

Abstract
Hypophosphatasia (HPP) is a rare metabolic disorder characterized by low tissue‐nonspecific alkaline phosphatase (TNSALP) typically caused by ALPL gene mutations. HPP is heterogeneous, with clinical presentation correlating with residual TNSALP activity and/or dominant‐negative effects (DNE). We measured residual activity and DNE for 155 ALPL variants by transient transfection and TNSALP enzymatic activity measurement. Ninety variants showed low residual activity and 24 showed DNE. These results encompass all missense variants with carrier frequencies above 1/25,000 from the Genome Aggregation Database. We used resulting data as a reference to develop a new computational algorithm that scores ALPL missense variants and predicts high/low TNSALP enzymatic activity. Our approach measures the effects of amino acid changes on TNSALP dimer stability with a physics‐based implicit solvent energy model. We predict mutation deleteriousness with high specificity, achieving a true‐positive rate of 0.63 with false‐positive rate of 0, with an area under receiver operating curve (AUC) of 0.9, better than all in silico predictors tested. Combining this algorithm with other in silico approaches can further increase performance, reaching an AUC of 0.94. This study expands our understanding of HPP heterogeneity and genotype/phenotype relationships with the aim of improving clinical ALPL variant interpretation.
Keywords: algorithms, genetic data bases, hypophosphatasia, rare disease, tissue‐nonspecific alkaline phosphatase, variant effect prediction
1. INTRODUCTION
Hypophosphatasia (HPP) is a rare, systemic, inherited, metabolic disorder characterized by low alkaline phosphatase (ALP) enzymatic activity and typically caused by pathogenic variants in the tissue‐nonspecific alkaline phosphatase (TNSALP) gene (ALPL; MIM #171760; Taillandier et al., 2018; Weiss et al., 1988). HPP is a clinically heterogeneous disease, with manifestations ranging from severe skeletal hypomineralization, seizures, and respiratory problems at birth (Baumgartner‐Sigl et al., 2007; Kozlowski et al., 1976; Leung et al., 2013; Silver, Vilos, & Milne, 1988) to predominantly recurrent fractures, pain, muscle weakness, and functional limitations later on (Berkseth et al., 2013; Seshia, Derbyshire, Haworth, & Hoogstraten, 1990; Weber, Sawyer, Moseley, Odrljin, & Kishnani, 2016; Whyte, 2017; Whyte et al., 2015). Historically, HPP has been classified into distinct clinical forms based on the age at which skeletal disease or other significant complications present: perinatal, prenatal benign, infantile, childhood, adult, and odonto‐HPP (Whyte, 2016). Perinatal and infantile HPP are generally considered the most severe forms and are associated with high mortality (Baumgartner‐Sigl et al., 2007; Leung et al., 2013; Silver et al., 1988; Whyte et al., 2016), especially before the availability of enzyme replacement therapy. Though these classifications are helpful in describing the disease, the clinical presentation and severity of HPP can vary widely between patients, regardless of form and even within a given form of the disease (Kishnani et al., 2017; Whyte, 2017).
The clinical variability of HPP is largely a consequence of its ALPL genetic heterogeneity (Mornet, 2018). To date, over 390 pathogenic variants have been identified, with the majority being missense variants (University of Versailles‐Saint Quentin, 2019). Missense ALPL variants can affect the expression, folding, modification, trafficking, and dimerization of the TNSALP protein, resulting in varying levels of residual enzymatic activity (Brun‐Heath et al., 2007; Lia‐Baldini et al., 2008; Makita et al., 2012). Further, ALPL variants can exhibit a dominant‐negative effect (DNE), potentially as a result of negative interactions between mutated and wild‐type (WT) monomers or because of sequestration of the WT protein by the mutated one, preventing transport to the membrane (Lia‐Baldini et al., 2008; Mornet, 2015). Some evidence suggests that the clinical presentation of HPP correlates with the in vitro enzymatic activity of the mutant protein and/or with the strength of the DNE (Fauvert et al., 2009; Lia‐Baldini et al., 2001; Zurutuza et al., 1999). However, the residual enzymatic activity and DNE are known for only a fraction of reported HPP variants (University of Versailles‐Saint Quentin, 2019).
The present study had two specific objectives, with the overarching goal of improving the understanding of ALPL genotype/phenotype relationships by (a) expanding the pool of ALPL variants whose residual enzymatic activity is characterized in vitro and (b) using the in vitro data to develop novel computational approaches to detect the presence or absence of low residual enzymatic activity of a particular variant.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
Patients included in this analysis were tested for diagnostic purposes, and the study was designed in accordance with the tenets of the Declaration of Helsinki. Informed consent was obtained from all patients and/or their parents for ALPL variant screening and testing for HPP‐related genes. All patients were of apparent European, Middle Eastern, or Japanese ancestry.
2.2. ALPL variant selection
With the goal of expanding the current catalog of functionally tested ALPL variants, 155 variants were selected and prioritized to represent those in general population data bases and in HPP patients (see details in the Supporting Information Materials). Genotypic data were assembled from a retrospective historical cohort of 345 patients with confirmed HPP diagnoses who underwent genetic testing between 1997 and 2016 at the Unité de Génétique Constitutionnelle Prénatale et Postnatale (formerly the SESEP Laboratory; Centre Hospitalier de Versailles, Le Chesnay, France).
Additional missense, frameshift, in‐frame indel, and nonsense ALPL variants were collected from the Genome Aggregation Database (gnomAD; Karczewski et al., 2019; Lek et al., 2016), the HPP patient cohort, and other sources (see details in the Supporting Information Materials). Together, the assembled set of variants included all nonsynonymous exonic gnomAD ALPL variants with an allele frequency of >1/25,000 and a random sample of rarer patient and population variants.
2.3. Plasmid preparation, transfection, and TNSALP activity measurement
Complete details regarding plasmid preparation, transfection, and TNSALP activity measurement can be found in the Supporting Information Materials. Briefly, MDCK II cells were transiently transfected with 100% of mutant plasmid or a mix corresponding to mutant plasmid:WT plasmid (ratio 50:50). The normalized TNSALP value was determined by dividing the measured TNSALP absorbance at 405 nm by the measured β‐galactosidase absorbance at 405 nm. Pure mutant and cotransfected WT:mutant data were calculated as the mean value over three transfections and expressed as a percentage of pure WT plasmid transfected on the same plate.
2.4. Predicting in vitro activity of patient genotypes
To predict the in vitro activity of variants from patients in the historical HPP cohort (n = 345) by genotype (i.e., homozygous, heterozygous, and compound heterozygous), TNSALP residual activity was estimated and expressed as a fraction of WT activity for each patient as follows:
The estimated activity for homozygous patients was the measured mutant in vitro activity. If a patient did not have a variant that had been tested in vitro but had a variant computationally predicted to be a complete loss of function (LoF; i.e., nonsense, splice, or frameshift variant), it was assumed that their TNSALP residual activity would be null.
The estimated activity for heterozygous patients was the measured in vitro activity of the 50/50 cotransfected mixture. For patients with a single heterozygous LoF variant with no measured TNSALP residual activity, the estimated TNSALP residual activity was 0.5, with the assumption that the LoF allele would not express any protein.
For compound heterozygous patients, if neither allele had a DNE (defined as any allele that had a measured in vitro activity from the mutant/WT mixture of <0.4 relative to WT), the TNSALP residual activity was the average activity of both alleles. However, if allele 1 showed a DNE in its in vitro assay, the total activity was estimated to be allele 2's activity divided by four (see details in the Supporting Information Materials).
Patients who did not have at least one of their ALPL variants tested in vitro were considered as having missing data and were removed from further evaluation.
2.5. TNSALP homology model and variant scoring based on stability/affinity changes
A protein homology model for human TNSALP was constructed using a template corresponding to the human placental alkaline phosphatase (PLAP; Le Du, Stigbrand, Taussig, Menez, & Stura, 2001). The template had a sequence identity to TNSALP of 57% and an X‐ray crystal structure resolution of 1.8 Å. The structure was prepared for residue scanning using the Schrödinger Suite Protein Preparation Wizard (Sastry, Adzhigirey, Day, Annabhimoju, & Sherman, 2013). Each possible ALPL missense variant was automatically generated, and its effect on protein stability and dimer binding affinity were predicted using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method, a physics‐based approach that scores single‐residue mutations based on predicted changes to protein stability and binding affinity (Beard, Cholleti, Pearlman, Sherman, & Loving, 2013). Complete details regarding the construction of the TNSALP model and residue‐scanning software can be found in the Supporting Information Materials.
For a particular variant or allele ai, we denote as Δs(ai) the computationally predicted change in protein stability, and we denote as Δa(a i) the change in binding affinity between both dimer complexes, using methods described by Beard et al. (2013). We also denote Δmax(ai) = max(Δs(ai), Δa(ai)) as the maximum of these two values. The Δs(a i), Δa(ai), and Δmax(ai) were computed for all possible TNSALP missense variants (see details in the Supporting Information Materials).
2.6. Using in vitro data to assess in silico algorithm performance
In vitro activity values were used as a reference data set to assess the performance of (a) existing in silico variant scoring algorithms and (b) a novel approach of scoring variants based on MM/GBSA stability/affinity changes. In assessments of the in silico approaches, “low” enzymatic activity was defined as ≤0.25 relative to WT and “high” enzymatic activity was defined as ≥0.5 relative to WT. Variants with activity between these thresholds were excluded from further evaluation, as their activity range makes their pathogenicity hard to ascertain.
Receiver operating characteristic (ROC) curves and area under the ROC curves (AUC) were computed for each of the following in silico variant prediction algorithms:
Combined Annotation Dependent Depletion (CADD; Kircher et al., 2014).
Deleterious Annotation of Genetic Variants Using Neural Networks (DANN; Quang, Chen, & Xie, 2015).
MutationTaster2 (Schwarz, Cooper, Schuelke, & Seelow, 2014).
PolyPhen‐2 (Adzhubei, Jordan, & Sunyaev, 2013).
Protein Variation Effect Analyzer (PROVEAN; Choi, Sims, Murphy, Miller, & Chan, 2012).
Sorting Intolerant From Tolerant (SIFT; Sim et al., 2012).
ROC and AUC scores were computed using the algorithm rank scores obtained from the dbNSFP variant annotation database (version 3.1; Liu, Wu, Li, & Boerwinkle, 2016), accessed through the Ensembl Variant Effect Predictor annotation tool (version 87; McLaren et al., 2016).
A novel approach was explored in which protein stability algorithm scores were combined with existing in silico algorithm scores to improve overall variant classification. For a particular ALPL variant or allele ai, we first denoted e(ai) = 1 if a variant had low activity, or 0 otherwise. The protein stability‐based estimator took the following form:
In this equation, T s = stability scoring threshold, above which Δs(ai) is deemed pathogenic, and T a = affinity scoring threshold, above which Δa(ai) is deemed pathogenic.
The reasoning behind the form of this estimator was that, if a variant had a high change in protein stability or binding affinity (estimator yields a value e(ai) = 1), it was likely to have low enzymatic activity and, thus, be deleterious. However, the converse (estimator yields a value e(ai) = 1) was not necessarily true, as there can be causes of low variant activity other than protein structural changes. In this case, no conclusions can be reached about variant pathogenicity and the existing in silico score should be used. Nevertheless, the classifications provided by the structural model can be used to improve any existing in silico variant scoring algorithm by setting the algorithm output as max(e(ai), f(ai)), where f(ai) is any particular in silico score normalized to (0, 1).
To determine the variability of AUC estimates and to assess the statistical significance of performance differences between the in silico algorithms, a classical bootstrap procedure was performed by sampling data with replacement, measuring resulting AUC for each in silico algorithm, and repeating the sampling 10,000 times.
2.7. Statistical analysis
All statistical tests were performed in the R language (R Core Team, 2017), version 3.6.0. Plots were generated using R package ggplot2, version 3.1.1. (Wickham, 2016).
2.8. Variant nomenclature
The Human Genome Variation Society recommendations were used to standardize the nomenclature of all analyzed variants. The reference sequences used to specify ALPL variants were RefSeq sequence NM_000478.6 (identical to Ensembl sequence ENST000000374840.8) for the coding region canonical transcript. Protein variants are specified relative to the amino acid sequence resulting from translation of this transcript, specified in NP_000469.3 (identical to Ensembl sequence ENSP00000363973.3).
3. RESULTS
3.1. ALPL functional assays, residual TNSALP activity, and presence of DNE
Overall, 94% (146/155) of the assayed variants were missense variants (Table 1). Fifty‐eight percent (90/155) of the variants showed residual activity ≤0.25, 34% (52/155) showed activity ≥0.5%, and 8% (13/155) showed activity between these two thresholds (Figure 1). As expected, LoF variants (i.e., frameshift, in‐frame deletions, and nonsense) showed null residual activity (relative to WT), whereas missense variants spanned the entire range of possible relative enzymatic activities (Figure 1).
Table 1.
Molecular consequence of the 155 ALPL variants
| Variant type | No. of variants assayed, n (%) |
|---|---|
| Missense | 146 (94.2) |
| Nonsense (stop‐gained) | 5 (3.2) |
| Frameshift | 2 (1.3) |
| In‐frame deletion | 2 (1.3) |
Figure 1.
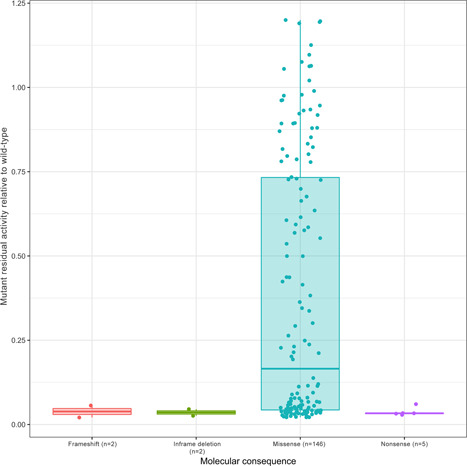
Residual activity of ALPL variants relative to wild‐type by variant type
Variants present only in HPP patients had significantly lower residual enzymatic activity than variants only in the gnomAD population or variants in both the gnomAD population and HPP patients (Figure 2). Of all variants tested, 24 showed presence of DNE (15 in HPP patients only, 8 in both HPP patients and in the gnomAD population, and 1 only in the gnomAD population) and 124 showed absence of DNE, where the presence of DNE is defined as a WT/mutant activity <0.4 and the absence of DNE is defined as a WT/mutant activity >0.45. These thresholds account for measurement variability and are consistent with previous reports on the classification of DNE (Taillandier et al., 2018). A complete listing of the residual enzymatic activity and DNE results for the 155 assayed variants is provided in Table S1.
Figure 2.
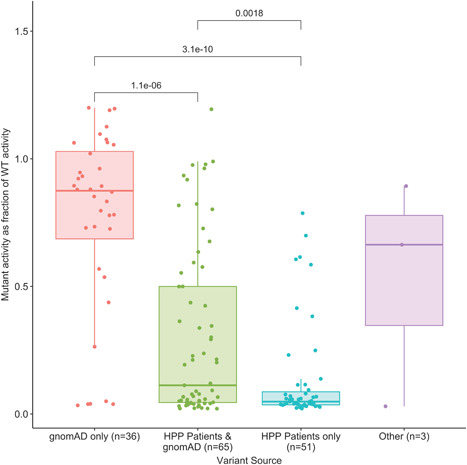
Residual activity of ALPL variants expressed as a fraction of wild‐type activity by the source of the variant. p Values from pairwise Kruskal–Wallis nonparametric tests are added on top of each pair of groups if they are below the significance threshold. gnomAD, Genome Aggregation Database; HPP, hypophosphatasia
Figure 3a shows the enzymatic activity (relative to WT) for all tested ALPL variants, including mutant plasmids and the 50:50 WT:mutant plasmid cotransfections, categorized by the protein domain in which the variant appears (Silvent, Gasse, Mornet, & Sire, 2014). The analysis of mutant residual values as a function of each domain shows that the active site or vicinity, crown domain, and homodimer interface are strongly associated with low TNSALP residual activity values (Figure 3b). For example, 94% (17/18) of tested variants that localized to the active site or vicinity showed low residual enzymatic activity, as opposed to the 53% (73/137) of tested variants outside this region that showed low activity (odds ratio: 11.74; Fisher's exact test for binary comparison p = .003). Similarly, of 18 variants that localized to the active site or vicinity, 10 showed DNE presence and 6 showed DNE absence (with two indeterminate results); conversely, of 137 variants that localized outside this site, 14 showed DNE presence (odds ratio: 13.61; Fisher's exact test p = 8.5 × 10−6).
Figure 3.
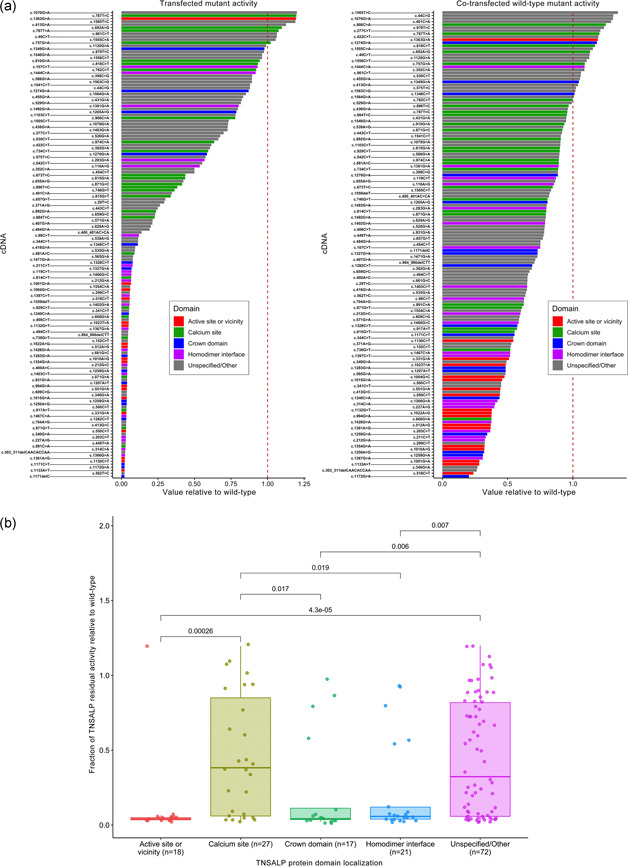
Residual activity of ALPL variants (mutant plasmids and 50:50 WT:mutant plasmid cotransfections) relative to WT (a) and fraction of residual activity of ALPL variants relative to WT (b) categorized by protein domain location. Each mutant's cDNA is listed relative to canonical RefSeq transcript NM_000478.6, and is colored according to the protein domain in which it appears, based on the descriptions and positions referenced in Silvent et al. (2014). p Values from pairwise Kruskal–Wallis nonparametric tests are added on top of each pair of groups if they are below significance threshold. cDNA, complementary deoxyribonucleic acid; TNSALP, tissue‐nonspecific alkaline phosphatase; WT, wild‐type
3.2. Patient characteristics and TNSALP activity
When the historical patient cohort is categorized based on clinical subtype, estimated in vitro residual activity is significantly reduced relative to WT in patients with the perinatal lethal and infantile HPP subtypes compared with the other subtypes and the overall cohort (pairwise p < .01; Figure 4 and Table S2). In contrast, there is no significant statistical difference in estimated in vitro residual activity between the childhood, adult, and odonto‐HPP groups (pairwise p > .25; not shown in the figure).
Figure 4.
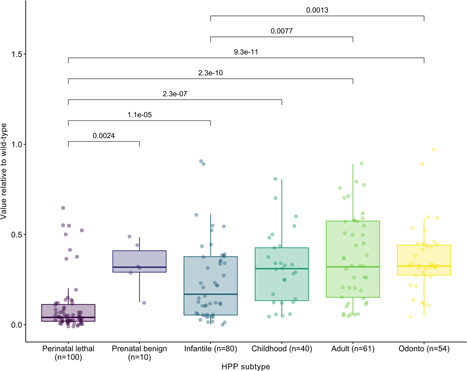
Residual activity of ALPL variants relative to wild‐type by HPP subtype. p Values from pairwise Kruskal–Wallis nonparametric tests are added on top of each pair of groups if they are below significance threshold. ALPL, tissue‐nonspecific alkaline phosphatase gene; HPP, hypophosphatasia
In vitro residual activity relative to WT of ALPL variants (n = 57), that overlap with ClinVar (Landrum et al., 2018), categorized by reported clinical classification (benign/likely benign, variant of uncertain significance, and pathogenic/likely pathogenic) is shown in Figure 5. As expected, pathogenic/likely pathogenic variants had lower residual enzymatic activity. Figure 5 also illustrates the relationship between in vitro activity relative to WT and total gnomAD allele frequency in the population. The majority of assessed variants are rare in the population, having an allele frequency of <10−4 regardless of the level of residual enzymatic activity; more common variants with allele frequencies >10−3 tend to have higher residual enzymatic activity. Interestingly, the variant c.571G>A/p.Glu191Lys was the only one with low residual enzymatic activity and a gnomAD allele frequency >10−3.
Figure 5.
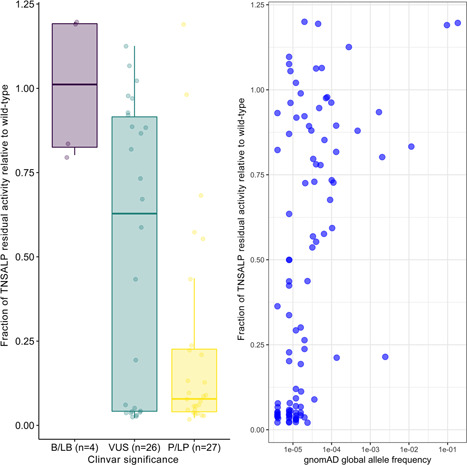
Fraction of residual activity relative to wild‐type based on ClinVar significance (left) and gnomAD global allele frequency (right). ALP, alkaline phosphatase; B, benign; gnomAD, Genome Aggregation Database; LB, likely benign; LP, likely pathogenic; P, pathogenic; TNSALP, tissue‐nonspecific alkaline phosphatase; VUS, variant of uncertain significance
3.3. Protein structural model and TNSALP activity predictions
Figure 6 shows the TNSALP dimer homology model depicted as a ribbon structure, with amino acid positions found to have a DNE from in vitro experiments displayed in red. Although DNE variants appear throughout all protein domains, the homodimer interface is enriched for DNE (odds ratio: 4.04; Fisher's exact test p = .0026) based on protein domain positions described in Silvent et al. (2014).
Figure 6.
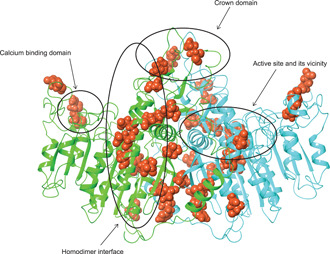
Location of variants with DNE within the ribbon structure of the TNSALP dimer homology model. Positions where a dominant‐negative variant was found, are shown as red atoms. Mirror residues on the opposite chain are not marked with arrows/circles. DNE, dominant‐negative effect; TNSALP, tissue‐nonspecific alkaline phosphatase
From the homology model, a total of 469 amino acid positions were included in the residue scanning process, with 16 excluded positions where side chains interacted via coordinate covalent bonds with metal cofactors or where disulfide bridges were present (see Supporting Information Materials for a list of the exclusions). Each was scored with all 19 possible standard amino acid changes to yield a total of 8,911 mutant predictions. A complete list of the resulting Δs, Δa, and Δmax values is shown in Table S3. Importantly, the presence of such coordinate bonds at a particular amino acid position is a strong predictor of disrupted enzymatic activity for a variant. Of the 155 variants assayed here, 16 were in these crosslinked positions and all showed low enzymatic activity (odds ratio: infinity; Fisher's exact test p = .001). Of these 16 assayed variants in crosslinked positions, seven showed a DNE, as opposed to 17 of 134 non‐crosslinked variants showing a DNE (odds ratio: 5.17; Fisher's exact test p = .005). Seven variants were excluded from testing as they showed measured DNE values between low and high thresholds (as defined above). Of note is the enrichment of variants showing a DNE in the homodimer interface. Whereas variants located in the homodimer interface did not show a statistically significant enrichment for low enzymatic activity (odds ratio: 2.26; Fisher's exact test p = .09), these variants were more enriched for the presence of a DNE (odds ratio: 3.48; Fisher exact test p = .017; Figure 6).
Using low/high enzymatic activity classification as the reference data, ROC curves for the six in silico algorithms assessed (i.e., CADD, DANN, MutationTaster, PolyPhen‐2, PROVEAN, and SIFT) and for the three approaches based on protein stability/affinity measurements (i.e., Prime_S, Prime_A, and Prime_max) are shown in Figure 7a, Scoring based on stability changes (Prime_S) achieved a significant true‐positive rate (0.63), while maintaining the false‐positive rate at 0, which is notably better than any of the other tested approaches. The composite score (Prime_max) performed slightly better than the stability score (Prime_S), and the affinity score (Prime_A) was the poorest performer.
Figure 7.
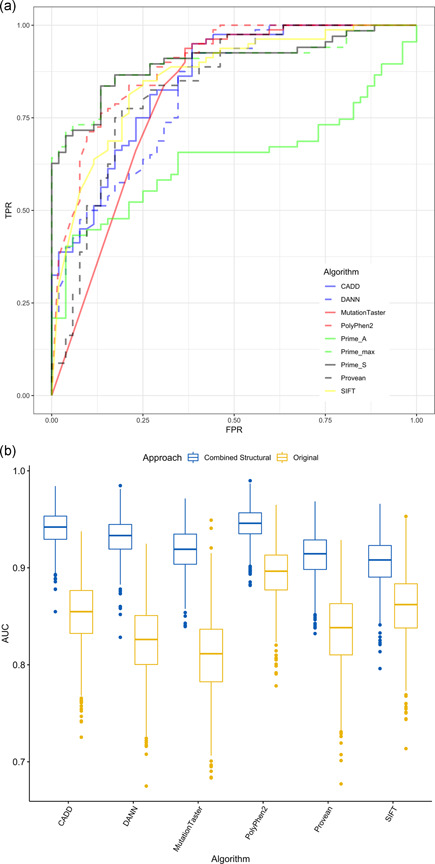
ROC curves showing TPR versus FPR for variant prediction algorithms (a) and bootstrap AUCs for in silico algorithms by original AUC values and AUC and ROC values obtained after combining in silico scores with Δs (b). AUC, area under the ROC curve; CADD, Combined Annotation Dependent Depletion; DANN, Deleterious Annotation of Genetic Variants Using Neural Networks; FPR, false‐positive rate; PROVEAN, Protein Variation Effect Analyzer; ROC, receiver operating characteristics; SIFT, Sorting Intolerant From Tolerant; TPR, true‐positive rate; Δs, computationally predicted change in protein stability
On the basis of these data, the scores from the in silico algorithms were combined with predictions based on the stability score (Prime_S). Figure 7b shows bootstrap AUCs obtained for all six algorithms tested, with both original AUC values and AUC and ROC values obtained after combining the in silico scores with stability score predictions. Scores were combined using a threshold value of T s = 36.9, which corresponded to the threshold at which Prime_S showed the highest true‐positive rate subject to a false‐positive rate of 0. Using the combined approach resulted in significant AUC performance increases, reaching a mean (95% confidence interval) of 0.945 (0.907–0.976) when combined with PolyPhen‐2, the best existing in silico algorithm assessed. Mean and 95% confidence intervals for AUC scores are summarized in Table S4.
4. DISCUSSION
In this study, we tested 155 ALPL variants, expanding the catalog of ALPL variants whose residual enzymatic activity is characterized. Functional testing was performed not only on variants in HPP patients, but also on variants in the general population, which allowed for a more comprehensive analysis. Low residual activity and/or DNEs were discovered in a number of previously unreported variants.
Overall, 94% of the assayed variants were missense; the remainder were LoF (i.e., frameshift, in‐frame deletions, and nonsense). As expected, variants present only in HPP patients and not in the gnomAD population (i.e., rare variants) had lower residual enzymatic activity than variants in the overall gnomAD population or variants in both HPP patients and the gnomAD population. This is consistent with evolutionary pressures keeping variants with lower enzymatic activity to a lower population frequency. The variant c.571G>A/p.Glu191Lys was a notable outlier, with a relatively high presence in both HPP patients and the gnomAD population while having a residual enzymatic activity of 0.214, close to the low activity threshold of 0.25. This was the only variant classified as having a low activity that had a gnomAD allele frequency >10−3 (Figure 5b). This variant is known to be the most common variant in HPP patients of European ancestry, with a previously reported allele frequency of 0.08 in an HPP cohort consisting of infantile, childhood, and odonto‐HPP patients (Whyte et al., 2015) and an allele frequency of 0.09 in the HPP cohort analyzed herein. However, this variant is, in all likelihood, not fully penetrant, as illustrated by the fact that gnomAD version 2.1 possesses five homozygous, presumably healthy subjects, and this variant reached a carrier frequency of >3% of the Finnish population in this data set (Karczewski et al., 2019). Lastly, it should be noted that some variants can be found with a higher frequency in a specific population. For example, the variant c.1001G>A/p.Gly334Asp was present only in HPP patients in our analysis (Table S1) but has an estimated carrier frequency of 1/25 in the Manitoba Mennonite population (Greenberg et al., 1990)
The analysis of residual enzymatic activity as a function of each protein domain showed that the active site, crown domain, and homodimer interface were strongly associated with low TNSALP residual activity. This finding is consistent with previous findings that pathogenic variants associated with the severe disease commonly occur around these three sites, which are critical functional domains of TNSALP (Mornet et al., 2001). Previous reports also note that variants associated with severe disease are located in the calcium‐binding site (Brun‐Heath, Taillandier, Serre, & Mornet, 2005; Mornet et al., 2001). Interestingly, in our analysis, TNSALP residual activity at this site was wide‐ranging and there was no significant enrichment of low enzymatic activity variants in this domain. Hence, variant pathogenicity in this domain may depend on other factors, such as the specific amino acid changes involved and more detailed positional interactions. Furthermore, our analysis found that though variants with DNE were located throughout all protein domains, the TNSALP homodimer interface, as well as the active site or vicinity, were enriched with variants with DNE. This was consistent with previous analyses that found that the majority of variants with DNE were located in the active site or its vicinity, the crown domain, or the homodimer interface (Fauvert et al., 2009; Taillandier et al., 2018).
Variants with severely depleted enzymatic activity tend to have very low frequency in the HPP population, consistent with the rare nature of HPP, especially in its perinatal and infantile forms (Mornet, Yvard, Taillandier, Fauvert, & Simon‐Bouy, 2011). Interestingly, while low residual activity predicts pathogenicity, the converse is not true, and a higher residual activity value does not necessarily preclude that a variant is pathogenic. This was observed in two instances. First, when predicting residual activity in the HPP cohort, we found that very low in vitro values were associated almost exclusively with perinatal lethal or infantile HPP (Figure 4). However, there was substantial overlap in predicted in vitro activity among HPP subtypes. In addition, there was an overlap between activity levels in HPP patients and activity levels theoretically corresponding to healthy carriers (≥50%). Hence, the predictive value of low in vitro TNSALP enzymatic activity is limited to very low values being associated with the most severe HPP subtypes. Second, a similar observation was noted when relating a variant's measured in vitro residual activity with ClinVar clinical significance (Landrum et al., 2018). Though we observed an expected concentration of low enzymatic activity values in the pathogenic/likely pathogenic variant classifications, higher activity values (>50% of WT reference) could be associated with any clinical significance (Figure 5). It is important to note that accurate prediction of milder HPP subtypes (e.g., perinatal benign, childhood, adult, odonto) based exclusively on ALPL variant information will likely not be improved only by collecting in vitro data from more variants. There is significant clinical heterogeneity among patients, even among family members with the same ALPL variants (Hofmann et al., 2014; Stevenson et al., 2008). This suggests that other genes, as well as epigenetic or environmental factors, may have an influence on the HPP phenotype (Mornet, 2018). As such, a better understanding of how other genes interact with TNSALP and how they might modify TNSALP expression and activity will be needed to improve clinical subtype prediction.
Collectively, these observations underscore the genetic complexity of HPP and support the classification of a variant as potentially disease‐causing if it has low in vitro residual activity. This is consistent with the published literature (Mornet, 2015; Zurutuza et al., 1999), in which very low values of residual enzymatic TNSALP activity were associated with the more severe clinical presentations of HPP (e.g., perinatal lethal, infantile). Conversely, higher frequency ALPL variants, as well as variants classified in ClinVar (Landrum et al., 2018) as “benign” or “likely benign,” have residual enzymatic activities closer to the WT reference value. However, high values of residual enzymatic activity relative to WT do not necessarily preclude HPP, as impairment of enzymatic activity may not be the only pathogenic mechanism by which a genetic variant causes this disease. Specifically, it has been suggested that particular alleles in patients with severe disease may not degrade enzymatic activity, but instead impair regular functionality through other mechanisms, such as disrupting the TNSALP dimer anchoring, transport, and localization in the extracellular domain, or compromising the creation of aggregates not correctly degraded in the proteasome (Brun‐Heath et al., 2007; Lia‐Baldini et al., 2008).
We calculated ROC curves for six in silico variant prediction algorithms (i.e., CADD, DANN, MutationTaster, PolyPhen‐2, PROVEAN, and SIFT) and for the three approaches based on protein stability/affinity measurements (Prime_S, Prime_A, and Prime_max). The Prime_S approach provided the best result, with a true‐positive rate of 0.63, while maintaining a false‐positive rate of 0. Additionally, by itself, Prime_S showed the best mean AUC of all assessed computational approaches (Figure 7 and Table S4). These results show that Prime_S is, by itself, the most specific ALPL variant pathogenicity predictor tested and that, for all tested variants, if Prime_S flags a variant as potentially pathogenic, this prediction will, in all likelihood, be correct. Importantly, we showed a simple way to improve performance scores even more for estimated residual TNSALP enzymatic activity by combining the scores from existing in silico algorithms with predictions based on the stability score (Prime_S). To our knowledge, this combined predictor is the best computational predictor of ALPL variant pathogenicity published so far.
Our study had several limitations. Our analysis is based on a homology model of human placental ALP as opposed to a direct 3D structural model of TNSALP. However, the template chosen had an X‐ray crystal structure of high‐resolution (1.8 Å) and high‐sequence identity with TNSALP (57%; Supporting Information Materials). As such, we believe that the computational methods used for this analysis would only improve further if a high‐resolution TNSALP structural model had been available. This study was not designed to capture mechanisms by which ALPL variants may become pathogenic other than disrupting TNSALP enzymatic activity. With regard to the estimation of TNSALP residual activity for compound heterozygous patients, this was determined based on the absence or presence of DNE, as described in Section 2.4 and the Supporting Information Materials. A better method, in principle, would be to produce a mutant/mutant mixture and measure its activity. However, the feasibility of this approach is limited in practice, as it would require testing all possible variant pairings (e.g., about 5,000 tests would be required for 100 variant pairings). Additionally, the plasmid‐based technology used for in vitro variant characterization cannot assess the pathogenicity of non‐exonic variants such as splicing, intronic or variants of the 5′/3′ untranslated region and is an involved and expensive process that is hard to scale to assess an ever‐increasing number of reported ALPL variants. The computational scoring of ALPL variants based on changes to protein stability could also be improved in the future, for example by using free‐energy perturbation methods (Ford & Babaoglu, 2017; Steinbrecher, Abel, Clark, & Friesner, 2017), which would allow simulating changes in protein backbone flexibility as well as explicit solvation effects. Additionally, whereas the focus of the computational aspect of this study was on predicting low residual enzymatic activity, further research should be done to improve the prediction of DNE from variants.
5. CONCLUSIONS
In total, 155 ALPL variants present in both the general population and HPP patients were assayed, expanding the catalog of known ALPL in vitro functional variation. Associations were observed between residual enzymatic activity and the source of the variant, type of variant, affected protein domain, and HPP subtype (only in terms of perinatal/infantile vs. others; there was no significant statistical difference in estimated in vitro residual activity between the childhood, adult, and odonto‐HPP groups). We described an approach combining structural model predictions with existing in silico algorithms that improved performance scores for estimating residual TNSALP enzymatic activity. Importantly, this study confirms that low in vitro residual activity supports classifying a variant as potentially disease‐causing. However, the predictive value of low in vitro TNSALP enzymatic activity is limited to very low values being associated with the most severe HPP subtypes (perinatal and infantile HPP). Although it is clear that TNSALP enzymatic activity alone cannot be used to assess HPP disease severity, this tool in combination with clinical assessments will further our understanding of the HPP phenotypic spectrum.
CONFLICT OF INTERESTS
G. d. A. and J. R. are employees of Alexion Pharmaceuticals, Inc., the study sponsor, and may own stock and/or stock options in the company. E. M. has received honoraria from Alexion Pharmaceuticals, Inc. C. N. is a former employee and T. S. is a current employee of Schrödinger and they may own stock and/or stock options in the company.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
In vitro functional assays were performed at RD‐Biotech (Besançon, France), under the joint direction of authors Guillermo del Angel and Etienne Mornet. Protein structural modeling and simulation were performed in collaboration with Schrödinger, Inc. (New York, NY). The authors wish to thank Mary Kunjappu and Meena Kathiresan from Alexion Pharmaceuticals, Inc., for their medical/scientific writing support, and Veruska Sena, Anna Petryk, Thomas Brown, and Sean Brugger from Alexion Pharmaceuticals, Inc., for their insights and suggestions. Editorial support was provided by Bina Patel from Peloton Advantage, LLC (Parsippany, NJ), an OPEN Health company, and funded by Alexion Pharmaceuticals, Inc. This study was sponsored by Alexion Pharmaceuticals, Inc., Boston, MA.
del Angel G, Reynders J, Negron C, Steinbrecher T, Mornet E. Large‐scale in vitro functional testing and novel variant scoring via protein modeling provide insights into alkaline phosphatase activity in hypophosphatasia. Human Mutation. 2020;41:1250–1262. 10.1002/humu.24010
DATA AVAILABILITY STATEMENT
Qualified academic investigators may request participant‐level, deidentified clinical data, and supporting documents (statistical analysis plan and protocol) pertaining to this study. Further details regarding data availability, instructions for requesting information and our data disclosure policy are available on the Alexion.com website (http://alexion.com/research‐development).
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 76, 7.20.1–7.20.41. 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner‐Sigl, S. , Haberlandt, E. , Mumm, S. , Scholl‐Burgi, S. , Sergi, C. , Ryan, L. , … Hogler, W. (2007). Pyridoxine‐responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T>C, p.M226T; c.1112C>T, p.T371I) of the tissue‐nonspecific alkaline phosphatase gene. Bone, 40(6), 1655–1661. 10.1016/j.bone.2007.01.020 [DOI] [PubMed] [Google Scholar]
- Beard, H. , Cholleti, A. , Pearlman, D. , Sherman, W. , & Loving, K. A. (2013). Applying physics‐based scoring to calculate free energies of binding for single amino acid mutations in protein‐protein complexes. PLOS One, 8(12), e82849 10.1371/journal.pone.0082849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkseth, K. E. , Tebben, P. J. , Drake, M. T. , Hefferan, T. E. , Jewison, D. E. , & Wermers, R. A. (2013). Clinical spectrum of hypophosphatasia diagnosed in adults. Bone, 54(1), 21–27. 10.1016/j.bone.2013.01.024 [DOI] [PubMed] [Google Scholar]
- Brun‐Heath, I. , Lia‐Baldini, A. S. , Maillard, S. , Taillandier, A. , Utsch, B. , Nunes, M. E. , … Mornet, E. (2007). Delayed transport of tissue‐nonspecific alkaline phosphatase with missense mutations causing hypophosphatasia. European Journal of Medical Genetics, 50(5), 367–378. 10.1016/j.ejmg.2007.06.005 [DOI] [PubMed] [Google Scholar]
- Brun‐Heath, I. , Taillandier, A. , Serre, J. L. , & Mornet, E. (2005). Characterization of 11 novel mutations in the tissue non‐specific alkaline phosphatase gene responsible for hypophosphatasia and genotype‐phenotype correlations. Molecular Genetics and Metabolism, 84(3), 273–277. 10.1016/j.ymgme.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Choi, Y. , Sims, G. E. , Murphy, S. , Miller, J. R. , & Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLOS One, 7(10), e46688 10.1371/journal.pone.0046688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvert, D. , Brun‐Heath, I. , Lia‐Baldini, A. S. , Bellazi, L. , Taillandier, A. , Serre, J. L. , … Mornet, E. (2009). Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Medical Genetics, 10, 51 10.1186/1471-2350-10-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, M. C. , & Babaoglu, K. (2017). Examining the feasibility of using free energy perturbation (FEP+) in predicting protein stability. Journal of Chemical Information and Modeling, 57(6), 1276–1285. 10.1021/acs.jcim.7b00002 [DOI] [PubMed] [Google Scholar]
- Greenberg, C. R. , Evans, J. A. , McKendry‐Smith, S. , Redekopp, S. , Haworth, J. C. , Mulivor, R. , & Chodirker, B. N. (1990). Infantile hypophosphatasia: Localization within chromosome region 1p36.1‐34 and prenatal diagnosis using linked DNA markers. American Journal of Human Genetics, 46(2), 286–292. [PMC free article] [PubMed] [Google Scholar]
- Hofmann, C. , Girschick, H. , Mornet, E. , Schneider, D. , Jakob, F. , & Mentrup, B. (2014). Unexpected high intrafamilial phenotypic variability observed in hypophosphatasia. European Journal of Human Genetics, 22(10), 1160–1164. 10.1038/ejhg.2014.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , … MacArthur, D. G. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv, 10.1101/531210 [DOI] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani, P. S. , Rush, E. T. , Arundel, P. , Bishop, N. , Dahir, K. , Fraser, W. , … Ozono, K. (2017). Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Molecular Genetics and Metabolism, 122(1‐2), 4–17. 10.1016/j.ymgme.2017.07.010 [DOI] [PubMed] [Google Scholar]
- Kozlowski, K. , Sutcliffe, J. , Barylak, A. , Harrington, G. , Kemperdick, H. , Nolte, K. , … Uniecka, W. (1976). Hypophosphatasia. Review of 24 cases. Pediatric Radiology, 5(2), 103–117. 10.1007/bf00975316 [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–d1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Du, M. H. , Stigbrand, T. , Taussig, M. J. , Menez, A. , & Stura, E. A. (2001). Crystal structure of alkaline phosphatase from human placenta at 1.8 Å resolution. Implication for a substrate specificity. Journal of Biological Chemistry, 276(12). [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, E. C. , Mhanni, A. A. , Reed, M. , Whyte, M. P. , Landy, H. , & Greenberg, C. R. (2013). Outcome of perinatal hypophosphatasia in Manitoba Mennonites: A retrospective cohort analysis. JIMD Reports, 11, 73–78. 10.1007/8904_2013_224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lia‐Baldini, A. S. , Brun‐Heath, I. , Carrion, C. , Simon‐Bouy, B. , Serre, J. L. , Nunes, M. E. , & Mornet, E. (2008). A new mechanism of dominance in hypophosphatasia: The mutated protein can disturb the cell localization of the wild‐type protein. Human Genetics, 123(4), 429–432. 10.1007/s00439-008-0480-1 [DOI] [PubMed] [Google Scholar]
- Lia‐Baldini, A. S. , Muller, F. , Taillandier, A. , Gibrat, J. F. , Mouchard, M. , Robin, B. , … Mornet, E. (2001). A molecular approach to dominance in hypophosphatasia. Human Genetics, 109(1), 99–108. 10.1007/s004390100546 [DOI] [PubMed] [Google Scholar]
- Liu, X. , Wu, C. , Li, C. , & Boerwinkle, E. (2016). dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita, S. , Al‐Shawafi, H. A. , Sultana, S. , Sohda, M. , Nomura, S. , & Oda, K. (2012). A dimerization defect caused by a glycine substitution at position 420 by serine in tissue‐nonspecific alkaline phosphatase associated with perinatal hypophosphatasia. FEBS Journal, 279(23), 4327–4337. 10.1111/febs.12022 [DOI] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. , Thormann, A. , … Cunningham, F. (2016). The Ensembl variant effect predictor. Genome Biology, 17(1), 122 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornet, E. (2015). Molecular genetics of hypophosphatasia and phenotype‐genotype correlations. Sub‐Cellular Biochemistry, 76, 25–43. 10.1007/978-94-017-7197-9_2 [DOI] [PubMed] [Google Scholar]
- Mornet, E. (2018). Hypophosphatasia. Metabolism: Clinical and Experimental, 82, 142–155. 10.1016/j.metabol.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Mornet, E. , Stura, E. , Lia‐Baldini, A. S. , Stigbrand, T. , Menez, A. , & Le Du, M. H. (2001). Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. Journal of Biological Chemistry, 276(33), 31171–31178. 10.1074/jbc.M102788200 [DOI] [PubMed] [Google Scholar]
- Mornet, E. , Yvard, A. , Taillandier, A. , Fauvert, D. , & Simon‐Bouy, B. (2011). A molecular‐based estimation of the prevalence of hypophosphatasia in the European population. Annals of Human Genetics, 75(3), 439–445. 10.1111/j.1469-1809.2011.00642.x [DOI] [PubMed] [Google Scholar]
- Quang, D. , Chen, Y. , & Xie, X. (2015). DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics, 31(5), 761–763. 10.1093/bioinformatics/btu703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry, G. M. , Adzhigirey, M. , Day, T. , Annabhimoju, R. , & Sherman, W. (2013). Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer‐Aided Molecular Design, 27(3), 221–234. 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Seshia, S. S. , Derbyshire, G. , Haworth, J. C. , & Hoogstraten, J. (1990). Myopathy with hypophosphatasia. Archives of Disease in Childhood, 65(1), 130–131. 10.1136/adc.65.1.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvent, J. , Gasse, B. , Mornet, E. , & Sire, J. Y. (2014). Molecular evolution of the tissue‐nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. Journal of Biological Chemistry, 289(35), 24168–24179. 10.1074/jbc.M114.576843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver, M. M. , Vilos, G. A. , & Milne, K. J. (1988). Pulmonary hypoplasia in neonatal hypophosphatasia. Pediatric Pathology, 8(5), 483–493. 10.3109/15513818809022304 [DOI] [PubMed] [Google Scholar]
- Sim, N. L. , Kumar, P. , Hu, J. , Henikoff, S. , Schneider, G. , & Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research, 40(W1), W452W457 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrecher, T. , Abel, R. , Clark, A. , & Friesner, R. (2017). Free energy perturbation calculations of the thermodynamics of protein side‐chain mutations. Journal of Molecular Biology, 429(7), 923–929. 10.1016/j.jmb.2017.03.002 [DOI] [PubMed] [Google Scholar]
- Stevenson, D. A. , Carey, J. C. , Coburn, S. P. , Ericson, K. L. , Byrne, J. L. , Mumm, S. , & Whyte, M. P. (2008). Autosomal recessive hypophosphatasia manifesting in utero with long bone deformity but showing spontaneous postnatal improvement. Journal of Clinical Endocrinology and Metabolism, 93(9), 3443–3448. 10.1210/jc.2008-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillandier, A. , Domingues, C. , Dufour, A. , Debiais, F. , Guggenbuhl, P. , Roux, C. , … Mornet, E. (2018). Genetic analysis of adults heterozygous for ALPL mutations. Journal of Bone and Mineral Metabolism, 36(6), 723–733. 10.1007/s00774-017-0888-6 [DOI] [PubMed] [Google Scholar]
- University of Versailles‐Saint Quentin . (2019). The tissue nonspecific alkaline phosphatase gene mutations database . Retrieved from http://www.sesep.uvsq.fr/03_hypo_mutations.php
- Weber, T. J. , Sawyer, E. K. , Moseley, S. , Odrljin, T. , & Kishnani, P. S. (2016). Burden of disease in adult patients with hypophosphatasia: Results from two patient‐reported surveys. Metabolism: Clinical and Experimental, 65, 1522–1530. 10.1016/j.metabol.2016.07.006 [DOI] [PubMed] [Google Scholar]
- Weiss, M. J. , Cole, D. E. , Ray, K. , Whyte, M. P. , Lafferty, M. A. , Mulivor, R. A. , & Harris, H. (1988). A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proceedings of the National Academy of Sciences of the United States of America, 85(20), 7666–7669. 10.1073/pnas.85.20.7666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte, M. P. (2016). Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nature Reviews: Endocrinology, 12(4), 233–246. 10.1038/nrendo.2016.14 [DOI] [PubMed] [Google Scholar]
- Whyte, M. P. (2017). Hypophosphatasia: An overview for 2017. Bone, 102, 15–25. 10.1016/j.bone.2017.02.011 [DOI] [PubMed] [Google Scholar]
- Whyte, M. P. , Rockman‐Greenberg, C. , Ozono, K. , Riese, R. , Moseley, S. , Melian, A. , … Hofmann, C. (2016). Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. Journal of Clinical Endocrinology and Metabolism, 101(1), 334–342. 10.1210/jc.2015-3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte, M. P. , Zhang, F. , Wenkert, D. , McAlister, W. H. , Mack, K. E. , Benigno, M. C. , … Mumm, S. (2015). Hypophosphatasia: Validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone, 75, 229–239. 10.1016/j.bone.2015.02.022 [DOI] [PubMed] [Google Scholar]
- Wickham, H. (2016). ggplot2: Elegant graphics for data analysis. New York, NY: Springer‐Verlag. [Google Scholar]
- Zurutuza, L. , Muller, F. , Gibrat, J. F. , Taillandier, A. , Simon‐Bouy, B. , Serre, J. L. , & Mornet, E. (1999). Correlations of genotype and phenotype in hypophosphatasia. Human Molecular Genetics, 8(6), 1039–1046. 10.1093/hmg/8.6.1039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Data Availability Statement
Qualified academic investigators may request participant‐level, deidentified clinical data, and supporting documents (statistical analysis plan and protocol) pertaining to this study. Further details regarding data availability, instructions for requesting information and our data disclosure policy are available on the Alexion.com website (http://alexion.com/research‐development).