

Abstract
Background
Alpha‐2‐antiplasmin (α2AP) is the main natural inhibitor of plasmin. The C‐terminus of α2AP is crucial for the initial interaction with plasmin(ogen) and the rapid inhibitory mechanism. Approximately 35% of circulating α2AP has lost its C‐terminus (non‐plasminogen binding α2AP/NPB‐α2AP) and thereby its rapid inhibitory capacity. The C‐terminal cleavage site of α2AP is still unknown. A commercially available monoclonal antibody against α2AP (TC 3AP) detects intact but not NPB‐α2AP, suggesting that the cleavage site is located N‐terminally from the epitope of TC 3AP.
Objectives
To determine the epitope of TC 3AP and then to localize the C‐terminal cleavage site of α2AP.
Methods
For epitope mapping of TC 3AP, commercially available plasma purified α2AP was enzymatically digested with Asp‐N, Glu‐C, or Lys‐N. The resulting peptides were immunoprecipitated using TC 3AP‐loaded Dynabeads® Protein G. Bound peptides were eluted and analyzed by liquid chromatography‐tandem mass spectometry (LC‐MS/MS). To localize the C‐terminal cleavage site precisely, α2AP (intact and NPB) was purified from plasma and analyzed by LC‐MS/MS after enzymatic digestion with Arg‐C.
Results
We localized the epitope of TC 3AP between amino acid residues Asp428 and Gly439. LC‐MS/MS data from plasma purified α2AP showed that NPB‐α2AP results from cleavage at Gln421‐Asp422 as preferred site, but also after Leu417, Glu419, Gln420, or Asp422.
Conclusions
The C‐terminal cleavage site of human α2AP is located N‐terminally from the TC 3AP epitope. Because C‐terminal cleavage of α2AP can occur after multiple residues, different proteases may be responsible for the generation of NPB‐α2AP.
Keywords: alpha‐2‐antiplasmin, epitope mapping, mass spectrometry, proteolysis, western blot
Essentials.
Due to proteolytic cleavage, part of circulating a2‐antiplasmin (α2AP) loses the C‐terminus.
C‐terminally cleaved α2AP is unable to inhibit plasmin rapidly.
We found that C‐terminal cleavage of α2AP occurs between residues Leu417 and Asp422.
Our study gives more insight into the regulation of C‐terminal heterogeneity of α2AP.
1. INTRODUCTION
Alpha‐2‐antiplasmin (α2AP) is the main natural inhibitor of the fibrinolytic enzyme plasmin.1, 2, 3
Native α2AP is produced by the liver and circulates in blood as an approximately 67‐kDa single‐chain glycoprotein of 464 amino acid residues, containing 11% to 14% carbohydrate.2, 4, 5 α2AP possesses unique N‐ and C‐terminal ends, as we extensively reviewed.6 The N‐terminus is involved in α2AP incorporation into a blood clot by activated factor XIII‐mediated crosslinking.7, 8 The C‐terminus is crucial for the initial interaction of α2AP with plasmin(ogen).9 α2AP contains six lysine residues at its C‐terminus, which are involved in the interaction with lysine binding sites in plasminogen.9, 10 Based on the interaction with these lysine binding sites, two molecular species of α2AP have been described in plasma.11 One molecular form is able to bind to plasminogen (plasminogen binding α2AP, PB‐α2AP), comprises approximately 65% of circulating α2AP, and is a fast‐acting inhibitor of plasmin by rapidly forming plasmin‐antiplasmin complexes. The other molecular form, which comprises approximately 35% of circulating α2AP, does not bind to plasminogen (non‐plasminogen binding α2AP, NPB‐α2AP) and inhibits plasmin at a much slower rate.11, 12 Kluft and Los demonstrated the presence of the two variants in plasma and in serum by modified crossed immunoelectrophoresis.13 They also showed that only PB‐α2AP is produced in the liver and that NPB‐α2AP is the result of posttranslational modification in the circulation.14 Furthermore, it was reported that the NPB‐α2AP fraction in plasma may contain some inactive or inactivated inhibitor.11, 15
In addition to the main form of NPB‐α2AP in vivo, it was shown that purified PB‐α2AP can spontaneously convert into an NPB‐α2AP form in vitro.16 Sasaki et al17 treated both purified PB‐α2AP and in vitro formed NPB‐α2AP after acetamidination with trypsin and analyzed the peptides by HPLC. A peptide representing the 26 C‐terminal amino acids of the molecule was found in PB‐α2AP, which was not present in NPB‐α2AP formed in vitro. This indicated the participation of this C‐terminal part in plasminogen binding. Some subsequent papers have mistakenly considered this trypsin cleavage site as the position where PB‐α2AP is truncated into NPB‐α2AP.18, 19 The study by Sasaki et al only showed that NPB‐α2AP is formed in vitro after a cleavage of a peptide smaller than 26 amino acid residues. To date, it is unknown where and how the conversion happens in the circulation. An actual in vivo cleavage site and a protease responsible have not yet been identified.
Leebeek et al20 have suggested that the in vivo and in vitro conversions of PB‐ α2AP to NPB‐α2AP in plasma may have different mechanisms and possibly involve different cleavage sites. Using a commercially available monoclonal antibody against the C‐terminus of α2AP (TC 3AP21) in a crossed immunoelectrophoresis technique, they showed that NPB‐α2AP formed in vivo lost its capacity to bind to both plasminogen and the antibody, whereas NPB‐α2AP formed in vitro lost the plasminogen binding capacity, but could still bind to the antibody. This indicated that in vivo and in vitro generated NPB‐α2AP are not the same molecules but may differ in their C termini or may be structurally different. We reckoned that the epitope recognized by TC 3AP in the α2AP protein can be used to obtain a crude localization of the in vivo C‐terminal cleavage site of α2AP; therefore, this study aimed to determine the epitope of TC 3AP and subsequently localize the C‐terminal cleavage site of α2AP by mass spectrometry.
2. METHODS
2.1. Materials
Pooled normal plasma was prepared from citrated apheresis plasma (Sanquin blood bank, Rotterdam, The Netherlands) of five healthy donors. Commercially available plasma‐purified α2AP (Calbiochem) was purchased from Merck Millipore. The mouse monoclonal antibody against the C‐terminus of α2AP (TC 3AP) was purchased from Technoclone. The custom made polyclonal rabbit Asn‐α2AP antibody (anti‐Asn‐α2AP, affinity purified IgG) was raised against a peptide corresponding to amino acids Gln14 to Pro30 of the α2AP protein (Charles River Laboratories, Squarix GmbH) and has been used previously.22 The polyclonal goat total α2AP antibody (affinity purified IgG) (anti‐total α2AP) directed to all forms of α2AP and the horseradish peroxidase‐conjugated anti‐total α2AP antibody were purchased from Affinity Biologicals. Cyanogen bromide‐activated‐Sepharose® 4B was purchased from GE Healthcare. SigmaFAST TM Protease Inhibitor Cocktail, ammonium bicarbonate, 2‐chloroacetamide, 1,4‐dithiothreitol (DTT), and 3,3′,5,5′‐tetramethylbenzidine were purchased from Sigma‐Aldrich. Magnetic Dynabeads® Protein G, GelCode® Blue Stain Reagent, and endoproteinase Glu‐C were purchased from Thermo Fisher Scientific. Endoproteinase Arg‐C was purchased from Protea. Endoproteinase Asp‐N was obtained from Roche. Endoproteinase Lys‐N was purchased from U‐Protein Express BV. XT Sample Buffer, XT reducing agent, and precast XT Criterion 10% polyacrylamide gels were purchased from BioRad.
2.2. Epitope mapping of antibody TC 3AP
Epitope mapping of TC 3AP was carried out according to an adapted epitope extraction method as described previously.23, 24 In detail, commercially available plasma‐purified α2AP, lyophilized in buffer (200 mmol/L NaCl, 20 mmol/L Bis‐Tris, pH 6.4), was resuspended in distilled water followed by addition of Tris‐HCl (pH 8.5) (50 mmol/L final concentration). Reduction and alkylation of α2AP was carried out by adding DTT (5 mmol/L final concentration), followed by incubation for 30 minutes at 50°C under constant motion. Next, 2‐chloroacetamide was added (10 mmol/L final concentration) followed by incubation for 30 minutes at room temperature (RT) in the dark. The reaction was quenched by the addition of extra DTT (2.5 mmol/L final concentration), followed by digestion of α2AP with Asp‐N, Glu‐C, or Lys‐N. A 1:20 final ratio (w/w) of Asp‐N or Glu‐C with α2AP, and a 1:1 final ratio of Lys‐N with α2AP was used (buffers: 50 mmol/L Tris‐HCl, 1 mmol/L CaCl2, pH 8.5 for Asp‐N; 50 mmol/L Tris‐HCl, pH 8.5 for Glu‐C; 100 mmol/L Tris‐HCl, 1 mmol/L CaCl2, pH 10 for Lys‐N). The mixtures were incubated overnight at 30°C. The digestion reactions were terminated by boiling the mixtures for 10 minutes at 95°C.
After enzymatic digestion, α2AP peptides containing the epitope were purified from the mixtures by immunoprecipitation with TC 3AP coupled magnetic Dynabeads® Protein G. The coupling procedure was carried out by diluting 20 µg TC 3AP with phosphate‐buffered saline with Tween (PBST; 137 mmol/L NaCl, 2.7 mmol/L KCl, 6.5 mmol/L Na2HPO4, 1 mmol/L KH2PO4, 0.02% Tween‐20, pH 7.4), followed by incubation of the diluted antibody solution with 3 mg magnetic Dynabeads® Protein G for 1 hour at 4°C under constant motion. The beads were separated from the solution using a magnet, followed by removal of the supernatant. The beads were washed with PBST, mixed with the digested α2AP, and incubated overnight at 4°C under constant motion. After three stringent wash steps with PBST and five wash steps with phosphate buffered saline (PBS), bound α2AP peptides were eluted from the beads using 100 mmol/L glycine buffer (pH 2.2) during incubation for 1 hour at RT under constant motion. Eluted peptides were collected, desalted using ZipTip C18 according to the manufacturer's instructions, dried, and analyzed by liquid chromatography‐tandem mass spectrometry.
2.3. Purification of α2AP
For the localization of the C‐terminal cleavage site, PB‐ and NPB‐α2AP were purified from normal pooled plasma by affinity chromatography. Briefly, 1 mg anti‐total α2AP was coupled to 1 mL cyanogen bromide‐activated‐Sepharose® 4B. Normal pooled plasma (6 mL) was diluted with 6 mL wash buffer (0.1 mol/L Tris‐HCl, 0.1% Tween‐20, 1 mol/L NaCl, pH 8.1). To prevent nonspecific cleavages in α2AP, Protease Inhibitor solution was added to the diluted plasma sample, wash buffer and elution buffer (100 mmol/L glycine, 0.01% Tween‐20, 0.1 mol/L NaCl, pH 2.2). The diluted plasma sample was incubated overnight with the anti α2AP‐IgG‐Sepharose 4B gel at 4°C with gentle stirring. After incubation, the gel was washed four times with wash buffer and placed in a column. α2AP was eluted with 4 mL elution buffer. The eluted fractions were neutralized with 1 mol/L Tris‐HCl, pH 9.0.
The α2AP concentration of the eluted fractions was determined by ELISA. 96‐Well microtiter plates (Nunc A/S, Roskilde, Denmark) were coated with 2 µg/mL (110 µL well‐1 in 0.05 mol/L carbonate buffer, pH 9.6) anti‐total α2AP antibody overnight at 4°C. Nonspecific sites were blocked with 200 µL 1% BSA in PBS containing 0.002% Tween‐20 for 1 hour at RT and plates were washed once with PBS/0.002% Tween‐20. Subsequently, eluted fractions were diluted 40 times and serial dilutions were made of normal pooled plasma (1:200‐1:12 800) for the standard curve. Plates were incubated for 2 hours at RT followed by four rounds of washing with PBS/0.002% Tween‐20. Next, a horseradish peroxidase‐conjugated anti‐total α2AP antibody (1:2000) was added and incubated for 1 hour at RT. After four wash steps with PBS/0.002% Tween‐20, enzyme activity was determined using 3,3′,5,5′‐tetramethylbenzidine as substrate and the reaction was stopped after 10 minutes by adding 100 µL/well of 1 mol/L H2SO4. The absorbance was read at 450 nm on a Victor3 1420 Multilabel Plate Counter (Perkin Elmer).
The eluted fractions with the highest α2AP concentrations were pooled and dialyzed against distilled water. Finally, the dialyzed pooled sample was dried by a SpeedVac procedure and resuspended in 60 µL distilled water.
2.4. SDS‐PAGE and Western blotting
To separate PB‐α2AP and NPB‐α2AP, 5 µg (for SDS‐PAGE) and 300 ng (for Western blot analyses) affinity‐purified α2AP in Tris‐buffered saline were boiled for 5 to 10 minutes at 95°C in the presence of XT Sample Buffer and XT reducing agent and resolved on a precast XT Criterion 10% polyacrylamide gel. After electrophoresis, the proteins were stained by Coomassie or transferred to a nitrocellulose membrane using a PowerPac™ HC power supply (BioRad, Richmond, CA) with transfer buffer (25 mmol/L Tris, 192 mmol/L glycine [pH 8.3], and 20% methanol) at 100V for 1 hour. After protein transfer, nonspecific sites on the nitrocellulose membrane were blocked with 5% milk in PBS, pH 7.4, followed by three wash steps with PBS/0.1% Tween‐20. Postblocking, the blot was incubated with a mixture of TC 3AP (1 mg/mL, 1:5000) and anti‐Asn‐α2AP (1 mg/mL, 1:5000) in PBS/0.1% Tween‐20 overnight at 4°C under constant motion. After three wash steps, IRDye® 680CW donkey anti‐mouse secondary antibody and IRDye® 800CW donkey anti‐rabbit secondary antibody were used for detection of TC 3AP and the anti‐Asn‐α2AP antibody, respectively. After incubation for 1 hour at RT, three wash steps were performed and the blot was scanned in the 680‐ or 800‐nm channel of an Odyssey® Imaging System (Lincoln, NE).
2.5. Mass spectrometry for epitope mapping and C‐terminal cleavage site analysis
For epitope mapping, the dried peptides containing the TC 3AP epitope were dissolved in 3% acetonitrile (ACN) with 0.5% formic acid (FA) and briefly sonicated. A fraction was injected onto a Waters nanoAcquity LC equipped with a Waters 20 mm x 180 µm nanoACQUITY UPLC Symmetry C18 Trap Column with 5‐µm particles and a homemade 40 cm x 75 µm fused silica analytical column with Waters 3.5‐µm XBridge BEH C18 particles. Peptides were separated using a 60‐minute gradient from 99%A/1%B to 65%A/35%B (A = 0.1% FA, B = 0.1% FA in ACN) at 0.3 µL/min and 50°C and analyzed on a Thermo Orbitrap Fusion mass spectrometer in positive mode using a nanoESI source and a top speed method with 3 seconds’ cycle time and HCD/ETD fragmentation, full scan detection at 120K resolution in the orbitrap, and fragment ion detection in the ion trap.
Peak lists were automatically created from raw data files using the Mascot Distiller software (version 2.3; MatrixScience). The Mascot algorithm was used for searching against a UniProt database (release 2015_03.fasta or newer, taxonomy: Homo sapiens). The peptide tolerance was set to 10 ppm and the fragment ion tolerance was set to 0.8 Da for fragmentation spectra. A maximum number of two missed cleavages were allowed. Carbamidomethylated cysteine was set as fixed modification and oxidized methionine and deamidated asparagine and glutamine were set as variable modifications.
Data were additionally analyzed with Proteome Discoverer (version 1.4; Thermo Scientific) and MaxQuant (version 1.5.2.8) using similar settings25 and appropriate digestion modes. For MaxQuant a false discovery rate of 0.01 for proteins and peptides and a minimum peptide length of seven amino acids were required. The tandem mass spectometry spectra were searched against the UniProt database concatenated with the reversed versions of all sequences.
For C‐terminal cleavage site analysis, a pilot experiment was performed in which gel bands corresponding to PB‐α2AP and NPB‐α2AP were cut from the gel, divided into equal parts, and destained, reduced/alkylated, and dehydrated. The protein bands were subjected to overnight in‐gel digestion at RT using 0.25 µg Arg‐C in 50 mmol/L ammonium bicarbonate, 1 mmol/L CaCl2 1 mmol/L DTT pH 8, 0.05 µg Asp‐N in 50 mmol/L Tris‐HCl 1mM CaCl2 pH 8, 0.1 µg Lys‐C in 25 mmol/L Tris‐HCl pH 8.5, or 0.2 µg trypsin in 50 mmol/L ammonium bicarbonate 1 mmol/L CaCl2 pH 8.0. Peptides were extracted from the gel with 30% ACN 0.5% FA and mixing for 30 minutes in a shaker followed by sonication in a water bath for 2 minutes. The extracts were dried in a SpeedVac and the residues were redissolved in 3% ACN 0.5% FA and desalted with a ZipTip C18 according to the manufacturer's instructions. The final eluates were dried in a SpeedVac and redissolved in 3% ACN 0.5% FA and sonicated briefly. Aliquots were injected into the mass spectrometer as described previously.
After the pilot experiment, the C‐terminal cleavage site analysis was repeated under optimized conditions in which bands on gel were quantified using a BioRad GS‐900 Calibrated Densitometer and the NPB‐α2AP band, calculated at 1 ug protein, was cut from the gel and pretreated as described previously. Protein was in‐gel digested at 30°C overnight with 0.5 µg Arg‐C in 50 mmol/L ammonium bicarbonate, 5 mmol/L CaCl2, and 5 mmol/L DTT. Peptides were extracted, desalted, dried, and redissolved as described. An aliquot was injected onto a Thermo Scientific Easy‐nLC 1200 equipped with a homemade 40 cm x 75 µm fused silica analytical column with Waters 3.5 µm XBridge BEH C18 particles without trapping column. Peptides were separated using a 60‐minute gradient from 99%A/1%B to 50%A/50%B (A = 0.1% FA, B = 0.1% FA in 80% ACN) at 0.3 µL/min and 50°C and analyzed on a Thermo Orbitrap Fusion Lumos Tribrid mass spectrometer in positive mode using a nanoESI source and a top speed method with 3 seconds’ cycle time and HCD/EThcD fragmentation, full scan detection at 60K resolution in the orbitrap, and fragment ion detection in the ion trap. Expected NPB‐α2AP C‐terminal peptide masses, based on results from the pilot experiment, were calculated assuming charge states 2+ and 3+, up to 4 deamidations and up to 1 missed cleavage using sequences ELKEQQ, ELKEQQD, NPNSAPRELKEQQ, and NPNSAPRELKEQQD. These in total 26 m/z masses were favored for MS2 fragmentation by HCD and EThcD over other masses using an AGC target of 1E4 and increased injection times of 200 ms versus 50 ms for other masses (Table S1).
Data were analyzed with Mascot and MaxQuant (version 1.5.4.1) using similar settings as described and searched against the UniProt human database supplemented with truncated versions of α2AP (UniProt identifier P08697‐1) starting from amino acid 300 up to 491 and concatenated with the reversed versions of all sequences. Numbering of α2AP amino acids was according to methionine in position 1.
3. RESULTS
3.1. Epitope mapping of antibody TC 3AP
To obtain a crude localization of the α2AP C‐terminal cleavage site, we first determined the epitope of TC 3AP. Because this antibody does not react with NPB‐α2AP, we hypothesized that the in vivo cleavage site is located N‐terminally from the TC 3AP epitope. The MS data of the Asp‐N digest identified two peptides that were bound to immobilized TC 3AP and that should contain the epitope of TC 3AP: Asp428‐Gly439 (ions score: 39 and 51) and Asp422‐Gly439 (ions score: 41, 57 and 61) (Table 1). Multiple ions scores were found for the same peptide, depending on their charge (2+, 3+, or 4+). The MS data of the Glu‐C and Lys‐N digests did not display any peptides, probably because of the presence of multiple protease specific cleavage sites in this region, resulting in small peptides that do not bind to immobilized TC 3AP or are undetectable by MS. Thus, we localized the epitope of TC 3AP to the region between Asp428 and Gly439.
Table 1.
Epitope mapping of antibody TC 3AP
| Sequence | Amino acids | Frequency | Mass | m/z | Dev | Score |
|---|---|---|---|---|---|---|
| K.DFLQSLKGFPRG.D | 428‐439 | 11 | 1363.7248 | 455.5815 | −1.57 | 39 |
| K.DFLQSLKGFPRG.D | ‐ | ‐ | ‐ | 682.8696 | −0.18 | 51 |
| Q.DSPGNKDFLQSLKGFPRG.D | 422‐439 | 12 | 1961.9959 | 491.5056 | −1.44 | 41 |
| Q.DSPGNKDFLQSLKGFPRG.D | ‐ | ‐ | ‐ | 655.0053 | −1.02 | 57 |
| Q.DSPGNKDFLQSLKGFPRG.D | ‐ | ‐ | ‐ | 982.0041 | −1.11 | 61 |
Dev, the difference between the observed and calculated mass (in ppm); frequency, the number of times a peptide is observed in the mass spectrometry analysis; mass: the theoretical monoisotopic mass of the neutral peptide (in Daltons); m/z, the observed mass/charge; score, the ions score given to the peptide.
Peptides identified by Mascot analysis in the immobilized TC 3AP‐bound α2AP fraction produced by enzymatic digestion of α2AP with Asp‐N. Best scoring spectra are reported.
3.2. C‐terminal cleavage site analysis
SDS‐PAGE of the affinity purified α2AP showed protein bands with molecular weights between 50 and 67 kDa (Figure 1A). Western blot analyses showed reactivity of the anti‐Asn‐α2AP antibody to apparently two bands with molecular weights of approximately 60 kDa and 67 kDa (Figure 1B). Multiple protein bands within the 60‐ to 67‐kDa range showed reactivity with TC 3AP (Figure 1C), indicating the presence of the C‐terminus. However, there was one protein band at approximately 63 kDa that did not react with TC 3AP, suggesting the absence of the C‐terminus, thus representing NPB‐α2AP (red band in Figure 1D). The two minor bands below the main band of PB‐α2AP in Figure 1C could possibly explained by deglycosylation, truncation at the N‐terminus, or by the presence of isoform 2 originating from alternative splicing (www.uniprot.org).
Figure 1.
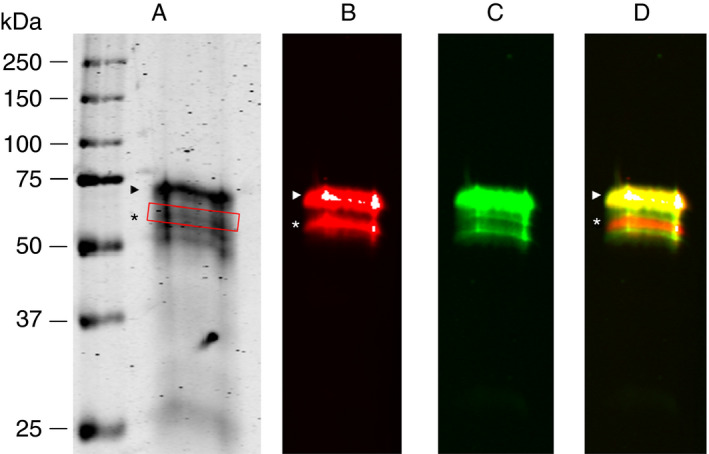
In‐house purified α2AP visualized by SDS‐PAGE and Western blot. (A) SDS‐gel of purified α2AP. (B) Immunoblot obtained with anti‐Asn‐α2AP. (C) Immunoblot obtained with TC 3AP. (D) Merged anti‐Asn‐α2AP and TC 3AP blots. The relative migration distances of molecular weight marker proteins are indicated on the left. PB‐α2AP is indicated by arrowhead. NPB‐α2AP is indicated by an asterisk, as well as by a red box in (A)
Mascot analysis data from the pilot experiment with Arg‐C displayed one peptide (Asn408‐Gln421) in the NPB‐α2AP protein band that did not end with the Arg‐C specific amino acid residue arginine (data not shown). This peptide was not present in PB‐α2AP. Therefore, we hypothesized that this C‐terminal end was already present in the purified α2AP sample and represents the in vivo cleavage site. The digestions with Asp‐N, Lys‐C, and trypsin did not result in any C‐terminal ends that were not the result of protease specific cleavages. The experiment with Arg‐C digestion of NPB‐α2AP was repeated under optimized conditions to confirm the Gln421‐Asp422 cleavage site. Mascot analysis data identified six peptides that could not be ascribed to the action of Arg‐C: Asn408‐Leu417, Asn408‐Glu419, Asn408‐Gln420, Asn408‐Gln421, deamidated Asn408‐Gln421, and Asn408‐Asp422 (Table 2). MaxQuant analysis data from the optimized experiment revealed four of these six peptides: Asn408‐Gln420, Asn408‐Gln421, deamidated Asn408‐Gln421, and Asn408‐Asp422 (Table 2; Figures S1‐S6). In both analysis methods, the Asn408‐Gln421 peptide displayed the highest ions scores and best spectra (Figure 2). Therefore, our data suggest that the Gln421‐Asp422 is a preferred cleavage site in the C‐terminus of α2AP.
Table 2.
NPB‐α2AP peptides identified by Mascot and MaxQuant analyses after Arg‐C digest (Figures S1‐S6)
| Sequence | Mascot analysis | MaxQuant analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino acids | Frequency | Mass | m/z | Dev | Score | Frequency | Mass | m/z | Dev | Score | |
| R.NPNPSAPREL.K | 408‐417 | 1 | 1093.5518 | 547.7832 | 0.18 | 35 | n.d. | n.d. | n.d. | n.d. | n.d. |
| R.NPNPSAPRELKE.Q | 408‐419 | 2 | 1350.6891 | 451.2374 | 0.89 | 34 | n.d | n.d | n.d | n.d | n.d |
| R.NPNPSAPRELKEQ.Q | 408‐420 | 5 | 1478.7477 | 740.3826 | 2.03 | 58 | 1 | 1478.7478 | 740.3826 | 1.44 | 185 |
| R.NPNPSAPRELKEQ.Q | ‐ | ‐ | ‐ | ‐ | ‐ | 1 | ‐ | 494.2582 | ‐2.63 | 101 | |
| R.NPNPSAPRELKEQQ.D | 408‐421 | 53 | 1606.8063 | 804.4133 | 3.62 | 71 | 25 | 1606.8063 | 536.60947 | ‐2.14 | 193 |
| R.NPNPSAPRELKEQQ.D | 49 | ‐ | 536.6099 | 0.91 | 55 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| R.NPNPSAPRELKEQQ.Da | 408‐421 | 24 | 1607.7903 | 804.9037 | 1.57 | 68 | 10 | 1607.7903 | 804.9047 | 2.21 | 234 |
| R.NPNPSAPRELKEQQ.Da | 19 | ‐ | 536.9383 | 1.71 | 49 | 15 | ‐ | 536.94424 | 1.96 | 187 | |
| R.NPNPSAPRELKEQQD.S | 408‐422 | 1 | 1721.8332 | 861.9233 | ‐0.71 | 33 | 1 | 1721.8333 | 861.9233 | 0.19 | 163 |
| R.NPNPSAPRELKEQQD.S | 8 | ‐ | 574.9523 | 1.01 | 61 | 6 | ‐ | 574.9523 | ‐1.27 | 213 | |
Dev, the difference between the observed and calculated mass (in ppm); frequency, the number of times a peptide is observed in the mass spectrometry analysis; mass: the theoretical monoisotopic mass of the neutral peptide (in Daltons); m/z, the observed mass/charge; n.d., not determined; score, the ions score given to the peptide.
Best scoring spectra are reported.
Deamidation.
Figure 2.
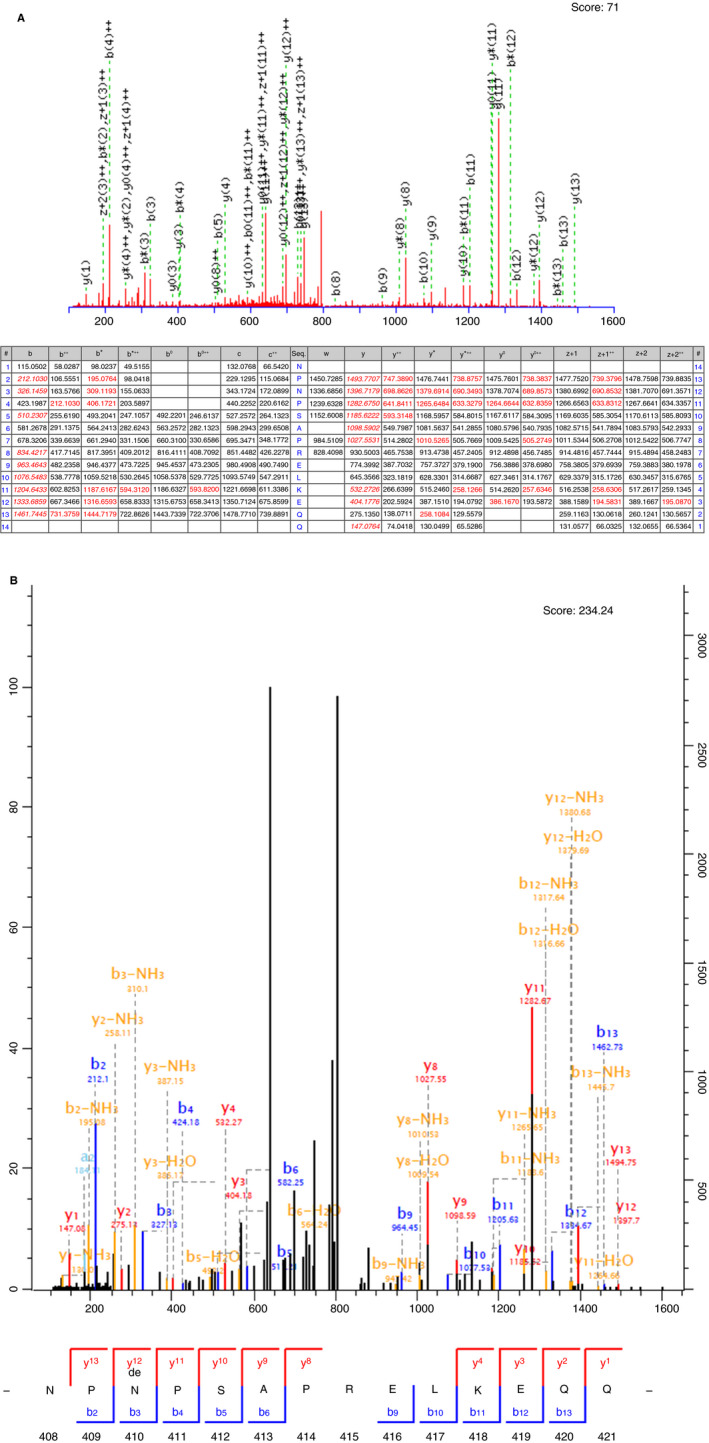
MS/MS fragmentation spectra of the (A) nondeamidated and (B) deamidated Asn408‐Gln421 peptide in an overnight Arg‐C digest of NPB‐α2AP as determined by Mascot and MaxQuant analysis, respectively. Best spectrum data are shown. The overlapping labels of the peaks between m/z 730 and 750 in (A) should be read as the following: b(13)++ m/z 731.3759; y0(13)++,y*(13)++, z + 1(13)**, m/z 738.3837, 738.8757, 739.3796; y(13)++ m/z 747.3890
Taken together, our results suggest that in vivo NPB‐α2AP results from cleavage after Leu417, Glu419, Gln420, Gln421, or Asp422, with Gln421 as preferred cleavage site (Figure 3). As expected, the Leu417‐Asp422 region is located N‐terminally from the TC 3AP epitope, explaining why NPB‐α2AP is not detected by TC 3AP, as summarized in Figure 4.
Figure 3.
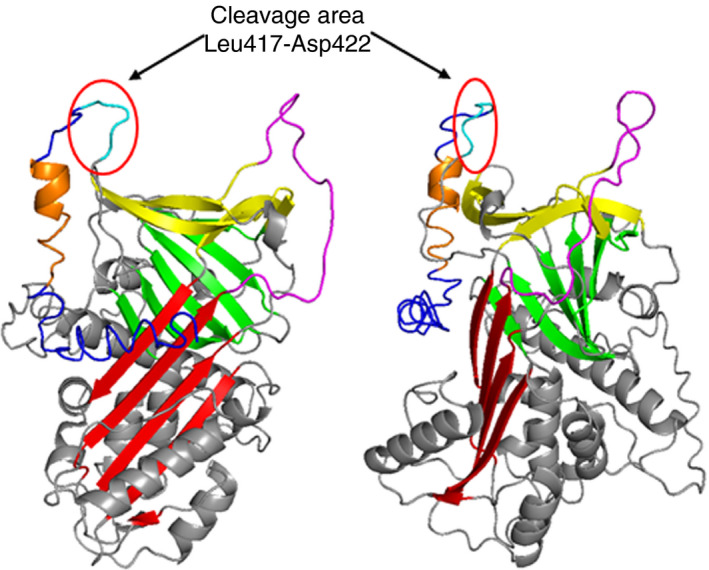
Three‐dimensional structure of human α2AP as predicted by I‐TASSER.27 Red, β‐sheet A; green, β‐sheet B; yellow, β‐sheet C; magenta, reactive center loop; cyan, C‐terminal cleavage area Leu417‐Asp422 (LKEQQD); blue, C‐terminus; orange, epitope of TC 3AP. Left panel: front view. Right panel: side view, 90°C clockwise rotation
Figure 4.

Schematic representation of the α2AP C‐terminus from Asn408 to Lys464. Arrows indicate the in vivo C‐terminal cleavage sites (preferred site between Gln421 and Asp422), the bold underlined sequence represents the area that includes the TC 3AP epitope, and the brace indicates the area that includes the in vitro C‐terminal cleavage site as described by Sasaki et al.17
4. DISCUSSION
In this study, we determined the epitope of monoclonal antibody TC 3AP, which detects PB‐α2AP but not in vivo formed NPB‐α2AP.20 Supported by this information, we specified where PB‐α2AP is truncated in vivo for the generation of NPB‐α2AP. We found that the TC 3AP epitope is located in a region between Asp428 and Gly439 in the α2AP C‐terminus and that NPB‐α2AP results from multiple cleavages in PB‐α2AP that occur in a region between Leu417 and Asp422.
The region of the TC 3AP epitope overlaps the region recognized by a commercial polyclonal antibody directed to α2AP (EB08777, Everest Biotech Ltd.). This commercial antibody was raised against the peptide Lys427‐Lys441. This suggests, that this C‐terminal region is highly immunogenic and strengthens the reliability of our found TC 3AP epitope.
We found that in vivo cleavage of PB‐α2AP occurs between Leu417 and Asp422. In a previous study, Clemmensen et al described the purification of PB‐α2AP from plasma.16 In that study, PB‐α2AP (67 kDa) was spontaneously converted in vitro into two different forms with lower molecular weights (65 and 60 kDa). Native PB‐α2AP was able to bind plasminogen and rapidly inactivate plasmin, whereas the 65‐kDa form did not bind plasminogen but could still slowly inactivate plasmin. The 60‐kDa form neither bound plasminogen nor inactivated plasmin. In our study, we focused on in vivo‐generated NPB‐α2AP. This NPB‐α2AP form still contains the reactive site, which is needed for plasmin inhibition. Our MS data revealed several possible C‐terminal cleavage sites, located C‐terminally from the reactive site, indicating that the NPB‐α2AP form still has inhibitory activity toward plasmin. Thus, although Sasaki et al showed that in vitro‐generated 65 kDa NPB‐α2AP lacks at most 26 amino acid residues (cleavage after Arg438),17 our MS data show that in vivo‐generated NPB‐α2AP lacks about 43 amino acid residues by cleavage in a region between Leu417 and Asp422.
Leebeek et al20 described that NPB‐α2AP formed in vivo lost its capacity to bind to both plasminogen and TC 3AP, which is in line with our MS data showing that NPB‐α2AP formed in vivo is cleaved in a region between Leu417 and Asp422, which is located N‐terminally from the TC 3AP epitope. Furthermore, they showed that NPB‐α2AP formed in vitro lost its plasminogen binding capacity, but could still bind to TC 3AP.20 This in vitro‐formed NPB‐α2AP might be similar to the 65‐kDa form of NPB‐α2AP described by Sasaki et al,17 which lacks at most 26 amino acid residues (Gly439‐Lys464), and still contains the TC 3AP epitope located in a region between Asp428 and Gly439.
Five potential cleavage sites located in close proximity of each other were obtained from MS data, indicating the possibility of multiple NPB‐α2AP forms and suggesting the presence of a proteolytic sensitive region (Figure 4). This makes the possibility of NPB‐α2AP being the result of cleavage by one specific regulating protease unlikely. However, the highest ions scores and best spectra were found for the Asn408‐Gln421 peptide. This peptide was also prominently found in the pilot experiment, suggesting that the Gln421‐Asp422 is a preferred cleavage site. The Peptidase Database MEROPS (release 12.1) contains 33 enzymes capable of cleaving Gln‐Asp bonds and might be helpful for finding candidate enzymes.
An alternative explanation for the five potential cleavage sites is that PB‐α2AP is cleaved by one endopeptidase at Asp422‐Ser423 and further truncated by different carboxypeptidases, such as thrombin‐activatable fibrinolysis inhibitor and carboxypeptidase N. It is, however, unknown whether all required carboxypeptidases exist in plasma. Mutagenesis of α2AP could be useful to further study the cleavage region.
To date, there is no three‐dimensional protein structure available for the complete C‐terminus of α2AP, because of its flexibility.26 Law et al26 showed that the first 10 amino acids of the C‐terminus of α2AP (Asn410‐Glu419) are tightly associated with the serpin body, because residues 416 and 417 are incorporated in β‐sheet C. The remainder of the C‐terminus (Gln420‐Lys464) could not be modeled into electron density. The C‐terminal cleavage sites described in the current study are situated between Leu417 and Asp422, which are located in the beginning of the flexible C‐terminus (Figure 3). Therefore, it is plausible that the found C‐terminal cleavage sites are located in a region sensitive for proteolysis.
To conclude, we mapped the epitope of antibody TC 3AP and localized the in vivo C‐terminal cleavage sites of PB‐α2AP. These results can be used to gain more insight into the regulation of C‐terminal heterogeneity of α2AP and thus the regulation of the fibrinolytic system.
CONFLICT OF INTERESTS
The authors state that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
S. Abdul performed laboratory analyses, analyzed and interpreted data, and wrote the paper. D.H.W. Dekkers performed proteomic analyses and analyzed and interpreted data. R.A.S. Ariëns and F.W.G. Leebeek designed the research and interpreted data. D.C. Rijken and S. Uitte de Willige designed the research, interpreted data, and wrote the paper. All authors critically reviewed the manuscript and gave their consent to the final version.
Supporting information
ACKNOWLEDGEMENTS
S. Uitte de Willige was awarded the EHA‐ISTH Joint Fellowship 2013 for this research.
Abdul S, Dekkers DHW, Ariëns RAS, Leebeek FWG, Rijken DC, Uitte de Willige S. On the localization of the cleavage site in human alpha‐2‐antiplasmin, involved in the generation of the non‐plasminogen binding form. J Thromb Haemost. 2020;18:1162–1170. 10.1111/jth.14761
Manuscript handled by: Ton Lisman
Final decision: Ton Lisman, 5 February 2020
REFERENCES
- 1. Mullertz S, Clemmensen I. The primary inhibitor of plasmin in human plasma. Biochem J. 1976;159:545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moroi M, Aoki N. Isolation and characterization of alpha2‐plasmin inhibitor from human plasma. A novel proteinase inhibitor which inhibits activator‐induced clot lysis. J Biol Chem. 1976;251:5956‐5965. [PubMed] [Google Scholar]
- 3. Collen D. Identification and some properties of a new fast‐reacting plasmin inhibitor in human plasma. Eur J Biochem. 1976;69:209‐216. [DOI] [PubMed] [Google Scholar]
- 4. Wiman B, Collen D. Purification and characterization of human antiplasmin, the fast‐acting plasmin inhibitor in plasma. Eur J Biochem. 1977;78:19‐26. [DOI] [PubMed] [Google Scholar]
- 5. Holmes WE, Nelles L, Lijnen HR, Collen D. Primary structure of human alpha 2‐antiplasmin, a serine protease inhibitor (serpin). J Biol Chem. 1987;262:1659‐1664. [PubMed] [Google Scholar]
- 6. Abdul S, Leebeek FW, Rijken DC. Uitte de Willige S. Natural heterogeneity of alpha2‐antiplasmin: functional and clinical consequences. Blood. 2016;127:538‐545. [DOI] [PubMed] [Google Scholar]
- 7. Sakata Y, Aoki N. Cross‐linking of alpha 2‐plasmin inhibitor to fibrin by fibrin‐stabilizing factor. J Clin Invest. 1980;65:290‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kimura S, Aoki N. Cross‐linking site in fibrinogen for alpha 2‐plasmin inhibitor. J Biol Chem. 1986;261:15591‐15595. [PubMed] [Google Scholar]
- 9. Wiman B, Lijnen HR, Collen D. On the specific interaction between the lysine‐binding sites in plasmin and complementary sites in alpha2‐antiplasmin and in fibrinogen. Biochim Biophys Acta. 1979;579:142‐154. [DOI] [PubMed] [Google Scholar]
- 10. Lijnen HR, Holmes WE, van Hoef B, Wiman B, Rodriguez H, Collen D. Amino‐acid sequence of human alpha 2‐antiplasmin. Eur J Biochem. 1987;166:565‐574. [DOI] [PubMed] [Google Scholar]
- 11. Wiman B. Affinity‐chromatographic purification of human alpha 2‐antiplasmin. Biochem J. 1980;191:229‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wiman B, Nilsson T, Cedergren B. Studies on a form of alpha 2‐antiplasmin in plasma which does not interact with the lysine‐binding sites in plasminogen. Thromb Res. 1982;28:193‐199. [DOI] [PubMed] [Google Scholar]
- 13. Kluft C, Los N. Demonstration of two forms of alpha 2‐antiplasmin in plasma by modified crossed immunoelectrophoresis. Thromb Res. 1981;21:65‐71. [DOI] [PubMed] [Google Scholar]
- 14. Kluft C, Los P, Jie AF, et al The mutual relationship between the two molecular forms of the major fibrinolysis inhibitor alpha‐2‐antiplasmin in blood. Blood. 1986;67:616‐622. [PubMed] [Google Scholar]
- 15. Christensen U, Clemmensen I. Purification and reaction mechanisms of the primary inhibitor of plasmin from human plasma. Biochem J. 1978;175:635‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clemmensen I, Thorsen S, Mullertz S, Petersen LC. Properties of three different molecular forms of the alpha 2 plasmin inhibitor. Eur J Biochem. 1981;120:105‐112. [DOI] [PubMed] [Google Scholar]
- 17. Sasaki T, Morita T, Iwanaga S. Identification of the plasminogen‐binding site of human alpha 2‐plasmin inhibitor. J Biochem. 1986;99:1699‐1705. [DOI] [PubMed] [Google Scholar]
- 18. Hortin GL, Gibson BL, Fok KF. Alpha 2‐antiplasmin's carboxy‐terminal lysine residue is a major site of interaction with plasmin. Biochem Biophys Res Commun. 1988;155:591‐596. [DOI] [PubMed] [Google Scholar]
- 19. Thomas L, Moore NR, Miller S, Booth NA. The C‐terminus of alpha2‐antiplasmin interacts with endothelial cells. Br J Haematol. 2007;136:472‐479. [DOI] [PubMed] [Google Scholar]
- 20. Leebeek FWG, Los P, Kluft C. A monoclonal antibody against the COOH‐terminal site of a2‐antiplasmin reveals heterogeneity in the non‐plasminogen binding form of a2‐antiplasmin. Fibrinolysis. 1996;10(Supplement 2):67‐70. [Google Scholar]
- 21. Hattey E, Wojta J, Binder BR. Monoclonal antibodies against plasminogen and alpha‐2‐antiplasmin: binding to native and modified antigens. Thromb Res. 1987;45:485‐495. [DOI] [PubMed] [Google Scholar]
- 22. Uitte de Willige S, Miedzak M, Carter AM, et al Proteolytic and genetic variation of the alpha‐2‐antiplasmin C‐terminus in myocardial infarction. Blood. 2011;117:6694‐6701. [DOI] [PubMed] [Google Scholar]
- 23. Zhao Y, Chalt BT. Protein epitope mapping by mass spectrometry. Anal Chem. 1994;66:3723‐3726. [DOI] [PubMed] [Google Scholar]
- 24. Hager‐Braun C, Tomer KB. Determination of protein‐derived epitopes by mass spectrometry. Expert Rev Proteomic. 2005;2:745‐756. [DOI] [PubMed] [Google Scholar]
- 25. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol. 2008;26:1367‐1372. [DOI] [PubMed] [Google Scholar]
- 26. Law RH, Sofian T, Kan WT, et al X‐ray crystal structure of the fibrinolysis inhibitor alpha2‐antiplasmin. Blood. 2008;111:2049‐2052. [DOI] [PubMed] [Google Scholar]
- 27. Roy A, Kucukural A, Zhang Y. I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials