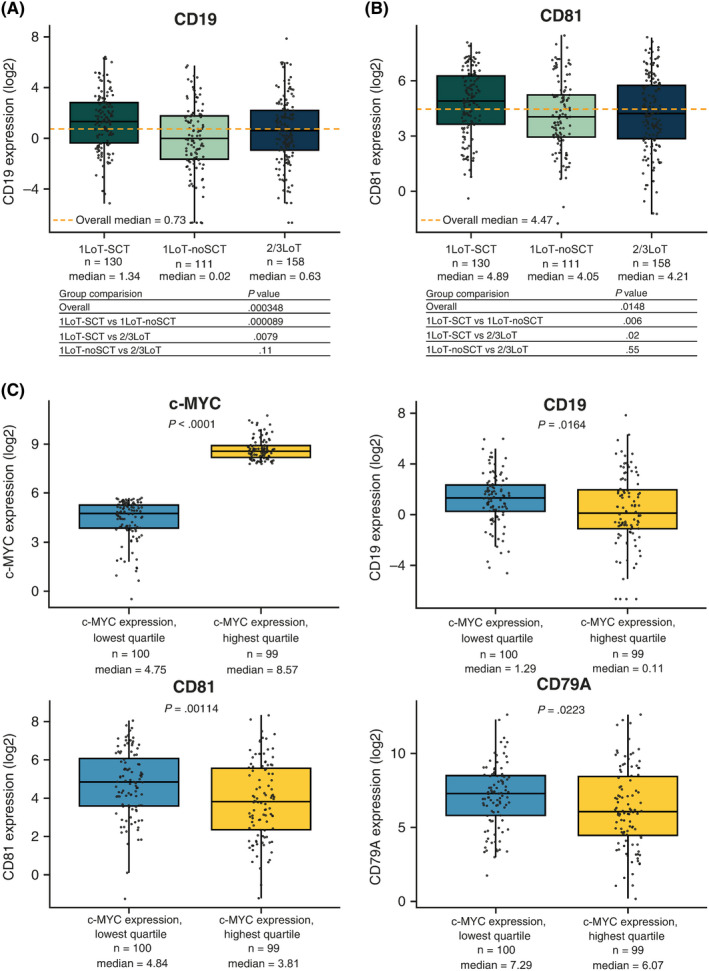
CD19 and CD81 expression by prior treatment exposure and by c‐MYC expression. Median levels of (A) CD19 and (B) CD81 were significantly higher in tumors from 1LoT‐SCT vs 1LoT‐noSCT or 2/3LoT patients. No difference was observed between 1LoT‐noSCT and 2/3LoT patients. C, CD19, CD81, and CD79A were highly expressed in tumors in the lowest quartile for c‐MYC expression. Solid black horizontal lines show the medians, dotted orange lines show the overall medians, boxes show interquartile range, lines show range excluding outliers. LoT, line of therapy; SCT, stem cell transplantation [Colour figure can be viewed at wileyonlinelibrary.com]
3.8. Lenalidomide and ixazomib appear to target tumors at different stages of maturation
To further understand how different molecular features influence the activity of IRd and placebo‐Rd, we stratified patients based on median c‐MYC, CD19, or CD81 expression and evaluated PFS with IRd and placebo‐Rd in each subgroup. PFS with IRd appeared longer among patients with c‐MYC‐high vs c‐MYC‐low tumors (median not reached vs 20.6 months; HR 0.72; 95% CI, 0.43, 1.2; P > .05) and those with CD19‐low vs CD19‐high (median not reached vs 21.4 months; HR 0.85; 95% CI, 0.51, 1.4; P > .05) or CD81‐low vs CD81‐high (median not reached vs 18.5 months; HR 1.5; 95% CI, 0.87, 2.5; P > .05), although the differences were not statistically significant (Figure 4). In contrast, PFS with placebo‐Rd appeared shorter among c‐MYC‐high vs c‐MYC‐low patients (median 11.3 vs 16.6 months; HR 1.3; 95% CI, 0.9, 2.0; P > .05) and longer among CD19‐high vs CD19‐low (median 17.5 vs 11.1 months; HR 0.6; 95% CI, 0.4, 0.91; P = .05) and CD81‐high vs CD81‐low (median not reached vs 11.1 months; HR 0.51; 95% CI, 0.33, 0.7); P = .01) patients, although the difference between c‐MYC groups was not significant (Figure 4). Taken together, these data suggest that IRd and placebo‐Rd target tumors at different stages of maturation, with placebo‐Rd performing better in less mature tumors (low levels of c‐MYC and high levels of CD19 or CD81 expression). Conversely, IRd appeared more beneficial in more mature tumors (high levels of c‐MYC and low levels of CD19 or CD81 expression).
FIGURE 4.
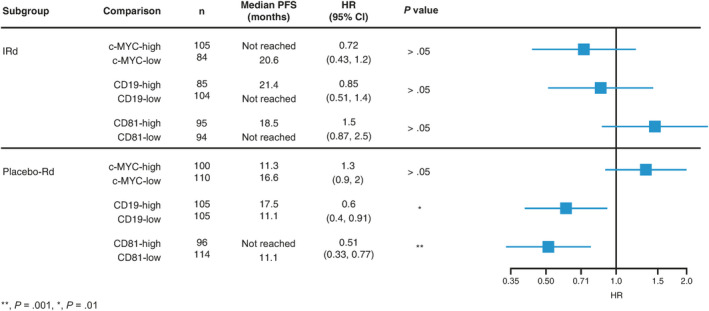
Differential activity of IRd and placebo‐Rd between patients with tumors with high vs low expression of c‐MYC, CD19, and CD81. While PFS with IRd did not differ significantly between these expression subgroups, PFS was significantly shorter with placebo‐Rd in patients with tumors expressing low vs high levels of CD19 and CD81. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; PFS, progression‐free survival; Rd, lenalidomide‐dexamethasone [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
MM is a complex disease demonstrating clear evidence of substantial clonal heterogeneity, with different clones coexisting in the same patient.18 Next‐generation sequencing studies have shown the complexity of MM at the genetic level, with changes in chromosome numbers, genetic translocations, and genetic mutations. However, evidence is emerging that the clonal cells that constitute the disease are not homogenously mature plasma cells.3
The MYC signaling pathway is of great interest in MM.19 MYC abnormalities are typically characterized by complex rearrangement of the proto‐oncogene c‐MYC, 20 which encodes a transcription factor that regulates cell proliferation, growth, protein translation, metabolism, and apoptosis.21 Several studies have also suggested an involvement of c‐MYC during the expansion of committed progenitors in the adult hematopoietic system,12 thereby playing a role in the differentiation of B cells.22, 23 During this process, c‐MYC appears to control the expression of specific integrins, regulating the balance between self‐renewal and differentiation by modulating the migration and/or adhesion of hematopoietic stem cells.12
Data from TOURMALINE‐MM1 have shown a different magnitude of PFS benefit between 1LoT and 2/3LoT patients.7 Using the RNAseq dataset from the study, we uncovered molecular differences between tumors of patients who relapsed post‐SCT vs those who relapsed after prior therapies not including SCT.
After excluding cereblon expression as responsible for the differential activity of IRd vs placebo‐Rd, we focused our attention on c‐MYC. The rationale for evaluating c‐MYC expression in tumors was provided by observations that c‐MYC plays a role in PI‐induced cell death24, 25 and by a prior analysis suggesting that patients with tumors expressing higher levels of c‐MYC derive relatively greater benefit from PI therapy with bortezomib.11 We initially demonstrated that median c‐MYC levels were significantly higher in 2/3LoT vs 1LoT patients and that within 1LoT patients the lowest levels were observed among 1LoT‐SCT patients (Figure 1). This observation was strengthened by results from a multivariate analysis that showed the best predictor for c‐MYC expression levels within 1LoT patients was indeed receiving SCT as part of frontline therapy.
Progression‐free survival in c‐MYC‐high vs c‐MYC‐low patients, with IRd and placebo‐Rd groups pooled, appeared similar. However, c‐MYC‐high patients had a larger magnitude of PFS benefit with IRd vs placebo‐Rd than c‐MYC‐low patients (Figure 2A), confirming the previous observation that tumors with high c‐MYC expression appear to benefit from PI‐based therapy.11 Baseline characteristics of c‐MYC‐high vs c‐MYC‐low patients appeared well balanced, except for a higher frequency of IgA MM, higher rates of refractory and relapsed and refractory disease, a higher percentage of 2/3LoT patients, and a higher rate of del(17p) in the c‐MYC‐high group (Table 1). These data suggest the presence of more aggressive disease within the high c‐MYC group, in agreement with other reports.26
The observation that c‐MYC expression level differed based on the number/type of prior LoTs and that PFS benefit was larger with IRd vs placebo‐Rd in c‐MYC‐high patients, supports the hypothesis that the differential PFS benefit observed in TOURMALINE‐MM1 could be due in part to differences in c‐MYC expression levels between patient subgroups. We therefore evaluated PFS with IRd vs placebo‐Rd in c‐MYC‐high and c‐MYC‐low patients and based on prior LoTs. The analysis showed that 1LoT‐noSCT and 2/3LoT patients had the largest magnitude of PFS benefit with IRd vs placebo‐Rd, while in 1LoT‐SCT patients PFS was similar (Figure 2B). This suggests that the smaller PFS benefit observed with IRd vs placebo‐Rd in 1LoT patients in TOURMALINE‐MM1 might have been driven by the 1LoT‐SCT subgroup (this group represented 65% of 1LoT patients). Additionally, while PFS with IRd appeared independent of c‐MYC level (c‐MYC‐high vs c‐MYC‐low: median 21.4 vs 20.6 months in 1LoT‐SCT, median 18.5 vs 17 months in 1LoT‐noSCT, not reached vs not reached in 2/3LoT patients), we observed differences in PFS with placebo‐Rd among 1LoT‐noSCT (c‐MYC‐high vs c‐MYC‐low: median 9.69 vs 12.2 months) and 2/3LoT (median 11.3 vs 14.1 months) patients. Notably, the 1LoT‐SCT subgroup appeared to have the longest PFS with placebo‐Rd, independent of c‐MYC expression, suggesting that the greater PFS benefit with IRd vs placebo‐Rd in TOURMALINE‐MM1 in 1LoT‐noSCT and 2/3LoT patients might have been driven by reduced activity of placebo‐Rd in these subgroups. This finding is supported by a recent report suggesting high c‐MYC expression as a potential mechanism of resistance to lenalidomide activity in MM.27
We next looked at the baseline characteristics of 1LoT‐SCT patients compared to 1LoT‐noSCT and 2/3LoT patients. We found that 1LoT‐SCT patients had better prognostic features, including a lower proportion of elderly patients, better performance status, a lower rate of high‐risk cytogenetics, and a higher rate of relapsed‐only (non‐refractory) disease. A high proportion of 1LoT‐SCT patients (71%) received SCT ≥ 24 months prior to TOURMALINE‐MM1 study entry, suggesting that they did not have an early, aggressive relapse post‐SCT (Table S2). It is well‐known that quality and durability of response post‐SCT are important factors associated with long‐term outcome.28 Patients with early relapse have more aggressive MM and shorter PFS and overall survival29, 30 compared to those with relapse >1 year post‐SCT, who have a more indolent disease biology.31
Although high c‐MYC levels are associated with increased tumor sensitivity to IRd, this did not appear to be the case in 1LoT‐SCT patients, as PFS was similar with IRd and placebo‐Rd (Figure 2B). We compared RNAseq data between 1LoT‐SCT patients and 1LoT‐noSCT and 2/3LoT patients, and the immunoglobulin heavy constant delta and epsilon genes, IGHD and IGHE, components of the BCR complex (CD19, CD81, CD79A),13 and genes involved in different steps of cell maturation (PAX5,32, 33 VPREB1,33 IGLL134) were among the 10 most differentially expressed genes. These genes were significantly overexpressed in 1LoT‐SCT patients’ tumors, while c‐MYC was downregulated (Table 2). The presence of high CD19 levels in 1LoT‐SCT patients was unexpected, although minor subsets of CD19‐positive cells have been observed previously35, 36 in a disease predominantly comprising CD19‐negative mature plasma cells. These CD19‐positive cells tend to be found in circulation; it is possible that these cells were collected with blood during screening bone marrow sampling. It has been reported that levels of CD19‐positive cells in the peripheral blood are significantly higher in MM patients vs healthy donors.37 Cell numbers are low during induction and high‐dose treatment but appear to increase post‐SCT, when a simultaneous rise in CD19 cells and CD19 mRNA is detected.38 Interestingly, these cells have less differentiated phenotypes and appear to make up a drug‐resistant, clonogenic disease reservoir.39, 40, 41
In our dataset, CD19 and CD81 expression was inversely correlated with CD138 expression, a marker of B cell maturity42 (Figure S2), further supporting the hypothesis of the presence of immature tumor cells in 1LoT‐SCT patients. While CD19, CD81, and CD79A were significantly overexpressed in 1LoT‐SCT patients’ tumors, no differences were observed between 1LoT‐noSCT and 2/3LoT patients (Figure 3A/B). Importantly, CD19 and CD81 expression appeared to be inversely correlated with c‐MYC expression across all subgroups analyzed (Figure 3C). This observation suggests that subsets of these less mature, CD19‐positive and CD81‐positive tumors might be present in all MM patients, independent of prior therapy, but that the size of these subsets depends on prior therapy received, with the largest proportion observed in 1LoT‐SCT patients.
Analyses of PFS with IRd and placebo‐Rd activity according to c‐MYC, CD19, or CD81 expression levels (Figure 4) showed prolonged PFS with placebo‐Rd in patients with tumors with higher CD19 and CD81 expression, as well as low c‐MYC expression. We could therefore hypothesize that in TOURMALINE‐MM1 the Rd component of therapy targeted tumors with a less mature phenotype, while ixazomib targeted clonal populations with a more mature/differentiated phenotype, consistent with higher proteasomal stress.43 The differential distribution of less mature tumor cells among patients with different prior therapies might in part explain the differential magnitude of PFS benefit with IRd vs placebo‐Rd in TOURMALINE‐MM1. Interestingly, previous reports have alluded to a preferential targeting of plasma cell precursors by lenalidomide, suggesting that resistance could be associated with the selection of mature plasma cell subclones.44 Similarly, it has been suggested that bortezomib preferentially kills mature cells and that resistance is associated with the emergence of dedifferentiated MM cells.45
It has been reported that immunoglobulin secretion increases with the differentiation of plasma cells, while proteasome activity decreases.46, 47 Following differentiation, cells have higher levels of ubiquitin‐conjugated proteins and lower free ubiquitin levels, suggesting that differentiated, more mature MM cells are under proteasomal stress as a consequence of the imbalance between greater workload and lower proteasomal capacity.43 This imbalance is thought to explain the exquisite sensitivity of MM cells to PIs.48, 49 In our study, a trend toward lower M‐protein levels in 1LoT‐SCT patients was observed (data not shown), supporting prior observations of less mature tumors in this subgroup.
While these data provide some explanations for the differential PFS benefit of IRd vs placebo‐Rd in TOURMALINE‐MM1, we recognize that our analyses have some limitations, including the small numbers of patients in each of the subgroups. Although we conducted a multivariate analysis incorporating various baseline disease characteristics, our analysis did not account for all the molecular subtypes, as these data were not collected in TOURMALINE‐MM1; for example, c‐MYC expression has been shown to be associated with hyperdiploidy.17 In addition, in the context of clonal evolution in MM, it would have been of value to incorporate data from bone marrow aspirates taken at diagnosis or earlier in the disease course; however, these data were not available. Thus, while our analyses have enabled the development of hypotheses to help understand TOURMALINE‐MM1 findings, it is important to emphasize that more detailed prospective studies, with larger sample sizes and longer follow‐up (to capture clonal changes), and addressing additional parameters of potential differential prognostic relevance, are required to validate and extend the clinical relevance of our findings.
CONFLICT OF INTEREST
ADB: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. NJB: Research funding: Celgene; Honoraria: Janssen, Celgene, Amgen. NCM: Consultancy: Celgene, Novartis, Takeda; Scientific founder: Oncopep. HA‐L: Research funding: Amgen, Celgene, Sanofi, Takeda; Membership of advisory committees: Abbvie, Amgen, Celgene, Janssen, Sanofi, Takeda. TM: Advisory board member, Janssen‐Cilag, Novartis, Takeda, Bristol‐Myers Squibb, Abbvie, Pfizer. LV: None. LP: None. PG: None. MC: Consultancy, honoraria: Janssen, Celgene, Takeda, Amgen, Bristol‐Myers Squibb, AbbVie. CL: Consultancy: Celgene, Takeda, Janssen, Amgen, Novartis, Bristol‐Myers Squibb. SKK: Institutional research funding for clinical trials: Celgene, Takeda, Janssen, BMS, Sanofi, KITE, Merck, Abbvie, Medimmune, Novartis, Roche‐Genentech, Amgen; Consultancy/Advisory board participation (no personal payments): Celgene, Takeda, Janssen, KITE, Merck, Abbvie, Medimmune, Genentech, Amgen; Consultancy/Advisory board participation (with personal payment): Oncopeptides, Adaptive; Honorarium for educational event: Dr Reddys Lab, Ono Pharmaceuticals. SVR: Institutional research funding for clinical trials: Takeda. JJK: None. DB: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. JL: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. BL: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. SB: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. LS: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. JZ: Employment, Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. D‐LE: Employment (at time of study/analyses), Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. KL: Employment, previously Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, currently TESARO, Waltham, MA. HvdV: Employment (at time of study/analyses), Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. PGR: Advisory Committee Member: Karyopharm, Oncopeptides, Celgene, Takeda, Janssen, Sanofi. Research support: Oncopeptides, Celgene, Takeda, BMS. PM: Advisory board member and honoraria: Takeda, Celgene.
AUTHOR CONTRIBUTIONS
ADB, NJB, NCM, HA‐L designed the analyses. ADB, DB, D‐LE, PGR, PM contributed to the design of the overall study. ADB, JL, BL, SB, LS, JZ performed the analyses. NJB, NCM, HA‐L, TM, LV, LP, PG, MC, CL, SKK, SVR, JJK, PGR, PM provided patient samples and/or experimental tools for the analyses. ADB, NJB, NCM, HA‐L, D‐LE, KL, HvdV analyzed and interpreted the data. ADB, KL, HvdV wrote the manuscript. All authors reviewed the manuscript and approved the final draft for submission.
Supporting information
Fig S1
Fig S2
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank all patients and their families, physicians, research nurses, study coordinators, and research staff participating in this study. The authors acknowledge Laura Webb, PhD, and Steve Hill, PhD, of FireKite, an Ashfield company, part of UDG Healthcare plc, for writing assistance during the development of this manuscript, which was funded by Millennium Pharmaceuticals Inc, and complied with Good Publication Practice 3 ethical guidelines (Battisti WP, et al Ann Intern Med 2015;163:461‐4), and Renda Ferrari, PhD, of Millennium Pharmaceuticals, Inc, Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, for editorial support. This study was sponsored by Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Di Bacco A, Bahlis NJ, Munshi NC, et al. c‐MYC expression and maturity phenotypes are associated with outcome benefit from addition of ixazomib to lenalidomide‐dexamethasone in myeloma. Eur J Haematol. 2020;105:35–46. 10.1111/ejh.13405
Affiliation at the time that the work was conducted: Katarina Luptakova, Tesaro, Waltham, MA, USA; Helgi van de Velde, Sanofi, Cambridge, MA, USA.
DATA AVAILABILITY STATEMENT
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
REFERENCES
- 1. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature. 2013;499(7457):214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100‐113. [DOI] [PubMed] [Google Scholar]
- 3. de Mel S, Lim SH, Tung ML, Chng W‐J. Implications of heterogeneity in multiple myeloma. Biomed Res Int. 2014;2014:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Majumder MM, Silvennoinen R, Anttila P, et al. Identification of precision treatment strategies for relapsed/refractory multiple myeloma by functional drug sensitivity testing. Oncotarget. 2017;8(34):56338‐56350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cornell RF, Kassim AA. Evolving paradigms in the treatment of relapsed/refractory multiple myeloma: increased options and increased complexity. Bone Marrow Transplant. 2016;51(4):479‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621‐1634. [DOI] [PubMed] [Google Scholar]
- 7. Mateos MV, Masszi T, Grzasko N, et al. Impact of prior therapy on the efficacy and safety of oral ixazomib‐lenalidomide‐dexamethasone vs. placebo‐lenalidomide‐dexamethasone in patients with relapsed/refractory multiple myeloma in TOURMALINE‐MM1. Haematologica. 2017;102(10):1767‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dang CV. MYC metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3(8):a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122(10):3456‐3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glitza IC, Lu G, Shah R, et al. Chromosome 8q24.1/c‐MYC abnormality: a marker for high‐risk myeloma. Leuk Lymphoma. 2015;56(3):602‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chng W‐J, Huang GF, Chung TH, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25(6):1026‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilson A, Murphy MJ, Oskarsson T, et al. c‐Myc controls the balance between hematopoietic stem cell self‐renewal and differentiation. Genes Dev. 2004;18(22):2747‐2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vences‐Catalán F, Kuo C‐C, Sagi Y, et al. A mutation in the human tetraspanin CD81 gene is expressed as a truncated protein but does not enable CD19 maturation and cell surface expression. J Clin Immunol. 2015;35(3):254‐263. [DOI] [PubMed] [Google Scholar]
- 14. Avet‐Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109(8):3489‐3495. [DOI] [PubMed] [Google Scholar]
- 15. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walker BA, Wardell CP, Brioli A, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weinhold N, Kirn D, Seckinger A, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116‐e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brioli A, Melchor L, Walker BA, Davies FE, Morgan GJ. Biology and treatment of myeloma. Clin Lymphoma Myeloma Leuk. 2014;14(Suppl):S65‐S70. [DOI] [PubMed] [Google Scholar]
- 19. Jovanovic KK, Roche‐Lestienne C, Ghobrial IM, Facon T, Quesnel B, Manier S. Targeting MYC in multiple myeloma. Leukemia. 2018;32(6):1295‐1306. [DOI] [PubMed] [Google Scholar]
- 20. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2(3):175‐187. [DOI] [PubMed] [Google Scholar]
- 21. Dang CV. c‐Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin Y, Wong K, Calame K. Repression of c‐myc transcription by Blimp‐1, an inducer of terminal B cell differentiation. Science. 1997;276(5312):596‐599. [DOI] [PubMed] [Google Scholar]
- 23. Murn J, Mlinaric‐Rascan I, Vaigot P, Alibert O, Frouin V, Gidrol X. A Myc‐regulated transcriptional network controls B‐cell fate in response to BCR triggering. BMC Genomics. 2009;10:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ravi D, Beheshti A, Abermil N, et al. Proteasomal inhibition by ixazomib induces CHK1 and MYC‐dependent cell death in T‐cell and Hodgkin lymphoma. Cancer Res. 2016;76(11):3319‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wirth M, Stojanovic N, Christian J, et al. MYC and EGR1 synergize to trigger tumor cell death by controlling NOXA and BIM transcription upon treatment with the proteasome inhibitor bortezomib. Nucleic Acids Res. 2014;42(16):10433‐10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Szabo AG, Gang AO, Pedersen MO, Poulsen TS, Klausen TW, Norgaard P. Overexpression of c‐myc is associated with adverse clinical features and worse overall survival in multiple myeloma. Leuk Lymphoma. 2016;57(11):2526‐2534. [DOI] [PubMed] [Google Scholar]
- 27. Franssen LE, Nijhof IS, Couto S, et al. Cereblon loss and up‐regulation of c‐Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematologica. 2018;103(8):e368‐e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jimenez‐Zepeda VH, Reece DE, Trudel S, Chen C, Tiedemann R, Kukreti V. Early relapse after single auto‐SCT for multiple myeloma is a major predictor of survival in the era of novel agents. Bone Marrow Transplant. 2015;50(2):204‐208. [DOI] [PubMed] [Google Scholar]
- 29. Kumar SK, Dingli D, Dispenzieri A, et al. Impact of additional cytoreduction following autologous SCT in multiple myeloma. Bone Marrow Transplant. 2008;42(4):259‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Venner CP, Connors JM, Sutherland HJ, et al. Novel agents improve survival of transplant patients with multiple myeloma including those with high‐risk disease defined by early relapse (<12 months). Leuk Lymphoma. 2011;52(1):34‐41. [DOI] [PubMed] [Google Scholar]
- 31. Fernández de Larrea C, Jiménez R, Rosiñol L, et al. Pattern of relapse and progression after autologous SCT as upfront treatment for multiple myeloma. Bone Marrow Transplant. 2014;49(2):223‐227. [DOI] [PubMed] [Google Scholar]
- 32. Lin P, Mahdavy M, Zhan F, Zhang HZ, Katz RL, Shaughnessy JD. Expression of PAX5 in CD20‐positive multiple myeloma assessed by immunohistochemistry and oligonucleotide microarray. Mod Pathol. 2004;17(10):1217‐1222. [DOI] [PubMed] [Google Scholar]
- 33. Kuo YH, Gerstein RM, Castilla LH. Cbfbeta‐SMMHC impairs differentiation of common lymphoid progenitors and reveals an essential role for RUNX in early B‐cell development. Blood. 2008;111(3):1543‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu X, Chu C‐S, Fang T, et al. MTA2/NuRD Regulates B Cell Development and Cooperates with OCA‐B in Controlling the Pre‐B to Immature B Cell Transition. Cell Rep. 2019;28(2):472‐485.e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajek R, Okubote SA, Svachova H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol. 2013;163(5):551‐564. [DOI] [PubMed] [Google Scholar]
- 36. Garfall AL, Maus MV, Hwang W‐T, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373(11):1040‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pilarski LM, Jensen GS. Monoclonal circulating B cells in multiple myeloma. A continuously differentiating, possibly invasive, population as defined by expression of CD45 isoforms and adhesion molecules. Hematol Oncol Clin North Am. 1992;6(2):297‐322. [PubMed] [Google Scholar]
- 38. Rasmussen T, Jensen L, Honore L, Johnsen HE. Frequency and kinetics of polyclonal and clonal B cells in the peripheral blood of patients being treated for multiple myeloma. Blood. 2000;96(13):4357‐4359. [PubMed] [Google Scholar]
- 39. Yaccoby S. The phenotypic plasticity of myeloma plasma cells as expressed by dedifferentiation into an immature, resilient, and apoptosis‐resistant phenotype. Clin Cancer Res. 2005;11(21):7599‐7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fuhler GM, Baanstra M, Chesik D, et al. Bone marrow stromal cell interaction reduces syndecan‐1 expression and induces kinomic changes in myeloma cells. Exp Cell Res. 2010;316(11):1816‐1828. [DOI] [PubMed] [Google Scholar]
- 41. Hideshima T, Mitsiades C, Ikeda H, et al. A proto‐oncogene BCL6 is up‐regulated in the bone marrow microenvironment in multiple myeloma cells. Blood. 2010;115(18):3772‐3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. 2001;2(12):1103‐1108. [DOI] [PubMed] [Google Scholar]
- 43. Gu J‐L, Li J, Zhou Z‐H, et al. Differentiation induction enhances bortezomib efficacy and overcomes drug resistance in multiple myeloma. Biochem Biophys Res Commun. 2012;420(3):644‐650. [DOI] [PubMed] [Google Scholar]
- 44. Jourdan M, Cren M, Schafer P, et al. Differential effects of lenalidomide during plasma cell differentiation. Oncotarget. 2016;7(19):28096‐28111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leung‐Hagesteijn C, Erdmann N, Cheung G, et al. Xbp1s‐negative tumor B cells and pre‐plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang H, Wu D, Fu J, et al. All‐trans retinoic acid can intensify the growth inhibition and differentiation induction effect of rosiglitazone on multiple myeloma cells. Eur J Haematol. 2009;83(3):191‐202. [DOI] [PubMed] [Google Scholar]
- 47. Cenci S, Mezghrani A, Cascio P, et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J. 2006;25(5):1104‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cascio P, Oliva L, Cerruti F, et al. Dampening Ab responses using proteasome inhibitors following in vivo B cell activation. Eur J Immunol. 2008;38(3):658‐667. [DOI] [PubMed] [Google Scholar]
- 49. Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus‐like disease from nephritis. Nat Med. 2008;14(7):748‐755. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Supplementary Material
Data Availability Statement
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.