

Abstract
Aims
Accumulating studies have suggested that base excision repair (BER) is the major repair pathway of oxidative DNA damage in neurons, and neurons are deficient in other DNA repair pathways, including nucleotide excision repair and homologous recombination repair. However, some studies have demonstrated that neurons could efficiently repair glutamate‐ and menadione‐induced double‐strand breaks (DSBs), suggesting that the DSB repair mechanisms might be implicated in neuronal health. In this study, we hypothesized that BER and nonhomologous end joining (NHEJ) work together to repair oxidative DNA damage in neurons.
Methods
Immunohistochemistry and confocal microscopy were employed to examine the colocalization of apyrimidinic endonuclease 1 (APE1), histone variant 2AX (γH2AX) and phosphorylated p53‐binding protein (53BP1). APE1 inhibitor and shRNA were respectively applied to suppress APE1 activity and protein expression to determine the correlation of APE1 and DSB formation. The neutral comet assay was used to determine and quantitate the formation of DSB.
Results
Both γH2AX and 53BP1 were upregulated and colocalized with APE1 in the nuclei of rat cortical neurons subjected to menadione‐induced oxidative insults. Phospho53BP1 foci were efficiently abolished, but γH2AX foci persisted following the suppression of APE1 activity. Comet assays demonstrated that the inhibition of APE1 decreased the DSB formation.
Conclusions
Our results indicate that APE1 can engage the NHEJ mechanism in the repair of oxidative DNA damage in neurons. These findings provide insights into the mechanisms underlying the efficient repair of oxidative DNA damage in neurons despite the high oxidative burden.
Keywords: apurinic/apyrimidinic endonuclease 1 (APE1), base excision repair (BER), neuron, nonhomologous end joining (NHEJ), oxidative DNA damage
Introduction
Genomic DNA is an unstable molecule that is easily and constantly modified and damaged by environmental factors (e.g. ionizing radiation, UV, chemical agents and heavy metals), normal metabolic products [e.g. reactive oxygen species (ROS) and 4‐hydroxynonenal] and cell replication (e.g. mitosis and meiosis). Accumulation of unrepaired DNA damage leads to cancer, neurodegeneration, ageing, apoptotic cell death and unhealthy offspring1, 2, 3, 4. Therefore, DNA repair is crucial for organismal survival and passing the genetic makeup on to the next generation. The mammalian DNA repair machinery has evolved to include various mechanisms activated in response to the different types of DNA lesions, including mismatch repair (adenosine‐guanosine mismatch), base excision repair (BER; oxidized base modification), nucleotide excision repair [UV‐induced cyclobutane pyrimidine dimers (CPDs)], homologous recombination (HR)/nonhomologous end joining [NHEJ; γ‐irradiation‐generated double‐strand breaks (DSBs)] and direct reversal repair (methyltransferase, guanosine methylation).
Oxidative stress can be induced by abnormal protein aggregation, hypoxia and inflammatory responses, and is implicated in neurodegenerative diseases, stroke and brain trauma. Accumulating evidence indicates that oxidative DNA damage is elevated in neurodegenerative diseases, stroke, brain trauma and psychological disorders5, 6, 7, 8 Hence, enhancing DNA repair efficiency is crucial for maintaining DNA integrity and improving neuronal survival. The BER pathway, which is highly conserved and involves multiple enzymes and sequential processes, is commonly thought to be the primary repair mechanism for DNA oxidation, deamination, alkylation and single‐strand breaks (SSBs) in neurons9. BER is categorized into two subpathways, that is, short‐patch BER (SP‐BER) and long‐patch BER (LP‐BER), according to the different processes of polymerase‐mediated gap‐filling: 5′‐flap structure removing and ligase‐mediated nick sealing. SP‐BER and LP‐BER share the same first two steps of BER: glycosylase recognizes and removes the injured/modified base, and apyrimidinic endonuclease 1 (APE1) then cleaves the sugar‐phosphate backbone at the AP site. Subsequently, SP‐BER finishes repair by DNA polymerase β and ligase III, but LP‐BER involves other proteins. Because several LP‐BER proteins, such as proliferating cell nuclear antigen, flap endonuclease 1 and ligase I, are associated with the cell cycle and are downregulated in nondividing cells, the SP‐BER pathway is likely to represent the predominant repair mechanism in nondividing cells10.
APE1 is an abundant multifunctional protein present in both the cytoplasm and nucleus. APE1 functions as a reductive activator in transcriptional regulation and as an endonuclease in DNA repair. APE1 plays an essential role in BER in response to DNA deamination, oxidative damage and SSBs. The nuclease function of APE1, a class II AP endonuclease, is located in the C‐terminal domain, which incises AP sites, creating a 5′‐dRP and a 3′‐OH nick11. APE1 also has phosphodiesterase activity to eliminate 3′‐blocking groups (phosphate or phospho‐α,β‐unsaturated aldehyde) from ROS‐generated SSBs12. More than a decade ago, Saparbaev et al. found APE1 involvement in an alternative repair pathway, nucleotide incision repair (NIR), for oxidative DNA damage13. The NIR mechanism does not involve a glycosylase, and APE1 directly cleaves the phosphoribose backbone to remove oxidatively damaged nucleotides. Multiple earlier studies demonstrated that the deficiency of AP endonuclease promoted sensitivity to oxidative and ionizing radiation insults14, 15, 16, and APE1 can be stimulated by oxidative stress17, neuronal peptides18, neurotransmitters (glutamate)19, brain‐derived neurotropic factor20 and glucagon‐like peptide‐121, resulting in increased DNA repair efficiency. In addition, the abrogation of APE1 expression increases arsenite‐ and menadione‐induced cytotoxicity as well as arsenite‐mediated mutagenesis20, 22. Therefore, APE1 might be a potential therapeutic target for enhancing oxidative DNA repair capacity and maintaining the integrity of genomic DNA.
DSBs represent the most cytotoxic type of DNA damage in eukaryotic cells. DSBs are common DNA lesions produced by endogenous DNA metabolic pathways including DNA replication and V(D)J recombination, as well as by exogenous factors including chemical agents, ionizing radiation (IR) and some anticancer drugs. Two repair mechanisms, HR and NHEJ, are involved in DSB repair. HR is an error‐free repair pathway using the identical sister chromatid as a template and is the predominant DSB repair mechanism during the S and G2 phases of the cell cycle23. NHEJ is an error‐prone repair process associated with small deletions at the site of repaired DSB. NHEJ repair occurs mainly during the G0/G1 phase and is the major DSB repair pathway in neurons24, but it is less efficient in proliferating astrocytes or tumour cells25, 26. NHEJ activities have been demonstrated in the developing and mature brains27, 28, 29. In the NHEJ repair process, the KU70/80 heterodimer recognizes and binds to broken DNA ends via the DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), followed by the sequential recruitment of repair complex proteins (including XRCC4, DNA ligase IV, Mre11, Rad50 and Xrs2) that remove damaged forms of DNA or nonligatable termini and seal the gaps23, 30.
Brain neurons are terminally differentiated cells. Therefore, replication‐produced DSBs and abducts (such as CPDs and 6‐4‐pyrimidine‐pyrimidone photoproducts) induced by UV irradiation should rarely occur in neuronal DNA. Consistent with this notion, multiple studies have demonstrated that global genome nucleotide excision repair and HR are deficient in neurons30, 31, 32, 33. However, because neurons are characterized by highly active metabolism and massive ROS production, oxidative DNA damage and SSBs are the major types of neuronal DNA lesions. Studies have demonstrated that oxidative DNA lesions are efficiently repaired after endogenous or exogenous oxidative stress in neurons via elevated APE1 levels19, 20, 21, and the BER pathway repairs oxidatively modified bases and SSBs to maintain the integrity of genomic DNA in neuronal cells3, 9, 34.
Our preliminary study showed that injured DNA was associated with foci of phosphorylated histone variant 2AX (γH2AX) and p53‐binding protein 1 (53BP1), two proteins proposed to be immediate and sensitive biomarkers of DSBs in mammalian cells35, 36, 37, 38, in the nuclei of neurons subjected to oxidative insults, suggesting that the DSB repair mechanisms might be implicated in neuronal health. However, to date, no studies have investigated NHEJ involvement in oxidative DNA repair in neuronal cells. In this study, we investigated whether both NHEJ and BER participate in oxidative DNA repair in neurons and examined the mechanism linking by which BER and NHEJ repair oxidative DNA damage. The present study indicates that APE1 not only cleaves AP sites but also generates DSBs, triggering the NHEJ repair machinery. The findings suggest that enhancing APE1 activity not only increases BER efficiency but also activates NHEJ, thereby accelerating oxidative DNA repair.
Materials and methods
Primary neuronal cultures and treatments
Primary cultures of cortical neurons were prepared from 17.5‐day‐old embryos of Sprague‐Dawley rats, as described previously19. All experimental procedures involving animals were approved by the Chang Gung Memorial Hospital Animal Care and Use Committee and were complied with the NIH guidelines.
All experiments were performed in 8‐ to 10‐day‐old primary neuron cultures that contained >90% of neurons (neuron‐specific class III beta‐tubulin/Tju1 positive) with few glial fibrillary acidic protein (GFAP, 1.9 ± 2%) and ionized calcium‐binding adaptor molecule 1 (IBA1, 3.9 ± 3.8%) positive cells (Figure S1). Menadione (2‐methyl‐1,4‐naphthoquinone; Sigma‐Aldrich St. Louis, MO, USA) is a synthetic compound that is widely used to induce oxidative stress via interrupting the mitochondrial electron transportation chain in cell culture39. Cultured neurons were pretreated with 10 μM APE1 inhibitor III (Cat. No. 262017, Millipore Corp. St. Louis, MO, USA) or 50 nM DNA‐PKcs inhibitor (NU7441, Cat. No. 3712, Tocris Bioscience, Minneapolis, MN, USA) 2 h before menadione treatment. Menadione was freshly prepared in plain Neurobasal medium (Thermo Fisher Scientific, Waltham, MA, USA) before each experiment. Neurons were transiently treated with 40 µM menadione for 10 min at 37°C. Cells were then washed with warm Neurobasal medium. The cultures were incubated for 1, 3, 6, 12 or 24 h, and the neurons were harvested and analysed at each time point.
Lentiviral shRNA Silencing
Lentiviral envelope (pMD2.G) and packaging (psPAX2) plasmids and scrambled control shRNA (5′‐CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAG GGCGACTTAACCTTAGG‐3′) plasmids were obtained from Addgene. The pLKO.1‐TRC vector incorporated with Apex1 shRNA (5′‐AAATTCAGCCACAATC ACCCG‐3′) was purchased from Thermo Scientific. HEK 293T cells were transfected with Apex1 shRNA, envelope and packaging plasmids together with FuGENE 6 (Promega, Madison, WI, USA) to produce lentiviral particles. The 4‐day‐old cultured cortical neurons were infected with Apex1 shRNA lentiviruses using the Addgene plasmid 10878 protocol (http://www.addgene.org/tools/protocols/plko/).
Western blotting
Cultured neurons were extracted in RIPA buffer [150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 50 mM Tris, protease inhibitor cocktail (Roche, Basel Switzerland), and phosphatase inhibitors II and III (Sigma‐Aldrich); pH 8.0], and protein concentrations of cell extracts were determined using a BCA™ protein assay kit (Thermo Fisher Scientific). The washing buffer was 0.1% Tween 20 in Tris‐buffered saline (20 mM Tris, 150 mM NaCl; pH 7.4), and the blocking buffer was 5% skim milk (Bio‐Rad, Hercules, CA, USA) in washing buffer. Precast 4–20% gradient SDS‐polyacrylamide gels were used for electrophoresis to separate 53BP1. Proteins obtained from menadione‐treated neurons were detected with the following primary antibodies: anti‐APE1 (Cat. No. 4128; Cell Signaling, Danvers, MA, USA; 1:1000), anti‐γH2AX (Cat. No. 9718; Cell Signaling; 1:1000), anti‐53BP1 (Cat. No. 2675; Cell Signaling; 1:500) and anti‐actin (Cat. No. A5441B; Sigma‐Aldrich; 1:10 000). The goat anti‐rabbit (Cat. No. AP132P; Millipore), rabbit anti‐goat (Millipore Corp.) and goat anti‐mouse (Cat. No. AP124P; Santa Cruz, Dallas, TX, USA) secondary antibodies were diluted 1:1000, 1:2000 and 1:20 000 respectively.
Immunofluorescent labelling
Neurons cultured for 8–10 days were briefly washed with cold phosphate‐buffered saline (PBS) and fixed with 4% formaldehyde at room temperature (22–25°C) for 10 min. The fixed cells were incubated with permeabilization buffer (0.5% Triton X‐100, 100 mM glycine, 1% BSA, 0.7 mM EDTA) for 10 min in ice, followed by incubation with blocking buffer (10% foetal bovine serum, 0.01% sodium azide in 1 × PBS) at 37°C for 1 h or overnight at 4°C. The cells were then incubated with diluted anti‐GFAP (Cat. No. 3670; Cell Signalling; 1:200), anti‐IBA1 (Cat. No. 01919741; Wako, Osaka, Japan; 1:1000), anti‐Tuj1 (Cat. No. T8578; Sigma‐Aldrich; 1:1000), anti‐APE1 (Cat. No. SC9919; Santa Cruz; 1:500), anti‐γH2AX (Cat. No. 80312; Cell Signaling; 1:500) or anti‐phosphorylated 53BP1 (Cat. No. 2675; Cell signaling; 1:500) antibodies for 1 h at 37°C. The samples were rinsed with washing buffer (0.05% Triton X‐100 in 1 × PBS) three times for 5 min each at room temperature, followed by incubation with fluorophore‐conjugated secondary antibodies (Alexa 488, Cat. No. A11034; Alexa 568, Cat. No. A11079; 1: 1000; ThermoFisher Scientific, Waltham, MA, USA) for 1 h at 37°C. The cells were rinsed with washing buffer (3 × 5 min) and mounted in ProLong Gold containing DAPI (ThermoFisher Scientific). Images of immunostained neurons (300–400 cells per time point) were captured using an Olympus FV 1000 confocal microscope system and analysed with the Olympus Fluoview version 2.1C software (Olympus Corporation, Tokyo, Japan).
Cell viability
Because neurons are postmitotic and terminally differentiated cells, we used direct neuron counting to determine the cell viability after various treatments. Rat cortical neurons were plated and maintained in chamber‐slide dishes to measure the cytotoxicity of oxidative insults. Neurons were pretreated with APE1 inhibitor (10 μM) or DNA‐PKcs inhibitor (50 nM) for 2 h or overnight before menadione treatment. The neurons were treated with 40 µM menadione for 10 min at 37°C. The cells were washed with warm Neurobasal medium and incubated in fresh media supplemented with inhibitors as appropriate. Images of neurons in designated microscope fields (20 × objective) were acquired at 0, 6, 12, 24, 48 and 72 h by an Olympus Cell R system with PALM module. The operating software of the Cell R microscopy system was set at multiple coordinate positions of each chamber, and images were acquired at different time points as described above. The viable neurons at each time point were counted and recorded by ImageJ. Neuronal survival was expressed as the percentage of viable neurons relative to the baseline.
Comet assay
The comet assay is a well‐developed technique for examining the nuclear DNA damage and repair efficiency. We employed the neutral comet assay to measure nuclear DNA DSBs and repair efficiency. Briefly, neuronal cultures were washed with cold PBS to remove dead cells and debris. Neurons (200–300 thousand per culture) were harvested in 1 ml PBS by gentle scraping. A 10‐μl aliquot of the sample, including approximately 2000 neurons, was mixed with 75 μl of prewarmed 0.5% low melt point agarose (Sigma Life Science, Darmstadt, Germany), and the cells were spread onto an agarose‐coated glass slide. A coverslip was placed on the sample, and the slide was placed in an ice‐chilled aluminium tray for 5 min. After the coverslip was removed, another 75 µl of low melt point agarose was added and covered with a new coverslip, and the slide was placed in the chilled aluminium tray for another 5 min. The coverslip was removed, and the slide was incubated in freshly prepared lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, and 1% Triton X‐100) for at least 4 h (or overnight). The slide was then washed (three times, 10 min each) with neutralization buffer (0.4 M Tris, pH 7.4). Electrophoresis was performed in TBE buffer (pH 7.4) at 25 V for 15 min at 4°C, and the slide was then dehydrated in 100% ethanol for 5 min. The dehydrated sample was stained with ethidium bromide (10 ng/ml) immediately before image acquisition. Images of nuclei were acquired under epifluorescence illumination using an Olympus Cell R system and were analysed using the Komet 6 software (Andor, Belfast, UK).
Statistical analysis
General statistical comparisons were performed using Student’s t‐test. Two‐way anova was used to evaluate the significance of differences between groups; the Bonferroni t‐test was used for multiple comparisons. Data were presented as means and standard errors obtained from four to six independent experiments.
Results
APE1 upregulation is associated with γH2AX and 53BP1phosphorylation after oxidative insults
Confocal imaging demonstrated that APE1 and γH2AX accumulated and colocalized in neuronal nuclei several minutes after an oxidative insult. APE1 and γH2AX staining intensities followed a similar time course, reaching a peak at 1 h, starting to decline at 3 h, and returning to near‐basal levels at 24 h (Figure 1 A,B). Western blotting showed that oxidative insults increased APE1 expression during the first 6 h, followed by recovery to the baseline level at 24 h (Figure 1 C, top panel). The γH2AX level was promptly elevated after oxidative insults, reaching a maximum at 1 h, and slowly decreased to the basal level at 24 h (Figure 1 C, bottom panel). The enlarged immunofluorescent images demonstrate the colocalization of APE1 and γH2AX in neurons from 1 h after oxidative insults (Figure 1 D). Therefore, both immunoblotting and immunofluorescent staining showed a transient increase in the APE1 and γH2AX levels following an oxidative insult. Our findings are consistent with those in the previous studies by Grosch et al., which indicate oxidative stress‐induced increases in mRNA and protein levels of APE1 in CHO cells17, 40.
Figure 1.
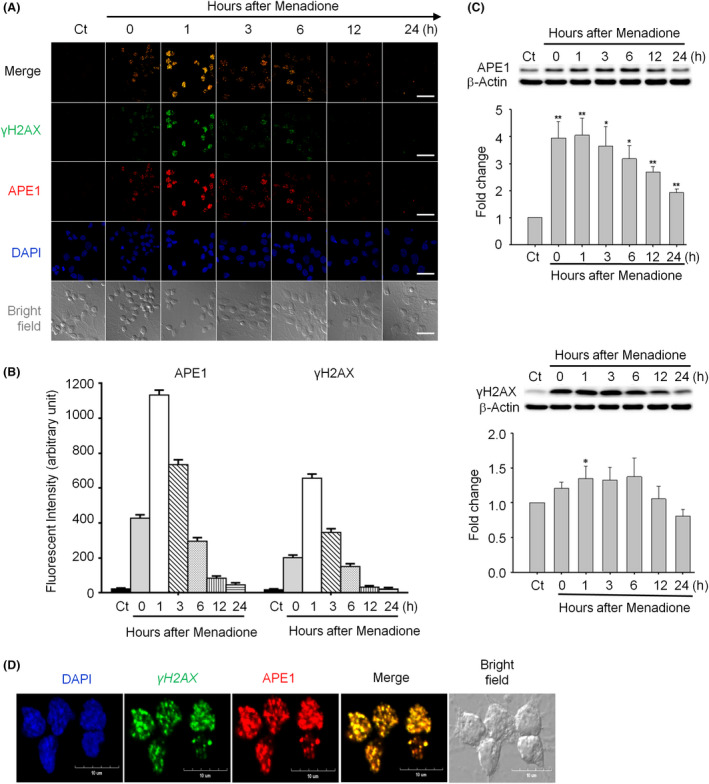
Apurinic/apyrimidinic endonuclease 1 (APE1) and phosphorylated histone 2AX (γH2AX) foci are colocalized and co‐induced in neurons subjected to an oxidative insult. (A) The levels of APE1 (red) and γH2AX (green) were rapidly elevated in the nuclei (DAPI, blue) of rat cortical neurons after transient exposure to 40 μM menadione and returned to the background levels at 12‐h post‐treatment (scale bar: 50 nm). (B) Quantitation of the data in (A). (C) Western blotting showed that both APE1 and γH2AX levels peaked at 1 h, started decreasing at 6 h and returned to the basal/control levels at 24 h after menadione treatment. (D) The enlarged image (1‐h postmenadione treatment) demonstrates colocalization of APE1 and γH2AX foci (scale bar: 10 nm). All statistics were based on comparison with the control group. Data are shown as means ± SE (n = 3). *P < 0.05; **P < 0.01.
Because γH2AX is a biomarker of DSBs36, 37, our results indicate that oxidative insults could generate DSBs in neurons. We used another DSB marker, 53BP1, to confirm the formation of DSBs after an oxidative insult in cultured rat cortical neurons. We examined the levels of APE1 and phosphorylated 53BP1 in neuronal nuclei under menadione‐induced oxidative stress by immunofluorescent staining. The accumulation pattern of APE1 was similar to that of γH2AX in neuronal cells (Figure 2 A,B), and phosphorylated 53BP1 was highly colocalized with APE1 (Figure 2 A,B). Phosphorylated 53BP1 rapidly accumulated in neuronal nuclei following an oxidative insult, and its level reached a peak at 1 h and decreased to the basal level at 24‐h postoxidative insults (Figure 2 B). The results of immunoblotting are similar to those of staining. Although almost no phosphorylated 53BP1 was found in control cells, 53BP1 was nearly instantly phosphorylated, and the phosphorylation level was sustained during the first hour after the oxidative insult (Figure 2 C). The level of phosphorylated 53BP1 started decreasing from 3 h and returned to the baseline level at 24‐h postoxidative insults. Magnified immunofluorescent images show that APE1 and 53BP1 are highly colocalized in neurons from 1‐h postoxidative insults (Figure 2 D). These findings suggest that nuclear DSBs appear shortly after the oxidative insult and are repaired within 24 h. Taken together, the results of immunofluorescent labelling and immunoblotting confirm that DSBs occur after oxidative stress and indicate that BER co‐occurs with NHEJ in cortical neurons.
Figure 2.
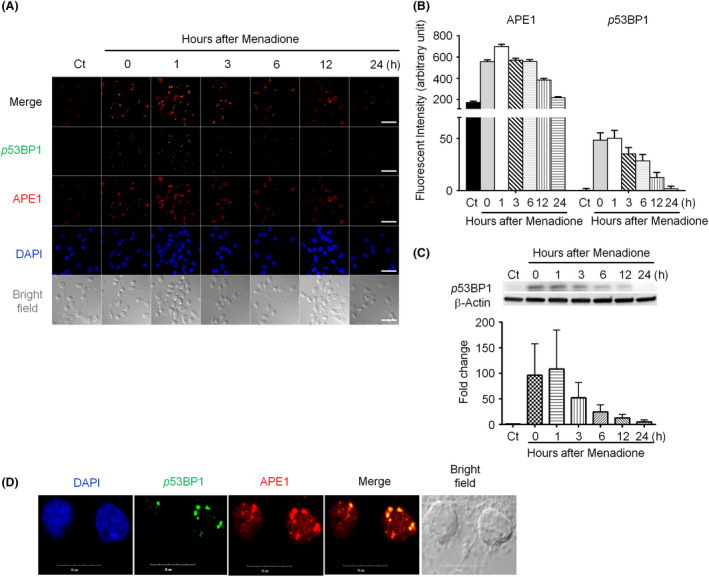
P53‐binding protein 1 (53BP1) colocalizes with apurinic/apyrimidinic endonuclease 1 (APE1) after menadione‐induced oxidative DNA damage in rat cortical neurons. (A) APE1 (red) and 53BP1 (green) accumulated in the nuclei (blue) immediately after transient exposure to 40 μM menadione, and colocalized 53BP1 and APE1 foci were observed. 53BP1 foci were smaller and less intense than APE1 foci (scale bar: 50 nm). (B) Both APE1 and 53BP1 focus intensities peaked at 1 h, started decreasing at 6 h, and returned to the basal/control levels at 24 h after the oxidative insult. (C) Immunoblotting demonstrated that the phosphorylated 53BP1 level immediately increased, peaked at 1 h after oxidative stress, and then gradually decreased. (D) The enlarged image of 1‐h postmenadione treatment shows colocalization of APE1 and 53BP1 foci (scale bar: 10 nm). Data are shown as means ± SE (n = 3).
Suppression of APE1 endonuclease activity or silencing of APE1 expression results in sustained γH2AX phosphorylation
APE1 is a multifunctional protein, with its N‐terminus displaying strong redox activity (through Cys65) and its C‐terminus functioning as an endonuclease at abasic DNA sites in the BER pathway41. Based on our findings that DSB markers (γH2AX and 53BP1) appear immediately following oxidative stress and colocalize with APE1, we hypothesized that APE1 plays a role in DSB generation. To test this hypothesis, we examined the effect of suppressing APE1 endonuclease activity on the formation of DSBs. The inhibition of APE1 activity resulted in persistent nuclear APE1 accumulation and suppressed APE1 expression as assessed by immunocytochemistry and Western blotting respectively (Figure 3 A,B). The persistent APE1‐positive foci suggest that DNA repair efficiency is significantly reduced. Interestingly, nuclear γH2AX foci also persisted, and the level of phosphorylated γH2AX remained largely stable during the 24‐h observation period in the presence of APE1 inhibitor (Figure 3 A,C). Furthermore, APE1 shRNA was applied to silence APE1 expression to validate the results of the APE1 inhibitor. Primary neurons were infected with lentiviral shRNA to knockdown APE1 (Figure 3 D) and were then transiently treated with an oxidative insult. Immunocytochemistry showed that γH2AX foci persisted in the nuclei up to 24 h, but the pattern was different from that induced by inhibition of APE1 activity (Figure 3 E,F). APE1‐knockdown neurons preserved a little repair capacity; therefore, γH2AX foci were reduced but not returned to the background. The repair capacity of APE1‐inhibited neurons was more thoroughly abolished; hence, γH2AX foci were not reduced and remained stable up to 24 h. γH2AX has been proposed to be an immediate and sensitive biomarker of DSBs in mammalian cells35. If our hypothesis that APE1 induces DSB formation is correct, the γH2AX foci would be expected to decrease following APE1 activity inhibition. However, γH2AX immunostaining and immunoblotting results do not support this hypothesis, suggesting that APE1 does not produce DSBs after oxidative DNA damage, or γH2AX is not a reliable DSB marker.
Figure 3.
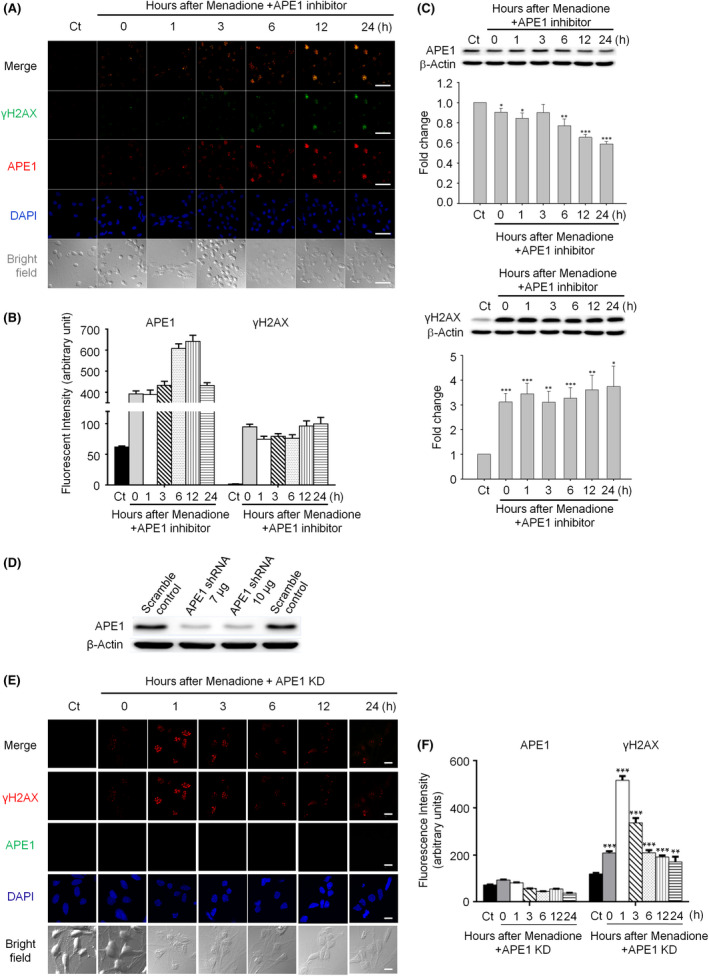
Suppression of apurinic/apyrimidinic endonuclease 1 (APE1) activity or knockdown of APE1 leads to persistent nuclear histone 2AX (γH2AX) foci in neurons subjected to an oxidative insult. (A) Both APE1 and γH2AX foci immediately accumulated and persisted in the nuclei for up to 24 h in APE1 inhibitor‐treated neurons after a transient menadione insult (scale bar: 50 nm). (B) Quantitation of the data in (A). (C) Western blotting demonstrated that the suppression of APE1 endonuclease activity resulted in high‐phosphorylated γH2AX and APE1 levels in the nuclei for at least 24 h after the oxidative insult. However, the total protein level of APE1 significantly decreased. (D) Western blotting showed that APE1 shRNA vector (7 μg)‐produced lentiviruses were sufficient to silence APE1 expression in primary cortical neurons. (E) In the APE1‐knockdown neurons, γH2AX foci promptly accumulated, peaked at 1 h and persisted in the nuclei for up to 24 h after a transient menadione insult (scale bar: 10 nm). (F) Quantitation of the data in (E). All statistics were based on comparison with the control group. Data are shown as means ± SE (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
Suppression of APE1 endonuclease activity or silencing of APE1 expression prevents 53BP1 phosphorylation
Another DSB biomarker, 53BP1, was examined to confirm the γH2AX results obtained under APE1 inhibition. Immunofluorescent APE1 nuclear foci persisted; however, phosphorylated 53BP1 foci were few and nearly undetectable (Figure 4 A). The immunofluorescent images of APE1‐shRNA silenced neurons demonstrated similar results: Formation of 53BP1 foci was robustly suppressed after transient oxidative insults (Figure 4 D,E). Western blotting also demonstrated that the level of phosphorylated 53BP1 significantly decreased compared with that in the absence of APE1 inhibitor (Figure 4 C). Both 53BP1 immunofluorescent labelling (Figure 4 A,D) and Western blotting (Figure 4 C) results indicate that APE1 not only is the critical component of the BER pathway but also induces DSBs and promotes NHEJ to repair oxidative DNA damage in neurons. The results also suggest that γH2AX might be a general marker of DNA damage, but not an accurate and specific marker of DSBs.
Figure 4.
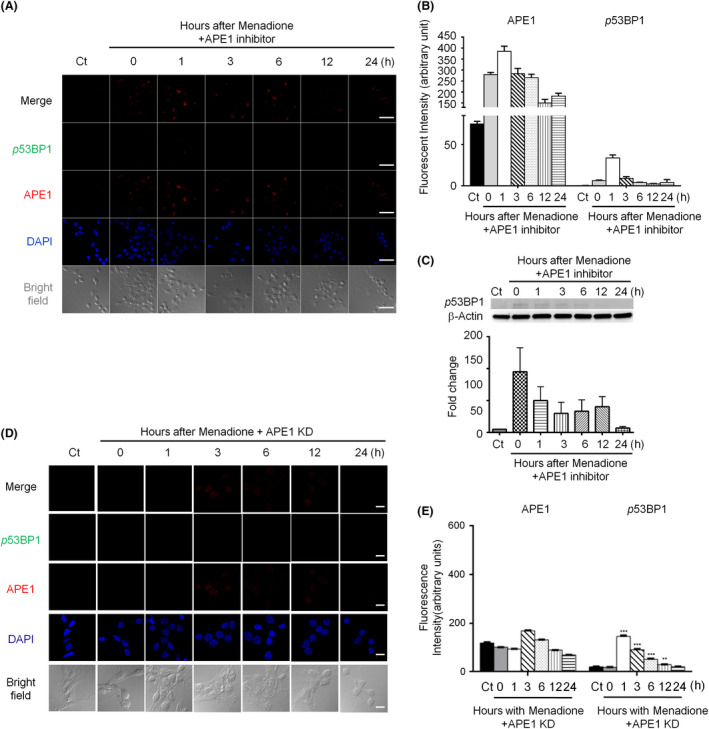
Suppression of apurinic/apyrimidinic endonuclease 1 (APE1) activity or knockdown of APE1 reduces phosphorylated p53‐binding protein 1 (53BP1) foci in the nuclei of rat cortical neurons subjected to oxidative stress. (A) Phosphorylated 53BP1 foci (green) were dramatically repressed when APE1 activity was inhibited following menadione treatment. APE1 (red) accumulation appeared immediately and persisted in the nuclei (blue) for at least 24 h (scale bar: 50 nm). (B) Quantitation of the data in (A). (C) Immunoblotting images showed that the levels of phosphorylated 53BP1 foci (green) significantly decreased in APE1‐knockdown neurons (scale bar: 10 nm). (D) Quantitation of the data in (C). Data are shown as means ± SE (n = 3).
DNA‐PKcs inhibition promotes DSB formation
After examining the indirect biomarkers of DNA DSBs, γH2AX and 53BP1, we employed the comet assay to assess double‐strand DNA fragments as direct evidence of DNA damage and a measure of repair efficiency. Double‐stranded DNA fragments can be preserved in neutral electrophoresis conditions compared with alkaline conditions, under which double‐stranded fragments are denatured into single strands. Comet tails of neurons increased at 1 and 6 h after menadione treatment, followed by reduction to the background level at 24 h (Figure 5 A, first column; Figure 5 B, black‐filled bars). These results correlated well with the immunocytochemistry results of APE1, γH2AX and 53BP1 described above (Figures 1 and 2). When neurons were treated with both menadione and an APE1 inhibitor, the formation of comet tails was suppressed (Figure 5 A, second column; Figure 5 B, white bars). The comet images of APE1‐silenced neurons showed reduced tails compared with those of scramble control neurons. Similar results were found in APE1 inhibitor‐treated neurons (Figure 5 C,D). These comet assay results suggest that APE1 is directly involved in the formation of nuclear DNA DSBs and confirm that 53BP1 foci decrease after an oxidative insult in APE1 inhibitor‐treated neurons (Figure 4 A,D). In addition, comet tails were significantly increased at 1 and 6 h in neurons treated with a DNA‐PKcs inhibitor and menadione compared with those treated with menadione with or without APE1 inhibitor (Figure 5 A, last column; Figure 5 B, grey‐filled bars). Thus, the DNA‐PKcs inhibitor increased the production of DNA double‐stranded fragments, suggesting that the double‐strand repair mechanism, NHEJ, is involved in the repair of oxidative DNA damage in neurons. Taken together, these results indicate that APE1 not only is involved in BER but also generates DSBs, and the NHEJ pathway is involved in the repair of DNA damage induced by oxidative stress.
Figure 5.
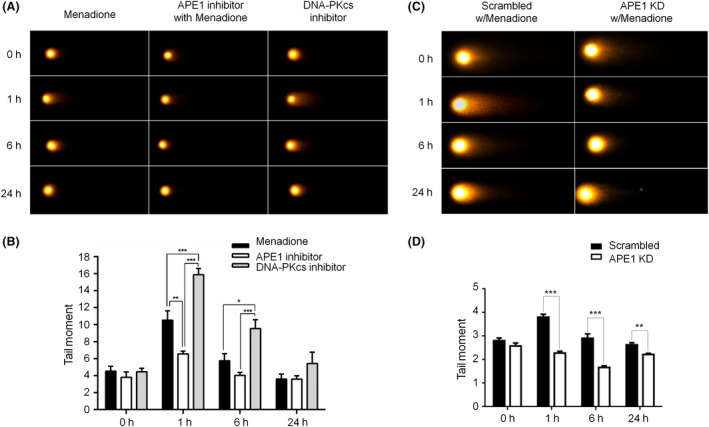
Inhibition of apurinic/apyrimidinic endonuclease 1 (APE1) activity or knockdown of APE1 suppresses double‐stranded break (DSB) formation in oxidatively damaged neurons. (A) Comet assay demonstrated that DSB formation was increased at 1 and 6 h and then decreased to the basal level at 24‐h posttreatment in cortical neurons after a menadione insult. Comet tails were reduced in APE1 inhibitor‐treated cortical neurons compared with both control (menadione only) and DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) inhibitor‐treated neurons. Neurons treated with the DNA‐PKcs inhibitor showed the largest comet tails at 1 and 6 h, which were reduced to the control levels at 24 h. (B) Quantitation of the data in (A) Comet images showed that DSB formation was significantly less in APE1 knockdown neurons than in scrambled control neurons at 1, 6 and 24 h after an oxidative insult. (D) Quantitation of the data in (C). Data are shown as means ± SE (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.
Suppression of APE1 or DNA‐PKcs enhances neuronal death after oxidative insults
ROS and various oxidative metabolites are continuously produced and cause oxidative DNA damage in living cells, especially in the highly metabolically active neurons. The suppression of APE1 activity was cytotoxic to neurons and induced a similar extent of neuronal death (34–35%) to that induced by menadione at 24‐h post‐treatment and an even higher neuronal death rate (58%) than that induced by menadione at 48 h (Figure 6 A), indicating that APE1 is essential for regular oxidative DNA repair under normal conditions. APE1 inhibitor treatment following a transient oxidative insult dramatically accelerated neuronal death (54% at 24 h and 71% at 48 h; Figure 6 A), suggesting that APE1 is crucial in repairing oxidative DNA damage induced by external agents. However, inhibition of DNA‐PKcs activity did not cause neuronal death (Figure 6 B), suggesting that NHEJ is not involved in regular, physiologic oxidative DNA repair. Furthermore, the suppression of DNA‐PKcs activity following an oxidative insult induced extensive neuronal death (54% at 24 h, 61% at 48 h; Figure 6 B). Thus, blocking the NHEJ repair process significantly reduces neuronal viability after an oxidative insult, indicating NHEJ involvement in the repair of oxidative DNA damage in neurons. Taken together, these results suggest that NHEJ is not involved in the repair of regular metabolite‐produced (mild) oxidative DNA damage but is engaged in the repair of exogenously induced (severe) oxidative DNA damage.
Figure 6.
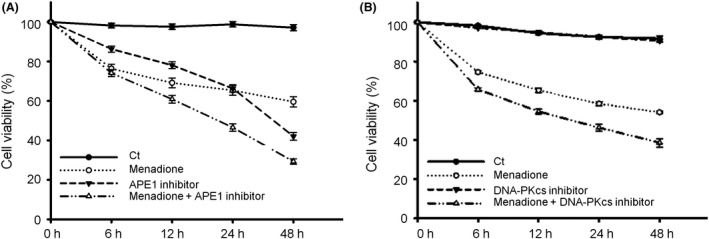
Suppression of apurinic/apyrimidinic endonuclease 1 (APE1) or DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) activity significantly increases neuronal death after oxidative insults. (A) Suppression of APE1 endonuclease activity induced a similar rate of neuronal death to that induced by a menadione oxidative insult at 24‐h post‐treatment and an even higher rate than that induced by menadione at 48 h. Neuronal viability dramatically decreased from 6 h after menadione and APE1 inhibitor treatments and continued to decrease for 48 h. (B) DNA‐PKcs inhibitor was not toxic to neurons under a normal condition; however, when neurons were treated with both menadione and DNA‐PKcs inhibitor, a higher rate of neuronal death was observed than that observed in cells treated with menadione alone. Data are shown as means ± SE (n = 4).
Discussion
Endogenous sources, including metabolite‐induced oxidative stress, rarely produce DSBs, except for recombination‐activating gene‐induced DSBs during V(D)J recombination in B cells. The two well‐known DSB‐inducing agents are IR, such as x‐rays and γ‐rays, and anticancer drugs42. Most metabolic by‐products result in abundant oxidative DNA lesions, especially in the highly active neurons. BER is widely thought to be the most active DNA repair mechanism in neurons and is a sequential process involving multiple enzymes to repair oxidized DNA damage and starting with removing damaged bases by glycosylases to form AP sites.
AP sites are frequent DNA lesions resulting from spontaneous loss of bases by ROS attacks. Using a novel aldehyde reactive probe, which sensitively reacts with the aldehyde groups of AP sites, Nakamura et al. demonstrated that AP sites occur at a rate of 1.54/106 nucleotides per day (approximately 9000 AP sites/cell/day) under physiological conditions 43. AP sites are genotoxic intermediates of BER and may block DNA replication and transcription. If bypassed by DNA polymerases, AP sites may lead to the incorporation of adenosines, causing mutations 44. Moreover, the aldehyde groups of AP sites are highly reactive, forming bulky DNA‐protein lesions 45. AP endonucleases, which are conserved in organisms including Escherichia coli, yeast, and mammals, play a central role in BER by incising the phosphate backbone and neutralizing the genotoxicity of AP sites. APE1 is the most abundant human AP endonuclease homologous to exonuclease III in E. coli and Apn1 in Saccharomyces cerevisiae 46.
NIR is an alternative repair pathway of oxidative DNA lesions that bypasses DNA glycosylase and involves AP endonuclease directly generating a nick with a 3ʹ‐hydroxyl terminus and a 5ʹ‐phosphate terminus13, 47. NIR has been suggested to occur through the FEN1‐dependent LP‐BER pathway in human cells48. However, LP‐BER activity is significantly attenuated in postmitotic neurons because of the reduced FEN1, PCNA and ligase I protein levels49. Because NIR can only be completed via the FEN1‐dependent long‐patch repair mechanism, FEN1 deficiency may cause the accumulation of APE1‐produced DNA nicks. Thus, neurons tend to generate numerous DNA nicks via the BER and NIR mechanisms, as well as high ROS production. However, BER may not be efficient enough to repair oxidized DNA damage because of the involvement of multiple proteins in the repair process13, whereas NIR may not play a role in the repair of oxidative DNA damage in neurons because of the deficiency of FEN149. Therefore, neurons might utilize various DNA repair mechanisms, including BER, to repair oxidative DNA damage. The present study indicates that APE1 promptly responds to abundant AP sites, occurring spontaneously or generated by glycosylases, by incising the DNA backbone, eventually resulting in the formation of DSBs and engagement of both the BER and NHEJ pathways to repair oxidative DNA damage (Figure 7). Milanese et al. found that γH2AX and 53BP1 foci significantly increased following intranigral delivery of AAV‐expressing α‐synuclein or an intrastriatal injection of α‐synuclein fibril in a mouse model of Parkinson’s disease50. The α‐synuclein aggregation and fibrillation induce mitochondrial dysfunction and oxidative stress, thereby causing oxidative DNA damage. Based on the findings and ours, we speculate that 53BP1 foci appear in the neurons of PD mice because of incision of APE1. These findings provide some insights into the pathology of PD and other neurodegenerative diseases. Because APE1 is an inducible protein via the PI3K‐AKT signalling axis20, 21, APE1 might be a potential target for repair of oxidative DNA damage and maintenance of DNA integrity, thus preventing further deterioration in neurodegenerative disorders.
Figure 7.
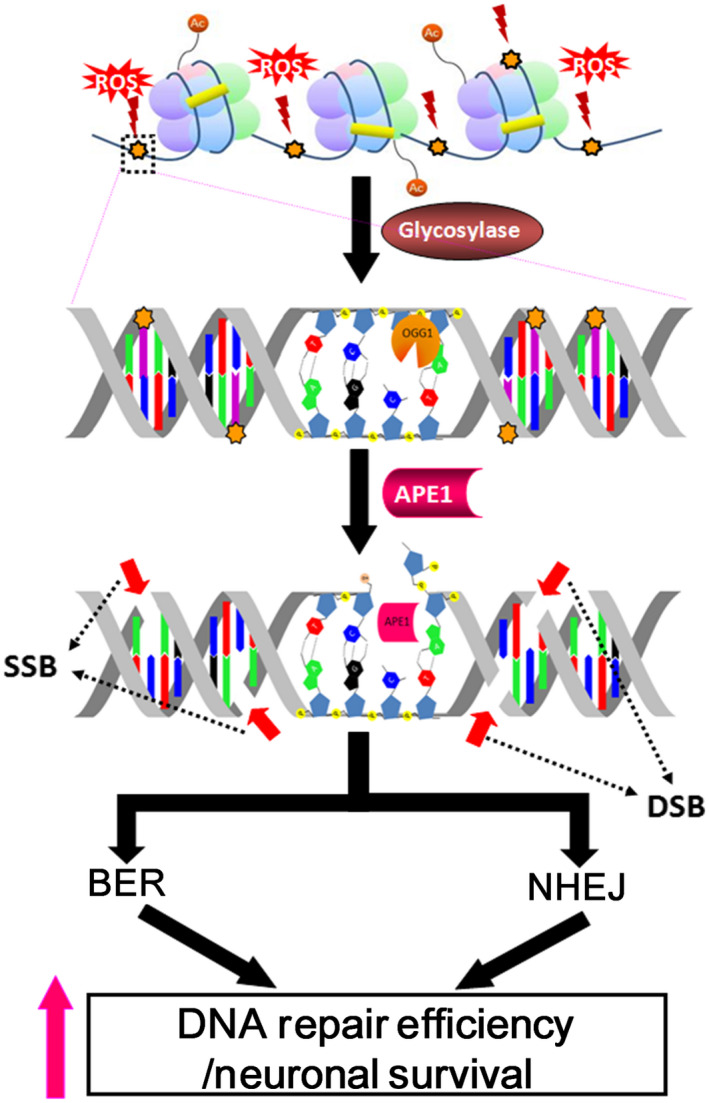
Proposed model of apurinic/apyrimidinic endonuclease 1 (APE1) involvement in both base excision repair (BER) and nonhomologous end joining (NHEJ) in response to oxidative DNA damage. After glycosylase removes the oxidised base, APE1, a key BER enzyme, incises the DNA backbone 5′ of the abasic site. The APE1‐produced nick results in spontaneous single‐strand breaks (SSBs) in the same strand as well as nearby nicks in the opposite strand, producing double‐strand break (DSB)‐like damage. Confocal microscopy and comet assay results suggest that APE1 plays a central role in the generation of DSBs that trigger the NHEJ repair pathway in the repair process of oxidative DNA damage in neurons. The concurrent activation of BER and NHEJ increases oxidative damage repair efficiency and promotes neuronal survival.
DNA is a vulnerable molecular substrate of cellular ROS and other superoxide molecules. H2AX is a specific variant of the histone 2A family that contributes to the formation of the nucleosome by wrapping around 140‐bp DNA segments along with histones 2B, H3 and H4. In response to DSB formation, activated kinases of the PI3K‐like family, including Ataxia telangiectasia mutated, Ataxia telangiectasia and Rad3‐related, and DNA‐PK, immediately phosphorylate serine 139 of H2AX at the DSB sites, and γH2AX foci are generated along the chromosome regions surrounding every DSB lesion35. 53BP1, Rad50, Mre11, Nbs1, Rad51 and BRCA1 have been reported to colocalize with γH2AX at DSB sites and used as biomarkers for assessing DSB damage and repair51, 52, 53, and γH2AX foci represent the most commonly used and sensitive biomarker of DSBs54. The present study showed that γH2AX foci developed following menadione‐induced oxidative damage and colocalized with APE1 (Figure 1). Nuclear APE1 and γH2AX foci persisted for up to 24 h after a transient oxidative insult when BER was suppressed by an APE1 inhibitor (Figure 3), indicating neurons are more vulnerable to oxidative stress under APE1 inhibition conditions (Figure 6). In contrast, few 53BP1 foci were detected after an oxidative insult followed by APE1 inhibition (Figure 4). Given that both γH2AX and phosphorylated 53BP1 are considered DSB biomarkers, the discordant results were unexpected. Therefore, we employed the neutral comet assay to examine DSB formation directly. DSB levels were robustly decreased when the APE1 inhibitor and APE1‐shRNA were applied (Figure 5). Based on the γH2AX results, we speculate that increased γH2AX phosphorylation reflects DNA damage other than DSBs. We also found that 53BP1 foci did not fully colocalize with γH2AX after an oxidative insult: while some major foci of 53BP1 staining overlapped with γH2AX staining, numerous 53BP1 microfoci appeared to be γH2AX‐negative (Figure 3). Consistent with this, a study by Cleaver et al. indicates that γH2AX may be associated with limited DSBs but not all DSBs55, and another study by Rybak et al. also suggests the lack of a link between γH2AX sites with low‐level phosphorylation and double‐strand DNA breaks in A549 human lung adenocarcinoma cells exposed to topoisomerase I or II inhibitors56. Taken together, our findings indicate that phosphorylated 53BP1 may be a more specific biomarker of DSBs, whereas γH2AX may be a general biomarker of DNA damage.
Packed chromatin presents a barrier to all DNA repair processes that require the recruitment of repair enzymes to the damage sites. To facilitate DNA repair, the chromatin must be remodelled into a loosened form, a process mediated in part by γH2AX following DSB formation57. Neurons are terminally differentiated cells with highly active metabolism; therefore, multiple neuronal genes need to be continuously transcriptionally active to initiate the DNA repair process. Because the chromatin is loosened for active gene transcription, exposed DNA is more vulnerable to various insults. Therefore, we speculate that oxidative stress‐induced 53BP1 microfoci are located at the damage sites in actively transcribed DNA regions that do not require γH2AX for chromatin unwinding signalling. The persistent γH2AX foci observed in this study (Figures 3 A,E and S2) indicate the presence of unrepaired DNA due to a deficit of the repair machinery, and our study demonstrated that 53BP1 foci only colocalized with intensive γH2AX foci (Figure S3). The findings suggest that stronger γH2AX signalling is able to recruit APE1 or repair proteins, further facilitating DNA repair in nontranscription regions. However, further study is needed to prove the hypothesis and elucidate the involved mechanisms.
Author contributions
Jenq‐Lin Yang and Shang‐Der Chen contributed to research idea, experimental design, troubleshooting and manuscript writing. Wei‐Yu Chen and Sujira Mukda contributed to data analysis, figure editing and manuscript review. Yun‐Ru Yang and Shu‐Fang Sun performed experiments and obtained data.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Eight‐day cultured cortical neurons were examined purity by immunostaining glial fibrillary acidic protein (astrocyte maker), ionized calcium binding adaptor molecule 1 (microglia marker) and neuron‐specific class III beta‐tubulin (Tuj1, neuron marker).
Figure S2. Knockdown of apurinic/apyrimidinic endonuclease 1 expression leads to persistence of phosphorylated histone 2AX (γH2AX), but robustly decreased 53BP1 foci in neurons subjected to an oxidative insult.
Figure S3. Phosphorylated P53‐binding protein 1 and histone 2AX (γH2AX) foci did not fully colocalize after menadione‐induced oxidative DNA damage in rat cortical neurons.
Acknowledgement
This study was supported by research grants from the Ministry of Science and Technology of Taiwan (103‐2320‐B‐182A‐011‐MY3) and the Chang Gung Medical Foundation (CMRPG8F1513, CMRPG8E0761 and CMRPG8C1223).
Yang J.‐L., Chen W.‐Y., Mukda S., Yang Y.‐R., Sun S.‐F. and Chen S.‐D. (2020) Neuropathology and Applied Neurobiology 46, 375–390 Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)‐initiated nonhomologous end joining (NHEJ) in cortical neurons
References
- 1. Bohr VA, Ottersen OP, Tonjum T. Genome instability and DNA repair in brain, ageing and neurological disease. Neuroscience 2007; 145: 1183–6 [DOI] [PubMed] [Google Scholar]
- 2. Mandavilli BS, Rao KS. Accumulation of DNA damage in aging neurons occurs through a mechanism other than apoptosis. J. Neurochem 1996; 67: 1559–65 [DOI] [PubMed] [Google Scholar]
- 3. Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J. Neuropathol Exp Neurol 2008; 67: 377–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin LJ, Liu Z, Pipino J, Chestnut B, Landek MA. Molecular regulation of DNA damage‐induced apoptosis in neurons of cerebral cortex. Cereb Cortex 2009; 19: 1273–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev 2012; 133: 157–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron 2014; 83: 266–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slemmer JE, Shacka JJ, Sweeney MI, Weber JT. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem 2008; 15: 404–14 [DOI] [PubMed] [Google Scholar]
- 8. Yang JL, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008; 7: 1110–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson DM III, McNeill DR. Base excision repair and the central nervous system. Neuroscience 2007; 145: 1187–200 [DOI] [PubMed] [Google Scholar]
- 10. Akbari M, Pena‐Diaz J, Andersen S, Liabakk NB, Otterlei M, Krokan HE. Extracts of proliferating and non‐proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 2009; 8: 834–43 [DOI] [PubMed] [Google Scholar]
- 11. Mol CD, Izumi T, Mitra S, Tainer JA. DNA‐bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature 2000; 403: 451–6 [DOI] [PubMed] [Google Scholar]
- 12. Izumi T, Hazra TK, Boldogh I, Tomkinson AE, Park MS, Ikeda S et al. Requirement for human AP endonuclease 1 for repair of 3′‐blocking damage at DNA single‐strand breaks induced by reactive oxygen species. Carcinogenesis 2000; 21: 1329–34 [PubMed] [Google Scholar]
- 13. Gros L, Ishchenko AA, Ide H, Elder RH, Saparbaev MK. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res 2004; 32: 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M et al. A murine AP‐endonuclease gene‐targeted deficiency with post‐implantation embryonic progression and ionizing radiation sensitivity. Mutat Res 1998; 409: 17–29 [DOI] [PubMed] [Google Scholar]
- 15. Ramotar D, Popoff SC, Gralla EB, Demple B. Cellular role of yeast Apn1 apurinic endonuclease/3'‐diesterase: repair of oxidative and alkylation DNA damage and control of spontaneous mutation. Mol Cell Biol 1991; 11: 4537–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cunningham RP, Saporito SM, Spitzer SG, Weiss B. Endonuclease IV (nfo) mutant of Escherichia coli . J Bacteriol 1986; 168: 1120–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grosch S, Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (Ape, Ref‐1) by oxidative stress requires CREB. Biochem Biophys Res Commun 1999; 261: 859–63 [DOI] [PubMed] [Google Scholar]
- 18. Stetler RA, Gao Y, Zukin RS, Vosler PS, Zhang L, Zhang F et al. Apurinic/apyrimidinic endonuclease APE1 is required for PACAP‐induced neuroprotection against global cerebral ischemia. Proc Natl Acad Sci U S A 2010; 107: 3204–09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate‐induced oxidative DNA damage by a process involving CREB‐mediated up‐regulation of apurinic endonuclease 1. J Biol Chem 2010; 285: 28191–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang JL, Lin YT, Chuang PC, Bohr VA, Mattson MP. BDNF and exercise enhance neuronal DNA repair by stimulating CREB‐mediated production of apurinic/apyrimidinic endonuclease 1. NeuroMolecular Med 2014; 16: 161–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang JL, Chen WY, Chen YP, Kuo CY, Chen SD. Activation of GLP‐1 receptor enhances neuronal base excision repair via PI3K‐AKT‐induced expression of apurinic/apyrimidinic endonuclease 1. Theranostics 2016; 6: 2015–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fung H, Liu P, Demple B. ATF4‐dependent oxidative induction of the DNA repair enzyme Ape1 counteracts arsenite cytotoxicity and suppresses arsenite‐mediated mutagenesis. Mol Cell Biol 2007; 27: 8834–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scharer OD. Chemistry and biology of DNA repair. Angew Chem Int Ed Engl 2003; 42: 2946–74 [DOI] [PubMed] [Google Scholar]
- 24. Vyjayanti VN, Rao KS. DNA double strand break repair in brain: reduced NHEJ activity in aging rat neurons. Neurosci Lett 2006; 393: 18–22 [DOI] [PubMed] [Google Scholar]
- 25. Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan PH. Response of postmitotic neurons to X‐irradiation: implications for the role of DNA damage in neuronal apoptosis. J Neurosci 1998; 18: 147–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang TS, Wheeler KT. Repair of X‐ray‐induced DNA damage in rat cerebellar neurons and brain tumor cells. Radiat Res 1978; 73: 464–75 [PubMed] [Google Scholar]
- 27. Culmsee C, Bondada S, Mattson MP. Hippocampal neurons of mice deficient in DNA‐dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Brain Res Mol Brain Res 2001; 87: 257–62 [DOI] [PubMed] [Google Scholar]
- 28. Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, Ferguson D et al. Defective embryonic neurogenesis in Ku‐deficient but not DNA‐dependent protein kinase catalytic subunit‐deficient mice. Proc Natl Acad Sci U S A 2000; 97: 2668–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oka A, Takashima S, Abe M, Araki R, Takeshita K. Expression of DNA‐dependent protein kinase catalytic subunit and Ku80 in developing human brains: implication of DNA‐repair in neurogenesis. Neurosci Lett 2000; 292: 167–70 [DOI] [PubMed] [Google Scholar]
- 30. Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res 2007; 614: 24–36 [DOI] [PubMed] [Google Scholar]
- 31. Brooks PJ. DNA repair in neural cells: basic science and clinical implications. Mutat Res 2002; 509: 93–108 [DOI] [PubMed] [Google Scholar]
- 32. Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurogibol 2011; 94: 166–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair 2002; 1: 59–75 [DOI] [PubMed] [Google Scholar]
- 34. Coppede F, Migliore L. DNA repair in premature aging disorders and neurodegeneration. Curr Aging Sci 2010; 3: 3–19 [DOI] [PubMed] [Google Scholar]
- 35. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273: 5858–68 [DOI] [PubMed] [Google Scholar]
- 36. Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. H2AX: functional roles and potential applications. Chromosoma 2009; 118: 683–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Geric M, Gajski G, Garaj‐Vrhovac V. gamma‐H2AX as a biomarker for DNA double‐strand breaks in ecotoxicology. Ecotoxicol Environ Saf 2014; 105: 13–21 [DOI] [PubMed] [Google Scholar]
- 38. Panier S, Boulton SJ. Double‐strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 2014; 15: 7–18 [DOI] [PubMed] [Google Scholar]
- 39. Criddle DN, Gillies S, Baumgartner‐Wilson HK, Jaffar M, Chinje EC, Passmore S et al. Menadione‐induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 2006; 281: 40485–92 [DOI] [PubMed] [Google Scholar]
- 40. Grosch S, Fritz G, Kaina B. Apurinic endonuclease (Ref‐1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Res 1998; 58: 4410–16 [PubMed] [Google Scholar]
- 41. Xanthoudakis S, Miao GG, Curran T. The redox and DNA‐repair activities of Ref‐1 are encoded by nonoverlapping domains. Proc Natl Acad Sci U S A 1994; 91: 23–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iliakis G, Murmann T, Soni A. Alternative end‐joining repair pathways are the ultimate backup for abrogated classical non‐homologous end‐joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 2015; 793: 166–75 [DOI] [PubMed] [Google Scholar]
- 43. Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res 1998; 58: 222–5 [PubMed] [Google Scholar]
- 44. Strauss BS. The 'A rule' of mutagen specificity: a consequence of DNA polymerase bypass of non‐instructional lesions? BioEssays 1991; 13: 79–84 [DOI] [PubMed] [Google Scholar]
- 45. Rieger RA, Zaika EI, Xie W, Johnson F, Grollman AP, Iden CR et al. Proteomic approach to identification of proteins reactive for abasic sites in DNA. Mol Cell Proteomics 2006; 5: 858–67 [DOI] [PubMed] [Google Scholar]
- 46. Wilson DM 3rd, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res 2001; 485: 283–307 [DOI] [PubMed] [Google Scholar]
- 47. Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002; 415: 183–7 [DOI] [PubMed] [Google Scholar]
- 48. Klungland A, Lindahl T. Second pathway for completion of human DNA base excision‐repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J 1997; 16: 3341–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sykora P, Yang JL, Ferrarelli LK, Tian J, Tadokoro T, Kulkarni A et al. Modulation of DNA base excision repair during neuronal differentiation. Neurobiol Aging 2013; 34: 1717–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Milanese C, Cerri S, Ulusoy A, Gornati SV, Plat A, Gabriels S et al. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson's disease. Cell Death Dis 2018; 9: 818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage‐signaling pathways. J Cell Biol 2001; 153: 613–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 2001; 21: 1719–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double‐strand breaks. J Cell Biol 2000; 151: 1381–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S et al. GammaH2AX and cancer. Nat Rev Cancer 2008; 8: 957–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Revet I, Feeney L, Bruguera S, Wilson W, Dong TK, Oh DH et al. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc Natl Acad Sci U S A 2011; 108: 8663–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rybak P, Hoang A, Bujnowicz L, Bernas T, Berniak K, Zarebski M et al. Low level phosphorylation of histone H2AX on serine 139 (gammaH2AX) is not associated with DNA double‐strand breaks. Oncotarget 2016; 7: 49574–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kruhlak MJ, Celeste A, Dellaire G, Fernandez‐Capetillo O, Muller WG, McNally JG et al. Changes in chromatin structure and mobility in living cells at sites of DNA double‐strand breaks. J Cell Biol 2006; 172: 823–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Eight‐day cultured cortical neurons were examined purity by immunostaining glial fibrillary acidic protein (astrocyte maker), ionized calcium binding adaptor molecule 1 (microglia marker) and neuron‐specific class III beta‐tubulin (Tuj1, neuron marker).
Figure S2. Knockdown of apurinic/apyrimidinic endonuclease 1 expression leads to persistence of phosphorylated histone 2AX (γH2AX), but robustly decreased 53BP1 foci in neurons subjected to an oxidative insult.
Figure S3. Phosphorylated P53‐binding protein 1 and histone 2AX (γH2AX) foci did not fully colocalize after menadione‐induced oxidative DNA damage in rat cortical neurons.