

Abstract
Peroxygenases are heme‐dependent enzymes that use peroxide‐borne oxygen to catalyze a wide range of oxyfunctionalization reactions. Herein, we report the engineering of an unusual cofactor‐independent peroxygenase based on a promiscuous tautomerase that accepts different hydroperoxides (t‐BuOOH and H2O2) to accomplish enantiocomplementary epoxidations of various α,β‐unsaturated aldehydes (citral and substituted cinnamaldehydes), providing access to both enantiomers of the corresponding α,β‐epoxy‐aldehydes. High conversions (up to 98 %), high enantioselectivity (up to 98 % ee), and good product yields (50–80 %) were achieved. The reactions likely proceed via a reactive enzyme‐bound iminium ion intermediate, allowing tweaking of the enzyme's activity and selectivity by protein engineering. Our results underscore the potential of catalytic promiscuity for the engineering of new cofactor‐independent oxidative enzymes.
Keywords: enzyme engineering, epoxidation, oxidative enzymes, peroxide, peroxygenase
No cofactor needed! An uncommon cofactor‐independent peroxygenase was engineered based on a promiscuous tautomerase that uses different hydroperoxides (t‐BuOOH and H2O2) to achieve enantiocomplementary epoxidations of various α,β‐unsaturated aldehydes, giving access to both enantiomers of the corresponding α,β‐epoxy‐aldehydes with high enantiopurity (up to 98 % ee).

Peroxygenases are oxidative enzymes catalyzing the insertion of an oxygen atom into a variety of substrates using peroxide‐borne oxygen instead of dioxygen.1 Earlier studies have mainly focused on the peroxygenase activity of chloroperoxidases that normally catalyze peroxide‐driven halogenation of organic compounds2 and engineered cytochrome P450 monooxygenases that are able to promote peroxide‐mediated hydroxylation reactions.3 However, since the discovery of the unspecific peroxygenase AaeUPO in 2004,4 which uses hydrogen peroxide to catalyze diverse oxyfunctionalization reactions including oxidations, sulfoxidations, epoxidations and hydroxylations,5 there has been an increasing interest to discover and develop novel peroxygenases. Note that peroxygenases are cofactor‐dependent enzymes, typically relying on heme to react with hydrogen peroxide, forming an oxoferryl‐heme as the actual oxygenating species to promote oxyfunctionalization reactions.1, 5a, 6
In this study, we report the engineering of an unusual cofactor‐independent peroxygenase based on the promiscuous peroxygenase activity of the enzyme 4‐oxalocrotonate tautomerase (4‐OT). This engineered non‐natural peroxygenase accepts different hydroperoxides (t‐BuOOH and H2O2) to achieve enantiocomplementary epoxidations of various α,β‐unsaturated aldehydes, providing access to both enantiomers of the resultant α,β‐epoxy‐aldehydes with high enantiomeric excess (up to 98 %). Our results showcase the potential of catalytic promiscuity for the creation of new cofactor‐independent oxidative enzymes.
We have recently demonstrated that 4‐OT can catalyze the Michael addition of nitromethane to cinnamaldehyde via an enzyme‐bound iminium ion intermediate, which is formed between cinnamaldehyde and the catalytic active site Pro‐1 residue.7 In this study, we examined if this catalytic mechanism of 4‐OT could be exploited for other useful synthetic applications, and envisioned that substituting nitromethane with t‐BuOOH could trigger a C−O bond formation between the reactive enzyme‐bound iminium ion species and t‐BuOOH (Supporting Information, Scheme S1). The resulting enamine intermediate may undergo ring closure to construct the final epoxide moiety. To our delight, when wild‐type 4‐OT (0.33 mg mL−1) was incubated at room temperature with 200 mm t‐BuOOH (1, Figure 1) and 1 mm cinnamaldehyde (2 a) in 20 mm sodium phosphate buffer (pH 7.3) containing 5 % (v/v) EtOH, 38 % of substrate 2 a was consumed within 48 h and the corresponding α,β‐epoxy‐aldehyde 3 a was identified by GC‐MS analysis. Chiral HPLC analysis of the alcohol derivative of 3 a revealed the product to be (2R,3S)‐3 a with an e.r. of 62:38 and a d.r. of 92:8 (syn/anti). The S configuration of the C3 carbon of 3 a indicates that t‐BuOOH selectively attacks cinnamaldehyde from the re‐face, similar to what was previously observed for the nitromethane addition reaction.7 These results demonstrated that 4‐OT has low‐level promiscuous peroxygenase activity and may possibly serve as a template to construct a cofactor‐independent peroxygenase.
Figure 1.
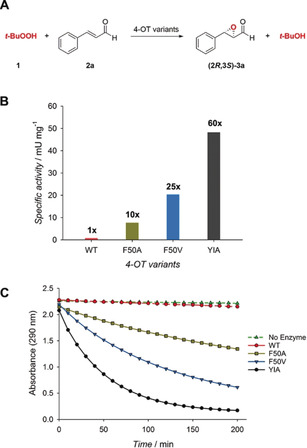
Peroxygenase activity of 4‐OT variants. A) Reaction scheme of 4‐OT‐catalyzed epoxidation of 2 a using t‐BuOOH as oxidant. B) Comparison of the peroxygenase activity of wild‐type 4‐OT (WT) and engineered 4‐OT variants. C) Reaction progress curves using different 4‐OT variants. The initial rate of the 4‐OT(YIA)‐catalyzed reaction corresponds to 16 μm min−1; specific activity is 48 mU per mg of protein.
Motivated by these initial findings, a mutability‐landscape‐guided protein engineering approach8 was applied to enhance the promiscuous peroxygenase activity of wild‐type 4‐OT. For this, a previously constructed collection of 4‐OT genes encoding nearly all possible single‐mutant variants of 4‐OT9 was screened to identify hotspot positions in the protein where mutations give improved peroxygenase activity. Improved variants were identified by monitoring the depletion of substrate 2 a in a spectrophotometric kinetic assay in multiwell plates (see Supporting Information). Interestingly, six mutations at position F50 significantly improved (>5‐fold) the peroxygenase activity, with F50A (10‐fold enhanced activity) and F50V (25‐fold enhanced activity) being the best mutant enzymes (Figure 1). Several mutations at positions Q4, I41, M45, and H49 also resulted in enzyme variants with 2‐ to 5‐fold enhanced activity compared to wild‐type 4‐OT, including mutants Q4Y, I41Y, M45I, and H49T. The most beneficial mutations, F50V and F50A, were subsequently combined with mutations at other hotspot positions by using site‐saturation mutagenesis (Table S2). Activity screening of the seven mutant libraries resulted in the identification of three double mutants (Q4Y/F50V, Q4F/F50A and M45I/F50A) with around 1.6‐ to 3.5‐fold enhanced activity compared to the parental enzymes. Based on these results, the mutant libraries Q4X/M45I/F50A, Q4Y/M45X/F50A, Q4F/M45X/F50A, and Q4Y/M45I/F50X were constructed (Table S2). Screening of these four libraries led to the discovery of the triple mutant Q4Y/M45I/F50A (YIA) that showed a remarkable 60‐fold enhancement in activity for the epoxidation of 2 a compared to the wild‐type enzyme (Figure 1). Notably, this mutant enzyme also has enhanced enantioselectivity, allowing the production of (2R,3S)‐3 a with high enantiopurity (e.r.=98:2). Replacement of Pro‐1 with an alanine in 4‐OT(YIA) led to a 210‐fold decrease in t‐BuOOH‐dependent epoxidation of 2 a (Figure S31), providing support for a reaction mechanism that proceeds via iminium ion formation between the active site Pro‐1 residue and the α,β‐unsaturated aldehyde substrate (Scheme S1).
With the improved 4‐OT mutant YIA in hand, the substrate scope of this enzyme was explored by testing a set of α,β‐unsaturated aldehydes using t‐BuOOH as oxidant. The results showed that 4‐OT(YIA) has a broad substrate scope, accepting ortho‐, meta‐, and para‐substituted cinnamaldehydes (2 a–i, Figure 2), and catalyzes their epoxidation with good conversions (94–97 %) to yield the corresponding products (3 a–i) with excellent enantiopurity (e.r. up to 99:1). The d.r. values (up to 92:8) obtained indicate that the enzyme has good stereocontrol over both carbons of the double bond of 2. The enzyme also accepts the aliphatic α,β‐unsaturated aldehyde citral (2 j) with good enantioselectivity, moderate diastereoselectivity, and excellent regioselectivity (Figure 2). All epoxide products have the syn configuration with 2R,3S being the major enantiomer, indicating that t‐BuOOH exclusively attacked from the re‐face of the enzyme‐bound iminium ion intermediate (Figure 3). Note that while the enzymatic α,β‐epoxy‐aldehyde products 3 have a 2R,3S configuration, the corresponding α,β‐epoxy‐alcohols have a 2S,3S configuration. This deviant configuration of the α,β‐epoxy‐alcohols is due to different prioritization of the substituents at the C2 chiral center relative to their α,β‐epoxy‐aldehyde precursors.
Figure 2.

Substrate scope of the epoxidation reactions catalyzed by 4‐OT(YIA) (Q4Y/M45I/F50A) using t‐BuOOH or H2O2 as the oxidant. Reaction conditions: 2 (1 mm), t‐BuOOH (100 mm) or H2O2 (25 mm), 4‐OT(YIA) (0.1 mg mL−1), NaPi buffer (20 mm), 5 % (v/v) EtOH (MeCN for 2 f, 2 g and 2 h). Conversions were calculated based on the depletion of the absorbance corresponding to substrate 2. Product identifications were performed by GC‐MS analysis. The enzymatic α,β‐epoxy‐aldehyde products 3 were reduced to the corresponding α,β‐epoxy‐alcohols with NaBH4 for chiral HPLC analysis. The e.r., d.r. and absolute stereochemistry of products were determined by chiral HPLC using authentic standards. The diastereomer ratio (d.r.) is defined as the syn/anti ratio. 2 j: E/Z=3:2; n.d.: not determined.
Figure 3.
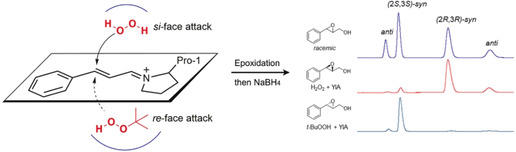
Enantiocomplementary epoxidation of 2 a catalyzed by 4‐OT(YIA) using t‐BuOOH or H2O2 as oxidant. The predominant enzymatic diastereomer has the syn configuration; the minor anti diastereomeric product presumably is formed by rotation of the C2‐C3 bond after addition of the peroxide, but before ring closure.
Given that t‐BuOOH is a relatively large hydroperoxide, we investigated if the use of a small hydroperoxide such as H2O2 would change the preference of attack to the si‐face (Figure 3). Again, compound 2 a was used as a substrate for initial testing with H2O2 as oxidant and mutant 4‐OT(YIA) as catalyst. Notably, the epoxidations with H2O2 showed relatively high non‐enzymatic (background) reaction rates when applying the same conditions as used with t‐BuOOH. To suppress the background reaction, without diminishing the enzymatic activity, the pH of the reaction was lowered from 7.3 to 6.3 (Figure S1). Gratifyingly, the results showed that the use of H2O2 not only inverted the stereochemistry of the reaction, but product 3 a was also obtained with good e.r. (92:8) and d.r. (84:16) values (Figure 2 and Figure 3). Thus, an enantiocomplementary epoxidation of 2 a was achieved by replacing t‐BuOOH by H2O2 as the oxidant. Next, we evaluated the same set of α,β‐unsaturated aldehydes 2 b–2 j for 4‐OT(YIA)‐catalyzed epoxidation reactions using H2O2 (Figure 2). Importantly, the stereo inversions occurred with all aromatic substrates (2 a–2 i), and high conversions and good enantio‐ and diastereoselectivity were observed. Notably, the epoxidation reaction with the aliphatic substrate citral (2 j) also showed an inversion of diastereoselectivity when using H2O2 instead of t‐BuOOH, resulting in the anti‐product as the major isomer; the syn‐product also showed an inverted enantiomer ratio (Figure S30).
Having achieved enantiocomplementary epoxidation reactions, we next investigated semi‐preparative scale chemoenzymatic synthesis of α,β‐epoxy‐alcohols 4 a–j using 4‐OT(YIA) as catalyst. With t‐BuOOH as oxidant, the reactions were performed with higher substrate concentrations (50 mm 2 a, 20 mm 2 b–g and 2 j, 5 mm 2 i), maintaining high conversions and stereoselectivity with good isolated product yields (Table 1). Only a minor decrease in enantio‐ and diastereoselectivity was observed, most likely due to the non‐enzymatic background reaction, since somewhat longer reaction times were needed. The semi‐preparative scale synthesis using H2O2 as oxidant was also attempted using 20 mm 2 a and 22 mm H2O2. However, it was observed that the e.r. of the product (isolated as 4 a) substantially decreased to 73:27 (92:8 when using 1 mm 2 a, Figure 2). We further explored this unexpected result by testing different concentrations of 2 a and H2O2 in analytical‐scale reactions (Table S3). The results revealed that an increasing concentration of 2 a has a negative effect on the e.r. of the final product 4 a, while the concentration of H2O2 has no effect (Table S3). This effect may be caused by the higher concentration of peroxyhydrate (S4) formed between the enzymatic aldehyde product (3 a) and H2O2 when higher substrate (2 a) concentrations were used (Scheme S2). This peroxyhydrate can be considered as a large hydroperoxide that, like t‐BuOOH, attacks the iminium ion on the re‐face during the reaction, thus lowering the e.r. of the product. In reactions with t‐BuOOH, the peroxyhydrate (S5) of the enzymatic aldehyde product (3 a) is not a hydroperoxide, and therefore cannot be accepted by the enzyme as an oxidant. Consequently, the negative effect of high substrate concentrations on the enantiopurity of the final product was not observed in 4‐OT(YIA)‐catalyzed epoxidation reactions with t‐BuOOH (Scheme S2).
Table 1.
Preparative‐scale 4‐OT(YIA)‐catalyzed epoxidations of α,β‐unsaturated aldehydes using t‐BuOOH as oxidant.[a]

|
Entry |
2 |
R1 |
R2 |
t [h] |
Conv. (Yield[%])[b] |
e.r.[c] |
d.r.[d] |
Abs. config.[e] |
|---|---|---|---|---|---|---|---|---|
|
1[f] |
a |
H |
Ph |
24 |
93 (52) |
98:2 |
92:8 |
2S,3S |
|
2 |
b |
H |
o‐Cl‐Ph |
48 |
85 (55) |
93:7 |
68:32 |
2S,3S |
|
3 |
c |
H |
m‐Cl‐Ph |
66 |
85 (68) |
94:6 |
85:15 |
2S,3S |
|
4 |
d |
H |
p‐Cl‐Ph |
48 |
91 (62) |
97:3 |
89:11 |
2S,3S |
|
5 |
e |
H |
p‐F‐Ph |
24 |
95 (50) |
96:4 |
83:17 |
2S,3S |
|
6[f] |
f |
H |
p‐Br‐Ph |
46 |
98 (80) |
98:2 |
92:8 |
2S,3S |
|
7 |
g |
H |
p‐NO2‐Ph |
48 |
92 (64) |
97:3 |
89:11 |
2S,3S |
|
8 |
i |
H |
o‐Me‐Ph |
45 |
90 (61) |
97:3 |
80:20 |
2S,3S |
|
9[g] |
j |
Me |
(CH3)2CCHCH2CH2 |
96 |
79 (50) |
96:4 |
60:40 |
2S,3S |
[a] General reaction conditions: 2 (20 mm), t‐BuOOH (100 mm), 4‐OT(YIA) (0.93 mg mL−1) in 20 mm NaPi (sodium phosphate, pH 7.3) buffer with 10 % (v/v) EtOH (MeCN for 2 f and 2 g), volume=10 mL (5 mL for 2 a, 40 mL for 2 f), enzymatic product 3 reduced with NaBH4 to 4 after completion of the reaction [b] Conversion determined by GC‐MS; yield of the diastereomer mixture. [c] syn diastereomer; determined by chiral HPLC. [d] syn/anti. [e] syn diastereomer; determined by comparison with authentic standards on chiral HPLC. [f] conc. 2 a=50 mm, conc. 2 f=5 mm, see supporting information for details. [g] 2 j: E/Z=3:2; 1.3 mg mL−1 4‐OT(YIA).
In summary, we have exploited the catalytic promiscuity of 4‐OT to engineer an effective peroxygenase (4‐OT‐YIA) that promotes peroxide‐driven asymmetric epoxidations of α,β‐unsaturated aldehydes. Unlike the peroxygenases offered by nature, the engineered non‐natural peroxygenase 4‐OT(YIA) does not require any cofactors, such as heme, for catalysis. While Candida antarctica lipase B (CALB) reportedly performs cofactor‐independent epoxidation reactions, the described enzymatic products were either racemic10 or reported without demonstrating any enantioselectivity.11 Notably, several amino acid and peptide organocatalysts, as well as bovine serum albumin (BSA), have been shown to catalyze asymmetric peroxide‐driven epoxidations of α,β‐unsaturated aldehydes and ketones, but these systems offer a more general mode of catalysis and, with the exception of BSA, often require the use of organic solvents.12 In contrast, peroxygenase 4‐OT(YIA) uses a distinct active‐site process to accomplish highly enantioselective epoxidations of α,β‐unsaturated aldehydes, and by using either t‐BuOOH or H2O2 to drive the reaction, access to both product enantiomers can be achieved. This may suggest that there are two binding pockets for hydroperoxides in the active site of 4‐OT(YIA); one relatively large and hydrophobic pocket at the re‐face of the enzyme‐bound iminium ion that preferably binds t‐BuOOH and one relatively small and probably more hydrophilic pocket at the si‐face of the iminium ion that selectively recruits H2O2 for asymmetric epoxidations (Figure 3). We have initiated structural studies of 4‐OT(YIA) aimed at elucidating the mechanistic origin of the enantiocomplementary epoxidation reactions. Furthermore, given that 4‐OT(YIA) exhibits a moderate specific activity of 48 mU mg−1 for the t‐BuOOH‐driven epoxidation of 2 a, current work in our group is focused on further improving the peroxygenase activity of this promising cofactor‐independent enzyme, making use of a recently developed selective colorimetric “turn‐on” probe for efficient engineering of iminium biocatalysis.13 The non‐natural peroxygenase activity of 4‐OT, together with its previously reported promiscuous C−C bond‐forming Michaelase7, 9, 14 and aldolase15 activities, emphasize the chemical versatility of the 4‐OT protein scaffold.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support from the Netherlands Organization of Scientific Research (VICI grant 724.016.002 and ECHO grant 713.015.003).
G. Xu, M. Crotti, T. Saravanan, K. M. Kataja, G. J. Poelarends, Angew. Chem. Int. Ed. 2020, 59, 10374.
References
- 1. Wang Y., Lan D., Durrani R., Hollmann F., Curr. Opin. Chem. Biol. 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Morris D. R., Hager L. P., J. Biol. Chem. 1966, 241, 1763–1768; [PubMed] [Google Scholar]
- 2b. Zaks A., Dodds D. R., J. Am. Chem. Soc. 1995, 117, 10419–10424; [Google Scholar]
- 2c. Littlechild J., Curr. Opin. Chem. Biol. 1999, 3, 28–34. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Joo H., Lin Z., Arnold F. H., Nature 1999, 399, 670–673; [DOI] [PubMed] [Google Scholar]
- 3b. Cirino P. C., Arnold F. H., Angew. Chem. Int. Ed. 2003, 42, 3299–3301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3421–3423; [Google Scholar]
- 3c. Chen J., Kong F., Ma N., Zhao P., Liu C., Wang X., Cong Z., ACS Catal. 2019, 9, 7350–7355. [Google Scholar]
- 4. Ullrich R., Nueske J., Scheibner K., Spantzel J., Hofrichter M., Appl. Environ. Microbiol. 2004, 70, 4575–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125; [DOI] [PubMed] [Google Scholar]
- 5b. Bassanini I., Ferrandi E. E., Vanoni M., Ottolina G., Riva S., Crotti M., Brenna E., Monti D., Eur. J. Org. Chem. 2017, 7186–7189. [Google Scholar]
- 6.
- 6a. Prier C. K., Arnold F. H., J. Am. Chem. Soc. 2015, 137, 13992–14006; [DOI] [PubMed] [Google Scholar]
- 6b. Arnold F. H., Angew. Chem. Int. Ed. 2018, 57, 4143–4148; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4212–4218. [Google Scholar]
- 7. Guo C., Saifuddin M., Saravanan T., Poelarends G. J., ACS Catal. 2019, 9, 4369–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van der Meer J. Y., Biewenga L., Poelarends G. J., ChemBioChem 2016, 17, 1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Der Meer J. Y., Poddar H., Baas B. J., Miao Y., Rahimi M., Kunzendorf A., Van Merkerk R., Tepper P. G., Geertsema E. M., Thunnissen A. M. W. H., et al., Nat. Commun. 2016, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Zhou P., Wang X., Yang B., Hollmann F., Wang Y., RSC Adv. 2017, 7, 12518–12523; [Google Scholar]
- 10b. Ranganathan S., Zeitlhofer S., Sieber V., Green Chem. 2017, 19, 2576–2586. [Google Scholar]
- 11. Svedendahl M., Carlqvist P., Branneby C., Allnér O., Frise A., Hult K., Berglund P., Brinck T., ChemBioChem 2008, 9, 2443–2451. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Juliá S., Masana J., Vega J. C., Angew. Chem. Int. Ed. Engl. 1980, 19, 929–931; [Google Scholar]; Angew. Chem. 1980, 92, 968–969; [Google Scholar]
- 12b. Colonna S., Manfredi A., Tetrahedron Lett. 1986, 27, 387–390; [Google Scholar]
- 12c. Colonna S., Manfredi A., Annunziata R., Spadoni M., Tetrahedron 1987, 43, 2157–2164; [Google Scholar]
- 12d. Baars S., Drauz K.-H., Krimmer H.-P., Roberts S. M., Sander J., Skidmore J., Zanardi G., Org. Process Res. Dev. 2003, 7, 509–513; [Google Scholar]
- 12e. Sundén H., Ibrahem I., Córdova A., Tetrahedron Lett. 2006, 47, 99–103. [Google Scholar]
- 13. Biewenga L., Crotti M., Saifuddin M., Poelarends G. J., ACS Omega 2020, 5, 2397–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Zandvoort E., Geertsema E. M., Baas B. J., Quax W. J., Poelarends G. J., Angew. Chem. Int. Ed. 2012, 51, 1240–1243; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1266–1269; [Google Scholar]
- 14b. Miao Y., Geertsema E. M., Tepper P. G., Zandvoort E., Poelarends G. J., ChemBioChem 2013, 14, 191–194; [DOI] [PubMed] [Google Scholar]
- 14c. Geertsema E. M., Miao Y., Tepper P. G., Dehaan P., Zandvoort E., Poelarends G. J., Chem. Eur. J. 2013, 19, 14407–14410; [DOI] [PubMed] [Google Scholar]
- 14d.Y. Miao, P. G. Tepper, E. M. Geertsema, G. J. Poelarends, Eur. J. Org. Chem 2016, 5350–5354; [DOI] [PMC free article] [PubMed]
- 14e. Biewenga L., Saravanan T., Kunzendorf A., Van Der Meer J., Pijning T., Tepper P. G., Van Merkerk R., Charnock S. J., Thunnissen A. W. H., Poelarends G. J., ACS Catal. 2019, 9, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Zandvoort E., Baas B. J., Quax W. J., Poelarends G. J., ChemBioChem 2011, 12, 602–609; [DOI] [PubMed] [Google Scholar]
- 15b. Rahimi M., van der Meer J. Y., Geertsema E. M., Poddar H., Baas B. J., Poelarends G. J., ChemBioChem 2016, 17, 1225–1228; [DOI] [PubMed] [Google Scholar]
- 15c. Rahimi M., Geertsema E. M., Miao Y., van der Meer J.-Y., van den Bosch T., de Haan P., Zandvoort E., Poelarends G. J., Org. Biomol. Chem. 2017, 15, 2809–2816. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary