

Abstract
Since the first description of galactosemia in 1908 and despite decades of research, the pathophysiology is complex and not yet fully elucidated. Galactosemia is an inborn error of carbohydrate metabolism caused by deficient activity of any of the galactose metabolising enzymes. The current standard of care, a galactose‐restricted diet, fails to prevent long‐term complications. Studies in cellular and animal models in the past decades have led to an enormous progress and advancement of knowledge. Summarising current evidence in the pathophysiology underlying hereditary galactosemia may contribute to the identification of treatment targets for alternative therapies that may successfully prevent long‐term complications. A systematic review of cellular and animal studies reporting on disease complications (clinical signs and/or biochemical findings) and/or treatment targets in hereditary galactosemia was performed. PubMed/MEDLINE, EMBASE, and Web of Science were searched, 46 original articles were included. Results revealed that Gal‐1‐P is not the sole pathophysiological agent responsible for the phenotype observed in galactosemia. Other currently described contributing factors include accumulation of galactose metabolites, uridine diphosphate (UDP)‐hexose alterations and subsequent impaired glycosylation, endoplasmic reticulum (ER) stress, altered signalling pathways, and oxidative stress. galactokinase (GALK) inhibitors, UDP‐glucose pyrophosphorylase (UGP) up‐regulation, uridine supplementation, ER stress reducers, antioxidants and pharmacological chaperones have been studied, showing rescue of biochemical and/or clinical symptoms in galactosemia. Promising co‐adjuvant therapies include antioxidant therapy and UGP up‐regulation. This systematic review provides an overview of the scattered information resulting from animal and cellular studies performed in the past decades, summarising the complex pathophysiological mechanisms underlying hereditary galactosemia and providing insights on potential treatment targets.
Keywords: animal models, cellular models, hereditary galactosemia, pathophysiology, treatment targets
Abbreviation
- ATP
adenosine triphosphate
- BiP
binding protein
- CG
classic galactosemia
- CHO
Chinese hamster ovary
- ER
endoplasmic reticulum
- Gal‐1‐P
galactose‐1‐phosphate
- GALE
UDP‐galactose 4′‐epimerase
- GALK
galactokinase
- GALM
galactose mutarotase
- GALT
galactose‐1‐phosphate uridylyltransferase
- Glc‐1‐P
glucose‐1‐phosphate
- HPLC
high‐performance liquid chromatography
- PI3/Akt
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase/protein kinase B
- PRISMA
Preferred Reporting Items for Systematic Reviews and Meta‐Analysis
- SIGN
Scottish Intercollegiate Guidelines Network
- SNA
Sambucus nigra agglutinin
- SOD
superoxide dismutase
- UDP‐Gal
uridine diphosphate‐galactose
- UDP‐GalNAc
UDP‐N‐acetylgalactosamine
- UDP‐Glc
uridine diphosphate‐glucose
- UDP‐GlcNAc
UDP‐N‐acetylglucosamine
- UGP
UDP‐glucose pyrophosphorylase
- UPR
unfolded protein response
1. INTRODUCTION
Despite that the first description of galactosemia that dates from more than 100 years ago (1908) and decades of research, the pathophysiology of this disease is complex and not yet fully understood. Studies in cellular and animal models in the past decades have led to an enormous progress and advancement of knowledge. Systematically summarising current evidence in the pathophysiology underlying hereditary galactosemia may contribute to the identification of treatment targets for alternative therapies that may successfully prevent the occurrence of long‐term complications.
Galactose is an aldohexose and is mainly metabolised via the Leloir pathway (Figure 1). In most organisms, the first step of the Leloir pathway involves epimerization of β‐d‐galactose to α‐d‐galactose by galactose mutarotase (GALM; EC 5.1.3.3), encoded by the GALM gene located on chromosome 2p22.1. The next step entails the phosphorylation of α‐d‐galactose into galactose‐1‐phosphate (Gal‐1‐P) by the action of galactokinase (GALK; EC 2.7.1.6), encoded by the GALK1 gene located on chromosome 17q25.1. Together with uridine diphosphate‐glucose (UDP‐Glc), Gal‐1‐P is converted to glucose‐1‐phosphate (Glc‐1‐P) and uridine diphosphate‐galactose (UDP‐Gal) by galactose‐1‐phosphate uridylyltransferase (GALT; EC 2.7.7.12) encoded by the GALT gene located on chromosome 9p13. UDP‐galactose 4′‐epimerase (GALE; EC 5.1.3.2) is encoded by the GALE gene located on chromosome 1p36.11 and catalyses the conversion of UDP‐Gal into UDP‐Glc.1 In mammals, GALE is also responsible for the interconversion of UDP‐N‐acetylglucosamine (UDP‐GlcNAc) and UDP‐N‐acetylgalactosamine (UDP‐GalNAc). GALE maintains the intracellular pools of UDP‐Gal/UDP‐Glc and UDP‐GlcNAc/UDP‐GalNAc.
Figure 1.
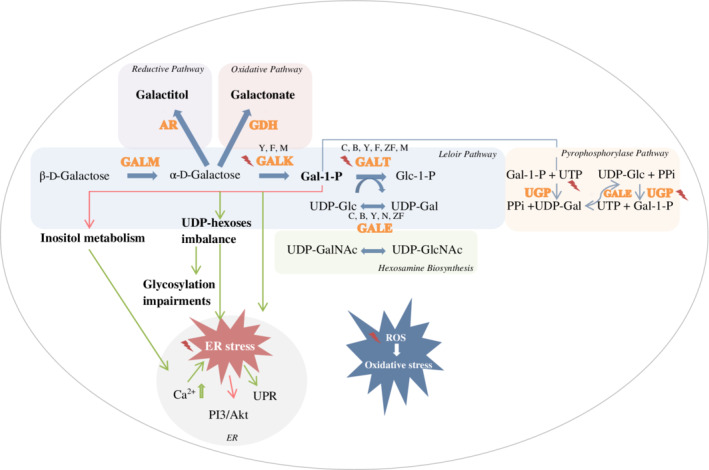
Involved pathways, pathophysiological agents and targets for treatment in hereditary galactosemia, studied in cellular and animal models , indicating a treatment approach that has been evaluated in the cellular and animal models.
, indicating a treatment approach that has been evaluated in the cellular and animal models.  , inhibition.
, inhibition.  , induction. GALK deficiency was studied in yeast (Y), fruitfly (F) and mouse (M). GALT deficiency was studied in patient cell cultures (C), bacteria (B), yeast, fruitfly, zebrafish (ZF) and mouse. GALE deficiency was studied in bacteria, yeast, nematode (N) and fruitfly. AR, aldose reductase, ER, endoplasmic reticulum, Gal‐1‐P, galactose‐1‐phosphate, GALE, UDP‐galactose 4′‐epimerase, GALK, galactokinase, GALT, galactose‐1‐phosphate uridylyltransferase, GALM, galactose mutarotase, GDH, galactose dehydrogenase, Glc‐1‐P, glucose‐1‐phosphate, PI3/Akt, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase/protein kinase B, PPi, inorganic pyrophosphate, ROS, reactive oxygen species, RP, ribosomal protein, UDP‐Gal, uridine diphosphate‐galactose, UDP‐GalNAc, UDP‐N‐acetylgalactosamine, UDP‐Glc, uridine diphosphate‐glucose, UDP‐GlcNAc, UDP‐N‐acetylglucosamine, UGP, UDP‐glucose pyrophosphorylase, UPR, unfolded protein response, UTP, uridine‐5′‐triphosphate
, induction. GALK deficiency was studied in yeast (Y), fruitfly (F) and mouse (M). GALT deficiency was studied in patient cell cultures (C), bacteria (B), yeast, fruitfly, zebrafish (ZF) and mouse. GALE deficiency was studied in bacteria, yeast, nematode (N) and fruitfly. AR, aldose reductase, ER, endoplasmic reticulum, Gal‐1‐P, galactose‐1‐phosphate, GALE, UDP‐galactose 4′‐epimerase, GALK, galactokinase, GALT, galactose‐1‐phosphate uridylyltransferase, GALM, galactose mutarotase, GDH, galactose dehydrogenase, Glc‐1‐P, glucose‐1‐phosphate, PI3/Akt, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase/protein kinase B, PPi, inorganic pyrophosphate, ROS, reactive oxygen species, RP, ribosomal protein, UDP‐Gal, uridine diphosphate‐galactose, UDP‐GalNAc, UDP‐N‐acetylgalactosamine, UDP‐Glc, uridine diphosphate‐glucose, UDP‐GlcNAc, UDP‐N‐acetylglucosamine, UGP, UDP‐glucose pyrophosphorylase, UPR, unfolded protein response, UTP, uridine‐5′‐triphosphate
Galactosemia is an inborn error of carbohydrate metabolism caused by a severe impairment of any of the enzymes involved in the Leloir pathway and comprises four subtypes. Type 1, also known as classic galactosemia (CG, OMIM 230400) is characterised by severe deficiency of the GALT enzyme. Glycosylation abnormalities have been described in CG, characterised by processing defects (reviewed by Maratha et al2). UDP‐hexoses abnormalities and inhibition of galactosyltransferases by high levels of Gal‐1‐P have been suggested as potential mechanisms.3, 4, 5, 6 Defective galactosylation has been described in patient fibroblasts7, 8 and serum glycoproteins of untreated CG patients.3, 9 CG is potentially lethal in the neonatal period, with multi‐organ involvement. The current standard of care, a galactose restricted diet, although life‐saving in the neonatal period, fails to prevent complications, such as cognitive impairments, neurological symptoms, and subfertility in female patients.10 Type 2 is characterised by deficiency of the GALK enzyme (OMIM 230200), in untreated patients presenting with cataract.11 Other symptoms have also been described in several patients: pseudotumor cerebri, hypoglycemia (asymptomatic), mental retardation, microcephaly, failure to thrive, seizures, deafness, hepatomegaly, and hypercholesterolemia.12, 13 Type 3 is caused by a deficiency of the GALE enzyme (OMIM 230350), presenting with acute symptoms similar to CG (14 updated in 2016). Long‐term outcomes reported in GALE deficient patients include physical and cognitive developmental delay and/or learning disabilities. Recently, the fourth type of galactosemia has been described, caused by biallelic pathogenic variants in GAL.15
When one of the Leloir pathway enzymes is deficient, galactose is disposed through alternative pathways. These include galactose reduction to galactitol, catalysed by the NADPH dependent aldose reductase (AR, EC 1.1.1.21), galactose oxidation, leading to the production of galactonate by galactose dehydrogenase (EC 1.1.1.48) and presumably activation of the pyrophosphorylase pathway, which converts galactose to UDP‐Glc by the sequential activities of GALK and UDP‐glucose pyrophosphorylase (UGP, EC 2.7.7.10).16
In the past decades, a myriad of research has taken place using cellular and animal models to gain insight on the complex pathophysiology of hereditary galactosemia. This study provides a comprehensive systematic review of the scattered information in the different studies, through long periods of time. This can be of great benefit to researchers in the field and provide insights on potential treatment targets.
2. MATERIALS AND METHODS
This study was carried out and reported according to the Preferred Reporting Items for Systematic Reviews and Meta‐Analysis (PRISMA) guidelines.17 The PRISMA checklist for this report can be found in the (Supplementary file 1). A protocol was developed prospectively that detailed the specific objectives, criteria for study selection and the approach to assess study quality and outcomes. The protocol was first published on October 16th, 2018. During the conduction of this systematic review, the protocol was adapted and updated on August 16th, 2019 (http://syrf.org.uk/protocols/).
2.1. Research question
The primary outcomes for this systematic review were disease complications in GALK, GALT, and/or GALE deficiency studied in animal and/or cellular models of the disease, reported as clinical signs and/or biochemical findings. Our aim was to answer the following question: “What have cellular and/or animal models contributed to unravel the pathophysiology of hereditary galactosemia and to identify targets/approaches for treatment?”
2.2. Search strategy
A computerised literature search was conducted in PubMed/MEDLINE, EMBASE, and Web of Science by two independent researchers (MH and AIC), using animal filters.18, 19 Databases were searched on October 24th, 2018. Additionally, reference lists from included studies were hand‐searched to identify relevant studies. The records retrieved were imported and de‐duplicated in ENDNOTE X8. The complete search strategies can be extracted from the (Supplementary file 2).
2.3. Inclusion and exclusion criteria for study selection
All original studies including animal and/or cellular models focusing on disease complications and/or treatment targets in hereditary galactosemia were included. Articles in a language other than English, case reports, conference abstracts, and reviews were excluded.
2.4. Study eligibility
Titles and abstracts were independently screened by M.H. and A.I.C. Studies not relating to the research question were excluded. Full‐text articles were independently reviewed by M.H. and A.I.C. for the inclusion in the systematic review. In case of exclusion, the reason was reported. In case of a discrepancy in the selection of studies, considerations were discussed with the other authors until consensus on eligibility was reached.
2.5. Data collection
Data were independently extracted by M.H. and A.I.C. according to predefined criteria in the protocol. Outcome measures of interest were disease complications, reported as clinical signs (mimicking the human phenotype: neurologic and cognitive impairment [motor abnormalities], gonadal damage [reduced fertility], cataract, as well as survival rate) and/or biochemical findings (increased metabolites [galactose, Gal‐1‐P, galactitol, galactonate], decreased enzyme activity [GALT, GALK, GALE], altered levels of inositol [and related metabolites], disrupted signalling pathways, glycosylation abnormalities/altered UDP‐hexoses levels, oxidative stress, activation of the unfolded protein response [UPR], endoplasmic reticulum [ER] stress, apoptosis).
2.6. Risk of bias and quality assessment in individual studies
Two independent researchers (M.H. and A.I.C.) performed and discussed risk of bias assessment using the SYRCLE's risk of bias tool for animal studies20 and the Scottish Intercollegiate Guidelines Network (SIGN, https://www.sign.ac.uk/checklists-and-notes.html) quality appraisal checklists for case‐control study designs for cellular studies. Articles were appraised as low, acceptable, or high quality. Those assessed as low quality were excluded from the review. Discrepancies were resolved by discussion with all authors.
3. RESULTS AND DISCUSSION
3.1. Included studies
The computerised literature search identified 3407 potential hits, from which 46 articles were included in this systematic review. Figure 2 shows the PRISMA flow diagram for inclusion of individual studies. Of the 46 included articles, three reported on GALK deficiency, 39 on GALT deficiency, and 10 on GALE deficiency. Synoptic tables of the included studies are presented in the supplementary material (Supplementary file 3).
Figure 2.
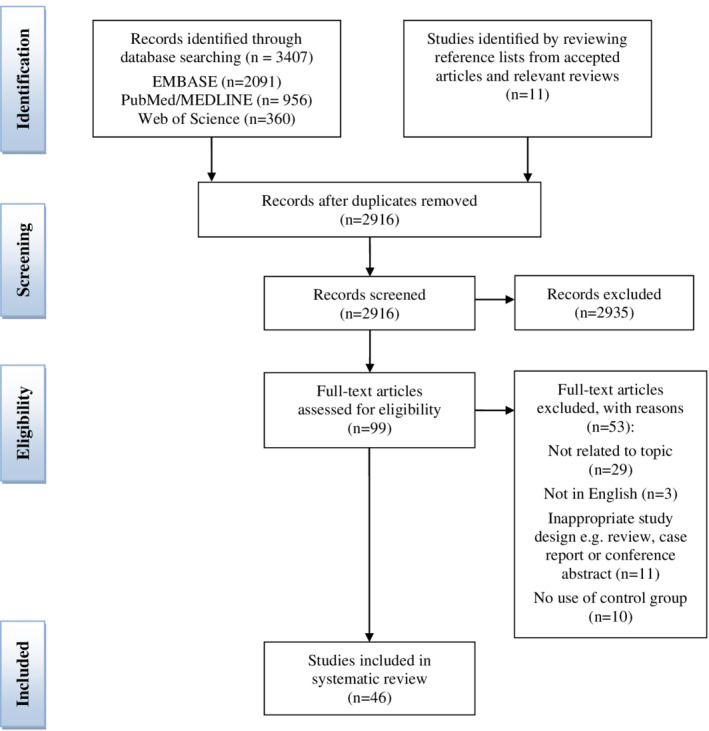
Preferred Reporting Items for Systematic Reviews and Meta‐Analysis flow diagram for the inclusion of studies
3.2. Quality assessment
The quality of the included individual studies was assessed using SYRCLE's risk of bias tool and SIGN quality appraisal checklist (Supplementary file 4).
We categorised the obtained data on pathophysiology (summary in Tables 1 and 2, and Figure 1) and treatment targets (summary in Table 3 and Figure 1) as follows: (a) accumulation of metabolites, (b) UDP‐hexoses abnormalities and glycosylation impairments, (c) signalling pathways alterations, UPR activation, ER stress, and oxidative stress, (d) structural and functional defects, and (e) treatment approaches.
Table 1.
UDP‐hexoses studies in different patient cell lines
| Cell line | Reference | GALT activity | Measurement method | UDP‐hexoses levels/ratiosb |
|---|---|---|---|---|
| Erythrocytes | Ng et al6 | Undetectable | Enzyme‐based method | Decreased UDP‐Gal levels
|
| 0.2%‐2% | Normal UDP‐Gal levels | |||
| Berry et al51 a | Absent/markedly reduced | HPLC |
Decreased UDP‐Gal levels
Higher UDP‐Glc to UDP‐Gal ratio
|
|
| Keevill et al52 | Negligible | HPLC |
Decreased UDP‐Gal levels
Higher UDP‐Glc to UDP‐Gal ratio |
|
| Gibson et al53 | Absent/markedly reduced | HPLC |
Decreased UDP‐Gal levels
Higher UDP‐Glc to UDP‐Gal ratio
|
|
| Xu et al54 | Undetectable | HPLC | Decreased UDP‐Gal levels
|
|
| 0.05‐2.5 μmol/h/g Hb (normal >20) |
Normal UDP‐Gal levels
Normal UDP‐Glc/UDP‐Gal ratio |
|||
| Lymphoblasts | Gibson et al55 | Unknown | HPLC | Normal UDP‐Gal levels
|
| Fibroblasts | Ng et al6 | Undetectable | Enzyme‐based method |
Decreased UDP‐Gal levels 36% of normal mean |
| 0%‐0.2% | Normal UDP‐Gal levels | |||
| Gibson et al55 | Unknown | HPLC |
Normal UDP‐Gal levels
Normal UDP‐Glc/UDP‐Gal ratio |
|
| Xu et al54 | Undetectable | HPLC | Decreased UDP‐Gal levels
|
|
| 0.6‐7.8 μmol/h/g Hb (normal >50) | Normal UDP‐Gal levels
|
|||
| SV40‐transformed fibroblasts | Lai et al5 | Undetectable | Enzyme‐based method | Decreased UDP‐Gal
|
Abbreviations: CI, confidence intervals; HPLC, high‐performance liquid chromatography; UDP‐Gal, uridine diphosphate‐galactose, UDP‐Glc, uridine diphosphate‐glucose.
Absolute levels four times lower compared to values reported by Ng et al.6
In GALT‐deficient cell lines compared to cell lines from normal controls.
Table 2.
Summary of the proposed pathophysiological mechanisms in systematically reviewed studies
| Type of galactosemia | Pathophysiological mechanism | Animal/cellular model | References |
|---|---|---|---|
| GALK deficiency | Galactitol accumulation | Mouse | Ai et al26 |
| GALT deficiency | Accumulation of metabolites (galactose, Gal‐1‐P, galactitol, galactonate) |
Patient cell lines (SV40‐transformed fibroblasts) Saccharomyces cerevisiae Mouse
Drosophila melanogaster Danio rerio |
Lai et al5
Ross et al33 Leslie et al25; Ning et al38, 39; Yager et al41; Wehrli et al40; Tang et al30 Kushner et al27; Ryan et al44; Daenzer et al34 Vanoevelen et al31 |
| Abnormalities in UDP‐Gal and UDP‐Glc levelsa | Saccharomyces cerevisiae | Chhay et al50 | |
| Aberrant glycosylation |
Patient cell lines (fibroblasts)
Patient cell lines (SV40‐transformed fibroblasts) Patient cell lines (lymphoblasts) Drosophila melanogaster |
Dobbie et al7; Ornstein et al8; Coss et al4; Staubach et al56 Lai et al5
Petry et al57 |
|
| Down‐regulation of PI3K/Akt pathway | Mouse | Balakrishnan et al60, 61; Chen et al62 | |
| ER stress/Unfolded protein response | Saccharomyces cerevisiae | Slepak et al63; De‐Souza et al64 | |
| Patient cell lines (fibroblasts) | Slepak et al65; Staubach et al56 | ||
| Mouse | Balakrishnan et al61; Chen et al62 | ||
| Oxidative stress | Drosophila melanogaster | Jumbo‐Lucioni et al66, 67 | |
| Mouse | Tang et al30 | ||
| Structural/functional defects | Saccharomyces cerevisiae | McCorvie et al68 | |
| Escherichia coli | Coelho et al22 | ||
| GALE deficiency | Gal‐1‐P accumulation | Saccharomyces cerevisiae | Wohlers and Fridovich‐Keil45; Ross et al33; Mumma et al46 |
| Abnormalities in UDP‐hexoses levels | ldlD cells | Schulz et al69 | |
| Saccharomyces cerevisiae | Ross et al33 | ||
| Caenorhabditis elegans | Brokate‐Llanos et al29 | ||
| Drosophila melanogaster | Daenzer et al70; Jumbo‐Lucioni et al59 | ||
| ER stress | Caenorhabditis elegans | Brokate‐Llanos et al29 | |
| Structural/functional defects | Saccharomyces cerevisiae | McCorvie et al71 | |
| Escherichia coli | Pey et al.72 |
Abbreviations: ER, endoplasmic reticulum; GALE, uridine diphosphate‐galactose 4′‐epimerase; GALK, galactokinase; UDP‐Gal, uridine diphosphate‐galactose, UDP‐Glc, uridine diphosphate‐glucose.
A separate table (Table 3) is provided for UDP‐hexoses studies in different patient cell lines.
Table 3.
Summary of the studied treatment approaches in systematically reviewed studies
| Type of galactosemia | Treatment approach | Animal/cellular model | References |
|---|---|---|---|
| GALT deficiency | GALK inhibition | Saccharomyces cerevisiae | Ross et al33; Mumma et al46; De‐Souza et al64; Machado et al73 |
| Patient cell lines (fibroblasts) | Tang et al74 | ||
| Drosophila melanogaster | Jumbo‐Lucioni et al58; Daenzer et al34 | ||
| Antioxidants | Drosophila melanogaster | Jumbo‐Lucioni et al,66, 67 | |
| Pharmacological chaperones | Escherichia coli | Coelho et al75 | |
| Patient cell lines (fibroblasts) | Haskovic et al76 | ||
| Antisense therapy | Escherichia coli | Coelho et al77 | |
| ER stress reducers | Saccharomyces cerevisiae | De‐Souza et al64 | |
| Mouse | Balakrishnan et al60, 61 | ||
| UGP up‐regulation | Saccharomyces cerevisiae | Lai and Elsas78 | |
| Patient cell lines (SV40‐transformed fibroblasts) | Lai et al5 | ||
| Uridine supplementation | Patient cell lines (erythrocytes) | Ng et al6 | |
| GALE deficiency | GALK inhibition | Saccharomyces cerevisiae | Ross et al33; Mumma et al46 |
| Uridine supplementation | ldlD cells | Schulz et al69 |
Abbreviations: ER, endoplasmic reticulum; GALE, uridine diphosphate‐galactose 4′‐epimerase; GALK, galactokinase.
In the past decades, studies in patient cell lines, yeast—Saccharomyces cerevisiae—21 bacteria—Escherichia coli—22, 23, 24 as well as studies in animal models have been performed. The first animal model of classic galactosemia, a Galt knockout mouse, was described in 1996.25 After several years, a mouse model for GALK deficiency was developed.26 Fruit fly—Drosophila melanogaster—models for both GALT and GALE deficiency followed in 2010.27, 28 Furthermore, a nematode—Caenorhabditis elegans—for GALE deficiency was developed.29 In addition to the mouse model described by Leslie et al, a Galt gene‐trapped mouse was reported in 2014.30 More recently, a zebrafish—Danio rerio—model for GALT deficiency was described.31
3.3. Accumulation of metabolites
3.3.1. GALK deficiency
In GALK deficiency, there is no accumulation of Gal‐1‐P.32
Ross et al showed that GALK‐deficient yeast demonstrated normal growth, no accumulation of Gal‐1‐P and normal UDP‐Gal and UDP‐Glc levels.33 Studies in fruit flies demonstrated that, despite low levels of Gal‐1‐P, loss of GALK slightly lowered survival rates, in both the absence and presence of dietary galactose. Loss of GALK had a larger negative impact on the climbing ability and fecundity, in the absence of galactose.34 Studies in a mouse model for GALK deficiency showed that increased galactitol synthesis, when overexpressing aldose reductase, led to cataract formation.26 A low expression of aldose reductase in the lens of mice has been reported.35 Moreover, galactitol accumulation depletes NADPH levels and leads to decreased glutathione reductase activity. As a result, free radicals accumulate, causing oxidative stress that can lead to cell death.36
3.3.2. GALT deficiency
Gal‐1‐P has been hypothesized as one of the key pathogenic agents in classic galactosemia37 and studied extensively.
Leslie's GALT‐deficient mouse demonstrated accumulated levels of Gal‐1‐P (in liver homogenates and erythrocytes) and galactose (in plasma) during the suckling period. Despite this metabolic abnormality, these mice did not exhibit the expected clinical phenotype,25 even after a high galactose diet.38 Substantial levels of Gal‐1‐P and galactitol in liver, kidney and brain of the adult mice were found38, 39 and only a trace of galactitol in plasma.38 Highest levels of galactitol were reported in muscle and heart,40, 41 in agreement with the tissue‐specific differences in aldose reductase expression levels.42 Measurable galactose, galactitol, and Gal‐1‐P levels in mice deprived of exogenous galactose for 4 weeks provided evidence of endogenous galactose production.38 Galactose level in the liver appeared to be lower than that in plasma, kidney and brain, likely due to its conversion to galactonate in the liver.43 Furthermore, a relatively low brain tissue galactose level was found which led to the hypothesis that the blood‐brain barrier may restrict galactose entry.38, 39, 41 Galactonate has been measured in different tissues (liver, kidney, and brain) in mice.38, 39 They observed that galactonate levels accumulated in all tissues after galactose exposure. In the unexposed situation, no galactonate was found in the brain. In another study, galactonate levels in mice muscle and heart were found 10 times higher than that in brain or kidney after galactose exposure.41 The combined accumulation of Gal‐1‐P and galactitol was suggested to cause the phenotype of human GALT deficiency.38, 39
In 2014, a Galt gene‐trapped mouse was developed by Tang et al. They showed that galactose challenge in homozygous Galt gene‐trapped lactating females affected the outcome of the newborn Galt gene‐trapped mice. The newborn mice showed a high mortality rate, insults in brain tissues and manifestations of oxidative stress in red blood cells (altered glutathione/oxidised glutathione ratio). Brain lesions in the galactose‐intoxicated pups were characterised by smaller, more acidophilic Purkinje cells and a thicker outer granular cell layer of the cerebellum. This finding led the authors to suggest a post‐natal role of galactose in the pathophysiology of the ataxia observed in patients. The mice accumulated only modest levels of Gal‐1‐P in red blood cells, even under galactose challenge. Furthermore, they observed growth restriction in unexposed mice and reduced fertility in female mice who were not exposed to galactose beyond the weaning period. Authors proposed that this finding may explain the higher galactose resistance in rodents compared to humans.30
Extensive studies in the GALT‐deficient D. melanogaster revealed that larvae showed more rapid lethality compared to adult flies, despite comparable levels of Gal‐1‐P. The authors concluded that this observation might result from stage‐specific ways in which the fruit flies responded to high Gal‐1‐P levels.27 Moreover, movement defects were demonstrated in adult flies, which did not exacerbate by early galactose exposure, despite a 20‐fold increase in Gal‐1‐P.44 Galactose exposure was sufficient to cause elevated levels of Gal‐1‐P, without impacting the larval survival or adult outcome.
More recently, a zebrafish (Danio rerio) model of CG was created resembling the human phenotype of neurological sequel and subfertility, despite the lack of exposure to exogenous galactose. High levels of Gal‐1‐P were found in 9‐day old larvae in the absence of exogenous galactose.31 These results further support the evidence that neurological and fertility impairments occur in zebrafish without exposure to exogenous galactose.
3.3.3. GALE deficiency
Studies in GALE‐deficient S. cerevisiae demonstrated accumulated Gal‐1‐P levels and growth arrest following galactose exposure.45 The role of Gal‐1‐P as mediator of galactose sensitivity was studied in GALE‐ as well as GALT‐deficient strains. GALE‐deficient cells were 10‐fold more sensitive to galactose compared to GALT‐deficient cells and showed to remain constant Gal‐1‐P levels after galactose exposure, while in GALT‐deficient cells the Gal‐1‐P levels gradually decreased over time.33 Loss of GALT was able to rescue the galactose‐induced growth restriction of GALE‐deficient cells, without impacting the Gal‐1‐P levels.33, 46 GALE‐deficient yeast also showed increased levels of UDP‐Gal and slightly decreased levels of UDP‐Glc, in parallel to Gal‐1‐P accumulation. Authors concluded that Gal‐1‐P alone could not explain the differential growth rates observed in galactose‐sensitive yeast.33, 46
Taken together, the results from different cellular and animal models suggest that Gal‐1‐P is not the sole pathophysiological agent responsible for the phenotype observed in GALT and GALE deficiency. Most probably a combination of events contributes to the observed human phenotype. We hypothesize that Gal‐1‐P is necessary but not sufficient in all tissues for expression of the human phenotype.
3.4. UDP‐hexoses abnormalities and glycosylation impairments
3.4.1. GALT deficiency
Several reactions are involved in maintaining UDP‐hexoses levels. There is evidence that in the absence of GALT, when UDP‐Gal levels are low, the GALE reaction shifts toward the formation of UDP‐Gal.47 Furthermore, UGP is responsible for the GALT‐independent formation of UDP‐Glc from Glc‐1‐P and UTP. However, UGP can also catalyse the conversion of Gal‐1‐P and UTP to form UDP‐Gal.48 Additionally, studies in mice showed that UDP‐Gal can possibly be formed by UGP in the absence of GALT activity.49
UDP‐hexose levels were studied in S. cerevisiae expressing patient GALT mutations, erythrocytes and cultured fibroblasts from patients with GALT deficiency.
Yeast strains with the lowest levels of GALT activity accumulated intracellular UDP‐Gal and UDP‐Glc levels after galactose exposure, possibly due to the UGP‐mediated conversion of accumulated Gal‐1‐P to UDP‐Gal. Given that the increase in UDP‐Gal levels parallel those in UDP‐Glc, the authors concluded that a more likely explanation might be the epimerization of the accumulated UDP‐Glc to UDP‐Gal by GALE, in absence of GALT activity.50
UDP‐hexoses levels measured in patient erythrocytes, lymphocytes, and normal or transformed fibroblasts show sometimes contradictory results (Table 1).51, 52, 53, 54, 55 Drawing any conclusion from the observed findings is therefore very difficult. Various techniques (high‐performance liquid chromatography [HPLC], indirect enzyme‐based techniques) have been applied to measure UDP‐hexoses levels, some of which are now recognised as susceptible to interference. In addition, different cell lines might behave differently and give distinct results. Furthermore, pre‐analytical conditions, such as culturing and incubation techniques can also affect the results.
Despite the fact that some groups report significantly lower UPD‐Gal and UDP‐Glc levels in erythrocytes and fibroblasts of CG patients (Table 1), the clinical relevance of the reduced levels remains questionable. Furthermore, UDP‐hexoses levels in the relevant target tissues of damage (brain and gonads) have not been studied. More studies using accurate cutting‐edge techniques to measure UDP‐hexoses preferably in these relevant tissues of damage would greatly improve our knowledge regarding the UDP‐hexoses levels in CG and its potential clinical relevance. The discrepancies in the data available hitherto do not unambiguously answer the question whether UDP‐hexoses alterations are relevant for the pathophysiology and warrant further investigation.
There is evidence of abnormal glycosylation in CG patients. Haberland et al described abnormal patterns of glycoproteins in autopsy brain tissue and suggested that this could be due to defective galactose incorporation.79 Studies in fibroblasts deriving from GALT‐deficient patients showed an aberrant glycosylation of glycoproteins and glycolipids.7, 8 Abnormal galactose to mannose and sialic acid plus galactose to mannose ratios were found. Sialic acid is a terminal residue and is normally attached to galactose within the oligosaccharide units of glycoproteins.7 Lai et al showed a reduced abundance of the high molecular weight Sambucus nigra agglutinin (SNA) positive glycoproteins in GALT‐deficient SV40‐transformed fibroblasts exposed to galactose, indicating an abnormal glycosylation.5
Coss et al demonstrated dys‐regulation of the glycosylation and inflammatory pathways at the gene and protein level in GALT‐deficient fibroblasts.4 Imbalance in glycolipids in GALT‐deficient lymphoblasts was found by Petry et al, hypothesized to be caused by deficient levels of UDP‐Gal and UDP‐GalNAc and thereby reduction of galactosylation products.57 In addition, Staubach et al showed a decrease in plasma membrane expression of N‐glycoproteins upon galactose exposure in GALT‐deficient fibroblasts, important in the regulation of cell signalling. Authors suggested that at least for some of the glycosylated membrane proteins a dys‐glycosylation might be the cause of decreased membrane expression.56
Jumbo‐Lucioni et al studied the synaptomatrix glycosylation in the GALT‐deficient D. melanogaster and showed that glycans play a primarily inhibitory role in modulating synapse morphogenesis. They suggested that neuromuscular junction development defects and associated impairments in coordinated movement in the GALT‐deficient fruit flies might result from disrupted UDP‐hexose balance and the consequently impaired glycosylation.58 Double mutant GALT‐, UGP‐deficient fruit flies showed exacerbated coordinated movement deficits. UGP played a particularly important role enabling coordinated movement and was found to be a strong negative modulator of neurobehavioral outcomes in the fruit flies.59
3.4.2. GALE deficiency
The GALE deficiency was studied in the cellular models, S. cerevisiae, the Caenorhabditis elegans and the D. melanogaster models.
GALE‐deficient S. cerevisiae accumulated UDP‐Gal and showed decreased levels of UDP‐Glc upon galactose exposure.33
Schulz et al showed that GALE‐deficient ldlD cells (GALE‐impaired mammalian cells utilising a strain of Chinese hamster ovary [CHO]) accumulated both Gal‐1‐P and UDP‐Gal when exposed to galactose, whereas depleted levels of UTP, UDP‐Glc, UDP‐GalNAc, and UDP‐GlcNAc were found. An explanation given by the authors is that absent of GALE activity prevented the production of UDP‐Gal into UDP‐Glc. The accumulation of UDP‐Gal resulted in a UDP‐sink, leading to UTP/uridine depletion. Because UTP is necessary for the de novo synthesis of UDP‐Glc by UGP and UDP‐GlcNAc by UDP‐N‐acetylglucosamine pyrophosphorylase, the cells were not able to synthesise these products. Therefore, UTP depletion might contribute to the galactose sensitivity in these cells. Accumulation of Gal‐1‐P and UDP‐Gal correlated with galactose‐induced compromised growth of GALE impaired cells, whereas deficiency of UDP‐GalNAc did not. Cells expressing E. coli GALE exhibiting activity exclusively toward UDP‐Gal/UDP‐Glc (lacking activity toward UDP‐GalNAc/UDP‐GlcNAc), grew normally in the presence of galactose, despite undetectable levels of UDP‐GalNAc.69
Studies in the GALE‐deficient C. elegans model showed increased levels of UDP‐Gal and reduction of UDP‐GalNAc. The reduction of GALE activity affected multiple aspects of development, most likely resulting from a general defect in the glycosylation process. Strong developmental defects were seen in gonad migration and vulva formation, possibly resulting from a decrease in the availability of UDP‐GalNAc.29
Studies in GALE‐deficient D. melanogaster showed acute complications regardless of galactose and galactose‐induced accumulation of Gal‐1‐P. Furthermore, they showed the importance of GALE activity for development.28 Fruit flies expressing GALE activity toward UDP‐Gal/UDP‐Glc and/or UDP‐GalNAc/UDP‐GlcNAc were studied and showed that both activities were necessary for survival.70 Loss of activity toward UDP‐Gal was shown to result in Gal‐1‐P and UDP‐Gal accumulation, a reduced lifespan when exposed to galactose and impaired fecundity in both male and female larvae, whereas loss of activity toward UDP‐GalNAc did not lead to metabolic abnormalities but resulted in compromised survival and impaired fecundity in female larvae. Authors suggested that nucleotide sugars imbalance rather than Gal‐1‐P was responsible for the compromised survival and fecundity in this model.70
Jumbo‐Lucioni et al showed that loss of neuronal GALE activity worsened the coordinated movement defects in GALT‐deficiency. GALE‐deficient fruit flies showed compromised coordinated movement as well as neuromuscular junction synaptogenesis defects. Furthermore, the GALE deficient fruit flies showed also loss of UDP‐GalNAc in the synaptomatrix.59
The results provided by these cellular and animal studies reveal UDP‐hexose impairments in GALE deficiency. The degree of severity of the clinical phenotype depends on the impairment of GALE activity (toward UDP‐Gal/UDP‐Glc and/or UDP‐GalNAc/UDP‐GlcNAc).
4. SIGNALLING PATHWAYS ALTERATIONS, UPR ACTIVATION, ER STRESS, AND OXIDATIVE STRESS
4.1. GALT deficiency
Signalling pathway alterations have been investigated in GALT‐deficient mouse fibroblasts and isolated ovaries and cerebella from GALT‐deficient mice. These studies demonstrated that the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K)/protein kinase B (Akt) signalling pathway was down regulated.60, 61 This pathway is essential for the growth, proliferation, and survival of many tissues, including neuronal and reproductive tissues.80, 81 Studies in GALT‐deficient mouse fibroblasts showed that the down regulation of the PI3K/Akt pathway occurred at both mRNA and protein levels. The authors proposed ER stress and aberrant glycosylation as candidate factors to connect GALT deficiency and the signalling pathway alterations.60 The increase in binding protein (BiP) abundance and the up regulation of Hspb1 that were found, indicated ER stress. This ER stress resulted in reduced mRNA level of pan‐Akt and its abundance, which could lead to decreased Akt availability for phosphorylation. Moreover, a decreased pan‐Akt level and diminished pAkt(thr308) and pAkt (Ser473) phosphorylation were found.60
Studies by Chen et al and Balakrishnan et al showed a down‐regulated PI3K/Akt signalling pathway in the cerebellum isolated from GALT‐deficient mice. They found a significantly reduced steady‐state protein abundance of pAkt (Ser473), pAkt (Thr308), pan‐Akt, and pGSK‐3β. Furthermore, an increased abundance of BiP was found in this tissue.61, 62 Decreased thicknesses of the molecular and granular layer in the cerebellum of mutant mice suggested cerebellar hypoplasia. These mice manifested ataxia‐related motor impairments with varying degrees of severity. A progressive nature of the impairments in older mice was observed.62 Additionally, studies by Balakrishnan et al showed down‐regulated of the PI3/Akt pathway in isolated GALT‐deficient ovaries. pAkt (Ser473), pGsk3β, and Hsp90 levels in the isolated GALT‐deficient ovaries were significantly reduced and higher levels of BiP were found. Moreover, less primordial follicles and Purkinje cells were seen in GALT‐deficient mice.61, 82 The PI3/Akt signalling is important in supporting the survival of immature ovarian follicles.82
ER stress and the unfolded protein response (UPR) activation were studied in GALT deficient S. cerevisiae and fibroblasts.
In mammalian cells, the UPR signal transduction pathway is designed to cope with ER stress, caused by accumulation of misfolded proteins in the ER.83, 84, 85 If the UPR activation is prolonged, it can trigger pro‐apoptotic pathways.86, 87
Slepak et al showed that accumulated levels of Gal‐1‐P exerted down regulation of ribosomal protein (RP) genes (eg, RPA12 and RRN11) and genes involved in the various aspects of RNA metabolism (eg, UTP6, SNR54 snRNA) in GALT‐deficient yeast exposed to galactose.63 De‐Souza et al showed that Gal‐1‐P synthesis was essential to induce ER stress by galactose and consequently UPR activation in a yeast model of CG. A defect in inositol metabolism and subsequently calcium homeostasis was proposed to contribute to the observed ER stress.64
Studies in human GALT‐deficient fibroblasts exposed to galactose showed that the accumulated Gal‐1‐P competitively inhibited human inositol monophosphatase, leading to inositol depletion. Furthermore, they showed that as a result, Ca2+ was released from the ER.65 Perturbation of calcium homeostasis in the ER is known to be an initiator of ER stress.85 Slepak et al found higher expression of ER stress related genes in GALT‐deficient fibroblasts (CHOP, calreticulin, BiP, GRP94, and XBP‐1) after galactose exposure.65
Staubach et al found increased levels of BiP and calreticulin in GALT‐deficient fibroblasts after galactose exposure.56 They are stress markers that indicate activation of the UPR and are involved in protein folding (GRP78) and protein quality control of N‐glycoproteins by binding dysglycosylated molecules (calreticulin).88 Sustained activation of the UPR has been implicated in the progression of several neurodegenerative diseases.89
Moreover, studies in D. melanogaster revealed that oxidative stress and the response to stress contributed to the mechanism of acute galactose toxicity in GALT deficiency. Galactose exposure led to oxidative stress in both GALT‐deficient and control fruit fly, but they responded differently. An induction of genes involved in glutathione metabolism and known to function in the response to oxidative stress (GSTD6 and GSTE7)90 were found in both GALT‐deficient and control larvae, however, GALT‐deficient larvae showed diminished response. The survival rates of galactose‐exposed mutant animals were affected by the presence of oxidants and antioxidants whereas the survival rates of control animals were unaffected.66 In 2014, Jumbo‐Lucioni et al demonstrated that GALT‐deficient larvae exposed to galactose showed increased protein oxidative damage, with almost twice the level of protein‐bound cysteine and glutathione compared to controls.67
Above presented results suggest that ER stress plays an important role in the pathophysiology of GALT deficiency. Subsequently, ER stress can induce UPR and signalling pathway alterations. Furthermore, oxidative stress also seems to be of influence in the observed phenotype.
4.2. GALE deficiency
The expression of C. elegans genes involved in the activation of the UPR pathway (hsp‐4 and xbp‐1) were altered in this GALE‐deficient animal model, thus inducing ER stress. Authors suggested that the observed ER stress might be caused by the incorrect glycosylation of proteins involved in ER functions or by alterations of the UDP‐Glc/UDP‐Gal ratio.29
4.3. Structural and functional defects
4.4. GALT deficiency
Several studies in S. cerevisiae and E. coli have shown that mutations in the human GALT gene resulted in a less stable variant of the protein which failed to fold correctly, was more susceptible to proteolysis or formed aggregates.22, 68 In particular, the p.Q188R variant (most common GALT mutation variant in the Caucasian population) affect GALT aggregation propensity.22 The yeast model (described by21) has been used to study the critical role of different mutations in determining the expression and functionality of the human GALT enzyme.50, 91, 92, 93, 94
4.5. GALE deficiency
McCorvie et al showed that altered cofactor binding can affect stability and activity of human GALE in an E. coli and S. cerevisiae expression systems. A certain level of flexibility was shown to be required for proper function. An increase or decrease in flexibility of GALE, as seen with p.K161 N and p.D175N, respectively, resulted in loss of function.71 Studies by Pey et al concluded that destabilisation and altered conformational dynamics of GALE dimer at physiological conditions (temperature and pH) were linked to loss of enzyme function in GALE deficient E. coli. The authors found that natural ligands (NAD+ or UDP‐Glc) could stabilise the protein.72
In conclusion, in both GALT‐ and GALE‐deficiency, numerous disease‐causing mutations are described be give rise to variants with higher propensity for aggregation and altered stability.
4.6. Treatment approaches
Since the description of the first infant with hypergalactosemia who responded well to a lactose‐restricted diet,95 galactose restriction is the cornerstone of treatment for the galactosemias. The understanding of the molecular basis underlying this group of diseases hitherto yields several other treatment approaches possibilities.
4.6.1. GALK inhibition
Inhibition of GALK, as a potential therapeutic strategy, has been studied. In GALT‐deficient yeast secondary deletion of GALK1 stabilised the levels of UDP‐Gal and UDP‐Glc and relieved the galactose‐induced lethality in both GALT‐ and GALE‐deficient cells.33 Furthermore, it alleviated the galactose‐mediated growth arrest of GALT‐ and GALE‐deficient yeast.46 GALK1 deletion completely inhibited the galactose‐dependent UPR activation and galactose toxicity in yeast,64 but did not increase tolerance to other ER stressors such as tunicamycin or dithiothreitol. Furthermore, GALK inhibitors had a positive influence on ER stress and on the phosphate levels in GALT‐deficient yeast.73 The effect of GALK inhibitors in GALT‐deficient fibroblasts showed a dose‐dependent effect on Gal‐1‐P accumulation, where levels were lowered to control levels. Tang et al provided a proof‐of‐concept that GALK inhibition can lower Gal‐1‐P in galactose‐challenged GALT deficient fibroblasts.74
GALK inhibition was studied in the D. melanogaster. Jumbo‐Lucioni et al showed that GALK1 co‐removal corrected the movement defects observed in GALT‐deficient fruit flies and restored the synaptomatrix glycosylation state, however, it did not fully reverse NMJ glycan changes.58 Deletion of GALK1 in the GALT‐deficient fruit flies was studied further and revealed to be beneficial in reducing the levels of Gal‐1‐P, but not in rescuing survival of larvae to adulthood, motor defects or fecundity. On the contrary, deletion of GALK1 had a significant negative impact on climbing ability and fecundity in GALT‐deficient fruit flies, in the absence of galactose.34
Taken together, GALK inhibitors are effective in rescuing the biochemical phenotype (eg, reducing Gal‐1‐P levels), however, the effect on the clinical phenotype remains not fully clear. Systematically summarising the current evidence in galactosemia's pathophysiology revealed that Gal‐1‐P is not the sole pathogenic agent responsible for the observed disease complications.
4.6.2. Antioxidants
Increased oxidative stress has been reported in the GALT‐deficient D. melanogaster. The antioxidants vitamin C and alpha‐mangostin increased the survival rates of mutant larvae to adulthood, whereas Gal‐1‐P remained elevated, suggesting that these antioxidants act downstream or independently of Gal‐1‐P.66 In addition, free radical scavengers that mimic the action of Mn2+‐based superoxide dismutase (SOD) have been shown to be effective in rescuing larval survival and reverse protein oxidative damage in GALT‐deficient flies in the presence of galactose.67 Reactive oxygen species (ROS), scavenged by the SOD mimics, contributed to the acute galactose toxicity and galactose‐independent long‐term complications in GALT‐deficient adult fruit flies. These SOD mimics are described to accumulate in critical intracellular compartments, including mitochondria, and promote binding to electron‐rich anionic reactive species enabling them to scavenge superoxide, peroxynitrite, and other free radicals.96 These compounds showed little toxicity, and thus have considerable potential as lead compounds for therapeutic strategies.67
Antioxidant therapy could be one of the possible co‐adjuvant therapies that needs further evaluation.
4.6.3. Pharmacological chaperones
In 2016, the crystal structure of human GALT was resolved97 that provided insights into the functional impact of mutations. The observation that many of the disease‐causing mutations of human GALT and GALE showed structural instability has led to the suggestion that pharmacological chaperones that stabilise and increase activity of the affected protein could be beneficial.68, 72 The effect of arginine, a well‐established suppressant of protein aggregation,98 has been studied in an E. coli model. Arginine has shown a mutation‐specific effect, with rescue of human GALT p.Q188R, p.K285 N, and p.G175D variants, suggesting that this amino acid could be of therapeutic benefit in patients carrying these mutations.75 However, studies in GALT‐deficient fibroblasts from patients carrying the p.Q188R exposed to arginine did not show changes in GALT activity. Thermal shift analysis of recombinant p.Q188R GALT protein in the presence of arginine did not exhibit a positive effect. The authors also evaluated this approach in four p.Q188R homozygous patients but it did not prove effective.76
4.6.4. Antisense therapy
Coelho et al employed two locked nucleic acid oligonucleotides to successfully restore the aberrant splicing profile caused by the intronic mutation c.820 + 13A > G (IVS8 + 13A > G), thus, establishing a proof of concept for the application of antisense therapy for mis‐splicing mutations in CG.77
4.6.5. ER stress reducers
Balakrishnan et al showed that diminished PI3K/Akt signalling, induced by ER stress, played an important role in subfertility and cerebellar ataxia in the GALT‐deficient mouse. Follicle loss due to insufficient Akt activity could be responsible for depletion of most of the primordial follicle pool very early in postnatal life. The effect of salubrinal, an experimental ER stress reducer, was evaluated in the mice. Treatment with salubrinal increased the numbers of primordial follicles. Furthermore, it significantly slowed down the loss of Purkinje cells in GALT‐deficient mice.60, 61
The chemical chaperone 4‐phenylbutyric acid was not effective in reducing ER stress in a S. cerevisiae model for CG.64 De‐Souza et al suggested the idea that molecules that interfere with ER stress might be good drug candidates to treat CG.
4.6.6. UGP up‐regulation
Lai and Elsas showed that UGP1 up‐regulation in isolated GALT revertant from S. cerevisiae cells succeeded to help overcome the galactose toxicity in the GALT revertant, reduced the level of Gal‐1‐P and could replenish UDP‐Gal. The over‐expression of the human homologue hUGP2 gene alone could rescue the growth defect of the GALT‐deficient S. cerevisiae exposed to galactose.78 Studies in GALT‐deficient SV40‐ transformed fibroblasts showed increased UDP‐hexoses levels and no accumulation of Gal‐1‐P in cells expressing human UGP2.5
Based on the studies hitherto described, human UGP2 up‐regulation could be a candidate as therapeutic approach and should also be evaluated in animal models.
4.6.7. Uridine supplementation
The effect of uridine was evaluated in erythrocytes from GALT‐deficient patients and demonstrated that UDP‐Glc and UDP‐Gal levels increased to control levels upon supplementation, without impacting the Gal‐1‐P level. When exposed to both uridine and galactose, UDP‐Gal increased even more compared to uridine alone. The fact that uridine increased UDP‐Gal levels in the absence of GALT activity suggested another mechanism, possibly through the UGP reaction.6
In GALE‐deficient ldlD cells, uridine supplementation relieved the galactose‐specific growth inhibition and corrected UDP‐Glc and to a lesser extent UDP‐GalNAc and UTP, without resucing the Gal‐1‐P and UDP‐Gal levels. A large pool of UDP‐Gal leading to uridine depletion and a potential spectrum of negative downstream effects in the GALE‐impaired cells exposed to galactose was suggested as a key mediator in GALE‐impaired mammalian cells.69 In 1997, Manis et al studied the effects of oral uridine supplementation (150 mg/kg/day for a mean of 3.1 years) in 35 patients (aged 1 week‐16.2 years) with GALT deficiency. Uridine supplementation did not prove to be effective in improving cognitive functioning in these patients compared with patient that were only on a galactose restricted diet.99
Taken together, results obtained from cellular studies and the clinical trial in patients with GALT deficiency show that uridine supplementation is not promising in rescuing the biochemical or clinical phenotype observed in patients.
This study has several limitations. First, we excluded original articles that were not written in English. Also, we have excluded the hypergalactosemia models from this study and focused only on hereditary galactosemia, thereby possibly missing insights on the pathophysiology that underlies galactosemia. Furthermore, results obtained from the cellular and animal models cannot directly be extrapolated to the human situation. The strengths of this study include the comprehensive search and a large number of included studies. Moreover, the inclusion and data extraction have been performed by independent researchers, thereby reducing the risk of bias. To the best our knowledge, this is the first study that provides a comprehensive overview of the scattered information regarding the pathophysiology of hereditary galactosemia based on studies in cellular and animal models performed in the past decades. This information can be of great benefit to researchers in the field and provides insights on potential treatment targets and approaches.
5. CONCLUSIONS
Cellular and animal models have considerably contributed to the pathophysiological insights underlying hereditary galactosemia. Results obtained from this systematic review of the hitherto performed cellular and animal studies reveal that Gal‐1‐P is not sole pathophysiological agent responsible for the phenotype observed in GALT and GALE deficiency. We hypothesize that Gal‐1‐P is necessary but not sufficient in all tissues for expression of the human phenotype. Other currently described contributing factors that underlie the pathophysiology of acute and long‐term outcomes in GALT and GALE deficiency include galactose metabolites accumulation, UDP‐hexoses alterations and subsequent impaired glycosylation, ER stress, signalling pathway alterations and oxidative stress.
ER stress plays an important role in the pathophysiology of GALT deficiency. Subsequently, ER stress can induce UPR and signalling pathway alterations. Furthermore, oxidative stress is another contributing factor in the observed phenotype. Structural and functional studies show that both GALT and GALE disease‐causing mutations give rise to variants with a higher propensity for aggregation and altered stability. The crystal structure of human GALT that was resolved in 2016, allows us to study new therapies enhancing GALT activity/stability, including pharmacological chaperones.
Several therapeutic approaches addressing the contributing factors in the pathophysiology of hereditary galactosemia have been subsequently evaluated in cellular and animal models. GALK inhibitors are found to be effective in rescuing the biochemical phenotype (eg, reducing Gal‐1‐P levels), however, the effects on the observed clinical phenotype remain unclear. Other treatment approaches that have shown to rescue the biochemical and/or clinical outcome include antioxidants, pharmacological chaperones, ER stress reducers, UGP up‐regulation, and uridine supplementation. Based on the hitherto performed studies, possible co‐adjuvant therapies that need further evaluation include antioxidant therapy and UGP up‐regulation. Results from cellular studies and studies in patients show that uridine supplementation is not promising in rescuing the biochemical phenotype. Studies investigating the utilisation of the optimum therapeutic window are crucial for the success of any treatment modality.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
M.H. is contributed in conception and design, acquisition of data, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content. A.I.C. is contributed in conception and design, acquisition of data, analysis and interpretation of data, revising the article critically for important intellectual content. J.B. is contributed in revising the article critically for important intellectual content. J.M.V. is contributed in revising the article critically for important intellectual content. L.K.M.S. is contributed in revising the article critically for important intellectual content. L.J.I.Z. is contributed in revising the article critically for important intellectual content. E.V.‐M. is contributed in conception and design, revising the article critically for important intellectual content. G.T.B. is contributed in analysis and interpretation of data, revising the article critically for important intellectual content. M.E.R.‐G. is contributed in conception and design, analysis and interpretation of data, drafting the article and revising it critically for important intellectual content.
Supporting information
SUPPLEMENTARY FILE 1 Preferred Reporting Items for Systematic Reviews and Meta‐Analysis (PRISMA) 2009 checklist
SUPPLEMENTARY FILE 2. Search strategies
SUPPLEMENTARY FILE 3. Synoptic tables of the included studies
SUPPLEMENTARY FILE 4. Quality assessment and risk of bias of individual studies
Table S1. Included cellular studies evaluating the accumulation of metabolites
Table S2. Included cellular studies evaluating UDP‐hexoses abnormalities and glycosylation impairments
Table S3. Included cellular studies evaluating signalling pathways alterations, UPR activation, ER stress and oxidative stress
Table S4. Included cellular studies evaluating structural and functional defects
Table S5. Included animal studies evaluating th accumulation of metabolites
Table S6. Included animal studies evaluating UDP‐hexoses abnormalities and glycosylation impairments
Table S7. Included animal studies evaluating signalling pathways alterations, UPR activation, ER stress and oxidative stress
Table S8. Risk of bias/quality assessment in cellular studies based on the SIGN checklist for case‐control studies.
Table S9. Risk of bias/quality assessment in animal studies based on the SYRCLE's risk of bias tool for animal studies.
ACKNOWLEDGEMENTS
The authors would like to thank Britt Derks for her contribution in this systematic review.
Haskovic M, Coelho AI, Bierau J, et al. Pathophysiology and targets for treatment in hereditary galactosemia: A systematic review of animal and cellular models. J Inherit Metab Dis. 2020;43:392–408. 10.1002/jimd.12202
Minela Haskovic and A. I. Coelho are co‐first authors.
G.T. Berry and M. E. Rubio‐Gozalbo are co‐last authors.
Communicating Editor: John H Walter
REFERENCES
- 1. Holden HM, Rayment I, Thoden JB. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem. 2003;278:43885‐43888. [DOI] [PubMed] [Google Scholar]
- 2. Maratha A, Colhoun HO, Knerr I, Coss KP, Doran P, Treacy EP. Classical Galactosaemia and CDG, the N‐glycosylation Interface. A review. JIMD Rep. 2017;34:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Defective galactosylation of serum transferrin in galactosemia. Glycobiology. 1998;8(4):351‐357. [DOI] [PubMed] [Google Scholar]
- 4. Coss KP, Treacy EP, Cotter EJ, et al. Systemic gene dysregulation in classical Galactosaemia: is there a central mechanism? Mol Genet Metab. 2014;113(3):177‐187. [DOI] [PubMed] [Google Scholar]
- 5. Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ. GALT deficiency causes UDP‐hexose deficit in human galactosemic cells. Glycobiology. 2003;13(4):285‐294. [DOI] [PubMed] [Google Scholar]
- 6. Ng WG, Xu YK, Kaufman FR, Donnell GN. Deficit of uridine diphosphate galactose in galactosaemia. J Inherit Metab Dis. 1989;12(3):257‐266. [DOI] [PubMed] [Google Scholar]
- 7. Dobbie JA, Holton JB, Clamp JR. Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann Clin Biochem. 1990;27(3):274‐275. [DOI] [PubMed] [Google Scholar]
- 8. Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S. Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose‐1‐phosphate uridyltransferase deficiency. Pediatr Res. 1992;31(5):508‐511. [DOI] [PubMed] [Google Scholar]
- 9. Sturiale L, Barone R, Fiumara A, et al. Hypoglycosylation with increased fucosylation and branching of serum transferrin N‐glycans in untreated galactosemia. Glycobiology. 2005;15(12):1268‐1276. [DOI] [PubMed] [Google Scholar]
- 10. Rubio‐Gozalbo ME, Haskovic M, Bosch AM, et al. The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J Rare Dis. 2019;14(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holton JB, Walter JH, Tyfield LA. Galactosemia In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw‐Hill; 2001:1553‐1583. [Google Scholar]
- 12. Bosch AM, Bakker HD, van Gennip AH, van Kempen JV, Wanders RJ, Wijburg FA. Clinical features of galactokinase deficiency: A review of the literature. J Inherit Metab Dis. 2002;25:629‐634. [DOI] [PubMed] [Google Scholar]
- 13. Hennermann JB, Schadewaldt P, Vetter B, Shin YS, Monch E, Klein J. Features and outcome of galactokinase deficiency in children diagnosed by newborn screening. J Inherit Metab Dis. 2011;34:399‐407. [DOI] [PubMed] [Google Scholar]
- 14. Fridovich‐Keil J, Bean L, He M, Schroer R. Epimerase Deficiency Galactosemia In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReview® Seattle. Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]
- 15. Wada Y, Kikuchi A, Arai‐Ichinoi N, et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med. 2019;21(6):1286‐1294. [DOI] [PubMed] [Google Scholar]
- 16. Berry GT, Walter JH. Disorders of galactose metabolism In: Saudubray JM, van den Berghe G, Walter JH, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. Heidelberg: Springer; 2012. [Google Scholar]
- 17. Moher D, Liberati A, Tetzlaff J, Altman DG, Prisma Group . Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. BMJ. 2009;339:b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hooijmans CR, Tillema A, Leenaars M, Ritskes‐Hoitinga M. Enhancing search efficiency by means of a search filter for finding all studies on animal experimentation in PubMed. Lab Anim. 2010;44(3):170‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vries de RB, Hooijmans CR, Tillema A, Leenaars M, Ritskes‐Hoitinga M. Updated version of the Embase search filter for animal studies. Lab Anim. 2014;48(1):88. [DOI] [PubMed] [Google Scholar]
- 20. Hooijmans CR, Rovers MM, de Vries RB, Leenaars M, Ritskes‐Hoitinga M, Langendam MW. SYRCLE's risk of bias tool for animal studies. BMC Med Res Methodol. 2014;14:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fridovich‐Keil JL, Jinks‐Robertson S. A yeast expression system for human galactose‐1‐phosphate uridylyltransferase. Proc Natl Acad Sci U S A. 1993;90(2):398‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coelho AI, Trabuco M, Ramos R, et al. Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol Genet Genomic Med. 2014;2(6):484‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lai K, Elsas LJ. Structure‐function analyses of a common mutation in blacks with transferase‐deficiency galactosemia. Mol Genet Metab. 2001;74(1–2):264‐272. [DOI] [PubMed] [Google Scholar]
- 24. Lai K, Willis AC, Elsas LJ. The biochemical role of glutamine 188 in human galactose‐1‐phosphate uridyltransferase. J Biol Chem. 1999;274(1):6559‐6566. [DOI] [PubMed] [Google Scholar]
- 25. Leslie ND, Yager KL, McNamara PD, Segal S. A mouse model of galactose‐1‐phosphate uridyl transferase deficiency. Biochem Mol Med. 1996;59(1):7‐12. [DOI] [PubMed] [Google Scholar]
- 26. Ai Y, Zheng Z, O'Brien‐Jenkins A, et al. A mouse model of galactose‐induced cataracts. Hum Mol Genet. 2000;9(12):1821‐1827. [DOI] [PubMed] [Google Scholar]
- 27. Kushner RF, Ryan EL, Sefton JM, et al. A Drosophila melanogaster model of classic galactosemia. Dis Model Mech. 2010;3(9–10):618‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanders RD, Sefton JM, Moberg KH, Fridovich‐Keil JL. UDP‐galactose 4′ epimerase (GALE) is essential for development of Drosophila melanogaster . Dis Model Mech. 2010;3(9–10):628‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brokate‐Llanos AM, Monje JM, Murdoch Pdel S, Munoz MJ. Developmental defects in a Caenorhabditis elegans model for type III galactosemia. Genetics. 2014;198(4):1559‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang M, Siddiqi A, Witt B, et al. Subfertility and growth restriction in a new galactose‐1 phosphate uridylyltransferase (GALT) ‐ deficient mouse model. Eur J Hum Genet. 2014;22(10):1172‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vanoevelen JM, van Erven B, Bierau J, et al. Impaired fertility and motor function in a zebrafish model for classic galactosemia. J Inherit Metab Dis. 2018;41(1):117‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gitzelmann R, Wells HJ, Segal S. Galactose metabolism in a patient with hereditary galactokinase deficiency. Eur J Clin Invest. 1974;4(2):79‐84. [DOI] [PubMed] [Google Scholar]
- 33. Ross KL, Davis CN, Fridovich‐Keil JL. Differential roles of the Leloir pathway enzymes and metabolites in defining galactose sensitivity in yeast. Mol Genet Metab. 2004;83(1–2):103‐116. [DOI] [PubMed] [Google Scholar]
- 34. Daenzer JM, Jumbo‐Lucioni PP, Hopson ML, Garza KR, Ryan EL, Fridovich‐Keil JL. Acute and long‐term outcomes in a Drosophila melanogaster model of classic galactosemia occur independently of galactose‐1‐phosphate accumulation. Dis Model Mech. 2016;9(11):1375‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wen Y, Bekhor I. Levels of expression of hexokinase, aldose reductase and sorbitol dehydrogenase genes in lens of mouse and rat. Curr Eye Res. 1993;12(4):323‐332. [DOI] [PubMed] [Google Scholar]
- 36. Lai K, Elsas LJ, Wierenga KJ. Galactose toxicity in animals. IUBMB Life. 2009;61(11):1063‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gitzelmann R. Galactose‐1‐phosphate in the pathophysiology of galactosemia. Eur J Pediatr. 1995;154(7 Suppl 2):S45‐S49. [DOI] [PubMed] [Google Scholar]
- 38. Ning C, Reynolds R, Chen J, et al. Galactose metabolism in mice with galactose‐1‐phosphate uridyltransferase deficiency: sucklings and 7‐week‐old animals fed a high‐galactose diet. Mol Genet Metab. 2001;72(4):306‐315. [DOI] [PubMed] [Google Scholar]
- 39. Ning C, Fenn PT, Blair IA, Berry GT, Segal S. Apparent galactose appearance rate in human galactosemia based on plasma [(13)C]galactose isotopic enrichment. Mol Genet Metab. 2000;70(4):261‐271. [DOI] [PubMed] [Google Scholar]
- 40. Wehrli S, Reynolds R, Segal S. Metabolic fate of administered [13C]galactose in tissues of galactose‐1‐phosphate uridyl transferase deficient mice determined by nuclear magnetic resonance. Mol Genet Metab. 2007b;90:42‐48. [DOI] [PubMed] [Google Scholar]
- 41. Yager C, Ning C, Reynolds R, Leslie N, Segal S. Galactitol and galactonate accumulation in heart and skeletal muscle of mice with deficiency of galactose‐1‐phosphate uridyltransferase. Mol Genet Metab. 2004;81(2):105‐111. [DOI] [PubMed] [Google Scholar]
- 42. Gui T, Tanimoto T, Kokai Y, Nishimura C. Presence of a closely related subgroup in the aldo‐ketoreductase family of the mouse. Eur J Biochem. 1995;227(1–2):448‐453. [DOI] [PubMed] [Google Scholar]
- 43. Cuatrecasas P, Segal S. Mammalian galactose dehydrogenase. I. Identification and purification in rat liver. J Biol Chem. 1966;241(24):5904‐5909. [PubMed] [Google Scholar]
- 44. Ryan EL, DuBoff B, Feany MB, Fridovich‐Keil JL. Mediators of a long‐term movement abnormality in a Drosophila melanogaster model of classic galactosemia. Dis Model Mech. 2012;5(6):796‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wohlers TM, Fridovich‐Keil JL. Studies of the V94M‐substituted human UDPgalactose‐4‐epimerase enzyme associated with generalized epimerase‐deficiency galactosaemia. J Inherit Metab Dis. 2000;23(7):713‐729. [DOI] [PubMed] [Google Scholar]
- 46. Mumma JO, Chhay JS, Ross KL, Eaton JS, Newell‐Litwa KA, Fridovich‐Keil JL. Distinct roles of galactose‐1P in galactose‐mediated growth arrest of yeast deficient in galactose‐1P uridylyltransferase (GALT) and UDP‐galactose 4′‐epimerase (GALE). Mol Genet Metab. 2008;93(2):160‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prodan‐Zitnik I, Karas‐Kuzelicki N, Lukac‐Bajalo J. Positive correlation between galactose‐1‐phosphate uridyltransferase (GALT) and UDP‐galactose‐4′‐epimerase (GALE) activities. Clin Biochem. 2009;42(15):1561‐1564. [DOI] [PubMed] [Google Scholar]
- 48. Isselbacher KJ. A mammalian uridinediphosphate galactose pyrophosphorylase. J Biol Chem. 1958;232(1):429‐444. [PubMed] [Google Scholar]
- 49. Wehrli S, Reynolds R, Segal S. Evidence for function of UDP galactose pyrophosphorylase in mice with absent galactose‐1‐phosphate uridyltransferase. Mol Genet Metab. 2007a;91(2):191‐194. [DOI] [PubMed] [Google Scholar]
- 50. Chhay JS, Openo KK, Eaton JS, Gentile M, Fridovich‐Keil JL. A yeast model reveals biochemical severity associated with each of three variant alleles of galactose‐1P uridylyltransferase segregating in a single family. J Inherit Metab Dis. 2008;31(1):97‐107. [DOI] [PubMed] [Google Scholar]
- 51. Berry GT, Palmieri MJ, Heales S, Leonard JV, Segal S. Red blood cell uridine sugar nucleotide levels in patients with classic galactosemia and other metabolic disorders. Metabolism. 1992;41(7):783‐787. [DOI] [PubMed] [Google Scholar]
- 52. Keevill NJ, Holton JB, Allen JT. The investigation of UDPGlucose and UDPGalactose concentration in red blood cells of patients with classical galactosaemia. Clin Chim Acta. 1994;221(1–2):135‐142. [DOI] [PubMed] [Google Scholar]
- 53. Gibson JB, Reynolds RA, Palmieri MJ, et al. Comparison of erythrocyte uridine sugar nucleotide levels in normals, classic galactosemics, and patients with other metabolic disorders. Metabolism. 1995;44(5):597‐604. [DOI] [PubMed] [Google Scholar]
- 54. Xu YK, Kaufman FR, Donnell GN, Giudici T, Alfi O, Ng WG. HPLC analysis of uridine diphosphate sugars: decreased concentrations of uridine diphosphate galactose in erythrocytes and cultured skin fibroblasts from classical galactosemia patients. Clin Chim Acta. 1995;240(1):21‐33. [DOI] [PubMed] [Google Scholar]
- 55. Gibson JB, Reynolds RA, Palmieri MJ, States B, Berry GT, Segal S. Uridine diphosphate hexoses in leukocytes and fibroblasts of classic galactosemics and patients with other metabolic diseases. Pediatr Res. 1994;36:613‐618. [DOI] [PubMed] [Google Scholar]
- 56. Staubach S, Muller S, Pekmez M, Hanisch FG. Classical Galactosemia: insight into molecular Pathomechanisms by differential membrane proteomics of fibroblasts under galactose stress. J Proteome Res. 2017;16(2):516‐527. [DOI] [PubMed] [Google Scholar]
- 57. Petry K, Greinix HT, Nudelman E, et al. Characterization of a novel biochemical abnormality in galactosemia: deficiency of glycolipids containing galactose or N‐acetylgalactosamine and accumulation of precursors in brain and lymphocytes. Biochem Med Metab Biol. 1991;46(1):93‐104. [DOI] [PubMed] [Google Scholar]
- 58. Jumbo‐Lucioni P, Parkinson W, Broadie K. Overelaborated synaptic architecture and reduced synaptomatrix glycosylation in a Drosophila classic galactosemia disease model. Dis Model Mech. 2014a;7(12):1365‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jumbo‐Lucioni PP, Parkinson WM, Kopke DL, Broadie K. Coordinated movement, neuromuscular synaptogenesis and trans‐synaptic signaling defects in Drosophila galactosemia models. Hum Mol Genet. 2016;25(17):3699‐3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Balakrishnan B, Chen W, Tang M, et al. Galactose‐1 phosphate uridylyltransferase (GalT) gene: a novel positive regulator of the PI3K/Akt signaling pathway in mouse fibroblasts. Biochem Biophys Res Commun. 2016;470(1):205‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Balakrishnan B, Nicholas C, Siddiqi A, et al. Reversal of aberrant PI3K/Akt signaling by Salubrinal in a GalT‐deficient mouse model. Biochim Biophys Acta Mol Basis Dis. 2017;1863(12):3286‐3293. [DOI] [PubMed] [Google Scholar]
- 62. Chen W, Caston R, Balakrishnan B, et al. Assessment of ataxia phenotype in a new mouse model of galactose‐1 phosphate uridylyltransferase (GALT) deficiency. J Inherit Metab Dis. 2017;40(1):131‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Slepak T, Tang M, Addo F, Lai K. Intracellular galactose‐1‐phosphate accumulation leads to environmental stress response in yeast model. Mol Genet Metab. 2005;86(3):360‐371. [DOI] [PubMed] [Google Scholar]
- 64. De‐Souza EA, Pimentel FS, Machado CM, et al. The unfolded protein response has a protective role in yeast models of classic galactosemia. Dis Model Mech. 2014;7(1):55‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Slepak TI, Tang M, Slepak VZ, Lai K. Involvement of endoplasmic reticulum stress in a novel classic Galactosemia model. Mol Genet Metab. 2007;92(1–2):78‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jumbo‐Lucioni PP, Hopson ML, Hang D, Liang Y, Jones DP, Fridovich‐Keil JL. Oxidative stress contributes to outcome severity in a Drosophila melanogaster model of classic galactosemia. Dis Model Mech. 2013;6(1):84‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jumbo‐Lucioni PP, Ryan EL, Hopson ML, et al. Manganese‐based superoxide dismutase mimics modify both acute and long‐term outcome severity in a Drosophila melanogaster model of classic galactosemia. Antioxid Redox Signal. 2014;20(15):2361‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McCorvie TJ, Gleason TJ, Fridovich‐Keil JL, Timson DJ. Misfolding of galactose 1‐phosphate uridylyltransferase can result in type I galactosemia. Biochim Biophys Acta. 2013;1832(8):1279‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schulz JM, Ross KL, Malmstrom M, Krieger M, Fridovich‐Keil JL. Mediators of galactose sensitivity in UDP‐galactose 4′‐epimerase‐impaired mammalian cells. J Biol Chem. 2005;280(14):13493‐13502. [DOI] [PubMed] [Google Scholar]
- 70. Daenzer JM, Sanders RD, Hang D, Fridovich‐Keil JL. UDP‐galactose 4′‐epimerase activities toward UDP‐Gal and UDP‐GalNAc play different roles in the development of Drosophila melanogaster . PLoS Genet. 2012;8:e1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCorvie TJ, Liu Y, Frazer A, Gleason TJ, Fridovich‐Keil JL, Timson DJ. Altered cofactor binding affects stability and activity of human UDP‐galactose 4′‐epimerase: implications for type III galactosemia. Biochim Biophys Acta. 2012;1822(10):1516‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pey AL, Padin‐Gonzalez E, Mesa‐Torres N, Timson DJ. The metastability of human UDP‐galactose 4′‐epimerase (GALE) is increased by variants associated with type III galactosemia but decreased by substrate and cofactor binding. Arch Biochem Biophys. 2014;562:103‐114. [DOI] [PubMed] [Google Scholar]
- 73. Machado CM, De‐Souza EA, De‐Queiroz ALFV, et al. The galactose‐induced decrease in phosphate levels leads to toxicity in yeast models of galactosemia. Biochim Biophys Acta Mol Basis Dis. 2017;1863(6):1403‐1409. [DOI] [PubMed] [Google Scholar]
- 74. Tang M, Wierenga K, Elsas LJ, Lai K. Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem Biol Interact. 2010;188(3):376‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Coelho AI, Trabuco M, Silva MJ, et al. Arginine functionally improves clinically relevant human Galactose‐1‐phosphate Uridylyltransferase (GALT) variants expressed in a prokaryotic model. JIMD Rep. 2015;23:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Haskovic M, Derks B, van der Ploeg L, et al. Arginine does not rescue p.Q188R mutation deleterious effect in classic galactosemia. Orphanet J Rare Dis. 2018;13(1):212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coelho AI, Lourenco S, Trabuco M, et al. Functional correction by antisense therapy of a splicing mutation in the GALT gene. Eur J Hum Genet. 2015a;23(4):500‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lai K, Elsas LJ. Overexpression of human UDP‐glucose Pyrophosphorylase rescues Galactose‐1‐phosphate Uridyltransferase‐deficient yeast. Biochem Biophys Res Commun. 2000;271(2):392‐400. [DOI] [PubMed] [Google Scholar]
- 79. Haberland C, Perou M, Brunngraber EG, Hof H. The neuropathology of galactosemia. A histopathological and biochemical study. J Neuropathol Exp Neurol. 1971;30(3):431‐447. [DOI] [PubMed] [Google Scholar]
- 80. Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46(6):372‐383. [DOI] [PubMed] [Google Scholar]
- 81. Yung HW, Charnock‐Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One. 2011;6(3):e17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reddy P, Adhikari D, Zheng W, et al. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet. 2009;18(15):2813‐2824. [DOI] [PubMed] [Google Scholar]
- 83. Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87(3):391‐404. [DOI] [PubMed] [Google Scholar]
- 84. Kaufman RJ, Scheuner D, Schroder M, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol. 2002;3(6):411‐421. [DOI] [PubMed] [Google Scholar]
- 85. Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569(1–2):29‐63. [DOI] [PubMed] [Google Scholar]
- 86. Kraskiewicz H, FitzGerald U. InterfERing with endoplasmic reticulum stress. Trends Pharmacol Sci. 2012;33(2):53‐63. [DOI] [PubMed] [Google Scholar]
- 87. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081‐1086. [DOI] [PubMed] [Google Scholar]
- 88. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase‐7 activation. J Biol Chem. 2003;278(23):20915‐20924. [DOI] [PubMed] [Google Scholar]
- 89. Scheper W, Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 2015;130:315‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Alias Z, Clark AG. Studies on the glutathione S‐transferase proteome of adult Drosophila melanogaster: responsiveness to chemical challenge. Proteomics. 2007;7(19):3618‐3628. [DOI] [PubMed] [Google Scholar]
- 91. Christacos NC, Fridovich‐Keil JL. Impact of patient mutations on heterodimer formation and function in human galactose‐1‐P uridylyltransferase. Mol Genet Metab. 2002;76(4):319‐326. [DOI] [PubMed] [Google Scholar]
- 92. Elsevier JP, Fridovich‐Keil JL. The Q188R mutation in human galactose‐1‐phosphate uridylyltransferase acts as a partial dominant negative. J Biol Chem. 1996;271(50):32002‐32007. [DOI] [PubMed] [Google Scholar]
- 93. Elsevier JP, Wells L, Quimby BB, Fridovich‐Keil JL. Heterodimer formation and activity in the human enzyme galactose‐1‐phosphate uridylyltransferase. Proc Natl Acad Sci. 1996;93(14):7166‐7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Riehman K, Crews C, Fridovich‐Keil JL. Relationship between genotype, activity, and galactose sensitivity in yeast expressing patient alleles of human galactose‐1‐phosphate uridylyltransferase. J Biol Chem. 2001;276:10634‐10640. [DOI] [PubMed] [Google Scholar]
- 95. Mason HH, Turner ME. Chronic galactosemia, report of case with studies on carbohydrates. Am J Dis Child. 1935;50(2):359. [Google Scholar]
- 96. Batinic‐Haberle I, Rajic Z, Tovmasyan A, et al. Diverse functions of cationic Mn(III) N‐substituted pyridylporphyrins, recognized as SOD mimics. Free Radic Biol Med. 2011;51:1035‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. McCorvie TJ, Kopec J, Al P, et al. Molecular basis of classic galactosemia from the structure of human galactose 1‐phosphate uridylyltransferase. Hum Mol Genet. 2016;25(11):2234‐2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Baynes BM, Wang DI, Trout BL. Role of arginine in the stabilization of proteins against aggregation. Biochemistry. 2005;44(12):4919‐4925. [DOI] [PubMed] [Google Scholar]
- 99. Manis FR, Cohn LB, McBride‐Chang C, Wolff JA, Kaufman FR. A longitudinal study of cognitive functioning in patients with classical galactosaemia, including a cohort treated with oral uridine. J Inherit Metab Dis. 1997;20(4):549‐555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY FILE 1 Preferred Reporting Items for Systematic Reviews and Meta‐Analysis (PRISMA) 2009 checklist
SUPPLEMENTARY FILE 2. Search strategies
SUPPLEMENTARY FILE 3. Synoptic tables of the included studies
SUPPLEMENTARY FILE 4. Quality assessment and risk of bias of individual studies
Table S1. Included cellular studies evaluating the accumulation of metabolites
Table S2. Included cellular studies evaluating UDP‐hexoses abnormalities and glycosylation impairments
Table S3. Included cellular studies evaluating signalling pathways alterations, UPR activation, ER stress and oxidative stress
Table S4. Included cellular studies evaluating structural and functional defects
Table S5. Included animal studies evaluating th accumulation of metabolites
Table S6. Included animal studies evaluating UDP‐hexoses abnormalities and glycosylation impairments
Table S7. Included animal studies evaluating signalling pathways alterations, UPR activation, ER stress and oxidative stress
Table S8. Risk of bias/quality assessment in cellular studies based on the SIGN checklist for case‐control studies.
Table S9. Risk of bias/quality assessment in animal studies based on the SYRCLE's risk of bias tool for animal studies.