

Abstract
The metalloradical activation of o‐aryl aldehydes with tosylhydrazide and a cobalt(II) porphyrin catalyst produces cobalt(III)‐carbene radical intermediates, providing a new and powerful strategy for the synthesis of medium‐sized ring structures. Herein we make use of the intrinsic radical‐type reactivity of cobalt(III)‐carbene radical intermediates in the [CoII(TPP)]‐catalyzed (TPP=tetraphenylporphyrin) synthesis of two types of 8‐membered ring compounds; novel dibenzocyclooctenes and unprecedented monobenzocyclooctadienes. The method was successfully applied to afford a variety of 8‐membered ring compounds in good yields and with excellent substituent tolerance. Density functional theory (DFT) calculations and experimental results suggest that the reactions proceed via hydrogen atom transfer from the bis‐allylic/benzallylic C−H bond to the carbene radical, followed by two divergent processes for ring‐closure to the two different types of 8‐membered ring products. While the dibenzocyclooctenes are most likely formed by dissociation of o‐quinodimethanes (o‐QDMs) which undergo a non‐catalyzed 8π‐cyclization, DFT calculations suggest that ring‐closure to the monobenzocyclooctadienes involves a radical‐rebound step in the coordination sphere of cobalt. The latter mechanism implies that unprecedented enantioselective ring‐closure reactions to chiral monobenzocyclooctadienes should be possible, as was confirmed for reactions mediated by a chiral cobalt‐porphyrin catalyst.
Keywords: carbene radicals, cobalt, dibenzocyclooctenes, metalloradicals, 8-membered diene rings
Back in the ring! An efficient strategy based on metalloradical catalysis leads to 8‐membered ring compounds. Both dibenzocyclooctenes and monobenzocyclooctadienes can be obtained in high yields under mild reaction conditions. DFT calculations indicate different ring‐closure pathways for the formation of these two structures.
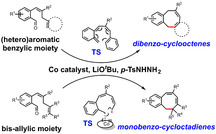
Introduction
The importance of medium‐sized cyclic compounds, especially 8‐membered rings, is exemplified by their prevalence in a broad variety of bio‐active natural products,1 fuels,2 fragrances3 and catalysts4 (Figure 1). Although many synthetic approaches exist to generate 5‐ and 6‐membered ring structures, ring‐closure reactions to form medium‐sized ring structures are still rather challenging in general.5 Due to entropic factors and transannular interactions, the flexible, linear precursor molecules typically employed in these procedures tend to react in an intermolecular way, leading to unwanted dimerization or polymerization instead of intramolecular cyclization. These side reactions are particularly problematic in the synthesis of 7‐ and 8‐membered ring compounds. Over the past years, several methods to form 8‐membered rings have been developed, including ring‐closing metathesis,6 metal complex assisted alkynyl/dienyl or carbene–carbene cyclizations,7 metal‐catalyzed [4+4],8 [m+n+o],9 and [6+2]10 cycloadditions, as well as oxabicyclic metal‐promoted ring‐opening reactions.11 However, the harsh reaction conditions, poor functional group tolerance, inevitable by‐product formation or the necessity of noble metal catalysts can put limitations on the general applicability of the existing methods in both academic and industrial settings. Therefore, developing efficient and broadly applicable strategies to synthesize 8‐membered ring systems, especially those employing inexpensive metal catalysts, remains an important challenge in the field of synthetic organic chemistry.
Figure 1.
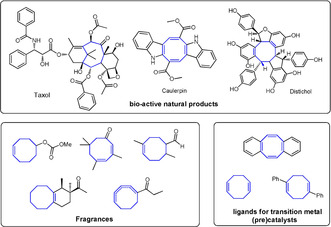
Natural products, fragrances and ligands containing 8‐membered rings.
Recently, we demonstrated that metalloradical catalysis based on the intrinsic radical‐type reactivity of cobalt(III)‐carbene radicals provides access to a particular class of 8‐membered ring compounds: dibenzocyclooctenes (Scheme 1 A).12 In these reactions, cobalt(II) complexes react with carbene precursors, such as N‐tosylhydrazones or diazo compounds, to produce cobalt(III)‐carbene radical intermediates that enable unusual catalytic radical‐type pathways.13 This metalloradical strategy has so far been employed in the synthesis of cyclopropanes,14 chromenes,15 furans,16 indenes,17 indolines,18 ketenes,19 butadienes,20 dihydronaphthalenes,20 piperidines,21 pyrrolidines22 and some dibenzocyclooctenes.12 Furthermore, for many reactions, the CoII system tolerates in situ generation of the carbene precursor from a readily available aldehyde via in situ generation of the N‐tosylhydrazone intermediate using p‐toluenesulfonyl hydrazide. Follow‐up conversion to the diazo compound upon deprotonation and subsequent trapping by the catalyst (Figure 2)23 significantly simplifies the synthetic methodology and makes the overall metalloradical strategy more widely applicable.21, 24
Scheme 1.
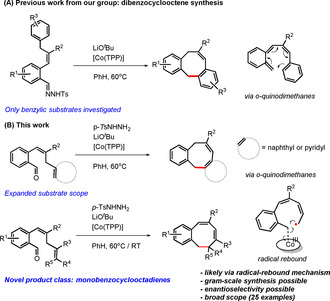
Metalloradical strategy for the synthesis of a variety of dibenzocyclooctenes and monobenzocyclooctadienes.
Figure 2.
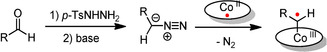
In situ generation of a cobalt(III)‐carbene radical intermediate from an aldehyde in a one‐pot sequence.
Herein, we report our efforts to advance the reactivity of cobalt(III)‐carbene radicals for the generation of 8‐membered rings. The method enables the synthesis of two types of ring systems: novel dibenzocyclooctenes and unprecedented monobenzocyclooctadienes. We first investigated the synthesis of novel dibenzocyclooctenes from substrates containing other aromatic carbocycles and heterocycles than previously reported,12 such as pyridyl and naphthyl moieties (Scheme 1 B). In addition, we found that the protocol can be extended to enable the synthesis of monobenzocyclooctadienes, for which no synthetic protocols currently exist. In this paper, we show that the latter products can be synthesized by ring‐closure of o‐aryl aldehydes containing bis‐allylic C−H bonds, and they seem to be formed via a different mechanism than previously reported for the dibenzocyclooctenes.12 While the dibenzocyclooctenes are most likely formed by dissociation of o‐quinodimethanes (o‐QDMs) from the catalyst and a subsequent non‐catalyzed 8π‐cyclization (Scheme 1 A), DFT calculations suggest that ring‐closure to the monobenzocyclooctadienes involves a direct radical‐rebound step within the coordination sphere of cobalt (Scheme 1 B). The latter mechanism implies that enantioselective ring‐closure reactions to chiral monobenzocyclooctadienes should be possible, as was confirmed by use of a chiral cobalt‐porphyrin catalyst. The reactions have a broad substrate scope, are operationally simple and can be carried out on a (near) gram scale. The details are disclosed in this paper.
Results and Discussion
In an attempt to expand the scope of 8‐membered ring synthesis via metalloradical catalysis, we first decided to examine the influence of changing the (hetero)aromatic benzyl moiety on the ring closure efficacy. We were particularly interested to explore the possibility of using substrates with naphthyl and pyridyl moieties (Table 1). These substrates proved to be surprisingly suitable for the synthesis of new dibenzocyclooctenes, as for substrates 1–3 the 8‐membered ring products were generated in good yields (Table 1, entries 1–3). The reaction with substrate 4 also worked well, but afforded structural isomers 4 a and 4 b in a 1:1 ratio (Table 1, entry 4). Each isomer could be isolated by column chromatography. The assigned structures of dibenzocyclooctenes 1–4, as determined by NMR spectroscopy (see Supporting Information, SI), are in accordance with previous work, with the C=C double bond in the product migrating from the 4‐ to the 5‐position, thus implying that the products are formed via o‐QDM intermediates, as reported previously.12 The isolated yields reported in Table 1 are corrected for the E/Z ratios of the substrates, because only the E‐isomer can undergo ring‐closure in these reactions.12 It is remarkable that these reactions are so tolerant to the use of naphthyl‐ and pyridyl moieties at the benzylic position, thus enabling the synthesis of interesting new 8‐membered ring product classes.
Table 1.
Substrate scope of dibenzocyclooctene synthesis, varying the (hetero)aromatic benzylic moiety of the substrate.[a]
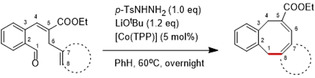
|
Entry |
Substrate |
Product |
Yield[b] |
|---|---|---|---|
|
1 |
|
|
82 % |
|
2 |
|
|
85 % |
|
3 |
|
|
75 % |
|
4 |
|
|
|
|
|
|
|
1:1 71 % in total |
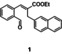
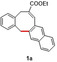
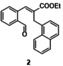
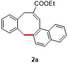
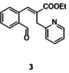
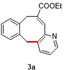
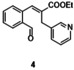
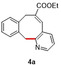
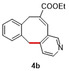
[a] Reaction conditions: 1–4 (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (5 mol %), benzene (1 mL), 60 °C, overnight. [b] Isolated yield, corrected for the E/Z ratios of the substrates.
Inspired by the flexibility and functional group tolerance of this protocol, we wondered if 8‐membered ring formation would still be possible when the benzylic moiety in the substrates is replaced by an allylic moiety. We therefore continued our investigations by testing bis‐allylic substrate 5 (Table 2, R1=H) in the metalloradical ring‐closure protocol. While we expected to observe 6‐membered ring formation in this case, to our surprise this substrate still produced the 8‐membered ring in a high yield, thus opening up the catalytic synthesis of a range of unprecedented 8‐membered ring products: monobenzocyclooctadienes.
Table 2.
Substrate scope of monobenzocyclooctadiene synthesis, varying the substituents of the aromatic moiety of the substrate.[a,b]

|
|
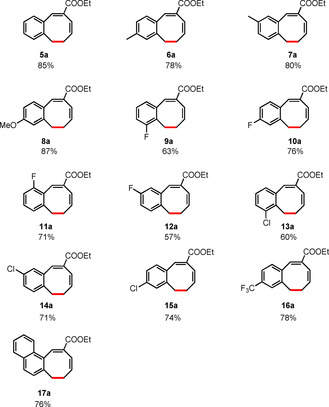
[a] Reaction conditions: For 5–12: (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (5 mol %), benzene (1 mL), 60 °C, overnight. For 13–16: (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (10 mol %), benzene (1 mL), room temperature, 48 h. [b] Isolated yield, corrected for the E/Z ratios of the substrates.
In contrast to the observed migration of the double bond from the 4‐ to the 5‐position during the synthesis of dibenzocyclooctenes, the position of the acrylic C=C double bond does not shift during the formation of monobenzocyclooctadienes (Table 2). No 6‐membered ring product formation was observed at all, which is remarkable in view of the fact that 6‐membered rings are generally formed more easily than 8‐membered rings.
We next explored the substrate scope for 8‐membered monobenzocyclooctadiene ring synthesis. We first looked at the effect of different aromatic ring substituents on the efficacy of the reaction (Table 2). In general, good yields were obtained, but we found that substrates containing electron‐withdrawing groups (13–16) are not very stable when heated in presence of base. This explains the lower yield of the corresponding products obtained under the standard reaction conditions (60 °C, 5 mol % catalyst). To solve this problem we lowered the reaction temperature while slightly increasing the catalyst loading to suppress base‐mediated substrate decomposition. With these slightly modified reaction conditions, monobenzocyclooctadienes 13 a–16 a could be obtained in good yields. For the most part, substrates with either electron‐withdrawing or electron‐donating groups on the aromatic ring afforded the desired products in good yields, but substrates with electron‐donating groups produced the monobenzocyclooctadienes in slightly higher yields. The experimental results also indicate that the substitution pattern on the phenyl ring or the presence of an extended aromatic system do not have a pronounced effect on the yield of the reaction. Notably, the experimental observations also indicated that heating is not a strict requirement to convert the N‐tosylhydrazones to the required diazo compounds, thus enabling us to expand the substrate scope to less stable substrates and simplifying the mode of operation of these reactions.
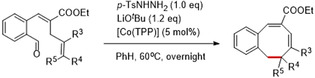
To further expand the scope, we explored the possibility of 8‐membered ring formation from substrates containing different substituents on the terminal allyl moiety (Table 3). Since the terminal vinyl group is directly involved in the ring‐closing step (regardless of the mechanism), the introduction of substituents on this C=C double bond was expected to have a large steric, electronic and geometric influence on the intermediates and transition states leading to the desired product, and hence low(er) yields were anticipated. However, quite unexpectedly, the desired 8‐membered structures were still obtained in decent to good yields (56–86 %, depending on the substituents). For substrate 20, several by‐products were generated upon heating the reaction mixture to 60 °C, but lowering the reaction temperature and increasing the catalyst loading allowed us to obtain the 8‐membered ring product in good yield (79 %). Substrate 24 did not cyclize to the desired monobenzocyclooctadiene, but this is not surprising considering the high torsional energy required to generate the intermediate and in view of the expected instability of the ring‐strained product.
Table 3.
Substrate scope of monobenzocyclooctadiene synthesis, varying the substituents at the terminal allylic position of the substrate.[a,b]
|
|
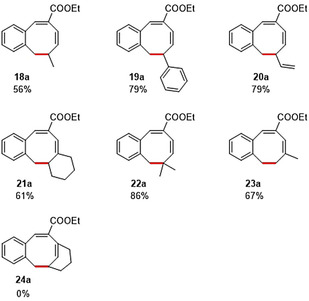
[a] Reaction conditions: For 18, 19, 21–24: (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (5 mol %), benzene (1 mL), 60 °C, overnight. For 20: (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (10 mol %), benzene (1 mL), room temperature, 48 h. [b] Isolated yield, corrected for the E/Z ratios of the substrates.
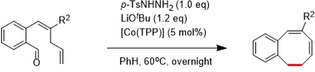
To further explore the generality of the reaction, substrates varying in substituents on the internal C=C double bond were synthesized (Table 4). While both substrates 25 and 26 afforded the desired monobenzocyclooctadienes, replacing the ester at this position for an acetyl or cyano group proved to have a damping effect on the obtained yields. This behavior seems to be related to the low thermal stability of 26 toward double bond isomerization, thus resulting in formation of other (yet unidentified) products in the catalytic reaction. Notably, double bond isomerization leading to formation of conjugated dienes also prevented us from synthesizing various substrates with different substituents on the internal C=C double bond (see caption of Table 4, with R2=‐Ph, ‐SO2Me and ‐C(O)NMe2. See SI for details). For 25, the accessibility of unwanted side reactions due to in situ carbene formation from the ketone might also play a role (see SI for details).
Table 4.
Substrate scope of monobenzocyclooctadiene synthesis, varying the substituent on the internal C=C double bond.[a,b]
|
|
[a] Reaction conditions: 25, 26 (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [Co(TPP)] (5 mol %), benzene (1 mL), 60 °C overnight. [b] Isolated yield, corrected for the E/Z ratios of the substrates.
Overall, the above observations show that these cobalt‐catalysed reactions are compatible with a wide variety of substituents and with overall excellent chemoselectivity. In addition, the reaction can be easily scaled‐up and was carried out in (near) gram scale. At this scale, lowering the catalyst loading to 1 mol % did not have an obvious influence on the yield (Scheme 2).
Scheme 2.
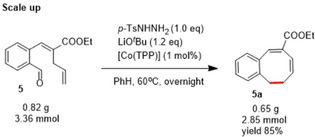
Catalytic monobenzocyclooctadiene synthesis performed at larger scale.
The molecular structures of monobenzocyclooctadiene products 13 a and 19 a were confirmed by single crystal X‐ray diffraction, as shown in Figure 3 (see SI for details).
Figure 3.
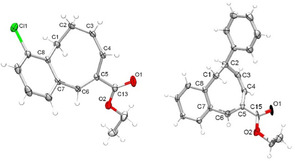
Molecular structures of 13 a (left) and 19 a (right), as determined by single crystal X‐ray diffraction. Selected bond distances [Å] for 13 a: C1–C2 1.530(2), C2–C3 1.501(2), C3–C4 1.334(2), C4–C5 1.478(2), C5–C6 1.374(1), C6–C7 1.471(2), C7–C8 1.405(2), C8–C1 1.506(2). For 19 a: C1–C2 1.536(2), C2–C3 1.504(2), C3–C4 1.328(2), C4–C5 1.473(2), C5–C6 1.341(2), C6–C7 1.469(2), C7–C8 1.399(2), C8–C1 1.501(2).
To gain a better understanding of the mechanism, the generation of 5 a from substrate 5 was investigated by DFT calculations (Scheme 3). First, aldehyde substrate 5 is in situ converted to the corresponding N‐tosylhydrazone and diazo compound 5′ by a non‐catalyzed process. Diazo compound 5′ is trapped and activated by the cobalt(II) catalyst (intermediate B) to produce cobalt(III)‐carbene radical intermediate C through irreversible, exergonic dinitrogen loss via low barrier (+11.3 kcal mol−1) transition state TS1. Subsequently, H‐atom transfer (HAT) from the terminal allylic position to the carbene radical carbon affords delocalized allyl radical intermediate D. This process is also exergonic with a low barrier (TS2, barrier +8.1 kcal mol−1).
Scheme 3.
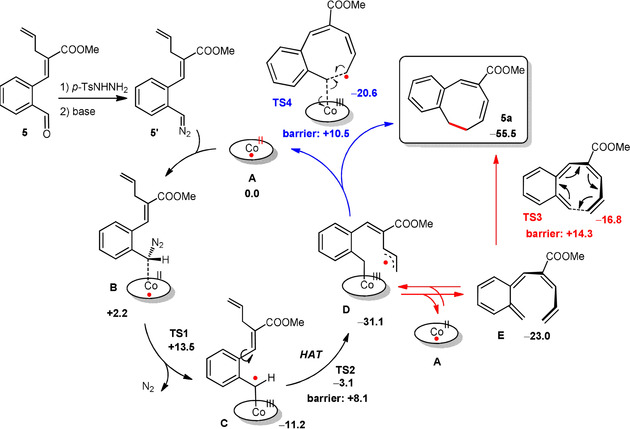
Proposed mechanism for the [Co(porphyrin)]‐catalyzed formation of monobenzocyclooctadienes, based on DFT calculations (BP86, def2‐TZVP, m4 grid, disp3). All Gibbs free energies (ΔG°333 K in kcal mol−1) including those of TS1–TS4, are reported relative to the energy of intermediate A. The ellipse represents the porphyrin ligand. To reduce computation time, a COOMe group and a simplified porphyrin without phenyl substituents on the meso‐positions were used.
The first steps of the mechanism (from A to D) are similar to those proposed for closely related ring‐closure reactions of carbene precursors, catalyzed by [Co(TPP)].12, 13, 17, 18, 19, 20, 21 For these reactions, PBN spin trapping experiments confirmed formation of carbene radical complexes of type C,12, 17 which have also been detected in relation to cyclopropanation reactions.13c, 13d
To gather more experimental data about the 1,6‐HAT process in TS2 we decided to perform kinetic H,D‐isotope competition experiments. Unfortunately, selective (mono)deuteration of substrate 5 is non‐trivial. Therefore we performed these studies on a related cyclization producing a 6‐membered (piperidine) ring.21 This reaction also proceeds via an intramolecular radical‐type 1,6‐HAT step, similar to TS2 connecting C and D in Scheme 3, as could be confirmed by a combination of intermolecular (k H/k D=1) and intramolecular (k H/k D=8.5) kinetic isotope effect (KIE) measurements (see ESI for details). Furthermore, the data show that the HAT step in these ring‐closure reactions is not rate‐limiting, in good agreement with the DFT calculations (see also ref. 21). The experimentally determined intramolecular KIE (k H/k D=8.5) is a bit larger than the DFT calculated value (k H/k D=4.1) based on the harmonic oscillator approximation in the gas phase (333 K), which is perhaps caused by solvation effects or a tunneling contribution to the 1,6‐HAT process. Similar inter‐ and intramolecular KIE values were measured for the [Co(TMP)]‐catalyzed cyclization of (4‐azidobutyl)benzene to the corresponding pyrrolidine product, which proceeds via a 1,5‐HAT process involving a nitrene radical intermediate.25
From D, two competitive pathways for formation of the final product 5 a are conceivable, each with similar barriers. One process is ring‐closure via a radical‐rebound step in the coordination sphere of cobalt, in which the terminal carbon atom of the allyl radical moiety of D attacks the antibonding orbital of the cobalt(III)–carbon bond, leading to simultaneous C−C bond formation and homolysis of the weak Co−C bond (TS4) to form product 5 a in an exergonic process (ΔG°333K=−24.4 kcal mol−1) with regeneration of the cobalt(II) catalyst. This process has a low barrier of only +10.5 kcal mol−1 (Scheme 3, blue arrows). An alternative pathway involves dissociation of o‐QDM intermediate E from intermediate D (and the regenerated cobalt(II) catalyst) by a homolysis reaction of the weak Co−C bond that is decoupled from the C−C bond forming ring‐closure step (Scheme 3, red arrows). The final product 5 a is then formed by a non‐catalyzed, exergonic and low‐barrier re‐aromatizing 8π‐cyclization step (Scheme 3, red arrows, TS3).
We also computed the energy barriers for the various mechanisms leading to formation of the thermodynamically more stable 6‐membered 1,2‐dihydronaphthalene ring product from intermediate D. However, in good agreement with the experimental results, the energy barriers for those reactions are all much higher than those leading to the 8‐membered ring product, thus explaining the experimentally observed formation of monobenzocyclooctadienes as the kinetically favored products (see SI for more details). The transition states for 6‐membered ring formation cannot adopt a favorable boat conformation in these reactions, which probably contributes to the kinetic preference for formation of 8‐membered rings. The reaction is highly chemoselective and no 6‐membered ring product is formed at all.
Relative to the energy of species D, the overall barrier of the 8π‐cyclization (TS3) is computed to be ≈4 kcal mol−1 higher than the barrier of the radical‐rebound process (TS4). The DFT data thus suggest that the reaction might proceed via a metal‐catalyzed ring‐closure step in the coordination sphere of cobalt, involving radical‐rebound transition state TS4, which is of relevance for the development of enantioselective reactions (see below). However, since the TS3 pathway via o‐QDM E is associated with a significant entropy change (which is difficult to compute), distinguishing between the TS3 and TS4 pathways solely on the basis of DFT is not reliable (also because the ring‐closing mechanism could very well be dependent on the applied reaction temperature).
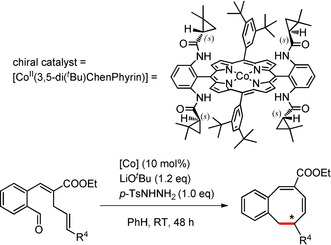
Hence, to get more information about the reaction mechanism and to explore the prospects for enantioselective formation of 8‐membered rings, we decided to perform the ring‐closure reaction of substrate 18 and 19 using Zhang's D2‐symmetric chiral catalyst [CoII(3,5‐di(tBu)ChenPhyrin)].26
Interestingly, initial results demonstrate that monobenzocyclooctadienes can be obtained in an enantioselective manner and with similar yields as with [CoII(TPP)] (Table 5). While the obtained enantiomeric excesses are low at present, the results clearly show that catalytic enantioselective 8‐membered ring synthesis via a carbene radical intermediate is possible. The mechanistic implications of these results currently remain a bit ambiguous. The data strongly suggest that ring‐closure proceeds via the TS4 pathway. However, we cannot fully exclude the TS3 pathway based on these results, because ring‐closure of o‐QDM intermediate E via TS3 could in principle also occur in an enantioselective manner via H‐bonding interactions with the catalyst in the second coordination sphere.26, 27 We are currently investigating the origin of the enantioselectivity and are trying to obtain the products in a higher enantiomeric excess.
Table 5.
First attempts towards enantioselective [Co(porphyrin)]‐catalyzed synthesis of monobenzocyclooctadienes.[a]
|
R |
Yield [%][b] |
ee [%][c] |
|---|---|---|
|
Me |
47 |
12 |
|
Ph |
70 |
18 |
[a] Reaction conditions: 18 or 19 (0.1 mmol, 1.0 equiv), LiOtBu (0.12 mmol, 1.2 equiv), p‐TsNHNH2 (0.1 mmol, 1.0 equiv), [CoII(3,5‐di(tBu)ChenPhyrin)] (10 mol %), benzene (1 mL), room temperature, 48 h. [b] Isolated yield, corrected for the E/Z ratios of the substrates. [c] Determined by chiral HPLC analysis.
Conclusion
In conclusion, cobalt(II) metalloradical catalysis enables the synthesis of two types of 8‐membered ring compounds. Both dibenzocyclooctenes and unprecedented monobenzocyclooctadienes are readily obtained. The method described in this paper has a broad substrate scope, thus allowing the synthesis of a variety of new 8‐membered ring compounds in good yields and with excellent functional group tolerance. The carbene precursor in this reaction is prepared by reacting the corresponding aldehyde with p‐toluenesulfonyl hydrazide in one pot, which greatly simplifies the experimental operation. The reactions proceed via initial hydrogen atom transfer from the bis‐allylic/benzallylic C−H bond to the carbene radical, followed by two different ring‐closure steps leading to the two types of 8‐membered ring products. While the dibenzocyclooctenes are most likely formed by dissociation of o‐QDMs that undergo a non‐catalyzed ring‐closure via 8π‐cyclization, DFT calculations suggest that the monobenzocyclooctadienes are more likely to be generated in a radical‐rebound step in the coordination sphere of cobalt, thus allowing enantioselective formation of chiral monobenzocyclooctadienes, as supported by our initial results.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
We thank Ed Zuidinga for HRMS measurements. Financial support from the Netherlands Organization for Scientific Research (NWO TOP‐Grant 716.015.001 and ARC‐CBBC), the University of Amsterdam (Research Priority Area Sustainable Chemistry) and the China Scholarship Council for a PhD fellowship (CSC 201806050112) is gratefully acknowledged.
M. Zhou, M. Lankelma, J. I. van der Vlugt, B. de Bruin, Angew. Chem. Int. Ed. 2020, 59, 11073.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.11871501.v1).
References
- 1. Petasis N. A., Patane M. A., Tetrahedron 1992, 48, 5757–5821. [Google Scholar]
- 2. Harvey B. G., Merriman W. W., Koontz T. A., Energy Fuels 2015, 29, 2431–2436. [Google Scholar]
- 3.
- 3a. Kraft P., Bajgrowicz J. A., Denis C., Fráter G., Angew. Chem. Int. Ed. 2000, 39, 2980–3010; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3106–3138; [Google Scholar]
- 3b. Fráter G., Bajgrowicz J. A., Kraft P., Tetrahedron 1998, 54, 7633–7703. [Google Scholar]
- 4.
- 4a. Yang J. F., Wang R. H., Wang Y. X., Yao W. W., Liu Q. S., Ye M., Angew. Chem. Int. Ed. 2016, 55, 14116–14120; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14322–14326; [Google Scholar]
- 4b. Kina A., Ueyama K., Hayashi T., Org. Lett. 2005, 7, 5889–5892; [DOI] [PubMed] [Google Scholar]
- 4c. Nakamura I., Chan C. S., Araki T., Terada M., Yamamoto Y., Adv. Synth. Catal. 2009, 351, 1089–5892. [Google Scholar]
- 5.
- 5a. Molander G. A., Acc. Chem. Res. 1998, 31, 603–609; [Google Scholar]
- 5b. Yet L., Chem. Rev. 2000, 100, 2963–3007.11749312 [Google Scholar]
- 6.“Synthesis of Natural Products Containing Medium-Size Carbocycles by Ring-Closing Alkene Metathesis”: Blanchard N., Eustache J. in Metathesis in Natural Product Synthesis (Eds.: J. Cossy, S. Arseniyadis, C. Meyer), Wiley, New York, 2010, pp. 1–43. [Google Scholar]
- 7.
- 7a. Frühauf H. W., Chem. Rev. 1997, 97, 523–596; [DOI] [PubMed] [Google Scholar]
- 7b. Kirillova M. S., Muratore M. E., Dorel R., Echavarren A. M., J. Am. Chem. Soc. 2016, 138, 3671–3674; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Xia Y., Liu Z., Xiao Q., Qu P., Ge R., Zhang Y., Wang J., Angew. Chem. Int. Ed. 2012, 51, 5714–5717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5812–5815. [Google Scholar]
- 8.
- 8a. Wender P. A., Ihle N. C., J. Am. Chem. Soc. 1986, 108, 4678–4679; [Google Scholar]
- 8b. Wender P. A., Ihle N. C., Correia C. R. D., J. Am. Chem. Soc. 1988, 110, 5904–5906; [Google Scholar]
- 8c. Kennedy C. R., Zhong H., Macaulay R. L., Chirik P. J., J. Am. Chem. Soc. 2019, 141, 8557–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Evans A. P., Robinson J. E., Baum E. W., Fazal A. N., J. Am. Chem. Soc. 2002, 124, 8782–8783; [DOI] [PubMed] [Google Scholar]
- 9b. Lautens M., Tam W., Lautens C. J., Edwards L. G., Crudden C. M., Smith C. A., J. Am. Chem. Soc. 1995, 117, 6863–6879; [Google Scholar]
- 9c. Wang Y., Yu Z.-X., Acc. Chem. Res. 2015, 48, 2288–2296. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Rigby J. H., Laurent S. B., Kamal Z., Heeg M. J., Org. Lett. 2008, 10, 5609–5612; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Rigby J. H., Henshilwood J. A., J. Am. Chem. Soc. 1991, 113, 5122–5123. [Google Scholar]
- 11. Lautens M., Fagnou K., Hiebert S., Acc. Chem. Res. 2003, 36, 48–58. [DOI] [PubMed] [Google Scholar]
- 12. te Grotenhuis C., van den Heuvel N., van der Vlugt J. I., de Bruin B., Angew. Chem. Int. Ed. 2018, 57, 140–145; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 146–151. [Google Scholar]
- 13.
- 13a. Dzik W. I., Zhang X. P., de Bruin B., Inorg. Chem. 2011, 50, 9896–9903; [DOI] [PubMed] [Google Scholar]
- 13b. Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279; [Google Scholar]
- 13c. Dzik W. I., Xu X., Zhang X. P., Reek J. N. H., de Bruin B., J. Am. Chem. Soc. 2010, 132, 10891–10902; [DOI] [PubMed] [Google Scholar]
- 13d. Lu H., Dzik W. I., Xu X., Wojtas L., de Bruin B., Zhang X. P., J. Am. Chem. Soc. 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Intrieri D., Caselli A., Gallo E., Eur. J. Inorg. Chem. 2011, 5071–5081; [Google Scholar]
- 14b. Chirila A., Das B. G., Paul N. D., de Bruin B., ChemCatChem 2017, 9, 1413–1421; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Zhu S., Ruppel V. J., Lu H., Wojtas L., Zhang X. P., J. Am. Chem. Soc. 2008, 130, 5042–5043; [DOI] [PubMed] [Google Scholar]
- 14d. Goswami M., de Bruin B., Dzik W. I., Chem. Commun. 2017, 53, 4382–4385; [DOI] [PubMed] [Google Scholar]
- 14e. Huang L., Chen Y., Gao G.-Y., Zhang X. P., J. Org. Chem. 2003, 68, 8179–8184. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Majumdar N., Paul N. D., Mandal S., de Bruin B., Wulff W. D., ACS Catal. 2015, 5, 2329–2366; [Google Scholar]
- 15b. Paul N. D., Mandal S., Otte M., Cui X., Zhang X. P., de Bruin B., J. Am. Chem. Soc. 2014, 136, 1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui X., Xu X., Wojtas L., Kim M. M., Zhang X. P., J. Am. Chem. Soc. 2012, 134, 19981–19984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Das B. G., Chirila A., Tromp M., Reek J. N. H., de Bruin B., J. Am. Chem. Soc. 2016, 138, 8968–8975. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Wen X., Wang Y., Zhang X. P., Chem. Sci. 2018, 9, 5082–5086; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Karns A. S., Goswami M., de Bruin B., Chem. Eur. J. 2018, 24, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Paul N. D., Chirila A., Lu H., Zhang X. P., de Bruin B., Chem. Eur. J. 2013, 19, 12953–12958; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Chirila A., van Vliet K. M., Paul N. D., de Bruin B., Eur. J. Inorg. Chem. 2018, 2251–2258. [Google Scholar]
- 20. te Grotenhuis C., Das B. G., Kuijpers P. F., Hageman W., Trouwborst M., de Bruin B., Chem. Sci. 2017, 8, 8221–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lankelma M., Olivares A. M., de Bruin B., Chem. Eur. J. 2019, 25, 5658–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y., Wen X., Cui X., Zhang X. P., J. Am. Chem. Soc. 2018, 140, 4792–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Bamford W. R., Stevens T. S., J. Chem. Soc. 1952, 4735–4740; [Google Scholar]
- 23b. Xia Y., Wang J., Chem. Soc. Rev. 2017, 46, 2306–2362. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Roy S., Das S. K., Chattopadhyay B., Angew. Chem. Int. Ed. 2018, 57, 2238–2243; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2260–2265; [Google Scholar]
- 24b. Reddy A. R., Zhou C. Y., Guo Z., Wei J., Che C.-M., Angew. Chem. Int. Ed. 2014, 53, 14175–14180; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14399–14404. [Google Scholar]
- 25. Kuijpers P. F., Tiekink M. J., Breukelaar W. B., Broere D. L. J., van Leest N. P., van der Vlugt J. I., Reek J. N. H., de Bruin B., Chem. Eur. J. 2017, 23, 7945–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Chen Y., Fields K. B., Zhang X. P., J. Am. Chem. Soc. 2004, 126, 14718–14719; [DOI] [PubMed] [Google Scholar]
- 26b. Lu H., Tao J., Jones J. E., Wojtas L., Zhang X. P., Org. Lett. 2010, 12, 1248–1251. [DOI] [PubMed] [Google Scholar]
- 27. van Leest N. P., Epping R. F. J., van Vliet K. M., Lankelma M., van den Heuvel E. J., Heijtbrink N., Broersen R., de Bruin B. in Advances in Organometallic Chemistry, Vol. 70 (Eds.: P. J. Pérez, F. G. A. Stone, R. West), Academic Press, New York, 2018, pp. 71–180. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary