

Abstract
Thioglycosides and C‐glycosides represent pharmacologically useful classes of glycomimetics that possess a high degree of biological stability. One emerging tool for the stereoselective synthesis of thioglycosides is the photoinitiated addition of thiols to unsaturated sugars. Moreover, thiyl radical‐mediated reactions of exo‐glycals and 1‐substituted endo‐glycals offer facile routes to β‐C‐glycosidic structures. This Concept article summarizes the thiol‐ene coupling strategies developed recently by our group and Somsák's group for the synthesis of several kinds of glycomimetics which are difficult to synthesize by conventional methods. One unusual characteristic of the thiol‐ene reactions of endo‐glycals is that heating inhibits, whereas cooling promotes the reaction. This unique temperature dependence as well as the effects of the enose structures and thiol configurations on the efficacy and stereoselectivity of the reactions are also discussed.
Keywords: addition, enose, stereoselective, thioglycoside, thiyl radical
UV‐light induced radical‐mediated addition of thiols onto endo‐ or exo‐cyclic double bonds of unsaturated sugars offers an easy and stereoselective route to a broad range of stable glycomimetics including 1,2‐cis‐α‐S‐glycosides and new types of β‐C‐glycosides.
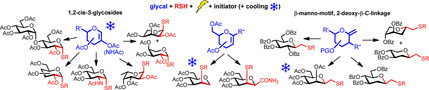
Introduction
Synthetic oligosaccharides and their mimetics are widely used for elucidating the essential roles that carbohydrates play in living organisms. Thio‐linked oligosaccharides and glycoconjugates are valuable glycomimetics, able to resist enzymatic and acidic hydrolysis, which significantly potentiate their application not only in molecular recognition studies but also in drug development.1, 2, 3, 4, 5, 6 Consequently, thioglycosides are favorable synthetic targets and many methodologies have been developed for their synthesis.7, 8 However, the stereoselective S‐glycosylation particularly the selective formation of the 1,2‐cis‐α‐S‐glycosidic bond still poses a major challenge.
The most common approaches for S‐glycosylation include nucleophilic displacement of the anomeric leaving group of glycosyl donors by thiol nucleophiles (Scheme 1 A)7, 8, 9, 10 and utilization of glycosyl thiols in various base‐promoted reactions (Scheme 1 B, a–c).11, 12, 13, 14 The classical nucleophilic substitution reaction at the anomeric carbon often proceeds, at least partly, through an oxocarbenium ion, resulting in complete loss of anomeric integrity of the donor, and leading to the formation of a mixture of α‐ and β‐glycosides (Scheme 1 A).7, 15 The α/β‐stereoselectivity in these reactions fundamentally depends on the identity and protecting groups of the donor and can be affected by the steric and stereoelectronic effects around the anomeric center. Although 1,2‐trans glycosides can be efficiently prepared by anchimeric assistance of the carbonyl‐based C2 protecting groups, this method has only limited efficacy in the stereoselective synthesis of 2‐deoxy glycosides and 1,2‐cis glycosides. Moreover, the anomeric effect, which can be exploited for the axial α‐O‐glycosylation, is subtle for S‐glycosides and its impact on the stereochemical outcome of the glycosylation is hardly to predict, further complicating the formation of the 1,2‐cis α‐S‐glycosidic bond.
Scheme 1.
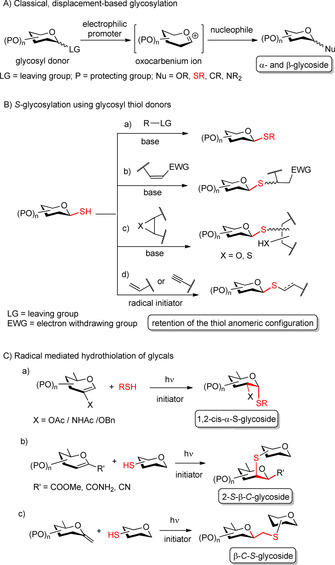
Approaches for S‐glycosylation.
Glycosyl thiols (1‐thiosugars) has been widely used in the synthesis of thioglycosides due to its high anomeric stability and the high nucleophilicity of sulfur.7, 8 Unlike saccharide hemiacetals which undergo mutarotation, glycosyl thiols retain their anomeric configuration under neutral and basic conditions,16 ensuring full steroselectivity in the thiolate‐mediated S‐glycosylation,11 conjugate addition,12 and epoxide or thiirane ring‐opening reactions,13, 14 as well as in the recently emerged thiyl radical‐mediated thiol‐ene17, 18 and thiol‐yne19 processes (Scheme 1 B, a–d). However, whereas the equatorial thiols are readily accessible by the treatment of acetohalo sugars with a sulphur nucleophile,20 stereoselective synthesis of the axial thiol congeners still remains a challenge. Existing methods for the synthesis of 1,2‐cis α‐thiols including treatment of 1,2‐ or 1,6‐anhydro sugars or glycosyl trichloroacetimidates with bis‐trimethylsilyl sulfide21 and epimerizations of β‐glycosyl thiols22 are generally characterized by narrow substrate scope or low efficacy.
The radical‐mediated addition of thiols to non‐activated double bonds23, 24 has been used in organic syntheses for a long time and with the advent of click chemistries25 this transformation has its renaissance in glycochemistry as a robust ligation tool.17, 18 However, it has become evident only in recent years that unsaturated sugars can efficiently be applied as the alkene partners in the thiol‐ene coupling reactions to provide a new, radical pathway for the stereoselective synthesis of a broad variety of thio‐linked glycomimetics (Scheme 1 C). We have demonstrated that photoinduced addition of thiols to 2‐substituted hexoglycals led to exclusive formation of 1,2‐cis‐α‐S‐glycosides independently of the sugar identity and the protecting groups applied (Scheme 1 C, a).26, 27, 28, 29, 30 Further studies by our group and Somsák's group revealed that radical‐mediated hydrothiolation of exo‐glycals31, 32, 33 or 1‐substituted endo‐glycals34 enabled stereoselective formation of C‐β‐d‐glycosidic thiodisaccharides, including the highly challenging β‐mannosidic and 2‐deoxy β‐C‐glycosidic structures (Scheme 1 C, b–c). These results show, that the thiyl radical‐mediated reactions of glycals represent a useful complementary strategy of the classical ionic thioglycosylation approaches. The mild conditions, high protecting group tolerance and the insensitivity to air and water further increase the practical utility of the thiol‐ene reactions.
During our optimization studies on endo‐glycals, we discovered that the reaction temperature plays a crucial role in the reaction efficacy in the hexose series and exerts great influence on both the efficacy and stereoselectivity in the pentose series.
This Concept article summarizes the thiol‐ene coupling strategies developed so far for the easy and stereoselective synthesis of biologically important carbohydrate mimetics which are otherwise very difficult to prepare. The effects of reaction temperature and the enose structures on the efficacy and stereochemical outcome of the reactions are discussed in detail and a plausible explanation for the exquisite stereoselectivities observed in most cases is provided.
Photoinduced Hydrothiolation of 2‐Substituted Glycals
Hydrothiolation of terminal alkenes was discovered by Posner in 190523 and the free free‐radical chain mechanism of the reaction was first described by Mayo and co‐workers in 1938.24 The process begins with the generation of a thiyl radical from the thiol by light‐irradiation and/or the presence of a radical initiator then the addition reaction proceeds in two steps. In the first, reversible propagation step the thiyl radical formed adds to the alkene, generating a carbon‐centered radical intermediate which then, in the chain transfer step, abstracts a hydrogen from another thiol to result in the formation of the thioether product along with a new thiyl radical.
The first application of glycals as alkene partners in the photoinduced thiol‐ene reaction was described by Kushida and co‐workers in 1973 (Scheme 2 A).35 The acetone‐sensitized photochemical addition of ethanethiol and propanethiol to 2‐acetoxy‐d‐glucal 1 proceeded efficiently to result in the 1‐thio‐α‐glucopyranosides 2 and 3 with full selectivity. However, the high thiol excess required prevented a wider application of the reaction, therefore, it has long been forgotten.
Scheme 2.
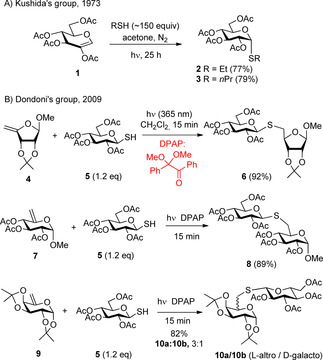
First results on photoinduced additions of thiols to unsaturated sugars: A) hydrothiolation of 2‐acetoxy glycal 1 with alkyl thiols, B) synthesis of thiodisaccharides by thiol‐ene click reaction using DPAP.
Over the last 15 years the radical‐mediated addition of thiols to terminal alkenes initiated by low‐energy UV‐light (365 nm) in the presence of the cleavable photoinitiator 2,2‐dimethoxy‐2‐phenylacetophenone (DPAP), referred as thiol‐ene coupling (TEC) or thio‐click reactions, have been intensively studied in various fields of chemistry as a robust metal‐free click process.36 Dondoni, Marra and co‐workers were the first who applied this mild and efficient method for the synthesis of thiodisaccharides (Scheme 2 B).37 They reported that reactions of furanoside‐ and pyranoside‐derived alkenes such as 4, 7 and 9 with a slight excess of 1‐thiosugar (e.g. 5) provided the corresponding S‐linked disaccharide mimetics with high yields and most cases with excellent diastereoselectivity.
On the basis of the above results, we envisaged that the thiol‐ene coupling of 2‐substituted glycals under the mild, DPAP‐mediated conditions can function as a powerful and generally applicable method for stereoselective formation of the challenging 1,2‐cis‐α‐S‐glycosidic bond. (Scheme 3).26 Initially, addition of ethanethiol to 2‐acetoxy‐glycals 1, 11 and 13 under UV‐light irradiation in the presence of DPAP were studied at room temperature in a glass vial by irradiation with a mercury lamp using different solvents, thiol excess and reaction time. It has been found that several short irradiation cycles, adding a new dose of initiator to the reaction mixture before each cycle, are more effective than longer term continuous irradiation. The best results were obtained in toluene using 3×5 equivalents of EtSH, 3×0.1 equivalents of initiator and 3×15 min irradiation. Under these conditions almost complete conversion of the glycals was observed resulting in the 1,2‐cis‐α 1‐ethylthio‐glycosides 2, 12 and 14 in full diastereoselectivity, independently of the configuration of the starting 2‐acetoxy glycal.
Scheme 3.
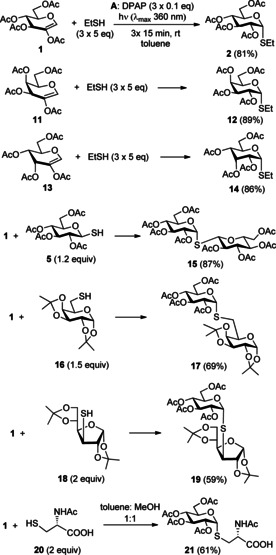
Hydrothiolation of 2‐acetoxy glycals proceeds with exclusive diastereoselectivity to provide 1,2‐cis‐α‐S‐glycosides and ‐S‐disaccharides (All reactions were performed under the A conditions).
The viability of the method in the synthesis of S‐linked glycoconjugates and disaccharides was demonstrated by reacting 2‐acetoxy‐d‐glucal 1 with a range of thiols including various sugar thiols (e.g. 5, 16, 18), amino acids (e.g. 20) and peptides using 1.2‐2:1 thiol:ene ratio.26 In all cases very clean reactions and exclusive formation of the corresponding 1,2‐cis‐α‐glucoside were observed. (In some cases, a small amount of disulfide formed from the thiol was observed as the only by‐product.) If only moderate yield was observed (e.g. 19 and 21), it was due to moderate conversion of the alkene which could be recovered from the reaction mixture. We have found that prolonged irradiation and the use of higher excess of sugar thiols hardly increase the conversion of the glycal. Moreover, the high excess of thiol can even be disadvantageous, since the unreacted thiol and the disulfide formed from the thiol make the chromatographic purification very difficult.
Our further study revealed that the nature of thiols greatly influences the efficacy of the reactions and some thiols exhibit no or very low reactivity towards the acetylated glycal 1 (Scheme 4).27 It was found that the high stability of a thiyl radical was detrimental to the conversion by shifting the equilibrium of the reversible propagation step backwards. As the thiol‐ene reaction is an electrophilic addition, the electron‐withdrawing ester substituents on the alkene are also disadvantageous to the reaction efficiency. Accordingly, the reactivity of the alkene partner could successfully be increased by changing its ester protecting groups to electron‐donating benzyl groups (22). In this way, the yields with benzyl mercaptan and 1‐thioglycerol could be doubled (24 a/b, 25 a/b) and a moderate reaction could also be elicited with the highly resonance stabilized phenylthiyl radical (23). It has to be noted, that in the absence of DPAP and thiol, the perbenzylated glycal 22 was completely resistant to UV irradiation. However, during the thiol‐ene coupling reaction, due to the easily abstractable benzylic hydrogen, side reactions also occurred which made difficult the purification and, in some cases, noticeably decreased the isolated yields
Scheme 4.
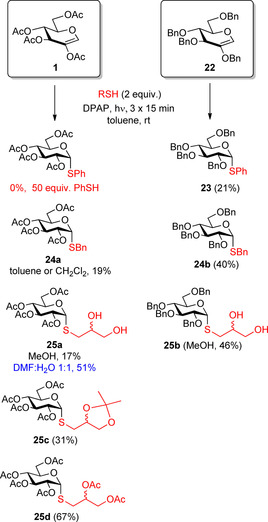
Effects of nature of thiols, solvents and protecting groups on the reaction efficacy.
We also demonstrated that the reactivity of a thiol toward a given alkene can be tuned by its substitution pattern. The synthesis of 25 a,c and d highlighted that electron withdrawing substituents on the thiol are most preferred for increasing its reactivity.
The effect of solvents was also studied. It is important to note that the reaction is compatible with all solvents, and the high concentration of reactants is crucial for the high conversion in the thiol‐ene coupling reaction. Therefore, the choice of solvent is primarily governed by the solubility of the reaction partners. We have found toluene the best solvent for the reactions with apolar thiols. Although the reactions proceeded with the same efficacy in dichloromethane, we observed deacetylation of the products in several experiments. Toluene–MeOH or MeOH have been used as the solvents for the thiol‐ene couplings with polar thiols; which, however, resulted in low conversions in some cases. In those cases, significantly enhanced yields were achieved in DMF–water (e.g. in the synthesis 25 a), presumably because the DMF–water system stabilized the polar carbon‐centered radical intermediate better than MeOH.
The method was utilized for the synthesis of S‐linked di‐ and trisaccharides by reacting 1‐thioglucose 5 with different 2‐substituted hexoglycals including 2‐acetoxy d‐ and l‐glycals (11, 13, 26, 27), 2‐acetamido‐d‐glucal (28), and maltose‐derived disaccharide glycal (29) as the alkene partners (Scheme 5).28, 29, 30 In all cases exclusively the expected 1,2‐cis α‐S‐glycoside was formed, highlighting that the identity of reactants does not affect the complete stereoselectivity of the reaction. On the other hand, the low yields of 30–33 obtained in the d‐galacto, ‐allo, ‐gulo, and l‐fuco cases at room temperature revealed the high impact of the configuration of glycals on the efficiency of the addition.28, 29, 30 Through further optimization experiments, we discovered the very unique temperature effect that heating inhibits, whereas cooling promotes the thiol‐ene coupling reaction.38 It was found that conversion of the starting glycals increased gradually by cooling and good to excellent yields were achieved at −80 °C in most cases (30–34).28, 29, 30 We formulated that the reaction temperature controls the equilibrium of the rapidly reversible thiyl addition (propagation) step of the radical chain process and conducting the reactions at low temperature is an adequate strategy to prevent the degradation of the intermediate carbon‐centered radical, formed in the thiyl addition step, hence allowing it to react with a thiol in the hydrogen abstraction step. Although enoses with all‐equatorial substitution patterns (28, 29) generally showed good reactivity at room temperature, the cooling was beneficial in those cases too.
Scheme 5.
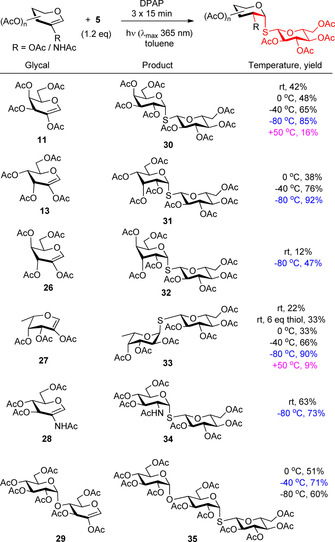
Configuration‐ and temperature‐dependent reactivity of glycals—Unique promoting effect of cooling.
The heating proved to be detrimental to the reaction as it was demonstrated by running the hydrothiolation of d‐galactal 11 and l‐fucal 27 at +50 °C.30
The low‐temperature thiol‐ene coupling reaction was exploited for the stereoselective synthesis of a series of 1,2‐cis‐α‐thio‐linked d‐glucoside (37–39), d‐galactoside (40–44), l‐fucoside (45, 47–50), and d‐GlcNAc (52–54) derivatives (Scheme 6).28, 29, 30 The reactions generally worked efficiently at −80 °C with various thiols, including 1‐thiomaltose 38, using a low, 1.2 equivalents thiol excess. In the case of alkyl thiols, however, high thiol excess and moderate cooling proved to be the adequate strategy to elicit efficient additions, probably because the excessive cooling inhibits the hydrogen abstraction from alkyl thiols. It is worth noting that though the reaction mixture was frozen at −80 °C in some cases, the reactions also took place in frozen mixtures.
Scheme 6.
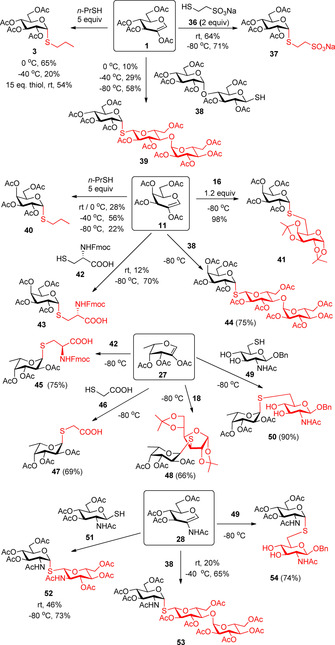
UV‐induced low‐temperature hydrothiolation of 2‐substituted glycals—Universal method for stereoselective 1,2‐cis‐α‐S‐glycosylation (Conditions: hν, 3×15 min; 3×0.1 equiv DPAP).
We and Somsák's group30, 39 demonstrated that the radical‐mediated S‐glycosylation method opens the way for expeditious synthesis of higher oligosaccharide mimetics possessing the challenging 1,2‐cis‐α‐S‐linkages (Scheme 7). Hydrothiolation of 2‐acetoxy glycal 29 with mono‐ and disaccharide thiols at low temperature provided the non‐reducing tri‐ and tetrasaccharides (57 and 55) with moderate to excellent yields (Scheme 7 A).30
Scheme 7.
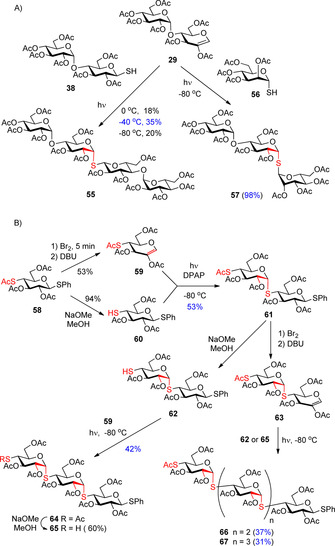
Synthesis of higher thio‐oligosaccharides by the low‐temperature thiol‐ene coupling.
Lázár et al. developed an iterative thiol‐ene coupling protocol for the synthesis of thio‐linked maltooligomers up to pentasaccharide (Scheme 7 B).39 The starting 1,4‐dithio‐glucopyranoside 58 was converted to both enose 59 and thiol 60, then the 1,2‐cis α‐configured anomeric sulfur atom was introduced in the radical‐mediated coupling step with full stereoselectivity to produce trithiomaltoside 61 as the only product. Toward higher oligomers 64–67, the neccessary 2‐acetoxy‐glycals and thiols were formed at oligosaccharide levels and coupled to each other by irradiating with UV‐light at −80 °C. All coupling steps proceeded with full 1,2‐cis α‐stereoselectivity, albeit the yields decreased when higher coupling partners were reacted.
It is well known from previous studies that radical addition of thiols onto substituted cyclohexene derivatives occurs preferentially in a trans‐diaxial manner as the result of a kinetically favored axial attack of the thiyl radical onto the cyclic alkene in its half‐chair conformation together with a stereoselective hydrogen abstraction from the thiol into an axial position.40, 41 However, complete diastereoselectivity has not been reported, because the nature of the thiol, the configuration of the alkene, the molar ratio of the reactants and also the temperature play a role in the stereochemical outcome of the reactions.37, 41, 42
In the case of 2‐substituted hexoglycals, the highly different stability of the possible carbon‐centered radical intermediates and the rapidly reversible nature of the thiyl addition step43 together lead to the complete 1,2‐cis‐α‐stereoselectivity. The reaction proceeds exclusively through the most stable chair conformational form of the intermediate C2‐centered radicals (4C1 in the d‐series and 1C4 in the l‐series), whereas the other C2‐centered radical intermediates are of high‐energy, therefore rapidly dissociate to the starting compounds in the first reversible step before reacting with a thiol in the H‐abstraction step (Figure 1).
Figure 1.
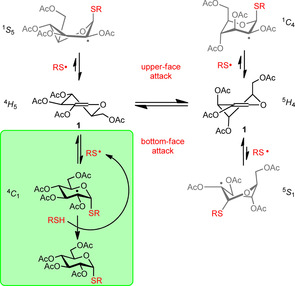
Plausible mechanistic pathway of free‐radical addition of thiols to 2‐substituted hexoglycals shown on the example of 1. The only productive pathway leading to complete 1,2‐cis‐α diastereoselectivity is highlighted in green.
Extending the hydrothiolation reactions to pentopyranosyl glycals, a lower level of diastereoselectivity was observed (Scheme 8).30 Addition reactions of 1‐thiosugars 5 and 56 to 2‐acetoxy d‐ and l‐pentoglycals 68–70 led to the formation of 1,2‐cis‐α‐ and 1,2‐cis‐β‐thioglycosides 71–78 in varying ratios depending on the temperature and the configuration of both reactants. Cooling had a double positive effect on the reactions, increased the yields and in most cases significantly raised the stereoselectivity. Starting from the d‐xylose‐derived glycal 68 the 1,2‐cis‐β‐S‐linked d‐lyxosides 71 and 73 were obtained at −80 °C with high yields and almost complete stereoselectivity, whereas hydrothiolation of d‐arabinal 69 proceeded with a moderate 1,2‐cis‐α‐d‐ribo selectivity at either −80 °C or −100 °C, and addition of 5 to the l‐arabinose‐derived glycal 70 occurred with very high 1,2‐cis‐α‐diastereoselectivity and excellent yield at −80 °C. Although the additions proceeded with exclusive 1,2‐cis selectivity in all cases, our results suggest that the exact stereochemical outcome of the reactions in the pentopyranosyl series is difficult to predict.
Scheme 8.
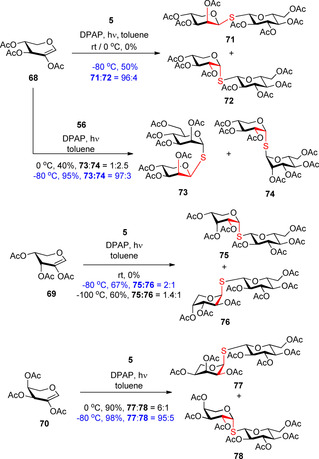
Extension of the reaction to 2‐acetoxy pentoglycals—The exclusive 1,2‐cis‐α‐selectivity turns to 1,2‐cis‐selectivity.
The lack of complete diastereoselectivity in the pentose series can be explained by the higher conformational flexibility of the pentopyranoses.44 Whereas in the hexose series, one of the chair conformations of the intermediate radical has exquisite stability due the bulky C6 group, in the case of pentopyranoses, the 1C4 and 4C1 forms of the radical intermediate have comparable stabilities; therefore, the reaction can proceed through both chair conformations. In these cases, the low‐reaction temperature proved to be a stereo controlling effect shifting the product ratio towards the stereoisomer which was formed through to a more stable radical.30
Photoinduced Hydrothiolation of Glycals and 1‐Substituted Glycals
The photoinduced thiol‐ene additions of unsubstituted and 1‐substituted glycals enabled regio‐ and stereoselective synthesis of 2‐thio‐linked saccharide mimetics (Scheme 9 and Scheme 10).27, 30, 34
Scheme 9.
Regioselective hydrothiolation of glycals—cooling increases the axial selectivity (except for the galacto case).
Scheme 10.
Hydrothiolation of 1‐substituted glycals occurs with exclusive 1,2‐cis‐β‐selectivity.
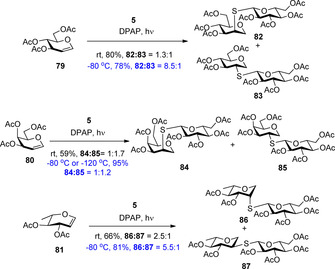
The room temperature hydrothiolation of peracetylated d‐glycals was first investigated by Dondoni and Marra.45 They reported that although regioselective addition occurred in all cases, mostly very low stereoselectivity and often low yields were observed. We studied the effect of cooling on the efficiency and diastereoselectivity of the hydrothiolation of various glycals, for example, d‐glucal 79, d‐galactal 80 and l‐rhamnal 81 (Scheme 9).30 Cooling always promoted the conversion and in most cases significantly increased the stereoselectivity. Performing the reactions at −80 °C, the additions occurred with excellent yields and, with the exception of the galacto case, with high to complete diastereoselectivity in favor of the axially C2‐S‐linked products.
The hydrothiolation reactions of glycals proceed through stable glycosyl radicals of different conformational forms (Figure 2). It has been demonstrated that the most preferred conformation of glycopyranosyl radicals is the one in which the C2 substituent adopts a quasi‐axial position, because this conformer is stabilized by the so‐called quasi‐homo‐anomeric effect.46 At low temperature, the reaction is shifted towards the product formed through the lower energy radical intermediate, as it is demonstrated by the increased ‘manno’ selectivity at −80 °C in the case of 79 and 81. The substantially low diastereoselectivity observed at any temperature upon hydrothiolation of galactal 80 can be explained by the very similar energy of the of the 4C1 talopyranosyl and 4H5 galactosyl radical intermediates.30
Figure 2.
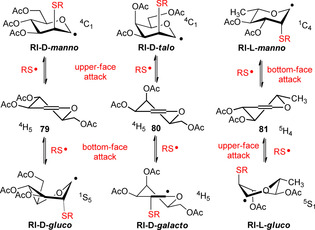
Stable glycopyranosyl radical intermediates (RI) formed upon upper‐ and bottom‐face attacks by the thiyl radicals to d‐ and l‐glycals 79–81.30
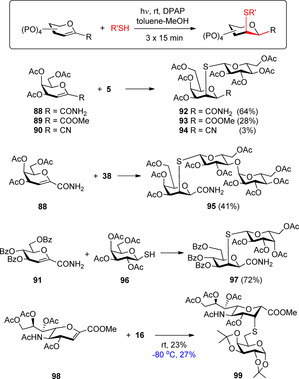
Somsák's group reported that the free‐radical hydrothiolation of amide‐, methoxycarbonyl‐ and cyano‐substituted glycal derivatives (e.g. 88–91) took place with full regio‐ and stereoselectivity due to the preferential axial attack of the thiyl radical to the less substituted C2 position of the pyranose ring and the preferred axial attack on the C1 radical in the hydrogen abstraction step (Scheme 10).34
The glycal configuration (88 vs. 91) had no effect on the stereochemical outcome of the reaction. However, the efficiency of the additions to 88–90 was highly variable and increased in line with the decreasing electron‐withdrawing capabilities of the 1‐C substituents (CN>COOMe>CONH2).
Remarkably, this approach allows the stereoselective synthesis of the challenging β‐mannosidic motif (97), and get an access to valuable glycomimetics such as 99 which is an S‐linked analogue of the natural α‐(2–6)‐linked sialyl‐galactoside structure.27
Photoinduced Hydrothiolation of exo‐Glycals
Our group and Somsák's group investigated the thiol‐ene reactions of hexo‐ and pentopyranosyl exo‐glycals as a stereoselective method for the synthesis of glycosylmethyl sulfide type glycomimetics (Schemes 11and 12)27, 31, 32, 33. Addition of thiols onto d‐gluco (100) and galacto (104) configured exo‐glycals furnished the β‐C‐S‐bonded glycosides (101–103, and 105) with exclusive regio‐ and stereoselectivity (Scheme 11).31, 32, 33 In most cases the reaction went to completion within 15 min at room temperature (rt), demonstrating the high reactivity of exo‐glycals in the thiol‐ene reactions. Addition of various thiols onto the d‐manno‐configured exo‐glycal 106 proceeded with the same exclusive stereoselectivity to provide C‐β‐d‐mannopyranosyl derivatives (e.g. 107, 108) as the sole products.47 The exclusive β‐selectivity in the ‘manno’ series is of particular significance because synthesis of β‐d‐manno configured C‐glycosides with high selectivity is notoriously difficult.48
Scheme 11.
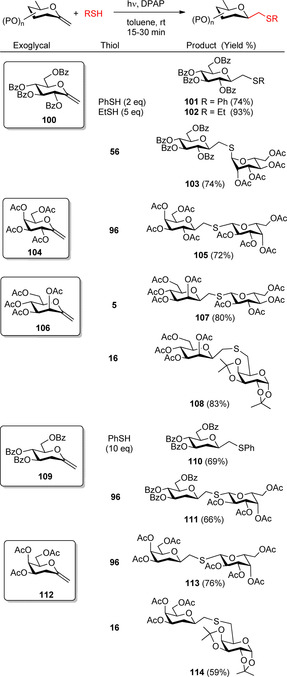
Thio‐click reaction of hexopyranosyl exo‐glycals including 2‐deoxy‐exo‐glycals—Synthesis of C‐S‐bridged disaccharides with full β‐C‐selectivity.
Scheme 12.
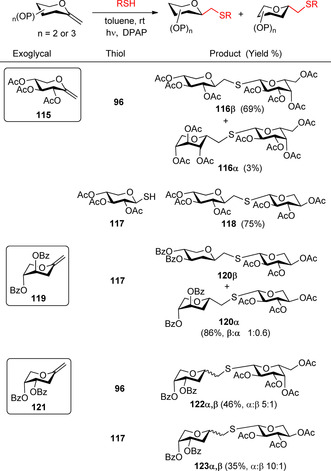
Hydrothiolation of pentopyranosyl exo‐glycals including 2‐deoxy‐exo‐glycals.
The thiol‐ene reactions of hexopyranosyl 2‐deoxy‐exo‐glycals 109 and 112 also proceeded with exclusive regio‐ and stereoselectivity providing the corresponding C‐β‐d‐glycopyranosyl derivatives 110, 111, 113 and 114 with good yields (Scheme 11). The results highlight that the radical‐mediated approach enables stereoselective construction of novel types of glycomimetics in the 2‐deoxy‐glycopyranosyl series49 of high biological relevance.
The exclusive β‐stereoselectivity of the reaction in the hexose series can be explained by the exquisite stability of the radical intermediate in the 4C1 conformation and by the preferred axial attack on this radical in the hydrogen abstraction step.
Hydrothiolation reactions of pentopyranosyl exo‐glycals took place with complete regioselectivity (Scheme 12). However, similarly to endo‐glycals, the total stereoselectivity of the additions ceased in the pentose series, and, in contrast to the hexose series, the presence and absence of the C2‐acyloxy group greatly influenced on both the efficacy and the stereochemical outcome of the reactions. Additions to pentopyranosyl exo‐glycals bearing C2‐acyloxy group (e.g.115) proceeded with high to complete stereoselectivity, providing the C‐β‐glycosides with good yields. In the case of 2‐deoxy‐exo‐glycals the β‐selectivity of the reaction drastically decreased, or even turned to α‐selectivity as shown in the reactions of 119 and 121. The strikingly different stereochemical results observed can be explained by the different conformational preferences of the intermediate glycosyl radicals, which is governed by the number and spatial arrangement of the substituents of glycals. In the case of 115, the reaction predominantly (or exclusively) occurs through the 4C1 conformational form, in the case of the 2‐deoxy glycal 119 the 1C4 and 4C1 forms of the glycosyl radical have comparable stabilities, whereas upon addition reaction of 121 the glycosyl radical dominantly exists in its 1C4 form. In these cases, the effect of temperature has not been studied in detail.
Photoinduced Hydrothiolation of Enoses other than Glycals
Extension of the thiol‐ene reaction to unsaturated sugars with endo‐ or exo‐cyclic double bond at a non‐anomeric position allowed the synthesis of further valuable glycomimetics.26, 31, 50, 51, 52, 53
We demonstrated that hydrothiolation of 2,3‐unsaturated glycosides, which are readily available by Ferrier rearrangement of commercially available glycals, is a useful strategy for introduction of a thio‐linkage to either C2‐ or C3‐position of pyranoses, depending on the substitution of the double bond (Scheme 13).26, 50, 51 Studying the reaction of 124 with functionalized anomeric thiols we have found that the thiyl radical added axially to position C2 of the sugar ring with complete regio‐ and stereoselectivity (e.g. 125).26 The synthetic utility of the method was demonstrated by the simple and efficient synthesis of 127, an α‐(1→2)‐thio‐linked mannobioside mimetic, which was converted to multivalent mannoclusters for lectin‐binding studies.50
Scheme 13.
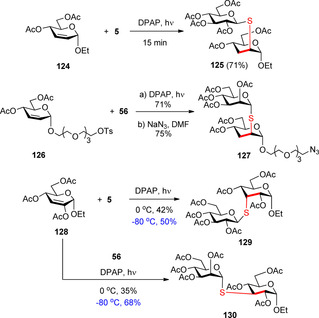
Regio‐ and stereoselective synthesis of 2‐S‐ or 3‐S‐linked disaccharide mimetics by photoinduced hydrothiolation of 2,3‐unsaturated glycosides.
Hydrothiolation of 2,3‐unsaturated glycoside 128 demonstrated that the regioselectivity of the reaction can be changed by substitution of position C2.51 Surprisingly, whereas addition of 5 led to the formation of the allo‐configured thiodisaccharide 129, reaction with 56 provided the gluco‐configured product 130. The different stereoselectivity observed with the different thiosugars can be explained by the steric congestion of 129 leading to different fitting to thiols of different configurations. This phenomenon of double stereodifferentiation, is well‐known in the field of chemical glycosylation.54
The reaction was extended to furanose exomethylene derivatives including glucofuranose 131 and nucleoside exomethylenes 133–135 (Scheme 14).31, 52, 53 Addition of various thiols onto 3‐exo‐methylene‐glucofuranose derivative 131 proceeded with high yield and complete diastereoselectivity to give the d‐allofuranosyl glycomimetics, such as 132, as the sole products.31
Scheme 14.
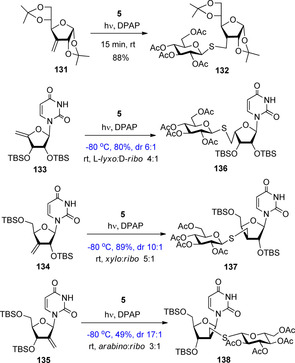
Hydrothiolation of furanosyl exomethylene derivatives (dr: diastereomeric ratio).
Upon hydrothiolation of nucleoside exo‐methylenes under the standard conditions at room temperature moderate yields and low level of stereoselectivity were observed. Cooling the reaction mixture was again advantageous on both the conversion and the stereoselectivity. This approach can be exploited for rapid synthesis of nucleoside analogues with non‐natural l‐lyxo (136), d‐xylo (137) and d‐arabino (138) configured sugar units.52, 53
Summary and Outlook
Development of efficient strategies for the stereoselective synthesis of stable analogues of biorelevant glycans and glycoconjugates for biomedical applications is an important task for organic chemists.
The results of our group and Somsák's group summarized in this Concept article demonstrate that photoinitiated thiol‐ene reactions of enoses represent a powerful tool for stereoselective synthesis of a broad range of stable glycomimetics including S‐glycosides and new types of C‐glycoside analogues.
The method can be applied on various unsaturated sugars such as pyranosyl and furanosyl structures in both the pentose and hexose series possessing endo‐ or exo‐cyclic double bonds. Our study revealed, that the reaction temperature profoundly influences the reaction efficacy in the hexose series and both the efficacy and stereoselectivity in the pentose series. The discovery and application of the unique promoting effect of cooling led to efficient conversions and complete or sufficient stereoselectivity independently of the glycal identity, allowing universal applicability of the method.
As radical reactions, contrary to ionic ones, are not sensitive to directing effects of neighbouring groups, the radical‐mediated thiol‐ene reactions can be utilized for efficient synthesis of 1,2‐cis‐α‐, 2‐deoxy‐β as well as β‐manno‐linked carbohydrate mimetics, structures which are very difficult to prepare by the classical ionic methods. This characteristic makes the thiol‐ene approach an excellent complementary strategy to the existing S‐ and C‐glycosylation methods.
In a recent Review on radical reactions Studer and Curran described: „The challenge in synthesis today is not so much discovering new elementary reactions of organic radicals; it is instead discovering how to conduct these reactions efficiently and selectively.“55 We have met this challenge in the centenary‐old thiol‐ene reaction by employing it as a novel S‐glycosylation method and by conducting the reaction at low temperature which led to excellent yields and high to complete stereoselectivity in most cases.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
The author thanks Dr. Pál Herczegh for the discussion. This work was supported by the EU and co‐financed by the European Regional Development Fund under the project GINOP‐2.3.2‐15‐2016‐00008. This research was also funded by the National Research, Development and Innovation Office of Hungary (K132870).
A. Borbás, Chem. Eur. J. 2020, 26, 6090.
References
- 1. Ernst B., Magnani J. L., Nat. Rev. Drug Discovery 2009, 8, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cipolla L., Araújo A. C., Bini D., Gabrielli L., Russo L., Shaikh N., Expert Opin. Drug Discovery 2010, 5, 721–737. [DOI] [PubMed] [Google Scholar]
- 3.S. Sattin, A. Bernardi, in Carbohydrate Chemistry, Vol. 41 (Eds.: A. P. Rauter, T. K. Lindhorst, Y. Queneau), RSC Publishing, Cambridge, 2015, pp. 1–25.
- 4. Driguez H., Topics Curr. Chem. 1997, 187, 85–116. [Google Scholar]
- 5. Witczak Z. J., Curr Med Chem 1999, 6, 165–178. [PubMed] [Google Scholar]
- 6. Driguez H., ChemBioChem 2001, 2, 311–318. [DOI] [PubMed] [Google Scholar]
- 7. Pachamuthu K., Schmidt R. R., Chem. Rev. 2006, 106, 160–187. [DOI] [PubMed] [Google Scholar]
- 8. Szilágyi L., Varela O., Current Org. Chem. 2006, 10, 1745–1770. [Google Scholar]
- 9. Andrews J. S., Pinto B. M., Carbohydr. Res. 1995, 270, 51–62. [DOI] [PubMed] [Google Scholar]
- 10. Blanc-Muesser M., Defaye J., Driguez H., Carbohydr. Res. 1978, 67, 305–328. [Google Scholar]
- 11. Rye C. S., Withers S. G., Carbohydr. Res. 2004, 339, 699–703. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Witczak Z. J., Lorchak D., Nguyen N., Carbohydr. Res. 2007, 342, 1929–1933; [DOI] [PubMed] [Google Scholar]
- 12b. Dada L., Manzano V. E., Varela O., Org. Lett. 2018, 20, 6225–6228. [DOI] [PubMed] [Google Scholar]
- 13. Manzano V. E., Uhrig M. L., Varela O., J. Org. Chem. 2008, 73, 7224–7235. [DOI] [PubMed] [Google Scholar]
- 14. Repetto E., Manzano V. E., Uhrig M. L., Varela O., J. Org. Chem. 2012, 77, 253–265. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Zhu X., Schmidt R. R., Angew. Chem. Int. Ed. 2009, 48, 1900–1934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1932–1967; [Google Scholar]
- 15b. Adero P. O., Amarasekara H., Wen P., Bohé L., Crich D., Chem. Rev. 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Caraballo R., Deng L., Amorim L., Brinck T., Ramström O., J. Org. Chem. 2010, 75, 6115–6121; [DOI] [PubMed] [Google Scholar]
- 16b. Deng L., Wang X., Uppalapati S., Norberg O., Dong H., Joliton A., Yan M., Ramström O., Pure Appl. Chem. 2013, 85, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Dondoni A., Angew. Chem. Int. Ed. 2008, 47, 8995–8997; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9133–9135; [Google Scholar]
- 17b. Dondoni A., Marra A., Chem. Soc. Rev. 2012, 41, 573–586; [DOI] [PubMed] [Google Scholar]
- 17c. Floyd N., Vijayakrishnan B., Koeppe J. R., Davis B. G., Angew. Chem. Int. Ed. 2009, 48, 7798–7802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7938–7942. [Google Scholar]
- 18.
- 18a. Witczak Z. J., Bielski R., Click chemistry in glycoscience: New developments and strategies. New York, NY, USA: Wiley, 2013; [Google Scholar]
- 18b. Witczak Z. J., Phosphorus, Sulfur Silicon Rel. Elem. 2013, 188, 413–417; [Google Scholar]
- 18c. McSweeney L., Dénès F., Scanlan E. M., Eur. J. Org. Chem. 2016, 2080–2095. [Google Scholar]
- 19.
- 19a. Lo Conte M., Staderini S., Marra A., Sanchez-Navarro M., Davis B. G., Dondoni A., Chem. Commun. 2011, 47, 11086–11088; [DOI] [PubMed] [Google Scholar]
- 19b. Semsarilar M., Ladmiral V., Perrier S., Macromolecules 2010, 43, 1438; [Google Scholar]
- 19c. Norberg O., Lee I. H., Aastrup T., Yan M., Ramström O., Biosens. Bioelectron. 2012, 34, 51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Csávás M., Herczeg M., Borbás A., Synlett 2013, 24, 719–722. [Google Scholar]
- 20.
- 20a. Horton D., Methods Carbohydr. Chem. 1963, 2, 433–437; [Google Scholar]
- 20b. Matta K. L., Girotra R. N., Barlow J. J., Carbohydr. Res. 1975, 43, 101–109. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Dere R. T., Wang Y., Zhu X., Org. Biomol. Chem. 2008, 6, 2061–2063; [DOI] [PubMed] [Google Scholar]
- 21b. Zhu X., Dere R. T., Jiang J., Zhang L., Wang X., J. Org. Chem. 2011, 76, 10187–10197; [DOI] [PubMed] [Google Scholar]
- 21c. Dere R. T., Kumar A., Kumar V., Zhu X., Schmidt R. R., J. Org. Chem. 2011, 76, 7539–7545; [DOI] [PubMed] [Google Scholar]
- 21d. Tardieu D., Céspedes Dávila M. F., Hazelard D., Compain P., Synthesis 2018, 50, 3927–3930. [Google Scholar]
- 22. Doyle L. M., O'Sullivan S., Di Salvo C., McKinney M., McArdle P., Murphy P. V., Org. Lett. 2017, 19, 5802–5805. [DOI] [PubMed] [Google Scholar]
- 23. Posner T., Ber. Dtsch. Chem. Ges. 1905, 38, 646–657. [Google Scholar]
- 24.
- 24a. Kharasch M. S., Read A. T., Mayo F. R., Chem. Ind. 1938, 57, 752; [Google Scholar]
- 24b. Griesbaum K., Angew. Chem. Int. Ed. Engl. 1970, 9, 273–287; [Google Scholar]; Angew. Chem. 1970, 82, 276–290. [Google Scholar]
- 25. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 26. Lázár L., Csávás M., Herczeg M., Herczegh P., Borbás A., Org. Lett. 2012, 14, 4650–4653. [DOI] [PubMed] [Google Scholar]
- 27. Lázár L., Csávás M., Hadházi Á., Herczeg M., Tóth M., Somsák L., Barna T., Herczegh P., Borbás A., Org. Biomol. Chem. 2013, 11, 5339–5350. [DOI] [PubMed] [Google Scholar]
- 28. Eszenyi D., Kelemen V., Balogh F., Bege M., Csávás M., Herczegh P., Borbás A., Chem. Eur. J. 2018, 24, 4532–4536. [DOI] [PubMed] [Google Scholar]
- 29. Eszenyi D., Lázár L., Borbás A., McCourt R. in Carbohydrate Chemistry: Proven Synthetic Methods, Vol. 4 (Eds.: P. Kováč, C. Vogel, P. Murphy), CRC Press, Weinheim, 2017, pp. 33–44. [Google Scholar]
- 30. Kelemen V., Bege M., Eszenyi D., Debreczeni N., Bényei A., Stürzer T., Herczegh P., Borbás A., Chem. Eur. J. 2019, 25, 14477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lázár L., Csávás M., Tóth M., Somsák L., Borbás A., Chem. Pap. 2015, 69, 889–895. [Google Scholar]
- 32. József J., Juhász L., Illyés T. Z., Csávás M., Borbás A., Somsák L., Carbohydr. Res. 2015, 413, 63–69. [DOI] [PubMed] [Google Scholar]
- 33. József J., Juhász L., Somsák L., New J. Chem. 2019, 43, 5670–5686. [Google Scholar]
- 34. Lázár L., Juhász L., Batta G., Borbás A., Somsák L., New J. Chem. 2017, 41, 1284–1292. [Google Scholar]
- 35. Araki Y., Matsuura K., Ishido Y., Kushida K., Chem. Lett. 1973, 2, 383–386. [Google Scholar]
- 36.
- 36a. Killops K. L., Campos L. M., Hawker C. J., J. Am. Chem. Soc. 2008, 130, 5062–5064; [DOI] [PubMed] [Google Scholar]
- 36b. Cramer N. B., Reddy S. K., Cole M., Hoyle C., Bowman C. N., J. Polym. Sci. Part A 2004, 42, 5817–5825; [Google Scholar]
- 36c. Hoyle C. E., Bowman C. N., Angew. Chem. Int. Ed. 2010, 49, 1540–1573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1584–1617. [Google Scholar]
- 37. Fiore M., Marra A., Dondoni A., J. Org. Chem. 2009, 74, 4422–4425. [DOI] [PubMed] [Google Scholar]
- 38.Thorough literature survey revealed that Bantchev and co-workers have reported that efficacy of UV-initiated addition of butanethiol to corn oil double bonds could be increased by cooling and the highest conversion of double bonds into sulfides was realized when the reaction mixture was irradiated at −78 °C. As it has been explained by the authors, at higher temperatures the dissociation of the thiyl double bond adduct radical was entropically favored. This shifted the equilibrium toward reactants before the carbon-centered radical can be trapped through butanethiol hydrogen abstraction, thus reducing the overall conversion. See in: Bantchev G. B., Kenar J. A., Biresaw G., Han M. G., J. Agric. Food Chem. 2009, 57, 1282–1290.19166316 [Google Scholar]
- 39. Lázár L., Borbás A., Somsák L., Carbohydr. Res. 2018, 470, 8–12. [DOI] [PubMed] [Google Scholar]
- 40. Dénès F., Pichowicz M., Povie G., Renaud P., Chem. Rev. 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. LeBel N. A., DeBoer A., J. Am. Chem. Soc. 1967, 89, 2784–2785; [Google Scholar]
- 41b. LeBel N. A., Czaja R. F., DeBoer A., J. Org. Chem. 1969, 34, 3112–3126; [Google Scholar]
- 41c. Bordwell F. G., Landis P. S., Whitney G. S., J. Org. Chem. 1965, 30, 3764–3768; [Google Scholar]
- 41d. Goering H. L., Relyea D. I., Larsen D. W., J. Am. Chem. Soc. 1956, 78, 348–353. [Google Scholar]
- 42. Igarashi K., Honma T., J. Org. Chem. 1970, 35, 606–609. [Google Scholar]
- 43. Northrop B. H., Coffey R. N., J. Am. Chem. Soc. 2012, 134, 13804–13817. [DOI] [PubMed] [Google Scholar]
- 44. Horton D., Durette P. L., J. Org. Chem. 1971, 36, 2658–2669. [Google Scholar]
- 45. Staderini S., Chambery A., Marra A., Dondoni A., Tetrahedron Lett. 2012, 53, 702–704. [Google Scholar]
- 46.
- 46a. Giese B., Dupuis J., Angew. Chem. Int. Ed. Engl. 1983, 22, 622–623; [Google Scholar]; Angew. Chem. 1983, 95, 633–634; [Google Scholar]
- 46b. Dupuis J., Giese B., Rüegge D., Fischer H., Korth H.-G., Sustmann R., Angew. Chem. Int. Ed. Engl. 1984, 23, 896–898; [Google Scholar]; Angew. Chem. 1984, 96, 887–888; [Google Scholar]
- 46c. Korth H.-G., Sustmann R., Dupuis J., Giese B., J. Chem. Soc. Perkin Trans. 2 1986, 1453–1459. [Google Scholar]
- 47.Unpublished results (L. Juhász, J. József, D. Eszenyi, N. Debreczeni, A. Borbás, L. Somsák, Eurocarb 18, Moscow, Russia, Aug 2–6, 2015).
- 48.
- 48a. Yang Y., Yu B., Chem. Rev. 2017, 117, 12281–12356; [DOI] [PubMed] [Google Scholar]
- 48b. Hashimoto Y., Tanikawa S., Saito R., Sasaki K., J. Am. Chem. Soc. 2016, 138, 14840–14843; [DOI] [PubMed] [Google Scholar]
- 48c. Crich D., Sharma I., Org. Lett. 2008, 10, 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bennett C. S., Galan M. C., Chem. Rev. 2018, 118, 7931–7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.
- 50a. Csávás M., Demeter T., Herczeg M., Timári I., Kövér K. E., Herczegh P., Borbás A., Tetrahedron Lett. 2014, 55, 6983–6986; [Google Scholar]
- 50b. Csávás M., Malinovská L., Perret F., Gyurkó M., Illyés Z. T., Wimmerová M., Borbás A., Carbohydr. Res. 2017, 437, 1–8. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a.V. Kelemen, M. Csávás, J. Hotzi, M. Herczeg, P. Singh, B. Rathi, P. Herczegh, N. Jain, A. Borbás, Chem. Asian J. doi.org/10.1002/asia.201901560. [DOI] [PMC free article] [PubMed]
- 52. Bege M., Bereczki I., Herczeg M., Kicsák M., Eszenyi D., Herczegh P., Borbás A., Org. Biomol. Chem. 2017, 15, 9226–9233. [DOI] [PubMed] [Google Scholar]
- 53. Bege M., Kiss A., Kicsák M., Bereczki I., Baksa V., Király G., Szemán-Nagy G., Szigeti Z., Herczegh P., Borbás A., Molecules 2019, 24, 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.
- 54a. Masamune S., Choy W., Petersen J. S., Sita L. R., Angew. Chem. Int. Ed. Engl. 1985, 24, 1–30; [Google Scholar]; Angew. Chem. 1985, 97, 1–31; [Google Scholar]
- 54b. Spijker N. M., van Boeckel A. A., Angew. Chem. Int. Ed. Engl. 1991, 30, 180–183; [Google Scholar]; Angew. Chem. 1991, 103, 179–182; [Google Scholar]
- 54c. Fraser-Reid B., López J. C., Gomez A. M., Uriel C., Eur. J. Org. Chem. 2004, 7, 1387–1395; [Google Scholar]
- 54d. Guillemineau M., Auzanneau F.-I., Carbohydr. Res. 2012, 357, 132–138. [DOI] [PubMed] [Google Scholar]
- 55. Studer A., Curran D. P., Angew. Chem. Int. Ed. 2016, 55, 58–102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 58–106. [Google Scholar]