

Abstract
Advances in genome sequencing have led to a tremendous increase in the discovery of novel missense variants, but evidence for determining clinical significance can be limited or conflicting. Here, we present Learning from Evidence to Assess Pathogenicity (LEAP), a machine learning model that utilizes a variety of feature categories to classify variants, and achieves high performance in multiple genes and different health conditions. Feature categories include functional predictions, splice predictions, population frequencies, conservation scores, protein domain data, and clinical observation data such as personal and family history and covariant information. L2‐regularized logistic regression and random forest classification models were trained on missense variants detected and classified during the course of routine clinical testing at Color Genomics (14,226 variants from 24 cancer‐related genes and 5,398 variants from 30 cardiovascular‐related genes). Using 10‐fold cross‐validated predictions, the logistic regression model achieved an area under the receiver operating characteristic curve (AUROC) of 97.8% (cancer) and 98.8% (cardiovascular), while the random forest model achieved 98.3% (cancer) and 98.6% (cardiovascular). We demonstrate generalizability to different genes by validating predictions on genes withheld from training (96.8% AUROC). High accuracy and broad applicability make LEAP effective in the clinical setting as a high‐throughput quality control layer.
Keywords: clinical genetics, genetic testing, machine learning, variant classification
LEAP, a machine learning model for variant classification, was developed with explainability in mind. The expected variant classification and supporting evidence are data‐driven, and are displayed in a web application to aid variant scientists in a clinical reporting workflow. Contributing evidence features are ranked based on overall significance, and contribution magnitude and direction (pathogenic or benign driver) are displayed and color coded. As additional context, percentiles are shown for numeric features with respect to the distribution observed in the training data, and past classifications for similar variants (based on gene, chromosome, and exon) are listed.
1. INTRODUCTION
The prevalence of genetic testing in clinical care is growing rapidly, with applications in the diagnosis, management, prevention, and treatment of a large variety of disorders. The utility of genetic testing depends on the accurate classification of genetic variants. Variant classification needs to be rigorous and reproducible between different individual scientists and institutions. To help facilitate this, the American College of Medical Genetics and Genomics (ACMG) issued guidelines for variant classifications (Richards et al., 2015). Early efforts (Nykamp et al., 2017) aimed to expand on this framework by establishing a rules‐based approach to the evaluation of evidence that increases the efficiency and consistency of variant scientists by systematizing the process. Rules such as population frequency cutoffs are known to be strong criteria for automatic classification. However, strong criteria like these only apply to a small minority of variants. For the majority of variants, classification involves reviewing many sources of evidence, some of which are structured (functional predictors and population frequency) and some unstructured (literature text and health history). As our scientific knowledge advances and availability of evidence increases, the interpretation process may become increasingly complex and new edge cases may arise that are not captured by existing rules.
The variant classification process shares many characteristics with other areas in which machine learning is effective. A database of variant classification labels determined by experts and a large number of input signals that drive these expert decisions exists and a mapping between the two is needed. To tackle this with a machine learning approach, meta‐predictors like REVEL (Ioannidis et al., 2016; Tavtigian et al., 2018), MetaSVM, and MetaLR (Dong et al., 2015) were developed, which integrate multiple features primarily from one evidence category (functional predictors) to predict the pathogenicity of missense variants. ClinPred (Alirezaie, Kernohan, Hartley, Majewski, & Hocking, 2018) was developed to incorporate population frequency in addition to functional predictors. However, these features only encompass a small part of the considerations in a variant scientist's workflow.
Here, we present Learning from Evidence to Assess Pathogenicity (LEAP), a machine‐learned approach that has access to many more types of underlying data used in manual variant classification, including functional prediction, splice prediction, evolutionary conservation, population frequency, protein domain, co‐occurring pathogenic (P/LP) variants, and individual and family health history. LEAP effectively predicts the classification that would be applied by variant scientists and outlines driving evidence weighted in order of contribution towards that prediction. We evaluate the predictive performance of LEAP with increasing evidence category inclusion and different model types across many genes and two disease areas and discuss its utility as an aid in the clinical interpretation process. Initial external validation for LEAP's performance resulted from a blind prediction challenge held by the Critical Assessment of Genome Interpretation (CAGI5) ENIGMA Consortium (Cline et al., 2019), in which variations of LEAP came in first, second, third and fourth place against competing models that were either published or newly developed.
2. MATERIALS AND METHODS
2.1. Training variants
A set of 14,226 missense variants in genes associated with elevated risk for hereditary cancer and 5,398 missense variants in genes associated with elevated risk for cardiovascular disorders were used to train models to predict a variant classification. Missense variants were identified using the Alamut Batch coding effect (Interactive Biosoftware, Rouen, France, v1.8). Variants were detected by next‐generation sequencing (NGS) multigene panel tests for hereditary cancer and cardiovascular disorders. Models were trained and assessed separately using cancer and cardiovascular variants but shared the same modeling and validation framework.
Training variants were previously observed and classified in routine clinical testing of individual samples. The cancer test included 24 genes associated with elevated risk for hereditary breast, ovarian, uterine/endometrial, colorectal, melanoma, pancreatic, prostate, and stomach cancer. These genes are APC, ATM, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDKN2A, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53. The cancer test included six other genes (CDK4, EPCAM, GREM1, MITF, POLD1, and POLE) that were excluded from this analysis because only specific variant types and/or preselected variants were tested. The cardiovascular test included 30 genes associated with elevated risk for a cardiovascular disorder. These genes are ACTA2, ACTC1, APOB, COL3A1, DSC2, DSG2, DSP, FBN1, GLA, KCNH2, KCNQ1, LDLR, LMNA, MYBPC3, MYH11, MYH7, MYL2, MYL3, PCSK9, PKP2, PRKAG2, RYR2, SCN5A, SMAD3, TGFBR1, TGFBR2, TMEM43, TNNI3, TNNT2, and TPM1. Laboratory procedures were performed as previously described (Neben et al., 2019). Our cardiovascular variant database may be more enriched for pathogenic (P/LP) variants related to familial hypercholesterolemia (FH) due to the inclusion of large research cohorts that were specifically enriched for this disease phenotype.
Variants were classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB), and benign (B) according to the ACMG 2015 guidelines for sequence variant interpretation (Richards et al., 2015), and all variant classifications were reviewed by a trained variant scientist and signed out by a board‐certified medical geneticist or pathologist. Table S1 provides missense variant counts by classification. To generate model training labels, P and LP variants were grouped and will be referred to as “P/LP” in this paper, and B and LB variants were grouped and will be referred to as “B/LB.” All variants were classified using our existing rules‐based protocol and were not auto‐classified or in any way influenced by LEAP predictions.
2.2. Features
A set of 245 total features were chosen to encompass a majority of the inputs that an expert variant scientist may consider for clinical variant interpretation as recommended by ACMG guidelines. Features were grouped in categories derived from the ACMG guidelines for variant interpretation and are listed in Table 1. These categories are used for comparison of variant evidence categories and their contribution to model performance. The computational features of functional impact at the protein level are GERP++ (Davydov et al., 2010), likelihood ratio test (Chun & Fay, 2009), phastCons100way (Siepel et al., 2005), Align GVGD (Tavtigian et al., 2006), MutationTaster2 (Schwarz, Cooper, Schuelke, & Seelow, 2014), PolyPhen‐2‐HVAR (Adzhubei, Jordan, & Sunyaev, 2013), and SIFT (Ng & Henikoff, 2003). The features with RNA splicing impact are Alamut (Interactive Biosoftware, Rouen, France) and Skippy (Woolfe, Mullikin, & Elnitski, 2010). The features based on the location of the variant are dbNSFP Interpro (Finn et al., 2017) and the gene annotation. Population minor allele frequency (MAF) was derived from gnomAD (Lek et al., 2016). The detailed feature list for both cancer and cardiovascular models are available in Tables S2 and S3.
Table 1.
Variant evidence features grouped by category, inspired by the ACMG variant interpretation guidelines
| Category | Source | Description |
|---|---|---|
| Computational predictions of functional impact (FUNC) | GERP++ | Nucleotide conservation |
| Likelihood ratio test | Evolutionary conservation | |
| phastCons100way | Nucleotide conservation | |
| Align GVGD | Protein functional impact and evolutionary conservation | |
| MutationTaster2 | Protein functional impact and evolutionary conservation | |
| Polyphen2‐HVAR | Protein functional impact and evolutionary conservation | |
| SIFT | Evolutionary conservation | |
| Splicing impact (SPLICE) | Human Splicing Finder | Position weight matrices |
| MaxEnt | Maximum entropy principle | |
| NNSplice | Neural networks | |
| SpliceSiteFinder‐like | Position weight matrices | |
| Skippy | Detection of exonic variants that modulate splicing: | |
| Distance from splice site | ||
| Regulatory Constraint Score: regulatory potential | ||
| Log odds ratio: changes in regulatory elements (predicted exon splicing enhancers and silencers) | ||
| Location | Interpro domain | Domain or conserved site of variant |
| Exon position relative to transcript | Exon position of variant, divided by the exon position at which the given transcript ends | |
| Gene | Gene annotation | |
| Homopolymer length | Homopolymer length | |
| Health condition | Used in cardiovascular disorders model only. Indicates FH or non‐FH cardiovascular disorders | |
| Population minor allele frequency (MAF) | gnomAD | MAF for overall population and subpopulations (African, Ashkenazi Jewish, East Asian, Finnish European, Latino, Non‐finnish European, and South Asian) |
| Aggregated individual‐level information | Covariants | Proportion of carriers with a known P/LP variant in the same health condition (cancer or cardiovascular) |
| Health history | Proportion of carriers diagnosed with a phenotype before age cutoff | |
| Proportion of family members of carriers diagnosed with a phenotype before age cutoff | ||
| If > 100 carriers, only 100 were randomly sampled and considered |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; FH, familial hypercholesterolemia; P/LP, pathogenic/likely pathogenic.
Numeric features were standardized by centering at the median and scaling to the interquartile range. Missing values were filled using the most frequent value for numeric features, or filled with a “missing” label for categorical features. Categorical features were binarized and one categorical level was dropped for each categorical feature to serve as the reference level in model training. A categorical feature with k levels resulted in k−1 binarized columns: the “missing” level was dropped as the reference level if it existed, but otherwise, the first alphabetically sorted level was dropped.
2.2.1. Feature engineering
Additional features were derived from those listed above to represent various classification criteria for modeling. Population frequency numeric features from gnomAD were grouped to create additional categorical features including: (a) log‐scale MAF groups (0%, 0.0001–0.001%, 0.001–0.01%, 0.01–0.1%, etc.), (b) linear‐scale MAF groups (in increments of 0.005%), and (c) custom MAF groups (<0.1%, <1%, <5%, and ≥5%). To represent a variant's position relative to a clinically relevant transcript, the exon position of the variant was divided by the exon position at which a given transcript ends. Finally, to capture potential splicing impact, four RNA splicing impact algorithms (Human Splicing Finder, MaxEnt, NNSplice, and SpliceSiteFinder) were assessed for significant difference between the variant and wild‐type scores. The proportion of these algorithms indicating a significant difference was calculated, and each algorithm was given equal weight.
2.2.2. Individual‐level clinical features
Two groups of features based on data about individuals from the Color database (Color Genomics, Burlingame, CA) were included: (a) co‐occurrence of P/LP variants (covariants) in genes associated with the same phenotype in the same individuals and (b) health history of the carriers and their family members.
Covariants were encoded as a feature by taking the proportion of total carriers of a given variant who also have another known P/LP variant in the same associated phenotype. We utilize the co‐occurrence of two variants in an individual when those variants are located either in the same gene or in different genes. Both of these may be used as supporting evidence for a benign classification, based on ACMG guideline criteria BP2 and BP5 (Richards et al., 2015). The consideration of co‐occurrence in different genes is not as commonly cited as that for the same gene. However, our rationale for including the former is based on the observation that having two pathogenic variants contributing to an individual's disease phenotype is a very rare event, and as such, a co‐occurring pathogenic variant may signal an alternate genetic explanation for the individual's disease. Our internal data show that the co‐occurrence of two pathogenic variants in the same gene (0.02% of Color samples) or different genes (0.43% of Color samples) is indeed very rare for cancer. During the manual practice of variant classification, we exercise caution in the application of these criteria by considering other mitigating factors, such as the disease spectrum for each gene, disease frequency in the population, penetrance, and the nature and severity of the observed phenotype of the carrier, and these may be additional features to add in the future. The current LEAP co‐occurrence features are simplified, based on the frequency of individuals with co‐occurring pathogenic variants in genes associated with the same phenotype. However, this is one of many types of features used in LEAP, and are taken into consideration with other features like genes that may still allow the model to learn some of the previously listed concepts such as penetrance.
Health histories were represented by features based on the following format for combinations of phenotypes and age cutoffs: (a) proportion of carriers diagnosed with a phenotype X before age cutoff Y and (b) weighted proportion of family members of carriers diagnosed with a phenotype X before age cutoff Y, where first degree relatives were weighted 1.0 while second‐degree relatives were weighted 0.5. In addition, the maximum number of carriers considered for a given variant was capped at 100. If a variant had more than 100 carriers, only 100 were randomly sampled and considered and a flag was added as a feature for “greater than 100 carriers.” The large majority of variants detected were rare (MAF < 0.001%), and only 2.3% of cancer missense variants have >100 carriers.
For cancer, phenotypes considered included breast cancer, colorectal cancer, endometrial cancer, fallopian tube cancer, gastric cancer, hematological malignancy, kidney cancer, melanoma, ovarian cancer, pancreatic cancer, primary peritoneal cancer, prostate cancer, and thyroid cancer. For cardiovascular disorders, phenotypes considered included xanthelasma, xanthoma, corneal arcus, FH, hypertrophic cardiomyopathy, dilated cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic cardiomyopathy, left ventricular noncompaction cardiomyopathy, Fabry disease, long QT syndrome, short QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, Ehlers–Danlos syndrome, Marfan syndrome, Loeys–Dietz syndrome, familial thoracic aortic aneurysms and dissections, thoracic aneurysm, and aorta dissection. Cancer age cutoffs were 40, 50, 60, and 100 years. Cardiovascular disorders age cutoffs were 30, 40, 50, and 100 years.
2.2.3. Feature comparison
For feature comparison, 245 unique features were considered and grouped into five feature categories: computational predictions of functional impact and evolutionary conservation (FUNC), splicing impact (SPLICE), variant location (LOC), population minor allele frequency (MAF), and aggregated individual‐level information (IND; Table 1). These categories were included additively in the specified order to LEAP model variations detailed in Table 2, with the goal of understanding conceptual patterns in model performance from increasing availability of evidence.
Table 2.
Descriptions of feature category subsets and model types used in the feature comparison and model selection analyses for cancer and cardiovascular disorders
| Model name | Features | Model | Variants |
|---|---|---|---|
| LEAP_FEATURE_1 | FUNC | Binary logistic regression | P/LP and B/LB variants |
| LEAP_FEATURE_2 | FUNC+SPLICE | ||
| LEAP_FEATURE_3 | FUNC+SPLICE+LOC | ||
| LEAP_FEATURE_4 | FUNC+SPLICE+LOC+MAF | ||
| LEAP_FEATURE_5 | FUNC+SPLICE+LOC+MAF+IND | ||
| LEAP_MODEL_1 | FUNC+SPLICE+LOC+MAF+IND | Binary logistic regression | P/LP and B/LB variants |
| LEAP_MODEL_2 | Binary random forest | ||
| LEAP_MODEL_3 | Multiclass logistic regression (one‐vs.‐rest) | P/LP, B/LB, and VUS variants | |
| LEAP_MODEL_4 | Multiclass logistic regression (multinomial) | ||
| LEAP_MODEL_5 | Multiclass random forest |
Note: See Table 1 for feature category descriptions. Models submitted to the CAGI5 ENIGMA prediction challenge include LEAP_FEATURE_4 (CAGI5 “LEAP 2”), LEAP_FEATURE_5 (CAGI5 “LEAP 1”), LEAP_MODEL_2 (CAGI5 “LEAP 3”), and LEAP_MODEL_3 (CAGI5 “LEAP 4”). The latter three differ in that the CAGI5 submissions included a set of HGMD features that were excluded in this analysis (see Figure S1 for model performance with HGMD features). Of note, LEAP_FEATURE_4 (CAGI5 “LEAP 2”) excluded HGMD features in both the CAGI5 submission and in this analysis. See the Materials and Methods section for more details.
Abbreviations: B/LB, benign/likely benign; FUNC, functional impact and evolutionary conservation; HGMD, Human Gene Mutation Database; IND, individual‐level information; LOC, variant location; MAF, minor allele frequency; P/LP, pathogenic/likely pathogenic; SPLICE, splicing impact; VUS, variants of uncertain significance.
REVEL and LEAP_FEATURE_1 were used as baseline models, and represent a class of widely utilized computational predictors that consider only functional and conservation scores as inputs. Next, variant location (which includes gene features) and population frequency feature categories were added in LEAP_FEATURE_2 and LEAP_FEATURE_3, respectively. When population frequency cutoffs are used in manual classification, the cutoffs are typically gene‐specific, which implies a hierarchy in which the gene precedes population frequency. Finally, features derived from individual‐level data (covariants and health history) were added in LEAP_FEATURE_5 as this information, while helpful in a few distinct cases, is not expected to be crucial to variant classification and is also not publicly available.
2.3. Models
Models were trained using Python's “scikit‐learn” (sklearn) library (Pedregosa, 2011) on version 0.19.1 to output a predicted probability of pathogenicity for each variant (hereby referred to as predictions). Table 2 provides descriptions of the models considered for feature comparison and model comparison. Default parameters were used from sklearn's LogisticRegression and RandomForestClassifier implementations unless otherwise specified.
An L2‐regularized logistic regression (linear) model and a random forest classification (nonlinear) model with “n_estimators” set to 1,000 trees were trained. In addition to comparing linear and nonlinear models, binary and multiclass models were also compared. Binary classification models excluded VUS and were trained to predict pathogenic or benign only. Multiclass classification models included all variants and were trained to predict P, B, or VUS. Multiclass logistic regression models included one using a “ovr” (one‐vs.‐rest) multiclass method and another using a “multinomial” multiclass method with the “newton‐cg” solver. For multiclass models, “class_weight” was set to “balanced.”
2.4. Model validation
2.4.1. Train‐test split
For assessment and comparison of models with variations on feature category inclusion and model selection outlined in Table 2, 10‐fold cross‐validated predictions were used to assess area under the receiver operating characteristic curve (AUROC) and precision‐recall curve (AUPRC). AUPRC was included as a more representative performance metric for datasets with imbalanced class labels; AUPRC penalizes false positives (FPs) more than AUROC would, because AUPRC assesses precision, defined as (FP/[FP+TP]) as opposed to specificity defined as (FP/FP+TN) in AUROC. Both AUPRC and AUROC assess sensitivity, also known as recall, which is defined as (TP/[TP+FN]). Our training labels were relatively imbalanced, with 1.7% pathogenic rate in cancer and 2.3% in cardiovascular including VUS, and 12.8% and 13.3% excluding VUS, respectively. VUS were used to train multiclass models (LEAP_MODEL_3, 4, and 5), but were excluded from all model area under the curve (AUC) plots (Figures 1, 2, 4, and 5), including for multiclass model predictions to yield a more commensurable comparison with binary classification model (LEAP_MODEL_1 and 2) predictions.
Figure 1.
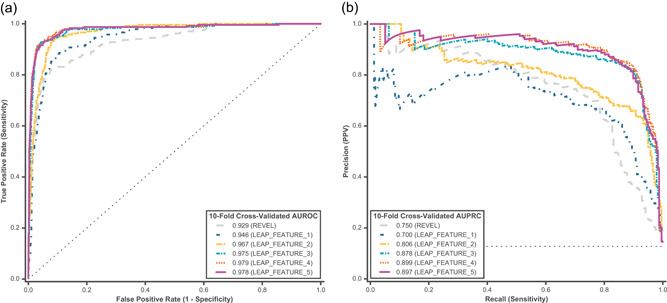
Ten‐fold cross‐validated predictions were assessed from a binary L2‐regularized logistic regression model for feature comparison. Predictions for P/LP and B/LB hereditary cancer variants were assessed using (a) AUROC and (b) AUPRC. Feature comparison models are described in Table 2. REVEL and LEAP_FEATURE_1 were used as baseline models, and represent a class of widely utilized computational predictors that consider only functional and conservation scores as inputs. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; P/LP, pathogenic/likely pathogenic
Figure 2.
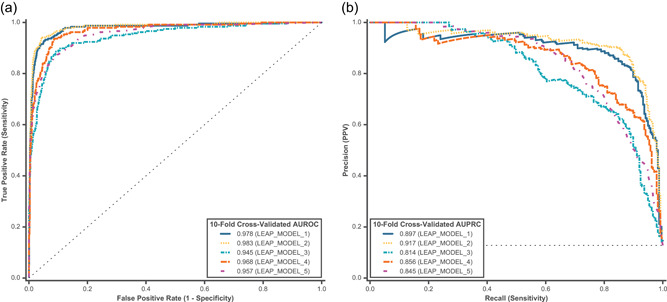
Ten‐fold cross‐validated predictions were assessed using all feature categories (FUNC+SPLICE+LOC+MAF+IND) for model comparison on hereditary cancer variants. Predictions for P/LP and B/LB variants were assessed for both binary and multiclass models using (a) AUROC and (b) AUPRC. Model comparison models are described in Table 2. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; FUNC, functional impact and evolutionary conservation; IND, individual‐level information; LOC, variant location; MAF, minor allele frequency; P/LP, pathogenic/likely pathogenic; SPLICE, splicing impact
Figure 4.
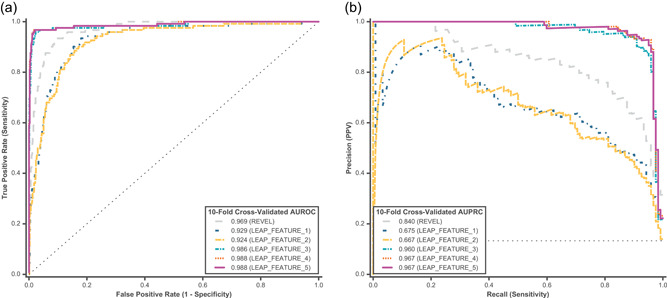
Ten‐fold cross‐validated predictions were assessed from a binary L2‐regularized logistic regression model for feature comparison on cardiovascular disorder P/LP and B/LB variants using (a) AUROC and (b) AUPRC. Feature comparison models are described in Table 2. REVEL and LEAP_FEATURE_1 were used as baseline models, and represent a class of widely utilized computational predictors that consider only functional and conservation scores as inputs. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; P/LP, pathogenic/likely pathogenic
Figure 5.
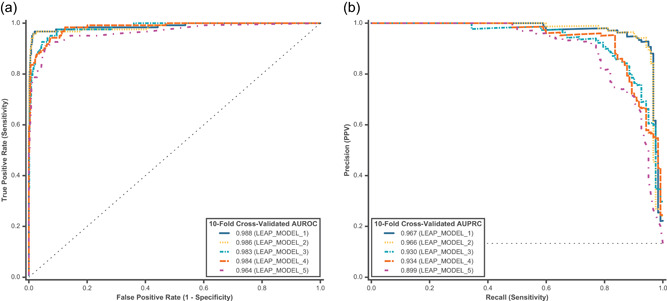
Ten‐fold cross‐validated predictions were assessed using all feature categories (FUNC+SPLICE+LOC+MAF+IND) for model comparison on cardiovascular disorder variants. Predictions for P/LP and B/LB variants were assessed for both binary and multiclass models using (a) AUROC and (b) AUPRC. Model comparison models are described in Table 2. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; FUNC, functional impact and evolutionary conservation; IND, individual‐level information; LOC, variant location; MAF, minor allele frequency; P/LP, pathogenic/likely pathogenic; SPLICE, splicing impact
As additional validation, model robustness to different genes or genes unseen by the model was assessed by using LEAP_MODEL_1 to generate “gene holdout” cross‐validated predictions, which were obtained for a given variant in each gene that was withheld from model training. For example, a model would be trained on 23 out of the 24 cancer genes and predictions would be made using this model for variants in the gene that was left out of training. This was done systematically for all genes, and resulting predictions were used for assessment. AUROC and AUPRC were similarly used to assess gene holdout predictions.
As external holdout validation, LEAP_FEATURE_4 and LEAP_FEATURE_5+HGMD (Figure S1) predictions were assessed on an external holdout set of 324 rare BRCA1 and BRCA2 variants newly classified by the CAGI5 ENIGMA blind prediction challenge and were referred to as “LEAP 2” and “LEAP 1”, respectively, in the assessment paper (Cline et al., 2019). LEAP_MODEL_2 and LEAP_MODEL_3 correspond to “LEAP 3” and “LEAP 4,” respectively, but exclude Human Gene Mutation Database (HGMD) features in this analysis. HGMD features used in three of the four CAGI5 submissions include the existence of HGMD entry for the variant, the existence of PubMed ID associated with the variant, assigned category of disease association (disease‐causing, disease‐associated, functional, etc.), and association of the variant with a cancer phenotype (Stenson et al., 2017). Ultimately, HGMD features were excluded from the main analysis in this paper for a couple of reasons: (a) they were not crucial for strong model performance in our assessment (Figure S1) or in the CAGI5 ENIGMA challenge, and (b) as a curated and proprietary database, presence in HGMD may bias against understudied genes or rare variants.
3. RESULTS
Using machine learning, we created a model that can classify missense variants with high precision, recall (sensitivity), and specificity. In a 10‐fold cross‐validation of missense variants in cancer‐associated genes, our best model (LEAP_MODEL_2) achieved 98.3% AUROC and 91.7% AUPRC. Here, we interrogate and report the results of the key factors that contributed to model performance.
3.1. Feature comparison
Our initial analysis aimed to assess which feature category components contributed the most to pathogenicity prediction in LEAP. We trained a binary logistic regression model on the cancer variant training set and evaluated model performance using both AUROC and AUPRC. Similar to those used in other published meta‐predictors, functional prediction features (Table 1, FUNC) were used in a baseline model (Table 2, LEAP_FEATURE_1). REVEL was also included as a baseline model because it is a best‐in‐class machine learning‐based pathogenicity meta‐predictor (Ghosh, Oak, & Plon, 2017) trained on functional predictors similar but not identical to those in LEAP_FEATURE_1. It is important to note that the REVEL training set (HGMD variants) and model choice (random forest) were different and distinct from LEAP. Thus, a head‐to‐head model comparison would not have been equitable and was not the intent of this analysis. The inclusion of REVEL is to compare differences in performance at varying levels of feature inclusion, as opposed to absolute model performance. The baseline model attained a reasonably high performance of 94.6% AUROC and 70.0% AUPRC (Figure 1, LEAP_FEATURE_1), and REVEL attained 92.9% AUROC and 75.0% AUPRC.
We next sought to improve the model by incorporating additional sets of evidence as features. We found that including additional evidence categories as features improved model performance at varying degrees depending on the category (Figure 1). Individual feature category contribution to cancer model performance is shown in Figure S2A. Initial performance improvement was already evident by simply adding splicing impact features in addition to functional predictors (Figure 1, LEAP_FEATURE_2 vs. LEAP_FEATURE_1). Overall, functional prediction, splicing prediction, and variant location features contributed to the most improvement in predictive performance (LEAP_FEATURE_3). Interestingly, population frequency contributed only slightly (LEAP_FEATURE_4) and aggregated individual‐level information (LEAP_FEATURE_5) actually decreased model performance slightly. The best model in the feature comparison (LEAP_FEATURE_4) achieved 97.9% AUROC and 89.9% AUPRC. Performance improvement was particularly pronounced in AUPRC between feature comparison models, with a 1.3× increase in LEAP_FEATURE_4 compared with the baseline LEAP_FEATURE_1 (Figure 1b). In addition, inclusion of additional feature categories not only improved AUROC and AURPC but also resulted in more confident predictions (Figure S3).
3.2. Model comparison
We next investigated whether model selection made a difference in performance. Using all feature categories (as in LEAP_FEATURE_5), the performance of five different models were compared: binary logistic regression, binary random forest, multiclass logistic regression (one‐vs.‐rest and multinomial methods), and multiclass random forest (Table 2). Nonlinear models like random forest were of interest as they may more adequately capture complex classification behavior and more closely reflect the hierarchical classification criteria that are employed in manual variant classification. Figure S4 is an example of nonlinearity or more complex interaction between features (in this case, population frequency and Polyphen2 functional prediction) that is automatically captured by a decision tree. To illustrate this example, two decision tree models were trained to predict pathogenicity probability in cancer variants using population frequency as the only feature in Figure S4A, and population frequency along with Polyphen2 as a feature in Figure S4B. High population frequency is commonly used as a strong standalone criterion for classifying a variant as benign. However, because most variants are rare (MAF < 0.001%, Figure S5), population frequency alone is not able to strongly differentiate many pathogenic and benign variants, so the model predicts all variants to be benign, albeit at differing low levels of probability that are directionally correct. By coupling population frequency with Polyphen2 HVAR, a decision tree is able to more confidently differentiate between pathogenic and benign variants. For example, the model automatically learned that a Polyphen2 HVAR label of “probably damaging” and a very low population frequency can be classified as pathogenic at higher probability (0.826, Figure S4B) than that based on population frequency alone (0.354, Figure S4A) or Polyphen2 HVAR alone (0.550, Figure S4B).
Separately, multiclass models were of interest because in some cases VUS may be the best classification for a given variant based on the current available evidence, and a model may learn to distinguish VUS as a distinct class from pathogenic and benign. In addition, because the majority of missense variants in our training sets are VUS, including VUS would increase the number of training instances and may, in turn, help a model discern different levels of pathogenicity more effectively. However, the inclusion of VUS in multiclass models also increases imbalance in the data (1.7% P/LP variants including VUS and 12.8% excluding VUS), which may make pathogenicity prediction more challenging.
Model comparisons using 10‐fold cross‐validated probability predictions in cancer are shown in Figure 2. When assessing both the AUROC and the AUPRC, random forest models performed better than logistic regression models, and the binary class models performed better than the multiclass models. Binary random forest performed slightly better than logistic regression in terms of AUPRC (Figure 2b, LEAP_MODEL_2 [91.7%] vs. LEAP_MODEL_1 [89.7%]). However, we did not observe a similar trend in multiclass models. This may be due to greater class imbalance in the multiclass model training set, which the random forest methodology is more sensitive to. Although multiclass models did not perform as well as binary models overall, there were optimizations that improved performance within multiclass models. For instance, using a multinomial method (LEAP_MODEL_4) was superior to a one‐versus‐rest method (LEAP_MODEL_3), perhaps because the former is better able to capture the ordinal structure that exists in the labels (B/LB<VUS<P/LP).
3.3. Model performance by gene
The LEAP models were trained on variants from 24 cancer genes. However, given the heterogeneous molecular functions of these genes, it is possible that the models were more adept at classifying variants in some genes but not others or less equipped to predict new genes unseen by the model. To explore this question, we assessed model performance on different genes using a “gene holdout” method (Figure 3).
Figure 3.
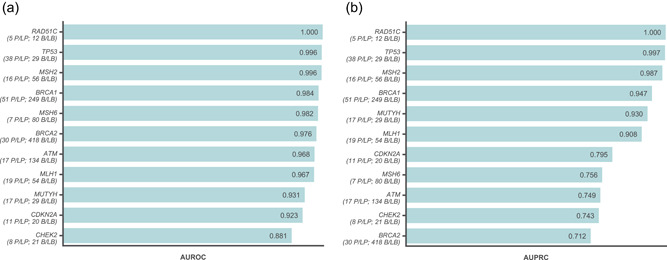
Gene‐holdout predictions from a binary L2‐regularized logistic regression model using all feature categories (LEAP_MODEL_1 or LEAP_FEATURE_5) were assessed for robustness across different hereditary cancer genes. Performance was assessed with (a) AUROC and (b) AUPRC on predictions for variants in each gene withheld from model training. The number of actual P/LP and B/LB variants detected in each gene are listed below the gene name. Genes in which at least five P/LP variants were detected were included in this figure. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; P/LP, pathogenic/likely pathogenic
Overall, the model was robust to the gene holdout procedure. Gene‐holdout predictions achieved 96.8% AUROC and 84.9% AUPRC, which is only slightly lower than that from 10‐fold cross‐validated predictions (Figure 1, LEAP_FEATURE_5). The frequency‐weighted average of these scores across genes was 93.0% and 76.7%, respectively. AUROC was high across most genes (Figure 3a), although AUPRC was slightly lower in a few genes due to more false positives (Figure 3b). Of note, performance was consistently high across genes when assessing per‐gene performance of 10‐fold cross‐validated predictions (data not shown).
3.4. Model performance on a holdout set
Finally, LEAP performance was assessed externally on a holdout set. We submitted four versions of LEAP to the CAGI5 ENIGMA challenge to classify variants in BRCA1 and BRCA2, all of which were in the top four when assessed by AUROC and AUPRC. The model that placed first included the HGMD literature category as a feature (Figure S1, LEAP_FEATURE_5+HGMD) achieved 98.8% AUROC and 82.0% AUPRC on the ENIGMA assessed variants, while another version of LEAP that only included publicly available information (LEAP_FEATURE_4) still placed ahead of all other competitors with 96.6% AUROC and 70.6% AUPRC on the ENIGMA assessed variants (Cline et al., 2019).
3.5. Application to other health conditions
Although LEAP was initially developed on genes associated with cancer risk, the model framework should theoretically be extensible to additional disease areas outside of cancer. To test this, we applied the same LEAP model framework developed for classifying cancer variants and trained on variants in genes associated with an entirely different phenotypic disease type: inherited cardiovascular disorders.
Directly mirroring the feature comparison framework used for cancer, a binary logistic regression model was used to test how different feature categories listed in Table 2 impacted performance in cardiovascular disorders (Figure 4). The best models in the feature comparison for cardiovascular disorders (tie between LEAP_FEATURE_4 and LEAP_FEATURE_5) achieved 98.8% AUROC and 96.7% AUPRC, which is higher than the corresponding models in cancer. In terms of relative feature contribution, functional predictors contributed the most (LEAP_FEATURE_1), followed by variant location (mainly gene; LEAP_FEATURE_3). For example, a given variant's location in LDLR is a strong predictor of pathogenicity, likely because this gene in our training data set was more enriched with P/LP variants as compared with other genes. Unlike in cancer, spliceimpact features do not contribute as much to model performance. The contributions of individual feature categories are shown in Figure S2B.
Next, we compared the model selection variations listed in Table 2 using 10‐fold cross‐validation in cardiovascular disorders (Figure 5). Unlike in the cancer models, the logistic regression models for cardiovascular disorders performed better than the random forest models (LEAP_MODEL_1, 3, and 4 vs. LEAP_MODEL_2 and 5). In addition, we saw no appreciable difference in performance between binary logistic regression and binary random forest models (LEAP_MODEL_1 vs. LEAP_MODEL_2). However, within the multiclass models, the linear models showed superior performance (LEAP_MODEL_3 and 4 vs. LEAP_MODEL_5) in cardiovascular disorder variants.
We performed a “gene holdout” validation using LEAP_MODEL_1 trained on cardiovascular disorder variants, and obtained consistently strong results across genes (Figure S6). Overall, gene holdout predictions across 29 cardiovascular genes achieved 98.3% AUROC and 94.1% AUPRC. The gene‐weighted average of these scores across genes was 88.8% and 62.4%, respectively.
4. DISCUSSION
Many published computational meta‐predictors have attempted to determine whether a variant is disease‐causing at the molecular or biological level. Here, we described the development of LEAP, a machine learning model developed to predict an expected variant classification utilizing the same evidence a variant scientist would in a clinical setting. Thus, this model does not try to determine the molecular pathogenesis of the disease, but simply aims to emulate the classifications and assertions that would be made by an expert variant scientist. We demonstrated that the model is highly accurate for the classification of missense variants using various forms of validation, including 10‐fold cross‐validation, gene holdout cross‐validation, and external holdout validation from the ENIGMA challenge.
A few novel approaches contributed to the strength and utility of the model. First, LEAP combines many different forms of evidence that would be used in expert manual variant classification based on ACMG guidelines. This differs from previous variant prediction models, which have used narrower categories of evidence such as functional predictors (Ioannidis et al., 2016), or functional predictors combined with population frequency (Alirezaie et al., 2018). These other meta‐predictors are ultimately solving different components of the variant classification problem, but are different from and not directly comparable with LEAP. Additional feature categories that represented different types of variant classification evidence were shown to improve the performance of LEAP. These feature categories included publicly available information such as functional impact predictors, splicing impact predictors, variant location and domain, and population frequency. Individual‐level information such as co‐occurring P/LP variants in individuals and individual phenotypic information was also assessed. However, the highest performing version of LEAP did not include individual‐level features (LEAP_FEATURE_4) and utilized publicly available features only. This suggests that LEAP's high performance is not due to any special individual‐level data that is in Color's database but is mostly driven by evidence that can be found in publicly available databases.
One potential pitfall of including many features in a model is multicollinearity, which may increase the risk of overfitting. We chose to incorporate correlated features as long as they were not 100% collinear, as they may still encode distinct inputs that are considered in variant classification. To manage multicollinearity without discarding valuable information, we selected models that are known to be more robust against strong correlation and automatically adjust. In the logistic regression case, L2 regularization was chosen because it penalizes large “double counted” coefficients resulting from highly correlated features by reducing coefficients and evenly distributing weight across correlated features, as opposed to reducing them completely to zero as in L1 regularization. In the random forest model, decision tree splits that minimize Gini impurity automatically prioritize features that are most informative for pathogenicity prediction. This optimization prevents redundancy and ensures that the most predictive feature within a group of correlated features has the most influence at the top of a given decision tree.
Although we found that increasing breadth of features improved model performance, clinical information such as the personal and family health history of carriers or covariant data did not meaningfully improve the performance of the models overall. This may have been due to the sparsity of data, especially in association with the many rare variants tested here. Separately, upon a case‐level investigation of variants that were discordant between LEAP prediction and manual classification, the latter was often based on functional study data extracted from published literature figures, tables, and text, which were not considered in LEAP's feature comparisons. This could be an avenue for future improvement. One approach would be to summarize primary evidence metadata such as “number of papers that reference a variant.” This may be a helpful start to building a more comprehensive model that mirrors the variant classification process and could continue to improve performance as functional study evidence becomes available across more genes. However, due to the sparsity of studies and open‐ended nature of free‐text interpretation, this would be a complex problem that requires more dedicated time that may not produce a proportional increase in model performance.
Another performance differentiator for LEAP included the availability of high‐quality variant classifications (ground truth labels) determined by trained variant scientists and approved by board‐certified medical geneticists at Color. One commonly cited pitfall for pathogenicity predictors is the lack of standardized and consistent classifications; many published models are trained on noisy public databases or inconsistently defined “consensus” classifications, which may be unreliable. Higher quality data will contain higher quality signals that result in higher quality predictions. Although the Color database is not comparable in volume with some public databases, it does reflect deep clinical testing experience in the set of genes evaluated here and the application of a consistent and critical variant classification protocol. A potential pitfall is that dependence on the classifications of one lab may run the risk of reinforcing inherent structural biases if they exist; any machine learning model, however, well‐developed, will only recapitulate those biases and not automatically correct for them. However, the variant classifications in Color's database are highly concordant with consensus ClinVar classifications, with a recent analysis within the All of Us consortium demonstrating a >98.5% concordance with participating genome centers (personal communication to S. T. from the All of Us Clinical Interpretation and Reporting Working Group, September 12, 2019). This suggests that the training set derived from the Color database represents current best practices.
Separately, a more extensive investigation into the overlap between the training sets of LEAP and its constituent variant predictors would help to assess the risk of overfitting. This is a known challenge for other meta‐predictors as well (Ghosh et al., 2017). Completely removing all overlap is challenging as many of the variant predictors do not publish their sets of variants used for training. With LEAP, we sought to represent the considerations of a variant scientist as closely as possible, which includes consideration of multiple variant predictors. To mitigate the risk of overfitting, other efforts include gene‐holdout validation to demonstrate model robustness to entirely new genes, as well as external validation on ENIGMA's hand‐picked and newly‐interpreted variants as a holdout set that was entirely separate from LEAP's training set.
Positive results from gene holdout validation indicate the generalizability of one model to different genes within one health condition. Initial results from extending the existing cancer framework to cardiovascular disorders suggest that machine learning can be useful in disease areas with less research understanding in genetics. In this paper, some differences between health conditions were found: population frequency was more predictive in cardiovascular than in cancer (Figure S2), and splicing impact seemed to be a stronger contributing feature in cancer than in cardiovascular (Figure S7). However, these differences may not represent true differences in the underlying biology, and may instead be due to limitations in genetics research and availability of evidence in cardiovascular relative to cancer. In the future, with greater research understanding, a more mature database, and further model tuning, one disease‐agnostic model could be trained to take into account disease‐specific complexities and may benefit from increased training size and generalizability.
In a clinical laboratory setting, the outputs of LEAP can be integrated into a clinical variant interpretation workflow to increase variant scientist efficiency and act as a quality control mechanism for variant classification. For example, LEAP can be used to prioritize variants, which are more likely to be pathogenic for human review to optimize clinical reporting efficiency. Similarly, LEAP can also be used for the prioritization of VUSs for reclassification (Figure 6). Indeed, our early exploratory analysis shows LEAP is able to discern VUS‐P (variants internally tagged as one additional piece of evidence away from an LP classification based on ACMG guidelines) from VUS with 86.3% AUROC in cancer (Figure S7). In addition, LEAP could serve as a quality control layer on top of existing variant interpretation processes, by flagging cases that are discordant between LEAP and an expert variant scientist for further review. These types of cases, over time, would also help to train and improve the model's accuracy. Finally, LEAP predictions could be used to automatically assert the “computational prediction” criterion in the ACMG classification guidelines (Richards et al., 2015).
Figure 6.
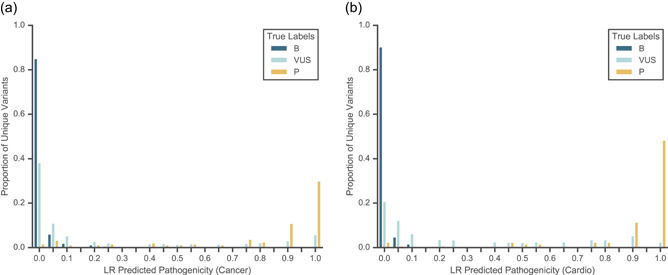
Distributions of 10‐fold cross‐validated predictions from a binary L2‐regularized logistic regression model using all feature categories (LEAP_MODEL_1 or LEAP_FEATURE_5) are shown for P/LP variants, B/LB variants, and VUS. (a) Predictions on all hereditary cancer variants with 0.978 AUROC and 0.897 AUPRC. (b) Predictions on all cardiovascular disorder variants with 0.988 AUROC and 0.967 AUPRC. AUROC, area under the receiver operating characteristic curve; AUPRC, area under the precision‐recall curve; B/LB, benign/likely benign; P/LP, pathogenic/likely pathogenic
Variant classification is a complex and evolving field. The implementation of the ACMG Guidelines in 2015 helped drive consistency and transparency by establishing a common language and standard process. It is this standard process, and its reliance on structured data, that has ultimately paved the way for computational models such as LEAP to be developed. Variant classification represents a conclusion that the available evidence is sufficient to prove the variant's role in the development of the disease. To that end, one of the major advantages of LEAP is its usability and interpretability. By making the contributions of each specific evidence type for each variant clear to the human eye, LEAP aims to unmask the “black box” nature of many machine learning models. This allows the expert scientist to more deeply understand and evaluate the underlying logic for LEAP's predictions. As we and others continue to make novel computational tools for these applications, we believe that tool usability will be as important as prediction accuracy towards the utility and adoption of these tools in the practice of clinical genetics.
CONFLICTS OF INTEREST
C. L., A. D. Z., R. O., S. K., R. C., Jvd. A., A. Y. Z., and S. T. are currently employed by and own equity interest in Color Genomics. S. T. was previously employed at Invitae. C. L. was previously employed at 23andMe. G. M. was previously employed at Color Genomics and Operator.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We would like to thank Will Stedden, Julian R. Homburger, Mark Berger, Elaine Chen, and Danny DeSloover for helpful discussions. This study was supported by Color Genomics. The CAGI Experiment Coordination was supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650.
Lai C, Zimmer AD, O'Connor R, et al. LEAP: Using machine learning to support variant classification in a clinical setting. Human Mutation. 2020;41:1079–1090. 10.1002/humu.24011
Contributor Information
Scott Topper, Email: scott@color.com.
Gilad Mishne, Email: gilad@mishne.org.
DATA AVAILABILITY STATEMENT
The data that support the findings in this study are available on request from the corresponding author (S. T.). The data are not publicly available as they contain information that could compromise research participant privacy or consent. All reported variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/505849/).
REFERENCES
- Adzhubei, I. , Jordan, D. M. , & Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics, 7(20), 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaie, N. , Kernohan, K. D. , Hartley, T. , Majewski, J. , & Hocking, T. D. (2018). ClinPred: Prediction tool to identify disease‐relevant nonsynonymous single‐nucleotide variants. American Journal of Human Genetics, 103, 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun, S. , & Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Research, 19, 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, M. S. , Babbi, G. , Bonache, S. , Cao, Y. , Casadio, R. , Cruz, X. , … Goldgar, D. E. (2019). Assessment of blind predictions of the clinical significance of BRCA1 and BRCA2 variants. Human Mutation, 40, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov, E. V. , Goode, D. L. , Sirota, M. , Cooper, G. M. , Sidow, A. , & Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Computational Biology, 6, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, C. , Wei, P. , Jian, X. , Gibbs, R. , Boerwinkle, E. , Wang, K. , & Liu, X. (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human Molecular Genetics, 24, 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D. , Attwood, T. K. , Babbitt, P. C. , Bateman, A. , Bork, P. , Bridge, A. J. , … Mitchell, A. L. (2017). InterPro in 2017‐beyond protein family and domain annotations. Nucleic Acids Research, 45, D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, R. , Oak, N. , & Plon, S. E. (2017). Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biology, 18, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis, N. M. , Rothstein, J. H. , Pejaver, V. , Middha, S. , McDonnell, S. K. , Baheti, S. , … Sieh, W. (2016). REVEL: An Ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics, 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neben, C. L. , Zimmer, A. D. , Stedden, W. , Akker, J. , , van den O'Connor, R. , Chan, R. C. , … Zhou, A. Y. (2019). Multi‐gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. Journal of Molecular Diagnostics, 0, 646–657. [DOI] [PubMed] [Google Scholar]
- Ng, P. C. , & Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykamp, K. , Anderson, M. , Powers, M. , Garcia, J. , Herrera, B. , Ho, Y. ‐Y. , … Topper, S. (2017). Sherloc: A comprehensive refinement of the ACMG‐AMP variant classification criteria. Genetics in Medicine, 19, 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa, F. (2011). Scikit‐learn: Machine learning in Python. Journal of Machine Learning Research, 12, 2825–2830. [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- Siepel, A. , Bejerano, G. , Pedersen, J. S. , Hinrichs, A. S. , Hou, M. , Rosenbloom, K. , … Haussler, D. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Research, 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , … Cooper, D. N. (2017). The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136(6), 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Deffenbaugh, A. M. , Yin, L. , Judkins, T. , Scholl, T. , Samollow, P. B. , … Thomas, A. (2006). Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics, 43, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Greenblatt, M. S. , Harrison, S. M. , Nussbaum, R. L. , Prabhu, S. A. , Boucher, K. M. , & Biesecker, L. G. , ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) . (2018). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genetics in Medicine, 20, 1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe, A. , Mullikin, J. C. , & Elnitski, L. (2010). Genomic features defining exonic variants that modulate splicing. Genome Biology, 11, R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Data Availability Statement
The data that support the findings in this study are available on request from the corresponding author (S. T.). The data are not publicly available as they contain information that could compromise research participant privacy or consent. All reported variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/505849/).