

Abstract
Background
Effective treatment for severe asthma is a significant unmet need. While eosinophilic inflammation caused by type 2 cytokines is responsive to corticosteroid and biologic therapies, many severe asthmatics exhibit corticosteroid‐unresponsive mixed granulocytic inflammation.
Objective
Here, we tested the hypothesis that the pro‐allergic cytokine, IL‐13, can drive both corticosteroid‐sensitive and corticosteroid‐resistant responses.
Results
By integration of in vivo and in vitro models of IL‐13–driven inflammation, we identify a role for the epidermal growth factor receptor (EGFR/ERBB1) as a mediator of corticosteroid‐unresponsive inflammation and bronchial hyperresponsiveness driven by IL‐13. Topological data analysis using human epithelial transcriptomic data from the U‐BIOPRED cohort identified severe asthma groups with features consistent with the presence of IL‐13 and EGFR/ERBB activation, with involvement of distinct EGFR ligands. Our data suggest that IL–13 may play a dual role in severe asthma: on the one hand driving pathologic corticosteroid‐refractory mixed granulocytic inflammation, but on the other hand underpinning beneficial epithelial repair responses, which may confound responses in clinical trials.
Conclusion and Clinical Relevance
Detailed dissection of those molecular pathways that are downstream of IL‐13 and utilize the ERBB receptor and ligand family to drive corticosteroid‐refractory inflammation should enhance the development of new treatments that target this sub‐phenotype(s) of severe asthma, where there is an unmet need.
Keywords: animal models, asthma, basic mechanisms, corticosteroid‐refractory, epithelium, neutrophils
1. INTRODUCTION
Asthma is a heterogeneous disease characterized by a diverse profile of symptoms, severity and responses to medications. In mild‐to‐moderate asthma, treatment with inhaled corticosteroids can significantly reduce inflammation and control symptoms. However, in patients with more severe disease, symptoms can persist despite receiving standard of care treatment, including antibody‐based biologics.1 This subgroup has been defined as “severe refractory” asthma.2, 3 Up to 10% of the asthmatic population are classed as severe; they have higher rates of asthma exacerbations, increased morbidity and account for a disproportionate use of healthcare resources, accounting for more than 60% of the economic burden.4 Much work has been done to advance the clinical understanding of severe refractory asthma, but there still remains a clear unmet clinical need.4
The recent focus on disease heterogeneity has raised the concept that asthma consists of multiple phenotypes with distinct underlying mechanisms (endotypes).4, 5, 6, 7 Initially, the focus was on identification of subgroups based on clinical presentation including exacerbations, persistent symptoms and reduced lung function. However, with the advent of 'omic technologies (transcriptomics, proteomics, metabolomics, lipidomics) and the recruitment of large patient cohorts,8, 9, 10, 11 molecular and biological processes can now be evaluated with the aim of identifying distinct endotypes. A key phenotypic characteristic that is used to define asthma subgroups is the presence or absence of biomarkers of type‐2 inflammation.4 Typical type‐2 biomarkers include bronchial epithelial gene expression of POSTN (Periostin), CLCA1 (Chloride Channel Accessory 1) and SERPINB2 (Serpin Family B Member 2),12 blood and sputum eosinophils,13 fractional exhaled nitric oxide (Feno),14 serum periostin,12, 15 serum IgE 13 and type‐2 gene expression (IL4, IL5, IL13) in sputum cells. 16 Elevated levels of these biomarkers are frequently associated with the presence of atopy and bronchial hyperresponsiveness (BHR)17 and are considered to be sensitive to inhibition by corticosteroids and monoclonal antibodies against IgE, IL‐5 or IL‐5 receptor.1, 12, 17 In contrast, patients who have “non‐type‐2” (also known as “type‐2–low”) severe asthma are frequently characterized by airway neutrophilia, a type‐17 immune signature involving genes such as CXCL1, CXCL2 and CSF3 18 and disease that is not effectively treated by inhaled corticosteroids or biologics.6
While this broad classification of corticosteroid responsiveness in relation to type‐2 inflammation is a reasonable generalization, studies with dupilumab, an antibody to IL‐4Rα, suggest it is an over‐simplification. Thus, treatment of uncontrolled persistent asthmatic patients with dupilumab as an add‐on therapy increased lung function and reduced severe exacerbations irrespective of baseline eosinophil count.19 Subsequent studies confirmed that dupilumab treatment resulted in a reduction in glucocorticoid use and severe exacerbations.20, 21 As these beneficial effects of dupilumab, which inhibits both IL‐4 and IL‐13 signalling,22 were seen in patients who were receiving medium‐to‐high‐dose inhaled corticosteroids plus a long‐acting β2 agonist, this suggests that these asthma patients have persistent type‐2 responses despite inhaled corticosteroid therapy. Consistent with this, it has been reported that airway type‐2 inflammation is not suppressed adequately in approximately half of asthmatic patients treated with inhaled corticosteroids23 and that there are many severe asthma patients with mixed eosinophilic and neutrophilic inflammation.24
Based on these clinical observations, the aim of this work was to model allergic airway inflammation using a transgenic IL‐13 mouse25 and to test the hypothesis that a subset of IL‐13–induced responses are corticosteroid‐unresponsive and contribute to ongoing airway symptoms. We show that IL‐13–induced airway inflammation involves a corticosteroid‐refractory component characterized by pro‐neutrophilic cytokine expression, airway neutrophilia and BHR. IL‐13–driven pro‐neutrophilic cytokine expression was mediated by EGFR activation which was corticosteroid unresponsive. To translate these findings into human disease, we investigated gene epithelial expression in the U‐BIOPRED cohort. Our analyses suggest that IL‐13 may have a dual role, on the one hand driving mixed granulocytic inflammatory responses, but on the other hand underpinning epithelial repair responses. Thus, targeting IL‐13 may have both beneficial and detrimental effects, confounding responses in clinical trials.
2. METHODS
2.1. Mice
Doxycycline (DOX)‐inducible conditional double‐transgenic mice expressing a (tetO)7‐CMV‐Il13 transgene under the regulation of the line 1 Club Cell Secretory Protein (Ccsp) promoter (Scgb1a1) have been previously described.25 Expression of IL‐13 was induced in 6‐week‐old double‐transgenic (Ccsp/Il13) mice by provision of Doxycycline (DOX) (Lab Diet, 5LOS W/625 ppm DOX; TestDiet) in the food ad libitum. Single transgenic littermate controls were fed the same diet. Where indicated, Ccsp/Il13 mice were given daily intraperitoneal injections of 3 mg/kg Dexamethasone (DEX) (Hameln Pharmaceuticals) or 0.1 mg/kg EGFR inhibitor AG1478 (Bio‐Techne) for up to 7 days, while control mice received saline. Experiments followed the 3R (Replacement, Reduction, and Refinement) principles, and experiments were conducted according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines26 and the local Southampton University ethical committee under project and personal licences from the Home Office, United Kingdom.
2.2. Mouse tracheal epithelial cell expansion
Tracheal epithelial cells were isolated and expanded as previously described.27 Isolated cells were grown on Transwells® (Corning) in submerged culture to allow formation of tight junctions, determined by trans‐epithelial electrical resistance (TER). Cells were cultured until the TER was >1000 Ω.cm2. IL‐13 was induced by addition of 2 µmol/L DOX (Sigma Aldrich) to the culture medium. When required, AG1478 (Sigma Aldrich) and/or Dexamethasone (Hameln) were added to the culture medium, each at a concentration of 10 µmol/L. Apical and basolateral secretions were collected at 48 and 72 hour for KC and CXCL2 protein quantification and cells removed from the transwells in TRIzol and pooled in duplicates for RNA extraction.
2.3. Assessment of lung function
Mice were anaesthetized with 100 μL of anaesthetic containing a 4:1:1 mixture of ketamine, acepromazine and xylazine by intraperitoneal injection. A FlexiVent system (SCIREQ) was used to assess lung function in the form of airway resistance (R) after aerosolized methacholine challenge to provide a measure of BHR, according to the manufacturer's instructions. Airway resistance was measured by forced oscillation technique, with increasing values indicating bronchoconstriction of the lungs. BHR measurements were obtained from individual animals using increasing stepwise concentrations of 0, 2.5, 5 and 10 mg/mL methacholine (Sigma Aldrich).
2.4. Inflammatory cell counts
Bronchoalveolar lavage (BALF) samples were collected by washing the lungs three times with 800 μL sterile PBS. The total volume of the combined fluids was measured and then centrifuged at 300 g for 5 minutes. The BALF supernatants were frozen for cytokine analysis. Red blood cells were lysed from the cell pellets, which were subsequently resuspended in 300 μL PBS. Cells were counted, and 100 000 cells were loaded into a cytospin funnel and centrifuged at 300 g for 5 minutes on to a glass slide. Slides were air‐dried, and the cells were stained using a Diff‐Quick stain (Siemens). The different inflammatory cell types were counted to a total of 300 cells and expressed as the differential cell count in cells/mL of BALF.
2.5. RNA extraction and analysis
Lung tissue was stored in RNAlater (Life Technologies) before homogenization in Qiazol® Reagent and RNA isolated on columns from miRNeasy Kits (Qiagen). mTEC RNA was isolated via TRIzol® extraction (Life Technologies) using standard protocols, and genomic DNA contamination was removed by digestion with DNase (Life Technologies). mRNA expression was measured by reverse transcription quantitative PCR. First‐strand cDNA was generated by reverse transcription using the NanoScript2 cDNA synthesis kit (PrimerDesign). qPCR was performed using a CFX96 qPCR machine (Bio‐Rad) for 40 cycles at 95°C for 5 seconds and 60°C for 20 seconds (fast protocol) or 40 cycles at 95°C for 15 seconds and 60°C for 60 seconds (standard protocol). Each reaction contained either Expression Master Mix (Life Technologies) or Precision Master Mix (PrimerDesign) and a Taqman primer/probeset for one of the following targets: Il‐13 Mm00434204_m1; Il‐17a Mm00439618_m1, Cxcl1/Kc Mm04207460_m1; Ccl11/Eotaxin Mm00441238_m1; Muc5ac Mm01276718_m1; Periostin/Postn Mm01284919_m1; SerpinB2 Mm00440905_m1; Cxcl2 Mm00436450_m1; Csf3 Mm00438334_m1; Egfr Mm01187858_m1;, Erbb2 Mm00658541_m1; Erbb3 Mm01159990_g1 Areg Mm01354339_m1; Btc Mm00432137_m1; Egf Mm00438696_m1; Ereg Mm00514794_m1; Hbegf Mm00439306_m1, Tgfa Mm00446232_m1; (all from Life Technologies) and Gapdh (PrimerDesign). mRNAs were measured by reverse transcription quantitative PCR. Relative mRNA expression was analysed using the ΔΔCt method28 with Gapdh as housekeeping gene.
2.6. Immunohistochemistry
The lungs were inflation‐fixed at constant pressure with 10% neutral buffered formalin and embedded in paraffin wax. 5μm sections were stained using haematoxylin and eosin (H&E).
2.7. Cytokine measurements
IL‐13, KC, CCL11, CXCL2 and CSF3 proteins were measured by ELISA (R&D Systems) according to the manufacturer's instructions.
2.8. Statistical analyses
Normal distribution of the numeric data was evaluated, and appropriate parametric or non‐parametric statistical tests applied. Parametric data are plotted as mean with one standard deviation (SD) while non‐parametric data are shown as boxes representing the 25 and 75% interquartile ranges, whiskers depicting the minimum and maximum values. Statistical significance was assessed by using Student's t test (parametric, unpaired data) with Welch's correction if SDs were not equal or Mann‐Whitney test (non‐parametric, unpaired data) for comparisons between two groups. For comparison of three or more groups, a one‐way ANOVA with Dunn's multiple comparison test (parametric data) or Kruskal‐Wallis test with Dunn's test for correction for multiple comparisons (non‐parametric data) was used. For comparison of two or more groups with two independent variables, a two‐way ANOVA with Tukey's multiple comparison test was used. * = P < .05, ** = P < .01, *** = P < .001.
2.9. The U‐BIOPRED cohort
U‐BIOPRED is a multi‐centre study that enrolled 311 severe, non‐smoker asthmatics, 88 mild/moderate non‐smoking asthmatics and 101 healthy controls.8 Among these participants, 61 severe asthmatics, 36 mild/moderate asthmatics and 44 healthy volunteers underwent fiberoptic bronchoscopy and epithelial brushing. Epithelial brushings were processed into RNAlater for subsequent analysis on Affymetrix U133 Plus 2.0 microarrays, and only those passing stringent quality control were analysed.10 All participants provided written informed consent to participate in the study which was approved by national ethics committees. The transcriptomic data are stored as GSE76226.29
Using this dataset, epithelial expression of EGFR and EGFR ligands was compared between groups using SPSS v24. Paired t tests were applied to the log2‐transformed transcriptomic data, while the clinical data were analysed by Kruskal‐Wallis test and Mann‐Whitney U or Student t tests depending on the type of data distribution; P < .05 was considered significant. Topological data analysis (TDA) was then applied to the epithelial transcriptomic data as described previously,30, 31 with some modification, using the Ayasdi Core software (Ayasdi, Menlo Park, CA) with a norm correlation metric and the neighbourhood lens (resolution, 20 bins; gain, 4.00). Clinical and pathobiological data were then overlaid as metadata onto the generated TDA network to look for associations.
3. RESULTS
3.1. IL‐13 induces BHR and a mixed inflammatory phenotype
IL‐13 was significantly elevated (Figure S1A, B) when Ccsp/Il13 mice were fed DOX to induce transgene expression. As previously reported,25 when challenged with methacholine, these IL‐13–expressing mice showed a significant increase in BHR (P < .002) (Figure 1A); furthermore, both eosinophils and neutrophils were significantly elevated in BALF (Figure 1B) compared to single transgenic littermate controls. Histochemical analysis of lung sections also demonstrated a significant increase in airway inflammation, as well as goblet cell metaplasia (Figure S2A,B) accompanied by Mucin 5AC (Muc5ac) mRNA expression (Figure S2C).
Figure 1.
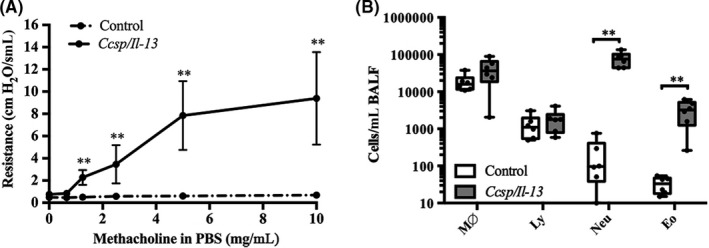
IL‐13 induces BHR and mixed inflammatory cell airway inflammation. A, Bronchial hyperresponsiveness to methacholine challenge of control and Ccsp/Il13 transgenic mice on DOX for 7 d. Resistance (R: cmH2O.s/mL) at methacholine concentrations from 0 to 10 mg/mL in phosphate‐buffered saline (PBS). Data are shown as mean ± SEM. B, Differential cell counts of BALF from Ccsp/Il13 and control mice (M∅ = macrophages; Ly = lymphocytes; Neu = neutrophils; and Eo = eosinophils). Box plots show medians and 25th to 75th percentiles, and whiskers represent minimum and maximum values; all data points are shown. N = 6 mice per group. Data are representative of three independent experiments. Statistical analysis was performed using two‐way ANOVA with Tukey's multiple comparison test. **P < .01
3.2. Both “type‐2” and “‐type‐17” biomarkers are induced by IL‐13
Markers of “type‐2”–mediated inflammation including eotaxin/Ccl11, periostin/Postn and SerpinB2 (Figure 2A‐C) mRNAs were all significantly elevated in lungs of IL‐13–expressing mice. Expression of mRNAs for genes including chemokine (C‐X‐C motif) ligand 1 (Cxcl1/Kc), Cxcl2 and Colony Stimulating Factor 3 (Csf3) (Figure 2E‐G) which are more usually associated with “type‐17” responses was also increased by IL‐13; however, expression of Il‐17a mRNA was undetectable in the lungs of IL‐13 expressing mice (C t values > 38). ELISAs for CCL11 and CXCL1/KC confirmed elevation of these cytokines in BALF of IL‐13–expressing mice (Figure 2D,H). BALF CCL11 protein levels were significantly correlated with eosinophil numbers (r 2 = 0.74, P = .02) (Figure S3A) but not neutrophil numbers, whereas CXCL1 levels were strongly correlated with neutrophil numbers (r 2 = 0.9, P = .003) (Figure S3B) but not with eosinophil numbers. Furthermore, BALF CXCL1 levels were correlated with the amount of IL‐13 in the BALF (r 2 = 0.74, P = .001) (Figure S3C) consistent with a role for IL‐13 in driving airway neutrophilia via CXCL1. CXCL2 protein was also significantly elevated in both the BALF and lung lysate (Figure S4A,B).
Figure 2.
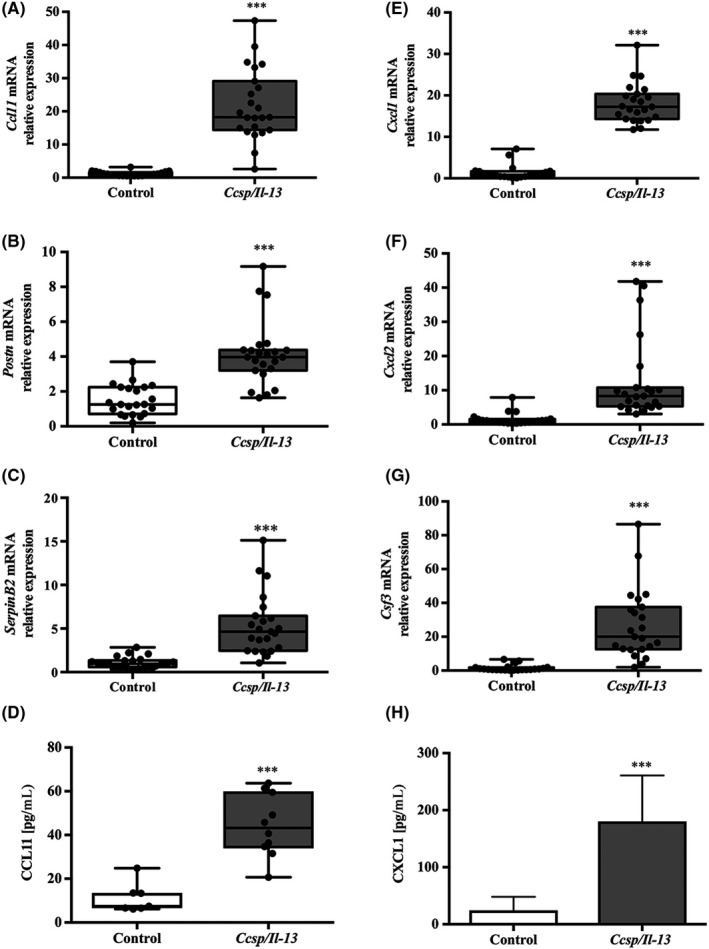
Expression of IL‐13 induces expression of both type‐2 and pro‐neutrophilic mediated markers. Relative mRNA expression in whole‐lung lobe lysates from littermate controls (white bars) vs Ccsp/Il13 mice (grey bars) after induction of IL‐13 for 7 d for A, Ccl11, B, Postn, C, Serpinb2, E, Cxcl1/KC, F, Cxcl2 and G, Csf3. ELISAs for D, CCL11 and H, CXCL1 protein levels in BALF. For mRNA expression, n = 22 for controls and n = 23 for Ccsp/Il13 mice and n = 7 and n = 10, respectively, for protein, from three independent experiments. Non‐parametric data are shown as box plots with medians and 25th to 75th percentiles, and whiskers representing minimum and maximum values with all data points shown; parametric data are shown as mean + SD. Statistical analyses were performed using Mann‐Whitney test or Student's t test. ***P < .001
3.3. Corticosteroid treatment does not influence neutrophilic inflammation and BHR
To determine how airway responses are modulated by corticosteroid treatment, IL‐13 was induced in Ccsp/Il13 mice for 7 days and dexamethasone (Dex) was given via intraperitoneal injection for the duration of IL‐13 induction (7 days) or for the final 5 or 3 days of induction. The control group received DOX to induce IL‐13 and were sham‐treated with saline for 7 days. Dex significantly reduced eosinophil numbers after 7 days of treatment; furthermore, even shorter treatments given during the final 3 or 5 days of IL‐13 induction significantly suppressed eosinophil numbers (Figure 3A). In contrast, neither infiltration of neutrophils (Figure 3B) nor BHR (Figure 3C) were affected by presence or duration of Dex treatment. Although long‐term treatment with corticosteroids can have metabolic effects,32 the dose and duration used had no effect on body weight (Figure S5). In addition, Dex treatment did not cause any inflammation in the airways of similarly treated sTg mice, nor did it affect body weight.
Figure 3.
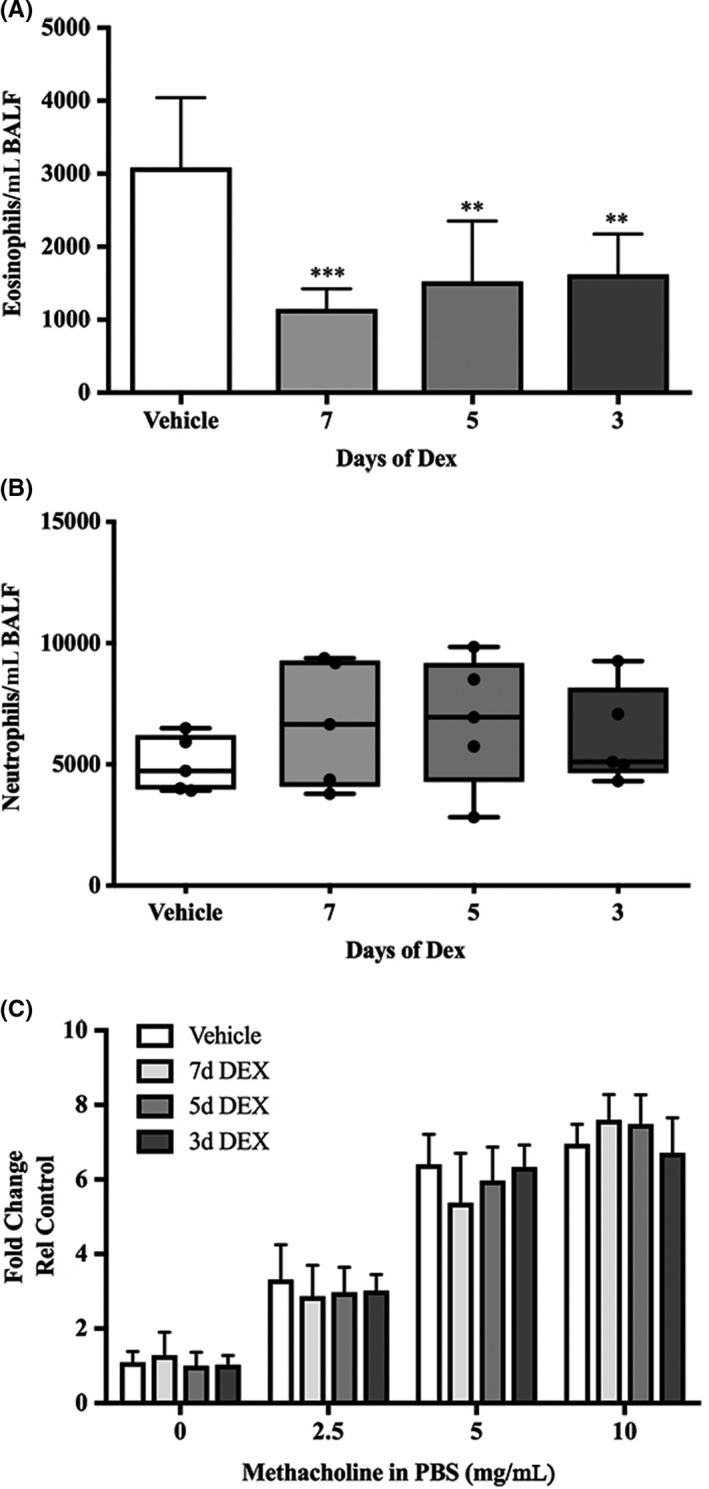
Dexamethasone reduces eosinophil count but does not affect neutrophils or suppress BHR. Differential inflammatory cell counts for A, Eo, eosinophils and B, Neu, neutrophils in BALF after a time‐course Dex treatment (grey bars) vs sham treatment (white bars) in Ccsp/Il13 mice. C, Airway resistance in response to increasing concentrations of methacholine (0‐10 mg/mL). Non‐parametric data are shown as box plots with medians and 25th to 75th percentiles, and whiskers representing minimum and maximum values with all data points shown; parametric data are shown as mean + SD. N = 5 per group from two independent experiments. Statistical analysis was performed using one‐way ANOVA or Kruskal‐Wallis test with Dunn's test for correction for multiple comparisons. **P < .01, ***P < .001
3.4. Corticosteroids dampen “type 2,” but not pro‐neutrophilic cytokine responses
Analysis of the effects of Dex on pulmonary gene expression revealed that Ccl11, Postn and SerpinB2 mRNAs were each suppressed by Dex treatment (Figure 4A‐C) and a significant suppression in CCL11 protein release was evident after only 3 days of Dex treatment (Figure 4D). Changes in Muc5ac mRNA were also evident after 7 days of corticosteroid treatment (Figure S6). In contrast, Cxcl1/Kc, Cxcl2 and Csf3 remained unchanged by Dex, regardless of duration of treatment (Figure 4E‐G). Similarly, protein levels of CXCL1/KC (Figure 4H) and CXCL2 in either BALF or lung lysate were unaffected by Dex treatment (data not shown).
Figure 4.
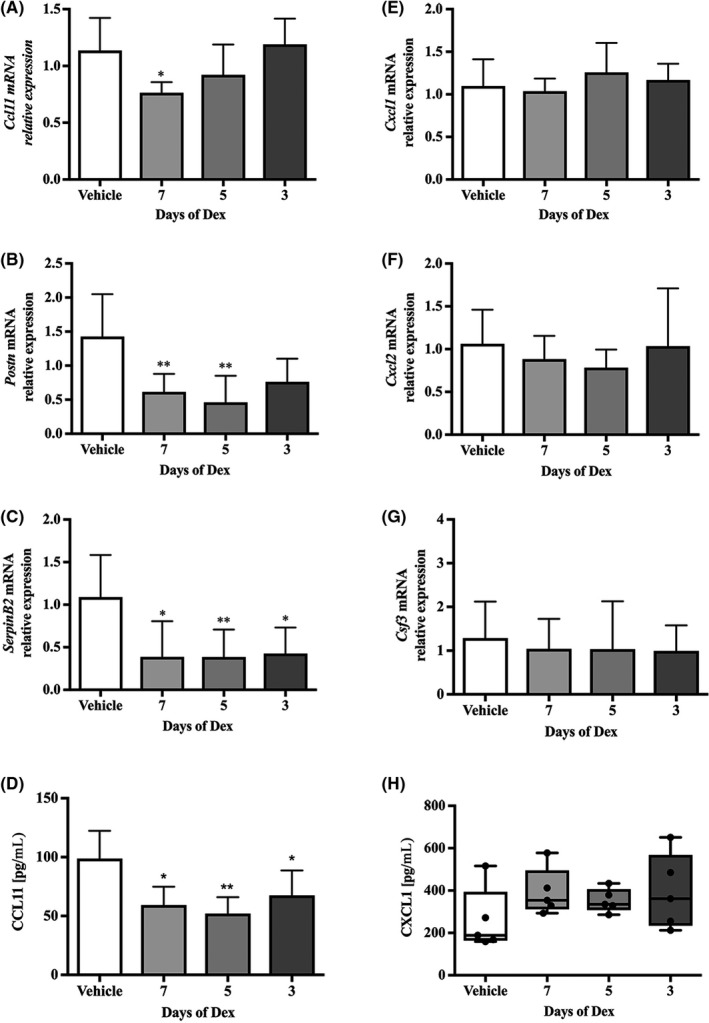
The type 2 lung inflammation signature is reduced by dexamethasone, but not the pro‐neutrophilic responses. Relative mRNA expression in whole‐lung lobe lysates from Ccsp/Il13 mice after induction of IL‐13 for 7 days and dexamethasone treatment for all 7, the final 5 or 3 days (grey bars) (n = 5) vs vehicle‐treated Ccsp/Il13 mice (white bars) (n = 5). A, Ccl11, B, Postn, C, Serpinb2, E, Cxcl1/KC, F, Cxcl2 and G, Csf3. ELISAs for D, CCL11 and H, CXCL1 protein levels in BALF. Parametric data are expressed as mean + SD and non‐parametric data as box plots showing medians and 25th to 75th percentiles, and whiskers representing minimum and maximum values with all data points shown. Data are from two independent experiments. Statistical analysis was performed using one‐way ANOVA or Kruskal‐Wallis test with Dunn's test with correction for multiple comparisons. *P < .05, **P < .01
3.5. The EGFR is a mediator of IL‐13–induced corticosteroid‐insensitive responses in vitro and in vivo
We have previously reported that EGFR activation drives corticosteroid‐insensitive release of IL‐8 from human bronchial epithelial cells.33, 34 As IL‐13 has been shown to drive epithelial proliferation via an EGFR/TGF‐α autocrine loop,35 we explored whether EGFR activation could contribute to the responses observed in the IL‐13 mice. Following 3Rs principles, we initially pursued in vitro mechanistic studies using cultures of murine tracheal epithelial cells (mTECs) from control and Ccsp/Il13 mice. Initial characterization showed that, regardless of genotype, the cultures developed a TER > 1000 Ω.cm2 (Figure S7). When DOX was applied to the cultures, it induced IL‐13 mRNA expression and IL‐13 protein release only in the Ccsp/Il13 mTECs (Figure S8A‐C) and this caused a significant drop in TER at 24h, consistent with the known effect of IL‐13 on ionic permeability 36 (Figure S8D). Induction of IL‐13 also resulted in an increase in Ccl11 mRNA expression which was suppressed by Dex (Figure S9).
Having validated the model, we evaluated the effect of IL‐13 on induction of the pro‐neutrophilic cytokine genes and compared the inhibitory effect of Dex or the highly selective EGFR tyrosine kinase inhibitor, AG1478.37 Expression of Cxcl1/Kc mRNA was significantly increased when IL‐13 was induced with DOX (Figure 5A) and this was accompanied by a fourfold induction of basolateral CXCL1/KC protein secretion (Figure 5B). In the control mTECs, DOX treatment failed to induce Cxcl1/Kc mRNA or CXCL1/KC protein; however in the presence of EGF, Cxcl1/CXCL1 expression was increased (Figure 5C,D), consistent with a role for the EGFR in driving expression of this neutrophilic chemokine. As observed in vivo, Dex had no significant effect on Cxcl1/Kc mRNA or CXCL1/KC protein release from the IL‐13–expressing cultures (Figure 5A,B). Similarly, EGF‐induced release of CXCL1/KC protein was insensitive to inhibition by Dex (Figure 5C,D). In contrast, the EGFR inhibitor AG1478 significantly reduced both apical and basolateral release of CXCL1/KC from IL‐13‐ or EGF‐stimulated cells (Figure 5B,D). Similar results were obtained for Cxcl2/CXCL2 mRNA and protein expression induced by IL‐13 expression or by EGF treatment of cells from control mice (Figure S10A‐D): these were not affected by Dex, but were suppressed by the EGFR inhibitor.
Figure 5.
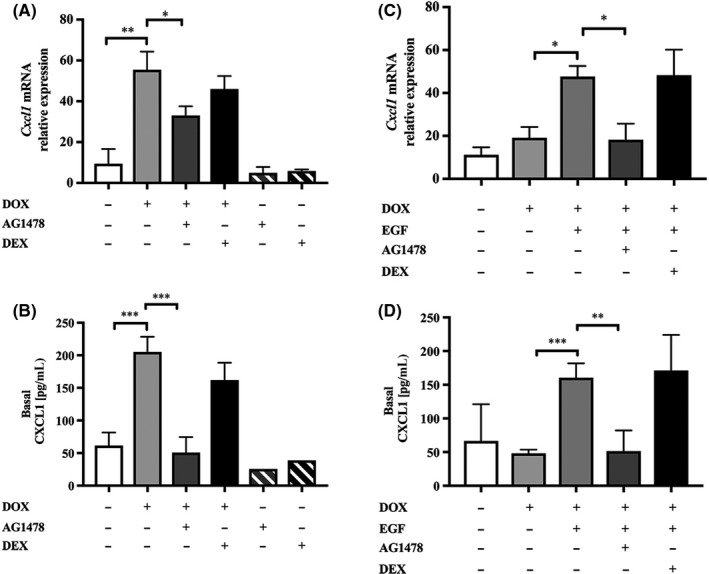
IL‐13 induces expression of Cxcl1/CXCL1 that can be blocked by the EGFR inhibitor AG1478 in mTEC cultures. A, Relative Cxcl1 mRNA expression and B, CXCL1 protein expression in Ccsp/Il13 mTECS treated, where indicated, with DOX (to induce IL‐13), AG1478 or Dex for 72 h. C, D, Relative Cxcl1/CXCL1 mRNA and protein expression, respectively, in control mTECs treated, where indicated, with DOX, EGF, AG1478 or Dex. Data are expressed as mean + SD. Experiments were performed in duplicate and are from three independent experiments. Statistical analysis was performed using one‐way ANOVA with Dunn's multiple comparison. *P < .05, **P < .01, ***P < .001
Based on our in vitro findings, we investigated whether EGFR inhibition in vivo had the ability to modulate the corticosteroid‐refractory responses observed in the Ccsp/Il13 transgenic mouse model. Thus, IL‐13 was induced with DOX and the animals were treated with AG1478 and/or Dex for 7 days. This revealed that AG1478 was able to suppress BHR, airway neutrophilia and pro‐neutrophilic cytokine expression (Figure 6A‐C), while it had no significant effect on airway eosinophilia and type‐2 biomarker expression (Figure 6D,E). In contrast, Dex while significantly suppressed the type 2 responses (eosinophils and CCL11 release), the combination of AG1478 and Dex significantly suppressed all responses.
Figure 6.
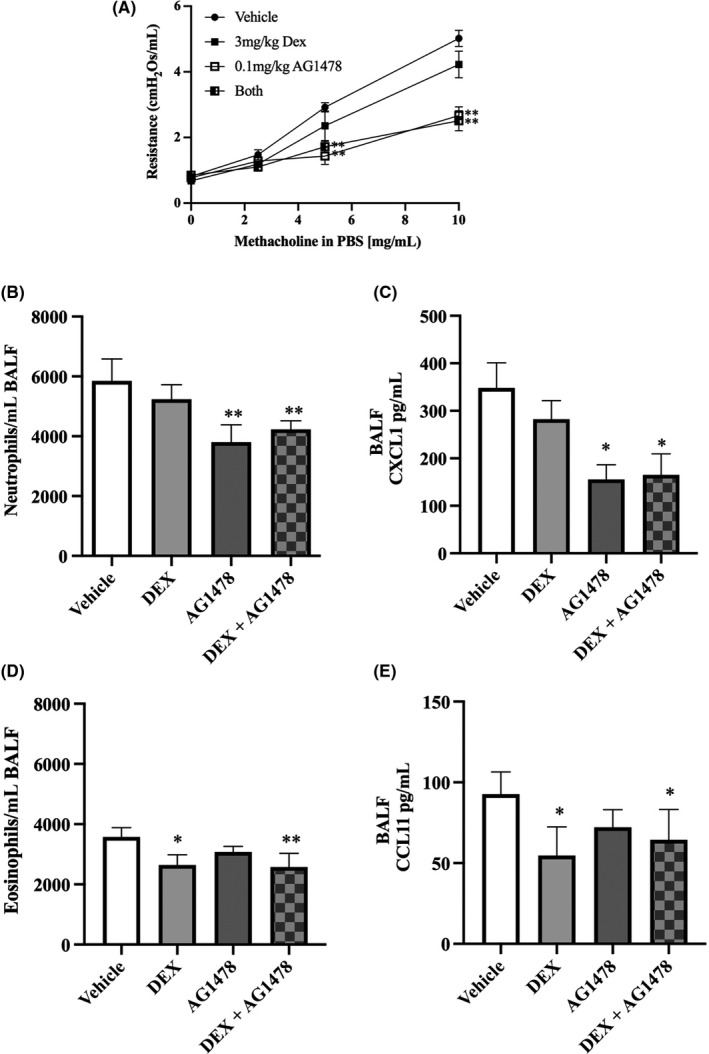
The EGFR inhibitor AG1478 blocks the pro‐neutrophilic responses caused by IL‐13 expression in vivo. Ccsp/Il13 mice were fed DOX for 7 d to induce IL‐13 and treated with dexamethasone (light grey bars), AG1478 (dark grey bars), dexamethasone + AG1478 (hatched bars) or vehicle control (white bars). A, Airway resistance in response to increasing concentrations of methacholine (0‐10 mg/mL). B and D, Differential inflammatory cell counts in BALF for neutrophils and eosinophils, respectively. C and E, CXCL1 or CCL11 protein expression, respectively, measured by ELISA in whole‐lung lobe lysates; data are shown as mean + SD. N = 6 per group from two independent experiments. Statistical analysis was performed using one‐way ANOVA. *P < .05, **P < .01
3.6. Transcriptomic analysis of U‐BIOPRED cohort
To explore the relevance of our findings in human asthma, epithelial transcriptomic data from healthy (n = 44), mild‐to‐moderate asthma (n = 36) and severe asthma (n = 61) were clustered by TDA which provides a general framework to analyse high dimensional data in a manner that is insensitive to the particular metric chosen and provides dimensionality reduction and robustness to noise.31, 38 It has the advantage over standard clustering methodologies in that it provides geometric representations of multi‐dimensional data and it is often possible to find subgroups in data sets that traditional methodologies fail to find. The clinical, pathobiological data and log2‐transformed expression levels of ERBB receptors and EGFR ligands were then applied as metadata (Figure 7A,B). This revealed a cluster of severe asthmatics with neutrophilia and varying numbers of eosinophils (Figure 7A). While EGFR expression was down‐regulated in this cluster, ERBB3 and several EGFR ligands, including heparin‐binding EGF (HB‐EGF), epiregulin (EREG) and EGF, were up‐regulated (Figure 7B). This severe asthma cluster was associated with increased expression of CSF3, CXCL2 and CXCL8, genes usually associated with an “IL‐17 signature”,39 although it was evident that expression of these genes was heterogeneous across the cluster. Furthermore, within this severe asthma cluster it was possible to identify two sub‐clusters: one was characterized by highest IL13 (Figure 7C), ERBB3 and HB‐EGF expression; the other sub‐cluster was characterized by highest amphiregulin (AREG) expression suggesting that there may be distinct sub‐phenotypes of severe asthma that can be defined by different ERBB receptor and ligand combinations. Further analysis using previously defined genes associated with the IL‐13 or IL‐17 signatures18 showed that EGFR and ERBB2 were significantly down‐regulated and ERBB3 up‐regulated in severe asthmatics with “IL‐13” or “IL‐17 high” phenotypes but not in those subjects with an “IL‐13/IL‐17 low” phenotype. In contrast, in the latter group, ERBB4 was the only ERBB family member to show significant modulation (Table 1). For the EGFR ligands, HBEGF was significantly increased in the “IL‐13 high” group whereas AREG, EGF and EREG were significantly up‐regulated in the “IL‐17 high” group.
Figure 7.
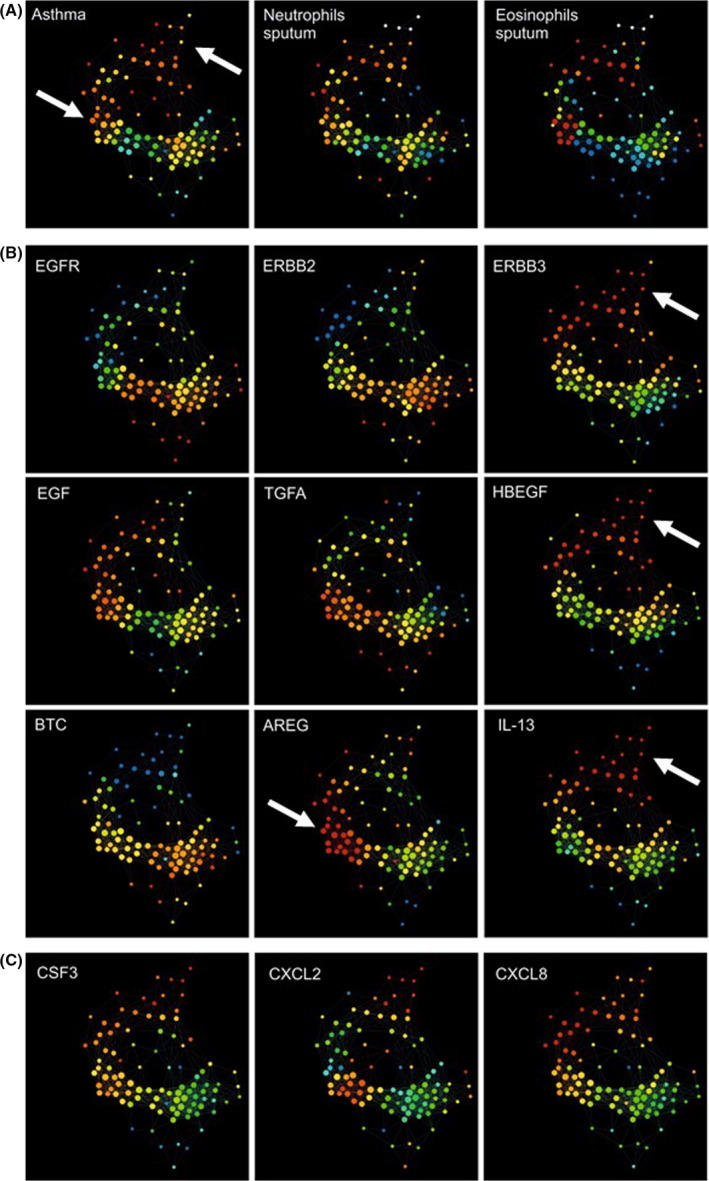
Topological data analysis of gene expression obtained from bronchial brushings from the U‐BIOPRED cohort. A TDA network was constructed using gene expression data obtained from bronchial brushings from non‐smoking severe asthmatics (n = 61), mild‐to‐moderate asthmatics (n = 36) and healthy controls (n = 44). As indicated, metadata were then applied for (A) asthma severity, sputum neutrophils or eosinophil counts; (B) EGFR, ERBB2, ERBB3, EGFR ligands or IL13; and (C) CSF3, CXCL2 or CXCL8. Nodes are coloured by intensity from blue (low) to red (high). Arrows point to the regions of interest referred to in the Results sections
Table 1.
Comparison of ERBB receptor and ligand expression in “IL‐13 high” and “IL‐17 high” asthma clusters relative to health in the U‐BIOPRED cohort
| n | “IL‐13 low/IL‐17 low” | “IL‐17 high” | “IL‐13 high” | |||
|---|---|---|---|---|---|---|
| 53 | 22 | 9 | ||||
| Log2 fold change | P | Log2 fold change | P | Log2 fold change | P | |
| EGFR | −0.017 | .1761 | −0.079 | .0001 | −0.097 | .0011 |
| ERBB2 | −0.007 | .2059 | −0.031 | .0006 | −0.026 | .0157 |
| ERBB3 | 0.034 | .0953 | 0.083 | .0043 | 0.098 | .0019 |
| ERBB4 | 0.096 | .0008 | −0.090 | .0501 | −0.059 | .2681 |
| EGF | 0.007 | .6470 | 0.076 | .0003 | 0.049 | .0621 |
| TGFA | 0.006 | .7253 | −0.008 | .7356 | −0.027 | .4088 |
| HBEGF | 0.015 | .2612 | 0.027 | .1056 | 0.081 | .0002 |
| BTC | −0.027 | .1971 | −0.124 | .0000 | −0.071 | .0359 |
| AREG | 0.040 | .3029 | 0.225 | .0011 | 0.043 | .5383 |
| EREG | 0.022 | .5274 | 0.344 | .0019 | 0.105 | .0863 |
Numbers are log2 fold change relative to expression in healthy participants; significant changes are highlighted in bold. The definition of “IL‐13 high” and “IL‐17 high” was based on that used in.18
The modulation of several ERBB receptors and ligands in human asthma led us to examine their expression in the mouse model. This revealed that corticosteroid treatment of the IL‐13–expressing mice increased expression of Egfr, Erbb3, Egf, Hbegf and Areg, whereas Erbb2 was unaffected (Figure 8A‐F). As the low levels of EGFR in the U‐BIOPRED cluster appeared paradoxical, we postulated that EGFR activation may cause a negative feedback response to lower its mRNA expression. Therefore, we investigated whether this happened in mTEC cultures by studying IL‐13–induced Muc5ac expression in vitro. Under these conditions, IL‐13 stimulated Muc5ac in an EGFR‐dependent way (Figure S11A) and this was accompanied by suppression of Egfr mRNA expression (Figure S11B); since inhibition of EGFR activation with AG1478 prevented the suppression of Egfr mRNA (Figure S11B), these results are consistent with maintenance of EGFR signalling even when mRNA levels are decreased via a feedback loop.
Figure 8.
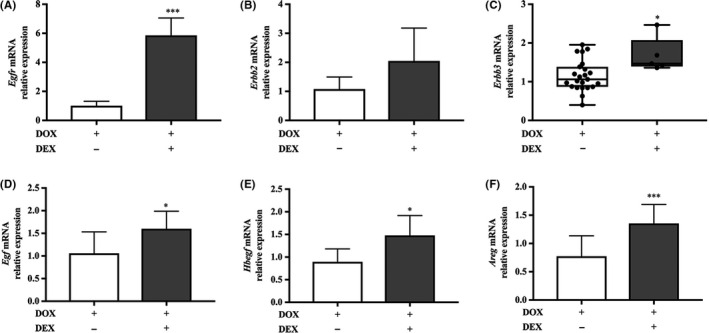
Erbb receptor and ligand mRNA expression in the IL‐13–expressing mouse model. Relative mRNA expression in whole‐lung lobe lysates from Ccsp/Il13 mice after induction of IL‐13 for 7 d (white bars) (n = 23) vs concurrent dexamethasone (grey bars) treatment for 7 d (n = 6) for A, Egfr, B, Erbb2, C, ErbB3 D, Egf, E, Hbegf and F, Areg. Parametric data are expressed as mean + SD and non‐parametric data as box plots showing medians and 25th to 75th percentiles, and whiskers representing minimum and maximum values with all data points shown. Data are from three independent experiments. Statistical analyses were performed using Student's t test or Mann‐Whitney test. *P<0.05, ***P < .001
4. DISCUSSION
Severe corticosteroid‐refractory asthma is a significant unmet medical. The disease is usually classified based on inflammatory cell profiles and related pathways, giving rise to the dichotomous definitions of “type‐2” and “non‐type 2” asthma with, by inference, distinct underlying mechanisms. In this study, we have shown that the lungs of transgenic mice expressing the classical type 2 pro‐allergic mediator, IL‐13, exhibited mixed eosinophilic and neutrophilic inflammation and increased expression of both type‐2 and non‐allergic, “type‐17” markers, even though Il‐17 was not elevated in the lungs of the mice. We also found that the characteristic type‐2 biomarkers can be significantly suppressed (but not ablated) by corticosteroid treatment whereas BHR, neutrophilia and pro‐neutrophilic biomarkers were corticosteroid‐refractory. Through in vitro mechanistic studies, we demonstrated that these corticosteroid‐refractory IL‐13–induced pro‐neutrophilic responses were sensitive to inhibition of the EGFR and that in vivo inhibition of EGFR signalling in the IL‐13 mouse model suppressed pro‐neutrophilic cytokine expression and reduced airway neutrophilia and BHR. To relate these findings to human asthma, transcriptomic analysis of epithelial brushings from human volunteers identified a cluster of severe asthmatics displaying eosinophilia and neutrophilia with increased expression several EGFR ligands and ERBB3 whose protein product is known to form heterodimers and signal with EGFR.40 Importantly, epithelial expression of IL13 was found within a sub‐cluster of these asthmatic subjects, suggesting that our transgenic mouse model, which expresses IL‐13 in the airway epithelium, mirrors a sub‐phenotype of the human disease. Together, our data suggest that epithelial expression of the pro‐allergic cytokine, IL‐13, can drive corticosteroid‐resistant asthma which is mediated by epithelial EGFR/ERBB signalling driving a gene signature and phenotypic responses that are more classically associated with IL‐17 and Th17 immune responses. Furthermore, our data suggest that distinct “ype 17” sub‐phenotypes of severe asthma may arise through involvement of different ERBB receptor and ligand combinations.
A range of pathways have been implicated in the pathogenesis of corticosteroid‐refractory asthma, including increased activity of kinases, which phosphorylate the glucocorticoid receptor (GR) and prevent its nuclear translocation, and oxidative stress which inhibits the activity of histone deacetylase 2 (HDAC2) which mediates the actions of corticosteroids on pro‐inflammatory cytokine expression.41, 42 Other studies have suggested that the neutrophil‐high severe asthma phenotype which is poorly responsive to high‐dose corticosteroids43 might be a consequence of the treatment itself, since corticosteroids promote neutrophil survival.44, 45 Other studies have associated the neutrophilic phenotype with bacterial colonization or infection 46 and activation of IL‐17–producing helper T cells.47 However, in a randomized, double‐blind, placebo‐controlled study of brodalumab, a human anti‐IL‐17 receptor monoclonal antibody, in moderate‐to‐severe asthma there was no significant therapeutic effect.48 Thus, the mechanisms of corticosteroid‐refractory disease remain poorly understood, and as a result, no specific treatment is available for this difficult‐to‐treat group of patients.42
Previous studies have shown that IL‐13 can promote airway neutrophilia. For example, direct instillation of IL‐13 into the tracheae of rats results in neutrophilia driven by chemokines including IL‐8 49 while transgenic mice expressing IL‐13 have been shown to exhibit chronic inflammation involving both eosinophils and neutrophils, as well as lung remodelling.25, 50, 51 Of note, while eosinophilic inflammation caused by expression of the IL‐13 transgene diminished rapidly after removal of doxycycline, macrophages, neutrophils, lymphocytes and remodelling changes all persisted in the lung 3‐4 weeks after IL‐13 expression ceased. This was accompanied by sustained expression of genes, including multiple chemokines, that are likely involved in regulating the inflammatory responses initially stimulated by IL‐13.25 Consistent with this, we have demonstrated that IL‐13 drives pro‐neutrophilic “type 17” inflammatory gene and protein expression including Cxcl1/Kc, Cxcl2 and Csf3 and that amounts of CXCL1 were strongly correlated with both IL‐13 levels and neutrophil numbers in BALF. Beyond this, in the current study, we have identified that these responses, as well as BHR, are refractory to corticosteroid treatment. This contrasts with other murine models of allergic airway disease which are highly sensitive to inhibition by corticosteroids unless specifically manipulated by transfer of immune cells or by infection,52 but is similar to observations made in asthmatic patients who exhibit poor glucocorticoid responsiveness and have higher levels of serum IL‐8.53
We have previously shown that EGFR protein expression is increased in the bronchial epithelium of asthmatic patients according to disease severity54; furthermore, EGFR expression correlates with epithelial IL‐8 levels and the extent of neutrophilic inflammation in severe asthma.33, 34 Although involvement of the EGFR in inflammatory responses has been demonstrated previously using murine models of ovalbumin55 or house dust mite‐induced allergic inflammation,56, 57 both of these models are corticosteroid sensitive and so cannot address those corticosteroid‐refractory responses that are important in severe asthma. Using the IL‐13 transgenic mouse model, we identified a subset of IL‐13–driven corticosteroid‐refractory pro‐neutrophilic responses, and by culturing murine epithelial cell cultures in vitro, we identified that IL‐13 can induce similar pro‐neutrophilic cytokine responses that are refractory to corticosteroids, yet they can be suppressed by EGFR inhibition. We then confirmed these findings in the IL‐13 transgenic mouse model by showing that EGFR inhibition with AG1478 in vivo can prevent IL‐13–induced corticosteroid‐refractory pro‐neutrophilic inflammatory responses, airway neutrophilia and BHR. One key difference between the corticosteroid‐sensitive models of allergic airways inflammation and the transgenic mouse model is that Il13 is expressed within the bronchial epithelium of the transgenic mouse, rather than in immune cells, as in the allergic models. As discussed below, analysis of the U‐BIOPRED cohort also identified IL13 expression in the bronchial epithelium of a subgroup of severe asthmatic subjects, suggesting that the cellular provenance of IL‐13 may be an important determinant of disease activity.
The relevance of our findings in human asthma was explored using bronchial epithelial cell transcriptomic data from the U‐BIOPRED cohort. TDA analysis revealed that EGFR and ERBB2 were significantly down‐regulated while ERBB3 and a number of EGFR ligands, as well as IL13, were up‐regulated in severe asthmatics with mixed eosinophilic and neutrophilic inflammation, and this was accompanied by up‐regulation of CSF3, CXCL2 and CXCL8.39 Although the low level of EGFR in this severe asthma cluster appears paradoxical, studies in cancer cells have shown that regulation of EGFR mRNA and protein expression is complex and can occur at multiple transcriptional and post‐transcriptional levels in a cell type–specific fashion.58, 59 In our in vitro studies, we showed that IL‐13–dependent EGFR activation suppressed Egfr mRNA expression suggesting feedback regulation of EGFR expression upon its activation in bronchial epithelial cells. As EGFR protein levels are increased in severe asthma,54 it would be of interest to study EGFR protein expression in the U‐BIOPRED cohort and to determine whether post‐translational mechanisms are also important for EGFR regulation.
A key finding from our analysis of the U‐BIOPRED data was that IL13 mRNA expression was present in bronchial epithelial cells within a sub‐cluster of the severe asthma cluster where genes usually associated with an “IL‐17” gene signature39 were also up‐regulated. IL‐13 expression is normally associated with immune cells such as type 2 innate lymphoid cells (ILC2s), T helper 2 (Th2) cells mast cells and basophils, with ILC2s and Th2 cells being considered major sources of this cytokine,60 especially in type 2 asthma.61 However, induction of IL‐13 mRNA and protein has also been observed in wounded bronchial epithelial cells in vitro,62 and in our own unpublished work, we also have observed a significant increase in IL‐13 protein release from primary bronchial epithelial cells in response to challenge with double‐stranded RNA (data not shown). In the published work, release of IL‐13 following wounding was shown to enhance epithelial repair via HB‐EGF.62 Thus, it is significant that IL13 expression in the U‐BIOPRED severe asthma cluster closely mirrored epithelial HBEGF expression suggesting a wound healing response in this subgroup of patients involving an IL‐13/HB‐EGF/EGFR axis. Of note, it has been shown that IL‐13 receptor α2 (IL‐13Rα2) can stimulate epithelial cell HB‐EGF production via TMEM219 and that TMEM 219 also contributes to optimal binding of IL‐13 to IL‐13Rα2.63 Unlike the Type II IL‐4 receptor complex which is a target Dupilumab 22 and binds both IL‐4 and IL‐13, IL‐13Rα2 is a high‐affinity receptor for IL‐13, but not IL‐4.64 Together, these observations may help to explain the so‐called “IL‐13 paradox”65 that while IL‐13 is involved in almost all aspects of asthma pathobiology, clinical trials using antibodies targeting IL‐13 have failed to demonstrate clinical benefit66 or any corticosteroid‐sparing effect.67 Thus, if IL‐13 is required for epithelial repair, potentially via IL‐13Rα2, neutralization of its effect on epithelial cells may prolong epithelial injury and impairment of epithelial barrier function, opposing any beneficial effects of the treatment on type 2 inflammation. Further work is required to investigate these possibilities.
In addition to the IL13 sub‐cluster, we also identified a second sub‐cluster within the neutrophilic and eosinophilic severe asthma cluster which was characterized by high expression of AREG, EGF and EREG. Given that previous studies have identified an “IL‐17 high” severe asthma phenotype which exhibits expression of genes that are reported as altered in psoriasis lesions,18 it is significant that transgenic expression of AREG in murine skin causes a psoriasis phenotype characterized by marked epidermal hyperplasia, accompanied by neutrophilia and significantly increased CD4+ T cell infiltration.68, 69 Both AREG and HB‐EGF interact with heparan sulphate proteoglycans and with members of the tetraspanin family of membrane‐associated proteins which can regulate their distribution, bio‐availability and action on target cells and can also serve as cell surface co‐receptors, facilitating ligand‐receptor interactions.70 Thus, differential regulation of these two EGFR ligands offers the potential to fine‐tune their interaction with key target cells to drive distinctive EGFR/ERBB mediated responses that, in turn, may give rise to differing sub‐phenotypes of severe asthma. It is also noteworthy that the “IL‐13 low/IL‐17 low” severe asthmatic group (Table 1) was the only group to show a significant up‐regulation of ERBB4, whose function appears critically involved in reactions that affect cell fate.71 Its role within this group of severe asthmatics merits further investigation.
Based on the findings from the U‐BIOPRED cohort, further analysis of the IL‐13 expressing mouse model revealed that corticosteroid treatment caused significant modulation of the EGFR ligand family including Hbegf and Areg, as well as Erbb3. Unlike other family members, ERBB3 is considered “kinase dead” and requires heterodimerization with another family member such as EGFR or ERBB2 for phosphorylation; otherwise, it is refractory to ligand‐induced activation.72 Phosphorylated ERBB3 has multiple binding sites for phosphatidyl inositol‐3 kinase (PI3K)73 whose pathway is central to the development of BHR and inflammation.74 Furthermore, activation of PI3Kδ has been implicated in corticosteroid resistance as it causes Akt activation and inactivation of HDAC2, one of the mechanisms associated with corticosteroid resistance.41 In support of this suggestion, EGFR signalling is associated with increased PI3Kδ/Akt activation in ovalbumin‐induced airways inflammation.55 Our novel finding that ERBB3 is up‐regulated in severe asthma requires further investigation as it suggests that EGFR/ ERBB3 heterodimers may contribute to corticosteroid‐refractory asthma. As EGFR and corticosteroids also have beneficial effects on epithelial repair and barrier function,54 discriminating between pro‐inflammatory and pro‐repair functions for these pathways, and the role(s) of individual ligands and ERBB receptor heterodimers, should help to tailor more effective therapies for severe asthma.45 Such studies would offer the potential of exploiting the array of small molecule drugs and antibodies that have been developed for cancer therapy.71, 75
In summary, our study suggests that the prototypic type‐2 mediator, IL‐13, can give rise to both eosinophilic type‐2 and neutrophilic “type‐17” stereotypic responses, the latter being corticosteroid insensitive and mediated by epithelial EGFR signalling rather than classical immunological Th17 signalling. Based on our current findings, the IL‐13 transgenic mouse model should enable further understanding and dissection of the molecular pathways involved in corticosteroid‐refractory pathways, especially those involving EGFR/ERBB signalling in promotion of mixed granulocytic or neutrophilic inflammation and distinguishing them from beneficial pro‐repair pathways within the epithelium. This should enhance the development of new treatments that target this sub‐phenotype(s) of severe asthma.
CONFLICT OF INTEREST
The authors declare the following conflicts of interest: DED and RD report personal fees from Synairgen, which is outside the submitted work. RD also reports receiving fees for lectures at symposia organized by Novartis, AstraZeneca and TEVA, consultation for TEVA and Novartis as member of advisory boards, and participation in a scientific discussion about asthma organized by GlaxoSmithKline. All are outside the submitted work. All other authors have nothing to declare.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
ACKNOWLEDGEMENTS
We would like to thank Professor Jeffrey A. Whitsett and Cincinnati Children's Hospital Medical Center for the transgenic IL‐13 mice. We want to thank the animal facility at the University of Southampton for husbandry of the mice. We also thank Professor Colin D. Bingle at the University of Sheffield for teaching JFCK how to extract murine tracheal epithelial cells (mTEC) and grow them in culture. This work was supported by a Medical Research Council UK Clinician Scientist Fellowship to HMH (G0802804), a grant from the Asthma, Allergy & Inflammation (AAIR) Charity to ERD & HMH and a Medical Research Foundation/Asthma UK grant to HMH and DED (MRFAUK‐2015‐322). JMP is funded by the European Respiratory Society (Fellowship LTRF 2017), Association Régionale pour l'Aide aux Insuffisants Respiratoires de Champagne‐Ardenne (ARAIRCHAR), Association Nationale de Formation Continue en Allergologie (ANAFORCAL), Association des Allergologues de Champagne‐Ardenne (ASALCAR) and Association des Pneumologues de Champagne‐Ardenne (APCA).
Davies ER, Perotin J‐M, Kelly JFC, Djukanovic R, Davies DE, Haitchi HM; the U‐BIOPRED Study Group . Involvement of the epidermal growth factor receptor in IL‐13–mediated corticosteroid‐resistant airway inflammation. Clin Exp Allergy. 2020;50:672–686. 10.1111/cea.13591
DED and HMH are joint senior authors.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available either in the GEO repository (GSE76226) or can be obtained from the corresponding author upon reasonable request.
REFERENCES
- 1. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet. 2018;391(10122):783‐800. [DOI] [PubMed] [Google Scholar]
- 2. Bel EH, Sousa A, Fleming L, et al. Diagnosis and definition of severe refractory asthma: an international consensus statement from the Innovative Medicine Initiative (IMI). Thorax. 2011;66(10):910‐917. [DOI] [PubMed] [Google Scholar]
- 3. Wener RR, Bel EH. Severe refractory asthma: an update. Eur Respir Rev. 2013;22(129):227‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Israel E, Reddel HK. Severe and difficult‐to‐treat asthma in adults. N Engl J Med. 2017;377(10):965‐976. [DOI] [PubMed] [Google Scholar]
- 5. Wenzel SE, Balzar S, Ampleford E, et al. IL4R alpha mutations are associated with asthma exacerbations and mast cell/IgE expression. Am J Respir Crit Care Med. 2007;175(6):570‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716‐725. [DOI] [PubMed] [Google Scholar]
- 7. Gauthier M, Ray A, Wenzel SE. Evolving concepts of asthma. Am J Respir Crit Care Med. 2015;192(6):660‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaw DE, Sousa AR, Fowler SJ, et al. Clinical and inflammatory characteristics of the European U‐BIOPRED adult severe asthma cohort. Eur Respir J. 2015;46(5):1308‐1321. [DOI] [PubMed] [Google Scholar]
- 9. Teague WG, Phillips BR, Fahy JV, et al. Baseline features of the Severe Asthma Research Program (SARP III) Cohort: differences with age. J Allergy Clin Immunol Pract. 2018;6(2):545–554.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuo CS, Pavlidis S, Loza M, et al. A Transcriptome‐driven analysis of epithelial brushings and bronchial biopsies to define asthma phenotypes in U‐BIOPRED. Am J Respir Crit Care Med. 2017;195(4):443‐455. [DOI] [PubMed] [Google Scholar]
- 11. Kuo CS, Pavlidis S, Loza M, et al. T‐helper cell type 2 (Th2) and non‐Th2 molecular phenotypes of asthma using sputum transcriptomics in U‐BIOPRED. Eur Respir J. 2017;49(2):1602135. [DOI] [PubMed] [Google Scholar]
- 12. Woodruff PG, Boushey HA, Dolganov GM, et al. Genome‐wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104(40):15858‐15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson D, Humbert M, Buhl R, et al. Revisiting Type 2‐high and Type 2‐low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy. 2017;47(2):161‐175. [DOI] [PubMed] [Google Scholar]
- 14. Ricciardolo FL. Revisiting the role of exhaled nitric oxide in asthma. Curr Opin Pulm Med. 2014;20(1):53‐59. [DOI] [PubMed] [Google Scholar]
- 15. Jia G, Erickson RW, Choy DF, et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol. 2012;130(3):pp. 647–54 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2‐high and TH2‐low subtypes of asthma. J Allergy Clin Immunol. 2014;133(2):388‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woodruff PG, Modrek B, Choy DF, et al. T‐helper type 2‐driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ostling J, van Geest M, Schofield JPR, et al. IL‐17‐high asthma with features of a psoriasis immunophenotype. J Allergy Clin Immunol. 2019. [DOI] [PubMed] [Google Scholar]
- 19. Wenzel S, Castro M, Corren J, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium‐to‐high‐dose inhaled corticosteroids plus a long‐acting beta2 agonist: a randomised double‐blind placebo‐controlled pivotal phase 2b dose‐ranging trial. Lancet. 2016;388(10039):31‐44. [DOI] [PubMed] [Google Scholar]
- 20. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate‐to‐severe uncontrolled asthma. N Engl J Med. 2018;378(26):2486‐2496. [DOI] [PubMed] [Google Scholar]
- 21. Rabe KF, Nair P, Brusselle G, et al. Efficacy and safety of dupilumab in glucocorticoid‐dependent severe asthma. N Engl J Med. 2018;378(26):2475‐2485. [DOI] [PubMed] [Google Scholar]
- 22. Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455‐2466. [DOI] [PubMed] [Google Scholar]
- 23. Ramsahai JM, Wark PAB. Appropriate use of oral corticosteroids for severe asthma. Med J Aust. 2018;209:18‐21. [DOI] [PubMed] [Google Scholar]
- 24. Taylor SL, Leong LEX, Choo JM, et al. Inflammatory phenotypes in patients with severe asthma are associated with distinct airway microbiology. J Allergy Clin Immunol. 2018;141(1):pp. 94–103 e15. [DOI] [PubMed] [Google Scholar]
- 25. Fulkerson PC, Fischetti CA, Hassman LM, Nikolaidis NM, Rothenberg ME. Persistent effects induced by IL‐13 in the lung. Am J Respir Cell Mol Biol. 2006;35(3):337‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage. 2012;20(4):256‐260. [DOI] [PubMed] [Google Scholar]
- 27. You Y, Brody SL. Culture and differentiation of mouse tracheal epithelial cells. Methods Mol Biol. 2013;945:123‐143. [DOI] [PubMed] [Google Scholar]
- 28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25(4):402‐408. [DOI] [PubMed] [Google Scholar]
- 29. Kuo CS. Expression data of epithelial brushing from Unbiased BIOmarkers in Prediction of REspiratory Disease outcomes (U‐BIOPRED) Project. Dec 2015 ed. NCBI GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76226.
- 30. Bigler J, Boedigheimer M, Schofield JPR, et al. A severe asthma disease signature from gene expression profiling of peripheral blood from U‐BIOPRED cohorts. Am J Respir Crit Care Med. 2017;195(10):1311‐1320. [DOI] [PubMed] [Google Scholar]
- 31. Lum PY, Singh G, Lehman A, et al. Extracting insights from the shape of complex data using topology. Sci Rep. 2013;3:1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poggioli R, Ueta CB, Drigo RA, Castillo M, Fonseca TL, Bianco AC. Dexamethasone reduces energy expenditure and increases susceptibility to diet‐induced obesity in mice. Obesity (Silver Spring). 2013;21(9):E415‐E420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamilton LM, Torres‐Lozano C, Puddicombe SM, et al. The role of the epidermal growth factor receptor in sustaining neutrophil inflammation in severe asthma. Clin Exp Allergy. 2003;33(2):233‐240. [DOI] [PubMed] [Google Scholar]
- 34. Uddin M, Lau LC, Seumois G, et al. EGF‐induced bronchial epithelial cells drive neutrophil chemotactic and anti‐apoptotic activity in asthma. PLoS ONE. 2013;8(9):e72502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Booth BW, Adler KB, Bonner JC, Tournier F, Martin LD. Interleukin‐13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor‐alpha. Am J Respir Cell Mol Biol. 2001;25(6):739‐743. [DOI] [PubMed] [Google Scholar]
- 36. Saatian B, Rezaee F, Desando S, et al. Interleukin‐4 and interleukin‐13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1(2):e24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267(5205):1782‐1788. [DOI] [PubMed] [Google Scholar]
- 38. Hinks TS, Brown T, Lau LC, et al. Multidimensional endotyping in patients with severe asthma reveals inflammatory heterogeneity in matrix metalloproteinases and chitinase 3‐like protein 1. J Allergy Clin Immunol. 2016;138(1):61‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choy DF, Hart KM, Borthwick LA, et al. T(H)2 and T(H)17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7(301):301ra129. [DOI] [PubMed] [Google Scholar]
- 40. Kennedy SP, Hastings JF, Han JZ, Croucher DR. The under‐appreciated promiscuity of the epidermal growth factor receptor family. Front Cell Dev Biol. 2016;4:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636‐645. [DOI] [PubMed] [Google Scholar]
- 42. Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy. 2014;69(7):817‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156(3 Pt 1):737‐743. [DOI] [PubMed] [Google Scholar]
- 44. Saffar AS, Ashdown H, Gounni AS. The molecular mechanisms of glucocorticoids‐mediated neutrophil survival. Curr Drug Targets. 2011;12(4):556‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nair P, Aziz‐Ur‐Rehman A, Radford K. Therapeutic implications of 'neutrophilic asthma'. Curr Opin Pulm Med. 2015;21(1):33‐38. [DOI] [PubMed] [Google Scholar]
- 46. Green BJ, Wiriyachaiporn S, Grainge C, et al. Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS ONE. 2014;9(6):e100645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakagome K, Matsushita S, Nagata M. Neutrophilic inflammation in severe asthma. Int Arch Allergy Immunol. 2012;158(Suppl 1):96‐102. [DOI] [PubMed] [Google Scholar]
- 48. Busse WW, Holgate S, Kerwin E, et al. Randomized, double‐blind, placebo‐controlled study of brodalumab, a human anti‐IL‐17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188(11):1294‐1302. [DOI] [PubMed] [Google Scholar]
- 49. Shim JJ, Dabbagh K, Ueki IF, et al. IL‐13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L134‐L140. [DOI] [PubMed] [Google Scholar]
- 50. Fulkerson PC, Fischetti CA, Rothenberg ME. Eosinophils and CCR3 regulate interleukin‐13 transgene‐induced pulmonary remodeling. Am J Pathol. 2006;169(6):2117‐2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin‐13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maltby S, Tay HL, Yang M, Foster PS. Mouse models of severe asthma: understanding the mechanisms of steroid resistance, tissue remodelling and disease exacerbation. Respirology. 2017;22(5):874‐885. [DOI] [PubMed] [Google Scholar]
- 53. Zhang J, Bai C. Elevated serum interleukin‐8 level as a preferable biomarker for identifying uncontrolled asthma and glucocorticosteroid responsiveness. Tanaffos. 2017;16(4):260‐269. [PMC free article] [PubMed] [Google Scholar]
- 54. Puddicombe SM, Polosa R, Richter A, et al. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14(10):1362‐1374. [DOI] [PubMed] [Google Scholar]
- 55. El‐Hashim AZ, Khajah MA, Renno WM, et al. Src‐dependent EGFR transactivation regulates lung inflammation via downstream signaling involving ERK1/2, PI3Kdelta/Akt and NFkappaB induction in a murine asthma model. Sci Rep. 2017;7(1):9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Le Cras TD, Acciani TH, Mushaben EM, et al. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L414‐L421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Habibovic A, Hristova M, Heppner DE, et al. DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma. JCI Insight. 2016;1(18):e88811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kesavan P, Mukhopadhayay S, Murphy S, Rengaraju M, Lazar MA, Das M. Thyroid hormone decreases the expression of epidermal growth factor receptor. J Biol Chem. 1991;266(16):10282‐10286. [PubMed] [Google Scholar]
- 59. Seth D, Shaw K, Jazayeri J, Leedman PJ. Complex post‐transcriptional regulation of EGF‐receptor expression by EGF and TGF‐alpha in human prostate cancer cells. Br J Cancer. 1999;80(5–6):657‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gurram RK, Zhu J. Orchestration between ILC2s and Th2 cells in shaping type 2 immune responses. Cell Mol Immunol. 2019;16(3):225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boonpiyathad T, Sozener ZC, Satitsuksanoa P, Akdis CA. Immunologic mechanisms in asthma. Semin Immunol. 2019;46:101333. [DOI] [PubMed] [Google Scholar]
- 62. Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL‐13 by airway epithelial cells enhances epithelial repair via HB‐EGF. Am J Respir Cell Mol Biol. 2008;38(2):153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee CM, He CH, Nour AM, et al. IL‐13Ralpha2 uses TMEM219 in chitinase 3‐like‐1‐induced signalling and effector responses. Nat Commun. 2016;7:12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andrews AL, Holloway JW, Puddicombe SM, Holgate ST, Davies DE. Kinetic analysis of the interleukin‐13 receptor complex. J Biol Chem. 2002;277(48):46073‐46078. [DOI] [PubMed] [Google Scholar]
- 65. Nair P, O'Byrne PM. The interleukin‐13 paradox in asthma: effective biology, ineffective biologicals. Eur Respir J. 2019;53(2):1802250. [DOI] [PubMed] [Google Scholar]
- 66. Zhang Y, Cheng J, Li Y, et al. The safety and efficacy of anti‐IL‐13 treatment with tralokinumab (CAT‐354) in moderate to severe asthma: a systematic review and meta‐analysis. J Allergy Clin Immunol Pract. 2019;7(8):2661–2671.e3 [DOI] [PubMed] [Google Scholar]
- 67. Busse WW, Brusselle GG, Korn S, et al. Tralokinumab did not demonstrate oral corticosteroid‐sparing effects in severe asthma. Eur Respir J. 2019;53(2):1800948. [DOI] [PubMed] [Google Scholar]
- 68. Cook PW, Piepkorn M, Clegg CH, et al. Transgenic expression of the human amphiregulin gene induces a psoriasis‐like phenotype. J Clin Invest. 1997;100(9):2286‐2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li Y, Stoll SW, Sekhon S, et al. Transgenic expression of human amphiregulin in mouse skin: inflammatory epidermal hyperplasia and enlarged sebaceous glands. Exp Dermatol. 2016;25(3):187‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Piepkorn M, Pittelkow MR, Cook PW. Autocrine regulation of keratinocytes: the emerging role of heparin‐binding, epidermal growth factor‐related growth factors. J Invest Dermatol. 1998;111(5):715‐721. [DOI] [PubMed] [Google Scholar]
- 71. Mota JM, Collier KA, Barros Costa RL, et al. A comprehensive review of heregulins, HER3, and HER4 as potential therapeutic targets in cancer. Oncotarget. 2017;8(51):89284‐89306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Steinkamp MP, Low‐Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo‐ and heterointeractions. Mol Cell Biol. 2014;34(6):965‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mujoo K, Choi BK, Huang Z, Zhang N, An Z. Regulation of ERBB3/HER3 signaling in cancer. Oncotarget. 2014;5(21):10222‐10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yoo EJ, Ojiaku CA, Sunder K, Panettieri RA Jr. Phosphoinositide 3‐kinase in asthma: novel roles and therapeutic approaches. Am J Respir Cell Mol Biol. 2017;56(6):700‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein‐tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395‐411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
Data Availability Statement
The data that support the findings of this study are available either in the GEO repository (GSE76226) or can be obtained from the corresponding author upon reasonable request.