

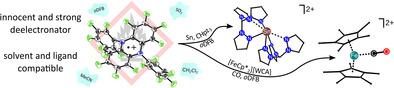
Abstract
The perfluorinated dihydrophenazine derivative (perfluoro‐5,10‐bis(perfluorophenyl)‐5,10‐dihydrophenazine) (“phenazineF”) can be easily transformed to a stable and weighable radical cation salt by deelectronation (i.e. oxidation) with Ag[Al(ORF)4]/ Br2 mixtures (RF=C(CF3)3). As an innocent deelectronator it has a strong and fully reversible half‐wave potential versus Fc+/Fc in the coordinating solvent MeCN (E°′=1.21 V), but also in almost non‐coordinating oDFB (=1,2‐F2C6H4; E°′=1.29 V). It allows for the deelectronation of [FeIIICp*2]+ to [FeIV(CO)Cp*2]2+ and [FeIV(CN‐tBu)Cp*2]2+ in common laboratory solvents and is compatible with good σ‐donor ligands, such as L=trispyrazolylmethane, to generate novel [M(L)x]n+ complex salts from the respective elemental metals.
Keywords: Iron complexes, main-group chemistry, oxidizing agents (deelectronators), radical ions, weakly coordinating anions
Innocence is bliss: A novel perfluorinated phenazine ammoniumyl radical cation is shown to be a very strong deelectronator (oxidant). It has broad solvent compatibility, can be used together with good sigma donor ligands, and allows access to tetravalent dicationic iron complexes in common laboratory solvents.
Introduction
Oxidation, or better deelectronation,1 represents one of the most fundamental transformations in synthetic chemistry. Very useful and extremely strong deelectronating agents, such as SbF5 or MF6 (M=Pd, Pt, …) show disadvantages, as they require the use of difficult to handle solvents, such as anhydrous HF (aHF), while MF6 species are best freshly prepared by treating the respective metals with F2. Both require appropriate facilities as well as trained personnel.2, 3 Well established and easy to generate and use deelectronating agents are solvent‐free nitrosonium and silver salts of weakly coordinating anions (WCAs). Since high performance WCAs, such as [B(C6F5)4]− or [B(C6H3(CF3)2)4]−, do not withstand high electrochemical potentials or even only solvent free silver and NO+ salts, the electrochemically robust [Al(ORF)4]− (ORF=OC(CF3)3), or related redox stable WCAs,4, 5 are of particular relevance. However, Ag+ as primary deelectronator is small and Lewis acidic, which opens unwanted side reactions, such as halide abstraction or coordination, rather than deelectronation. In addition, the influence of solvation on the formal potential of Ag+ is large and spans, for example, a range of 0.6 V between CH2Cl2 and MeCN.4 This dependence also prohibits the use of good σ‐ and π‐donating ligands, as they will effectively quench the deelectronation potential of silver(I) by forming coordinatively saturated complexes. The use of halogens together with weakly coordinated silver salts can boost their deelectronation potential tremendously. We have isolated the molecular equivalents of the Ag+/X2 couple as a series of [Ag(X2)]+ (X=Cl2, Br2, I2) as well as [Ag2(I2)n]2+ (n=2,4,6) complexes.6 Side reactions of this class of compounds are yet to be investigated. However, preliminary studies with transition‐metal carbonyl complexes have shown that they may act as a source for electrophilic X+. Nitrosyl salts, with a published formal potential versus Fc+/Fc0 (Fc=[Cp2Fe]; Cp=η‐C5H5) of 1.00 V7 in CH2Cl2, often act as irreversible deelectronating agents, if the gaseous reaction byproduct NO can be removed. However, the generated neutral NO is likely to undergo further ligand‐substitution, for example, with transition‐metal complexes.8 Moreover, nitrosyl salts show a noticeable solvent dependency of their formal potential and are known to form colored charge transfer complexes with arenes as the first step towards electrophilic substitution, thus lessening their applicability.9
Another class of deelectronating agents, and part of the work documented herein, are ammoniumyl radical cations, first observed over 100 years ago by Wieland.10 The intense blue color of phenylamine derivatives in deelectronating media was later assigned by Weitz and Schwechten to the formation of an ammoniumyl radical cation.11 When protecting the para positions on the aryl moieties, these radicals can be isolated and by variation of the substitution pattern on the aryl ring, the formal potentials can be tuned over a range of 1.5 V.12 The most prominent and commercial example is [N(C6H4Br‐4)3][SbCl6], colloquially called “magic blue” with E°′=0.70 V versus Fc+/Fc. In addition, the solvent dependency of the formal potential of ammoniumyl radical cations compared to silver(I) or nitrosonium salts is very small.4 Furthermore, ammoniumyl radical cations were shown to generally undergo innocent electron transfer (ET), and can be used in a wide variety of common laboratory solvents. For these reasons the ammoniumyl radical cations have found wide application in organic synthesis, catalysis, as mediators in electrochemistry or as doping agents in polymer sciences.13 Interestingly, reported uses in organometallic or inorganic chemistry are comparatively scarce;14 the most pertinent example being the generation of a variety of fullerene and azo‐fullerene radical cations by Reed and co‐workers with magic blue amine derivatives.15
Results and Discussion
The use of ammoniumyl radical cations as deelectronating agents appeared to offer advantages over the commonly used reagents described above. We have therefore replaced the poor [SbCl6]− WCA in the commercial “magic blue” salt for the much better performing WCA [Al(ORF)4]−. However, it soon became evident that this straightforward and in good yield accessible ET agent (see Supporting Information) is not compatible with the use of σ‐donor ligands, such as dimethylaminopyridine, carbenes (e.g, IPr) or phosphines (see Scheme 1).
Scheme 1.
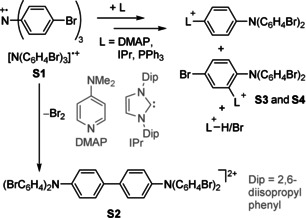
Reactivity of [N(C6H4Br)3][Al(ORF)4] (S1) towards σ‐donor ligands and the degradation of S1. In all cases the counterion is [Al(ORF)4]−. For the molecular structures of S1, S2, S3, and S4 see Supporting Information.28
The molecular structures of several reaction products suggest that these ligands are deelectronated with formation of the very sharp σ‐ligand‐radical cations, which rapidly undergo a series of H/Br. abstraction on the neutral magic blue amine to give a plethora of substitution products, which have been characterized by single‐crystal X‐ray diffraction (scXRD) and mass spectrometry (Figure S4–S7 in the Supporting Information). Decomposition by H/Br. abstraction has also been reported before to occur in the solid state upon simple storage of “magic blue” salt, for example, formation of S2.16 As all of the established and commonly used ammoniumyl radical cations always contain C−H and often also C−Br bonds, comparable reactivities are expected for these radicals.17 A potent alternative, the perchlorinated trisarylammoniumyl radical cation [N(C6Cl5)3]+., was only reported as a personal communication; its use in the literature is scarce and no preparations for either neutral amine nor the radical cation could be found.18
This situation motivated us to look for more stable alternatives with suitably high formal potential. To hinder the homolytic cleavage of C−X bonds (X=H, Br), we set out to design amines only employing very strong and weakly nucleophilic C−F bonds. The precursor for our system, the known bis‐perfluoroarylamide [NArF 2]− (ArF=‐C6F5) can be easily prepared from the cheap starting materials LiNH2 and C6F6 on a multigram scale as described by Sundermeyer and co‐workers, who used this amide as WCA in electrochemical applications.19, 20 The weak coordination ability underlines the very poor nucleophilicity of the amide nitrogen, which explains numerous own and others failed attempts to generate the corresponding trisarylamine derivative N(ArF)3 in satisfying yields. Yet, Sundermeyer et al. noticed the decomposition of solvent‐free Li[NArF 2] 2 above 180 °C to yield a perfluorinated dihydrophenazine derivative 3 by intermolecular abstraction of two equivalents of LiF [see Eq. 1].20
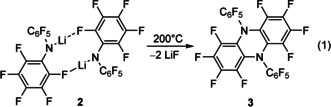
First own synthesis attempts were carried out in analogy to the method described by Sundermeyer et al.20 Solvent‐free 2 was generated by the reaction between HNArF 2 1 and Li{N(SiMe3)2} (LiHMDS) in toluene. The isolated product was then charged into a Schlenk tube equipped with a cool finger and heated in a vertical tube oven to 220 °C under dynamic vacuum. When the set temperature was reached, the reaction proceeded quickly and the brown reaction product could be washed from the cool finger with toluene. NMR spectroscopy investigations of the crude product, however, show equimolar amounts of the amine 1 and target compound 3. Suspected reprotonation by leftover HN(SiMe3)2 in the heat was ruled out by use of tBuLi as base. Closer analysis of the starting material 2 revealed the presence of 1 as determined by 1H‐19F NOESY NMR experiments. The non‐quantitative deprotonation of 1 is presumably attributable to strong intermolecular [(F5C6)2N−H−N(C6F5)2]− hydrogen bonding between 1 and 2 explaining the NOESY cross correlation between the acidic proton and the ortho‐fluorine atom in the single resonance set observed. This prevents complete deprotonation even in strongly coordinating solvents like acetonitrile or THF. Compound 2 free of solvent coordination and with only trace amounts (less than 1 %) of 1 could be generated by deprotonation with LiHMDS and switching from non‐polar toluene to ortho‐difluorobenzene (oDFB). Amine‐free 2, crystallizing as 2⋅2 oDFB (molecular structure in Supporting Information), was then charged into a Schlenk‐tube and submitted to controlled heating to 200 °C under dynamic vacuum on a hot‐stirrer plate with a commercially available aluminum heating block. Purification by sublimation of the obtained crudes at 170 °C in dynamic vacuum gave only poor yields of pure 3 as the reaction side products form a highly viscous oil, from which 3 is not readily liberated. Precipitation of 3 from MeCN solution with H2O followed by additional sublimation has resulted in acceptable multi‐gram yields of 3 (2.8 g, 38 %; molecular structure of 3 in Figure 1 a).
Figure 1.
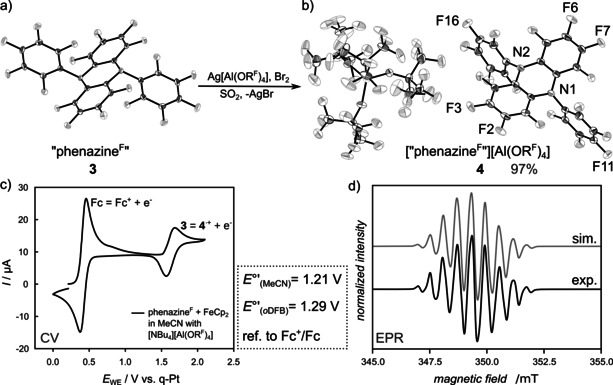
a) Molecular structure of “phenazineF” (3) and b) molecular structure of [“phenazineF”][Al(ORF)4] (4). Data collected at 100 K. Co‐crystallized CH2Cl2 omitted for clarity. Thermal displacement ellipsoids set at 50 % probability.28 c) Cyclic voltammogram of 3 (10 mm) in MeCN with Fc+/Fc as internal reference and [NBu4][Al(ORF)4] (100 mm) as conducting salt. The half‐wave potential is independent of the sweep rate. d) X‐band continuous‐wave‐EPR spectrum of 4 in SO2 at ambient temperature (black). The spectrum was recorded at a microwave frequency of 9.7973 GHz on an Elexsys E580 (Bruker Biospin) spectrometer equipped with a 4119HS‐W1 (Bruker) cavity using a microwave power of 0.04743 mW (35 dB attenuation, 200 mW source power), a modulation frequency of 100 kHz and a modulation amplitude of 0.05 mT. The corresponding spectral simulation is shown in gray. Two sets of six and two equivalent fluorine atoms with isotropic hyperfine coupling constants were used for the simulation. The isotropic hyperfine constants were determined as 15.44 and 20.35 MHz, respectively. The Gaussian linewidth was determined to be 0.267 mT (full width at half maximum).
Compound 3 was examined by cyclovoltammetry experiments (see Figure 1 c and Supporting Information) and gave a reversible oxidation wave at 1.21 V against Fc+/Fc in MeCN and at 1.29 V in oDFB with external reference to N(C6H4Br)3 (E 1/2=0.70 V, ref. Fc+/Fc). Thus, the formal potential of 3 exceeds that of both, silver and nitrosonium salts. Chemical deelectronation of 3 was successful by reaction with the synergistic Ag+/ X2 deelectronator pair. When employing iodine, non‐quantitative turnover to the desired radical species was observed, as even after prolonged reaction times crystallization attempts showed aside from the desired needle shaped deep purple crystals of 4, a thin film of off‐white powder in the crystallization ampoule; presumably from continuously formed AgI. When switching to Ag+/ Br2 however, quantitative turnover was achieved, with an isolated yield of 4 of 97 %. While solvent‐free 4 is best isolated from SO2 as solvent for synthesis, the radical was also successfully generated and is stable in both oDFB and CH2Cl2 as solvent. In analogy to the described procedure, the respective [SbF6]− salt can be accessed, when employing Ag[SbF6] in SO2 solution giving stable deeply purple colored solutions. Pure 4 is also stable in MeCN and can be indefinitely stored in the solid state and does not decompose, when exposed to a dry ambient atmosphere. However, introduction of moisture to a solution of 4 proceeds with ready color loss. Crystals suitable for single crystal diffraction analysis could be obtained by layering of an oDFB or CH2Cl2 solution with pentane. For the first solvent combination, a super‐structure in the analyzed deep purple crystal was found. A reliable molecular structure of 4 was gathered from crystals grown in CH2Cl2 as shown in Figure 1 b.
The central nitrogen atoms here are almost perfectly planar coordinated with a bond angle sum of 358.2° compared to more pyramidal neutral 3 with an angle sum of 342.2°. The isolated crystals of 4 were analyzed by ESI‐mass spectrometry and show mass envelopes for the desired radical at m/z 658.08 (calcd N2C24F18; 657.98 m/z) as well as little of an iminium cation at m/z 491.17 (calcd N2C18F13N2; m/z 490.99) which is formed by fragmentation processes as verified by collision induced decomposition experiments (CID). Continuous‐wave (cw) electron paramagnetic resonance (EPR) measurements were carried out at X‐band frequency to investigate a sample of 4 in SO2 at ambient temperature (see Figure 1 d). The EPR spectrum unambiguously shows an organic radical, centered at g iso=2.0031. The EPR spectrum exhibits a well‐resolved pattern with multiple hyperfine lines. The hyperfine pattern could be reproduced by a spectral simulation of the EPR spectrum in the isotropic limit using the EasySpin simulation package17 in combination with non‐linear least‐squares optimization algorithms and initial parameter sampling. The obtained fit result indicates two sets of six and two magnetically equivalent hyperfine couplings to I= nuclei. These hyperfine couplings can be assigned to the coupling between the unpaired electron spin and the fluorine atoms (I= ). While the EPR simulation is able to reproduce the experimental data, it is noteworthy that in analogy to NMR spectra of neutral 3 no six magnetically equivalent fluorine atoms can be present in the molecular ion 4+. DFT studies have shown that the largest coupling should come from 4:2:2 equivalent fluorine atoms. The two very similar calculated median coupling constants for F2, F3, F6, F7 of 9.7 MHz and F13, F18 of 10.5 MHz (for numbering scheme see Figure 1 b) may therefore be indistinguishable in the experiment. Together with the larger calculated coupling to the para‐fluorine in C6F5 moieties (F11, F16) of 15.6 MHz, these calculations support the simulated 6:2 coupling Scheme.
To verify the measured very high formal potentials of 1.21/1.29 V against Fc+/Fc, the deelectronation of model substrates was investigated (see Figure 2). As such, 4 was reacted with sheeted tin metal in MeCN. The reaction proceeds via complete loss of the intense purple color after 1 h at ambient temperature. NMR spectra of the dried reaction mixture in oDFB revealed the intact counterion and neutral 3 in a 1:1 ratio; underlining both stability and purity of employed 4. 119Sn resonances were not detectable, probably due to the low sample concentration. However, the ratio between MeCN and counterion determined by NMR integration is 6:2, suggesting the known species [Sn(MeCN)6][Al(ORF)]2 as product.21 Yet, the pentasolvate [Sn(MeCN)5][Al(ORF)]2 (5) crystallized from oDFB (see Figure 2). To confirm the ligand compatibility of 4, it was used together with trispyrazolylmethane (CHpz3) in the deelectronation of elemental tin to yield the respective biscoordinated complex [Sn(CHpz3)2][Al(ORF)4]2 (6) with each of the chelating ligands binding strongly κ2 (d Sn‐N=2.448(3) Å) and an additional third longer Sn‐N contact at 2.715(3) Å (see Figure 2).
Figure 2.
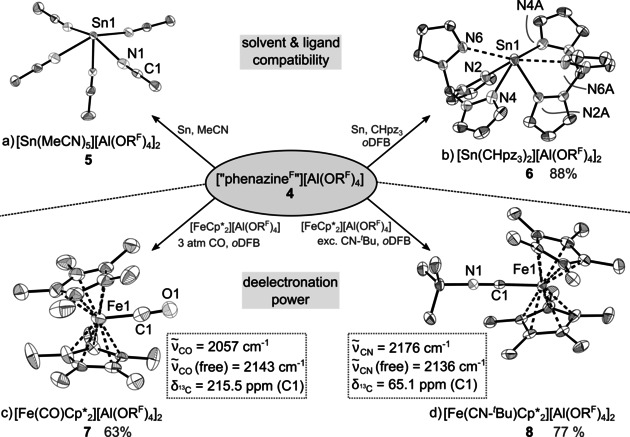
Reactivity of the deelectronator 4 towards different substrates and the molecular structures of a) [Sn(MeCN)5[Al(ORF)4]2 (5), b) [Sn(CHpz3)2][Al(ORF)4]2 (6), c) [Fe(CO)Cp*2][Al(ORF)4]2 (7), and d) [Fe(CN‐tBu)Cp*2][Al(ORF)4]2 (8). Data collected at 100 K. H atoms, counterions and (co‐crystallized) oDFB were omitted for clarity. Thermal displacement ellipsoids set at 50 % probability.28 Some selected spectroscopic data is included.
As upper limit of possible ET reactions, the deelectronation of monocationic [FeIIICp*2]+ (Cp*=η‐C5Me5) to the only reported dicationic [FeIVCp*2]2+ was chosen, a compound so far only accessible by deelectronation of the decamethylferrocenium ion with SbF5 in SO2 solution.3 While reactions in both CH2Cl2 and oDFB proceeded with very slow loss of the intense purple color of 4, only the starting material [FeCp*2][Al(ORF)4] and traces of neutral 3 could be identified by scXRD respectively NMR spectroscopy. When switching to SO2 as reaction medium, the purple color of 4 seems persistent indefinitely. When repeating the reaction between [FeCp*2][Al(ORF)2] and 4 under 3 atm of CO gas in oDFB, a ready color change from deep purple to red‐brown was observed allowing access to the 18 VE dicationic FeIV complex [Fe(CO)Cp*2][Al(ORF)4]2 7 in 63 % crystalline yield. The [Fe(CO)Cp*2]2+ dication was hitherto only accessible in aHF.22 Compound 7 was crystallized from concentrated oDFB solution by either vapor diffusion or layering of the reaction mixture with n‐pentane. Structure determination by scXRD experiments has however proven difficult. With both crystallization methods, 7 crystallized in large cells with underlying super‐structure. However, the identification of the crystals as 7 was unambiguous (molecular structure, see Figure 2). Presence of the assigned cationic species was verified by IR spectroscopic analysis showing a strong vibrational band of the single coordinated carbonyl moiety with a stretching frequency of 2057 cm−1, which is, as expected due to the better WCA performance of our counterion, slightly blue‐shifted towards the literature (cf. [Fe(CO)Cp*2][X]2; X=[AsF6] 2042 cm−1, X=[SbF6] 2034 cm−1) and fits very well to the DFT‐calculated IR spectrum ( 2058 cm−1 at the BP86/D3(BJ)/def‐SV(P) level of theory). Furthermore, bulk purity was confirmed by powder diffraction analysis and NMR spectra of isolated crystalline material. Internal referencing with solvent oDFB in both 1H and 19F NMR spectra shows a 2:2 ratio between Cp* and [Al(ORF)4]−, underlining purity of diamagnetic 7.
A related dicationic FeIV complex was synthesized, when excess of tert‐butylisonitrile (tBuNC) was used instead of CO allowed for isolation of unprecedented [Fe(CN‐tBu)Cp*2]2+ (8) as the [Al(ORF)4]− salt in the form of deep red crystals in 77 % crystalline yield (see Figure 2). The central iron is distorted η5‐coordinated by the Cp*− moieties with an average Fe–C distance of 2.1674(4) Å (d Fe‐C=2.142(4)– 2.194(4) Å). The two Cp* rings are tilted by 29° against each other forcing the methyl groups opposite of coordinated tBuNC out of the ring plane by up to 15°. The tBuNC ligand is coordinated to iron at d Fe1‐C1=1.874(4) Å. This lies between reported Fe‐(CN‐R) bond lengths of divalent [FeIICp(CO)2(CN‐Me)][BF4] (d Fe‐C=1.900(9) Å23) and zero‐valent Fe(CO)n(CN‐tBu)m (d Fe‐C=1.89 {n=4, m=1}24 1.865(2) {n=3, m=2}25 1.813(7) to 1.837(7) {n=0, m=5}26 Å) and is by 0.09 Å longer than in the related [Fe(CO)Cp*2][AsF6]2 (cf. d Fe‐C=1.785(9) Å).22 Its C≡N stretching frequency is slightly blue‐shifted to 2176 cm−1 (cf. free tBuNC at 2136 cm−1[27]) and in good agreement to its DFT‐calculated frequency at 2195 cm−1 (at the BP86/D3(BJ)/def‐SV(P) level of theory). Bulk purity was confirmed by powder diffraction analysis and NMR spectra of isolated crystalline material and revealed with both methods a contamination of the sample with less than 20 % of the starting material [FeCp*2][Al(ORF)4]. This appears to be due to the instability of the isonitrile ligand in the presence of 4. Thus, part of the employed deelectronator reacts with tBuNC to give a series of undefined degradation products resulting in partial re‐isolation of starting material.
Conclusion
In conclusion, we prepared and isolated the radical cation salt of the perfluorinated dihydrophenazine derivative [“phenazineF”][Al(ORF)4] (4) as a stable, weighable, and very strong deelectronating agent with broad solvent compatibility. The intermediate yields of parent neutral “phenazineF” 3 are attributable to the nature of the synthesis, but are counterbalanced by the ready and cheap availability of the starting materials as well as its easy isolation. The during the ET reactions regenerated neutral 3 shows negligible nucleophilicity and does not interact with the deelectronated substrates, allowing for new synthesis pathways in the generation of highly reactive cationic species. The promising synthetic scope of this reagent was shown by deelectronation of [FeCp*2]+ to organometallic tetravalent iron in [Fe(CO)Cp*2]2+ (7) and [Fe(CN‐tBu)Cp*2]2+ (8) in oDFB solution. Thus, even removal of an electron from an already cationic and evidently very stable species such as [FeCp*2]+ proceeds readily. Future investigations will focus on the applicability of this reagent to other main‐group and transition‐metal species to gain access to hitherto inaccessible or only in aHF accessible reactive cations.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Albert‐Ludwigs‐Universität Freiburg, the Deutsche Forschungsgemeinschaft DFG in the Normalverfahren and the Carl‐Zeiss‐Stiftung. We thank Dr. H. Scherer and F. Bitgül for performing NMR measurements, Dr. T. Ludwig and Dr. M. Daub for measuring pXRD, Dr. D. Kratzert for help with scXRD measurements, and the Magres Core Facility for EPR and NMR instrument support.
M. Schorpp, T. Heizmann, M. Schmucker, S. Rein, S. Weber, I. Krossing, Angew. Chem. Int. Ed. 2020, 59, 9453.
References
- 1.
- 1a.Here and in the following we use the particle-based view on the classical oxidation=deelectronation and reduction=electronation processes. Thus, an oxidant is a deelectronator and a reductant an electronator. This evolved from our work on the protoelectric potential map (PPM) for keeping with the successful and self-explaining acid–base picture. Thus, the equivalent to a deprotonation reaction is a deelectronation reaction. This is in keeping with earlier textbook suggestions by Bockris and Reddy and follows our concept paper on the PPM (Ref. [1 b]) and the recent Review (Ref. [1 c]);
- 1b. Radtke V., Himmel D., Putz K., Goll S. K., Krossing I., Chem. Eur. J. 2014, 20, 4194; [DOI] [PubMed] [Google Scholar]
- 1c. Himmel D., Radtke V., Butschke B., Krossing I., Angew. Chem. Int. Ed. 2018, 57, 4386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4471; [Google Scholar]
- 1d. Bockris J. O.’M., Reddy A. K. N., Modern Electrochemistry 1. Ionics, Kluwer Academic Publishers, Boston, MA, 2002. [Google Scholar]
- 2.
- 2a. Seppelt K., Chem. Rev. 2015, 115, 1296; [DOI] [PubMed] [Google Scholar]
- 2b. Shorafa H., Mollenhauer D., Paulus B., Seppelt K., Angew. Chem. Int. Ed. 2009, 48, 5845; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5959. [Google Scholar]
- 3. Malischewski M., Adelhardt M., Sutter J., Meyer K., Seppelt K., Science 2016, 353, 678. [DOI] [PubMed] [Google Scholar]
- 4. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Riddlestone I. M., Kraft A., Schaefer J., Krossing I., Angew. Chem. Int. Ed. 2018, 57, 13982; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14178; [Google Scholar]
- 5b. Niemann M., Neumann B., Stammler H.-G., Hoge B., Angew. Chem. Int. Ed. 2019, 58, 8938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9033; [Google Scholar]
- 5c. Hoffmann K. F., Wiesner A., Subat N., Steinhauer S., Riedel S., Z. Anorg. Allg. Chem. 2018, 644, 1344. [Google Scholar]
- 6.
- 6a. Malinowski P. J., Himmel D., Krossing I., Angew. Chem. Int. Ed. 2016, 55, 9262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9408; [Google Scholar]
- 6b. Malinowski P. J., Himmel D., Krossing I., Angew. Chem. Int. Ed. 2016, 55, 9259; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9405. [Google Scholar]
- 7. Kochi J. K., Acc. Chem. Res. 1992, 25, 39. [Google Scholar]
- 8.
- 8a. Bohnenberger J., Derstine B., Daub M., Krossing I., Angew. Chem. Int. Ed. 2019, 58, 9586; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9687; [Google Scholar]
- 8b. Bohnenberger J., Krossing I., Angew. Chem. Int. Ed. 2020, 59, 5581; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5629. [Google Scholar]
- 9.
- 9a. Brownstein S., Gabe E., Lee F., Piotrowski A., Can. J. Chem. 1986, 64, 1661; [Google Scholar]
- 9b. Lee K. Y., Kuchynka D. J., Kochi J. K., Inorg. Chem. 1990, 29, 4196; [Google Scholar]
- 9c. Kim E. K., Kochi J. K., J. Am. Chem. Soc. 1991, 113, 4962. [Google Scholar]
- 10. Wieland H., Ber. Dtsch. Chem. Ges. 1907, 40, 4260. [Google Scholar]
- 11.
- 11a. Wieland H., Wecker E., Ber. Dtsch. Chem. Ges. 1910, 43, 699; [Google Scholar]
- 11b. Weitz E., Schwechten H. W., Ber. Dtsch. Chem. Ges. 1926, 59, 2307. [Google Scholar]
- 12.
- 12a. Walter R. I., J. Am. Chem. Soc. 1955, 77, 5999; [Google Scholar]
- 12b. O'Connor A. R., Nataro C., Golen J. A., Rheingold A. L., J. Organomet. Chem. 2004, 689, 2411; [Google Scholar]
- 12c. Wu X., Davis A. P., Lambert P. C., Kraig Steffen L., Toy O., Fry A. J., Tetrahedron 2009, 65, 2408. [Google Scholar]
- 13.
- 13a. Bauld N. L., Tetrahedron 1989, 45, 5307; [Google Scholar]
- 13b. Mattay J., Trampe G., Runsink J., Chem. Ber. 1988, 121, 1991; [Google Scholar]
- 13c. Mirafzal G. A., Liu J., Bauld N. L., J. Am. Chem. Soc. 1993, 115, 6072; [Google Scholar]
- 13d. Steckhan E., Angew. Chem. Int. Ed. Engl. 1986, 25, 683; [Google Scholar]; Angew. Chem. 1986, 98, 681; [Google Scholar]
- 13e. Zhang N., Samanta S. R., Rosen B. M., Percec V., Chem. Rev. 2014, 114, 5848. [DOI] [PubMed] [Google Scholar]
- 14. Baker P. K., Connelly N. G., Jones B. M. R., Maher J. P., Somers K. R., J. Chem. Soc. Dalton Trans. 1980, 579. [Google Scholar]
- 15.
- 15a. Bolskar R. D., Mathur R. S., Reed C. A., J. Am. Chem. Soc. 1996, 118, 13093; [Google Scholar]
- 15b. Reed C. A., Science 2000, 289, 101; [DOI] [PubMed] [Google Scholar]
- 15c. Kim K.-C., Hauke F., Hirsch A., Boyd P. D. W., Carter E., Armstrong R. S., Lay P. A., Reed C. A., ChemInform 2003, 34. [Google Scholar]
- 16. Talipov M. R., Hossain M. M., Boddeda A., Thakur K., Rathore R., Org. Biomol. Chem. 2016, 14, 2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steckhan E., Top. Curr. Chem. 1987, 142, 1. [Google Scholar]
- 18.The formal potential of N(C6Cl5)3 was mentioned as a personal communication in Table 7 of Ref. [4].
- 19.
- 19a. Koppang R., Hedlund K., Rasmussen S. E., Svensson S., Koskikallio J., Kachi S., Acta Chem. Scand. 1971, 25, 3067; [Google Scholar]
- 19b. Miller A. O., Furin G. G., J. Fluorine Chem. 1995, 75, 169; [Google Scholar]
- 19c. Linder T., Sundermeyer J., Chem. Commun. 2009, 2914. [DOI] [PubMed] [Google Scholar]
- 20. Huber B., Linder T., Hormann K., Frömling T., Sundermeyer J., Roling B., Z. Phys. Chem. 2012, 226, 377. [Google Scholar]
- 21. Schleep M., Hettich C., Kratzert D., Scherer H., Krossing I., Chem. Commun. 2017, 53, 10914. [DOI] [PubMed] [Google Scholar]
- 22. Malischewski M., Seppelt K., Sutter J., Munz D., Meyer K., Angew. Chem. Int. Ed. 2018, 57, 14597; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14806. [Google Scholar]
- 23. Callan B., Manning A. R., Stephens F. S., J. Organomet. Chem. 1987, 331, 357. [Google Scholar]
- 24. Sattler W., Parkin G., Chem. Commun. 2009, 7566. [DOI] [PubMed] [Google Scholar]
- 25. Halbauer K., Dönnecke D., Görls H., Imhof W., Z. Anorg. Allg. Chem. 2006, 632, 1477. [Google Scholar]
- 26. Bassett J.-M., Berry D. E., Barker G. K., Green M., Howard J. A. K., Stone F. G. A., J. Chem. Soc. Dalton Trans. 1979, 1003. [Google Scholar]
- 27.National Institute of Advanced Industrial Science and Technology, accessed 17/02/2020 (SDBSWeb: https://sdbs.db.aist.go.jp).
- 28.CCDC 1985536, 1985539, 1985540, 1985541, 1985542, 1985543, 1985544, 1985545, 1985546, 1985547 and 1985548 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary