

Abstract
Gaining an understanding of the conformational behavior of fluorinated compounds would allow for expansion of the current molecular design toolbox. In order to facilitate drug discovery efforts, a systematic survey of a series of diversely substituted and protected fluorinated piperidine derivatives has been carried out using NMR spectroscopy. Computational investigations reveal that, in addition to established delocalization forces such as charge–dipole interactions and hyperconjugation, solvation and solvent polarity play a major role. This work codifies a new design principle for conformationally rigid molecular scaffolds.
Keywords: conformational behavior, fluorine, NMR analysis, piperidines, solvation effect
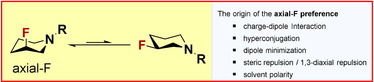
Fluorine ax‐plained! The axial‐F preference of fluorinated piperidines can be attributed to delocalization forces such as charge–dipole interactions and hyperconjugation. In addition to these established forces, solvation and solvent polarity were found to play a major role in the stabilization of these entities, which was supported by experimental and computational investigations of a variety of fluorinated piperidine derivatives (see scheme).
The introduction of fluorine atoms into molecules and materials across many fields of academic and industrial research is now commonplace, owing to their unique properties and effects.1 Therefore, the incorporation of fluorine into drug lead candidates has been recognized as a powerful strategy to improve their pharmacokinetic and physicochemical properties.2 For example, the high C−F bond energy increases metabolic stability2 and the electronic effects of fluorine allow for modification of critical properties such as the pK a.2 Significantly, a fine‐tuning of polarity and lipophilicity can increase solubility and membrane permeability, which may in turn increase the likelihood of success in clinical trials (Scheme 1).
Scheme 1.
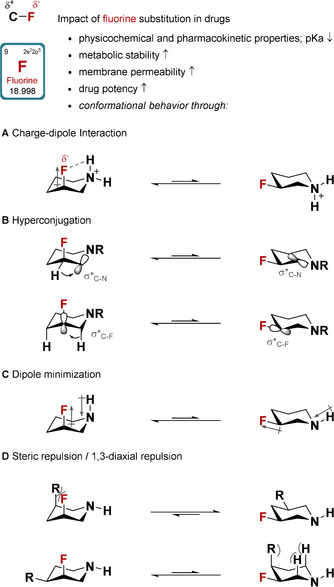
The conformational preferences of fluorinated piperidine derivatives can be attributed to A) charge‐dipole interactions, B) hyperconjugation, C) dipole minimization, and D) steric repulsion.
A particularly striking feature of fluorine substitution is its impact on the relative orientation of a C−F bond when incorporated into aliphatic carbocyclic and acyclic systems, which allows for the design of highly polar compounds.3 For aliphatic, heterocyclic systems, these effects can lead to more rigid structures, which enable the stabilization of well‐defined conformers. Fluorinated piperidines represent an exceptionally interesting case for these phenomena, since the piperidine moiety and related saturated N‐containing heterocycles are frequently present in bioactive compounds.4 Owing to limited synthetic access, typically via tedious, multi‐step synthesis, the study of their conformational behavior has been the subject of few reports, mainly focusing on 3‐fluoropiperidine (1) derivatives. For instance, the axial orientation of fluorine in the protonated 3‐fluoropiperidinium cation was mainly attributed to the occurrence of strong charge–dipole interactions (C−F⋅⋅⋅ HN+) (Scheme 1 A).5, 6 In addition, hyperconjugative interactions, often referred to as the fluorine gauche effect, can contribute to the stabilization of the axial orientation of the fluorine atom, mainly through electron donation from anti‐periplanar C−H bonds into the low‐lying σ*C−F and σ*C−N orbitals (Scheme 1 B).3, 7 Additional factors such as dipole minimization, steric repulsion and solvation effects have been described to partially contribute to the conformational behavior, but were considered to be the least competitive (Schemes 1 C–D).6, 8
While most studies are limited to examples of 3‐fluoropiperidine (1) derivatives, an extensive and systematic evaluation of the conformational effects of a wide range of substitution patterns has not been carried out until now. We believe that such a study would be highly valuable to the scientific community, in particular since even slight changes in the three‐dimensional structure might dramatically change the likelihood of success of lead compounds in therapeutic applications.2
We recently described a straightforward process for the preparation of fluorinated piperidines.9 In this reaction, fluoropyridine precursors underwent a catalytic dearomatization‐hydrogenation sequence to furnish a plethora of substituted, all‐cis‐(multi)fluorinated piperidines in a highly diastereoselective fashion. Within the course of this study we became interested in the conformational behavior of the newly accessed fluorinated piperidines (1–12), obtained as the trifluoroacetamide (1A–12A) or HCl salts (1B–12B). Analysis of the 3 J(19F,1H) coupling in NMR experiments allowed us to determine the relative orientation of the fluorine atom(s), which were often found to adopt either axial or equatorial orientations exclusively.10 In addition to the TFA and HCl analogues, we prepared an additional library of unprotected fluorinated piperidines (NH‐analogues, 1C–12C) and studied their conformational behavior. To rationalize the conformational behavior of the fluorinated piperidine derivatives (1–12), we performed a systematic computational analysis (M06‐2X/def2‐QZVPP). Individual DFT calculations were performed in the gas phase and in solution using a polarizable continuum model (PCM, TFA analogues in CHCl3, HCl‐ and NH‐analogues in water). Pleasingly, the experimentally observed conformer could be predicted computationally in almost all cases. For instance, the free enthalpy differences ΔG between the two conformers in 3‐ fluoropiperidine (1) and 3,5‐difluoropiperidine (2) derivatives indicate a strong preference towards the Faxial conformation in solution (Scheme 2). Interestingly, while the axial preference in solution for HCl‐analogues (1B, 2B) is mainly attributed to electrostatic interactions (ΔE elect,a‐e for 1B and 2B is +12.6, +14.7 kcal mol−1, respectively), hyperconjugative interactions are found to play a significant role in TFA‐ (1A, 2A) and NH‐analogues (1C, 2C) (ΔE hyperc,a‐e for 1A, 1C, 2A, and 2C is +3.3, +5.1, +11.7, and +10.8 kcal mol−1, respectively—for more details see the Supporting Information). It should be noted that the axial preference of fluorinated piperidine analogues of 1 and 2 was confirmed experimentally by NMR studies (Scheme 2). Along these lines, we also performed the same analysis on 3‐fluoro‐4‐methylpiperidine (3) (Scheme 3). Both computational and experimental studies showed high axial preference for all variants (TFA‐, HCl‐, and NH‐ analogues). In this particular case, we believe that in addition to the abovementioned forces (ΔE elect,a‐e of 3A, 3B and 3C is −0.8, +8.5, and +13.5 kcal mol−1, respectively; ΔE hyperc,a‐e of 3A, 3B and 3C is +5.0, +3.0, and +5.8 kcal mol−1, respectively), the steric influence of the methyl substituent (A Me=1.7 kcal mol−1) plays a major role in promoting the axial preference of the fluorine atom (ΔΔE steric,a‐e of 3A, 3B and 3C is +7.5, +6.4, and +0.3 kcal mol−1, respectively, relative to 1A‐C).
Scheme 2.
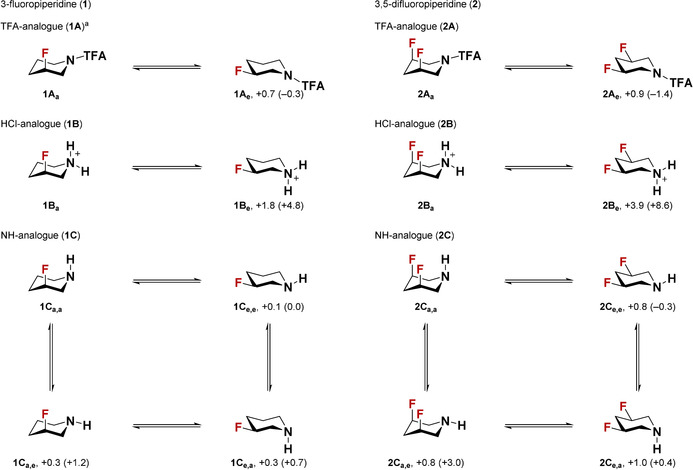
The conformational preferences of 3‐fluoropiperidine (1) and 3,5‐difluoropiperidine (2) and their TFA‐(A), HCl‐(B), and NH‐(C)‐analogues. The free enthalpy differences between the equatorial conformer to the axial conformer (ΔG) are presented as follows: ΔG Solvent (ΔG Gas Phase). The ΔG values for TFA‐, and for both HCl‐, and NH‐analogues are given in chloroform and water, respectively. All values are given in kcal mol−1. Experimentally, all analogues of 1 and 2 showed high axial preference. In NH‐analogues 1C and 2C, we were unable to determine the orientation of the N−H bond because of a fast H/D exchange in solution. [a] Both computational analysis and experimental observation were carried out in toluene.
Scheme 3.
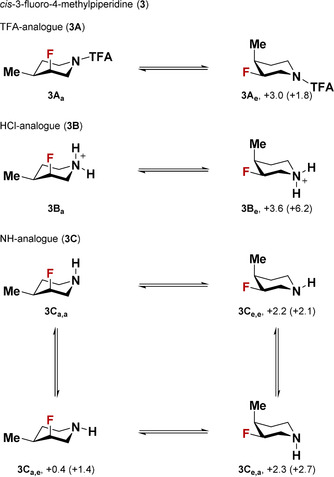
The conformational preferences of cis‐3‐fluoro‐4‐methylpiperidine (3) and its TFA‐(A), HCl‐(B), and NH‐(C)‐ analogues. All values are given in kcal mol−1. The experimental observation is based on 3 J(19F,1H) values. See the Supporting Information for more details.
Inspired by these preliminary results, we conducted the same systematic analysis for all of the newly accessed fluorinated piperidine derivatives, including all different analogues (1–12) (Table 1). The free enthalpy differences (ΔG), electrostatic, hyperconjugation and steric contributions including dipole moments and geometries for all conformers are presented in detail in the Supplementary Information.
Table 1.
Conformational behavior of all‐cis‐(multi)fluorinated piperidines.[a]
|
| ||||
|---|---|---|---|---|
|
|
[kcal mol−1] |
|
||
|
Compd. |
Gas phase |
H2O |
CHCl3 |
Exptl. |
|
1, A |
+0.1 |
– |
−0.4[a] |
axial |
|
B |
+4.8 |
+1.8 |
– |
axial |
|
C |
0.0 |
+0.1 |
– |
axial |
|
2, A |
−1.4 |
– |
+0.9 |
axial |
|
B |
+8.6 |
+3.9 |
– |
axial |
|
C |
−0.3 |
+0.8 |
– |
axial |
|
3, A |
+1.8 |
– |
+3.0 |
axial |
|
B |
+6.2 |
+3.6 |
– |
axial |
|
C |
+2.1 |
+2.2 |
– |
axial |
|
4, A |
−1.9 |
– |
−1.2 |
equatorial |
|
B |
+2.9 |
−0.4 |
– |
equatorial |
|
C |
−1.9 |
−2.0 |
– |
equatorial |
|
5, A |
−4.3 |
– |
−3.7 |
equatorial |
|
B |
+6.2 |
+3.3 |
– |
axial |
|
C |
+2.5 |
+2.1 |
– |
axial |
|
6, A |
−3.7 |
– |
−3.3 |
equatorial |
|
B |
+6.8 |
+3.5 |
– |
axial |
|
C |
+2.5 |
+2.7 |
– |
axial |
|
7, A |
−6.0 |
– |
−4.4 |
equatorial |
|
B |
+7.7 |
+5.2 |
– |
axial |
|
C |
+1.3 |
+2.1 |
– |
axial |
|
8, A |
+0.2 |
– |
+0.6 |
axial |
|
B |
+4.2 |
+1.1 |
– |
axial |
|
C |
+0.4 |
+0.3 |
– |
axial |
|
9, A |
+0.1 |
– |
+2.3 |
axial |
|
B |
+9.5 |
+5.1 |
– |
axial |
|
C |
+1.7 |
+2.8 |
– |
axial |
|
10, A |
+0.7 |
– |
+0.4 |
axial |
|
B |
+3.0 |
+1.0 |
– |
equatorial |
|
C |
−0.9 |
−0.4 |
– |
equatorial |
|
11, A |
+1.5 |
– |
+1.4 |
axial |
|
B |
+3.9 |
+2.3 |
– |
axial |
|
C |
+0.5 |
+1.1 |
– |
axial |
|
12, A |
+3.7 |
– |
+5.4 |
axial |
|
B |
+0.4 |
−1.7 |
– |
equatorial |
|
C |
−3.7 |
−3.7 |
– |
equatorial |
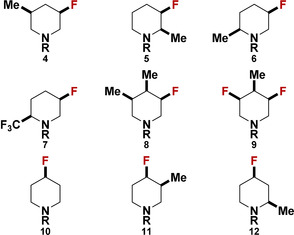
[a] The conformational preferences of fluorinated piperidine (1–12) and its R=TFA‐(A), HCl‐(B), and NH‐(C)‐analogues. The ΔG values for TFA‐ and for both HCl‐, and NH‐analogues are given in chloroform and water, respectively. All values are given in kcal mol−1. [b] This compound was measured in toluene.
As mentioned above, in the vast majority of cases the computed conformer free enthalpy differences in both TFA‐, HCl‐, and NH‐analogues (in solution) are in qualitative agreement with the experimentally observed conformational preferences (Table 1). In a singular event however, the supposedly less stable conformer, as derived from computational analysis, was observed experimentally. The free enthalpy difference of 4‐fluoropiperidinium salt (10B) in the gas phase and in aqueous solution (+3.0, +1.0 kcal mol−1, respectively) suggests that the axial orientation of the fluorine atom should be more favored. In aqueous solution, the equatorial conformer was observed to be dominant. This puzzling observation suggests that additional factors might play a major role in predicting the conformational preference. While examining all computational results, we realized that that the molecular dipole moment μ has a significant impact on the stabilization energy of conformers in polar solution. In the case of the 4‐fluoropiperidinium salt (10B), the equatorial conformer has a significantly larger dipole moment (μ e,gas=8.0 D) than the axial conformer (μ a,gas=6.4 D) and can therefore be significantly stabilized in aqueous solution. Such an effect can be observed particularly for charged species in highly polar solvents and is presumably underestimated computationally by the simple PCM.
Consequently, we became interested in examining whether solvent polarity can affect conformational behavior, as suggested by Abraham for the rotamers of ethane derivatives in the 1960s.11 We initially investigated whether the axial preference is preserved in 3,5‐difluoropiperidine (2C) in different solvents (See the Supplementary Information for more details). Both computational and experimental analyses showed that the fluorine atoms adopt an exclusively axial orientation in all cases (see the Supporting Information for more details). The computational analysis however suggests an increasing stability of the more polar Faxial conformer with increasing solvent polarity (ΔG a–e=+0.2, +0.5, +0.6, +0.8, and +0.8 kcal mol−1 in C6H6, CHCl3, CH2Cl2, DMSO and H2O respectively).
To further explore these phenomena, we conducted the same analysis on 3,5‐difluoropiperidine (2), employing different N‐protecting groups (13–15) (Table 2). Initially, we examined the conformational behavior of the TFA‐analogue (2A) in different solvents and identified the same clear correlation between solvent polarity and the preference for the Faxial conformation; the higher the solvent polarity, the higher the preference for axial orientation (3 J(3‐Fa,4‐Ha) values in C6H6, CHCl3, CH2Cl2, and DMSO are 34.1, 36.1, 38.8, 44.4 Hz, respectively). The same observation was made while studying acetyl‐protected 3,5‐difluoropiperidine (13): in both chloroform and DMSO an axial preference was obtained with significantly higher values of ΔG and 3 J(3‐Fa,4‐Ha) in the more polar solvent (DMSO). Encouraged by these results, we considered applying this technique to promote the formation of the Faxial conformer in further 3,5‐difluoropiperidine analogues (Table 2). Computational investigations in the gas phase, as well as the experimental observation in chloroform, suggest that in both Pivaloyl‐ (Piv) and tert‐butoxycarbonyl (Boc)‐protected 3,5‐difluoropiperidine (14, 15), the fluorine atoms adopt an equatorial orientation (3 J(3‐Fa,4‐Ha)=7.3, 12.5 Hz for 14 and 15 respectively). By increasing the solvent polarity from chloroform (ϵ=4.81) to DMSO (ϵ=46.7), the conformational behavior of both species can be inverted, favoring the Faxial conformation orientation (3 J(3‐Fa,4‐Ha)=38.5, 40.4 Hz for 14 and 15 respectively).
Table 2.
The conformational preferences of 3,5‐difluoropiperidine derivatives.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
Compd. |
Solvent |
[Kcal mol−1] |
μ (a) |
μ (e) |
Exptl. |
|
|
R=TFA (2A) |
none |
−1.4 |
6.58 |
2.20 |
– |
|
|
|
C6H6 |
+0.1 |
8.06 |
2.73 |
axial |
|
|
|
CHCl3 |
+0.9 |
8.94 |
3.04 |
axial |
|
|
|
CH2Cl2 |
+1.0 |
9.35 |
3.18 |
axial |
|
|
|
DMSO |
+2.0 |
9.79 |
3.33 |
axial |
|
|
R=AC (13) |
none |
−1.5 |
– |
– |
– |
|
|
|
CHCl3 |
+0.3 |
– |
– |
axial |
|
|
|
DMSO |
+2.0 |
– |
– |
axial |
|
|
R=Piv (14) |
none |
−2.4 |
– |
– |
– |
|
|
|
CHCl3 |
−0.9 |
– |
– |
equatorial |
|
|
|
DMSO |
+2.0 |
– |
– |
axial |
|
|
R=Boc (15) |
none |
−2.4 |
– |
– |
– |
|
|
|
CHCl3 |
−0.9 |
– |
– |
equatorial |
|
|
|
DMSO |
+2.0 |
– |
– |
axial |
|

[a] All values are given in kcal mol−1. The experimental observation is based on 3 J(19F,1H) values. See the Supporting Information for more details.
These results suggest that C−F bonds, although often considered to be a bioisostere of C−H bonds,2 can significantly alter the conformational behavior of fluorinated heterocycles such as piperidines. To illustrate how this concept could potentially be applied in the context of molecular design, we investigated the behavior of 4‐methylpiperidine (16) and its fluorinated analogue cis‐3,5‐difluoro‐trans‐4‐methylpiperidine (17) computationally (Scheme 4).
Scheme 4.
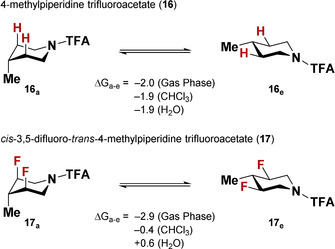
Tuning the conformational behavior of 4‐methylpiperidine analogues by fluorine substitutions. All free enthalpy values are given in kcal mol−1.
As expected for the sterically demanding methyl group, the Meequatorial conformer is preferred in both compounds, both in the gas phase and in chloroform (Scheme 4). By switching to more polar solvents such as water, the conformational equilibrium can be significantly shifted. Whilst compound 16 retains its Meequatorial conformer in polar solution, the fluorine atoms in 17 induce a conformational inversion, directing the methyl group into the sterically hindered axial position—showcasing how fluorine substitution can be utilized to manipulate the conformational behavior of polar molecules.12
In conclusion, the conformational behavior of fluorinated piperidines is influenced by the interplay of different forces such as electrostatic interactions, hyperconjugation and steric factors. In this Communication we provide, for the first time, a detailed and systematic overview of the major parameters that can control the conformational behavior of fluorinated piperidine derivatives while covering a wide range of substitution patterns on the piperidine ring. The fluorinated piperidines were analyzed experimentally (through NMR studies) and computationally (through DFT computations). Interestingly, in addition to the common forces that contribute to the stabilization of a specific conformer, we realized that the dipole moment can be used to further manipulate the orientation of the fluorine atoms, particularly in polar solutions. These forces may eventually be used to fine‐tune the conformational structure of lead compounds which can dramatically affect their likelihood of success in therapeutic applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
All computations were carried out on the high‐performance computing system PALMA II (WWU Münster). We thank Prof. Ryan Gilmour (WWU Münster), Prof. Alexander Hillisch (Bayer Pharma AG) and Professor Peer Kirsch (Merck KGaA) for helpful discussions. Financial support by the European Research Council (ERC Advanced Grant Agreement no. 788558) and Deutsche Forschungsgemeinschaft (SPP2102) is gratefully acknowledged.
Z. Nairoukh, F. Strieth-Kalthoff, K. Bergander, F. Glorius, Chem. Eur. J. 2020, 26, 6141.
Dedicated to Professor Bernd Giese on the occasion of his 80th birthday
Contributor Information
Dr. Zackaria Nairoukh, Email: z.nairoukh@mail.huji.ac.il.
Prof. Dr. Frank Glorius, Email: glorius@uni-muenster.de.
References
- 1.
- 1a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 1b. Hird M., Chem. Soc. Rev. 2007, 36, 2070–2095; [DOI] [PubMed] [Google Scholar]
- 1c. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 1d. Lowe P. T., O'Hagan D., J. Fluor. Chem. 2020, 230, 109420. [Google Scholar]
- 2.
- 2a. Shah P., Westwell A. D., J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540; [DOI] [PubMed] [Google Scholar]
- 2b. O′Hagan D., Chem. Soc. Rev. 2008, 37, 308–319; [DOI] [PubMed] [Google Scholar]
- 2c. Wang J., Sánchez-Roselló M., Aceña J. L., del Pozo C., Sorochinsky A. E., Fustero S., Soloshonok V. A., Liu H., Chem. Rev. 2014, 114, 2432–2506; [DOI] [PubMed] [Google Scholar]
- 2d. Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A., J. Med. Chem. 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Durie A. J., Slawin A. M. Z., Lebl T., Kirsch P., O′Hagan D., Chem. Commun. 2011, 47, 8265–8267; [DOI] [PubMed] [Google Scholar]
- 3b. Durie A. J., Slawin A. M. Z., Lebl T., Kirsch P., O′Hagan D., Chem. Commun. 2012, 48, 9643–9645; [DOI] [PubMed] [Google Scholar]
- 3c. Keddie N. S., Slawin A. M. Z., Lebl T., Philp D., O′Hagan D., Nat. Chem. 2015, 7, 483–488; [DOI] [PubMed] [Google Scholar]
- 3d. Fang Z., Al-Maharik N., Slawin A. M. Z., Bühl M., O′Hagan D., Chem. Commun. 2016, 52, 5116–5119; [DOI] [PubMed] [Google Scholar]
- 3e. Aiguabella N., Holland M. C., Gilmour R., Org. Biomol. Chem. 2016, 14, 5534–5538; [DOI] [PubMed] [Google Scholar]
- 3f. Molnár I. G., Gilmour R., J. Am. Chem. Soc. 2016, 138, 5004–5007; [DOI] [PubMed] [Google Scholar]
- 3g. Wiesenfeldt M. P., Nairoukh Z., Li W., Glorius F., Science 2017, 357, 908–912; [DOI] [PubMed] [Google Scholar]
- 3h. Thomson C. J., Zhang Q., Al-Maharik N., Bühl M., Cordes D. B., Slawin A. M. Z., O'Hagan D., Chem. Commun. 2018, 54, 8415–8418; [DOI] [PubMed] [Google Scholar]
- 3i. Scheidt F., Schäfer M., Sarie J. C., Daniliuc C. G., Molloy J. J., Gilmour R., Angew. Chem. Int. Ed. 2018, 57, 16431–16435; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16669–16673; [Google Scholar]
- 3j. Sadurní A., Gilmour R., Eur. J. Org. Chem. 2018, 3684–3687; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3k. Fang Z., Cordes D. B., Slawin A. M. Z., O′Hagan D., Chem. Commun. 2019, 55, 10539–10542. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. O′Hagan D., Nat. Prod. Rep. 2000, 17, 435–446; [DOI] [PubMed] [Google Scholar]
- 4b. Vitaku E., Smith D. T., Njardarson J. T., J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 5.The strong axial preference in 3-fluoropiperidinium cation and analogues in the gas phase was dictated by electrostatic interactions, which can be both charge dipole (C−F⋅⋅⋅HN+) and hydrogen bond (F⋅⋅⋅H(N+)) interactions. In aqueous solution, charge dipole interactions were strongly attenuated and hyperconjugation was calculated to be at least competitive with Lewis-type interactions. For more information, see ref. [6].
- 6.
- 6a. Lankin D. C., Chandrakumar N. S., Rao S. N., Spangler D. P., Snyder J. P., J. Am. Chem. Soc. 1993, 115, 3356–3357; [Google Scholar]
- 6b. Snyder J. P., Chandrakumar N. S., Sato H., Lankin D. C., J. Am. Chem. Soc. 2000, 122, 544–545; [Google Scholar]
- 6c. Sun A., Lankin D. C., Hardcastle K., Snyder J. P., Chem. Eur. J. 2005, 11, 1579–1591; [DOI] [PubMed] [Google Scholar]
- 6d. Silla J. M., Silva W. G. D. P., Cormanich R. A., Rittner R., Tormena C. F., Freitas M. P., J. Phys. Chem. A 2014, 118, 503–507. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Gooseman N. E. J., O'Hagan D., Peach M. J. G., Slawin A. M. Z., Tozer D. J., Young R. J., Angew. Chem. Int. Ed. 2007, 46, 5904–5908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6008–6012; [Google Scholar]
- 7b. Thiehoff C., Schifferer L., Daniliuc C. G., Santschi N., Gilmour R., J. Fluor. Chem. 2016, 182, 121–126; [Google Scholar]
- 7c. Molnár I. G., Holland M. C., Daniliuc C. G., Houk K. N., Gilmour R., Synlett 2016, 27, 1051–1055; [Google Scholar]
- 7d. Santschi N., Thiehoff C., Holland M. C., Daniliuc C. G., Houk K. N., Gilmour R., Organometallics 2016, 35, 3040–3044; [Google Scholar]
- 7e. Scheidt F., Selter P., Santschi N., Holland M. C., Dudenko D. V., Daniliuc C. G., Mück-Lichtenfeld C., Hansen M. R., Gilmour R., Chem. Eur. J. 2017, 23, 6142–6149; [DOI] [PubMed] [Google Scholar]
- 7f. Thiehoff C., Rey Y. P., Gilmour R., Isr. J. Chem. 2017, 57, 92–100; [Google Scholar]
- 7g. Cormanich R. A., O'Hagan D., Bühl M., Angew. Chem. Int. Ed. 2017, 56, 7867–7870; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7975–7978; [Google Scholar]
- 7h. Aufiero M., Gilmour R., Acc. Chem. Res. 2018, 51, 1701–1710; [DOI] [PubMed] [Google Scholar]
- 7i. Teschers C. S., Daniliuc C. G., Kehr G., Gilmour R., J. Fluorine Chem. 2018, 210, 1–5. [Google Scholar]
- 8. Freitas M. P., Org. Biomol. Chem. 2013, 11, 2885–2890. [DOI] [PubMed] [Google Scholar]
- 9. Nairoukh Z., Wollenburg M., Schlepphorst C., Bergander K., Glorius F., Nat. Chem. 2019, 11, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.It should be noted that the vicinal 3 J(19F,1Ha) coupling constants provide useful insight into the conformational structure, since large values of 3 J(19F,1Ha) indicate axial preference and small values of 3 J(F,Ha) indicate equatorial preference (for more details, see Supporting Information, Section 4, ref. [9]).
- 11.
- 11a. Abraham R. J., Cavalli L., Pachler K. G. R., Mol. Phys. 1966, 11, 471–494; [Google Scholar]
- 11b. Abraham R. J., Cooper M. A., J. Chem. Soc., Chem. Commun. 1966, 588–589; [Google Scholar]
- 11c. Abraham R. J., J. Phys. Chem. 1969, 73, 1192–1199; [Google Scholar]
- 11d. Abraham R. J., Gatti G., J. Chem. Soc. B 1969, 961–968; [Google Scholar]
- 11e. Abraham R. J., Parry K., J. Chem. Soc. B 1970, 539–545; [Google Scholar]
- 11f. Cavalli L., Abraham R. J., Mol. Phys. 1970, 19, 265–274; [Google Scholar]
- 11g. Abraham R. J., Kemp R. H., J. Chem. Soc. B 1971, 1240–1245. [Google Scholar]
- 12.Examples on chameleon molecules:
- 12a. Carrupt P. A., Testa B., Bechalany A., El Tayar N., Descas P., Perrissoud D., J. Med. Chem. 1991, 34, 1272–1275; [DOI] [PubMed] [Google Scholar]
- 12b. El Tayar N., Mark A. E., Vallat P., Brunne R. M., Testa B., van Gunsteren W. F., J. Med. Chem. 1993, 36, 3757–3764; [DOI] [PubMed] [Google Scholar]
- 12c. Fielding L., Clark J. K., McGuire R., J. Org. Chem. 1996, 61, 5978–5981; [Google Scholar]
- 12d. Fox S. J., Gourdain S., Coulthurst A., Fox C., Kuprov I., Essex J. W., Skylaris C.-K., Linclau B., Chem. Eur. J. 2015, 21, 1682–1691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary