

Abstract
Medulloblastoma (MB) is the most common CNS embryonal tumor. While the overall cure rate is around 70%, patients with high‐risk disease continue to have poor outcome and experience long‐term morbidity. MB is among the tumors for which diagnosis, risk stratification, and clinical management has shown the most rapid advancement. These advances are largely due to technological improvements in diagnosis and risk stratification which now integrate histomorphologic classification and molecular classification. MB stands as a prototype for other solid tumors in how to effectively integrate morphology and genomic data to stratify clinicopathologic risk and aid design of innovative clinical trials for precision medicine. This review explores the current diagnostic and classification of MB in modern neuropathology laboratories.
Keywords: classification, diagnosis, Group 3, Group 4, histology, medulloblastoma, neuropathology, non‐WNT/non‐SHH, SHH, WNT
Introduction
Medulloblastoma (MB) is the most common malignant pediatric brain tumor (57), where it accounts for around a quarter of all intracranial neoplasms and around half of posterior fossa tumors (19). The majority of MB arise in children with a median age of 9 years, and a peak in incidence between the ages of 3 and 7 years (65). However, a second peak is seen in adults accounting for around 25% of cases (48). The 5‐year overall survival for MB is approximately 75%, however, long‐term therapy‐related morbidity remains a significant concern (36, 50, 55, 71).
Medulloblastoma was first in described 1925, in a series of 31 cases first considered to be an unusual type of glioma arising primarily in the cerebellum of children (4). MB was included in the original histogenetic classification scheme introduced by Baily and Cushing, in which brain tumors were designated based on the morphologic similarity to cell types in the developing brain (5). The primitive embryonic tumor was believed to derive from an undifferentiated cell type termed the “medulloblast,” thought to arise around the ependymal lining of the fourth ventricle (reviewed in (20)). Although the developmental origins have proven more complicated than the original conceptualization (26, 27), the diagnostic entity has persisted over 95 years of revisions to brain tumor classification. In modern classification, MB represents a heterogeneous tumor with multiple subtypes. MBs share a primitive embryonal phenotype, composed of malignant tumor cells which are dominated by neuronal antigen expression. Stereotypic histologic patterns under light microscopy formed the basis of the histologic classification schemes. However, advances in our understanding of the biology and genomics of MB has led to significant changes in the pathologic diagnosis and classification of MB, making it a prototype for modern clinicopathologic diagnosis. This review will focus on the histology, pathologic diagnosis, and classification of MB with a specific emphasis on current neuropathologic practice.
Classification of MB by the WHO
The 2007 edition of the WHO Classification of Tumors of the Central Nervous System first recognized that histologic variants of MB carried variable clinical risk (43). In the 2016 update, a layered approach is employed integrating morphologic and genomic data in accordance with the Haarlem guidelines for nervous system tumor classification and grading (44). As such, the granularity of the diagnosis is made, dependent on the integration of tissue‐based information available to the pathologist (see Table 1). The newest classification scheme separates MB into two separate general designations, MB, histologically defined and MB, genetically defined (45). Histologically, MB can be separated into variants including classic, desmoplastic/nodular (DN), and large cell/anaplastic (LCA). Due to important clinicopathologic correlates, a special group of DN tumors in the infant population is designated MB with extensive nodularity (MBEN) (23, 45). A second general categorization is used for MB taking into account the molecular group of the tumor. Medulloblastoma, genetically defined is separated into WNT‐activated, SHH‐activated and TP53‐mutant, SHH‐activated and TP53‐wild‐type, and non‐WNT/non‐SHH groups. The latter group can be further subdivided to provisional subclasses, group 3 (G3) and group 4 (G4) when the ability to distinguish the two is possible. In the rare event that material available for review is inadequate or available testing prevents classification of the tumor into one of the genetically or histologically defined categories, the category of MB, NOS is designated (Table 1).
Table 1.
Medulloblastoma classification system in the WHO Classification of Tumors of the Central Nervous System 2016.
| Medulloblastoma, genetically defined |
| WNT‐activated |
| SHH‐activated, TP53‐wild‐type |
| SHH‐activated, TP53‐mutant |
| Non‐WNT/non‐SHH |
| Group 3 † |
| Group 4 † |
| Medulloblastoma, histologically defined |
| Medulloblastoma, classic |
| Desmoplastic/nodular medulloblastoma (DN) |
| Medulloblastoma with extensive nodularity (MBEN) |
| Large cell/Anaplastic medulloblastoma (LCA) |
| Medulloblastoma, NOS |
Provisional category.
Adapted from reference (45).
Canonical (WHO) histologic classification
The diagnosis of MB should be considered in the context of any embryonal brain tumor in the cerebellum, cerebellar peduncle, or fourth ventricle. Rarely other malignant tumors with small cell morphology can be encountered in this region, including atypical teratoid/rhabdoid tumor (ATRT), embryonal tumor with multilayered rosettes (ETMR), small cell glioblastoma, Ewing sarcoma (EWS), or high‐grade neuroepithelial tumor with BCOR alteration (HGNET‐BCOR) (79). These can typically be excluded by a combination of subtle histologic findings or immunohistochemistry (IHC), using lineage markers or entity specific stains. For instance, the presence of rhabdoid cells or ependymoblastic rosettes should alert the pathologist to the possibility of ATRT or ETMR, respectively. In the absence of these specific histologic features, IHC can aid proper classification. Apart from ETMR and rarely ATRT, the mimics of MB fail to express neuronal markers such as synaptophysin or NeuN. ATRTs typically demonstrate a polyimmunophenotype (co‐expressing EMA, smooth muscle actin, and GFAP among other antigens) and show loss of INI1 and Brg1, whereas ETMRs express high levels of LIN28A (41). Distinguishing small cell glioblastoma from MB can be challenging, but in addition to the absence of neuronal antigen expression, widespread expression of GFAP, Olig2, or SOX10 favors the diagnosis of astrocytoma. While Olig2 or SOX10 expression can be encountered in a subset of MB tumor cells, typically this is restricted to a minority population. Finally, EWS and HGNET‐BCOR are rarely encountered, but in most instances can be differentiated from MB by EWSR1 FISH or BCOR immunohistochemistry, respectively.
Classic variant
Classic variant MBs are by far the most frequent encountered in clinical practice, accounting for 72% of MB (45). Classic variant MBs are characterized by relatively round nuclei, the absence of increased cell size (defined as less than 4 times the size of a red blood cell), and the absence of frequent mitotic activity or mitoses (Figure 1A–D). Homer Wright rosettes are frequently encountered in classic MB (Figure 1B). Intrinsic desmoplasia is rare in classic variant tumors, and when desmoplasia presents it is typically associated with involvement of the leptomeninges by tumor. Similarly, nodules of differentiation are rare, and when present are not outlined by pericellular collagen as detected by reticulin staining.
Figure 1.
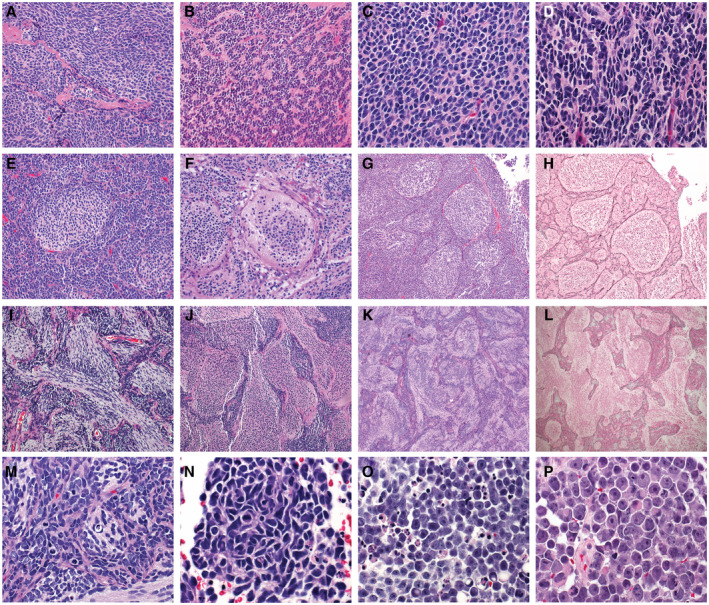
Medulloblastoma, histologically defined groups consist of four histologic variants including the classic variant (A‐D) characterized by small cells with round to ovoid nuclei (A), frequent Homer Wright rosettes (B), and no significant cytologic pleomorphism or cell molding (C). A slight increase in cell size and cytologic pleomorphism (D) are still within the spectrum of histologies in the classic variant. The desmoplastic/nodular variant (E‐H) is characterized by nodules of neurocytic differentiation surrounded by more primitive internodular areas (E and G). The differentiated nodules show desmoplasia surrounding the nodules which can be detected by pericellular reticulin deposition (H). Medulloblastoma with extensive nodularity (MBEN) (I‐L) is characterized by a high proportion of differentiated elements compared to primitive internodular elements (F‐J). The nodules in the MBEN variant often coalesce together forming irregular patterns accompanied by a pattern of linear “streaming” between nodules. Similar to other desmoplastic nodular tumors, MBENs show reticulin deposition in the internodular regions (L). The large cell/anaplastic variant (LCA) is a combination of two variant the anaplastic variant and the large cell variant (M‐P). The anaplastic variant is characterized by increased cell size, cytologic pleomorphism, cell molding and wrapping, frequent mitotic activity, and apoptotic bodies (M and N). The large cell variant is characterized by large discohesive cells with prominent nucleoli (O and P).
Desmoplastic/nodular variant and medulloblastoma with extensive nodularity
The Desmoplastic/nodular (DN) variant of MB is characterized by nodules of neurocytic differentiation with intervening embryonal elements. The “desmoplasia” associated with desmoplastic/nodular tumors refers to the propensity of these tumors to have pericellular collagen deposition, which is detectable by reticulin deposition, but which is absent from the nodules of differentiation (Figure 1E–H). There is variability in the proportion of tumors showing nodules or pure desmoplasia, and in fact historic controversy was centered on whether examples dominated by desmoplasia were in fact sarcomas of the cerebral arachnoid (70). All brain tumors elicit a desmoplastic reaction when they involve the leptomeninges, and this should be distinguished from pericellular reticulin deposition in tumor lying within the cerebellar parenchyma of DN tumors. In our practice, we typically try to assess the presence or absence of pericellular reticulin deposition in the intraparenchymal component to avoid this pitfall. Identification of the thick‐walled vessels of the leptomeninges can be used as a guide in histologic sections. Recognition of the DN variant is important because tumors with this morphology generally are associated with intermediate clinical risk and uniformly associate with the SHH molecular group for which targeted agents may be available to some patients (47, 63).
A special variant of desmoplastic/nodular MB is termed medulloblastoma with extensive nodularity (MBEN). This histologic variant typically presents in infants and is frequently more midline than more conventional DN tumors (Figure 1I–L). As the name implies, MBENs are dominated by nodules rather than primitive elements. The nodules tend to be irregular and coalesce together in many examples. It is typical to identify a “streaming” pattern in MBENs, in which linear arrays of neurocytic cells connect adjacent nodules.
Large cell/anaplastic histology
Large cell and anaplastic MBs represent two distinct histologic variants, combined in the latest histologic classification scheme. Giangaspero et al defined the poor prognosis Large cell MB subtype in 1992 (24) while a more comprehensive grading scheme for anaplasia was later established by Eberhart et al (14). Anaplasia in MB is defined by increased cell size, increased cytologic pleomorphism, nuclear wrapping/molding, frequent mitotic activity, and frequent apoptotic bodies (Figure 1M,N) (14, 46). There is no current consensus on the amount of mitotic activity or apoptosis that is sufficient for designation as frequent, but some authors have suggested >10 mitoses per 10 high‐power field in the case of mitoses (14). The large cell variant represents a histologic variant in itself which shows increased cell size, round cell morphology, and prominent nucleoli (Figure 1O,P). The large cell variant lacks the overt pleomorphism seen in the anaplastic variant, but often associates with anaplastic features such as frequent mitotic activity and apoptosis. In previous versions of the WHO Classification of Tumors of the Central Nervous System, large cell and anaplastic variants of MB were considered separately; however, the 2016 classification groups them as a single histologic category. The rationale for this grouping is that most large cell medulloblastomas also contain an element of anaplastic variant MB, at least focally (9, 14, 24, 46).
The prognostic significance for the large cell and anaplastic variants was initially established in two related cohorts. The relationship between large cell morphology and clinical risk was first established in a cohort of six Pediatric Oncology Group (POG) frontline trials (9). A more comprehensive grading scheme for anaplasia in MB was later established and applied to an augmented cohort containing seven POG frontline trials by Eberhart and Burger (14) who established a relationship between the large cell variant and severe anaplasia on event‐free survival (EFS) and overall survival (OS) (9, 14). The significance of the combined LCA variant was confirmed in the SIOPII and SIOP PNET3 cohorts for both OS and EFS (6, 25, 46, 80). Variability in the proportion of tumors demonstrating LCA morphology is observed between studies. For instance, in the POG cohort, moderate or severe anaplasia was encountered in 24% of samples with 10% demonstrating severe anaplasia (14). Whereas, McManamy and Ellison reported LCA in 19% of tumors (2% large cell and 17% anaplastic) (46). This likely represents natural enrollment variability as well as slight differences in application of the histologic criteria for anaplasia between central reviewers over time. The overall survival at 3 years for LCA tumors in the SIOP PNET3 study was 67% compared to 81% in children with classic tumors, and the prognostic significance of LCA morphology held up as an independent predicator of risk after multivariate analysis which included known clinical risk factors such as metastatic disease and extent of resection (46).
Some MBs demonstrate overall classic morphology, with focal severe anaplasia. We typically designate these as classic variant but notate the presence of focal anaplasia. This designation is important in that it alerts the clinical team to the possibility for more aggressive tumor elements in the context of subtotal resection, metastasis, or sampling bias. However, the presence of focal anaplasia does not definitively associate with poor outcome. For instance, in the POG cohort, focal anaplasia was identified in 17% of cases and the outcomes were comparable to tumors with classic histology with no anaplasia (14).
The association of LCA histology with poor outcome probably relates, at least partially, to its close association with other high‐risk molecular features. For instance, amplification of MYC and MYCN is more frequent in tumors with LCA histology (15, 17). Similarly, LCA tumors are also enriched in both the high‐risk G3 molecular subtype and SHH tumors with TP53 abnormalities, which are also associated with dismal prognosis (38).
The association of LCA histology with clinical risk is largely based on historic cohorts which failed to stratify by molecular group. There is some indication that the LCA morphology maintains its prognostic significance on progression‐free survival within molecular subgroup, however, whether anaplasia alone should dictate high‐risk disease alone in the absence of other high‐risk clinical features such as metastasis remains controversial (16, 61).
Non‐WHO designated histologic variants
Several histologic patterns recognized in clinical practice are not officially recognized in the MB, histologically defined scheme outlined by the 2016 WHO Classification of Tumors of the Central Nervous System (45). Despite their rarity and lack of specific diagnostic assignment many are important to recognize clinically because of their special association with specific molecular features or their propensity to affect testing interpretation. These include tumors with divergent differentiation as well as patterns termed “classic biphasic” pattern, “ganglioneuroblastoma” pattern (GNB), and “paucinodular” patterns (47).
Medulloblastoma can present with divergent differentiation, often presenting as myogenic or melanocytic differentiation (previously known as medullomyoblastoma and melanocytic medulloblastoma). On the extreme end of the histologic spectrum, myogenic differentiation presents as “strap cells” with true visible muscle striations (Figure 2), however, less differentiated cells can present as rhabdomyoblasts. Melanocytic differentiation can sometimes be recognized by the presence of melanin pigment within the tumor cells or merely clusters of epithelioid cells with prominent nucleoli. Occasionally, divergent differentiation is occult and can only be identified using IHC, of which desmin and HMB45 are typically reliable for detection of myogenic and melanotic differentiation, respectively. Divergent differentiation carries some prognostic significance in some studies and can be associated with focal or overt anaplasia. Definitive evidence that MB with divergent differentiation represents distinct molecular entities has not been established, and they both contain many of the same molecular findings typical of medulloblastomas without divergent differentiation (31, 42, 78).
Figure 2.
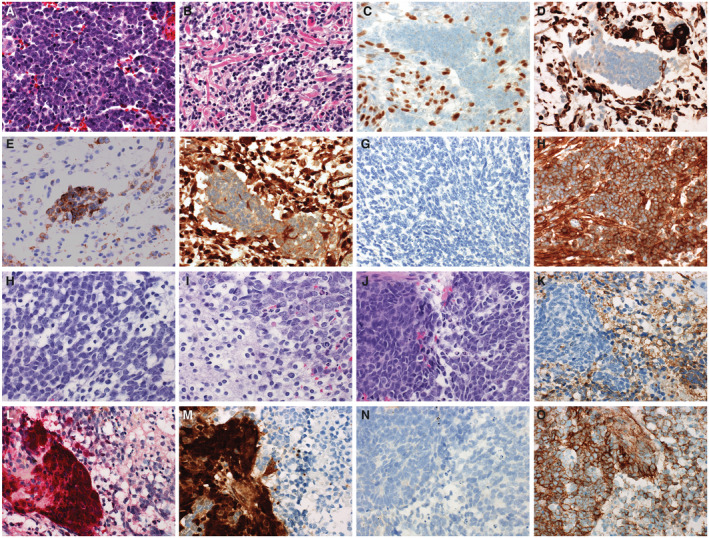
Medulloblastomas with divergent differentiation most often are characterized by myogenic. Here, a case of medulloblastoma with myogenic differentiation is presented (A‐H). These tumors typically show histology in regions that are typical of other medulloblastomas with primitive elements, often with some anaplastic features (A). In some, overt myogenic differentiation can be observed and can be detected by the presence of “strap” cells (B). Myogenic elements in medulloblastoma with myogenic differentiation can be detected by immunohistochemistry to myogenin (C) or desmin (D). The neuronal elements of medulloblastoma with myogenic differentiation is immunoreactive for synaptophysin (E). The panel of stains used for molecular subgrouping yield a discordant phenotype, showing immunoreactivity for YAP1 (F), no staining for GAB1 (G), and cytoplasmic only staining for beta‐catenin (H). Divergent differentiation can also take the form of melanotic differentiation (I‐P). This case demonstrates the more typical primitive elements of medulloblastoma (I) and neurocytic differentiation (J). A mixed population of the neuronal and melanotic elements can be seen in (K). The neuronal element is immunopositive for synaptophysin (L), whereas the melanotic element is detectable with antibodies to HMB45 (M). Medulloblastomas with divergent melanotic differentiation also yield a discordant pattern on the molecular subgrouping stains, demonstrating immunoreactivity for YAP1 in the melanotic element (N), no significant immunoreactivity for GAB1 (O), and cytoplasmic only staining for beta‐catenin (P).
Classic biphasic MB are characterized by alternating sheets of tumor cells with nodules of neurocytic differentiation (Figure 3). Often the nodules are more irregular in their contour than those encountered in the desmoplastic/nodular variant. Also, in contrast to the DN variant, the nodules of classic biphasic tumors are not outlined by reticulin (Figure 3). The relevance of this histologic variant is two‐fold. First, tumors with a classic biphasic pattern are more frequently associated with poor‐risk cytogenetic abnormalities such as isochromosome 17q, MYC amplification, or MYCN amplification compared to tumors with true desmoplastic nodular histology (47). Second, classic biphasic MB represents a clinically relevant mimic of DN medulloblastomas. As true DN tumors all fall into the SHH molecular subgroup and classic biphasic tumors typically fall into the non‐WNT/non‐SHH molecular immunophenotypic subgroup, the distinction may have therapeutic implications. Comprehensive evaluation to determine whether classic biphasic tumors primarily represent G3 or G4 molecular groups has not been performed.
Figure 3.
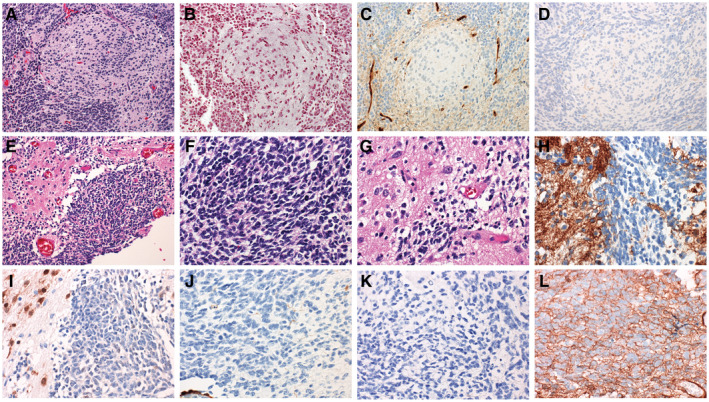
Histologic variants outside of those in the medulloblastoma, include the classic biphasic variant (A‐D) and the ganglioneuroblastoma variant (E‐L). Classic biphasic tumors are characterized by nodules of differentiation with primitive internodular areas (A). These tumors can be differentiated from desmoplastic/nodular tumors by reticlulin stain (B) which fails to outline the nodules. On the molecular subgrouping stains, classic biphasic tumors fall into the non‐WNT/non‐SHH molecular group based on negativity for YAP1 (C) and GAB1 (D), and staining for beta‐catenin that is restricted to the cytoplasm (not shown). The ganglioneuroblastoma variant is characterized by a mixture of primitive elements mixed with areas ganglion cell differentiation (E‐G). The ganglion cell differentiation is marked by especially high intensity staining for synaptophysin (H) and immunoreactivity for NeuN (I). Tumors showing ganglioneuroblastoma histology typically fall in the non‐WNT/non‐SHH molecular subgroup, showing negative staining for YAP1 (J), negative staining for GAB1 (K), and cytoplasmic only staining for beta‐catenin (L).
Some non‐desmoplastic MBs contain clusters of ganglion cells and neurocytic cells and have been designated the ganglioneuroblastoma (GNB) variant (Figure 3) based on their similarities to the CNS GNB encountered in the supratentorial compartment. These represent a small proportion of MB overall, likely less than 1% (47). GNB arise primarily in children and adolescents, most before the age of 12 years of age (47). Due to the rarity of this histomorphologic variant, a specific association with clinical risk or molecular subtype has not been established.
Special designation is given to a histologic pattern within the DN MB designated paucinodular MB which consists of tumors that are dominated by primitive cells with desmoplastic stroma, with only rare nodules of differentiation (47) (Figure 3). While not officially recognized as a variant by the WHO, this pattern represents an important clinical consideration in practice because it may not be easily recognized without the aid of special stains such as reticulin. In clinical diagnostic practice, the distinction of paucinodular tumors is important to distinguish from classic tumors which have invaded the leptomeninges because the latter typically represent non‐WNT/non‐SHH tumors. In many trial cohorts, MBEN and paucinodular tumors are combined with other DN tumors as a single histologic entity designated “desmoplastic medulloblastoma.”
Molecular Classification of Medulloblastoma
Medulloblastoma represents one of the earliest and best characterized examples of molecular subtypes within solid tumors. While the first molecular subtypes were based on transcription profiling using expression arrays (12, 40, 83), additional methods including those employing protein expression by IHC (16), DNA methylation signatures and RNA expression patterns have also been developed and implemented in clinical laboratories (10, 32, 54)}. The major molecular groups are summarized below with additional discussion of specific methodologies for molecular grouping.
Medulloblastoma, WNT‐activated
Medulloblastoma, WNT‐activated tumors account for about 10%‐15% of MBs (82) and typically present in older children, between the ages of 7 and 14 (peak age = 10–12), with a slight female predominance. WNT MB can be seen in approximately 15% of adults with MB (64). Tumors in the WNT group characteristically have classic morphology, though rare examples of anaplastic WNT tumors have been described. WNT tumors arise near midline but often involve the cerebellar peduncle and brainstem and protrude through the Foramen of Luschka (59). WNT tumors are associated with a high degree of hemorrhage which may be partially explained by the lack of a well‐developed blood‐brain barrier (60). WNT tumors are characterized by expression of WNT pathway genes, contain mutations in exon 3 of the CTTNB1 gene in approximately 85%–90% of cases, and exhibit loss or partial loss of chromosome 6 in 85%–90% of cases (1, 13, 17, 34, 39, 68). APC mutations can be identified in a high proportion of WNT MB lacking CTTNB1 mutations (84). Other genes commonly mutated in WNT MB include TP53, SMARCA4, KMT2D and DDX3X (34, 51, 68). Of note, TP53 mutations do not appear to carry the same poor prognosis in WNT tumors as they do in the SHH molecular group (86). Amplifications of MYC or MYCN are almost never detected in WNT MB, and the presence of such findings should raise clinical suspicion of misclassification. Additional substructure has been reported within the WNT molecular groups, separating it into the WNT‐α (70%) subtype seen predominantly in children and WNT‐β (30%) subtype seen predominantly in adults (11). WNT MB are associated with a favorable prognosis in the pediatric population, however, the prognosis in adults is less established (3, 13, 16, 28). Current methods of molecular classification available in most clinical laboratories are restricted to detection of a single WNT molecular group, and do not distinguish the WNT‐α and WNT‐β groups.
Medulloblastoma, SHH‐activated
Medulloblastoma, SHH‐activated tumors account for about 30% of MB and show a bimodal distribution of presentation with the first peak occurring in infants and a later peak occurring in adults and children greater than age 16 (45). In adults, SHH MB represent the predominant molecular group making up 60% of all cases (64). SHH MB arise predominantly in the cerebellar hemisphere, but also can arise in the cerebellar vermis. All histologic variants of MB can be found in SHH MB, but the DN variant is most common at slightly greater than 50%, followed by classic and LCA (16). LCA histology is predominantly seen in SHH MB with TP53 mutations. SHH MB are characterized by activation of SHH pathway transcriptional programs and recurrent mutations in SHH pathway genes including PTCH1, SMO, and SUFU (34, 68). Amplifications involving key downstream mediators of the SHH pathway such as GLI2 are also encountered (51, 76). MB, SHH‐activated and TP53 wild‐type tumors are associated with intermediate risk disease with an 5‐year overall survival of around 76%, whereas TP53 abnormalities in SHH MB are associated with especially high‐risk clinical disease, with dismal prognosis of 41% at 5‐years (38, 86). MB, SHH‐activated and TP53‐mutant tumors are associated with high rates of chromothrypsis which leads to amplification of oncogenes including MYC, MYCN, and GLI2 (62).
Additional substructure has been described by the MB Advanced Genomics International Consortium (MAGIC) dividing SHH MB in four molecular groups (SHH‐α, β, γ, δ) (11). These more granular molecular groups of SHH are enriched for specific genomic abnormalities and have distinct clinical associations. For instance, the δ SHH MB group is predominated by adults whereas most children fall into the SHH‐α group (11, 69). Infantile tumors are comprised almost entirely of SHH‐β and SHH‐γ tumors, and the SHH‐β group is associated with slightly worse prognosis (67% compared to 88% 5‐year survival) (69). To date, the granular subclasses of SHH MB can only be detected by methylation profiling. Although IHC‐based methods for molecular subgrouping are feasible in most clinical laboratories, the method is not able to distinguish the more granular molecular groupings and instead are restricted to designation of generic SHH MB (10, 16, 54).
Medulloblastoma, non‐WNT/non‐SHH and medulloblastoma G3 and G4
Medulloblastoma, non‐WNT/non‐SHH represents the largest molecular group of MB. It is comprised of the provisional G3 and G4 tumors, representing 20 and 40% of all cases. Most present in children, though up to 25% of adult MB fall in the non‐WNT/non‐SHH group (64). When present in adults, G4 tumors are typical while G3 tumors are exceedingly rare. Non‐WNT/non‐SHH tumors present in the midline, typically filling the fourth ventricle. G3 tumors present across the pediatric age spectrum and represent approximately 45% of tumors in the infant population, whereas G4 tumors typically present in older children. A male predominance is seen in non‐WNT/non‐SHH MB, and especially high in G4 tumors (3:1). Non‐WNT/non‐SHH tumors exhibit frequent metastatic disease, and this is frequently encountered at presentation. Non‐WNT/non‐SHH tumors have the worst prognosis overall, compared to SHH and WNT MB. This is largely driven by outcomes in MB G3 which have an overall survival of <60% with more intermediate survival in MB G4 tumors. While the majority of non‐WNT/non‐SHH tumors demonstrate classic histology, LCA histology is also seen, most often in G3 tumors. DN and MBEN histologic variants are not encountered in non‐WNT/non‐SHH tumors. The biology of MB in the non‐WNT/non‐SHH subtype is less established due to a paucity of recurrent single nucleotide variants (SNVs). A subset of non‐WNT/non‐SHH tumors are driven by enhancer hijacking, with both G3 and G4 tumors demonstrating activation of the GFI1 and GFI1B loci, and a subset of MB G4 tumors targeting activation of the PRDM6 locus (53). Recurrent copy number abnormalities are present in G3 and G4 tumors. MYC amplification is a hallmark of G3 tumors, but other events such as OTX2 amplification can also be observed (51). MYCN amplification is more typical of G4 tumors, though it can be seen in a small proportion of G3 tumors (51). More recent manuscripts have described molecular substructure in the non‐WNT/non‐SHH group of MB, separating these tumors into 4–8 additional subclasses (11, 51, 73). A recent consensus study established eight subclasses for the G3 and G4 tumors, designated I‐VIII (75). Methods currently available in most clinical laboratories are unable to distinguish MB groups to this level of granularity, and risk stratification based on these groups is still in its infancy.
Methods of Molecular Classification
Several different methods have emerged to perform molecular classification of MB in the clinical setting. The methods show variable performance related to their ability to resolve all molecular groups. This is especially true as more granular substructure has been established outside the four consensus MB molecular groups. The methods also differ in their accessibility, capital expense, and their ability to scale easily to other tumor types. An overview of the different methods is provided below and an in Table 2.
Table 2.
Comparison of methods for MB classification in the clinical laboratory
| Method | Description | Pros | Cons | References |
|---|---|---|---|---|
| Immunohistochemistry | Description: Classification based on pattern of expression of three proteins as detected by immunohistochemistry (YAP1, GAB1, and beta‐catenin) |
|
|
(17) |
|
Classification:
| ||||
| Transcription profiling | Description: Classification based on shared RNA expression signature as detected by transcription array, RNA sequencing, or nanostring TM |
|
|
(12, 40, 54, 83) |
|
Classification:
| ||||
| Methylation |
Description: Classification based on methylation signature using unsupervised or supervised classification models. Typically utilize Illumina Human Infinium 450K/EPIC arrays |
|
|
(10, 32) |
|
Classification:
| ||||
| DNA Sequencing | Description: Classification based on sequencing of class specific recurrent mutations |
|
|
(1, 51, 68) |
|
Classification:
|
Transcription‐based molecular classification
The earliest description of molecular subtypes within MB were derived from large cohorts in which cluster analysis was performed on transcription profiles from expression arrays (12, 40, 52, 83). While transcriptional profiling using expression arrays has led to important research advances in our understanding of the disease, widespread implementation into clinical laboratories has not been established. This is largely based on the relative instability of RNA compared to DNA, historic dependence of fresh or frozen material for transcriptional profiling which limited collection of a large‐scale repository of reference tumors, and variability in transcriptional profiling platforms making comparison across datasets challenging. In 2012, Northcott et al applied nanostring technology to molecular classification of MB using a set of 22 genes (54). The method relies on the capture of subtype‐specific transcripts which is measured on the nCounter machine (22). Because the input RNA is only 100 ng and the assay can be performed readily out of FFPE material, it is more easily translated into the clinical laboratory workflows than methods relying on fresh or frozen tumor material. There is some drop off in accuracy when evaluating cohorts greater than 8–10 years old, likely due to poor RNA fidelity, but overall the accuracy of classification is very high in prospective cases. Additionally, the per sample cost of the nanostring method is favorable compared to other methods. The primary limitations of the nanostring method is that comprehensive classification of all brain tumors cannot be performed using the same platform and it can be prone to some misclassification in event histologic mimics of medulloblastomas are encountered (2). The nanostring method also generates only a single data modality, that is expression of selected genes, and ancillary data such as genome‐wide copy number must be obtained using orthogonal methods.
Immunohistochemistry‐based molecular classification
In 2011, David Ellison and colleagues established a method for molecular subtyping MB using IHC (16). The original panel included four immunostains: Filamin A, YAP1, GAB1, and beta‐catenin (16). In its original design, tumors in the WNT and SHH molecular groups demonstrated expression of both Filamin A and YAP1, whereas G3 and G4 tumors were negative for these marks. The combined immunoreactivity for Filamin A, YAP1, and GAB1 was specific for SHH molecular subtype, whereas expression of Filamin A, YAP1, and nuclear beta‐catenin was specific for the WNT molecular subtype (Figure 4) (16). In modern practice, Filamin A is often omitted from the panel because YAP1 is a relatively robust stain and Filamin A serves a redundant role in subtyping.
Figure 4.
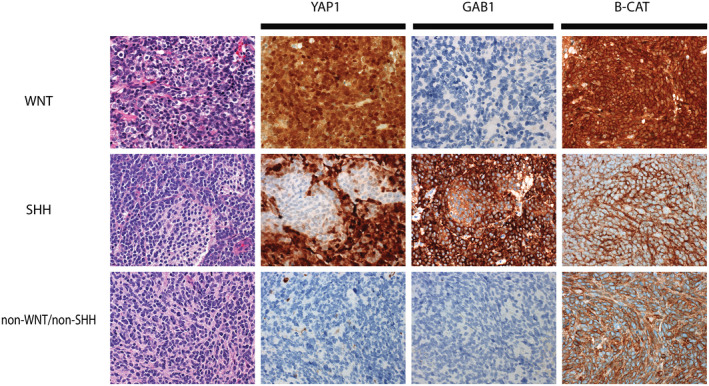
Example staining patterns for molecular subgrouping medulloblastoma using the panel of YAP1, GAB1, and beta‐catenin. WNT‐activated medulloblastomas typically demonstrate immunoreactivity for YAP1, but are immunonegative for GAB1. Immunoreactivity for beta‐catenin is positive in both the cytoplasm and nucleus of WNT‐activated tumors (top panels). SHH‐activated medulloblastomas are immunoreactive for YAP1 and GAB1, but show reactivity for beta‐catenin that is restricted to the cytoplasm (middle panels). Non‐WNT/non‐SHH medulloblastomas show no immunoreactivity for YAP1 or GAB1, and immunoreactivity for beta‐catenin that is restricted to the cytoplasm only (bottom panels).
Attempts to use a two‐stain panel, including only GAB1 and beta‐catenin, have been utilized in some trial designs; but in our experience this reduced panel is plagued by interpretation difficulties (B.A.O and David Ellison personal observations). Inclusion of the YAP1 stain also serves some underappreciated utility in the panel. In addition to being absent in the G3 or G4 tumors, it often can serve as a safeguard against misclassification of non‐MB embryonal tumors that can histologically mimic MB. For instance, rarely ATRT or ETMR occur in the cerebellum, and in the absence of appropriate downstream molecular interrogation can be mistaken for MB. These tumors typically demonstrate a discordant immunohistochemical pattern characterized by immunoreactivity for YAP1, negativity for GAB1, and only cytoplasmic expression of beta‐catenin (B.A.O and David Ellison personal observations).
The ease of use and relative low cost of the IHC panel make introduction into the clinical laboratory relatively simple and has made this method the mostly widely adopted of all methods for MB molecular classification and is currently utilized for molecular stratification in trial NCT01878617. However, IHC‐based classification has some important limitations. First, the IHC method requires proper tuning of the immunostains, and careful lot to lot validation is necessary to prevent misinterpretation. Second, the IHC method cannot distinguish between G3 and G4 tumors. Although reports of additional panels that include NPR3 for G3 and KCNA1 for G4 tumors have been reported, these have not been widely adopted (52). Third, rhabodomyoblasts, melanocytes, and muscle cells are all inherently positive for YAP1. Therefore, an indeterminant subtype (YAP1 positive, negative for GAB1, and cytoplasmic beta‐catenin) often is observed in the presence of divergent myogenic or melanotic differentiation (29) (Figure 2).
One additional pitfall of note is the loss of YAP1 expression in areas of heavy neuronal differentiation. It has been established that loss of YAP1 expression, and activation of the hippo pathway in general, are necessary for neuronal differentiation (21). The result is that the nodules of differentiation in the MBEN or DN MB are typically negative for YAP1. This phenomenon can also be observed in selected WNT MB in which clusters of neurocytic differentiation can sometimes be identified.
Methylation‐based classification
Methylation of cytosine at CpG sites across the genome represents an important method of epigenetic regulation. With specific enrichment at promoters and enhancer regions, CpG methylation state has important implications with regard to gene expression, cell specification, and differentiation state. Tumors have an inherent methylation signature that is thought to represent a combination of their cell of origin and their specific driver mutations and these signatures can be exploited for clinical tumor classification, including for MB (32, 74). Several methods exist to interrogate the methylation state within the genome including array‐based methods, whole‐genome bisulfite sequencing, and methylation‐specific PCR. Most methods used for molecular classification currently rely on bisulfite treatment of DNA allowing for single base resolution of methylated versus unmethylated cytosine after amplification. The mostly widely adopted platform for methylation profiling has been the Illumina Human Infinium Bead Array (450K and 850K Epic Array) (49, 72). This array platform uses the methylation state at specific CpG sites across the tumor genome as the input for downstream analysis including molecular classification. This method has several advantages. First, it takes DNA as its input which is an inherently stable macromolecule compared to RNA. Second, the method works equally well in frozen or FFPE tissue, making it well suited for the typical clinical workflows. Third, the DNA input is relatively low, 125–300 ng, amounts achievable off most clinical FFPE samples. These features have allowed for very large datasets to be accumulated of MB from retrospective clinical archives and clinical trials (11, 51, 73, 75) which has uncovered additional substructure within the individual molecular subgroups, some of which have either prognostic or therapeutic implications (See review by Northcott and colleagues in this issue).
Methylation‐based methods can differentiate all subtypes, including most G3 and G4 tumors. This method has been used to build supervised classification models, some of which have been implemented clinically (10). While the number of clinical laboratories currently offering clinical methylation profiling remains small, those that do typically have implemented a random forest algorithm trained on a comprehensive reference series of most WHO brain tumor entities, including MB (10). The comprehensive nature of this model offers significant advantage over more restricted classification models, because it has the potential to identify rare instances of misdiagnosis. Subtyping by methylation array also has the added advantage that genome‐wide copy number data can be extracted informatically, capable of detecting abnormalities in clinically relevant loci, including amplification of the MYC, MYCN, and GLI2 oncogenes (32). The primary limitation of methylation‐based classification is that the capital expense and expertise necessary for implementation prohibits introduction into many low‐volume centers. Additionally, some material, particularly low volume biopsy specimens or those with significant contamination by normal brain parenchyma may have insufficient DNA yields for classification or may lead to misleading normal brain class calls, respectively.
Other Ancillary Testing Modalities
Clinical sequencing
Clinical sequencing can provide important ancillary information, augmenting the diagnosis and prognosis of MB. Some gene variants relate to specific molecular subtypes. For instance, the presence of CTNNB1 mutations are pathognomonic for the WNT molecular subtype, and have diagnostic utility (51, 68). CTNNB1 mutations cannot be used in isolation for diagnosis of WNT MB as many as 10% harbor non‐CTNNB1 abnormalities such as APC mutations (69). APC variants in the WNT molecular subgroup can be especially important findings, because they often present as germline events and warrant additional genetic evaluation for Turcot syndrome.
SHH tumors demonstrate mutations in mediators of the SHH pathway including SUFU, PTCH1, or SMO (68). Mutations in upstream elements of the SHH pathway such as PTCH1 can be predicative for response to SMO inhibitors such as vismodegib (GDC‐0449) or sonidegib (LDE225), however, the use of such therapy is limited to skeletally mature patients due to the risk of growth plate fusion (35, 66). In contrast, alterations in the SHH pathway downstream of SMO, such as GLI2 amplification, are associated with resistance (67). Of note, while certain SHH pathway gene mutations are relatively selective, such as PTCH1 and SUFU mutations, mutations in SHH pathway genes alone are not diagnostic of the SHH molecular group, and can be detected in a small proportion of WNT MB (33, 51). TP53 mutations carry important clinical implications in MB, in which they are associated with a dismal prognosis in the SHH molecular subgroup and can sometimes be seen in the setting of germline mutations (86). The association of TP53 mutations with the SHH molecular subgroup was first established in a cohort of Li‐Fraumeni patients (62), but these can also arise as somatic variants (86). On histology, p53 immunohistochemistry is an effective method to screen for TP53 mutations in most cases. Tumors demonstrate wild‐type patterns on immunohistochemistry are associated with positive staining in the minority of tumor cells (Figure 5A–D), whereas in the presence of a dominant‐negative TP53 mutation, the majority of tumor cells demonstrate strong, diffuse staining (Figure 5E–H). Inactivating mutations are difficult to distinguished from failed immunohistochemistry tests and are best identified using sequencing.
Figure 5.
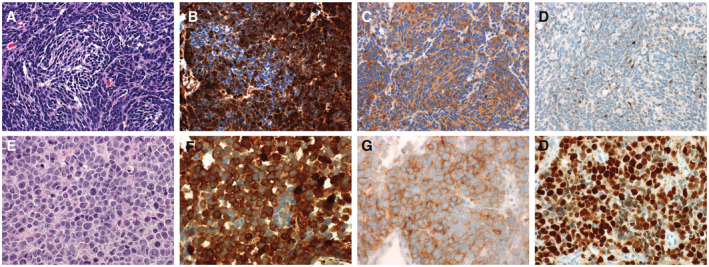
The presence of a TP53‐mutation is an important prognostic feature in SHH‐activated medulloblastomas. A typical wild‐type tumor is depicted in panels A‐D. The tumor demonstrates the typical pattern of immunoreactivity for YAP1 and GAB1 (B and C, respectively). Immunoreactivity for p53 restricted to weak expression in the minority of tumor cells in TP53‐wild‐type tumors (D). A tumor from a patient with Li‐Fraumeni syndrome is shown in panels E‐H. SHH‐activated, TP53‐mutant tumors often show large cell/anaplastic histology (E). This case demonstrates the typical immunophenotype of a SHH‐activated tumor with immunoreactivity for YAP1 and GAB1 (F and G). Strong, diffuse immunoreactivity for p53 is present (H) indicative of a dominant‐negative TP53‐mutation. Because many SHH‐activated tumors with TP53 mutations are associated with germline TP53 mutations, genetic testing is recommended for all cases showing a mutant pattern.
Although somatic sequencing can be used as an ancillary test to characterize MB, sequencing alone cannot be reliably used for comprehensive molecular classification of MB due to the high proportion of tumors that lack subgroup defining SNVs. Nevertheless, sequencing may uncover targetable abnormalities in some instances, particularly in SHH MB where sensitivity for SMO inhibitors associates with PTCH1 mutations (67).
Fluorescence in situ hybridization and copy number analysis
Fluorescence in situ hybridization (FISH) provides low‐level molecular classification or risk stratification but is not typically used as a primary method for molecular classification in isolation. For instance, clinical laboratories frequently evaluate for MYC and MYCN amplification, as both have been demonstrated to associate with reproducible poor clinical outcome in MB (15). The method has the advantage of quick turnaround time and can typically be reported within 3–6 working days. Compared to array‐based methods to evaluate copy number, FISH is capable of detecting focal subclonal cell populations with MYC or MYCN amplification in otherwise copy neutral tissue sections and can be performed even with diagnostic tissue is limited. FISH testing cannot definitively yield molecular subgroup in most cases. For instance, complete or partial loss of chromosome 6 is typically used to confirm the diagnosis of the WNT molecular group, but would not be used in isolation as diagnostic of the WNT molecular subtype as 10%–15% of WNT tumors are devoid of chromosome 6 abnormalities (1, 17). Additionally, chromosome 6 abnormalities are not restricted to WNT molecular group, especially in adult patients where a small proportion are also encountered in SHH or G4 tumors (39). Similarly, isochromosome 17q is also evaluated in some contexts and typically is observed in G3 or G4 tumors but is not diagnostic of either in isolation. Isochromosome 17q is associated with poor prognosis in some studies, but the association has not been uniformly confirmed (17, 58, 76). The strong association of isochromosome 17q with the non‐WNT/non‐SHH groups, is likely an important confounding factor. We sometimes use FISH for isochromosome 17q when faced with a challenging embryonal tumor presenting in the cerebellum or in the context of widely disseminated disease, often with limited or suboptimal material. Because of the relatively high specificity, a positive FISH result for isochromosome 17q may help place the tumor more definitively in the category of MB. In addition to a diagnostic or prognostic role, FISH can be predictive for therapeutic efficacy in select circumstances (67). We have observed that within the SHH molecular subgroup, PTCH1 deletion was associated with response to the smoothened inhibitor vismodegib (67).
Genome‐wide copy number analysis through sequencing or array‐based platforms are utilized in some centers and have identified additional copy number abnormalities with clinical import (8, 32). For instance, chromosome 11 loss portends a good prognosis in non‐metastatic G4 tumors (61). The primary limitation of using array‐based methods for copy number profiling is that they rely on bulk tumor for analysis, and therefore, may miss subclonal events.
Germline predisposition and testing in MB
While most MB are sporadic, several germline predisposition syndromes have been associated with an increased propensity for MB and awareness of the specific associations is an important consideration for neuropathologists (81). Among the first described MB predispositions syndrome includes Gorlin syndrome, caused by germline mutations in PTCH1 and SUFU and familial brain tumor polyposis syndromes associated with germline mutations in APC (18, 30, 77, 85). Li‐Fraumeni syndrome, associated with germline TP53 abnormalities, show predisposition to MB as well as high‐grade astrocytomas (37, 56). A recent study on a large cohort of MB found that germline abnormalities could be identified in nearly 6% of patients (84). The percentage of patients with detectable germline abnormalities is variably distributed across molecular subgroups with an incidence of 20% in SHH MB and 7% of WNT tumors (84). Germline predisposition was rarely identified in G3 or G4 tumors. Six consensus MB germline predisposition genes identified were APC, BRCA2, PTCH1, SUFU, TP53, and PALB2. As expected, germline predisposition to WNT MB was nearly exclusively associated with APC abnormalities and was especially common in WNT MB lacking CTNNB1 mutations. The causative abnormality in SHH tumors was more variable, being associated with abnormalities in PTCH1, SUFU, BRCA2, PALB2, and TP53. The age of onset for SHH MB with predisposition is variable, with SUFU and PTCH1 mutations occurring primarily in infants, and tumors with TP53 germline events presenting in children. Identification of TP53 abnormalities in SHH MB has important clinical implications as these tumors are associated with an especially aggressive clinical course and dismal outcomes on standard therapies (38, 86). Rare predisposition genes continue to be uncovered, including the SHH MB predisposing gene GPRI161 (7). Nearly 50% of the MB patients with predisposing mutations are not identified based on family cancer histories, suggesting that genetic counseling and testing should be considered in MB, specifically those arising in SHH MB in the pediatric population and WNT MB for which CTTNB1 mutations are not identified (84).
Combined Testing Recommendations
Combining our accumulated knowledge of MB, the standard of care for neuropathologic evaluation has changed dramatically over recent years. Initial review should be performed to establish the diagnosis of MB and assign a histologic variant. If tissue is adequate, molecular group designation should be determined using established methods in a CLIA certified laboratory. Laboratories should work toward establishing methods in time that can separate all molecular groups, including G3 and G4, by transcriptome or methylation‐based platforms. While many laboratories lack the capabilities to perform subgrouping by these methods, most are capable of implementing IHC‐based molecular subgrouping, separating tumors in to the WNT, SHH, or non‐WNT/non‐SHH groups. In low‐volume centers, for which introduction of MB immunostains for molecular grouping is impractical or validation is infeasible, secondary consultation should be explored. Ancillary FISH testing is important for risk stratification, particularly for MYC and MCYN. In WNT MB, evaluation for monosomy 6 by FISH or sequencing of CTTNB1 are important confirmatory tools. In SHH MB, PTCH1 deletion can predict response to smoothened inhibitors and should be considered, especially in the setting of recurrent disease for which the patient has not seen that drug class previously. Testing for TP53 abnormalities, by IHC or sequencing has important prognostic role in SHH MB and should be routinely performed in tumors falling in the SHH molecular group. While TP53 abnormalities can be seen in WNT MB, the significance is less clear and not uniformly required. Germline testing should be considered in all tumors falling into the SHH group, but especially in the infant population. For WNT MB, germline testing is important for CTTNB1 mutation‐negative cases. All pertinent findings related to the histologic classification, genetic definitions, risk‐stratifying molecular findings, and genomic abnormalities should be combined into an integrated report according the standards proposed by the Haarlem consensus conference, incorporating all tissue‐based information (44).
Conclusions
International collaboration has led to advanced understanding of the biology and molecular underpinnings of MB. These findings are now being translated to the clinical laboratory as a new standard of care and have made MB a prototype for modern tumor classification. Real progress will come from integration of molecular testing into risk‐adapted or targeted clinical trials, to improve survival and reduce long‐term treatment‐related morbidity by matching molecular groups to appropriate therapies.
Conflicts of Interest
The author has not conflict of interest to declare.
Acknowledgments
I thank Dr. David Ellison for thoughtful review of this manuscript. The manuscript is supported by the American Lebanese Syrian Associated Charities (ALSAC).
References
- 1. Andrey K, Felix S, Olga Z, Andrey G, Damian S, Daniel S et al (2019) DNA methylation profiling is a method of choice for molecular verification of pediatric WNT‐activated medulloblastomas. Neuro‐Oncology 21:214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrey K, Lukas C, Northcott PA, Tanvi S, Marina R, Jones DTW et al (2017) DNA‐methylation profiling discloses significant advantages over NanoString method for molecular classification of medulloblastoma. Acta Neuropathol 134:965–967. [DOI] [PubMed] [Google Scholar]
- 3. Arceci RJ (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. Yearb Oncol 28:203–204. [DOI] [PubMed] [Google Scholar]
- 4. Bailey P (1925) Medulloblastoma cerebelli. Arch Neurol Psychiatry 14:192–224. [Google Scholar]
- 5. Bailey P, Cushing H (1926) A Classification of the Tumors of the Glioma Group on a Histogenetic Basis With a Correlated Study of Prognosis. Lippincott: Philadelphia. [Google Scholar]
- 6. Bailey CC, Gnekow A, Wellek S, Jones M, Round C, Brown J et al (1995) Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International society of paediatric oncology (SIOP) and the (German) society of paediatric oncology (GPO): SIOP II. Med Pediatr Oncol 25:166–178. [DOI] [PubMed] [Google Scholar]
- 7. Begemann M, Waszak SM, Robinson GW, Jäger N, Sharma T, Knopp C et al (2020) Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol 38:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bourdeaut F, Grison C, Maurage CA, Laquerriere A, Vasiljevic A, Delisle MB et al (2013) MYC and MYCN amplification can be reliably assessed by aCGH in medulloblastoma. Cancer Genetics 206:124–129. [DOI] [PubMed] [Google Scholar]
- 9. Brown HG, Kepner JL, Perlman EJ, Friedman HS, Strother DR, Duffner PK et al (2000) “Large cell/anaplastic” medulloblastomas: a pediatric oncology group study. J Neuropathol Exp Neurol 59:857–865. [DOI] [PubMed] [Google Scholar]
- 10. Capper D, Jones DT, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation‐based classification of central nervous system tumours. Nature 555:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cavalli FM, Remke M, Rampasek L, Peacock J, Shih DJ, Luu B et al (2017) Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31:737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H et al (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Ellison DW (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub‐group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–2670. [DOI] [PubMed] [Google Scholar]
- 14. Eberhart CG, Kepner JL, Goldthwaite PT, Kun LE, Duffner PK, Friedman HS et al (2002) Histopathologic grading of medulloblastomas. Cancer 94:552–560. [DOI] [PubMed] [Google Scholar]
- 15. Eberhart CG, Kratz J, Wang Y, Summers K, Stearns D, Cohen K et al (2004) Histopathological and molecular prognostic markers in medulloblastoma. J Neuropathol Exp Neurol 63:441–449. [DOI] [PubMed] [Google Scholar]
- 16. Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G et al (2011) Medulloblastoma: clinicopathological correlates of SHH, WNT, and non‐SHH/WNT molecular subgroups. Acta Neuropathol 121:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ellison DW, Kocak M, Dalton J, Megahed H, Lusher ME, Ryan SL et al (2011) Definition of disease‐risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 29:1400–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Evans DGR, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM (1991) The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64:959–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farwell JR, Dohrmann GJ, Flannery JT (1984) Medulloblastoma in childhood: an epidemiological study. J Neurosurg 61:657–664. [DOI] [PubMed] [Google Scholar]
- 20. Ferguson S, Lesniak MS (2005) Percival Bailey and the classification of brain tumors. Neurosurg Focus 18:1–6. [DOI] [PubMed] [Google Scholar]
- 21. Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S et al (2009) YAP1 is amplified and up‐regulated in hedgehog‐associated medulloblastomas and mediates Sonic hedgehog‐driven neural precursor proliferation. Genes Dev 23:2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway D et al (2008) Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol 26:317–325. [DOI] [PubMed] [Google Scholar]
- 23. Giangaspero F, Perilongo G, Fondelli MP, Brisigotti M, Carollo C, Burnelli R et al (1999) Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91:971–977. [DOI] [PubMed] [Google Scholar]
- 24. Giangaspero F, Rigobello L, Badiali M, Loda M, Andreini L, Basso G et al (1992) Large‐cell medulloblastomas. Am J Surg Pathol 16:687–693. [PubMed] [Google Scholar]
- 25. Giangaspero F, Wellek S, Masuoka J, Gessi M, Kleihues P, Ohgaki H (2006) Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112:5–12. [DOI] [PubMed] [Google Scholar]
- 26. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilbertson Richard J, Ellison David W (2008) The origins of medulloblastoma subtypes. Annu Rev Pathol 3:341–365. [DOI] [PubMed] [Google Scholar]
- 28. Goschzik T, Schwalbe EC, Hicks D, Smith A, zur Muehlen A, Figarella‐Branger D et al (2018) Prognostic effect of whole chromosomal aberration signatures in standard‐risk, non‐WNT/non‐SHH medulloblastoma: a retrospective, molecular analysis of the HIT‐SIOP PNET 4 trial. Lancet Oncol 19:1602–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gupta K, Jogunoori S, Satapathy A, Salunke P, Kumar N, Radotra BD, Vasishta RK (2018) Medulloblastoma with myogenic and/or melanotic differentiation does not align immunohistochemically with the genetically defined molecular subgroups. Hum Pathol 75:26–33. [DOI] [PubMed] [Google Scholar]
- 30. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM et al (1995) The molecular basis of Turcot's syndrome. N Engl J Med 332:839–847. [DOI] [PubMed] [Google Scholar]
- 31. Helton KJ, Fouladi M, Boop FA, Perry A, Dalton J, Kun L, Fuller C (2004) Medullomyoblastoma: a radiographic and clinicopathologic analysis of six cases and review of the literature. Cancer 101:1445–1454. [DOI] [PubMed] [Google Scholar]
- 32. Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R et al (2013) Robust molecular subgrouping and copy‐number profiling of medulloblastoma from small amounts of archival tumour material using high‐density DNA methylation arrays. Acta Neuropathol 125:913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iorgulescu JB, Van Ziffle J, Stevers M, Grenert JP, Bastian BC, Chavez L et al (2018) Deep sequencing of WNT‐activated medulloblastomas reveals secondary SHH pathway activation. Acta Neuropathol 135:635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M et al (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kieran MW, Chisholm J, Casanova M, Brandes AA, Aerts I, Bouffet E et al. (2017) Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro‐Oncology 19:1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. King AA, Seidel K, Di C, Leisenring WM, Perkins SM, Krull KR et al (2017) Long‐term neurologic health and psychosocial function of adult survivors of childhood medulloblastoma/PNET: a report from the Childhood Cancer Survivor Study. Neuro Oncol 19:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kleihues P, Schäuble B, zur Hausen A, Estève J, Ohgaki H. (1997) Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 38. Kool M, Jones DT, Jäger N, Northcott PA, Pugh TJ, Hovestadt V et al (2014) Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P et al (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 3:e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Korshunov A, Ryzhova M, Jones DTW, Northcott PA, van Sluis P, Volckmann R et al (2012) LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 124:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leonard JR, Cai DX, Rivet DJ, Kaufman BA, Park TS, Levy BK, Perry A (2001) Large cell/anaplastic medulloblastomas and medullomyoblastomas: clinicopathological and genetic features. J Neurosurg 95:82–88. [DOI] [PubMed] [Google Scholar]
- 43. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A et al (2014) International Society of Neuropathology‐Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 24:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. [DOI] [PubMed] [Google Scholar]
- 46. McManamy CS, Lamont JM, Taylor RE, Cole M, Pearson AD J, Clifford SC, Ellison DW (2003) Morphophenotypic variation predicts clinical behavior in childhood non‐desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62:627–632. [DOI] [PubMed] [Google Scholar]
- 47. McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE et al (2007) Nodule formation and desmoplasia in medulloblastomas—defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol 17:151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Merchant TE, Pollack IF, Loeffler JS (2010) Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol 20:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moran S, Arribas C, Esteller M (2016) Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mulhern RK, Palmer SL, Merchant TE, Wallace D, Kocak M, Brouwers P et al (2005) Neurocognitive consequences of risk‐adapted therapy for childhood medulloblastoma. J Clin Oncol 23:5511–5519. [DOI] [PubMed] [Google Scholar]
- 51. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole‐genome landscape of medulloblastoma subtypes. Nature 547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D et al (2014) Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Northcott PA, Shih DJH, Remke M, Cho YJ, Kool M, Hawkins C et al (2012) Rapid, reliable, and reproducible molecular sub‐grouping of clinical medulloblastoma samples. Acta Neuropathol 123:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Olivier TW, Bass JK, Ashford JM, Beaulieu R, Scott SM, Schreiber JE et al (2019) Cognitive implications of ototoxicity in pediatric patients with embryonal brain tumors. J Clin Oncol 37:1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Orr BA, Clay MR, Pinto EM, Kesserwan C (2020) An update on the central nervous system manifestations of Li–Fraumeni syndrome. Acta Neuropathol 139:669–687. [DOI] [PubMed] [Google Scholar]
- 57. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz‐Sloan JS (2018) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro‐Oncology 20(suppl_4):iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pan E, Pellarin M, Holmes E, Smirnov I, Misra A, Eberhart CG et al (2005) Isochromosome 17q is a negative prognostic factor in poor‐risk childhood medulloblastoma patients. Clin Cancer Res 11:4733–4740. [DOI] [PubMed] [Google Scholar]
- 59. Patay Z, DeSain LA, Hwang SN, Coan A, Li Y, Ellison DW (2015) MR imaging characteristics of wingless‐type–subgroup pediatric medulloblastoma. Am J Neuroradiol 36:2386–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT et al (2016) Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell 29:508–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, Kool M (2016) Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 131:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rausch T, Jones DTW, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J et al (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Remke M, Hielscher T, Korshunov A, Northcott PA, Bender S, Kool M et al (2011) FSTL5 is a marker of poor prognosis in non‐WNT/non‐SHH medulloblastoma. J Clin Oncol 29:3852–3861. [DOI] [PubMed] [Google Scholar]
- 64. Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A et al (2011) Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 29:2717–2723. [DOI] [PubMed] [Google Scholar]
- 65. Roberts RO, Lynch CF, Jones MP, Hart MN (1991) Medulloblastoma: a population‐based study of 532 cases. J Neuropathol Exp Neurol 50:134–144. [DOI] [PubMed] [Google Scholar]
- 66. Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A et al (2017) Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8:69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I et al (2015) Vismodegib exerts targeted efficacy against recurrent sonic hedgehog‐subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC‐025B and PBTC‐032. J Clin Oncol 33:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS et al (2018) Risk‐adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 19:768–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rubinstein LJ, Northfield DWC (1964) The medulloblastoma and the so–called “arachnoidal cerebellar sarcoma”. Brain 87:379–412. [DOI] [PubMed] [Google Scholar]
- 71. Salloum R, Chen Y, Yasui Y, Packer R, Leisenring W, Wells E et al (2019) Late morbidity and mortality among medulloblastoma survivors diagnosed across three decades: a report from the childhood cancer survivor study. J Clin Oncol 37:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sandoval J, Heyn H, Moran S, Serra‐Musach J, Pujana MA, Bibikova M, Esteller M (2011) Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6:692–702. [DOI] [PubMed] [Google Scholar]
- 73. Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D et al (2017) Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol 18:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schwalbe EC, Williamson D, Lindsey JC, Hamilton D, Ryan SL, Megahed H et al (2013) DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin‐fixed biopsies. Acta Neuropathol 125:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek M et al (2019) Second‐generation molecular subgrouping of medulloblastoma: an international meta‐analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 138:309–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M et al (2014) Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32:886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith MJ, Beetz C, Williams SG, Bhaskar SS, O'Sullivan J, Anderson B et al (2014) Germline mutations in SUFU cause Gorlin syndrome‐associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32:4155–4161. [DOI] [PubMed] [Google Scholar]
- 78. Stefanits H, Ebetsberger‐Dachs G, Weis S, Haberler C (2014) Medulloblastoma with multi‐lineage differentiation including myogenic and melanotic elements: a case report with molecular data. Clin Neuropathol 33:122–127. [DOI] [PubMed] [Google Scholar]
- 79. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D et al (2016) New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 164:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taylor RE, Bailey CC, Robinson K, Weston CL, Ellison D, Ironside J et al (2003) Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children's Cancer Study Group PNET‐3 Study. J Clin Oncol 21:1581–1591. [DOI] [PubMed] [Google Scholar]
- 81. Taylor MD, Mainprize TG, Rutka JT (2000) Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery 47:888–901. [DOI] [PubMed] [Google Scholar]
- 82. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC et al (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931. [DOI] [PubMed] [Google Scholar]
- 84. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S et al (2018) Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G (1997) Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 57:2581–2585. [PubMed] [Google Scholar]
- 86. Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC et al (2013) Subgroup‐specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31:2927–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]