

Abstract
Mouse models of medulloblastoma have proven to be instrumental in understanding disease mechanisms, particularly the role of epigenetic and molecular drivers, and establishing appropriate preclinical pipelines. To date, our research community has developed murine models for all four groups of medulloblastoma, each of which will be critical for the identification and development of new therapeutic approaches. Approaches to modeling medulloblastoma range from genetic engineering with CRISPR/Cas9 or in utero electroporation, to orthotopic and patient‐derived orthotopic xenograft systems. Each approach or model presents unique advantages that have ultimately contributed to an appreciation of medulloblastoma heterogeneity and the clinical obstacles that exist for this patient population.
Keywords: Group 3, Group 4, medulloblastoma, mouse models, MYC, MYCN, patient‐derived xenografts, Sonic hedgehog, WNT
Introduction
Medulloblastoma (MB) is the most common pediatric malignant brain tumor and occurs mainly in children aged between 3 and 9 years 48. Medulloblastoma is characterized by four major molecular groups, with different pathologies, outcomes and genetics: two with mutations in developmental pathways, Wingless (WNT) and Sonic Hedgehog (SHH), and two with less well‐defined molecular alterations, Group 3 (G3) and Group 4 (G4) 53, 71. Recently, each of the four groups has been further subdivided into subgroups by methylation profiling 14, 52. However, it is not yet clear whether specific subgroups are associated with distinct outcomes.
Since the generation of the first genetically engineered mouse model of Sonic Hedgehog medulloblastoma, in which the Sonic Hedgehog receptor, Patched (Ptch1), was conditionally deleted 26, a large number of mouse models recapitulating each of the four groups has been developed. As medulloblastomas are separated into more subgroups, the development of accurate models recapitulating each of these is warranted if these models are to be used for preclinical studies. Several approaches have been employed over the years, but the discovery of CRISPR‐Cas9 enzymes has accelerated the development and the precision with which mouse models are generated. This review will discuss all mouse models developed to date and how they have already been used to inform the development of clinical trials.
Approaches to Modeling Medulloblastoma in Mice
The molecular analysis of human medulloblastoma has been instrumental in the development of mouse models that closely resemble their human counterparts. Several characteristics must be met for medulloblastoma mouse models to recapitulate human tumors. They must represent disease heterogeneity, recapitulate developmental origins and mimic molecular, epigenetic and genetic landscapes. Several approaches have been used over the years to develop accurate models, many of which fulfill most, if not all, of these requirements. These models have helped investigators better understand the disease.
Approaches to the development of mouse models or genetically engineered mouse models (GEMMs) include genetic engineering, in utero electroporation, viral gene transfer by the replication‐competent avian sarcoma leukosis virus and its receptor Tva (RCAS‐Tva) system and the Sleeping Beauty transposase, orthotopic transplant models using mouse or human cerebellar and neuronal progenitors, and the development of established mouse and human cell lines and patient‐derived orthotopic xenografts (PDOXs) (Figure 1). Each of these methods has advantages and disadvantages but all have facilitated the validation of genetic alterations found in human medulloblastoma groups, with the goal of finding new therapies and improving existing ones.
Figure 1.
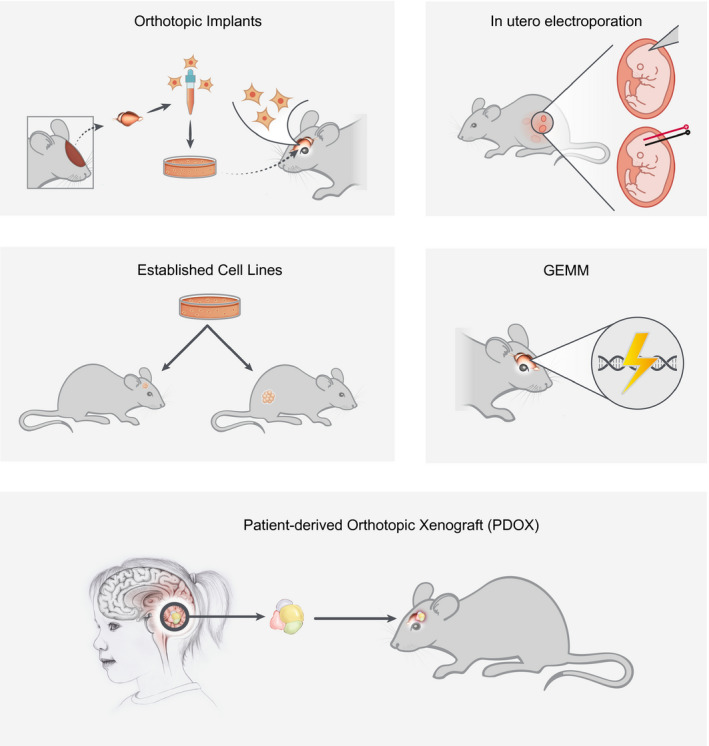
Summary of technical methods and approaches to medulloblastoma modeling.
In vitro established cell lines
Several human medulloblastoma cell lines (44) have been established in vitro over the years and have been well described 34. Not surprisingly, most were generated using aggressive Group 3 MBs with MYC amplification. Although these established human tumor lines are easy to grow in culture, as monolayers or spheres, and are commonly used to assess the effect of drug treatments in preclinical trials, recent molecular analysis by next generation sequencing (NGS) has revealed that they do not always faithfully mimic primary tumors and in some cases these lines have acquired additional mutations and/or have lost genetic material. In our own experience, while mouse Group 3 tumors are easily expandable in vitro and recapitulate the disease when re‐introduced into the cortex or cerebellum from naïve animals 37, we have been unable to generate stable lines from primary patient samples or PDOXs at low passage < 2 (Roussel, personal communication). Mouse or human SHH tumors fail to maintain a SHH signaling pathway signature when grown in vitro, and fail to generate tumors when cultured cells are implanted in the brain of mice, as previously reported 67. However, we have successfully grown PDOXs, both SHH tumors with MYCN amplification and TP53 mutations as well as MYC‐driven Group 3 tumors, transiently for 3–7 days in neural stem cell culture conditions. These transient cultures allow high throughput drug screens and the evaluation of specific compounds for their ability to induce perturbations of cell proliferation, cell death, cell cycle arrest or differentiation. We find that these culture conditions maintain tumor stem‐like properties and a tumor's genomic landscape (Roussel, personal communication).
Genetically engineered mouse models
Some of the most accurate models of medulloblastoma have used conventional knock‐out technology to generate genetically engineered mouse models (GEMMs). The Patched 1 model (Ptch1+/−), the first mouse model of medulloblastoma to be developed, has been extensively used to assess the role of genes that drive tumorigenesis in SHH MBs. This includes mutations in genes of the SHH signaling pathway, from the cell surface to the nucleus, that drive G1 progression and impact DNA repair and apoptotic pathways (Table 1). Realizing that some of the gene deletions were deleterious to embryonic development, many conditional and inducible knock‐out models have been developed, enabling precise temporal and spatial gene deletion. This required the development of mouse lines in which Cre recombinase expression is driven in specific cerebellar cell types. In many examples, such transgenic mice express a Cre recombinase fused to the mutated estrogen receptor (ERTM) allowing its expression by addition of tamoxifen. When fused to the mutated estrogen receptor, the Cre‐ERTM fusion protein is sequestered to the cytoplasm, but upon tamoxifen treatment is translocated to the nucleus enabling the expression of the Cre recombinase 20, 39. Cre technology permits the deletion of genes at specific times and in specific cell types in the mouse brain during embryonic development and also in adult mice. A number of Cre and Cre‐ERTM lines have been developed over the years and are now available to investigators. These lines have been well characterized and successfully used to study the role of gene deletion upon Cre excision in specific cerebellar cell types (Table 2).
Table 1.
Models of SHH medulloblastoma
| SHH GEMM | Reference |
|---|---|
| Ptch1+/−, LacZ | 26 |
| Ptch1+/−, Trp53−/− | 76 |
| Ptch1+/−, Cdkn2c−/− | 73 |
| Ptch1+/−, Math1‐Cre | 68, 78 |
| Ptch1+/−. hGFAP‐Cre | 68, 78 |
| Ptch1+/−, Math1‐CreER | 47 |
| SmoA2 | 5, 16 |
|
NeuroD2‐SmoA1 W539L |
27, 30 |
| Sufu+/−, Trp53−/− | 42 |
| Trp53−/−, H3K27M | 40 |
| Trp53−/−. XRCC4−/− | 77 |
| Trp53−/−, LigIV‐/‐ | 43 |
| Nestin‐Cre+/−, Trp53−/−, Brca2fl/Fl | 22 |
| Ptch1+/−, Cdkn1b−/− | 7 |
| Ptch1+/−, Ptch2+/− | 45 |
| KU80−/−, Trp53−/− | 43 |
| Parp−/−, Trp53−/− | 72 |
| PTEN floxed × RCAS‐Cre + RCAS‐Shh + radiation | 28 |
| Trp53−/−, PTEN−/− | 79 |
| Trp53−/−, RB−/− | 49 |
| Ink4d−/−, Kip1−/−, Trp53−/− | 44 |
| Ink4d−/−, Ink4c−/−, Trp53−/− | 44 |
| Ptch1+/−, Hic+/− | 12 |
| RCAS‐Shh + RCAS‐Mycn in Ntv‐a mice | 13 |
| RCAS‐Shh + RCAS‐MycnT48A in Ntv‐a mice | 13 |
| RCAS‐Shh + RCAS‐IGF2 in Ntv‐a mice | 60 |
| RCAS‐Shh + RCAS‐BCL2 in Ntv‐a mice | 50 |
| RCAS‐Shh, RCAS‐MYC, RCAS‐BCL2 in Ntv‐a mice | 35 |
| RCAS‐Shh + RCAS‐WIP1 in Ntv‐a mice | 18 |
| RCAS‐Shh + RCAS‐HGF in Ntv‐a mice | 10 |
| RCAS‐Shh + RCAS‐MYC | 60 |
| Math1‐Cre; Nfia+/+; Ptch1+/Lox | 24 |
| Nestin‐Cre/T2‐ONC × PTENFlox/Flox × Rosa26LslSB11/+ | 9 |
| SHH Orthotopic | |
| Trp53−/−, MYCN | 31, 37 |
| Cdk6 | 59 |
| Atoh1 + Gli1 | 6 |
| Trp53−/−, Cdn2c−/−, MYCN | 37 |
| MYCN in human iPSC‐derived NES | 33 |
| SHH In utero electroporation | |
| CRISPR‐Cas9 Ptch1 deletion | 64 |
Table 2.
Models of medulloblastoma
| WNT GEMM | Reference |
|---|---|
| Blbp‐Cre, Ctnnb1+/fl (ex3), Trp53+/fl | 25 |
| Blbp‐Cre, Ctnnb1+/fl (ex3), Trp53+/fl, PI3CAOE | 25 |
| Blbp‐Cre, DDX3Xfl/fl | Gilbertson, personal communication |
| G3 GEMM | |
| Gtl1‐tTA:TRE‐MYCN‐Trp53−/− (GTML) | 70 |
| Nestin‐Cre‐MLL4 | 17 |
| GMYC MYC GLT1 Tet‐OFF System | Swartling, personal communication |
| GTML Trp53KI/KI p53ER, TAM | 31 |
| RCAS‐TVA MYC + BCL2 in Ntv‐a mice | 35 |
| RCAS‐TVA MYC + Trp53 | 35 |
| Nestin‐Cre/T2‐ONC × Trp53LslR270H × Rosa26LslSB11/+ | 9 |
| G3 Orthotopic | |
| Myc OE, Trp53−/− in GNPs | 37 |
| Myc OE, Trp53DN in GNPs and NSCs CD133+.Lin‐ | 57 |
| Dox inducible MYC, DNTrp53 | 57 |
| MycT58A OE, Trp53DN in GNPs and NSCs | 57 |
| Myc OE, GFI1 in CD133 + Lin‐ NSC | 54 |
| Myc OE, GFI1OE in GNPs | 75 |
| Myc T58A OE, GFI1Flox in NSCs from CAG‐CreERTM mice | 46 |
| MycT58A OE, GFI1WT in NSCs | 46 |
| CRISPR‐dCas9‐MYC | 74 |
| G3 In utero electroporation | |
| conditional MYC, DNTrp53 in different neuronal progenitors | 36 |
| G4 In utero electroporation | |
| constitutively activated Src, Trp53DN | 21 |
GEMMs of medulloblastoma have been developed by somatic gene transfer using polyethylenimine (PEI)‐mediated transfection 2 and in utero electroporation 65 of plasmids carrying the genes of interest in the fourth ventricle of embryos (E) at 13.5 days post coitus. The advantage of these approaches is that they do not suffer the limitations imposed by the size of the retroviral and lentiviral vectors which cannot be much bigger than 10–15 kilobases and could provide tissue and cell‐type specificity by combination with CRISPR‐Cas9 or with specific Cre‐mice. For example, somatic CRISPR‐Cas deletion of Ptch1 by in utero electroporation of wild type E13.5 mouse embryos (“CRISPR‐Ptch1”) induced SHH medulloblastoma with complete penetrance by 16 weeks of age. Tumor onset is accelerated when these experiments are performed in Trp53‐null embryos. These tumors have the hallmarks of human SHH medulloblastomas 64 including overexpression of Gli1 and Sfrp1.
Several GEMMs of SHH or Group 3 medulloblastoma made use of the RCAS‐TVA system 4. This technique relies on the use of an avian retroviral vector, RCAS, to target gene expression to neuronal progenitors in transgenic mice in which the Nestin gene promoter drives expression of the viral receptor, tva 3, 23, 41, 55. Several SHH models were developed by infection with RCAS virus expressing Shh, alone or in combination with genes that include MYCN, activated Akt, HGF, WIP1, BCL2 all of which accelerate the onset of SHH medulloblastoma (Table 1). GEM models of Group 3 MB were also generated in which the chicken retrovirus expressed the oncogenes Myc and Bcl2 in addition to RCAS‐Shh which when transferred into the brain of mice induces Group 3 medulloblastoma 35 with large cell/anaplastic (LCA) features.
Another approach involves insertional mutagenesis with the transposase Sleeping Beauty that when conditionally expressed in Nestin‐Cre mice together with a Trp53 mutation (Nestin‐Cre/T2‐ONC × Trp53LslR270H × Rosa26LslSB11/+) or PTEN knock‐out (Nestin‐Cre/T2‐ONC × PTEN Flox/Flox × Rosa26LslSB11/+) induces SHH or Group 3/Group 4 medulloblastoma 9.
Orthotopic transplant of genetically modified precursors
Whereas GEMMs were the only mouse models of medulloblastoma initially developed for the SHH and WNT groups, for which mutations of Ptch1 and β‐catenin had been identified in human tumors, a lack of obvious genetic drivers was initially responsible for the delay in developing GEMMs for the other two MB groups. As a result, we and other investigators initially decided to use purified granule neuronal progenitors (GNPs) 37 or neural stem cells (NSCs/CD133+/Prominin+, Lin−) 56, to assess the role of potential drivers of medulloblastoma, including Myc and Gfi1 53, 56. This evaluation made use of retroviral or lentiviral vectors that could conditionally express or repress genes of interest to modify mouse neuronal progenitors or human induced pluripotent stem cells (iPSC)‐derived neuroepithelial stem cells (NSC) 33. Marked progenitors are then implanted into the cortices or cerebella of naïve immune‐compromised NSG or CD1‐nude mice or of naïve syngeneic animals, giving rise to tumors consistent with Group 3 based on histopathological and molecular analyses.
Patient‐derived xenograft models
GEMMs of MB provide valuable tools for in vitro and in vivo testing but fall short of capturing the heterogeneity or microenvironment of human tumors. Patient‐derived orthotopic xenograft (PDOX) models address these limitations and have become increasingly prevalent in MB preclinical research. PDOXs are generated by implanting tissue from a patient's tumor into immune‐compromised mice. After initial processing of the tumor sample, there are no intermediate in vitro steps, eliminating the risk of artifact or genetic drift that can arise during cell culture. Most groups working with MB PDOXs utilize orthotopic models, amplifying tumors intracranially in vivo. The presence of stromal environmental components and the heterogeneity of the tumor cell population provide a significant advantage, particularly with regard to preclinical evaluation of small molecules or other interventions 11, 66, 69. Research from St. Jude Children's Research Hospital, the German Cancer Research Center and the Fred Hutchinson Cancer Research Center suggests that the genetic, epigenetic and molecular signatures of MB tumors are maintained from patient sample to PDOX models (11, 32, 66, Roussel, personal communication). Unlike the artificially homogenous cell lines generated from genetic manipulation of progenitor cells, PDOX models retain intra‐ and inter‐tumoral heterogeneity that facilitates more accurate interrogation of disease mechanism and therapeutic response. Currently, PDOX models exist for each of the major MB groups, some from primary tumor biopsy and others from disease relapse or metastasis. Single cell RNA sequencing of PDOX models continues to provide insights into spatial and temporal tumor evolution, while analysis of the genetic and epigenetic landscape reveals new insights into tumorigenesis and progression 32.
Mouse models of the four major medulloblastoma groups
The wealth of data on human medulloblastomas from multiple “omics” approaches has facilitated the development of mouse models for each medulloblastoma group and some of the recently described subgroups (Figure 2).
Figure 2.
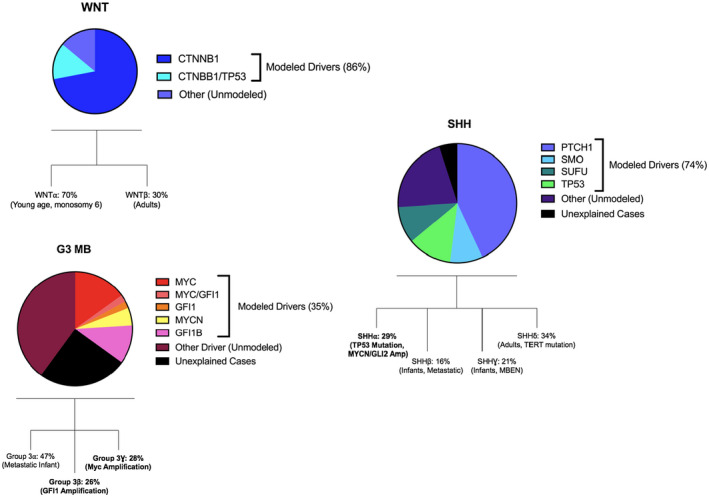
For each subgroup of medulloblastoma, the proportions of driver events as determined by Next‐Generation Sequencing (NSG) are displayed; Copy Number Variants (CNV) and mutation analysis. Modeled Drivers: of all driver events identified by NGS for this subgroup, the proportion represented by established in vivo models. Other: Driver events identified by NGS which have not been modeled in vivo. Unexplained Cases: cases of this MB group for which no events have been identified through NGS approaches. Recently, defined subdivisions of groups into subgroups are also displayed 14, with those in bold having corresponding in vivo models.
WNT
The molecular landscape of Wingless (WNT) group medulloblastomas is relatively well understood, and as such the genetically engineered mouse model (GEMM) developed by Gibson and collaborators has proved ideal to study this disease group 25. The majority (~85%) of patients with WNT MB harbor mutations in β‐catenin (CTNNB1) leading to over‐activation of the WNT signaling pathway, the nuclear localization of β‐catenin, and accelerated cell division and proliferation 19. Initial experiments targeted overexpression of a mutated CTNNB 1S37F in mouse Atoh1‐positive granule neural progenitors with or without Trp53 loss. Whereas β‐catenin protein was clearly overexpressed, no tumors ever formed, suggesting that GNPs were not the cell of origin of WNT MBs. It was only by mapping genes expressed in WNT MBs during embryonic cerebellar development that Gibson and collaborators discovered the cell of origin of WNT MBs in the floor of the fourth ventricle. Crossing the Blbp‐Cre line into Ctnnb1lox (ex3) × Trp53flx /flx mice drove the initiation of tumors closely resembling WNT MB in anatomic features, chromosomal changes and transcriptomic landscape. The penetrance of the disease was further increased by enforced expression of PI3KCA to activate the PI3K pathway. Importantly, analysis of this WNT model revealed an aberrant tumor blood–brain barrier, consistent with increased propensity for hemorrhage at surgery and improved response to chemotherapy 58. Because patients with WNT MBs respond well to current therapy and recognizing that intensive craniospinal irradiation causes long‐term neurological sequelae, this model has facilitated the evaluation of CSI dose reduction (ongoing SJ clinical trial with lower radiation doses—NCT02724579). Recently, a new model of WNT MB has been created based on recurrent DDX3X mutation found in a subset of WNT MB tumors 52 (Richard Gilbertson, personal communication).
SHH
Since the development of the first Ptch1+/− GEM model in 1997 26, a plethora of SHH medulloblastoma mouse models has been developed using multiple approaches (Table 1). Many investigators generated these models by initially testing the role of mutations and overexpressed genes found in human SHH medulloblastomas. Initial GEMMs all led to tumors mimicking the SHH group 16, 42, 68. This was the first set of data pointing to GNPs as the cell of origin of SHH medulloblastoma. Subsequent experiments confirmed that SHH medulloblastomas arose from GNPs that were blocked in their exit from the cell cycle and differentiation to post‐mitotic granule cells, thus remaining in the external granule layer 6. Recognizing that GNPs are highly susceptible to DNA damage, the McKinnon laboratory and others tested the role of DNA repair enzymes responsible for homologous recombination (HR) and non‐homologous end joining (NHEJ) 43, 77. These investigators found that loss of enzymes, including Ligase IV, Xrcc4 and Brca2 led to embryonic lethality, which was rescued by the loss of Trp53. While mice developed into adulthood, they all succumbed to SHH medulloblastomas. These studies emphasized the importance of intact p53 and DNA repair pathways in maintaining a healthy cerebellum during development. Whereas SHH MB models recapitulate the human tumors, they rely on the modification of mouse cells. Recent humanized models have been developed using human neuroepithelial stem cells (NSC) from iPSCs that are representative of cerebellar progenitors. Transduction of MYCN in human NSC induced tumors of the SHH medulloblastoma subgroup 33. Interestingly, these tumors did not lose TP53 function nor the PTCH1 gene. The same group showed that orthotopic transplantation of NSC cells from a Gorlin patient caring a germline PTCH1 mutation also induced SHH medulloblastoma. It is likely that the use of human NSC will be important to model mutations and gene fusions found in human medulloblastoma.
Group 3
Overexpression of the MYC oncogene typifies Group 3 medulloblastomas. MYC is amplified in about 17% of cases and correlates with frequent metastasis at diagnosis, an aggressive clinical behavior and poor prognosis. While MYC overexpression is seen in most human Group 3 MBs, rare tumors in this group display MYCN amplification.
The first mouse model of G3 MB was developed in a GEMM in which MYCN expression was driven by the glutamate transporter 1 (Glt1) promoter expressed in hindbrain progenitors (GTML) 70. In this model, most tumors exhibit a classic or large cell/anaplastic (LCA) morphology and a G3 MB molecular profile, although some more closely resemble other groups. Crossing the Glt1‐tTA; TRE‐MYCN/luciferase model to mice lacking Trp53 accelerates tumorigenesis. Because many recurrent cases of human G3 MB have deletion of one copy of TP53 as part of an isochromosome 17q, this transgenic model might represent a more efficient tool to examine MB relapse 31. Despite the histological relevance of the GTML model, MYCN amplification is far less common than MYC amplification in G3 MB. To overcome this criticism, the Swartling group recently developed a derivative model (GMYC), in which MYC is driven from the hindbrain‐specific glutamate transporter 1 (Glt1) promoter. Demonstrating differential expression of key features between MYC‐ and MYCN‐driven tumors, this new GMYC model highlights a role for CDKN2A in MB pathogenesis (Swartling, Personal Communication).
In 2012, our group and Wechsler‐Reya's laboratory independently developed mouse models of G3 MB that mimicked the histology and gene expression signatures of human tumors. Both groups leveraged enforced MYC overexpression by retroviral gene transfer and the loss of Trp53 function to reprogram granule cell neuronal progenitors (GNPs) or Prominin/CD133‐positive neural stem cells 37, 57. These two models relied on orthotopic transplants of modified neuronal progenitors in the cerebellum or cortices of naïve recipient mice. To better define the cell of origin of Group 3 medulloblastomas, Kawauchi and colleagues conditionally enforced MYC co‐expression with luciferase and a dominant form of Trp53 (DNp53) by in utero electroporation in embryonic cerebellar progenitor cells using several specific Cre lines. Irrespective of the cerebellar lineage targeted, all tumors were Group 3 suggesting that the combination of the two genetic insults, rather than a specific cell of origin, drive this MB group This conclusion was recently confirmed by single cell sequencing of primary tumor samples from patients with human Group 3 MBs 32. Because these G3 MB mouse models grow in vitro as spheres in stem cell media conditions, they have been instrumental for conducting high‐throughput drug screens and identifying novel therapies, some of which are currently in clinical trials (SJMB12‐NCT01878612, SJDAWN‐NCT03434262 and SJELiOT‐NCT04023669.
Most existing models of G3 MB rely on predetermined genetic perturbation and complex preimplantation protocols. In order to mimic the sporadic genetic changes that occur during tumor initiation in somatic cells, Jenkins et al developed a retroviral transfer system whereby MYC is delivered to and ectopically expressed in Nestin‐positive neural progenitor cells in postnatal mice 35. Similar to previous models, this RCAS/tv‐a model of G3 MB requires Trp53 loss or overexpression of Bcl‐2 to escape Myc‐induced apoptosis. Uniquely, this model develops within an intact host immune system which enables interrogation of the native microenvironment and immunotherapeutic testing.
In most of the murine models of G3 MB developed to date, MYC overexpression is driven by strong retroviral promoter elements. While this approach is effective at generating G3 MB tumors, it presents a limitation in investigating the signals that regulate MYC transcription. To circumvent this challenge, Vo and collaborators developed CRISPR/dCas9 gene activation in which endogenous MYC is activated by dCas9‐VP160 in Trp53‐null neural progenitors 74. Many of these tumors recapitulate G3 MB by histology and microarray analysis and importantly provide a useful model to interrogate endogenous Myc regulation and signaling.
Since the development of GEMM and orthotopic mouse models of G3 MB that all rely on the loss of Trp53, recent studies have demonstrated that enforced MYC expression can collaborate with GFI1 or GFI1B overexpression in neural progenitors, bypassing the requirement for Trp53 loss 54, 75. Prevalent genomic structural variants have been identified in human G3 and G4 MBs that result in the activation of the transcription factor repressors GFI1 and GFI1B by rearranging coding sequences to juxtapose them to active enhancer elements. These novel mouse models recapitulate one of the recently described subgroups of MYC‐driven Group3 medulloblastoma 14. Because medulloblastomas have a paucity of mutations but many chromosomal anomalies, it is likely that currently undiscovered rearrangements may lead to new targets and mouse models.
Recent sequencing studies have revealed mutations in epigenetic regulators in all medulloblastoma groups, including Group 3, but it is unclear whether these mutations drive tumor development. One epigenetic regulator Mll4 mutated in Group 3 MB was recently found to be a driver mutation 17. Whether other genetic alterations in chromatin regulators are driving medulloblastoma development will require further studies including CRISPR or/and shRNA screens of libraries of epigenetic regulators.
Group 4
Phosphoproteomic studies of human medulloblastomas have provided a better understanding of potential drivers in G4 tumors and facilitated the development of a G4 medulloblastoma model that has eluded investigators up to now. Investigators showed that G4 tumors harbor a phosphoproteomic landscape distinct from their RNA signature and characterized by activation of a tyrosine kinase program highlighting a unique receptor tyrosine signaling network (ERBB4‐SRC) specific to this group 21. Using these insights, the first mouse model of G4 MB was created using in utero electroporation to deliver a dominant negative form of Trp53 (DNp53) and a constitutively active form of SRC, with a truncated C‐terminal domain (SRC‐CA), into the fourth ventricle of E13.5 mouse embryos, an age selected to target the nuclear transitory zone (NTZ) progenitors of the developing cerebellum. Comparative analysis by cross‐species comparison confirmed that gene expression most closely resembled G4 MB. This model for the first time recapitulates the landscape integrity of G4 MBs and suggests that G4 MBs could be susceptible to kinase inhibitors, many of which are widely available and used in treating adult cancers.
Preclinical Use of Mouse models of MB
A key impetus for the development of accurate and representative mouse models of medulloblastoma was to create preclinical testing tools. The requisite characteristics of preclinical models are now represented across the spectrum of mouse models. Tumors driven by specific genetic perturbations are useful for assessing response to targeted therapies (SMO inhibitors, BET inhibitors, etc), while syngeneic orthotopic models will be invaluable to explore the immune landscape in medulloblastoma. PDOXs are increasingly becoming the gold standard for preclinical testing, consistent with a trend away from established cell lines that fail to recapitulate the primary tumors from which they are derived. The heterogeneity of MB warrants testing interventional approaches in multiple models, particularly to define relevant patient populations for trial stratification. Most of the preclinical work in MB has focused on SHH and Group 3, due in part to the development of multiple representative models and the need for improved therapies for the high‐risk subsets of these groups. However, major challenges are the blood–brain and blood–tumor barriers. These have limited the number of drugs that can be used to treat MB patients. While several drugs and small molecules have been tested in mouse models and found to be efficacious in suppressing medulloblastoma proliferation, most used established cell lines and flank tumors. Thus, ideally preclinical trials should be performed in multiple mouse models in vitro and in vivo and should require that all drugs be investigated for their brain and tumor penetration.
SHH
Some of the earliest preclinical studies for MB capitalized on PTCH and SMO mutations in SHH tumors and explored the potential of SMO inhibitors like Vismodegib and Sonidegib 63. The most common model to evaluate efficacy of SHH pathway inhibitors has been the Ptch1−/−; Trp53−/− mouse model, followed closely by SHH PDOXs. Our understanding of the signaling pathways involved in SHH MB, and the availability of a GEMM model that so closely recapitulates the genetic signature of these tumors has been immensely valuable in translating preclinical testing into Phase II clinical trials PBTC‐025B and PBTC‐032 (Vismodegib for recurrent or refractory MB with a 41% response in SHH patients) 62. However, as suggested by mouse studies, treatment of children with Vismodegib experienced growth plate fusion, and this has stimulated the identification of alternative therapeutics for this patient population 38, 61.
Molecular analysis of tumor propagating cells in the Math‐GFP/Ptch1 +/− mouse model revealed increased expression of G2/M regulators and led to the evaluation of Aurora Kinase (AurkA) and Polo‐like kinase 1 (Plk1) inhibitors as targeted therapies 29. Whether these small molecules effectively cross the blood–brain barrier is unknown. Preliminary studies from collaborative teams at St. Jude Children's Research Hospital, Telethon Kids Institute in Perth, Australia and the German Cancer Research Center (DKFZ) in Heidelberg, Germany are utilizing high‐risk SHH PDOX models of MB to test the preclinical efficacy of checkpoint inhibitors in combination with traditional chemotherapy and the DNA damage agent cyclophosphamide. The clinical trial based on this work (SJELiOT–NCT04023669) was recently approved by the FDA and began enrolling in 2019.
Group 3
Despite the relatively quiet mutational landscape of Group 3 MBs, early mouse models faithfully recapitulated the most common perturbation of MYC overexpression. Many groups use the Myc‐driven, Trp53−/− mouse model to investigate the utility of small molecule inhibitors. Because this model is amenable to in vitro culture, high‐throughput screening can be conducted to identify novel small molecules. Pei and collaborators identified a cooperative effect between HDAC inhibitors and PI3K inhibitors in this, and other PDOX models, while Morfouace and collaborators demonstrated a new combination approach of pemetrexed and gemcitabine 1, 51, 56. From these, preclinical efforts have emerged early stage clinical trials including SJMB12‐NCT0187861 (Pemetrexed and Gemcitabine for Newly diagnosed patients with Non‐WNT, Non‐SHH, MB). A more recent St. Jude clinical trial, SJDAWN‐NCT03434262 include the use of a CDK4/6 inhibitor (ribociclib) in combination with gemcitabine for recurrent G3 MB. Inhibition of CDK4/6 with Palbociclib (IBRANCE) was found to significantly extend the survival of mice harboring a MYC‐amplified Group3 medulloblastoma patient‐derived orthotopic xenograft, Med‐311FH 15. These studies stimulated a phase 1 study with Palbociclib in children with recurrent, progressive or refractory CNS tumors including medulloblastoma (PBTC‐042). More recently, inhibitors of DNA repair enzymes Check 1 and Check 2 were found to efficiently suppress the proliferation of mouse and human Group 3 medulloblastomas, with or without TP53 mutation, in combination with the DNA‐damaging agents, gemcitabine or cyclophosphamide (Roussel and Gottardo, personnal communication). The clinical trial designed based on this work (SJELiOT, NCT04023669 was recently approved by the FDA and began enrolling in July 2019.
Several groups have used the Myc‐amplified GEMM model and various PDOX models to demonstrate the efficacy of epigenetic regulation perturbation via BET bromodomain inhibitors 8, 74. Early G3 models were created using exogenous plasmids with foreign promoters to drive overexpression of Myc. Although they have provided preclinical value, Vo and collaborators recently developed a CRISPR‐Myc‐driven system that leveraged the endogenous Myc promoter and created a better tool to interrogate inhibitors that impact epigenetic regulation at the MYC promoter 74.
Conclusion
Since the development of the SHH medulloblastoma Ptch1 +/− mouse model, all four medulloblastoma groups have been modeled in mice, and many have been used in preclinical studies that have led to clinical trials. It is likely that in the near future new mouse models will be developed that will accurately recapitulate MB subgroups. This should strengthen the preclinical pipeline and contribute to finding novel therapies for the most aggressive forms of medulloblastoma.
Conflict of Interest
The authors have no conflict of interest to declare.
Acknowledgments
We thank Drs William A. Weiss, Frederick Swartling and Richard J. Gilbertson for providing their unpublished data. This work was funded in part by NIH grant CA‐96832, a Cancer Center Core grant CA‐21765 (MFR) and the American Lebanese‐Associated Charities (ALSAC) of St. Jude Children's Research Hospital.
References
- 1. Aaboud M, Aad G, Abbott B, Abdallah J, Abdinov O, Abeloos B et al (2017) Reconstruction of primary vertices at the ATLAS experiment in Run 1 proton‐proton collisions at the LHC. Eur Phys J C Part Fields 77:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdallah B, Hassan A, Benoist C, Goula D, Behr JP, Demeneix BA (1996) A powerful nonviral vector for in vivo gene transfer into the adult mammalian brain: polyethylenimine. Hum Gene Ther 7:1947–1954. [DOI] [PubMed] [Google Scholar]
- 3. Ahronian LG, Lewis BC (2014) In vivo delivery of RCAS virus to mice. Cold Spring Harb Protoc 2014:1167–1169. [DOI] [PubMed] [Google Scholar]
- 4. Ahronian LG, Lewis BC (2014) Using the RCAS‐TVA system to model human cancer in mice. Cold Spring Harb Protoc 2014:1128–1135. [DOI] [PubMed] [Google Scholar]
- 5. Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G et al (2016) Small cell lung cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J Thorac Oncol 12:704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF (2010) Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma‐initiating cells. Cancer Res 70:5618–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF (2009) Two tumor suppressors, p27Kip1 and patched‐1, collaborate to prevent medulloblastoma. Mol Cancer Res 7:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y et al (2014) BET bromodomain inhibition of MYC‐amplified medulloblastoma. Clin Cancer Res 20:912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beckmann PJ, Larson JD, Larsson AT, Ostergaard JP, Wagner S, Rahrmann EP et al (2019) Sleeping beauty insertional mutagenesis reveals important genetic drivers of central nervous system embryonal tumors. Cancer Res 79:905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Binning MJ, Niazi T, Pedone CA, Lal B, Eberhart CG, Kim KJ et al (2008) Hepatocyte growth factor and sonic Hedgehog expression in cerebellar neural progenitor cells costimulate medulloblastoma initiation and growth. Cancer Res 68:7838–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brabetz S, Leary SES, Gröbner SN, Nakamoto MW, Şeker‐Cin H, Girard EJ et al (2018) A biobank of patient‐derived pediatric brain tumor models. Nat Med 24:1752–1761. [DOI] [PubMed] [Google Scholar]
- 12. Briggs KJ, Corcoran‐Schwartz IM, Zhang W, Harcke T, Devereux WL, Baylin SB et al (2008) Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev 22:770–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Browd SR, Kenney AM, Gottfried ON, Yoon JW, Walterhouse D, Pedone CA, Fults DW (2006) N‐myc can substitute for insulin‐like growth factor signaling in a mouse model of sonic hedgehog‐induced medulloblastoma. Cancer Res 66:2666–2672. [DOI] [PubMed] [Google Scholar]
- 14. Cavalli FM, Remke M, Rampasek L, Peacock J, Shih DJ, Luu B et al (2017) Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31:737–754 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cook Sangar ML, Genovesi LA, Nakamoto MW, Davis MJ, Knobluagh SE, Ji P et al (2017) Inhibition of CDK4/6 by palbociclib significantly extends survival in medulloblastoma patient‐derived xenograft mouse models. Clin Cancer Res 23:5802–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA et al (2012) A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol 32:4104–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ et al (2018) MLL4 Is required to maintain broad H3K4me3 peaks and super‐enhancers at tumor suppressor genes. Mol Cell 70:825–841.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doucette TA, Yang Y, Pedone C, Kim JYH, Dubuc A, Northcott PD et al (2012) WIP1 enhances tumor formation in a sonic hedgehog–dependent model of medulloblastoma. Neurosurgery 70(4):1003–1010; discussion 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eberhart CG, Tihan T, Burger PC (2000) Nuclear localization and mutation of beta‐catenin in medulloblastomas. J Neuropathol Exp Neurol 59:333–337. [DOI] [PubMed] [Google Scholar]
- 20. Feil S, Valtcheva N, Feil R (2009) Inducible Cre mice. Methods Mol Biol 530:343–363. [DOI] [PubMed] [Google Scholar]
- 21. Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D et al (2018) Aberrant ERBB4‐SRC signaling as a hallmark of group 4 medulloblastoma revealed by integrative phosphoproteomic profiling. Cancer Cell 34:379–395.e7. [DOI] [PubMed] [Google Scholar]
- 22. Frappart PO, Lee Y, Lamont J, McKinnon PJ (2007) BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J 26:2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fults D, Pedone C, Dai C, Holland EC. (2002) MYC expression promotes the proliferation of neural progenitor cells in culture and in vivo. Neoplasia 4:32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Genovesi LA, Ng CG, Davis MJ, Remke M, Taylor MD, Adams DJ et al (2013) Sleeping Beauty mutagenesis in a mouse medulloblastoma model defines networks that discriminate between human molecular subgroups. Proc Natl Acad Sci U S A 110:E4325–E4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodrich LV, Milenković L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277:1109–1113. [DOI] [PubMed] [Google Scholar]
- 27. Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA et al (2004) The SmoA1 mouse model reveals that ntch signaling is critical for the growth and survival of sonic hedgehog‐induced medulloblastomas. Cancer Res 64:7794–800. [DOI] [PubMed] [Google Scholar]
- 28. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova‐Todorova K, Holland EC. (2008) PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 22:436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harris PS, Venkataraman S, Alimova I, Birks DK, Donson AM, Knipstein J et al (2012) Polo‐like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer 12:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B et al (2008) The Smo/Smo model: hedgehog‐induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 68:1768–1776. [DOI] [PubMed] [Google Scholar]
- 31. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K et al (2015) Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 27:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw ML, Baumgartner A et al (2019) Resolving medulloblastoma cellular architecture by single‐cell genomics. Nature 572:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang M, Tailor J, Zhen Q, Gillmor AH, Miller ML, Weishaupt H et al (2019) Engineering genetic predisposition in human neuroepithelial stem cells recapitulates medulloblastoma tumorigenesis. Cell Stem Cell 25:433–446.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ivanov DP, Coyle B, Walker DA, Grabowska AM. (2016) In vitro models of medulloblastoma: choosing the right tool for the job. J Biotechnol 236:10–25. [DOI] [PubMed] [Google Scholar]
- 35. Jenkins NC, Rao G, Eberhart CG, Pedone CA, Dubuc AM, Fults DW (2016) Somatic cell transfer of c‐Myc and Bcl‐2 induces large‐cell anaplastic medulloblastomas in mice. J Neurooncol 126:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawauchi D, Ogg RJ, Liu L, Shih DJH, Finkelstein D, Murphy Bl et al (2017) Novel MYC‐driven medulloblastoma models from multiple embryonic cerebellar cells. Oncogene 36:5231–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C et al (2012) A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21:168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kimura H, NG JM, Curran T. (2008) Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13:249–260. PMID:18328428. [DOI] [PubMed] [Google Scholar]
- 39. Lakso M, Sauer B, Mosinger B, Lee E j, Manning RW, Yu SH et al (1992) Targeted oncogene activation by site‐specific recombination in transgenic mice. Proc Natl Acad Sci U S A 89:6232–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH et al (2019) Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35:140–155.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lau J, Schmidt C, Markant SL, Taylor MD, Wechsler‐Reya RJ, Weiss WA (2012) Matching mice to malignancy: molecular subgroups and models of medulloblastoma. Childs Nerv Syst 28:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ (2007) Loss of suppressor‐of‐fused function promotes tumorigenesis. Oncogene 26:6442–6447. [DOI] [PubMed] [Google Scholar]
- 43. Lee Y, McKinnon PJ (2002) DNA ligase IV suppresses medulloblastoma formation. Cancer Res 62:6395–6399. [PubMed] [Google Scholar]
- 44. Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C et al (2003) A molecular fingerprint for medulloblastoma. Cancer Res 63:5428–5437. [PubMed] [Google Scholar]
- 45. Lee Youngsoo, Miller Heather L, Russell Helen R, Boyd Kelli, Curran Tom, McKinnon Peter J (2006) Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res 66:6964–6971. [DOI] [PubMed] [Google Scholar]
- 46. Lee C, Rudneva VA, Erkek S, Zapatka M, Chau LQ, Tacheva‐Grigorova SK et al (2019) Lsd1 as a therapeutic target in Gfi1‐activated medulloblastoma. Nat Commun 10:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li P, Du F, Yuelling LW, Lin T, Muradimova RE, Tricarico R et al (2013) A population of Nestin‐expressing progenitors in the cerebellum exhibits increased tumorigenicity. Nat Neurosci 16:1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marino S, Vooijs M, van der Gulden H, Jonkers J, Berns A (2000) Induction of medulloblastomas in p53‐null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 50. McCall TD, Pedone CA, Fults DW (2007) Apoptosis suppression by somatic cell transfer of Bcl‐2 promotes Sonic hedgehog‐dependent medulloblastoma formation in mice. Cancer Res 67:5179–85. [DOI] [PubMed] [Google Scholar]
- 51. Morfouace M, Shelat A, Jacus M, Freeman BB, Turner D, Robinson S et al (2014) Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 25:516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole‐genome landscape of medulloblastoma subtypes. Nature 547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D et al (2014) Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Orsulic S (2002) An RCAS‐TVA‐based approach to designer mouse models. Mamm Genome 13:543–547. [DOI] [PubMed] [Google Scholar]
- 56. Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA et al (2016) HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC‐Driven Medulloblastoma. Cancer Cell 29:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ et al (2012) An animal model of MYC‐driven medulloblastoma. Cancer Cell 21:155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Phoenix Timothy N, Patmore Deanna M, Boop Scott, Boulos Nidal, Jacus Megan O, Patel Yogesh T et al (2016) Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell 29:508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Raleigh DR, Choksi PK, Krup AL, Mayer W, Santos N, Reiter JF. (2018) Hedgehog signaling drives medulloblastoma growth via CDK6. J Clin Invest 128:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW (2003) c‐Myc enhances sonic hedgehog‐induced medulloblastoma formation from nestin‐expressing neural progenitors in mice. Neoplasia 5:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robinson GW, Kaste SC, Chemaititly W, Bowers DC et al (2017) Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8:69295–69302. PMID: 29050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I et al (2015) Vismodegib exerts targeted efficacy against recurrent sonic hedgehog‐subgroup medulloblastoma: results from phase II Pediatric brain tumor consortium studies PBTC‐025B and PBTC‐032. J Clin Oncol 33:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Romer JT, Kimura H, Magdaleno S, Sasai K et al (2004) Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell 6:229–240. PMID: 15380514. [DOI] [PubMed] [Google Scholar]
- 64. Roussel MF, Korshunov A, Reifenberger G, Pfister SM, Lichter P, Kawauchi D, Gronych J (2015) Somatic CRISPR/Cas9‐mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun 6:7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Saito T (2006) In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc 1:1552–1558. [DOI] [PubMed] [Google Scholar]
- 66. Sandén E, Dyberg C, Krona C, Gallo‐Oller G, Olsen TK, Enríquez Pérez J et al (2017) Establishment and characterization of an orthotopic patient‐derived Group 3 medulloblastoma model for preclinical drug evaluation. Sci Rep 7:46366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ et al (2006) Shh pathway activity is down‐regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res 66:4215–4222. [DOI] [PubMed] [Google Scholar]
- 68. Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG et al (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh‐induced medulloblastoma. Cancer Cell 14:123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shu Q, Wong KK, Su JM, Adesina AM, Yu LT, Tsang YT et al (2008) Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 26:1414–1424. [DOI] [PubMed] [Google Scholar]
- 70. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD et al (2010) Pleiotropic role for MYCN in medulloblastoma. Genes Dev 24:1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ (2003) Null mutation of DNA strand break‐binding molecule poly(ADP‐ribose) polymerase causes medulloblastomas in p53(‐/‐) mice. Am J Pathol 162:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S et al (2005) The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev 19:2656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vo BHT, Kwon JA, Li C, Finkelstein D, Xu B, Orr BA et al (2018) Mouse medulloblastoma driven by CRISPR activation of cellular Myc. Sci Rep 8:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vo BHT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S et al (2017) Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc‐Driven Group 3 Medulloblastoma. Cell Rep 18:2907–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wetmore C, Eberhart DE, Curran T (2001) Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 61(2):513–6. [PubMed] [Google Scholar]
- 77. Yan CT, Kaushal D, Murphy M, Zhang Y, Datta A, Chen C et al (2006) XRCC4 suppresses medulloblastomas with recurrent translocations in p53‐deficient mice. Proc Natl Acad Sci U S A 103:7378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M et al (2008) Medulloblastoma can be initiated by deletion of Patched in lineage‐restricted progenitors or stem cells. Cancer Cell 14:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhu G, Rankin SL, Larson JD, Zhu X, Chow LML, Qu C et al (2017) PTEN signaling in the postnatal perivascular progenitor niche drives medulloblastoma formation. Cancer Res 77:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]