

Abstract
T cells are crucial for the success of immune‐based cancer therapy. Reinvigorating antitumor T cell activity by blocking checkpoint inhibitory receptors has provided clinical benefits for many cancer patients. However, the efficacy of these treatments varies in cancer patients and the mechanisms underlying these diverse responses remain elusive. The density and status of tumor‐infiltrating T cells have been shown to positively correlate with patient response to checkpoint blockades. Therefore, further understanding of the heterogeneity, clonal expansion, migration, and effector functions of tumor‐infiltrating T cells will provide fundamental insights into antitumor immune responses. To this end, recent advances in single‐cell RNA sequencing technology have enabled profound and extensive characterization of intratumoral immune cells and have improved our understanding of their dynamic relationships. Here, we summarize recent progress in single‐cell RNA sequencing technology and current strategies to uncover heterogeneous tumor‐infiltrating T cell subsets. In particular, we discuss how the coupling of deep transcriptome information with T cell receptor (TCR)‐based lineage tracing has furthered our understanding of intratumoral T cell populations. We also discuss the functional implications of various T cell subsets in tumors and highlight the identification of novel T cell markers with therapeutic or prognostic potential.
Keywords: cancer microenvironment, exhausted T cells, memory T cells, regulatory T cells, single‐cell RNA sequencing, TCR‐based lineage tracing, tumor‐infiltrating T cells
Review on single‐cell RNA sequencing, coupled with TCR‐based lineage tracing analysis, with implications for biomarker identification and next‐generation cancer immunotherapies.
Abbreviations
- CPI
checkpoint inhibitor
- CyTOF
cytometry by time‐of‐flight
- MHC
major histocompatibility complex
- MSI
microsatellite‐instable
- NSCLC
non‐small‐cell lung cancer
- PBMC
peripheral blood mononuclear cells
- PD‐1
programmed cell death protein 1
- scRNA‐seq
single‐cell RNA sequencing
- STARTRAC
single T cell analysis by RNA sequencing and TCR tracking
- TME
tumor microenvironment
- Tem
effector memory T cell
- Texh
exhausted T cell
- Tmem
memory T cell
- Treg
regulatory T cell
- Trm
residential memory T cell
- UC
ulcerative colitis
1. INTRODUCTION
T cells play key roles in immune defense against tumor development and metastasis. Cytotoxic CD8+ T cells, CD4+ T helper (Th) cells (especially Th1 cells), and regulatory T cells (Treg) cells orchestrate antitumor T cell responses to fight malignancies in coordination with the rest of immune system. Harnessing antitumor T cell responses to fight malignancies has been the major focus of cancer immunotherapy. The essential antitumor effects of T cells are seen in the fraction of patients who develop long‐lasting complete responses after treatment with checkpoint inhibitors (CPIs) targeting CTLA4 and programmed cell death protein 1 (PD‐1). 1 , 2 , 3 However, response rates to CPIs are not uniform, and a substantial fraction of cancer patients do not respond at all. 1 , 2 Better understanding of tumor‐infiltrating T cell populations and their differential responses to CPIs will be fundamental to the development of novel therapeutic strategies to enhance and broaden T cell‐mediated antitumor immune responses.
Transcriptome‐scale analysis of tumor samples is a powerful tool to reveal the molecular pathways and cellular composition of cancers. Bulk RNA sequencing of tumors can reveal whether a given tumor type or a specific cancer sample contains a high degree of T cell infiltration, and whether these T cells express higher effector or exhaustion markers. 4 However, this type of approach is not sensitive enough to fully elucidate the factors underlying the success or failure of CPI treatments. The tumor microenvironment (TME) includes multiple heterogeneous T cell types, among many other immune and non‐immune cells, so novel multiplex and high throughput technologies are necessary to better dissect its cellular and molecular compositions. Here, we describe the recent development of single‐cell RNA sequencing (scRNA‐seq) technologies, which has become a key tool in efforts to unravel the complexities of the TME, and discuss how its applications are deepening our understanding of tumor‐infiltrating T cell populations in various human cancers.
2. SINGLE‐CELL SEQUENCING TECHNOLOGIES
Single‐cell resolution of cellular diversities and trajectories is critical to our understanding of immune responses to different pathogens or antigens. 5 Over time, major technologies, including flow cytometry, in situ histological assays, and microscopy, have been developed and broadly used to categorize immune cell subsets, as well as characterize their functional phenotypes and spatial distributions. 6 , 7 , 8 , 9 Although these technologies provide invaluable insights when analyzing limited samples with a few prior selected markers, they are not suitable for dissecting heterogenous cell population in tissue and tumor in a comprehensive and unbiased manner. More recently, mass cytometry (also known as Cytometry by Time‐Of‐Flight, CyTOF) was developed to simultaneously detect more than 40 protein markers in millions of individual cells. 10 , 11 CyTOF has been instrumental to our understanding of TME complexity. 12 For example, three recent studies employed CyTOF to study immune cells from patients with non‐small cell lung cancer (NSCLC), renal cancer, and breast cancer, revealing diverse lymphoid and myeloid cell populations and linking specific immune signatures with clinical features (Table 1). 13 , 14 , 15 Another recent study showed that mass cytometry can be used in combination with MHC‐tetramers to analyze antigen (Ag)‐specific T cells, elucidating phenotypic differences between tumor‐ and viral‐specific CD8+ T cells. 16 However, like the traditional technologies, CyTOF also has notable limitations: it requires prior knowledge to select markers, there is a lack of high‐quality reagents for certain markers, and it is relatively low‐throughput compared to genome‐wide analyses. Despite these limitations, CyTOF and emerging technologies like imaging mass cytometry 17 , 18 , 19 can rapidly provide essential information on protein expression in the context of anatomical location. Data from these approaches will continue to be widely used to confirm cellular subsets, delineate cell‐cell interactions and spatial relationships, and explore clinical biomarkers.
TABLE 1.
Summary of single cell studies of human tumor‐infiltrating immune cells/T cells
| Tissues | Single‐cell technologies | Platforms | Targeting cells | References |
|---|---|---|---|---|
| Melanoma | scRNA‐seq | Smart‐seq2 | Pan‐immune and non‐immune cells | 79 |
| Renal cancer | Mass cytometry | CyTOF | Innate immune cells | 14 |
| Breast cancer | scRNA‐seq | Smart‐seq2 | Pan‐immune and non‐immune cells | 127 |
| Lung cancer | Mass cytometry | CyTOF | Innate immune cells | 13 |
| Head and neck cancer | scRNA‐seq | Smart‐seq2 | Pan‐immune and non‐immune cells | 130 |
| Liver cancer | scRNA‐seq | Smart‐seq2 | T cells | 59 |
| Breast cancer | scRNA‐seq | InDrop & 10x Genomics | Pan‐immune cells | 43 |
| Lung cancer | scRNA‐seq | Smart‐seq2 | T cells | 60 |
| Renal cancer | scRNA‐seq | 10x Genomics | Pan‐immune and non‐immune cells | 45 |
| Lung cancer Colorectal cancer | Mass cytometry | CyTOF | CD8+ T cells | 12 |
| Breast cancer | scRNA‐seq | 10x Genomics | Pan‐immune cells | 128 |
| Lung cancer | scRNA‐seq | 10x Genomics | Pan‐immune and stroma cells | 44 |
| Colorectal cancer | scRNA‐seq | Smart‐seq2 | T cells | 61 |
| Melanoma | scRNA‐seq | MARS‐seq | T cells | 102 |
|
Melanoma Therapy |
scRNA‐seq | Smart‐seq2 | Pan‐immune and non‐immune cells | 169 |
|
Melanoma Therapy |
scRNA‐seq | Smart‐seq2 | Pan‐immune cells | 81 |
| Breast cancer | Mass cytometry | CyTOF | Pan‐immune and non‐immune cells | 15 |
Abbreviations: CyTOF, cytometry by time‐of‐flight.
2.1. The advantages and limitations of scRNA‐seq approaches
In 2009, scRNA‐seq was established to obtain unbiased appreciation of the whole‐transcriptome from a single mouse blastomere. 20 Since then, the technology has been used to probe cellular populations as varied as differentiating embryonic cells, intracranial neurons, malignant tumor cells, and individual immune cells. 9 , 21 , 22 , 23 , 24 , 25 , 26 All the while, the sensitivity, scale, and accuracy of scRNA‐seq has expanded and improved exponentially. Today, there are many single‐cell sequencing protocols available to researchers, each offering distinct advantages and disadvantages. Plate‐based approaches can profile hundreds or thousands of single cells using full‐length sequencing protocols (e.g., STRT‐seq, Smart‐seq, and Smart‐seq2), 27 , 28 , 29 or pooled 3′ end sequencing approaches (e.g., CEL‐seq and MARS‐seq). 30 , 31 , 32 Droplet‐based protocols (e.g., Drop‐seq, InDrop and 10× Chromium Genomics) 33 , 34 , 35 and other massively parallel approaches (e.g., Seq‐well, sci‐RNA‐seq, and SPLiT‐seq) have also been developed to increase throughput. 36 , 37 , 38 Comprehensive comparisons of these approaches have been reviewed elsewhere and thus will not be the focus here. 25 , 39 , 40
As scRNA‐seq technologies advance, we can expect that such technologies will be widely used to uncover key processes and critical pathways in different immune cells under steady state or disease conditions. Thus, it is paramount to select an appropriate single‐cell protocol for each individual study. The choice depends on the specific biological questions being addressed, and is influenced by several factors, including the depth of gene information needed, the number of cells to profile, and cost. 4 , 41 Protocols that sequence full‐length transcripts capture more comprehensive transcriptomes, including highly variable genes like TCRs. Such approaches facilitate the in‐depth functional interpretation of particular cell types (e.g., malignant cells and tumor‐infiltrating T cells), alternative splicing dynamics, and somatic mutation patterns. 42 In contrast, massively parallel 3′ sequencing protocols reduce costs and raise throughput, albeit at the expense of sequencing depth. These approaches facilitate broad surveys of cellular components from complex tissues. 43 , 44 , 45 The combined utility of these platforms will allow for a wider variety of biological questions to be probed by this technology.
Despite the advances of scRNA‐seq technologies, substantial limitations and challenges remain. One molecular limitation is the lack of unbiased identification of noncoding RNAs. This is due to the nature of the sequencing strategy, which specifically targets polyadenylated mRNA transcripts. Another limitation is that scRNA‐seq only provides a snapshot of transcriptomic information, while the genomic, epigenetic, and proteomic components of the cells are not captured. This hinders comprehensive understanding of the molecular mechanisms of cellular processes. Additionally, compared to single‐cell DNA genomics, scRNA‐seq exhibits limited sensitivity and is, therefore insufficient to reconstruct the clonal evolution of tumor cells in the TME. This limitation has only been somewhat overcome by applying gene expression‐based estimation of genomic alterations to define malignant cells. 22 , 26 Finally, because of the tissue sampling and disassociation processes, scRNA‐seq cannot yet map expression data to a precise anatomical location or cytoarchitecture. However, emerging spatially resolved transcriptomic methods can link cellular localization to molecular typing in neuronal tissues, and we expect these methods to be applied to immunological settings. 46 Thus, scRNA‐seq technology is beginning to overcome initial limitations, and future advancements may allow us to concurrently examine genomic, epigenomic, proteomic, and spatial information from single cells.
2.2. Lineage tracing by scRNA‐seq
Identifying the lineages and relationships between cell types will provide detailed insights into tissue development and homeostasis, as well as how the dysregulation of these pathways contributes to pathologies like cancer. Lineage reconstruction with scRNA‐seq data has its origins in embryonic development research, where scRNA‐seq has been used to infer the trajectories based on pseudotemporal ordering of sequenced cells according to their similarity in gene expression. 47 Monocle and related algorithms have since been applied to reveal the relationships of different immune cells and their progenitors during hematopoiesis, infection, and tumorigenesis. 8 , 47 , 48 Although these trajectory inferences can connect developmental pathways, the biological interpretation of these data is limited by the need for prior knowledge and the assumption that pseudotemporal ordering is largely based on similarity. 49 In some cases, these inferences may reflect the continuum of cellular states, rather than real developmental relationships.
Another way to trace cellular lineages is the analysis of genetic marks or scars. Such molecular identifiers can be induced via techniques such as CRISPR recombination. This methodology has been used to mark embryos and the juvenile brain of zebrafish. 50 , 51 However, due to technical limitations such as low labeling efficiency, this approach is restricted to specific experimental conditions. Another way to use genetic scars as lineage tracing tags is to measure mitochondrial genome mutations from human scRNA‐seq or scATAC‐seq data. 52 This approach consistently traced T cell clonal expansion in multiple sample types, but its ability to delineate dynamic relationships of different T cell subsets has not yet been determined. For the lineage tracing of immune cells in human tumors, a more feasible approach has been to assess the endogenous genetic scars that exist in lymphocytes. For T cell and B cells, germline DNA recombination results in a vast repertoire of gene sequences for TCRs and B cell receptors (BCRs; also known as immunoglobulin). The high diversity of these repertoires makes it unlikely that two unrelated cells would exhibit identical TCR or BCR sequences. Thus, TCR‐ or BCR‐based sequencing could be used to define the clonality and track the dynamic relationships of these lymphocytes.
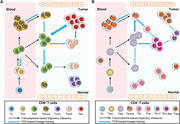
Previous studies based on bulk TCR‐α or TCR‐β sequencing revealed divergent T cell clonality in different tissues, as shown in an analysis of tumor‐infiltrating versus peripheral blood T cells from glioma patients. 53 However, such bulk sequencing methods were not able to capture the underlying phenotypic differences among individual T cell clones. In contrast, the simultaneous detection of TCRs and other transcripts in single cells have started to unmask such differences. One pioneering study used quantitative RT‐PCRs to simultaneously detect TCRs and selected transcripts in single CD4+ T cells to reveal the clonal ancestry and differentiation of these T cells. 54 This approach provided an avenue to link TCR identities and phenotypes, but was limited by its relatively low throughput. More recently, emerging scRNA‐seq technologies began to depict the clonality and developmental trajectories of individual T cells in the steady state, as well as in diseases such as infections and cancers, using integrated transcriptome and TCR analyses. 55 , 56 , 57 , 58 , 59 , 60 These studies greatly advanced our understanding of T cell dynamics in many tissues and disease states. However, further analysis of T cell dynamics across different tissues or subtypes required a more quantitative analytical platform. To this end, our group developed a new framework, called STARTRAC (single T cell analysis by RNA sequencing and TCR tracking). 61 STARTRAC analyses provide additional insights into the properties of T cell subsets, especially in cancer (Fig. 1). For example, previous studies based on bulk TCR‐β sequencing or single‐cell inferred trajectories showed connections between effector, effector memory, and exhausted CD8 T cells in multiple cancer types, indicating a linear developmental differentiation of these T cells. However, our STARTRAC analyses revealed limited connections of effector and exhausted T cells in colorectal cancer (CRC), suggesting an underappreciated effector‐independent development of exhausted T cells. 61 This observation was independently confirmed by a recent study of chronic infection, which showed that the effector versus exhausted fate decision occurred in a precursor subset, and not within the effector lineage. 62 More detailed dissection of exhausted and other T cell lineages by STARTRAC and other single cell‐based analyses will be discussed in the following sections.
FIGURE 1.

STARTRAC analyses define the dynamic status of T cell subsets in tumor. The dynamic status and relationship of CD8+ (A) and CD4+ (B) T cell subsets are inferred based on STARTRAC indices. Number of cells indicates the degree of clonal expansion. Black arrows show the potential developmental trajectory inferred by monocle. Blue arrows indicate the developmental relationship based on TCR sharing. The thickness of the arrows predicts the strength of the relationship
One limitation of the aforementioned approaches is the lack of directionality of the established trajectory. To overcome this obstacle, levels of unspliced precursor mRNA and spliced mRNA from scRNA‐seq data have been used to infer the future state of a cell on a timescale of hours. 63 This framework, termed RNA velocity, has been successfully applied to reconstruct the neural crest lineage, mouse hippocampus development, human embryonic neurogenesis, and even a whole planarian. 64 Since RNA metabolism is a relatively short‐term biological process, whether this approach can be applied to T cell lineage tracing in chronic conditions, such as cancer, remains to be seen.
3. TUMOR‐INFILTRATING T CELL CLUSTERS IDENTIFIED BY SCRNA‐SEQ
It has long been known that various leukocyte subsets, especially T cells, infiltrate human solid tumors. Studies of animal cancer models established that the immune system constantly surveils and eliminates primary transformed tumors, and that tumors evolve to establish an equilibrium and eventual escape from immune surveillance through cancer immunoediting. 65 Animal models with deficient T cell functions were particularly susceptible to tumor outgrowth. Supporting these preclinical observations, it is also known that many human cancers with higher T cell infiltration have better prognostic outcomes. 66 More importantly, T cell infiltration and activation status have been used as predictive markers to select for patients that respond to CPI treatment in many cancer types. 67 Thus, a better understanding of T cell composition and regulatory pathways is key for the improvement of cancer immunotherapies. Previously, characterization of tumor‐infiltrating T cell subsets using immunohistochemistry and multicolor flow cytometry techniques demonstrated the presence of multiple T cell subsets in tumors. However, it was only with the recent advancement of scRNA‐seq technology that researchers have had the opportunity to uncover detailed characteristics of tumor‐infiltrating T cell subsets in an unbiased manner (Table 1). Using this technology, heterogeneous T cell subsets have been identified in various types of human tumors. Whereas the T cell clusters identified in cancer patient blood are largely consistent across different tumor types and are similar to those found in healthy donor peripheral blood mononuclear cells, tumor‐infiltrating T cell clusters defined by scRNA‐seq analysis vary depending on tumor origin, type, location, and disease stage. Nevertheless, several major intratumoral CD4+ and CD8+ T cell populations are identified across different tumors, including exhausted T (Texh) cells, effector memory T (Tem) cells, effector or effector memory re‐expressing CD45RA T (Teff/Temra) cells, tissue‐resident is fine memory T (Trm) cells, and regulatory (Treg) cells. Herein, we will focus our discussion on up‐to‐date key findings of their properties and their role in antitumor immunity.
3.1. Exhausted CD8+ T cells in tumors
3.1.1. Overview
The majority of intratumoral CD8+ T cells are functionally impaired and stably express inhibitory co‐receptors such as PD‐1, Tim‐3, and LAG3. 68 , 69 , 70 Interestingly, these receptors are up‐regulated upon T cell activation and function to repress excessive T cell proliferation and effector functions. Stable expression of these receptors has since come to define a distinct lineage of dysfunctional T cells called exhausted CD8+ T cells (Texh cells). 71 , 72 , 73 The concept of T cell exhaustion originated in the 1960s but detailed examination of the lineage accelerated in the 1990s and 2000s with the implementation of mouse models of chronic infection, especially lymphocytic choriomeningitis virus (LCMV). 74 , 75 These cells are associated with a unique developmental pathway that arises due to repeated antigen exposure during chronic viral infection or cancer. Exhaustion is theorized to be a mechanism of avoiding autoreactivity and immunopathology while limiting tumor growth or chronic infection. Texh cell properties include progressive hyporesponsiveness to TCR or cytokine stimulation, inability to form memory T cell pools, and a reprogrammed metabolic and epigenetic circuitry. 76 Exhaustion is separated both phenotypically and developmentally from 2 other dysfunctional T cell fates, senescence, and anergy, with the primary difference being that Texh cells have previously undergone initial activation. 71 Blockade of PD‐1, perhaps the most well characterized inhibitory co‐receptor, can enhance the function and block the terminal differentiation of these cells to control chronic viral infection. 62 , 77 And in cancer settings, PD‐1 expressing intratumoral T lymphocytes are a predictive biomarker of cancer patients who may benefit from CPI treatments. Many studies have suggested that one of the major mechanisms of anti‐PD‐1 therapies is to revert the dysfunctional phenotypes of tumor‐infiltrating exhausted CD8+ T cells and enable them to better kill and control cancer cells. 67 More recently, single cell‐based transcriptomics has been applied to dissect this surprisingly heterogeneous lineage in even greater detail, with a major goal being to decipher just how CPI treatments manipulate intratumoral CD8+ T cell lineages. Below, we discuss 5 areas of Texh cell biology where single‐cell technologies have been particularly useful in furthering our understanding: molecular markers, properties, heterogeneity, developmental trajectory, and reactivity.
3.1.2. Biomarker genes for Texh cells
A major limitation within Texh cell biology has been the lack of specific molecular identifiers of these cells. The inhibitory receptors traditionally used to identify the Texh subset are broadly induced by TCR signaling on CD8+ T cells, and some are constitutively expressed on NK cells. 78 Similarly, the reported transcription factors governing Texh cells, including NFAT, Eomes, and TCF1, are also shared with other lymphocyte populations. The depth and resolution provided by single‐cell technologies repesent a powerful tool to identify more specific and actionable markers of exhaustion. Indeed, several new candidate genes have come out of such studies. For example, in a study of Texh cells in liver cancer and melanoma, 59 , 79 novel markers such as MYO7A, WARS, CXCL13, TOX, LAYN, PHLDA1, and SNAP47 were identified alongside well‐known Texh genes such as HAVCR2, PDCD1, ENTPD1 (CD39), CTLA4, TIGIT, TNFRSF9, and CD27. Some of these newly identified marker genes were further shown to regulate the function or the development of CD8+ Texh cells. For example, overexpression of LAYN inhibited effector functions of CD8+ T cells in in vitro systems. 59 CD39, which had been initially identified as a specific Texh cell marker during chronic infection 80 was found to mark Texh cells in several cancers. CD39 has since become a relevant cancer immunotherapeutic candidate. 16 , 81 , 82 Finally, beyond transcript‐level markers of exhaustion, it has been argued that an epigenetic signature may be the most robust marker of exhaustion. 83 , 84 It has also been theorized that the epigenetic state of exhaustion would hinder any attempt to invigorate Texh cells with lasting effect and therefore, reversing Texh epigenetics would be key to any therapy targeting these cells. To that end, TOX, which is preferentially expressed in Texh cells compared to other intratumoral subsets, was recently identified as a key mediator of chromatin remodeling and transcription in Texh cells. 85 , 86 , 87 , 88 , 89 TOX was shown to enhance Texh cell survival and inhibitory receptor expression, although it does not alter the effector functions of Texh cells. TOX is already known to play an important role in the development of several homeostatic leukocyte populations, but it is an intriguing exhaustion marker in cancer context given its pleiotropic effects within tumor Texh cells. 90
3.1.3. In vivo phenotypes of Texh cells
In addition to overexpressing various inhibitory cell surface markers, tumor‐infiltrating CD8+ Texh cells have been defined by their inferior ability to respond to antigen stimulation, resulting in reduced effector functions, such as cytokine secretion, and lower proliferative potential. Most of these phenotypes were observed from ex vivo analysis of TILs isolated from human and mouse tumors. 91 , 92 , 93 Texh cells are not completely inert but maintain a suboptimal functionality that limits viral replication or tumor progression. 73 , 94 , 95 Most importantly, blocking CPI pathways such as PD‐1 and Tim3 can invigorate these cells to control viral infections or eradicate tumors. However, the exact status of these cells inside tumors remained elusive until the recent emergence of scRNA‐seq. Based on TCR clonality analyses, such as STARTRAC, and combined transcriptome analyses, a surprising finding was that Texh cells were the most clonally expanded and proliferative (KI67high) T cell subset in multiple cancer types. In addition, these cells highly expressed various effector molecules such as IFN‐γ and Granzyme B, despite their expression of inhibitory receptors. These data suggest that tumor‐infiltrating CD8+ Texh cells are genuinely and constantly activated, probably through TCR engagement with tumor‐associated antigens presented by class I MHC. This would imply that their “exhaustion” state may in fact be normal T cell signaling in response to continuous antigen exposure within the highly immunosuppressive TME, providing a rationale for how CPI treatments reduce inhibitory signaling enough to reinvigorate Texh cells. 94 , 95 , 96 This premise is consistent with the finding that PD‐1+ intratumoral CD8+ T cells are the predominant tumor reactive CD8+ T cell clones. 97 Relatedly, STARTRAC analysis showed that Texh cells were less mobile compared to other T cell subsets, especially Teff cells, and that these cells highly express some cell surface markers associated with Trm cells, such as ITGAE. 61 This implies a closer relationship of the Texh lineage to memory T cell subsets than was previously appreciated.
3.1.4. Heterogeneity of tumor‐infiltrating Texh cells
Several studies have shown that Texh cells can be further subdivided into distinct populations: early Texh cells and terminal Texh cells. 94 , 98 , 99 , 100 Early Texh cells possess a stem‐like and memory‐like phenotype, express lower levels of effector transcripts, and possess a proliferative capacity that can seed the terminal Texh cell population independent of other CD8 subsets. By contrast, terminal Texh cells do not exhibit multipotency or memory‐like abilities, express high levels of effector transcripts, and are less proliferative. Interestingly, TOX expression and function provide a link between the Texh subsets. 85 , 86 , 87 , 88 , 89 In preclinical models and patient tumor biopsies, TOX expression is up‐regulated in early Texh cells, where it establishes epigenetic signatures required for cell persistence. TOX expression is sustained in terminal Texh cells, and genetic deletion of TOX in T cells results in reduced overall numbers of Texh cells. Therefore, early and terminal Texh cell subsets, which exhibit notable phenotypic and transcriptional divergence, are linked early in their developmental trajectory by epigenetic reorganization. Going forward, it will be important to determine the broad functionality of TOX‐dependent reprogramming in human intratumoral Texh cells.
Given the abundance of Texh cells in many tumor types, as well as mounting evidence of their role in immunotherapy responses, it is essential to better understand the molecular dynamics of this heterogeneous subset. Early Texh cells express several markers, including CXCR5 and TCF1, that are not associated with Teff cells. Subsequent single cell‐based analyses have since offered contrasting conclusions on precisely when and where TCF1 helps to establish Texh cells. In cancers, these cells were first identified in NSCLC by high‐dimensional FACS analysis, and they expressed other signature genes shared by follicular helper T (TFH) cells in addition to TCF1. 101 In another study of melanoma, TCF1 was overrepresented in a human effector/memory‐like tumor‐infiltrating T cell population that was predictive of checkpoint blockade responsiveness. 81 Expression of TCF1 was, in fact, a marker used to separate Teff/Tmem cells from Texh cells. However, in other recent studies, trajectory analyses of CD8+ T cells showed that TCF1 was both a marker and driver of early Texh lineage establishment. 98 , 100 In one such study, done in mice chronically infected with LCMV, TCF1 specifically inhibited effector programs and promoted the establishment of early Texh cells. 62 Relatedly, another study of melanoma TILs showed TCF1 to be a marker of a highly replicative, transitional CD8+ population that resembles early Texh cells. 102 Notably, TCF1, a transcription factor, is also highly expressed in naïve T cells and regulates memory T (Tmem) cell formation. 103 Thus, it is becoming apparent that, although its precise expression pattern may be context‐dependent, TCF1 can promote memory functionalities broadly within activated CD8+ T cells. TCF1 may help multiple lineages of memory and memory‐like CD8+ T cells to maintain stem‐like abilities, including self‐renewal, proliferative capacity, and differentiation into terminal subsets. In several scRNA‐seq studies with human tumors, the early Texh cells were classified as Tem populations because they share markers previously used to define effector memory cells. 59 , 60 , 61 STARTRAC analysis revealed that some Tem cells are developmentally connected to Texh cells and are potential precursors of the Texh lineage (Fig. 1). Interestingly, we found that, at least in CRC samples, both TCF1+ and TCF1− cells are embedded in the Tem cell population, and both populations are developmentally connected with Texh cells 61 (unpublished observation), suggesting that multiple developmental pathways can establish intratumoral Texh cells.
Although CD8+ FOXP3+ Tregs have been observed in CRC and pancreatic cancer patients by flow cytometry, 104 scRNA‐seq analyses have also identified these cells in various cancer types including HCC, lung cancer, and CRC. 59 , 60 , 61 More importantly TCR lineage tracing studies revealed that some of these T cells shared TCRs with CD8+FOXP3− Texh cells, indicating a possible conversion between CD8+FOXP3− Texh cells and FOXP3+ T cells. Another study identified a subpopulation of cells that express both hallmarks of Treg cells (e.g., FOXP3 and CTLA4) and cytotoxic molecules (e.g., PRF1 and NKG7) within the exhausted CD8+ T cell subtype. 59 Such analyses further the hypothesis that Texh cells share and acquire a transcriptomic program with Treg cells. It also remains to be studied if these cells can exert suppressive functions like bona fide CD4+ Treg cells.
3.1.5. Development of the Texh lineage
The developmental pathway leading to terminally differentiated Texh cells remains incompletely understood. Initial experiments led to the hypothesis that Texh cells derived from Teff cell precursors in a linear and progressive fashion during chronic antigen exposure. 105 This trajectory was contrasted with the differentiation Teff to Tmem cell transition that occurs after acute antigen exposure. Importantly, studies in animal models suggested the development of tumor‐specific CD8+ Texh cells was a dynamic process dependent on continuous antigen exposure during early tumorigenesis. 92 More recent work utilizing single cell transcriptome‐level analyses, has indicated that Texh cell development may be partially or completely independent of the cytotoxic Teff cell lineage and may exhibit more characteristics of memory formation than previously appreciated.
Gene expression and TCR‐indexing analyses have shown that a Tem cell population may be a transitional population preceding the lineage fate of Teff and Texh cells in tumors. 59 , 60 , 61 Analyses of TIL populations from 3 types of cancer show that a CD8+GZMK+ T cell population exhibits characteristics of Tem cells and appears to share a lineage with both Texh and Teff cells. 61 Previous studies based on bulk TCR‐β sequencing or single‐cell inferred trajectories showed connections between Teff, Tem/Trm, and Texh cells in multiple cancer types, indicating a linear developmental differentiation of these T cells. However, STARTRAC analysis revealed limited connections between Teff and Texh cells in CRC. 61 Further analysis of the clonotypes of GZMK+ Tem cells in CRC revealed that the subpopulation of these cells shared TCRs with effector T cells were mutually exclusive with those shared TCRs with exhausted T cells, indicating a TCR‐based fate decision. It is unclear whether such a mutually exclusive pattern can be generalized to different cancer types.
As discussed earlier, studies of preclinical models and human tumors suggested that TCF1+ Texh precursor or stem‐like cells can develop into Texh cells. However, lineage tracing analysis with STARTRAC reveals both TCF1+ and TCF1− subpopulations reside in the Tem clusters and are comparably connected with Texh cells developmentally 61 (unpublished observation). Also of note, analysis of NSCLC indicates that Trm cells share a direct link to the Texh cell lineage, indicating that the TME may broadly divert Tem cell recall responses to induce Texh cells. 60 Altogether, these data suggested potentially diverse origins of tumor‐infiltrating CD8+ Texh cells.
3.1.6. Reactivity of tumor Texh cells
Texh cells have been shown in multiple settings to actively contribute to immune responses. 77 , 85 However, it remains unclear if they can be reinvigorated in a clinical setting to provide long‐term cures against chronic infections or cancers. It is further unknown if broad reactivation of Texh cells could elicit cures without immunopathological side‐effects. And in the context of PD‐1/PDL1 blockade, the relevant CD8+ population being activated (or reactivated) in clinical responders has not been convincingly identified. To address these questions, several groups have applied single cell analyses to patient tumor samples to identify highly active and reactive CD8+ T cells. A study of CRC indicates that Texh cells exhibit high TCR clonality and are by far the most actively proliferating cell type in these tumors, suggesting that Texh cells possess at least some responsiveness that could be further manipulated for therapeutic benefit. 61 However, studies based on mouse models suggest that not all Texh cells are reactive. Terminal Texh cells with high PD‐1 expression could not be reinvigorated, but rather Texh precursor/stem‐like cells with low or intermediate expression of PD‐1 are reactive to CPI treatment. 94 , 95
Analyses of melanomas have been particularly insightful, even if the studies’ conclusions have important contrasts. One such study highlighted the heterogeneity and proliferative capacity of the Texh compartment, going so far as to correlate Texh cell abundance with reactivity to autologous tumor cells ex vivo. 102 In agreement with this, analysis of circulating CD8+ T cells following checkpoint blockade found that the most proliferative cells had an exhausted phenotype. 106 However, a separate study showed that responsiveness to checkpoint blockade was predicated on a low ratio of Texh cells within melanoma lesions. 81 Another recent study tracked site‐matched melanoma lesions before and after checkpoint blockade to better understand the origins and phenotypes of tumor‐reactive CD8+ clonotypes. 82 Surprisingly, while checkpoint blockade preferentially expanded Texh‐like cells, those clones were mostly not matched in pretreatment infiltrates. This suggests that checkpoint blockade elicits clonal replacement of CD8+ T cells, and the new infiltrates then become exhausted themselves. Combined, these studies show that there are highly heterogeneous Texh and Teff/mem populations in tumors, all of which remain potential targets of immunotherapy. More detailed studies of tumor‐reactive CD8+ T cells, and large‐scale identification of their tumor‐associated Ags, is needed to determine when and where each cell population could be manipulated.
3.2. Characterization of Treg cells by scRNA‐seq
Treg cells are a subset of CD4+ T cells that play a central role in maintaining immune homeostasis. Treg cells function by suppressing responses of other immune cells thereby maintaining peripheral tolerance and limiting host damage that can result from an exaggerated or unchecked immune response. 107 Importance of Treg cells is well recognized both in context of autoimmunity, where they can be protective, as well as in cancer, wherein their presence impedes tumor clearance. Transcription factor Foxp3 plays a major role in controlling all facets of Treg cell biology ranging from lineage specification and stability to function with a substantial proportion of Treg cell transcriptional signature being Foxp3 dependent. While Foxp3 expression is generally restricted to Treg cells, it can be transiently expressed upon activation in conventional T cells in human. Furthermore, Treg cells can lose Foxp3 expression under certain inflammatory conditions and convert into pathogenic effector cells.
Treg cells have been reported to exhibit considerable heterogeneity based on developmental origin (thymic derived vs. peripherally induced), expression of activation markers (such as CD62L and CD44 that mark central and effector Treg subsets), 108 expression of distinct transcription factors (Tbet, Gata3, RORγt, and Bcl6 that corresponds to functional Treg subsets that are specialized toward suppression of particular T helper responses) 109 presence in certain anatomical locations such as skin, intestine, and fat that can imprint tissue specific features (like expression of genes such as PPARγ and IL‐33R) 110 as well as mechanisms utilized for suppression of other immune cells (sink for growth factors like IL‐2, expression of suppressive molecules such as CTLA4 and granzyme mediated direct cytotoxicity). 111 scRNA‐seq therefore provides a useful approach to understand the diversity in Treg cell origin and function.
3.2.1. Regulatory T cells in normal tissues and inflammatory diseases
ScRNA‐seq analysis of splenic CD4+ conventional T cells and Treg cells sorted from unperturbed mice has revealed that they mostly cluster separately with a small degree of overlap. 56 , 112 Profiling of Treg cells by scRNA‐seq in non‐lymphoid tissues such as skin and colon has identified some interesting features. 113 As compared to lymphoid tissues, Treg cells in these two compartments showed substantial enrichment of genes that are part of the TNFR‐NF‐kB signaling axis such as TNFRSF4, TNFRSF9, TNFRSF18, and Pim1/2, suggesting that the TNFR pathway might play a role in modulating Treg cell homeostasis and function in these tissues.
Analysis of two discrete thymic Treg cell precursors (CD25+Foxp3− and CD25−Foxp3lo) by scRNA‐seq has shown that they have unique transcriptional signatures that were reflective of distinct modes of differentiation. 114 The CD25+ precursors were more enriched in genes associated with stronger TCR signaling while the Foxp3lo subset had increased expression of adapter genes that could enhance signaling via TNFRs and TCR. These differences in progenitor cells in turn resulted in production of Treg cells that were qualitatively different as measured by their ability to suppress experimental autoimmune encephalomyelitis.
Treg cells play a prominent role in suppressing autoimmunity and scRNA‐seq analysis of biopsy samples from animal models and inflamed tissues of patients suffering from autoimmune disorders has provided unique insights into their phenotype and function. During ulcerative colitis (UC), a subtype of inflammatory bowel disease, an enrichment of Treg cells has been previously reported in the colonic mucosa. In this setting, scRNA‐seq has uncovered that TNF expression, which is one of the prominent pathogenic drivers in this disease, shifts dramatically toward Treg cells. 115 In tissues from healthy patients and non‐inflamed tissue from UC patients, TNF is mostly expressed by activated CD4+ T cells and tissue resident CD8+ T cells, but in inflamed tissue, Treg cells are one of the major sources of TNF suggesting that they might have converted into effector‐like cells. However, these Treg cells maintain expression of characteristic genes (FOXP3, CTLA4, IL10), so more work is needed to identify whether they promote pathogenesis or resistance to anti‐TNF therapy. Furthermore, up‐regulation of IL18 by enterocytes correlates with this increased presence of Treg cells (that express IL18R1) in inflamed colonic mucosa indicating that Treg cell recruitment may be regulated by the epithelial cells during UC.
3.2.2. Regulatory T cells in cancer
Beyond providing a better understanding of Treg cell diversity during homeostasis, scRNA‐seq technology has helped elucidate their role during cancer. Relevance of Treg cells in cancer is highlighted by the fact that their increased presence often predicts poor prognosis and several therapeutic strategies designed to deplete them show efficacy. Analysis of infiltrating cells isolated from several different human tumors (liver, lung, breast, skin, and colon) by scRNA‐seq has identified a Treg cell gene signature that is distinct from normal tissue‐associated Treg cells. 43 , 59 , 60 , 61 , 79 Comparing all these studies has yielded a common set of genes such as CTLA4, TNFRSF4, TNFRSF18, TIGIT, ICOS, and CCR8 whose expression is higher in tumor‐associated Treg cells as compared to Treg cells from other tissues. Along with these genes whose function in Treg cells have previously been characterized, several other genes such as LAYN, CD177, IGFLR1, and IL1R2 that are not well studied are also up‐regulated in tumor Treg cells. A more detailed examination has revealed patterns of heterogenous gene expression in tumor Treg cells. Expression of TNFRSF9 (encoding CD137; 4‐1BB) demonstrated a bimodal distribution in tumor Treg cells and as TNFRSF9 is known to be uniquely up‐regulated in Treg cells upon TCR stimulation 116 , this subset might represent Ag‐activated Treg cells. Genes highly enriched in CD137hi Treg cells, as compared to tumor Treg gene signature, corelated with worse patient prognosis in the TCGA lung adenocarcinoma dataset suggesting that CD137hi Treg cells correspond to suppressive tumor Treg cells. 60
Co‐variance in gene expression has also been described in tumor Treg cells with co‐expression of genes such as CTLA4, TNFRSF18, and TIGIT in certain Treg cell clusters with mutually exclusive expression of these genes in other Treg cell clusters indicating that they may occupy distinct spatial or functional niches. 43 Interestingly, a small subset of genes enriched in tumor Treg cells such as CTLA4, TIGIT, TNFRSF9, CD27, and LAYN are also highly expressed by exhausted tumor‐infiltrating CD8+ T cells reflecting a shared program of activation and exhaustion in these cells. Along with CD8+ T cells, tumor‐infiltrating Treg cells are among the most highly clonally expanded population suggesting that they undergo local expansion after recognizing tumor‐associated Ags. Lineage tracking analysis using TCR repertoire has revealed that the source of these tumor‐infiltrating Treg cells is mostly recruitment from other lymphoid tissues with migration from adjacent tissues and conversion of CD4+ T cells to induced Treg cells providing only a minor component. Based on TCR sharing analysis, the induced Treg (iTreg) cells could be developmentally linked to either Th1‐like (BHLHE40+CXCL13+) or Th17 cells with BACH2 being selectively expressed in Th1‐like iTreg cells and RORC and SATB1 preferentially enriched in Th17 linked iTreg cells suggesting that different Treg cells subsets are present with in the TME. 61 Although gene expression profile of tumor Treg cells and their derivation from lymphoid tissues has previously been reported using bulk RNA‐seq, 117 , 118 scRNA‐seq has provided a clearer picture of tumor Treg cells and identified diverse subsets whose function is not yet well defined.
Overall, scRNA‐seq has been very informative in providing a better understanding of Treg cell diversity during various aspects of their development, tissue residence and function during inflammation and cancer.
3.3. Other memory T cell subsets in tumor
Besides tumor‐enriched Texh and Treg cells, scRNA‐seq analysis also identified additional T cell clusters that showed various cross‐tissue distribution between tumor and blood and/or normal tissues. These include naïve T cell (Tn), central memory T cell (Tcm), Tem, and Temra or Teff for both CD4+ and CD8+ T cells (Table 2). Within memory CD4+ T cells, different T helper (Th) subsets, including Th1, Th2, Th17, and T follicular helper (TFH) can also be identified. The signature genes identified by scRNA‐seq for these T cell clusters are largely consistent with previous studies that utilized microarray or bulk RNA‐seq and show parallel patterns in human CD4+ and CD8+ T cell lineages. 119 , 120 CyTOF analysis at protein level has confirmed the presence of these T cell subsets in tumor. 13 , 14 Previous studies with combined phenotypic, functional, epigenetic, and gene expression properties of these T cell subsets suggest a linear T cell progression model (Tn‐Tcm‐Tem‐Temra) along these T cell clusters at the quiescent state. 121 , 122 , 123 , 124 , 125 Similarly, inferred developmental trajectory of these T cell clusters in scRNA‐seq datasets based on either transcriptome or incorporated with TCRs also exhibited a continuous structure of these cells with Tem tending to be the intermediate cells. 59 , 60 , 61 However, scRNA‐seq revealed that these different subsets inside tumors may not display as discrete clusters, instead, in vivo isolated T cells demonstrate broad continuum of activation and differentiation transcriptome spectrums surrounding the core subset defining gene signatures, which is likely dictated by both TCR/Ag specificity and environmental factors. 43 , 59 , 60 , 61 , 79
TABLE 2.
CD8+ T cell subsets identified based on the integrated single‐cell studies
| T cell subset/state a | Gene signature | Annotation | Tumor type | Tissue enrichment b | Clonality | Mobility | Transition | Other tumor c | Functional interpretation |
|---|---|---|---|---|---|---|---|---|---|
| LEF1+ Tn | TCF7, SELL, LEF1, CCR7 | Naive |
CRC HCC NSCLC |
Blood b | Low | Low | Low | Melanoma (Triosh) | |
| GPR183+ Tcm |
GZMK, low GZMA/PRF1/NKG7/TCF7/EOMES/GPG183 |
Central memory/naïve‐like/memory |
CRC HCC NSCLC |
Blood b | Intermediate | Low | Intermediate | Lung cancer (Clarke) Melanoma (Li; Sade‐Feldman) | Correlated with response to ICB in melanoma (Sade‐Feldman) |
| CX3CR1+ Temra | GZMA, GNLY, PRF1, GZMB, NKG7, TBX21, ZEB2, HOPX | Effector memory recently activated /Effector/Cytotoxic |
CRC HCC NSCLC |
Blood b | High | High | High | Melanoma (Triosh, Li; Sade‐Feldman) Breast cancer (Savas) HNSCC (Puram) | |
| GZMK+ Tem | GZMK, EOMES, TOX, others similar to Tcm | Effector memory/Transitional |
CRC HCC NSCLC |
Tumor, Normal | Intermediate | Intermediate | High | Melanoma (Li; Sade‐Feldman) Breast cancer (Azizi, Savas) | |
| ZNF683+ Trm | IL2, ZNF683, HOPX, ID2, low effector molecules | Tissue‐resident memory/Pre‐exhausted | NSCLC | Tumor, Normal | Intermediate | Intermediate | Intermediate | ||
| LAYN+ Tex | LAG3, TIGIT, PDCD1, HAVCR2, CTLA4 | Exhausted/Dysfunctional |
CRC HCC NSCLC |
Tumor | High | Low | Intermediate | Melanoma (Triosh, Li; Sade‐Feldman) Breast cancer (Azizi, Savas) HNSCC 9Puram), Lung cancer (Clarke) | Highly proliferative (Melanoma, breast cancer, CRC, lung cancer), correlated with response to ICB in melanoma (Sade‐Feldman), correlated with tumor‐reactivity (Li) |
| CD6+ Trm | XCL1, XCL2, MYADM, CD6 | Tissue‐resident memory | CRC | Normal | Intermediate | Intermediate | Intermediate | ||
| CD160+ IEL | KLRC1/2/3, IKZF2, NR4A3, CD69, NR4A1/2, CD160 | Intraepithelial lymphocyte | CRC | Normal | Intermediate | Low | Low | ||
| SLC4A10+ MAIT | SLC4A10, ZBTB16 | Mucosal associated invariant |
CRC HCC NSCLC |
Tumor, Normal, Blood | Intermediate | Intermediate | Low |
The T cell subsets are summarized based on the integrated analysis of single cell transcriptome of T cells isolated from CRC, HCC, and NSCLC. All of these T cell transcriptomes were generated from the same scRNA‐seq platform (Smart‐seq2), and T cell subsets were obtained by re‐clustering analysis of these three datasets.
The tissue enrichment of each T cell subset is summarized based on the calculation of their distribution in blood, tumor and adjacent normal tissue. For T cell subsets enriched in blood, such as GPR183+ Tcm, these cells were also identified in the tumor with less abundance.
The T cell subsets identified by the above integrated analysis were aligned with other T cells based on the similar signature genes. Other tumor types/datasets with similar T cell subsets are listed here. The different annotations of these T cell subsets are also listed in the column “Annotation.”
3.3.1. Tumor T cell clusters shared with blood and normal tissues
Among these T cell clusters, Tn and Tcm subpopulations are mainly found in patient blood but very rarely in tumor, consistent with their biological property as circulating T cells that traffic between blood and secondary lymphoid organs. 59 , 60 , 61 , 79 , 102 , 126 , 127 Both Tem and Temra cells are present in blood, normal tissue, and tumor, with Tem cells being relatively abundant in tumor as well as normal tissue whereas Temra cells are predominantly observed in blood. 59 , 60 , 61 Temra cluster expresses high level of S1PR5 and distinct cell adhesion molecules and chemokine receptors than other T cell clusters in tumor, rendering these cells high mobility. 60 , 61 We and others have also identified Trm and mucosal‐associated invariant T (MAIT) cells in tumor. 59 , 61 , 128 The importance and function of MAIT cells in cancer immunity has been reviewed recently and will not be discussed here. 129
3.3.2. Tumor‐infiltrating Temra cells
Both CD4+ and CD8+ Temra clusters represent a small proportion of tumor‐infiltrating T cells in various cancer types. 59 , 60 , 61 Similar CD4+ and CD8+ clusters expressing high cytotoxic signature genes (e.g., GZMA, GZMB, PRF1, GNLY, NKG7) but low or no exhaustion signatures (PDCD1, LAG3, and CTLA4) are also reported in scRNA‐seq studies for melanoma 79 and head and neck cancer 130 , 131 (Table 2). The less abundance of Temra cells in tumors based on scRNA‐seq analysis is consistent with previous observations by traditional flow cytometry analysis (identified as CD45RA+CCR7− CD4+ or CD8+ T cells) in multiple cancer types. 131 , 132 , 133 , 134
Temra cells in healthy donor PBMC are found more frequently in CD8+ compartments and represent Ag‐experienced terminally differentiated memory T cells that are capable of immediate cytokine production and cytotoxicity without proliferation. 135 , 136 CD4+ Temra cells share similar phenotypes as CD8+ Temra cells with drastic variability in their frequency between individuals. 137 Both CD4+ and CD8+ Temra cells have been implicated in protective immunity against pathogens and contain expanded viral‐specific clones. 135 , 136 , 137 Similarly, both CD4+ and CD8+ Temra populations in cancer patients are found to be clonally expanded. 59 , 60 , 61 Although the functional role of CD4+ Temra population in cancer is still unclear, a recent scRNA‐seq study revealed that in humans, these cells might have developed from the precursors that express IL‐7 receptor, and the TCRs from these cells were clonally expanded and recognized dengue virus if the donors had been previously infected, supporting their role in viral control.
It is still controversial whether CD8+ Temra cells contain clonotypes that are specific for tumor Ags. Li et al. identified Temra‐like cluster in melanoma tumor with high expression of cytotoxic‐related genes (e.g., FGFBP2, GZMH, KLF2) and low expression of exhaustion signature genes. However, T cells from those tumors with high intensity of cytotoxic signature were associated with low tumor reactivity in an ex vivo assay, suggesting tumor Ag‐specific T cell clones were not enriched in such tumor‐infiltrating Temra cluster. 102 In our CRC scRNA‐seq study, we found that Temra cells were highly clonally expanded. 61 The STARTRAC‐expansion analysis in this study showed that the degree of clonal expansion of Temra cells in CRC patients was as high as Texh cells and higher than other memory T cell clusters. Among clonally expanded Temra cells, nearly half of them shared TCRs with tumor Tem cells, whereas only a small fraction shared TCRs with blood Tem cells. By introducing another 2 indices, STARTRAC‐transition and STARTRAC‐migration, for the measurement of state transition and tissue migration of T cell clusters, respectively, this study also showed that Temra exhibited significantly higher developmental connection with Tem than with other T cell clusters and had the highest capability to migrate. 61 Together, these findings suggest that at least some Temra cells may have differentiated from tumor‐specific memory T cells and these cells can circulate between peripheral blood and tumor. Indeed, blood Temra cells have been demonstrated to contain tumor Ag‐specific TCR clonotypes in breast cancer patients. 138 , 139 Percentage of blood Temra cells was significantly higher in those NSCLC patients that partially responded to anti‐PD1 (Nivolumab) treatment than in non‐responders at baseline. 140 In contrast, high blood Temra cells in melanoma patients were found to associate with worse outcome in these cancer patients, 141 suggesting a context‐dependent effect of Temra cells.
Nevertheless, NSCLC patients who responded to Nivolumab had increased frequency and activity of tumor Ag‐specific Temra cells, suggesting the antitumor activity of these cells at least in some tumor types. 138 , 140 Temra cells express low levels of Texh signature genes (e.g., PDCD1 and CTLA4) but high levels of NK‐associated receptors (e.g., CD94/NKG2A, KIRs, LILRB1, and KLRG1), indicating distinct functional regulatory pathways for Temra cells. Recently, we have demonstrated that PD1 blockade and LILRB1 blockade can synergistically enhance CD8+ T cell activity and cytolytic function in vitro. 142 Given the high potency of cytolytic activity of Temra cells, 142 understanding how to promote tumor‐specific Tem cell differentiation to Temra and how to improve their antitumor activity will be beneficial for immunotherapy.
3.3.3. Tumor‐infiltrating Tem cells
Tem cells are also Ag‐experienced T cells that can rapidly elicit effector responses during Ag re‐encounter. 143 Tem cells have better survival and proliferation capability than Temra cells. 136 , 144 These cells differentiate from naïve T cells during primary immune response to Ag, with a distinct phenotypic and functional profile that allows them to migrate to blood, secondary lymphoid organs, and tissues. 125 , 145 Prior gene expression profiling by microarray or bulk RNA‐seq studies of human memory T cell subsets as defined by cell surface markers CD45RA and CCR7 showed a continuous transcriptomic change for these memory T cell subpopulations. 119 , 120 , 146 Recent scRNA‐seq studies provide additional granularity and unbiased analysis for tumor‐infiltrating memory T cell subsets in cancer patients.
Our scRNA‐seq data identified that both CD4+ and CD8+ Tem cells were characterized by high expression of GZMK and intermediate expression of PD1. 59 , 60 , 61 Similar GZMK+ T cell cluster has also been identified in almost all tumor types based on different scRNA‐seq approaches (Table 2). Both pseudotime trajectory and TCR sharing analysis suggest Tem cluster as the center of developmental path with one end linked to Tn‐Tcm and the other end linked to Temra or Texh. However, as mentioned before, scRNA‐seq analysis in CRC revealed that Tem clones linked to Temra and Texh are mutually exclusive. 61 Such diverge developmental pattern of Temra and Texh cells is also observed in other cancer types like melanoma. 102 Diametric linkage of Tem with Temra or Texh in tumor suggests that tumor Tem cells may take distinct differentiation paths toward Texh or Temra and that TCR specificity may have a role to determine their developmental trajectories. However, it is unclear whether this observation can be applied broadly to other cancer types or situations. Recently, CXCR5+ Tem‐like CD8+ T cells possessing stem‐like properties were identified in NSCLC tumor by CyTOF and scRNA‐seq analyses. 101 These cells are precursors of Texh cells that were initially identified during chronic viral infection and are the primary cell types responding to CPIs. 98 , 100 Upon ex vivo stimulation, these cells isolated from tumor proliferate and gradually acquire Texh‐like phenotype and property. 101 As discussed above, we noticed that these CXCR5+/TCF1+ cells are embedded inside the Tem population and are equally connected with Texh cells as those TCF1− Tem cells in multiple cancers 59 , 60 , 61 (unpublished observation). These data support the hierarchical differentiation of Tem cells to become Texh cells in the context of TME.
It has been shown that Tem cells contain tumor‐specific subsets in cancer. 147 , 148 In agreement with these findings, we found that tumor Tem cells had significantly higher STARTRAC‐expansion index in CRC (Fig. 1), suggesting that these cells underwent clonal expansion in response to local tumor‐associated Ag stimulation. 61 Given the emerging evidences of tumor‐reactivity of Texh cells, high STARTRAC‐transition index between Tem and Texh cells also indicated the presence of tumor‐specific clonotypes in the Tem cluster. 61 Moreover, positive correlation of Tem gene signature with better outcome in cancer patients 60 suggests potential beneficial effects of therapeutic strategies to expand and activate Tem cells in tumor. A previous study using flow cytometry showed that the most prominent response to anti‐PD1 treatment was the expansion of intratumoral CD8+ memory T cells. 149 In line with this observation, a recent scRNA‐seq study showed that there were more expanded clones in memory CD8+ T cell cluster than newly emerged clones under anti‐PD‐1 treatment. 150 Nevertheless, further investigation is needed to validate the tumor‐reactivity of these tumor Tem cells and explore the extrinsic and intrinsic factors that allow their discrimination from bystander T cells.
Intratumor CD4+ Tem cell cluster is highly complex. Beside the major GZMK+ Tem clusters, Th17 cells, TFH, and T follicular regulatory subsets have also been identified in CRC samples. 61 Th17 cells are characterized by the marker genes such as RORC and IL‐23R. STARTRAC analysis has also revealed these intestine related Th17 cells are developmentally connected to Treg cells, supporting the conversion of these 2 cell types in this tissue. The majority of cells in CD4+ Tem cluster are IFN‐γ positive with high expression of EOMES and RUNX3, suggesting these cells may be the bona fide Th1 cells. Interestingly, in CRC, another IFN‐γ+ Th1‐like cluster was identified that also expressed higher BHLHE40 than traditional CD4+ Tem cells. 61 Intriguingly, microsatellite‐instable (MSI) CRC patients have higher BHLHE40+ Th1‐like CD4+ T cells than microsatellite‐stable (MSS) patients. 61 Given previous observation of better responses in MSI CRC patients, 151 the preferential enrichment of CD4+ Th1‐like T cells in these patients indicate a positive correlation of these T cells with response to anti‐PD1 treatment. Furthermore, IGFLR1 was found to be highly expressed on Th1‐like CD4+ T cells in CRC, which may represent a novel costimulatory pathway to enhance IFN‐γ production from these cells. 61 Although previous studies based on bulk RNA detection inferred the association between increased Th1 signature and the MSI status in CRC patients, 152 scRNA‐seq analysis further illustrated that only CXCL13+BHLHE40+ Th1‐like cluster but not the classical CD4+ Tem cluster in CRC was preferentially enriched in MSI patients and could be accountable for the favorable response of these patients to CPIs.
3.3.4. Tumor‐infiltrating Trm cells
Tissue‐resident memory T cells (Trm) are memory CD4+ or CD8+ T cells retained in peripheral tissues to patrol nonlymphoid organs for protection from pathogen infection. 153 These cells can be found in normal tissues, expressing CD69 and CD103 and devoid of CCR7 or CD62L. 154 , 155 Several transcription factors are involved in Trm generation and maintenance, including the up‐regulation of RUNX3, Blimp‐1, and Hobit (encoded by ZNF683), and down‐regulation of EOMES and KLF2. 156 , 157 , 158 , 159 Emerging evidence has shown that Trm cells are important for antitumor immunity mainly based on the positive correlation of CD103+ Trm level with favorable prognosis in cancer patients. 160 , 161 , 162 , 163 , 164 , 165 , 166 Although CD103 is a hallmark of Trm cells, especially for those of epithelial origin, liver‐resident Trm cells are characterized by the up‐regulation of integrin LFA‐1 instead of CD103. 167 Moreover, for CD103+ T cells, recent studies have also identified different phenotypic populations within these cells in the TME. One example is the scRNA‐seq analysis of Trm cells in lung cancer. In this study, multiple clusters were identified from sorted CD103+ Trm cells, including Texh‐like cluster and classical normal tissue Trm cluster. 168 Another example is that in breast cancer, tumor‐infiltrating CD103+ CD8+ T cells were also separated into 2 clusters by scRNA‐seq analysis, with 1 cluster expressing higher level of both effector molecules and inhibitory receptors (such as PRF1 and GZMB; HAVCR2 and LAG3) and the other cluster expressing genes related to cell proliferation (such as CCNA2 and TUBB). 128 Therefore, these studies suggest that different clusters of tumor‐infiltrating CD103+ CD8+ T cells may have distinct phenotypic and functional properties and scRNA‐seq technology provides granularity for us to better understand the heterogeneity of these cells in tumor.
The recent scRNA‐seq studies of T cells from blood, normal tissue, and tumor of HCC, NSCLC, and CRC patients comprehensively characterize diverse T cell clusters enriched in tissues and revealed the heterogeneity of Trm cell subsets in different cancer types. 59 , 60 , 61 First, multiple T cell clusters were identified in tumor and the adjacent normal tissue, including Tem, Texh, and Trm CD8+ T cell clusters, expressed Trm signature genes as observed in aforementioned studies. 132 , 168 However, detailed gene expression analysis revealed distinct expression patterns of these CD8+ T cell clusters: Tem cluster expressed low‐level ITGAE (encoding for CD103) but high‐level PDCD1; Trm exhibited the opposite pattern; and Texh showed high‐level for both genes. 60 Second, the classical Trm cells were present primarily in normal tissue than in tumor. The proportion of intratumoral Trm cells was different in distinct tumor types. For example, the integrated analyses of T cells in HCC, NSCLC, and CRC revealed that although ZNF683 + CD8+ Trm cells were present in these 3 cancer types, their frequency was significantly higher in NSCLC than in HCC and CRC, suggesting potential function of these Trm cells in NSCLC immunity. 60 , 61 Moreover, the Trm cluster in different tumor types displayed diverse gene expression properties, mainly associated with the tissue origin of a given tumor. As an example, CD160+ IEL cluster, as characterized by high expression of both NK and inhibitory receptors like ENTPD1 (encoding CD39) was found in the adjacent normal tissue from CRC but not NSCLC or HCC patients. 61
One critical question about Trm is whether they have different differentiation paths in diverse tissues or tumors. Pseudotime‐based analysis showed that in NSCLC ZNF683 + Trm located more centrally as Tem in the trajectory plot, suggesting these cells were at “pre‐exhaustion” state. 60 Moreover, these ZNF683 + Trm cells were also found to share TCRs with Texh cells and to a less extent with Tem cells. 60 In CRC, CD6+ Trm cluster showed dominant linkage with Tem but limited linkage with Texh clusters, while CD160+ IEL cells were barely linked to other T cells based on STARTRAC‐transition analysis, indicating the different origins of Trm cells in CRC. 61 Finally, STARTRAC based mobility analyses also revealed that Tem cells are more mobile than both Trm subsets and Texh cluster 61 (Fig. 1). Together, these findings support previous observation that Tem can give rise to Trm in some tissues, 154 but the Texh cells may be derived from Tem and/or Trm compartments in tumor. It will be of great interest to further explore the factors that drive the different location, phenotype, and differentiation of Trm cells in various tissues and tumors.
Based on these newly generated scRNA‐seq data and bulk RNA‐seq data from TCGA researchers identified that higher ratio of Trm to Texh signature gene expression associated with better overall survival in NSCLC patients. 60 Similarly, Trm signature genes defined by another scRNA‐seq analysis in breast cancer samples also correlated with better prognosis in these patients. 132 Notably, Trm signature genes in breast cancer also included several inhibitory receptors that are hallmarks of Texh cells. 132 It needs to be explored whether the favorable prognosis stemmed from Trm or Texh cells. Nevertheless, these scRNA‐seq studies provide evidences for the important roles of Trm cells in the control of solid tumor.
4. CONCLUDING REMARKS
Single cell transcriptomes have revolutionized the way to study the highly complex TME and provide a better understanding of various immune cell populations in this context. As illustrated in this review, scRNA‐seq based studies have unraveled the detailed characteristics of heterogeneous tumor‐infiltrating T cells in various human cancer types. By incorporating the transcriptomes and paired TCR analysis, single cell lineage tracing models, such as STARTRAC, now allow us not only to track the developmental trajectories but also the dynamic relationships of these T cells with different cellular states or tissue origins. Nevertheless, the snapshot of transcriptomic information captured by scRNA‐seq may make it less feasible for the analysis of other phenotypic parameters, such as spatial organization and epigenetic regulators. With continued advances in single cell transcriptomics and other omics technologies, we anticipate that the integrated single‐cell multi‐omics will broaden the scope of their applications in the immune system, such as providing cues that regulate the fate and cellular localization of tumor‐infiltrating T cells. With the accumulation of scRNA‐seq and other single cell omics data for baseline tumors and drug‐treated tumors, it is promising to discover tumor‐infiltrating T cell subtypes associated with treatment responses and explore their functions and relationships with other immune and stromal cells. Such information will further provide opportunities for the identification of improved biomarker strategy in the clinic to predict patient response to CPIs and the development of novel immunotherapeutic strategies to treat cancer.
The heterogeneous response of cancer patient to checkpoint blockades warrants urgent needs to address the mechanisms underlying resistance to these therapies. It remains elusive whether anti‐PD1 treatment can reinvigorate intratumoral T cells that recognize neoantigens or promote the development of new tumor Ag‐specific T cell clones. With above‐mentioned approach, one can address this question with tumor‐infiltrating immune cells collected before and on treatment. Applying scRNA‐seq analysis to characterize intratumoral immune cell changes to anti‐PD‐1 have started to emerge. 126 Alternatively, T cells in a given cancer type but with different outcomes to checkpoint blockades can be compared at single cell level. For example, in our CRC study, we compared intratumoral T cell clusters in MSI patients versus MSS patients given these patients exhibit distinct responses to anti‐PD‐1 treatment. 151 A new CXCL13 + BHLHe40 + Th1‐like CD4+ subset was found to be present at higher proportion in MSI tumor and may explain higher IFN‐γ level in MSI tumor and better response of these patients to anti‐PD‐1 treatment. 151 , 152 Other studies along similar lines are expected in the near future and these data will provide critical insight into mechanisms rendering tumors refractory to checkpoint blockade.
It is known that T cell functions can be greatly impacted by other factors and cells in TME. With further advancement of scRNA‐seq technology, combining with other single cell level techniques (such as scATAT‐seq), we foresee a great expansion of our understanding about tumor‐infiltrating immune cell heterogeneity, the intercellular interactions between intratumoral immune cell clusters, and the key regulatory pathways controlling cell fate decision in this context. These findings will shed light on the development of novel immunotherapeutic strategy to ultimately benefit cancer patients.
Yu X, Zhang L, Chaudhry A, Rapaport AS, Ouyang W. Unravelling the heterogeneity and dynamic relationships of tumor‐infiltrating T cells by single‐cell RNA sequencing analysis. J Leukoc Biol. 2020;107:917–932. 10.1002/JLB.6MR0320-234R
Contributor Information
Xin Yu, Email: xiyu@amgen.com.
Wenjun Ouyang, Email: wouyang@amgen.com.
REFERENCES
- 1. Gong J, Chehrazi‐Raffle A, Reddi S, Salgia R. Development of PD‐1 and PD‐L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long‐term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17): 1889‐1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang L, Zhang Z. Recharacterizing tumor‐infiltrating lymphocytes by single‐cell RNA sequencing. Cancer Immunol Res. 2019;7(7):1040‐1046. [DOI] [PubMed] [Google Scholar]
- 5. Proserpio V, Lonnberg T. Single‐cell technologies are revolutionizing the approach to rare cells. Immunol Cell Biol. 2016;94(3):225‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen‐colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4(8):648‐655. [DOI] [PubMed] [Google Scholar]
- 7. Bajenoff M, Germain RN. Seeing is believing: a focus on the contribution of microscopic imaging to our understanding of immune system function. Eur J Immunol. 2007;37(Suppl 1):S18‐S33. [DOI] [PubMed] [Google Scholar]
- 8. Kunz DJ, Gomes T, James KR. Immune cell dynamics unfolded by single‐cell technologies. Front Immunol. 2018;9:1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stubbington MJT, Rozenblatt‐Rosen O, Regev A, Teichmann SA. Single‐cell transcriptomics to explore the immune system in health and disease. Science. 2017;358(6359):58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bandura DR, Baranov VI, Ornatsky OI, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time‐of‐flight mass spectrometry. Anal Chem. 2009;81(16):6813‐6822. [DOI] [PubMed] [Google Scholar]
- 11. Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell. 2016;165(4):780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simoni Y, Chng MHY, Li S, Fehlings M, Newell EW. Mass cytometry: a powerful tool for dissecting the immune landscape. Curr Opin Immunol. 2018;51:187‐196. [DOI] [PubMed] [Google Scholar]
- 13. Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single‐cell analyses. Cell. 2017;169(4):750‐765 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chevrier S, Levine JH, Zanotelli VRT, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169(4):736‐749 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagner J, Rapsomaniki MA, Chevrier S, et al. A single‐cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. 2019;177(5):1330‐1345 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simoni Y, Becht E, Fehlings M, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575‐579. [DOI] [PubMed] [Google Scholar]
- 17. Giesen C, Wang HA, Schapiro D, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11(4):417‐422. [DOI] [PubMed] [Google Scholar]
- 18. Wang YJ, Traum D, Schug J, et al. Multiplexed in situ imaging mass cytometry analysis of the human endocrine pancreas and immune system in type 1 diabetes. Cell Metab. 2019;29(3):769‐783 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang Q, Ornatsky OI, Siddiqui I, Loboda A, Baranov VI, Hedley D. Imaging mass cytometry. Cytometry A. 2017;91(2):160‐169. [DOI] [PubMed] [Google Scholar]
- 20. Tang F, Barbacioru C, Wang Y, et al. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377‐382. [DOI] [PubMed] [Google Scholar]
- 21. Ofengeim D, Giagtzoglou N, Huh D, Zou C, Yuan J. Single‐cell RNA sequencing: unraveling the brain one cell at a time. Trends Mol Med. 2017;23(6):563‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single‐cell sequencing. Nat Rev Cancer. 2017;17(9):557‐569. [DOI] [PubMed] [Google Scholar]
- 23. Potter SS. Single‐cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018;14(8):479‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ren X, Kang B, Zhang Z. Understanding tumor ecosystems by single‐cell sequencing: promises and limitations. Genome Biol. 2018;19(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papalexi E, Satija R. Single‐cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2018;18(1):35‐45. [DOI] [PubMed] [Google Scholar]
- 26. Suva ML, Tirosh I. Single‐cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75(1):7‐12. [DOI] [PubMed] [Google Scholar]
- 27. Islam S, Kjällquist U, Moliner A, et al. Characterization of the single‐cell transcriptional landscape by highly multiplex RNA‐seq. Genome Res. 2011;21(7):1160‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramskold D, Luo S, Wang YC, et al. Full‐length mRNA‐Seq from single‐cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc. 2014;9(1):171‐181. [DOI] [PubMed] [Google Scholar]
- 30. Hashimshony T, Wagner F, Sher N, Yanai I. CEL‐Seq: single‐cell RNA‐Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666‐673. [DOI] [PubMed] [Google Scholar]
- 31. Hashimshony T, Senderovich N, Avital G, et al. CEL‐Seq2: sensitive highly‐multiplexed single‐cell RNA‐Seq. Genome Biol. 2016;17:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jaitin DA, Kenigsberg E, Keren‐Shaul H, et al. Massively parallel single‐cell RNA‐seq for marker‐free decomposition of tissues into cell types. Science. 2014;343(6172):776‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single‐cell transcriptomics applied to embryonic stem cells. Cell. 2015;161(5):1187‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gierahn TM, 2nd WadsworthMH, Hughes TK, et al. Seq‐Well: portable, low‐cost RNA sequencing of single cells at high throughput. Nat Methods. 2017;14(4):395‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cao J, Packer JS, Ramani V, et al. Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science. 2017;357(6352):661‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosenberg AB, Roco CM, Muscat RA, et al. Single‐cell profiling of the developing mouse brain and spinal cord with split‐pool barcoding. Science. 2018;360(6385):176‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haque A, Engel J, Teichmann SA, Lonnberg T. A practical guide to single‐cell RNA‐sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svensson V, Vento‐Tormo R, Teichmann S. Exponential scaling of single‐cell RNA‐seq in the past decade. Nat Protoc. 2018;13(4):599‐604. [DOI] [PubMed] [Google Scholar]
- 41. See P, Lum J, Chen J, Ginhoux F. A single‐cell sequencing guide for immunologists. Front Immunol. 2018;9:2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Enge M, Arda HE, Mignardi M, et al. Single‐cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 2017;171(2):321‐330 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Azizi E, Carr AJ, Plitas G, et al. Single‐cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174(5):1293‐1308 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277‐1289. [DOI] [PubMed] [Google Scholar]
- 45. Young MD, Mitchell TJ, Vieira Braga FA, et al. Single‐cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science. 2018;361(6402):594‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lein E, Borm LE, Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017;358(6359):64‐69. [DOI] [PubMed] [Google Scholar]
- 47. Kester L, van Oudenaarden A. Single‐cell transcriptomics meets lineage tracing. Cell Stem Cell. 2018;23(2):166‐179. [DOI] [PubMed] [Google Scholar]
- 48. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tritschler S, Büttner M, Fischer DS, et al. Concepts and limitations for learning developmental trajectories from single cell genomics. Development. 2019;146(12). [DOI] [PubMed] [Google Scholar]
- 50. Raj B, Wagner DE, McKenna A, et al. Simultaneous single‐cell profiling of lineages and cell types in the vertebrate brain. Nat Biotechnol. 2018;36(5):442‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM. Single‐cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018;360(6392):981‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ludwig LS, Lareau CA, Ulirsch JC, et al. Lineage tracing in humans enabled by mitochondrial mutations and single‐cell genomics. Cell. 2019;176(6):1325‐1339 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kirsch I, Vignali M, Robins H. T‐cell receptor profiling in cancer. Mol Oncol. 2015;9(10):2063‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Han A, Glanville J, Hansmann L, Davis M. Linking T‐cell receptor sequence to functional phenotype at the single‐cell level. Nat Biotechnol. 2014;32(7):684‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patil VS, Madrigal A, Schmiedel BJ, et al. Precursors of human CD4(+) cytotoxic T lymphocytes identified by single‐cell transcriptome analysis. Sci Immunol. 2018;3(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single‐cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19(3):291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stubbington MJT, Lönnberg T, Proserpio V, et al. T cell fate and clonality inference from single‐cell transcriptomes. Nat Methods. 2016;13(4):329‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lonnberg T, Svensson V, James KR, et al. Single‐cell RNA‐seq and computational analysis using temporal mixture modelling resolves Th1/Tfh fate bifurcation in malaria. Sci Immunol. 2017;2(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single‐cell sequencing. Cell. 2017;169(7):1342‐1356 e16. [DOI] [PubMed] [Google Scholar]
- 60. Guo X, Zhang Y, Zheng L, et al. Global characterization of T cells in non‐small‐cell lung cancer by single‐cell sequencing. Nat Med. 2018;24(7):978‐985. [DOI] [PubMed] [Google Scholar]
- 61. Zhang L, Yu X, Zheng L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564(7735):268‐272. [DOI] [PubMed] [Google Scholar]
- 62. Chen Z, Ji Z, Ngiow SF, et al. TCF‐1‐centered transcriptional network drives an effector versus exhausted CD8 T cell‐fate decision. Immunity. 2019;51(5):840‐855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. La Manno G, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018;560(7719):494‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Plass M, Solana J, Wolf FA, et al. Cell type atlas and lineage tree of a whole complex animal by single‐cell transcriptomics. Science. 2018;360(6391). [DOI] [PubMed] [Google Scholar]
- 65. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fridman WH, Pagès F, Sautès‐Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:9. [DOI] [PubMed] [Google Scholar]
- 67. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541(7637):321‐330. [DOI] [PubMed] [Google Scholar]
- 68. Ahmadzadeh M, Johnson LA, Heemskerk B, et al. Tumor antigen‐specific CD8 T cells infiltrating the tumor express high levels of PD‐1 and are functionally impaired. Blood. 2009;114(8):1537‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matsuzaki J, Gnjatic S, Mhawech‐Fauceglia P, et al. Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc Natl Acad Sci USA. 2010;107(17):7875‐7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289‐295. [DOI] [PubMed] [Google Scholar]
- 72. Hashimoto M, Kamphorst AO, Im SJ, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. 2018;69:301‐318. [DOI] [PubMed] [Google Scholar]
- 73. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gallimore A, Glithero A, Godkin A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus‐specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I‐peptide complexes. J Exp Med. 1998;187(9):1383‐1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zajac AJ, Blattman JN, Murali‐Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188(12):2205‐2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McLane LM, Abdel‐Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37:457‐495. [DOI] [PubMed] [Google Scholar]
- 77. Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682‐687. [DOI] [PubMed] [Google Scholar]
- 78. Blackburn SD, Shin H, Haining WN, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science. 2016;352(6282):189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gupta PK, Godec J, Wolski D, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015;11(10):e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sade‐Feldman M, Yizhak K, Bjorgaard SL, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175(4):998‐1013 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med. 2019;25(8):1251‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD‐1 blockade. Science. 2016;354(6316):1160‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sen DR, Kaminski J, Barnitz RA, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alfei F, Kanev K, Hofmann M, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571(7764):265‐269. [DOI] [PubMed] [Google Scholar]
- 86. Khan O, Giles JR, McDonald S, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Scott AC, Dündar F, Zumbo P, et al. TOX is a critical regulator of tumour‐specific T cell differentiation. Nature. 2019;571(7764):270‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Seo H, Chen J, González‐Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci USA. 2019;116(25):12410‐12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yao C, Sun HW, Lacey NE, et al. Single‐cell RNA‐seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20(7):890‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol. 2012;24(2):173‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med. 2010;207(10):2187‐2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schietinger A, Philip M, Krisnawan VE, et al. Tumor‐specific T cell dysfunction is a dynamic antigen‐driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45(2):389‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Baitsch L, Baumgaertner P, Devêvre E, et al. Exhaustion of tumor‐specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350‐2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Paley MA, Kroy DC, Odorizzi PM, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Thommen DS, Koelzer VH, Herzig P, et al. A transcriptionally and functionally distinct PD‐1(+) CD8(+) T cell pool with predictive potential in non‐small‐cell lung cancer treated with PD‐1 blockade. Nat Med. 2018;24(7):994‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Utzschneider DT Legat A, Fuertes Marraco SA, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14(6):603‐610. [DOI] [PubMed] [Google Scholar]
- 97. Gros A, Robbins PF, Yao X, et al. PD‐1 identifies the patient‐specific CD8(+) tumor‐reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246‐2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature. 2016;537(7620):417‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Utzschneider DT, Charmoy M, Chennupati V, et al. T cell factor 1‐expressing memory‐like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. 2016;45(2):415‐427. [DOI] [PubMed] [Google Scholar]
- 100. He R, Hou S, Liu C, et al. Follicular CXCR5‐ expressing CD8(+) T cells curtail chronic viral infection. Nature. 2016;537(7620):412‐428. [DOI] [PubMed] [Google Scholar]
- 101. Brummelman J, Mazza CEM, Alvisi G, et al. High‐dimensional single cell analysis identifies stem‐like cytotoxic CD8. J Exp Med. 2018;215:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li H, van der Leun AM, Yofe I, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176(4):775‐789 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W. Essential role of the Wnt pathway effector Tcf‐1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci USA. 2010;107(21):9777‐9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chaput N, Louafi S, Bardier A, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut. 2009;58(4):520‐529. [DOI] [PubMed] [Google Scholar]
- 105. Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19(4):408‐415. [DOI] [PubMed] [Google Scholar]
- 106. Huang AC, Postow MA, Orlowski RJ, et al. T‐cell invigoration to tumour burden ratio associated with anti‐PD‐1 response. Nature. 2017;545(7652):60‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Smigiel KS, Richards E, Srivastava S, et al. CCR7 provides localized access to IL‐2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med. 2014;211(1):121‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Campbell DJ, Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. 2011;11(2):119‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016;34:609‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fan X, Moltedo B, Mendoza A, et al. CD49b defines functionally mature Treg cells that survey skin and vascular tissues. J Exp Med. 2018;215(11):2796‐2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Miragaia RJ, Gomes T, Chomka A, et al. Single‐cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity. 2019;50(2):493‐504 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Owen DL, Mahmud SA, Sjaastad LE, et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol. 2019;20(2):195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Smillie CS, Biton M, Ordovas‐Montanes J, et al. Intra‐ and inter‐cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178(3):714‐730 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bacher P, Heinrich F, Stervbo U, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell. 2016;167(4):1067‐1078 e16. [DOI] [PubMed] [Google Scholar]
- 117. Plitas G, Konopacki C, Wu K, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. De Simone M, Arrigoni A, Rossetti G, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor‐infiltrating T regulatory cells. Immunity. 2016;45(5):1135‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:19. [DOI] [PubMed] [Google Scholar]
- 120. Chtanova T, Newton R, Liu MS, et al. Identification of T cell‐restricted genes, and signatures for different T cell responses, using a comprehensive collection of microarray datasets. J Immunol. 2005;175:11. [DOI] [PubMed] [Google Scholar]
- 121. Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell‐like properties. Nat Med. 2011;17:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lugli E, Dominguez M H, Gattinon L, et al. Superior T memory stem cell persistence supports long‐lived T cell memory. J Clin Invest. 2013;123:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Durek P, Nordström K, Gasparoni G, et al. Epigenomic profiling of human CD4. Immunity. 2016;45:14. [DOI] [PubMed] [Google Scholar]
- 124. van Aalderen MC, Remmerswaal EB, Verstegen NJ, et al. Infection history determines the differentiation state of human CD8+ T cells. J Virol. 2015;89:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. van Aalderen MC, van den Biggelaar M, Remmerswaal EBM, et al. Label‐free analysis of CD8. Cell Rep. 2017;19:12. [DOI] [PubMed] [Google Scholar]
- 126. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med. 2019;25:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chung W, Eum HH, Lee HO, et al. Single‐cell RNA‐seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Savas P, Virassamy B, Ye C, et al. Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nat Med. 2018;24(7):986‐993. [DOI] [PubMed] [Google Scholar]
- 129. Stolk D, van der Vliet HJ, de Gruijl TD, van Kooyk Y, Exley MA. Positive & negative roles of innate effector cells in controlling cancer progression. Front Immunol. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Puram SV, Tirosh I, Parikh AS, et al. Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611‐1624 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Poschke I, De Boniface J, Mao Y, Kiessling R. Tumor‐induced changes in the phenotype of blood‐derived and tumor‐associated T cells of early stage breast cancer patients. Int J Cancer. 2012;131:10. [DOI] [PubMed] [Google Scholar]
- 132. Savas P, Virassamy B, Ye C, et al. Single‐cell profiling of breast cancer T cells reveals a tissue‐resident memory subset associated with improved prognosis. Nat Med. 2018;24:8. [DOI] [PubMed] [Google Scholar]
- 133. Attig S, Hennenlotter J, Pawelec G, et al. Simultaneous infiltration of polyfunctional effector and suppressor T cells into renal cell carcinomas. Cancer Res. 2009;69:8. [DOI] [PubMed] [Google Scholar]
- 134. Ye SW, Wang Y, Valmori D, et al. Ex‐vivo analysis of CD8+ T cells infiltrating colorectal tumors identifies a major effector‐memory subset with low perforin content. J Clin Immunol. 2006;26:10. [DOI] [PubMed] [Google Scholar]
- 135. Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T‐cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:7. [DOI] [PubMed] [Google Scholar]
- 136. Henson SM, Riddell NE Akbar AN. Properties of end‐stage human T cells defined by CD45RA re‐expression. Curr Opin Immunol. 2012;24:6. [DOI] [PubMed] [Google Scholar]
- 137. Tian Y, Babor M, Lane J, et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re‐expressing CD45RA. Nat Commun. 2017;8(1):1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bernal‐Estévez D, Sánchez R, Tejada RE, Parra‐López C. Chemotherapy and radiation therapy elicits tumor specific T cell responses in a breast cancer patient. BMC Cancer. 2016;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kuznetsova M, Lopatnikova J, Shevchenko J, Silkov A, Maksyutov A, Sennikov S. Cytotoxic activity and memory t cell subset distribution of in vitro‐stimulated CD8+ T cells specific for HER2/neu epitopes. Front Immunol. 2019;10:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kunert A, Basak EA, Hurkmans DP, et al. CD45RA+CCR7‐ CD8 T cells lacking co‐stimulatory receptors demonstrate enhanced frequency in peripheral blood of NSCLC patients responding to nivolumab. J Immunother Cancer. 2019;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wistuba‐Hamprecht K, Martens A, Heubach F, et al. Peripheral CD8 effector‐memory type 1 T‐cells correlate with outcome in ipilimumab‐treated stage IV melanoma patients. Eur J Cancer. 2017;73:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kim A, Han CJ, Driver I, et al. LILRB1 blockade enhances bispecific T cell engager antibody‐induced tumor cell killing by effector CD8. J Immunol. 2019;203:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Abdelsamed HA, Moustaki A, Fan Y, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017;214(6):1593‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Romero P, Zippelius A, Kurth I, et al. Four functionally distinct populations of human effector‐memory CD8+ T lymphocytes. J Immunol. 2007;178:8. [DOI] [PubMed] [Google Scholar]
- 145. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. 2005;175:9. [DOI] [PubMed] [Google Scholar]
- 147. Beckhove P, Feuerer M, Dolenc M, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest. 2004;114:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Pagès F, Berger A, Camus M, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353:13. [DOI] [PubMed] [Google Scholar]
- 149. Ribas A, Shin DS, Zaretsky J, et al. PD‐1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sade‐Feldman M, Yizhak K, Bjorgaard SL, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mlecnik B, Bindea G, Angell HK, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:14. [DOI] [PubMed] [Google Scholar]
- 153. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:7. [DOI] [PubMed] [Google Scholar]
- 154. Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue‐resident memory T cells of skin. Nat Immunol. 2013;14:8. [DOI] [PubMed] [Google Scholar]
- 155. Kumar BV, Ma WMiron M, et al. Human tissue‐resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Mackay LK, Minnich M, Kragten NA, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:5. [DOI] [PubMed] [Google Scholar]
- 157. Milner JJ, Toma C, Yu B, et al. Runx3 programs CD8. Nature. 2017;552:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Mackay LK, Wynne‐Jones E, Freestone D, et al. T‐box transcription factors combine with the cytokines TGF‐β and IL‐15 to control tissue‐resident memory T cell fate. Immunity. 2015;43:11. [DOI] [PubMed] [Google Scholar]
- 160. Nizard M, Roussel H, Diniz MO, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. 2017;8:15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Djenidi F, Adam J, Goubar A, et al. CD8+CD103+ tumor‐infiltrating lymphocytes are tumor‐specific tissue‐resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. 2015;194:12. [DOI] [PubMed] [Google Scholar]
- 162. Ganesan AP, Clarke J, Wood O, et al. Tissue‐resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. 2017;18:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Murray T, Fuertes Marraco SA, Baumgaertner P, et al. Very late antigen‐1 marks functional tumor‐resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. 2016;7:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Webb JR, Milne K, Nelson BH. PD‐1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res. 2015;3:10. [DOI] [PubMed] [Google Scholar]
- 165. Wang B, Wu S, Zeng H, et al. CD103+ tumor infiltrating lymphocytes predict a favorable prognosis in urothelial cell carcinoma of the bladder. J Urol. 2015;194:7. [DOI] [PubMed] [Google Scholar]
- 166. Amsen D, van Gisbergen KPJM, Hombrink P, van Lier RAW. Tissue‐resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19:9. [DOI] [PubMed] [Google Scholar]
- 167. McNamara HA, Cai Y, Wagle MV, et al. Up‐regulation of LFA‐1 allows liver‐resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Clarke J, Panwar B, Madrigal A, et al. Single‐cell transcriptomic analysis of tissue‐resident memory T cells in human lung cancer. J Exp Med. 2019;216:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jerby‐Arnon L, Shah P, Cuoco MS, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984‐997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]