

Summary
Background
Chronic psoriasis may require medication adjustments over time.
Objectives
To evaluate the efficacy/safety of tildrakizumab in subgroups from the reSURFACE studies (N = 1862) that received continuous dosing, higher/lower dosing, treatment interruption/reinitiation and initiation.
Methods
Responders [Psoriasis Area and Severity Index (PASI) ≥ 75%] and partial responders (PASI ≥ 50% to < 75%) in Part 3 of the reSURFACE studies (weeks 28–52 or week 64) who received tildrakizumab 200 mg or 100 mg were rerandomized to the same dosage (T100/T100 or T200/T200), a higher/lower dosage (T100/T200 or T200/T100) or placebo (PBO) (T100/PBO or T200/PBO). Patients receiving PBO who relapsed were reinitiated to tildrakizumab. Etanercept (ETN) week‐28 partial responders and nonresponders (PASI < 50%) received tildrakizumab 200 mg (ETN/T200).
Results
Among T100/T100 and T200/T200 week‐28 partial responders, the proportion of patients who achieved as‐observed PASI 75 responses increased over time. Among T100/T200 week‐28 partial responders, PASI 75 responses increased from week 32 (38·5%) to week 52 (63·2%) and remained consistent in T200/T100 week‐28 responders. Among patients who relapsed in the T100/PBO and T200/PBO groups, 86% and 83% of those who reinitiated tildrakizumab achieved PASI 75 by week 64, respectively. Among ETN/T200 week‐28 partial responders, PASI 75 responses (nonresponder imputation) increased from week 32 (24·1%) to week 52 (74·7%). PASI 90, PASI 100 and Physician's Global Assessment responses were consistent with PASI 75 results. Treatment was well tolerated.
Conclusions
Patients generally fared well with tildrakizumab maintenance, reinitiation, dose adjustment or initiation.
What's already known about this topic?
Tildrakizumab demonstrated significant efficacy vs. placebo with a positive safety profile during the first 28 weeks of treatment in two randomized double‐blind trials.
What does this study add?
Treatment scenarios with tildrakizumab, such as long‐term continuous dosing (up to 64 weeks), treatment interruption/reinitiation and switching from another biologic, can be part of the management of plaque psoriasis with a reasonable expectation of efficacy and tolerability.
Short abstract
Plain language summary available online
Recent advances in the treatment of chronic plaque psoriasis have focused on targeting the interleukin (IL)‐23/T helper (Th)17 immunological pathway using the IL‐17A antagonists secukinumab and ixekizumab and the IL‐23p19 antagonists guselkumab and tildrakizumab.1, 2, 3, 4 Tildrakizumab, recently approved by the U.S. Food and Drug Administration and the European Medicines Agency for the treatment of moderate‐to‐severe plaque psoriasis, is a humanized monoclonal antibody that selectively inhibits IL‐23p19.5, 6 Large phase IIb and phase III trials have demonstrated the efficacy and safety of tildrakizumab.4, 7 The reSURFACE 1 and reSURFACE 2 studies were three‐part, randomized controlled phase III studies in which tildrakizumab 100 mg (T100) and 200 mg (T200) were evaluated compared with placebo; reSURFACE 2 also included an active comparator, etanercept (ETN).4 Both tildrakizumab dosages demonstrated significant efficacy vs. placebo (PBO) with a positive safety profile during Part 1 (initial 12 weeks) and Part 2 (subsequent 16 weeks).4
Long‐term treatment and medication adjustments may be needed to maintain disease control of chronic plaque psoriasis. Some medication adjustments that occur in real‐world settings include interruption of treatment/reinitiation of treatment, higher/lower dosing, or switching from an older biologic with an inadequate response to a newer biologic with a different mechanism of action.8, 9 In recognition of these real‐world biologic dosing practices for chronic psoriasis, the objectives of Part 3 of the reSURFACE studies (ClinicalTrials.gov NCT01722331, NCT01729754) were to evaluate (i) the maintenance of efficacy and safety with continuous tildrakizumab dosing, (ii) relapse after treatment interruption and retreatment effect upon relapse, (iii) the impact that adjusting doses (higher or lower) has on efficacy and safety, and (iv) the efficacy and safety of tildrakizumab after switching from ETN.
Materials and methods
Study designs
Data were obtained from two international multicentre, three‐part, double‐blinded, randomized controlled phase III studies [reSURFACE 1 (ClinicalTrials.gov NCT01722331; Merck Protocol 010) and reSURFACE 2 (ClinicalTrials.gov NCT01729754; Merck Protocol 011)].4 Details of both studies have been previously described.4 Briefly, eligible participants were adults aged ≥ 18 years with moderate‐to‐severe chronic plaque psoriasis [body surface area involvement ≥ 10%, Physician's Global Assessment (PGA) score ≥ 3 and Psoriasis Area and Severity Index (PASI) ≥ 12] at baseline who were candidates for phototherapy or systemic therapy. In Part 1 (weeks 0–12) of reSURFACE 1, patients were randomized (2 : 2 : 1) to receive tildrakizumab 200 mg, tildrakizumab 100 mg or PBO subcutaneously. In Part 1 (weeks 0–12) of reSURFACE 2, patients were randomized (2 : 2 : 1 : 2) to receive tildrakizumab 200 mg, tildrakizumab 100 mg, PBO or twice‐weekly ETN 50 mg subcutaneously (Fig. 1). In Part 2 (weeks 12–28) of both studies, patients who received tildrakizumab (or ETN in reSURFACE 2) in Part 1 of the studies continued the same originally randomized treatment (except ETN decreased to once weekly) and patients in the PBO group were rerandomized (1 : 1) to receive either tildrakizumab 200 mg or 100 mg (Fig. 1). At week 28, nonresponders to tildrakizumab and responders to ETN (reSURFACE 2 only) were discontinued (Fig. 1). In Part 3 (weeks 28–64, reSURFACE 1; weeks 28–52, reSURFACE 2) of the studies, responders (PASI ≥ 75%) and partial responders (PASI ≥ 50% to < 75%) who received tildrakizumab 200 mg or 100 mg were rerandomized to continue the same dosage of tildrakizumab, a different tildrakizumab dosage or PBO (reSURFACE 1 only). Tildrakizumab treatment was administered every 12 weeks. Responders in reSURFACE 1 rerandomized to PBO in Part 3 who relapsed were reinitiated to their originally randomized tildrakizumab dosage. ETN partial responders (PASI ≥ 50% to < 75%) and nonresponders (PASI < 50%) in reSURFACE 2 received tildrakizumab 200 mg (Fig. 1). Patients switched from ETN received tildrakizumab at weeks 32, 36 and 48.
Figure 1.

Study designs for (a) reSURFACE 1 and (b) reSURFACE 2. Source of patients from reSURFACE 1 and reSURFACE 2 contributing to pooled subgroups are indicated by blue (T200/T200), green (T100/T100) and red (T100/T200). D/C, discontinued; ETN, etanercept; NR, nonresponders; PBO, placebo; PR, partial responders; R, responders; TIL, tildrakizumab.
The studies were conducted in accordance with the Declaration of Helsinki. Local institutional review boards or ethics panels reviewed and approved the study protocols; written informed consent was signed by all participants prior to conducting study‐related procedures.
Study end points
Results for the efficacy and safety end points in Parts 1 and 2 of the individual reSURFACE studies have been previously reported.4 Efficacy end points included the proportion of patients achieving ≥ 75%, 90% and 100% improvement in PASI (i.e. PASI 75, PASI 90 and PASI 100 responses, respectively), and the proportion of patients achieving PGA response (defined as a PGA score of ‘clear’ or ‘minimal’ with at least a two‐grade reduction from baseline) at week 40, week 52, and/or week 64. Baseline for all efficacy end points was week 0. Safety and tolerability were assessed by clinical review of adverse events (AEs), laboratory tests and vital signs.
Continuous long‐term maintenance dosing was defined as patients who received the same dosage of tildrakizumab from baseline through the end of Part 3 (week 64 in reSURFACE 1 and week 52 in reSURFACE 2); data from the two studies were pooled according to the respective 100 mg or 200 mg tildrakizumab dosage. Relapse was defined as a 50% reduction in maximum PASI response following withdrawal of tildrakizumab treatment. Rebound was defined as worsening of psoriasis over baseline values (PASI > 125%) or new pustular, erythrodermic or more inflammatory psoriasis occurring within 2 months after stopping tildrakizumab.
Prespecified analyses of efficacy and safety end points were conducted in the subpopulations of patients with continuous dosing (T100/T100 and T200/T200, data pooled from both studies), patients switched to PBO (T100/PBO and T200/PBO) in reSURFACE 1, and in patients with dosage adjustment in either study [T100/T200 (data pooled from both studies) and T200/T100 (reSURFACE 2 only) (Table S1; see Supporting Information)]. End points were analysed post hoc in partial responders and nonresponders switched from ETN to tildrakizumab (ETN/T200) in reSURFACE 2 (Fig. 1).
Statistical analysis
The efficacy population included the full analysis set population, and the safety population included the all‐participants‐as‐treated population (i.e. patients who entered Part 3 and received at least one dose of study medication). In the efficacy population, patients were grouped based on allocated treatment, whereas patients were grouped based on the actual treatment in the safety population. Descriptive summary statistics were calculated for all end points. As per the protocols, data for Part 3 study end points were as‐observed (i.e. no imputation for missing data or nonresponders), except in the analysis of efficacy outcomes in ETN partial responders and nonresponders who switched to tildrakizumab 200 mg where nonresponder imputation was used. Post hoc nonresponder imputation analyses of Part 3 end points from the individual studies are provided in Table S2 (see Supporting Information).
Results
Patient disposition
Overall, 1862 patients were randomized to the studies; 1770 patients completed Part 1 and 1671 patients completed Part 2.4 A total of 676 patients and 794 patients participated in Part 3 of reSURFACE 1 and reSURFACE 2, respectively; 638 patients (94·4%) in reSURFACE 1 and 756 patients (95·2%) in reSURFACE 2 completed Part 3 of each study. Baseline demographic and disease characteristics for patients entering Part 3 were generally similar among the dosing subgroups and to the full randomized patient populations in reSURFACE 1 and 2 (Table 1).4
Table 1.
Baseline demographics and disease characteristics of all randomized patients and patients who entered Part 3
| reSURFACE 1 | reSURFACE 2 | Patients who entered Part 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All patients, N = 772 | All patients, N = 1090 | Continuous TIL dosinga | TIL withdrawalb | TIL dose adjustment | Switch to TIL from ETNc | ||||
| T100/T100, n = 40 | T200/T200, n = 102 | T100/PBO, n = 114 | T200/PBO, n = 119 | T100/T200, n = 40a | T200/T100, n = 110c | ETN/T200, n = 120d | |||
| Male sex, n (%) | 533 (69·0) | 779 (71·5) | 32 (80) | 69 (67·6) | 73 (64·0) | 95 (79·8) | 24 (60) | 90 (81·8) | 92 (76·7) |
| Mean age, years (SD) | 46·9 (13·2) | 45·2 (13·5) | 48·5 (14) | 46·9 (13·1) | 45·9 (12·4) | 45·9 (12·2) | 45·2 (12) | 43·5 (13·8) | 45·9 (13·0) |
| Age range, years | 18–82 | 19–81 | 22–76 | 22–76 | 20–81 | 19–69 | 18–70 | 19–80 | 19–72 |
| Race, n (%) | |||||||||
| White | 527 (68·3) | 996 (91·4) | 28 (70) | 78 (76·5) | 88 (77·2) | 85 (71·4) | 30 (75) | 101 (91·8) | 114 (95·0) |
| Asian | 195 (25·3) | 36 (3·3) | 11 (28) | 21 (20·6) | 20 (17·5) | 29 (24·4) | 10 (25) | 2 (1·8) | 2 (1·7) |
| Other | 50 (6·5) | 58 (5·3) | 1 (3) | 3 (2·9) | 6 (5·3) | 5 (4·2) | 0 | 7 (6·3) | 4 (3·3) |
| Mean weight, kg (SD) | 88·5 (24·4) | 88·6 (21·8) | 82·5 (22) | 90·3 (24·0) | 89·5 (23·4) | 86·6 (21·8) | 85·0 (22) | 90·3 (23·0) | 91·6 (22·0) |
| Mean body surface area, % (SD) | 30·2 (17·5) | 32·4 (17·1) | 36·6 (20) | 33·1 (19·4) | 28·1 (18·0) | 29·9 (17·3) | 36·0 (21) | 32·4 (18·3) | 31·2 (16·8) |
| Mean PASI score (SD) | 20·1 (8·0) | 20·1 (7·5) | 21·0 (8) | 20·2 (8·8) | 19·5 (8·2) | 20·4 (8·1) | 23·2 (11) | 20·1 (8·4) | 19·2 (6·7) |
| Prior exposure to biologics for psoriasis, n (%) | 177 (22·9) | 134 (12·3) | 5 (13) | 18 (17·6) | 21 (18·4) | 26 (21·8) | 7 (18) | 7 (6·4) | 16 (13·3) |
ETN, etanercept; T100, tildrakizumab 100 mg; T200, tildrakizumab 200 mg; TIL, tildrakizumab; PBO, placebo. aPooled data from reSURFACE 1 and reSURFACE 2. Includes only partial responders at week 28. breSURFACE 1 only. Includes only responders at week 28. creSURFACE 2 only. The T200/T100 group includes only responders at week 28. The ETN/T200 group includes only partial or nonresponders at week 28. dOne patient did not enter Part 3 and one patient entered Part 3 but was not treated.
Efficacy
Continuous long‐term tildrakizumab dosing
In reSURFACE 1 and reSURFACE 2, a subgroup of patients received the same dosage of tildrakizumab from baseline of Part 1 until the end of Part 3 (T100/T100 and T200/T200); data from the respective dosages in the two studies were pooled. Among patients in the T100/T100 (n = 40) and T200/T200 (n = 102) subgroups who were partial responders at week 28, the proportion of patients who achieved PASI 75 response at week 52 increased over time to 64% and 52·5%, respectively (Fig. S1; see Supporting Information). Results with continuous long‐term tildrakizumab dosing for PASI 90, PASI 100 and PGA in week‐28 partial responders were generally consistent with PASI 75 results (Fig. S1; see Supporting Information). As previously reported, among patients in the T100/T100 and T200/T200 subgroups who were responders at week 28 in both studies, PASI 75 responses remained consistent over time.4
Relapse or rebound of disease after tildrakizumab withdrawal
In reSURFACE 1, a subgroup of patients was withdrawn from tildrakizumab treatment and rerandomized to PBO (T100/PBO or T200/PBO). The median time to relapse in the T100/PBO and T200/PBO subgroups was 24 weeks in each group. The proportions of patients in the T100/PBO subgroup who were PASI 75 responders at week 28 and who experienced relapse at each time point were 0·9% (week 32), 5·5% (week 40), 10·8% (week 52) and 11·8% (week 64). The proportions of patients in the T200/PBO subgroup who were PASI 75 responders at week 28 and who experienced relapse at each time point were 0·8% (week 32), 3·5% (week 40), 14·1% (week 52) and 10·0% (week 64). In general, the proportion of patients in the T100/PBO and T200/PBO groups who were PASI 75 responders decreased over time in Part 3 (Fig. 2). Similar results were observed for PASI 90, PASI 100 and PGA responses (Fig. S2; see Supporting Information). Among those who relapsed and who had reinitiated tildrakizumab for at least 12 weeks, 86% who reinitiated tildrakizumab 100 mg (T100/PBO/T100, n = 35) and 83% who reinitiated tildrakizumab 200 mg (T200/PBO/T200, n = 30) achieved PASI 75 again by week 64. PASI 90, PASI 100 and PGA responses also increased with reinitiation of tildrakizumab treatment.
Figure 2.

Proportion of week 28 responders in reSURFACE 1 with Psoriasis Area and Severity Index (PASI) 75 response over time after withdrawal of tildrakizumab (T100/PBO and T200/PBO). PBO, placebo; T100, tildrakizumab 100 mg; T200, tildrakizumab 200 mg.
There were no patients who experienced rebound of disease after tildrakizumab withdrawal.
Dosage adjustment
In both studies, a subgroup of week‐28 partial responders receiving tildrakizumab 100 mg was rerandomized to 200 mg (T100/T200, n = 40); data from the two studies were pooled. The proportion of patients in the T100/T200 subgroup who were PASI 75 responders increased from week 32 (39%) to week 52 (63%) (Fig. 3). PASI 90, PASI 100 and PGA responses for the T100/T200 subgroup were generally consistent with the PASI 75 results (Fig. 3).
Figure 3.
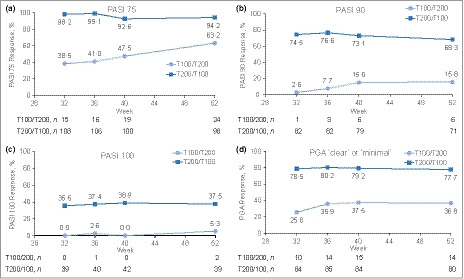
Proportion of patients with (a) Psoriasis Area and Severity Index (PASI) 75, (b) PASI 90, (c) PASI 100 and (d) Physician's Global Assessment (PGA) of ‘clear’ or ‘minimal’ response over time after dose adjustment of tildrakizumab 100 mg to 200 mg (T100/T200; week 28 partial responders) and tildrakizumab 200 mg to 100 mg (T200/T100; week 28 responders). PBO, placebo; T100, tildrakizumab 100 mg; T200, tildrakizumab 200 mg.
In reSURFACE 2, a subgroup of week‐28 responders receiving tildrakizumab 200 mg was rerandomized to tildrakizumab 100 mg (T200/100, n = 110). The proportions of patients in this T200/T100 subgroup who were PASI 75 responders remained consistent from week 32 (98·2%) to week 52 (94·2%) (Fig. 3). PASI 90, PASI 100 and PGA responses for the T200/T100 subgroup were generally consistent with the PASI 75 results (Fig. 3).
Treatment switch from etanercept
In reSURFACE 2, patients who were partial or nonresponders to ETN at week 28 were switched to tildrakizumab 200 mg following a 4‐week washout period (ETN/T200, n = 120). The proportion of patients in this ETN/T200 subgroup who achieved PASI 75 response increased from week 32 (24·1%, ETN partial responders; 10·3%, ETN nonresponders) to week 52 (74·7%, ETN partial responders; 53·8%, ETN nonresponders) (Fig. S3; see Supporting Information). PASI 90, PASI 100 and PGA responses for the ETN/T200 subgroup were generally consistent with the PASI 75 results (Fig. S3; see Supporting Information).
Safety
Overall, at least one treatment‐emergent AE was reported by 50·9–75·0% of the patient dosing subgroups in Part 3 (Table 2). The most common AE in all dosing subgroups was nasopharyngitis (11·7–20·6%). The rates of serious AEs were low (0·9–7·8%), except in the T100/T100 continuous dosing group, where six serious AEs were reported (15%); one of the serious AEs in the T100/T100 group was related to treatment. Rates of discontinuation owing to AEs were generally low (0–5·0%). Five deaths in patients receiving tildrakizumab occurred during Part 3 of the studies. None of the deaths were considered related to study medication.
Table 2.
Summary of adverse events (AEs) in Part 3 of reSURFACE 1 and reSURFACE 2 dosing subgroups
| AE, n (%) | Continuous TIL dosinga | TIL withdrawalb | TIL dose adjustment | Switch to TIL from ETNc | |||
|---|---|---|---|---|---|---|---|
| T100/T100, n = 40 | T200/T200, n = 102 | T100/PBO, n = 114d | T200/PBO, n = 119e | T100/T200, n = 40a | T200/T100, n = 110c | ETN/T200, n = 120f | |
| Any AE | 21 (53) | 62 (60·8) | 81 (71·1) | 82 (68·9) | 30 (75) | 56 (50·9) | 62 (51·7) |
| Serious AEs | 6 (15) | 8 (7·8) | 6 (5·3) | 4 (3·4) | 1 (3) | 1 (0·9) | 5 (4·2) |
| Discontinuations owing to AEs | 2 (5) | 2 (2·0) | 1 (0·9) | 0 | 0 | 1 (0·9) | 1 (0·8) |
| Any AE in ≥ 5% of patients | |||||||
| Nasopharyngitis | 5 (13) | 21 (20·6) | 20 (17·5) | 14 (11·8) | 8 (20) | 17 (15·5) | 14 (11·7) |
| Bronchitis | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% | 7 (5·8) |
| URTI | 2 (5) | < 5% | 11 (9·6) | 13 (10·9) | 3 (8) | < 5% | 6 (5·0) |
| Cough | < 5% | < 5% | 8 (7·0) | < 5% | < 5% | < 5% | < 5% |
| Psoriasis | < 5% | < 5% | 8 (7·0) | < 5% | < 5% | < 5% | < 5% |
| Influenza | < 5% | < 5% | < 5% | 7 (5·9) | 3 (8) | < 5% | < 5% |
| Limb injury | 3 (8) | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% |
| Arthralgia | 3 (8) | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% |
| Ligament sprain | 2 (5) | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% |
| Pain in extremity | 2 (5) | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% |
| Eczema | 2 (5) | < 5% | < 5% | < 5% | < 5% | < 5% | < 5% |
| Sinusitis | < 5% | < 5% | < 5% | < 5% | 2 (5) | < 5% | < 5% |
| Sciatica | < 5% | < 5% | < 5% | < 5% | 2 (5) | < 5% | < 5% |
| Contact dermatitis | 3 (8) | < 5% | < 5% | < 5% | 2 (5) | < 5% | < 5% |
| Hypertension | < 5% | < 5% | < 5% | < 5% | 2 (5) | < 5% | < 5% |
ETN, etanercept; T100, tildrakizumab 100 mg; T200, tildrakizumab 200 mg; TIL, tildrakizumab; PBO, placebo; URTI, upper respiratory tract infection. aPooled data from reSURFACE 1 and reSURFACE 2. Includes only partial responders at week 28. breSURFACE 1 only. Includes only responders at week 28. creSURFACE 2 only. The T200/T100 group includes only responders at week 28. The ETN/T200 group includes only partial or nonresponders at week 28. dA total of 72 of 114 patients (63·2%) who were randomized to TIL 100 mg in Part 1 and rerandomized to PBO in Part 3 had an adverse event while they took PBO. eIn total, 73 of 119 patients (61·3%) who were randomized to TIL 200 mg in Part 1 and rerandomized to PBO in Part 3 had an adverse event while they took PBO. fOne patient did not enter Part 3 and one patient entered Part 3 but was not treated.
Discussion
As spontaneous remission of long‐standing chronic plaque psoriasis is uncommon, long‐term treatment and medication adjustment may be needed to maintain disease control. The current analysis demonstrates that long‐term continuous dosing of tildrakizumab, not only maintains efficacy, but also improves efficacy over time in some patients; 63·9% of patients who were partial responders at week 28 with the 100‐mg dosage achieved PASI 75 by week 52. Durability of efficacy for psoriasis has also been observed with the IL‐23p19 antagonist guselkumab. In the phase III VOYAGE 1 trial (N = 837), the coprimary end point of a PGA score of ‘clear’ or ‘minimal’ was achieved with guselkumab treatment by 85·1% of patients at week 16 and 82·4% of patients at week 100; PASI 75 was achieved by 91·2% of patients at week 16 and 94·8% of patients at week 100.3, 10 In contrast, a review of long‐term studies of biologics for psoriasis indicated that up to one‐third of patients receiving anti‐tumour necrosis factor (TNF)‐α agents experience a loss of efficacy over time.11 Loss of efficacy with the IL‐17A antagonist secukinumab was reported for 21% of patients before 32 weeks of treatment in one study, although the investigators speculated that the patients in the study may have been subject to a selection bias towards treatment‐resistant psoriasis.12, 13 Other long‐term studies have demonstrated sustained efficacy for up to 5 years with secukinumab and 4 years with ixekizumab.14, 15
Interruption of treatment may occur for various reasons such as pregnancy, insurance coverage or surgery. The group in reSURFACE 1 who were PASI 75 responders and were rerandomized to PBO after 28 weeks of tildrakizumab treatment mimicked treatment interruption. As expected, disease control declined after tildrakizumab withdrawal. However, of the patients who relapsed and were reinitiated to tildrakizumab, approximately 85% achieved PASI 75. These data provide reassurance that patients may regain response after a treatment interruption of tildrakizumab. Regaining efficacy after treatment interruption has also been demonstrated for secukinumab in a withdrawal trial of clinical responders, where 181 patients were rerandomized to PBO at week 52.16 Overall, 75% of these patients relapsed and more than 90% were able to achieve PASI 75 after secukinumab reinitiation. In contrast, response rates with ETN were reduced after a treatment interruption, with only 52% of initial responders achieving PASI 75 after reinitiation.17
In real‐world practice, it is common for patients to adjust dosages of biologics.8 Occasionally, the dosage is increased because of a lack of efficacy, whereas a lengthy remission may prompt a decrease in dosage.8 Analysis of tildrakizumab dosage adjustments revealed that for patients receiving the 200‐mg dosage who were responders at week 28, adjusting the dosage to 100 mg had little impact on efficacy. For patients on the 100‐mg dosage who were only partial responders at week 28, increasing the dosage to 200 mg resulted in continued improvement, but there was no meaningful difference compared with those who continued to receive the 100‐mg dosage.
Access to biologics with different mechanisms of action provides patients with treatment options to achieve the best disease control possible. A majority of patients in reSURFACE 2 who were nonresponders or partial responders after 28 weeks of ETN achieved PASI 75 within 20 weeks of switching to tildrakizumab. Many of the trials of biologics for psoriasis that target the IL‐23/Th17 immunological pathway have used anti‐TNF‐α agents as active comparators, and clinical responses to guselkumab and ixekizumab in patients who are partial or nonresponders to anti‐TNF‐α agents have been demonstrated.18, 19
The safety and tolerability of tildrakizumab was demonstrated in Parts 1 and 2 of the reSURFACE studies and in a pooled analysis of phase II and III studies.4, 20 In Part 3 of the studies, tildrakizumab remained well tolerated among all the dosing subgroups examined, and treatment up to 64 weeks did not suggest a risk for important new safety events occurring later in treatment or a change in the overall pattern of AEs.
The portions of the analysis involving partial responders were limited by small sample size. Another potential limitation of the analysis was, as mentioned in the Materials and methods section above, that different statistical methods were used to handle dropouts and nonresponders in different cohorts. As‐observed data were used for analysis of some cohorts, which did not include patients who may have left the study owing to lack of efficacy.
The current analysis indicates that patients who receive long‐term continuous dosing with tildrakizumab for up to 64 weeks can maintain or improve clinical outcomes. Furthermore, the results provide reassurance that efficacy can be regained after a treatment interruption and reinitiation, and that reinitiation is well tolerated. Lastly, the majority of patients who do not respond or who only partially respond to ETN can achieve improved clinical responses with tildrakizumab. In sum, data from these clinical studies demonstrate the efficacy and tolerability of tildrakizumab for patients under various changing treatment scenarios.
Supporting information
Table S1 Source of patients from reSURFACE 1 and reSURFACE 2 contributing to pooled subgroups.
Table S2 Proportion of patients with Psoriasis Area and Severity Index (PASI) 75, PASI 90, PASI 100 and Physician's Global Assessment of ‘clear’ or ‘minimal’ at the end of Part 3 (week 52 for reSURFACE 1 and week 64 for reSURFACE 2) analysed as‐observed or by nonresponder imputation (NRI) when available.
Fig S1. Pooled analysis from reSURFACE 1 and reSURFACE 2 of proportion of patients with (a) Psoriasis Area and Severity Index (PASI) 75, (b) PASI 90, (c) PASI 100, and (d) Physician's Global Assessment of ‘clear’ or ‘minimal’ over time in week 28 partial responders who received continuous tildrakizumab dosing.
Fig S2. Proportion of week 28 responders in reSURFACE 1 with (a) Psoriasis Area and Severity Index (PASI) 90, (b) PASI 100 and (c) Physician's Global Assessment of ‘clear’ or ‘minimal’ over time after withdrawal of tildrakizumab [T100/placebo (PBO) and T200/PBO].
Fig S3. Proportion of week 28 etanercept (ETN) partial responders (PR) and nonresponders (NR) with (a) Psoriasis Area and Severity Index (PASI) 75, (b) PASI 90, (c) PASI 100 and (d) Physician's Global Assessment responses over time after switching to tildrakizumab 200 mg (ETN/T200). *N's include randomized patients who were partial or nonresponders at week 28 who completed Part 2; one patient did not enter Part 3 of the study.
Acknowledgments
Medical writing and/or editorial assistance were provided by Anish Mehta, of Merck & Co., Inc., (Kenilworth, NJ, U.S.A) and Erin P. Scott of Scott Medical Communications, LLC. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Conflicts of interest: A.B.K. is a consultant and investigator for AbbVie, Janssen, Lilly, Novartis and UCB, a consultant for Pfizer, and has fellowship funding from Janssen and AbbVie. K.A.P. has received honoraria for being a speaker, consultant and/or investigator for the following companies: AbbVie, Active Biotech, Akesis, Allergan, Amgen, Anacor, Astellas, AstraZeneca, Basilea, Baxter, Bayer, Biogen Idec, Boehringer Ingelheim, Bristol‐Myers Squibb, CanFite, Cato, Cepheid, Celgene, Centocor, Cipher, Coherus, Dow Pharma, Eli Lilly, Endocyte, Ferring Pharma, Forward Pharma, Galderma, Genentech, Gilead, GlaxoSmithKline, Janssen, Kyowa Hakko Kirin, Kythera, Leo Pharma, MedImmune, Meiji Seika Pharma, Merck Sharp & Dohme, Merck Serono, Mylan, Novartis, Pfizer, Regeneron Pharmaceuticals, Inc., Rigel, Roche, Sanofi‐Aventis, Sosei, Sun Pharma, Takeda, UBC and Vertex. K.R. has served as advisor and/or paid speaker for and/or participated in clinical trials sponsored by AbbVie, Affibody, Almirall, Amgen, Biogen Idec, Boehringer Ingelheim, Celgene, Centocor, Covagen, Forward Pharma, Fresenius Medical Care, GlaxoSmithKline, Janssen‐Cilag, Kyowa Kirin, Leo, Lilly, Medac, Merck Sharp & Dohme, Novartis, Miltenyi Biotec, Ocean Pharma, Pfizer, Regeneron, Samsung Bioepis, Sanofi, Takeda, UCB, Valeant and Xenoport. M.G. has been an investigator, advisor and/or speaker for Amgen, AbbVie, Akros, Arcutis, BMS, Boehringer Ingelheim, Celgene, Dermira, Galderma, GSK, Janssen, Kyowa Kirin, Leo Pharma, Lilly, Merck, Merck Serono, Novartis, Pfizer, Regeneron, Sun Pharma, Sanofi Genzyme, Sienna Pharmaceuticals, Takeda, Valeant and UCB. A.B. has served as a scientific advisor and/or clinical study investigator for AbbVie, Aclaris, Akros, Allergan, Almirall, Amgen, Arena, Athenex, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Dermavant, Dermira, Inc., Eli Lilly, FLX Bio, Forte, Galderma, Genentech/Roche, GlaxoSmithKline, Janssen, Leo, Meiji, Merck Sharp & Dohme, Novartis, Pfizer, Purdue Pharma, Regeneron, Revance, Sandoz, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, UCB Pharma, Valeant and Vidac, and as a paid speaker for AbbVie, Regeneron and Sanofi Genzyme. Q.L., N.C. and C.L.R. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, U.S.A., who may own stock and/or hold stock options in the Company.
All authors are responsible for the work described in this paper. All authors were involved in at least one of the following: conception, design of work or acquisition, analysis, or interpretation of data. All authors were involved in revising/reviewing the manuscript for important intellectual content and provided final approval of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding sources Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, U.S.A.
Conflicts of interest See Appendix.
Plain language summary available online
References
- 1. Gordon KB, Blauvelt A, Papp KA et al Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016; 375:345–56. [DOI] [PubMed] [Google Scholar]
- 2. Langley RG, Elewski BE, Lebwohl M et al Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 2014; 371:326–38. [DOI] [PubMed] [Google Scholar]
- 3. Blauvelt A, Papp KA, Griffiths CE et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double‐blinded, placebo‐ and active comparator‐controlled VOYAGE 1 trial. J Am Acad Dermatol 2017; 76:405–17. [DOI] [PubMed] [Google Scholar]
- 4. Reich K, Papp KA, Blauvelt A et al Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet 2017; 390:276–88. [DOI] [PubMed] [Google Scholar]
- 5. Ilumya® (tildrakizumab‐asmn) [prescribing information]. Whitehouse Station, NJ: Merck & Co., Inc., 2018.
- 6. Ilumetri . Summary of opinion. Tildrakizumab. European Medicines Agency, 2018. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/ilumetri (last accessed 29 August 2019).
- 7. Papp K, Thaci D, Reich K et al Tildrakizumab (MK‐3222), an anti‐interleukin‐23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo‐controlled trial. Br J Dermatol 2015; 173:930–9. [DOI] [PubMed] [Google Scholar]
- 8. Esposito M, Gisondi P, Conti A et al Dose adjustment of biologic therapies for psoriasis in dermatological practice: a retrospective study. J Eur Acad Dermatol Venereol 2017; 31:863–9. [DOI] [PubMed] [Google Scholar]
- 9. Feldman SR, Zhao Y, Navaratnam P et al Patterns of medication utilization and costs associated with the use of etanercept, adalimumab, and ustekinumab in the management of moderate‐to‐severe psoriasis. J Manag Care Spec Pharm 2015; 21:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Griffiths CEM, Papp KA, Kimball AB et al Long‐term efficacy of guselkumab for the treatment of moderate‐to‐severe psoriasis: results from the phase 3 VOYAGE 1 trial through two years. J Drugs Dermatol 2018; 17:826–32. [PubMed] [Google Scholar]
- 11. Levin EC, Gupta R, Brown G et al Biologic fatigue in psoriasis. J Dermatolog Treat 2014; 25:78–82. [DOI] [PubMed] [Google Scholar]
- 12. Huang YYM, Hsu S. Loss of efficacy of secukinumab for psoriasis at 24 to 32 weeks: update and commentary. J Am Acad Dermatol 2017; 76:e221. [DOI] [PubMed] [Google Scholar]
- 13. Huang YYM, Ruth JS, Hsu S. Loss of efficacy of secukinumab for psoriasis at 24 to 32 weeks. J Am Acad Dermatol 2016; 75:e169. [DOI] [PubMed] [Google Scholar]
- 14. Bissonnette R, Luger T, Thaci D et al Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate‐to‐severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol 2018; 32:1507–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zachariae C, Gordon K, Kimball AB et al Efficacy and safety of ixekizumab over 4 years of open‐label treatment in a phase 2 study in chronic plaque psoriasis. J Am Acad Dermatol 2018; 79:294–301.e6. [DOI] [PubMed] [Google Scholar]
- 16. Blauvelt A, Reich K, Warren RB et al Secukinumab re‐initiation achieves regain of high response levels in patients who interrupt treatment for moderate to severe plaque psoriasis. Br J Dermatol 2017; 177:879–81. [DOI] [PubMed] [Google Scholar]
- 17. Gordon KB, Gottlieb AB, Leonardi CL et al Clinical response in psoriasis patients discontinued from and then reinitiated on etanercept therapy. J Dermatolog Treat 2006; 17:9–17. [DOI] [PubMed] [Google Scholar]
- 18. Blauvelt A, Papp KA, Griffiths CEM et al Efficacy and safety of switching to ixekizumab in etanercept non‐responders: a subanalysis from two phase III randomized clinical trials in moderate‐to‐severe plaque psoriasis (UNCOVER‐2 and ‐3). Am J Clin Dermatol 2017; 18:273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reich K, Armstrong AW, Foley P et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76:418–31. [DOI] [PubMed] [Google Scholar]
- 20. Blauvelt A, Reich K, Papp KA et al Safety of tildrakizumab for moderate‐to‐severe plaque psoriasis: pooled analysis of three randomized controlled trials. Br J Dermatol 2018; 179:615–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Source of patients from reSURFACE 1 and reSURFACE 2 contributing to pooled subgroups.
Table S2 Proportion of patients with Psoriasis Area and Severity Index (PASI) 75, PASI 90, PASI 100 and Physician's Global Assessment of ‘clear’ or ‘minimal’ at the end of Part 3 (week 52 for reSURFACE 1 and week 64 for reSURFACE 2) analysed as‐observed or by nonresponder imputation (NRI) when available.
Fig S1. Pooled analysis from reSURFACE 1 and reSURFACE 2 of proportion of patients with (a) Psoriasis Area and Severity Index (PASI) 75, (b) PASI 90, (c) PASI 100, and (d) Physician's Global Assessment of ‘clear’ or ‘minimal’ over time in week 28 partial responders who received continuous tildrakizumab dosing.
Fig S2. Proportion of week 28 responders in reSURFACE 1 with (a) Psoriasis Area and Severity Index (PASI) 90, (b) PASI 100 and (c) Physician's Global Assessment of ‘clear’ or ‘minimal’ over time after withdrawal of tildrakizumab [T100/placebo (PBO) and T200/PBO].
Fig S3. Proportion of week 28 etanercept (ETN) partial responders (PR) and nonresponders (NR) with (a) Psoriasis Area and Severity Index (PASI) 75, (b) PASI 90, (c) PASI 100 and (d) Physician's Global Assessment responses over time after switching to tildrakizumab 200 mg (ETN/T200). *N's include randomized patients who were partial or nonresponders at week 28 who completed Part 2; one patient did not enter Part 3 of the study.