

Abstract
Background
Fluorogenic thrombin generation (TG) assays are commonly used to determine global coagulation phenotype in plasma. Whole blood (WB)‐TG assays reach one step closer to physiology by involving the intrinsic blood cells, but erythrocytes cause variable quenching of the fluorescence signals, hampering its routine application.
Objective
To develop a new assay for continuous WB‐TG measurement.
Methods
In the new WB‐TG assay, the erythrocyte‐caused distortion of signal was solved by continuously mixing the sample during the measurement. The assay was validated by evaluating the reproducibility and comparing with the paper‐based WB‐TG assay. Reconstituted human blood and WB from 119 healthy donors was tested to explore the influences of hematocrit and platelet count on TG.
Results
This novel WB‐TG assay showed good reproducibility while being less affected by contact activation compared with the previous paper‐based assay. Reconstitution experiments showed that the lag time of TG was shortened by the addition of platelets but not erythrocytes. Increasing hematocrit strongly augmented the peak thrombin, even in the presence of high platelet counts. The lag time and peak of WB‐TG of 119 healthy donors were positively related to erythrocyte count after adjusting for age, sex, and oral contraceptive use with multiple linear regression analyses. The reference range and interindividual variation of WB‐TG were determined in the healthy cohort.
Conclusions
A novel WB‐TG assay was developed, which is a straightforward tool to measure the involvement of platelets and erythrocytes in TG and may assist the research of blood cell‐associated coagulation disorders.
Keywords: blood cells, blood coagulation disorders, erythrocytes, platelet activation, thrombin
Essentials.
Thrombin generation in whole blood includes the influence of all blood cells.
We developed a novel whole blood thrombin generation (WB‐TG) assay with good reproducibility.
Reference range and inter‐individual variation of the WB‐TG were tested in 119 healthy donors.
The influences of platelet and hematocrit on TG were measured with this assay.
1. INTRODUCTION
There is increasing awareness that thrombin generation (TG) provides a global coagulation profile of the complex interplay between pro‐ and anticoagulant drivers, whereas conventional clotting time tests only provide limited information about the initiation of coagulation. 1 , 2 , 3 Modern TG assays, such as the calibrated automated thrombinography (CAT), allow continuous and high‐throughput TG measurements in plasma samples by using a thrombin‐specific fluorogenic substrate. 4 , 5 TG assays have shown great potential in elucidating the mechanisms behind coagulation disorders, predicting bleeding/thrombotic risk, and managing hemophilia/thrombosis treatment. 2 , 3
Although TG assays give more comprehensive information about coagulation compared with conventional clotting time assays, they still have some nonphysiological factors, including the source of procoagulant surfaces. In vivo TG requires physiological phospholipids (PL), which is regulated by the interplay between coagulation factors and blood cells. 6 , 7 Currently, TG is often tested in platelet poor plasma (PPP‐TG), with artificial PL supplemented at saturation concentration to serve as surface for the coagulation factors. Thrombin generation tested in platelet‐rich plasma (PRP‐TG) reflects the influence of platelets, 8 however, it does not give insights about the impacts of erythrocytes and leukocytes. Abnormal functions of blood cells have been suggested to contribute to coagulation disorders. 8 Importantly, both abnormally high hematocrit and abnormal function of erythrocytes are associated with an increased thrombosis risk. 9 Therefore, measuring TG in whole blood (WB) is more physiologically relevant than in plasma, especially when studying coagulation disorders that are likely blood cell‐originated. Additionally, unlike plasma‐TG assays, WB‐TG does not require centrifugation to prepare plasma, thus avoids many preanalytical variations and allows the development of point‐of‐care applications.
Several methods have been published for WB‐TG measurements; however, most of them are too time‐consuming because these methods either rely on tedious sampling at interval time points 10 , 11 , 12 or can only measure one sample per run. 13 Fluorogenic assays on a microplate reader can allow continuous and high‐throughput WB‐TG measurements; however, the hemoglobin in erythrocytes severely quenches fluorescence signals and this quenching effect varies over time as a result of erythrocyte sedimentation between reading rounds. For example, in a typical CAT setting the interval time between two reading rounds is 20 seconds, which is often longer than the time needed for an ascent fluorometer to move the assay plate to read the fluorescence signals in all sample wells. Consequently, erythrocyte sedimentation takes place during the idle time when the assay plate stops moving. Clot retraction induced by activated platelets may further worsen the uneven erythrocyte distribution. Two methods have been published trying to solve this problem. 14 , 15 Tappenden et al 15 , 16 , 17 reported a method in which an orbital shake was applied to the assay plate during the idle time between readings, but this method still gave rather high variations and was not used in any further studies. Restraining the erythrocytes in filter paper matrix avoids erythrocyte sedimentation by forming a thin layer of blood and this technique gives reproducible WB‐TG results, 14 but only if executed by an experienced operator. 18 Another disadvantage of the paper‐based WB‐TG assay is the strong contact activation induced by the filter paper. 14
In the present study, we developed and validated a novel assay for continuous WB‐TG measurement. The erythrocyte‐caused distortion of fluorescence signals was solved by continuously mixing the whole blood sample during the entire course of measurement. This assay was compared with the previously published paper‐based assay regarding the disturbance from contact activation. In addition, it was also used to explore the influences of hematocrit, platelet count and platelet activation on WB‐TG.
2. MATERIALS AND METHODS
2.1. Study subjects and blood sample preparation
Our study protocol was evaluated by the local medical ethical board (Medical Ethical Committee of Maastricht University Medical Center). Blood was collected into vacutainer tubes (with 3.2% sodium citrate; from BD Vacutainer System) from healthy adults who gave full informed consent according to the Helsinki declaration and had not taken any anticoagulants or platelet inhibitors for at least 2 weeks and had no history of thrombosis or bleeding. Blood was kept at room temperature and used within 4 hours after collection. Cell counts were measured on a Coulter Counter analyzer (Beckman Coulter).
2.2. Reconstitution of human blood
To determine the contribution of platelet count and hematocrit in the novel WB‐TG assay, samples with varying platelet counts and hematocrits were prepared.
PRP was prepared by centrifugation of blood at 220g for 15 minutes; PPP was prepared by double centrifugation of blood at 2840g for 10 minutes. To vary platelet count in plasma, PRP was mixed with autologous PPP. In some experiments synthetic PL was added to the plasma, which were from Avanti Polar Lipids and prepared as previously described. 4
To yield a platelet pellet, PRP was centrifuged at 890g for 15 minutes with Prostacyclin I2 (Sigma‐Aldrich) and resuspended in autologous PPP. Erythrocytes were washed three times in HEPES buffer (10 mmol/L HEPES, 136 mmol/L NaCl, 2.7 mmol/L KCl, 2 mmol/L MgCl2, 0.1% w/v glucose, and 0.1% w/v bovine serum albumin, PH 7.4) and centrifuged at 330g for 10 minutes after the first two washing steps and at 890g for 10 minutes after the third washing step. Autologous PPP, platelets, and washed erythrocytes were reconstituted to obtain samples with different hematocrits and platelet counts as indicated.
2.3. Platelet activation or inhibition in WB
To study the effect of platelet activation and inhibition on TG in WB, citrated WB samples were incubated with a platelet activator (50 μg/mL Convulxin) or inhibitor (30 μg/mL Reopro, 10 μmol/L Iloprost, or 36 μmol/L Cangrelor) at 37°C for 10 minutes before TG was triggered at 1 pmol/L recombinant human tissue factor (TF; Siemens Healthcare). The concentrations of these molecules were chosen as the concentration resulting in the maximum effect on PRP‐TG. 19 Reopro was from Janssen Biologics. Iloprost, Cangrelor, and Convulxin were from Sigma‐Aldrich.
2.4. WB‐TG measured with the novel assay
This novel WB‐TG assay was performed following the procedure of Ninivaggi et al 14 with major modifications. Citrated WB was firstly mixed with the substrate (ZGGR)2‐Rhodamine 110 (P2Rho; Diagnostica Stago) solution. Subsequently, a solution containing TF and CaCl2 was added to the WB and mixed. The volume ratio of WB, substrate solution, and TF‐containing solution is 3:1:2. Of the resulting mixture, 65 μL was transferred into the detection wells. The final concentrations in the well were 50% WB, 0‐5 pmol/L TF, 16.7 mmol/L CaCl2, and 300 μmol/L P2Rho. Each blood sample was calibrated by replacing the TF‐containing solution with α2‐macroglobulin‐thrombin complex (α2M‐T, corresponding with 300 nmol/L thrombin activity). 4 Measurements were performed at 37°C and each condition was tested in triplicate. Fluorescence signals were recorded with a Fluoroskan Ascent microplate fluorometer (Thermolabsystems) with λ ex = 485 nm and λ em = 538 nm using Fluoroskan Ascent Software (version 2.6). For the measurement of reconstituted blood samples, an independent calibration experiment was performed for each hematocrit level as this influences the quenching of the fluorescent signal. Fluorescence data were corrected using the H‐transform when necessary. 14 , 20
To prevent the light distortion caused by erythrocyte sedimentation, we optimized the assay plate and the settings of plate movement in the Ascent software so the WB sample was adequately mixed during the whole measurement course. In the optimized setting, the interval time was set as 6 seconds, and 36 wells were always measured, which ensures that the assay plate moves continuously without noticeable stop (idle time) between two rounds of reading. Different microplates were tested to find the best structure of the reaction wells, and the round‐cornered 96‐well assay microplate from Corning (type number 2595) resulted in the lowest variation (Table S1).
A dedicated preprogrammed spreadsheet template was used to calculate the WB‐thrombogram parameters from the experimental fluorescence data as described previously. 13 , 21 , 22 , 23 The template is available upon request; the algorithms used in the calculation template is described in the Supplementary information. In brief, an extended Chapman‐Richards growth equation, F = a(1 − e − b · t)c + d(1 − e − f · t)g was fitted on the sigmoidal part of the fluorescence data, where F is the fluorescence intensity at time t, and a, b, c, d, f, and g are the parameters that determine the shape of the fitted curve. The sigmoidal part of the fluorescence data was estimated after using a simple moving average technique to smooth out the noise within the fluorescence data. 5 From the fitted curve, WB‐TG parameters were calculated, including the lag time (minutes), time‐to‐peak (minutes), peak thrombin (nmol/L), and endogenous thrombin potential until the thrombin peak (ETPp; nmol x min/L). This algorithm used in the calculation template renders that the WB‐TG parameters are derived objectively from the fluorescence data, and are not disturbed by the subjectivity of the operator. To illustrate this, we let three operators each independently do the calculation with a same set of fluorescence data, using the programmed calculation template. As shown in Figure S2, the TG parameters obtained were identical among three operators, meaning that the calculation method was free of the subjectivity of the operator.
2.5. Fluorescence signal of Rhodamine 110 fluorophore in clotting WB over time
The fluorescence signal of Rhodamine 110 fluorophore (Rho; the end product of [ZGGR]2‐Rho cleavage by thrombin) in clotting WB was monitored over time to assess the stability of the quenching effect of clotting WB on fluorescence signal. For this purpose, 2.5 μmol/L Rho (Sigma‐Aldrich) was either added into, or replaced, the substrate solution in WB‐TG. Fluorescence signal was monitored as described previously.
2.6. Paper‐based WB‐TG assay
The paper‐based WB‐TG assay was performed as originally described. 14 Fluorescence data were transformed into TG parameters using the CAT‐method as originally described. 5 , 14
2.7. Statistics
Statistical analyses were done using GraphPad 5.0 (GraphPad Software, San Diego, CA) and SPSS 25 (IBM, New York, NY). Data were checked for normality and are shown as median and interquartile range or mean ± standard deviation. Independent groups were compared using the Mann‐Whitney test. Paired samples were compared using paired t test or Wilcoxon matched‐pairs test depending on normality. Pearson correlation was calculated between the parameters from the two WB‐TG techniques. Reference intervals of the WB‐TG parameters of healthy donors were calculated as 2.5th to 97.5th percentile according to the Clinical & Laboratory Standards Institute guideline. 24 Multiple linear regression analyses were performed with the ETPp, peak thrombin, or lag time of the 119 healthy donors as dependent variable and cell counts, age, sex, and oral contraceptive (OC) use as independent variables. For the sex parameter, male and female were coded as 0 and 1 in the analyses, respectively. Effect modification of sex on OC use and cell counts were also considered in the regression analysis and the backward method was used to find the best model. Any P values below .05 were considered statistically significant.
3. RESULTS
3.1. Continuous mixing resulted in a stable light transmission in WB‐TG
The efficacy of this continuous mixing method in preventing erythrocyte‐caused variable quenching on fluorescence signal transmission was first tested in calibration experiments, in which the cleavage of the thrombin substrate P2Rho by the calibrator was monitored in citrated WB. Figure 1A shows the mean fluorescence tracing of 12 replicate experiments over 20 minutes. The first derivatives (i.e., calibrator slope) of the tracings had a coefficient of variation (CV) of 3.7% and showed a horizontal trend when plotted vs time, indicating stable light transmission and no disturbance from substrate consumption or inner filter effect.
Figure 1.
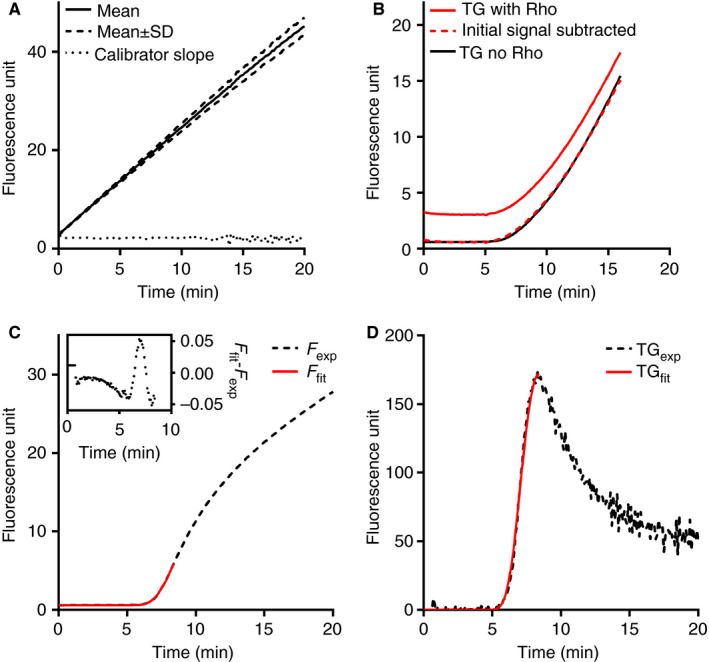
The performance of this novel WB‐TG assay and the CRG‐based calculation method. (A) The reaction between calibrator α2M‐T (100 nmol/L thrombin activity) and substrate P2Rho (300 μmol/L) in citrated whole blood. Mean (solid line) and mean ± standard deviation (dashed lines) of 12 replicate experiments are shown. The dotted line represents the first derivative of the average fluorescence tracing. (B) The fluorescence tracing of two parallel WB‐TG experiments at 2.5 pmol/L TF in the absence (black line) or presence (red line) of exogenously added 2.5 μmol/L Rhodamine (Rho) fluorophore. The red dashed line is the residual signal after subtracting the background Rho signal from the total signal. (C) An extended Chapman‐Richards growth (CRG) equation was fitted on the experimental fluorescence data from t = 0 until t = time‐to‐peak; the correctness of the fitting, as shown by the small difference between the fitted data (F fit, red solid line) and the experimental data (F exp, black dashed line) is shown in the insert. (D) The calculated thrombograms from the fitted data (TGfit, red solid line) and the experimental data (TGexp, black dashed line) overlap well with each other
The performance of the mixing method in clotting WB was evaluated by comparing the fluorescence signal of two parallel TF‐induced WB‐TG experiments in the presence and absence of exogenously added Rho. The added Rho induced a stable background signal during the lag phase (Figure 1B). From t = 0 until the time when signal increased the most (i.e., time‐to‐peak), the signal difference between the sample with exogenous Rho and the parallel sample without Rho addition remained relatively stable (Figure 1B), as shown by the overlap between the curves after subtracting the Rho background signal. This was repeated in WB samples from three donors, and the deviations of WB‐TG parameters ETPp and peak thrombin between with and without Rho fluorophore addition were on average 2.2 ± 1.6% and 4.9 ± 3.1% (n = 4), respectively. In addition, the fluorescence tracings of Rho added to clotting blood was monitored in the absence of P2Rho substrate, so the influence of the clot on the signal was judged independently of the amount of product generated. As shown in Figure S3, the signal of added Rho remained relatively stable in the first 0 to 10 minutes (during which TG reaches the peak), although a slight increase in signal as well as higher variation were observed in the later part (20‐30 minutes) of the experiments. Collectively, these results imply that this assay setting maintained the quenching effect of erythrocytes at a stable level at least until the thrombin peak is reached.
3.2. Conversion from fluorescence signals to WB‐TG parameters
The time‐dependent substrate cleavage in WB‐TG (Figure 1C; Figure S4) showed strong similarities with plasma‐TG; however, the tail part (thrombin decay) of the former had higher variation between replicates. Consequently, the estimation of the α2M‐T end level was less reliable. The first half of the WB‐thrombogram (from the lag phase until thrombin peak), however, was highly reproducible (average CV of 18 replicate measurements was 4.6%). Therefore, we calculated the endogenous thrombin potential until the thrombin peak (i.e., ETPp) after fitting an extended Chapman‐Richards growth (CRG) curve on the sigmoidal part of the experimental fluorescence signal as described in the Methods section. 13 , 22 Correctness of the fitting was shown by the high agreement between the experimental data and the fitted data (Figure 1C). This calculation mode resulted in a thrombogram that correlated well with the original one (Figure 1D; Figure S5).
3.3. Response of the WB‐TG parameters to varying TF concentrations
The response of the WB‐TG parameters to different TF concentrations is shown in Table S2. TF dose dependently altered thrombogram parameters. The lag time decreased from 14.8 ± 0.6 to 2.3 ± 0.1 minutes when increasing TF concentrations from 0 to 5 pmol/L; a similar trend was found with the time‐to‐peak. The peak thrombin increased from 193 ± 5 nmol/L to 279 ± 1 nmol/L with increasing TF concentration but the ETPp hardly changed.
3.4. Precision of the novel WB‐TG assay
The intra‐assay variation of this WB‐TG assay was assessed by 15 replicate measurements of a citrated WB sample at 1 pmol/L TF. As shown in Table 1, the CVs were below 6% for all TG parameters. Moreover, when this assay was measured in a population of 119 healthy volunteers the average intra‐assay CVs between triplicate measurements were 1.8%, 2.2%, 4.6%, and 4.6% for the lag time, time‐to‐peak, peak, and ETPp, respectively.
Table 1.
Intra‐ and interassay variations of this novel WB‐TG assay
| Intra‐assay variation (n = 15) | Interassay variation (n = 18) | |||
|---|---|---|---|---|
| Mean ± SD | CV, % | Mean ± SD | CV, % | |
| Lag time (min) | 5.7 ± 0.1 | 1.8 | 4.3 ± 0.3 | 6.2 |
| Time‐to‐peak (min) | 8.4 ± 0.2 | 2.0 | 6.7 ± 0.3 | 4.1 |
| Peak (nmol/L) | 173 ± 10 | 5.6 | 239 ± 14 | 6.0 |
| ETPp (nmol min/L) | 260 ± 9 | 3.6 | 308 ± 20 | 6.5 |
Abbreviations: ETPp, endogenous thrombin potential until thrombin peak; WB‐TG, whole blood thrombin generation.
The interassay precision could not be determined over a period longer than 1 day because of the storage limitation of WB. Therefore, it was determined by 18 independent experiments testing a single WB sample in triplicate resulting in CVs less than 7% for all thrombogram parameters. The interoperator variation of this assay, determined by letting four operators each independently test a same WB sample, was <13% for all WB‐TG parameters (Table S3).
3.5. Comparison between this novel WB‐TG assay and the paper‐based assay
Thrombin generation in WB of 10 donors was tested with the previously published paper‐based assay 14 and this novel assay at 0 and 1 pmol/L TF. Lag time was significantly shorter in the paper‐based assay, both without TF (Figure 2A; 9.3 ± 2.0 vs 20.0 ± 4.8 minutes; P < .0001) and at 1 pmol/L TF (Figure 2B; 3.7 ± 0.3 vs 4.5 ± 0.6 minutes; P < .001). Shorter time‐to‐peak was also observed in the paper‐based assay irrespective of the TF concentrations.
Figure 2.
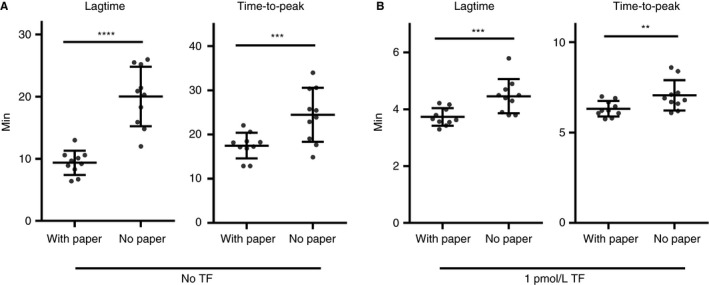
Comparisons between the novel WB‐TG assay and the paper‐based assay. For both assays, TG was triggered at (A) 0 or (B) 1 pmol/L TF in citrated whole blood from 10 healthy volunteers. Same reagents were used in both assays. Thrombogram parameters were calculated using the CRG assay for the novel assay without filter paper (no paper) or with the CAT assay for the filter paper‐based assay (with paper). Mean and standard deviation are indicated as bars in the figures. Comparisons between groups were done using the paired t test, **P < .01; ***P < .001; ****P < .0001
The TG parameters from these two assays showed significant correlations. The Pearson correlation coefficients were 0.79 (95% confidence interval [CI]: 0.32‐0.95; P = .007), 0.75 (95% CI: 0.22‐0.94; P = .013), and 0.82 (95% CI: 0.38‐0.96; P = .004) for the lag time, time‐to‐peak, and ETPp, respectively. The peak thrombin values showed weaker correlation (r = .5997; 95% CI: −0.05 to 0.89; P = .067).
3.6. Influences of hematocrit and platelet count on WB‐TG
We determined the effect of platelet count and hematocrit on the WB‐TG assay in PRP and reconstituted blood samples. Increasing the platelet count in plasma shortened the lag time from 11.7 minutes at 0 × 109/L platelets to 7.4 minutes at 25 × 109/L platelets, and further to 5.0 minutes at 360 × 109/L platelets (Figure 3A). Gradual increments in the thrombin peak were found with increasing platelet counts. Addition of 4 μmol/L synthetic PL markedly augmented the peak thrombin, even in the presence of 360 × 109/L platelets (Figure 3B).
Figure 3.
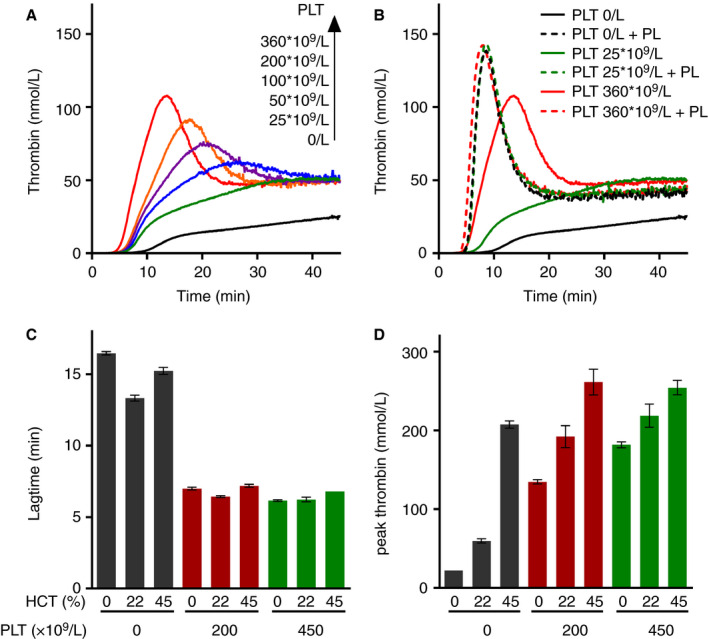
TG in reconstituted blood samples as measured by the novel WB‐TG assay. Human whole blood was separated into erythrocytes, platelets, and PPP and then reconstituted into the indicated levels. TG was measured in reconstituted PRP with varying platelet (PLT) count in the (A) absence or (B) presence of 4 μmol/L phospholipids (PL). (C, D) TG was measured in reconstituted samples with varying platelet counts (0, 200, or 450 × 109/L) and hematocrit (HCT; 0%, 22%, or 45%) as indicated in the figures. The (C) lag time and (D) peak thrombin of TG are shown. Each condition was measured in triplicate
In reconstituted blood samples, platelets shortened the lag time markedly irrespective of the hematocrit level, whereas increasing the hematocrit level hardly shortened the lag time (Figure 3C; Table 2). Interestingly, increasing hematocrit in the presence of 450 × 109/L platelets prolonged the lag time. The peak thrombin values showed dose‐dependent increase with platelet count and hematocrit (Figure 3D; Table 2). The ETPp of the reconstituted samples did not show dose‐dependent change.
Table 2.
Thrombin generation measured in reconstituted blood using the novel WB‐TG assay
| Platelet (×109/L) | 0 | 200 | 450 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Hematocrit (%) | 0 | 22 | 45 | 0 | 22 | 45 | 0 | 22 | 45 |
| Lag time (min) | 16.5 ± 0.1 | 13.3 ± 0.2 | 15.2 ± 0.3 | 7.0 ± 0.1 | 6.4 ± 0.1 | 7.2 ± 0.1 | 6.2 ± 0.1 | 6.3 ± 0.2 | 6.8 |
| Time‐to‐peak (min) a | NA | NA | 18.5 ± 0.2 | 12.2 ± 0.2 | 10.9 ± 0.3 | 10.0 ± 0.1 | 10.0 ± 0.2 | 10.2 ± 0.2 | 9.4 ± 0.1 |
| Peak (nmol/L) | 22.3 ± 0.1 | 59.7 ± 2.7 | 207.5 ± 4.6 | 134.7 ± 2.6 | 185.6 ± 13.6 | 261.3 ± 16.4 | 181.8 ± 3.9 | 211.5 ± 14.2 | 254.3 ± 9.1 |
| ETPp (nmol min/L) | NA | NA | 363.9 ± 18.2 | 390.8 ± 5.2 | 450.4 ± 12.0 | 381.3 ± 39 | 380.9 ± 13 | 446.0 ± 34.1 | 346.1 ± 16.1 |
Abbreviations: ETPp, endogenous thrombin potential until thrombin peak; NA, not available; WB‐TG, whole blood thrombin generation.
TG in 2 groups (platelet count = 0 and hematocrit = 0 or 22%) did not reach peak thrombin level even after 50 minutes; consequently, the ETPp could not be calculated and the thrombin concentration at t = 50 minutes was registered as peak thrombin value. Each condition was measured in triplicate. Data are expressed as mean ± SD (n = 3).
3.7. Influence of platelet activation/inhibition on WB‐TG
To study the influence of platelet on WB‐TG in more detail, different platelet agonists and antagonists were tested. As shown in Figure S6, Convulxin, a platelet agonist that induces phosphatidylserine exposure, shortened the lag time by 19.9 ± 3.7% (n = 5), but did not affect the ETPp and peak thrombin. Addition of Reopro (inhibitor of platelet integrins αIIbβ3 and αvβ3) prolonged the lag time by 15.8 ± 8.5% (n = 5) and reduced the peak thrombin by 17.1 ± 11.7%, but hardly affected the ETPp. The effect of Reopro was further confirmed in WB samples from 88 healthy donors in which Reopro hardly changed the ETPp (P = .06) but prolonged the lag time by 24.9% and reduced the peak thrombin by 22% (P < .0001 for both) (Figure S7). The addition of Iloprost and Cangrelor showed weaker inhibitory effect on WB‐TG compared with Reopro.
3.8. WB‐TG tested in a healthy population
Citrated WB samples from 119 healthy adult volunteers were tested to study the characteristics of WB‐TG in a normal population. The reference intervals and interindividual CVs of the WB‐thrombogram parameters were determined and are shown in Table 3 together with the cell counts. Compared with WB‐TG triggered at 1 pmol/L TF, similar ETPp was seen when triggered at 2.5 pmol/L TF (P = .88), along with significantly higher peak thrombin, shorter lag time, and time‐to‐peak (Table 3; P < .0001 for all).
Table 3.
Interindividual variations and reference intervals of the WB‐TG parameters
| n | % CV | Median | Reference Intervals (2.5th to 97.5th Percentiles) | |
|---|---|---|---|---|
| Female, number (%) | 60 (50.4%) | – | – | – |
| Age | 119 | – | 31.0 | 20.0‐63.0 |
| Blood counts | ||||
| White blood cell (×109/L) | 119 | 23.5% | 5.8 | 4.1‐8.8 |
| Erythrocyte (×1012/L) | 119 | 8.9% | 4.9 | 4.1‐6.0 |
| Hemoglobin (mmol/L) | 119 | 8.7% | 8.8 | 7.0‐10.4 |
| Hematocrit (%) | 119 | 8.1% | 42.9 | 35.4‐48.3 |
| Platelet (×109/L) | 119 | 20.9% | 263.3 | 151.1‐397.8 |
| Mean platelet volume (fL) | 119 | 11.1% | 7.5 | 6.0‐9.8 |
| WB‐TG 1 pmol/L TF | ||||
| Lag time (min) | 119 | 16.6% | 4.2 | 3.2‐6.1 |
| Time‐to‐peak (min) | 119 | 14.8% | 7.0 | 5.9‐10.6 |
| Peak (nmol/L) | 119 | 18.0% | 203.2 | 122.5‐297.7 |
| ETPp (nmol min/L) | 119 | 21.1% | 315.6 | 218.9‐522.9 |
| WB‐TG 2.5 pmol/L TF | ||||
| Lag time (min) | 119 | 15.5% | 2.8 | 2.1‐4.1 |
| Time‐to‐peak (min) | 119 | 12.3% | 5.3 | 4.5‐7.1 |
| Peak (nmol/L) | 119 | 15.2% | 230.7 | 161.3‐316.9 |
| ETPp (nmol min/L) | 119 | 19.8% | 307.6 | 222.6‐489.7 |
Abbreviations: ETPp, endogenous thrombin potential until the thrombin peak;TF, tissue factor; WB‐TG, whole blood thrombin generation.
The volunteers consisted of 60 females and 59 males. Females had significantly higher platelet counts (median [interquartile range], 282.8 [164.1‐417.8] vs 247.8 [149.4‐372.8] × 109/L, P < .01) and lower erythrocyte counts (4.7 [3.9‐5.9] vs 5.1 [4.5‐6.3] × 1012/L, P < .0001) than males, while having similar age, white blood cell count, and mean platelet volume. Significantly shorter lag time and higher ETPp were observed in females than in males (P < .01 for both) (Figure 4), but the thrombin‐peak was comparable.
Figure 4.
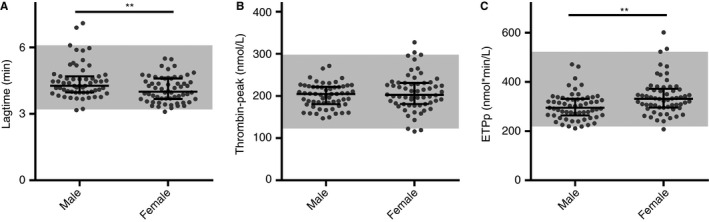
The WB‐TG parameters of males and females in 119 healthy volunteers. TG was triggered at 1 pmol/L TF in citrated WB samples. WB‐TG parameters (A) lag time, (B) thrombin‐peak, (C) and ETPp are shown for the different groups, including males (n = 59) and females (n = 60). Medians and interquartile ranges are indicated as bars; the gray areas represent the reference intervals of the total population (2.5th percentile to 97.5th percentile). Statistical significance was determined using the Mann‐Whitney U‐test, **P < .01
Age correlated with lag time (Spearman r = −.212, P < .05), peak thrombin (r = .215, P < .05), and ETPp (r = .218; P < .05). Multiple linear regression analyses were performed to explore the effect of blood cell counts on WB‐TG. After adjusting for age, gender and OC use, erythrocyte count significantly impacted the lag time (standardized Beta = 0.188, P = .034) and thrombin‐peak (standardized Beta = 0.205, P = .015) (Table S4). The effect of platelet count on the ETPp was borderline significant (standardized Beta = 0.159, P = .065).
4. DISCUSSION
Fluorogenic plasma‐TG assays are commonly used to model the global coagulation phenotype because these assays provide an efficient tool for assessing the complex interplay between pro‐ and anticoagulant factors. 2 , 4 , 25 WB‐TG is one step closer to physiology because it also includes the influence of the circulating blood cells on TG. The erythrocytes in WB can cause distortion of the fluorescence signal transmission, as a result of erythrocyte sedimentation during measurements. Previous reported fluorogenic methods for WB‐TG measurement suffered from high imprecision 15 or strong interference of contact activation, 14 hampering the routine applications of WB‐TG in routine applications.
In this study, we developed a novel fluorogenic assay for continuous WB‐TG measurement. Stable light transmission during WB‐TG was achieved by continuous mixing of the assay plate during the entire measurement course, using a microtiter plate with round‐cornered wells. We obtained reproducible fluorescence signals in both the calibration and TG experiments, and we showed that erythrocyte dependent light distortion was adequately controlled in clotting blood, because exogenously added rhodamine gave a stable fluorescence signal during a whole WB‐TG course.
Although the ascending part of the WB‐TG curve was stable and reproducible, the descending part of a WB‐TG curve had higher variation between measurements. Therefore, we used a CRG‐based calculation method to calculate the endogenous thrombin potential until the thrombin peak (ETPp). Similar to previous studies, 13 , 22 we found that the WB‐TG parameters from the CRG‐based calculation method correlates well with that from the classic CAT method (Spearman r > .95 for all TG parameters). The thrombin peak calculated by the CRG method is systematically higher than that by the regular CAT method, because no correction is foreseen for the α2M‐T that still cleaves the substrate. 13 The advantage of the CRG‐based calculation is that the measurement time is significantly shorter (from 50 to 20 minutes), although it may fail to give correct calculation for certain samples (e.g., samples with extremely slow TG or if the shape of the TG curve deviates from normal as is observed in the presence of direct FXa inhibitors). Of note, the mixing technique described here is optimized for the Fluoroskan Ascent fluorometer, therefore the user of the plasma‐CAT assay can readily adopt this new assay, but additional optimization will be needed for other fluorometers.
We tested the reproducibility of our novel WB‐TG assay and showed that the intra‐ and interassay variations of all parameters were below 7%. We also compared our novel WB‐TG assay with the paper‐based WB‐TG assay. The paper‐based assay had a shorter lag time (2.15‐fold, on average) than our novel WB‐TG when TG was not triggered with TF or another initiator of coagulation. This difference indicates that our novel WB‐TG assay is less disturbed by contact activation than the paper‐based assay. 14 , 18 Although the use of CTI in the paper‐based assay can reduce the effect of contact activation, this will increase the cost and increase the number of handling steps of the assay. 14 Another advantage of our novel WB‐TG is that there is no need for preparation of paper disks or addition of oil, making it less laborious and less dependent on the skills of the operator.
The interindividual variations of the WB‐TG parameters in a group of 119 healthy volunteers was between 12% and 22%, which is comparable to the values previously reported in PPP, PRP, and WB. 26 Previously identified prothrombotic risk factors (i.e., higher age and OC use) 27 , 28 were confirmed by our novel WB‐TG assay; higher age and OC use were associated with shorter lag time, higher ETPp, and peak thrombin. In agreement with previous observations, 26 females have a shorter lag time and higher ETPp than males. The reference ranges of the WB‐TG parameters were determined to pave the way for further studies.
Thrombin generation in WB depends on a complex interplay between coagulation factors and blood cells. 6 , 7 In a widely accepted cell‐based model of hemostasis, TG is initiated by factor (F)VIIa binding to surface‐bound TF, leading to the activation of trace amounts of FIX, FX, and, subsequently, thrombin. Low concentrations of thrombin activates platelets and cofactors FV and FVIII, which form a complex with FXa and FIXa, respectively to increase the efficiency of thrombin formation (i.e., propagation) on phosphatidylserine‐positive surfaces. 6 , 7 Platelets are equipped to create a procoagulant environment, via secretion of coagulation (co)factors, 6 expression of membrane receptors, and phosphatidylserine exposure to form the surface for the coagulation cascade. 7 The effects of platelet count and function on the onset and velocity of ex vivo TG was previously characterized with a PRP‐TG assay, 4 , 19 and was also confirmed in our WB‐TG assay.
The impact of erythrocyte on TG and coagulation is less well established and has gained interest. 10 , 11 , 29 , 30 Using our novel WB‐TG assay, we studied the influence of erythrocytes and platelets on TG in detail with reconstituted blood samples and in WB from 119 healthy donors. We found that the fast onset of TG (i.e., a short lag time) relied on the presence of platelets and was also affected by the activation or inhibition state of platelets. Conversely, high erythrocyte numbers prolonged the lag time, whereas erythrocyte count dose dependently augmented the peak thrombin of TG, which persisted in the presence of high platelet numbers. The negative impact of high erythrocyte numbers on the onset of TG could be attributed to steric hindrance because of its large size and high abundance in WB. Although a portion (approximately 0.5%) of erythrocytes in the circulation of normal individuals exhibited phosphatidylserine (PS) exposure, 10 , 11 this portion may be too low to reverse the previously mentioned effect. Erythrocytes may augment TG in the propagation phase once they are trapped inside the clot. Besides the PS exposure and their impact on blood rheology and endothelium function, erythrocytes have also been suggested to directly affect platelet function. 31 A recent finding showed that erythrocyte‐platelet interaction via the Fas ligand‐receptor increases the PS exposure on both cells, 32 which could potentially facilitate TG.
In healthy subjects, the normal variation in platelet count and in hematocrit seems to have minimal effects on the ETPp. This was also observed in previous studies in PRP‐TG, which showed that the endogenous thrombin potential was insensitive to platelet numbers, if the platelet numbers were higher than 35 × 109/L. 4 , 19 Furthermore, our experiments studying the influences of platelet agonist and antagonists on WB‐TG also showed that these molecules mainly influenced the initiation and velocity of TG but the influence on the ETPp was neglectable. Probably, the effects of platelet inhibitors in preventing venous thrombosis stem from delaying the onset of TG on platelets. However, there are several diseases in which the high thrombotic risk is likely induced by the blood cells and not by the plasma coagulation factors, including sickle cell disease and hematologic malignancies such as multiple myeloma and leukemia. 33 , 34 For example, patients with sickle cell disease are recognized to have a hypercoagulable state, resulting in higher amounts of in vitro thrombin‐antithrombin complex formation in WB than normal controls 11 ; however, their endogenous thrombin potential tested in plasma was similar 12 , 13 or even lower 11 compared with normal controls, suggesting that blood cells may play an important role for the hypercoagulability.
In conclusion, we have developed a novel assay for continuous WB‐TG measurement. This assay is a straightforward approach to measure the involvements of platelets and erythrocytes in TG and may assist the research of blood cell‐associated coagulation disorders.
CONFLICT OF INTEREST
All authors state that they have no conflict of interest related to this study.
ADDENDUM
J. Wan, J. Konings, B. de Laat, M. Roest designed the study; H. Kelchtermans and R. Kremers organized donors and collected donor characteristics; J. Wan, J. Konings, and Q. Yan performed the experiments and interpreted data; J. Wan, J. Konings, and M. Roest wrote the manuscript; H. Kelchtermans critically reviewed the manuscript. All authors approved the final version of the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank all the volunteers that participated in this study and the technical supports from colleagues in Synapse. Dana Huskens is especially acknowledged for critically reviewing the manuscript. Jun Wan is supported by a scholarship (No. 201606130068) from the China Scholarship Council.
Wan J, Konings J, Yan Q, et al. A novel assay for studying the involvement of blood cells in whole blood thrombin generation. J Thromb Haemost. 2020;18:1291–1301. 10.1111/jth.14786
Manuscript handled by: Robert Gosselin
Final decision: Robert Gosselin and 20‐Feb‐2020
REFERENCES
- 1. Pagano MB, Chandler WL. Thrombin generation assay: are we ready for prime time? [Editorial] JALM. 2017;2:135‐137. [DOI] [PubMed] [Google Scholar]
- 2. Tripodi A. Thrombin generation assay and its application in the clinical laboratory. Clin Chem. 2016;62:699‐707. [DOI] [PubMed] [Google Scholar]
- 3. Al Dieri R, de Laat B, Hemker HC. Thrombin generation: what have we learned? Blood Rev. 2012;26:197‐203. [DOI] [PubMed] [Google Scholar]
- 4. Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4‐15. [DOI] [PubMed] [Google Scholar]
- 5. Hemker HC, Kremers R. Data management in thrombin generation. Thromb Res. 2013;131:3‐11. [DOI] [PubMed] [Google Scholar]
- 6. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93:327‐358. [DOI] [PubMed] [Google Scholar]
- 7. Hoffman M, Monroe DM. A cell‐based model of hemostasis. Thromb Haemost. 2001;85:958‐965. [PubMed] [Google Scholar]
- 8. Regnault V, Beguin S, Wahl D, de Maistre E, Hemker HC, Lecompte T. Thrombinography shows acquired resistance to activated protein C in patients with lupus anticoagulants. Thromb Haemost. 2003;89:208‐212. [PubMed] [Google Scholar]
- 9. Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. 2017;130:1795‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood. 2012;120:3837‐3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peyrou V, Lormeau JC, Herault JP, Gaich C, Pfliegger AM, Herbert JM. Contribution of erythrocytes to thrombin generation in whole blood. Thromb Haemost. 1999;81:400‐406. [PubMed] [Google Scholar]
- 12. Kessels H, Béguin S, Andree H, Hemker HC. Measurement of thrombin generation in whole blood ‐ the effect of heparin and aspirin. Thromb Haemost. 1994;72:78‐83. [PubMed] [Google Scholar]
- 13. Kelchtermans H, Pelkmans L, Bouwhuis A, et al. Simultaneous measurement of thrombin generation and fibrin formation in whole blood under flow conditions. Thromb Haemost. 2016;116:134‐145. [DOI] [PubMed] [Google Scholar]
- 14. Ninivaggi M, Apitz‐Castro R, Dargaud Y, de Laat B, Hemker HC, Lindhout T. Whole‐blood thrombin generation monitored with a calibrated automated thrombogram‐based assay. Clin Chem. 2012;58:1252‐1259. [DOI] [PubMed] [Google Scholar]
- 15. Tappenden KA, Gallimore MJ, Evans G, Mackie IJ, Jones DW. Thrombin generation: a comparison of assays using platelet‐poor and ‐rich plasma and whole blood samples from healthy controls and patients with a history of venous thromboembolism. Br J Haematol. 2007;139:106‐112. [DOI] [PubMed] [Google Scholar]
- 16. Al Dieri R, Hemker CH. Thrombin generation in whole blood. Br J Haematol. 2008;141:896‐897. [DOI] [PubMed] [Google Scholar]
- 17. Giesen PL, Tappenden K, Jones W. Method for determining the course of proteolytic activity. U.S. Patent US20100105091. [Google Scholar]
- 18. Prior SM, Mann KG, Freeman K, Butenas S. Continuous thrombin generation in whole blood: new applications for assessing activators and inhibitors of coagulation. Anal Biochem. 2018;551:19‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanschoonbeek K, Feijge MAH, Van Kampen RJW, et al. Initiating and potentiating role of platelets in tissue factor‐induced thrombin generation in the presence of plasma: subject‐dependent variation in thrombogram characteristics. J Thromb Haemost. 2004;2:476‐484. [DOI] [PubMed] [Google Scholar]
- 20. Hemker HC, Hemker PW, Al Dieri R. The technique of measuring thrombin generation with fluorescent substrates: 4. the H‐transform, a mathematical procedure to obtain thrombin concentrations without external calibration. Thromb Haemost. 2009;101:171‐177. [PubMed] [Google Scholar]
- 21. Wagenvoord R, Hemker PW, Hemker HC. The limits of simulation of the clotting system. J Thromb Haemost. 2006;4:1331‐1338. [DOI] [PubMed] [Google Scholar]
- 22. Moorlag M, Schurgers E, Krishnamoorthy G, et al. Near patient thrombin generation in patients undergoing elective cardiac surgery. JALM. 2017;1:613‐625. [DOI] [PubMed] [Google Scholar]
- 23. Schurgers E, Moorlag M, Hemker C, Lindhout T, Kelchtermans H, de Laat B. Thrombin generation in zebrafish blood. PLoS One. 2016;11:e0149135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horowiz G, Altaie S, Boyd J, et al. Defining, establishing, and verifying reference intervals in the clinical laboratory. Wayne, PA: Clinical Laboratory and Standards Institute (CLSI); 2008. [Google Scholar]
- 25. Hemker HC, Al Dieri R, De Smedt E, Beguin S. Thrombin generation, a function test of the haemostatic‐thrombotic system. Thromb Haemost. 2006;96:553‐561. [PubMed] [Google Scholar]
- 26. Bloemen S, Huskens D, Konings J, et al. Interindividual variability and normal ranges of whole blood and plasma thrombin generation. JALM. 2017;2:150‐164. [DOI] [PubMed] [Google Scholar]
- 27. Haidl H, Cimenti C, Leschnik B, Zach D, Muntean W. Age‐dependency of thrombin generation measured by means of calibrated automated thrombography (CAT). Thromb Haemost. 2006;95:772‐775. [PubMed] [Google Scholar]
- 28. Rotteveel RC, Roozendaal KJ, Eijsman L, Hemker HC. The influence of oral contraceptives on the time‐integral of thrombin generation (thrombin potential). Thromb Haemost. 1993;70:959‐962. [PubMed] [Google Scholar]
- 29. Walton BL, Lehmann M, Skorczewski T, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129:2537‐2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horne MK 3rd, Cullinane AM, Merryman PK, Hoddeson EK. The effect of red blood cells on thrombin generation. Br J Haematol. 2006;133:403‐408. [DOI] [PubMed] [Google Scholar]
- 31. Weisel JW, Litvinov RI. Red blood cells: the forgotten player in hemostasis and thrombosis. J Thromb Haemost. 2019;17:271‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klatt C, Krüger I, Zey S, et al. Platelet‐RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J Clin Invest. 2018;128:3906‐3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sparkenbaugh E, Pawlinski R. Prothrombotic aspects of sickle cell disease. J Thromb Haemost. 2017;15:1307‐1316. [DOI] [PubMed] [Google Scholar]
- 34. Mukai M, Oka T. Mechanism and management of cancer‐associated thrombosis. J Cardiol. 2018;72:89‐93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material