

ABSTRACT
Epstein-Barr Virus (EBV) is etiologically associated with multiple human malignancies including Burkitt lymphoma and Hodgkin disease as well as nasopharyngeal and gastric carcinoma. Entry of EBV into target cells is essential for virus to cause disease and is mediated by multiple viral envelope glycoproteins and cell surface associated receptors. The target cells of EBV include B cells and epithelial cells. The nature and mechanism of EBV entry into these cell types are different, requiring different glycoprotein complexes to bind to specific receptors on the target cells. Compared to the B cell entry mechanism, the overall mechanism of EBV entry into epithelial cells is less well known. Numerous receptors have been implicated in this process and may also be involved in additional processes of EBV entry, transport, and replication. This review summarizes EBV glycoproteins, host receptors, signal molecules and transport machinery that are being used in the epithelial cell entry process and also provides a broad view for related herpesvirus entry mechanisms.
Keywords: Epstein–Barr virus, gamma-herpesviruses, epithelial cells
Overall the mechanism of EBV entry into B cells and epithelial cells is becoming clearer; numerous receptors have been implicated in this process and may also be involved in additional processes of EBV entry, transport, and replication.
Epstein–Barr virus (EBV) belongs to the herpesviridae family and specifically to gamma-herpesvirus subfamily. EBV infects >90% of the total population and there is no vaccine available (Longnecker, Kieff and Cohen 2013). EBV infection in childhood is generally asymptomatic; however, infection during adolescence or as an adult may result in mononucleosis, also called the kissing disease. EBV was the first identified human oncogenic herpesvirus and the infection is associated with Burkitt lymphoma and Hodgkin disease (B lymphocyte origin) as well as nasopharyngeal and gastric carcinoma (epithelial cell origin), reflecting the cell tropism of EBV (Burkitt 1961; Burkitt and O'Conor 1961; Epstein, Achong and Barr 1964; Gunven et al. 1970). The virus was found in nasopharyngeal carcinoma (NPC) in 1970 (Gunven et al. 1970; zur Hausen et al. 1970; Nonoyama and Pagano 1973; Wolf, zur Hausen and Becker 1973). The risk of NPC is higher for those with elevated anti-EBV DNase antibodies or anti-EBV VCA (viral capsid antigen) IgA and even higher when both antibodies are elevated indicating the importance of EBV infection in disease development (Henle et al. 1977). EBV is also present in gastric carcinoma. EBV is also commonly detected in gastric carcinoma with ∼9% being EBV positive (Burke et al. 1990; Murphy et al. 2009). EBV anti-VCA and anti-EBNA antibody titers are higher in persons with dysplasia on gastric biopsy, suggesting that EBV reactivation could be related to an early phase of gastric carcinoma (De Paschale and Clerici 2012). EBV may also occasionally infect other cell types such as T/natural killer cells (Isobe et al. 2004; Coleman et al. 2015).
The EBV life cycle starts with salivary transmission of shed virus from an infected person. The virus then replicates in or is transcytozed across epithelial cells and the virus then spreads to naive B cells (Yao, Rickinson and Epstein 1985; Tugizov, Herrera and Palefsky 2013). Lifelong latency is established in memory B cells (Babcock et al. 1998). In terminally differentiated plasma cells, the lytic virus life cycle is reactivated (Laichalk and Thorley-Lawson 2005). Epithelial cell infection in vitro usually results in active replication and lysis of the cells. EBV DNA can be routinely detected in epithelial cells of the oropharynx from acute infectious mononucleosis patients, suggesting that in vivo, EBV replicates lytically in epithelial cells (Sixbey et al. 1983). Normal nasopharyngeal epithelial cells are not readily permissive for latent EBV infection. Instead, infection typically results in growth arrest (Tsang et al. 2012). However, overexpression of cyclin D1 and/or Bmi-1 as well as the inactivation of p16 can overcome the growth arrest to support stable and latent EBV infection in nasopharyngeal epithelial cells (Tsang et al. 2010, 2012, 2014; Yip et al. 2013)
EBV INFECTION OF TARGET CELLS
EBV infection of target epithelial cells is a complex multistep process (Fig. 1). In the first step (Step I), EBV binds to target cells using variable host cell surface receptors and multiple viral envelope glycoproteins. In some cases, binding of EBV virions can induce signaling pathway activation (Step II). After binding to the cell surface receptors, the viral and host membranes merge (either by direct membrane fusion or fusion with the endosomal membrane) (Step III). The viral capsid is then transported in the cytosol to the nuclear periphery (Step IV). Once at a nuclear pore, the viral genome is released into the nucleus through a nuclear pore (Step V).
Figure 1.
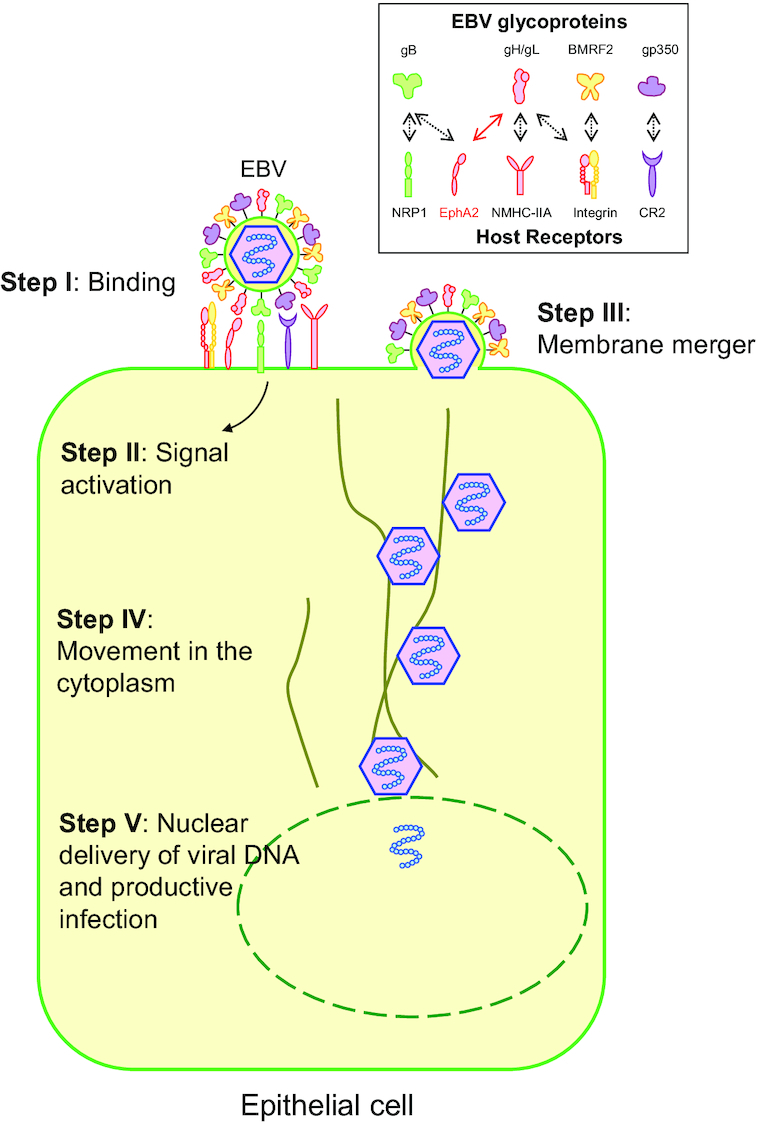
Model illustrating the steps of EBV infection of epithelial cells. In the first step (Step I), EBV binds to target cells using variable host cell surface receptors and multiple viral envelope glycoproteins. In some cases, binding of EBV virions can induce signaling pathway activation (Step II). After binding to the cell surface receptors, (either by direct membrane fusion or fusion with the endosomal membrane) (Step III), the viral capsid is then transported in the cytosol to the nuclear periphery (Step IV). Once at a nuclear pore, the viral genome is released into the nucleus through a nuclear pore (Step V). Integrins (αvβ5, αvβ6 and αvβ8), NRP1 and NMHC-IIA interact with corresponding glycoproteins indicated as black-dashed arrows. EphA2 binds to both gH/gL (red solid arrow) and gB (black-dashed arrow). EphA2 is the most important entry receptor for EBV epithelial cell infection and is therefore indicated in red.
EBV ENTRY STEP I: EBV BINDING TO TARGET CELLS INVOLVES MULTIPLE VIRAL ENVELOP GLYCOPROTEINS AND HOST CELL SURFACE RECEPTORS
Initiation of EBV infection of two major target cells, B lymphocytes and epithelial cells, is substantially different, involving different viral envelope glycoproteins and cell receptors for entry into each cell type. Entry into B cells occurs via endocytosis followed by fusion of the viral membrane with the membrane of the endocytic vesicle (Miller and Hutt-Fletcher 1992). Entry into epithelial cells occurs through direct fusion of the viral membrane with the host cell plasma membrane (Miller and Hutt-Fletcher 1992), although some studies suggest that entry of epithelial cells by EBV is via lipid raft-dependent endocytosis and macropinocytosis (Wang et al. 2015). It is likely that both pathways of entry, endocytosis or direct fusion, are used to infect cells, but what pathway is used is dependent on a variety of factors including expression of relevant EBV receptors.
The entry process of EBV into B cells and the receptors involved in this process are well studied compared to the entry process of epithelial cells; the B cell receptor was identified very early, allowing for extensive functional and structural studies of the B cell entry complex (Spriggs et al. 1996; Mullen et al. 2002; Connolly et al. 2011; Sathiyamoorthy et al. 2014; Sathiyamoorthy et al. 2016). Thus, this review will focus on EBV epithelial cell infection to provide a comprehensive review of what is currently understood regarding the process of EBV epithelial cell entry.
EBV GLYCOPROTEINS IMPORTANT FOR VIRAL ENTRY
The core fusion machinery for EBV includes glycoproteins gB and the gH/gL complex, which are required for both B cell fusion and epithelial cell fusion (Connolly et al. 2011). gB is a class III viral fusogen that activates membrane fusion of virus and host cell membranes. The crystal structure of EBV gB without its transmembrane or cytoplasmic domains was identified in 2009 in a presumed post-fusion form. It is a 16 nm spike-like trimer composed of five domains (Backovic, Longnecker and Jardetzky 2009).
While gB activates fusion, gH/gL regulates fusion; upon binding to a host cell receptor, it is thought to trigger the conformational change of gB from pre-fusion to post-fusion form, resulting in membrane fusion (Connolly et al. 2011; Gallagher et al. 2014). The crystal structure of EBV gH/gL is an elongated rod-like shape ∼100 Å in length and 30–60 Å in width. gH/gL is divided into four major domains, with domain I composed of gL and the N terminus of gH (1–66). The rest of gH (66–672) folds into three sequential globular domains (Matsuura et al. 2010). There is a large groove between domain I (D-I) and domain II (D-II) of gH/gL, which may be important for epithelial cell receptor binding (Chen, Jardetzky and Longnecker 2013). Previous mutagenesis studies, as well as studies performed by the Hutt-Fletcher laboratory, identified gH/gL mutations that decreased epithelial cell fusion but did not alter B cell fusion, indicating that gH/gL is also an important determinant for EBV cell tropism. These mutants are R152A (D-II), disulfide bond C278/C335 (D-II), as well as mutations located in D-V (Wu, Borza and Hutt-Fletcher 2005; Chen, Jardetzky and Longnecker 2013; Mohl et al. 2014). These mutants may provide new tools to study differences in the mechanism of epithelial cell and B cell fusion.
There are several anti-gH/gL monoclonal antibodies that target different regions of gH/gL that have been tested in fusion including CL40, CL59 and E1D1 (Fig. 2) (Molesworth et al. 2000). CL40 binds to a site occupied by the gp42 receptor-binding domain (D-II). CL59 binds to the C-terminal domain IV of gH. E1D1, however, engages a distinct surface of gH/gL compared to CL59 and CL40: the tip of D-I (Sathiyamoorthy et al. 2016; Sathiyamoorthy et al. 2017). Previously, it was reported that all three mAbs block epithelial but not B cell infection (Molesworth et al. 2000; Chesnokova and Hutt-Fletcher 2011). In a more recent study, CL40 and CL59 were shown to block membrane fusion with both B cells and epithelial cells (Sathiyamoorthy et al. 2017). Interestingly, E1D1 selectively inhibits epithelial cell fusion but not B cell fusion (Sathiyamoorthy et al. 2016). One explanation for this phenomenon is that the tip of D-I binds to the epithelial cell receptor, but not to gp42.
Figure 2.
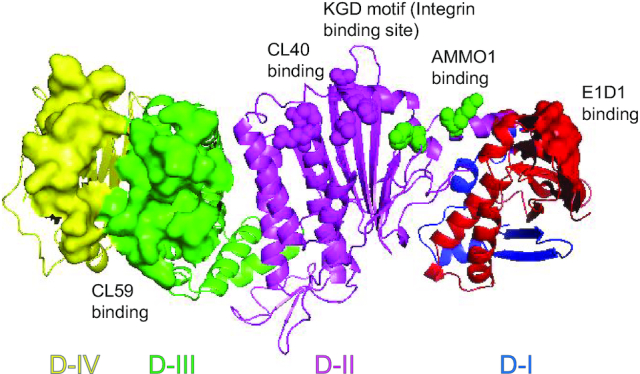
Different anti-gH/gL antibodies target different regions of gH/gL and inhibit EBV epithelial cell infection. The structure of gH/gL is shown as cartoon (Matsuura et al. 2010; Connolly et al. 2011). gH consists of four domains: D-I (blue), D-II (magenta), D-III (green) and D-IV (yellow). gL is colored red and interacts with gH in D-I. The E1D1 antibody binding region on gH/gL is shown as red surface representation in D-I gL (gL 27–33, gL 72–79 and gL 127–131; D-I is also the potential EphA2 binding region). The most important AMMO1 antibody-binding residues on EBV gH/gL are represented by green spheres in D-I (gH K73 and gH Y76). The CL40 binding amino acids on gH/gL are represented by magenta spheres (gH R184, gH H239, gH V243, gH D284 and gH E286). The CL59 antibody binding regions on gH/gL are shown as green and yellow surface representations in D-III and D-IV (gH406–415, gH456–468, gH494–503, gH623–626 and gH645–656).
A human monoclonal anti-gH/gL antibody, AMMO1, was recently isolated from EBV-infected humans. AMMO1 binds opposite to the large groove formed by both gH and gL at the D-I/D-II interface. It also binds on the same side as the gp42 binding region, partially overlapping the CL40 binding site. AMMO1 potently neutralizes infection of both B cells and epithelial cells (Snijder et al. 2018). These data together indicate that there are multiple functional regions on gH/gL that may bind to different host receptors and participate in EBV epithelial cell infection.
gB and gH/gL are sufficient for epithelial cell fusion. An additional glycoprotein, gp42, is required for B cell fusion and binds to HLA class II (Li, Turk and Hutt-Fletcher 1995; Wang and Hutt-Fletcher 1998; Shaw et al. 2010). Virus generated in B cells expresses less gp42 since gp42 is sequestered and degraded in HLA class II-positive B cells. Virus generated in epithelial cells expresses more gp42, which inhibits epithelial cell entry and fusion by binding to gH/gL. Thus, gp42 is the tropism switch for EBV infection (Borza and Hutt-Fletcher 2002; Kirschner et al. 2006).
Interestingly, soluble gp42 can inhibit viral fusion with epithelial cells. This suggests that the gp42 binding site on gH/gL is an important site for epithelial cell entry (Borza et al. 2004; Kirschner et al. 2006). Further mutational studies of the integrin-binding KGD motif on gH/gL have shown the mutation of the KGD motif to AAA decreases fusion with both epithelial cells and B cells and reduces gH/gL binding to both epithelial cells and gp42 (Chen et al. 2012). These results indicated that the KGD motif is a bifunctional region for both epithelial cell and B cell fusion.
HOST CELL PROTEINS IMPORTANT FOR VIRUS ATTACHMENT (A) AND VIRUS ENTRY (B) IN EPITHELIAL CELLS
Unlike B cell infection, which is initiated by attachment of gp350, the most abundant EBV glycoprotein in virions, to the complement receptor type 2 (CR2) or CD35(Fingeroth et al. 1984; Nemerow et al. 1987; Ogembo et al. 2013), EBV uses different glycoproteins for attachment to epithelial cells depending on cell types and expression of CR2. EBV can use gp350 for attachment to CR2-positive epithelial cells (Nemerow et al. 1987) (Fig. 1). In CR2-negative epithelial cells, EBV may use multispanning transmembrane envelope protein BMRF-2 (Tugizov, Berline and Palefsky 2003) to attach to integrin αvβ1 or it may use gH/gL to attach to integrin αvβ6 or αvβ8 (Fig. 1). It has also been reported that BMRF-2 can form a complex with the type II membrane protein BDLF2, which participates in rearrangement of cellular actin to increase intercellular contacts and thereby promote virus cell-to-cell spreading (Loesing et al. 2009).
Several host receptors important for EBV epithelial cell infection have been identified, including multiple integrins, neuropilin-1, non-muscle myosin heavy chain IIA (NMHC-IIA) and the recently identified primary entry receptor: ephrin receptor A2 (EphA2).
INTEGRINS AS EBV ATTACHMENT/TETHERING RECEPTORS
The receptor for EBV B cell infection was identified in 1996 by a gp42 ligand-binding screen. However, the first EBV epithelial cell receptor was not identified until 2009 (by the Hutt-Fletcher lab) due to less efficient EBV infection of epithelial cells compared to B cells (Spriggs et al. 1996; Chesnokova, Nishimura and Hutt-Fletcher 2009; Chesnokova and Hutt-Fletcher 2011). Integrins are widely used by many viruses, including herpesviruses, for virus attachment, virus endocytosis and cellular activation, which facilitates virus entry into host cells (Stewart and Nemerow 2007). It was proposed that EBV may also use integrins as epithelial cell entry receptors because an integrin-binding KGD motif was identified in gH/gL (Chesnokova, Nishimura and Hutt-Fletcher 2009; Chen et al. 2012). EBV gH/gL can bind to integrin αvβ5, αvβ6 and αvβ8 (Fig. 1). Soluble integrin (a peptide including residues 184–196 of gH that contains the KGD motif) and the natural ligands of integrins such as fibronectin and vitronectin can all partially reduce EBV binding and infection (Chesnokova, Nishimura and Hutt-Fletcher 2009; Chesnokova and Hutt-Fletcher 2011). Together, these results indicated that integrins play a role in EBV infection.
However, in more recent studies, integrin αv knockout HEK293 cells were generated using the CRISPR-Cas9 system and no difference between WT HEK293 and integrin αv knockout HEK293 cells was found for viral infection and fusion activity indicating that αvβ5, αvβ6 and αvβ8 integrins are not primary EBV entry receptor(s) in HEK293 cells but likely act as a tethering receptor (Chen et al. 2018). There have been similar studies in the context of KSHV infection that identified entry mechanisms independent of integrins α3β1, αvβ3 and αvβ5 (TerBush et al. 2018) even though integrins were also thought to be the primary entry receptor for KSHV (Garrigues et al. 2008).
NEUROPILIN-1(NRP1) AS AN EBV ENTRY FACTOR FOR EPITHELIAL CELLS
The neuropilins (NRPs) are multifunctional proteins that play important roles in development, immunity and cancer. NRPs were initially found to be expressed in neuronal cells and play a role in axonal growth and guidance (Takagi et al. 1987; Schwarz et al. 2008).
NRP1 has diverse functions in different cell types and has a particular role in signaling by enhancing the activity of receptor tyrosine kinases (RTKs) as a co-receptor for class III semaphorins and multiple growth factors (Zachary 2011). NRP1 is also involved in human T-cell lymphotropic virus type 1 entry (Ghez et al. 2006; Lambert et al. 2009). Many growth factors and other signaling molecules bind to NRPs through a carboxy (C)-terminal basic sequence motif (C-end Rule or CendR motif) (Pang et al. 2014). NRP1 can bind to peptides containing a CendR motif, which has a consensus sequence R/K/XXR/K for internalization (Pang et al. 2014). EBV gB is highly conserved within the herpesvirus family and a number of gB homologs contain the cleavage motif R-X-K/R-R recognized by the cellular protease furin (Sorem and Longnecker 2009). This cleavage site could also be a potential cryptic C-end Rule (CendR) motif.
It is hypothesized that NRP1 can serve as an entry factor for EBV gB infection (Wang et al. 2015). The interaction of gB with NRP1 was examined by an in vitro binding assay. It was found that NPR1 directly interacts with EBV gB23–431 (Fig. 1). A CendR motif-deletion mutant (gB23–427) had decreased interaction with NRP1. Further analysis showed that deletion of both gB23–88 and gB428–431 abolished the interaction between NRP1 and gB, indicating that both regions are important for binding. Knockdown of NRP1 or treatment with soluble NRP1 decreased EBV infection to ∼50% of control infection. Overexpression of NRP1 significantly increased the efficiency of EBV infection. Interestingly, the role of NRP2 was opposite to that of NRP1 reducing infection.
Previously, our lab reported that an EBV gB deletion mutant lacking the furin cleavage motif was expressed well in cell culture but was not cleaved. The fusion activity was reduced by 52% in epithelial cells and 28% in B cells compared to WT gB (Sorem, Jardetzky and Longnecker 2009). This data supports NRP1 as an epithelial cell entry factor for gB since the furin cleavage site is also the NRP1 binding site (Sorem, Jardetzky and Longnecker 2009). Oral squamous cell carcinoma (OSCC) is the most common subset (90%) of oral cancer with a global incidence of 275 000 cases annually. It results from the outgrowth of the mucosal epithelium. It has been shown that that EBV DNA, mRNAs and EBV proteins were expressed in the majority of OSCC cells (Shamaa et al. 2008; Sinevici and O'Sullivan 2016). Interestingly, NRP1 is also overexpressed in OSCC (Chu et al. 2014). This may explain the reason that EBV infection is associated with an increased risk of OSCC (She et al. 2017).
Since NPR1 is the gB receptor, the level of gB on EBV virion is related to the infection efficiency.
EBV virions that express high levels of gB (gBhigh) infect target cells more efficiently than virions that express lower levels of gB (gBlow) (Neuhierl et al. 2002). Interestingly, gBhigh can also infect cells that are normally resistant to EBV infection (Neuhierl et al. 2002). Previous studies identified EBV gB truncations or point mutations in the carboxy-terminal tail that have higher cell-surface expression of gB, allowing cell-cell fusion independent of other viral proteins (McShane and Longnecker 2004). It was proposed that this gH/gL-independent fusion was due to the increased gB cell surface expression. However, more recent studies using a comprehensive library of mutants with truncations of the C-terminal cytoplasmic tail domain (CTD) of EBV gB found that the higher level of gB cell surface expression did not correlate with higher fusion activity (Garcia, Chen and Longnecker 2013). More recent studies have shown that the gB CTD may also participate in fusion by maintaining gB in an inactive pre-fusion form prior to activation by receptor binding (Chen et al. 2014). One possibility is that mutation of the EBV gB CTD may release this restriction and cause gB to more readily change to a post-fusion conformation. In regard to identification of NRP1 as an EBV entry factor that specifically interacts with gB (Wang et al. 2015), there may be other explanations for its role in EBV infection that may include that the EBV gB CTD mutant may adopt a conformation that interacts better with NRP1 to facilitate fusion.
NON-MUSCLE MYOSIN HEAVY CHAIN IIA (NMHC-IIA) MEDIATES EPSTEIN–BARR VIRUS INFECTION OF NASOPHARYNGEAL EPITHELIAL CELLS
The identification of NMHC-IIA as EBV receptor comes from the differential infection efficiency on different NPEC culture. The major obstacle to identify factors that are important for EBV infection of NPECs is the inefficiency of EBV infection for primary or immortalized NPECs (Tsang et al. 2014). During the optimization of growth of the immortalized NPECs, the Zeng lab found 10-fold higher density of cells can cause the formation of ‘sphere-like cells’ (SLCs) compared to monolayer growth. EBV infection of these SLCs is increased ∼10-fold when compared to the same cells when grown as monolayer cells. Interestingly, in EBV-associated nasopharyngeal carcinoma, there are cancer stem-like cells (CSCs) that have the ability to self-renew, differentiate and sustain propagation. They are also chemo-resistant and can form spheres similar to these SLCs in anchorage-independent environments (Lun, Cheung and Lo 2014). As previously mentioned, there are four antibodies targeting different domains of gH/gL that can inhibit epithelial cell fusion, indicating that gH/gL may interact with more than one host factor for efficient infection. To identify host factors that may play an important role in this increased infection, a myc-tagged gH/gL pull-down assay was performed using EBV-infected SLC lysates followed by liquid chromatography-tandem MS (LC-MS/MS) proteomic analysis. The 250 kDa NMHC-IIA was identified to be the gH/gL binding protein (Fig. 1). This result was also confirmed by co-immunoprecipitation (Wang et al. 2015).
NMHC-IIA is an actin-binding protein that has actin cross-linking and contractile properties and is regulated by the phosphorylation of its light and heavy chains (Vicente-Manzanares et al. 2009). It has been shown that NMHC-IIA is important for many virus infections including porcine reproductive and respiratory syndrome virus, herpes simplex virus-1 and thrombocytopenia syndrome virus (Arii et al. 2010; Sun et al. 2014; Gao et al. 2016).
NMHC-IIA is mainly located in the cell cytoplasm in normal cultured cells. Using immunofluorescence staining and membrane fractionation methods, it was found that there is aggregated NMHC-IIA in apical surfaces of SLCs, which is associated with gH/gL (Wang et al. 2015). Knockdown of NMHC-IIA and NMHC-IIA antibody blocking resulted in both reduced EBV binding and SLC infection, but with no change for adenovirus infection (Wang et al. 2015). Overexpression of NMHC-IIA in the cytoplasm did not increase EBV infection. Infection was only increased when NMHC-IIA was redistributed to the cell membrane (Wang et al. 2015). Thus, the increased EBV infection efficiency is due to NMHC-IIA and gH/gL localization on the cell surface. However, the mechanism of how NMHC-IIA is redistributed is not known. Interestingly, herpes simplex virus 1 (HSV-1) was also reported to utilize NMHC-IIA as an entry co-receptor associating with gB, indicating that HSV-1 may use a similar mechanism for entry and infection (Arii et al.2010).
EPHRIN RECEPTOR A2 (EPHA2) AS THE EBV EPITHELIAL CELL ENTRY RECEPTOR
Previous results by the Hutt-Fletcher laboratory demonstrated that integrins αvβ5, αvβ6 and αvβ8 are host binding factors for EBV gH/gL (Chesnokova and Hutt-Fletcher 2011). However, after complete knockout of integrin αv in HEK293 cells using the CRISPR–Cas9 system, there was no difference between WT and integrin αv knockout cells in fusion or infection. These results indicated that integrins are not major host factors for EBV infection (Chen et al. 2018). To identify the major receptor for EBV epithelial cell fusion, a novel and more rapid approach using readily available RNA-seq databases was used to identify potential epithelial cell receptors (Chen et al. 2018). Potential receptors were determined by the ratio of membrane protein RNAs that were only expressed in permissive epithelial cells to that of B cells that are non-permissive for epithelial fusion from high to low. EphA2 was ranked as the number one candidate through this analysis. EphA2 belongs to the largest receptor tyrosine kinase (RTK) family, with 14 known human members that play roles in boundary formation, cell migration, axon guidance, synapse formation, angiogenesis, proliferation and cell differentiation (Park, Son and Zhou 2013; Kania and Klein 2016). EphA2 is also a receptor for Kaposi's sarcoma-associated herpesvirus (KSHV), another human gammaherpesvirus (Hahn et al. 2012). It has also been shown that other pathogens, including the hepatitis C virus, Cryptococcus neoformans, and the fungal pathogen Candida albicans use EphA2 as entry factors (Lupberger et al. 2011; Aaron et al. 2018; Swidergall et al. 2018). Overexpression of EphA2 but not EphA4 can promote the fusion and infection of EBV in HEK293 cells. Knockout of EphA2 can reduce EBV fusion and infection by up to 90% and 80%, respectively, and rescue of infection or fusion in EphA2 knockout cells is readily observed with overexpression of EphA2 but not EphA4 (Chen et al. 2018). Using label-free surface plasmon resonance (SPR) binding studies, we also confirmed that EphA2 but not EphA4 specifically bound to EBV gH/gL through the EphA2 extracellular domain (Chen et al. 2018) (Fig. 1).
Previous KSHV studies indicated that EphA2 regulates clathrin-mediated KSHV endocytosis through its kinase domain (Dutta et al. 2013). Results with EBV demonstrated that EphA2 kinase activity is not required for fusion activity, likely due to the different routes of entry between EBV and KSHV (Chen et al. 2018).
Interestingly, the Zeng laboratory also identified EphA2 as an EBV epithelial cell receptor using a different approach. It was found that EGF pre-treatment greatly increases EBV infection (Wang et al. 2015). To identify the genes that were upregulated after EGF treatment, an integrated approach using microarray and RNA interference screen analyses was used to identify plasma membrane proteins that were highly induced after EGF treatment. The membrane proteins AREG, NT5E, EPHA2, F3, EGFL5 and DCBLD2 were highly induced in two EGF-treated NPEC lines. However, only knockdown of EphA2 resulted in decreased EBV infection. This result is similar to what was found using CRISPR–Cas9 and overexpression of EphA2 in EBV epithelial cell infection. It was also found that soluble EphA2 protein, EphA2 antibodies, ephrinA1 (a soluble EphA2 ligand) and the EphA2 inhibitor 2,5-dimethylpyrrolyl benzoic acid derivative all efficiently inhibited EBV epithelial cell infection (Zhang et al. 2018).
The binding region of EphA2 on EBV gH/gL is not known. However, the binding region for gH/gL on EphA2 has been identified. EphA2 is a membrane protein with four different ectodomain regions including a ligand binding domain (LBD), a cysteine rich region (CYS) and two fibronectin regions (FBN). Interestingly, EphA2 binds to both EBV gB and EBV gH/gL at nM levels. The binding region for gH/gL is the LBD and the binding regions for EBV gB are the LBD and FBN (Zhang et al. 2018). Thus, the results of both laboratories are consistent and complementary.
EBV ENTRY STEP II: SIGNALING PATHWAY ACTIVATED BY BINDING OF THE EBV VIRUS PARTICLE TO EPITHELIAL CELLS
Virus interaction with cellular receptors often activates intracellular signaling pathways that consequently facilitate virus uptake. Multiple members of the herpesvirus family have been shown to activate such pathways. Previous studies of HSV showed that early virus–cell interactions at the plasma membrane may induce rapid phosphorylation of focal adhesion kinase (FAK) in several human target cells important for HSV entry post-binding (Cheshenko et al. 2005). KSHV interacts with cell surface integrin α3β1 of human endothelial cells and fibroblasts and activates the FAK that is immediately downstream in the outside-in signaling pathway by integrins, leading to the activation of several downstream signaling molecules (Krishnan et al. 2006). EBV has two glycoproteins including BMRF2 (Xiao et al. 2007) and gH (Chesnokova, Nishimura and Hutt-Fletcher 2009) that have integrin binding motifs and can bind to integrins. Upon ligand binding to integrins, downstream signaling pathways are activated resulting in the recruitment of adaptor proteins that regulate many cellular activities (Giancotti and Ruoslahti 1999). There is direct interaction between integrin cytoplasmic tails and specific actin-binding proteins (Morse, Brahme and Calderwood 2014). Thus, integrins may regulate actin dynamics. Inhibition of Src, ROCK, Rho and p38/MAPK, which are all involved in integrin signal transduction in SVKCR2 epithelial cells, decreases transcription from incoming virus genomes, indicating the role of integrin signaling in EBV infection (Valencia and Hutt-Fletcher 2012).
EphA2 is the entry receptor for both KSHV and EBV. Both KSHV and the natural EphA2 ligand ephrinA1 recombinantly expressed and fused to Fc (ephrinA1-Fc) increased EphA2 phosphorylation. Overexpression of full-length EphA2, but not EphA2ΔICMycHis (EphA2 without the intracellular kinase domain), enhanced KSHV infection by >70% indicating the importance of the EphA2 kinase domain (Hahn et al. 2012). The kinase domain of EphA2 is responsible for its downstream signaling. KSHV infection activates EphA2 and, in turn, EphA2 associates with phosphorylated c-Cbl, myosin IIA, FAK, Src and PI3-K as well as clathrin and its adaptor AP2 and effector Epsin-15 proteins (Dutta et al. 2013). EphA2 knockdown significantly reduced these signal inductions, virus internalization and gene expression (Dutta et al. 2013). For EBV, the kinase activity is not important for fusion, since the EphA2 kinase-dead mutants that were mutated in the EphA2 kinase domain have the same fusion activity as WT EphA2 (Chen et al. 2018). It has also been shown that upon overexpression of WT EphA2 (EphA2WT) or intracellular domain truncation of EphA2 (EphA2∆IC) in HNE1 cell knockout of EphA2, there is no difference in EBV infection, again indicating that the EphA2 intracellular domain is dispensable for EBV internalization (Zhang et al. 2018). The different requirement of EphA2 kinase activity for EBV and KSHV is probably due to the different routes of entry for these two viruses.
Moreover, cell-free EBV binding to NRP1 activates NRP1-dependent epidermal growth factor receptor (EGFR) signaling pathways as well as its downstream signaling components AKT and ERK (Wang et al. 2015). As a co-receptor of RTKs, NRP1 enhances the affinity of multiple growth factors to RTKs, such as EGF, HGF, VEGF, PIGF and PDGF-BB, and thus augments RTK signaling (Zachary 2011). Knockdown of NRP1 partially suppressed the phosphorylation of EGFR, AKT and ERK activated by EBV infection, suggesting that NRP1 was associated with EBV activation of EGFR/AKT and EGFR/ERK pathways.
In addition to cell-free virus infection, cell-to-cell contact is a more efficient mode of EBV infection of diverse human epithelial cells (Imai, Nishikawa and Takada 1998). Epidermal growth factor (EGF) increases cell-to-cell infection of EBV from infected Akata cells to uninfected HNE1 cells. This effect is partially dependent on the expression of NRP1 (Wang et al. 2015), confirming that NRP1 is also important for cell-to-cell contact-mediated infection that involves the downstream signaling components AKT and ERK.
EBV ENTRY STEP III: MERGER OF THE VIRAL AND HOST MEMBRANES
After gH/gL binding to a host receptor and induction of host cell signaling pathways, gH/gL may regulate fusion through interactions with gB. Using chimeric gL molecules composed of EBV and rhesus lymphocryptovirus sequences, a species-specific functional interaction between gH/gL and gB was mapped to EBV gL residues 54 and 94 and regions from 456 to 807 on EBV gB (Plate et al. 2009; Plate et al. 2011). The EBV fusion protein, gB, has surprising structural homology to the post-fusion form of vesicular stomatitis virus glycoprotein G (VSV G), the sole fusion protein of VSV that is necessary and sufficient for cell entry (Backovic, Longnecker and Jardetzky 2009). It is proposed that EBV gB may undergo large conformational changes from pre-fusion form to post-fusion form to bring the host cell membrane and viral membrane together. However, there is lack of the EBV gB pre-fusion structure and evidence of EBV gB refolding transition during fusion. A recent study of HSV gB in which fluorescent proteins (FP) were genetically inserted throughout the gB ectodomain revealed that CFP and YFP dual-labeled HSV gB had a significantly different FRET signal than the construct containing CFP alone thus allowing the monitoring of gB conformations (Gallagher et al. 2014).
The fusion loops of gB are crucial for membrane fusion and are located in Domain I, which is close to the expected location of the transmembrane region in the crystal structure (Backovic, Jardetzky and Longnecker 2007; Backovic, Longnecker and Jardetzky 2009). During the fusion process, the fusion loops are inserted into the cell membrane. gB then refolds to a post-fusion conformation, thereby driving the merger of the viral and host membranes. Mutation of the putative gB fusion loops WY (112–113) and WLIW (193–196) to alanine or insertion mutations in all five of the gB domains decreased EBV gB fusion in both epithelial and B cells, consistent with the hypothesis that EBV gB undergoes a large conformational change to facilitate membrane fusion (Backovic, Jardetzky and Longnecker 2007).
EBV ENTRY STEP IV AND V: TRANSPORT OF THE VIRUS PARTICLE IN THE CYTOSOL TO THE NUCLEAR PERIPHERY AND PRODUCTIVE INFECTION
Like other herpesviruses, EBV replicates in the nucleus of target cells (Hammerschmidt and Sugden 2013). Thus, EBV must transit from the membrane to the nucleus through the cytosol. Transport of virus particles is different for B cells and epithelial cells based on the sensitivity of B cell infection to the effects of chlorpromazine and actin remodeling inhibitors whereas epithelial cell infection is not altered (Borza et al. 2004; Valencia and Hutt-Fletcher 2012). This difference in inhibitor sensitivity is due to the different routes of B cell and epithelial cell infection: endocytosis (B cells) versus direct fusion of the membrane (epithelial cells) (Nemerow and Cooper 1984; Miller and Hutt-Fletcher 1992). As discussed earlier, lipid raft-dependent endocytosis and macropinocytosis of epithelial cell infection has also been reported (Wang et al. 2015). Endocytosis provides a mechanism through which viruses can pass through the actin cortex by exploiting the intrinsic migratory properties of endocytic vesicles (Grove and Marsh 2011). The transport of virus in B cells is more efficient compared to epithelial cells since virus endocytozed by the B cell is protected in the vesicle before fusion out of the vesicle. Delivery of EBV DNA into the infected cell nucleus peaks at 4 hours without being degraded. However, virus DNA is lost following internalization into epithelial cells, which reaches its peak at 15–30 minutes and then starts to degrade (Valencia and Hutt-Fletcher 2012).
Leupeptin, a serine protease inhibitor, stabilizes viral DNA in epithelial cells but has no effect on transcription of the viral genome. Wortmannin or LY294002 pre-treatment, which both inhibit class III phosphoinositide 3-kinase (PI3K) and reduce autophagy, can greatly increase the transcription of the virus genome. These results indicate that viral DNA loss in epithelial cells is due to shuttling of the entire particle to a degradative compartment rather than premature uncoating of virus and subsequent exposure of DNA to digestion in the cytoplasm (Valencia and Hutt-Fletcher 2012).
Nuclear delivery of viral DNA and infection of other herpesviruses usually requires the microtubule network. For EBV epithelial cell infection, both the actin and the microtubule networks are required. Reagents that disrupt actin remodeling and microtubules reduce transcription of the incoming virus genome (Valencia and Hutt-Fletcher 2012).
While comparing efficiency of virus binding and infection of CR2-positive and CR2-negative epithelial cells, it was found that virus binding is five times lower in CR2-negative cells. Virus transport is 100 times less in CR2-negative cells (Borza et al. 2004). Thus, CR2 may play a role in virus transport after cross-linking by EBV and this effect might occur through its cytoplasmic tail domain binding to actin nucleator formin FHOS/FHOD (Valencia and Hutt-Fletcher 2012). However, the infection rate remained the same when CR2-negative epithelial cells were transfected with either WT CR2 or CR2 lacking the cytoplasmic domain, indicating that the cytoplasmic domain of CR2 is not important (Valencia and Hutt-Fletcher 2012).
Additionally, EBV can traverse polarized human oral epithelial cells without causing productive infection. This process occurs bidirectionally from both the apical to the basolateral membranes (initial EBV infection) or vice versa (EBV secretion into saliva) (Tugizov, Herrera and Palefsky 2013). Inhibitors of macropinocytosis can reduce apical to basolateral virus transcytosis (Tugizov, Herrera and Palefsky 2013). Inhibitors of caveolin can also greatly reduce the basolateral entry (Tugizov, Herrera and Palefsky 2013). EBV infects oropharyngeal cells at their apical surface by direct cell-to-cell contact with infected lymphocytes. Cell-free EBV virions enter at the basolateral membrane of the epithelial cell lines HSC-3 sort, Detroit sort and OCO cells (Tugizov, Berline and Palefsky 2003). BMRF-2 and integrins are all expressed at the basolateral membranes of polarized cells, where virion attachment occurs. Anti-EphA2 but not anti-EphA4 antibody can inhibit EBV infection of Detroit 562 cells (Chen et al. 2018). Previous studies showed that in normal oral mucosa, immunostaining of EphA2 was detected in the basal cells and parabasal cells (Shao et al. 2008). Thus, EBV may use BMRF-2 and integrins for attachment and EphA2 for virus entry on the basolateral membrane.
In summary, EBV is an excellent model to study viral entry into different host cell types because it requires the coordination of multiple cellular molecules. The nature and mechanism of EBV entry is different for B cells and epithelial cells, requiring different glycoprotein complexes to bind to specific receptors on target cells. Overall the mechanism of EBV entry into epithelial cells is becoming clearer. Numerous receptors have been implicated in this process and may also be involved in additional processes of EBV entry, transport and replication.
EphA2 is the most important entry receptor for epithelial cells since infection of EphA2 knockout HEK 293T cells by EBV is reduced by 85% (Chen et al. 2018). It has been shown that EphA2 may bind gH/gL through its LBD (Chen et al. 2018; Zhang et al. 2018); however, the detailed binding region on EBV gH/gL is not known. For EBV, the intracellular kinase activity is not important for fusion (Chen et al. 2018). Further research such as CryoEM of gH/gL interacting with EphA2 might be helpful to determine the exact binding interactions of EBV gH/gL with EphA2. Moreover, comparative studies of EBV epithelial cell triggering complexes and B cell triggering complexes will be useful to understand how infection of these two cell types is orchestrated by EBV fusion glycoproteins.
ACKNOWLEDGEMENTS
We appreciate the help and advice received from the members of the Longnecker Laboratories, especially Samantha Schaller.
Contributor Information
Jia Chen, Department of Microbiology and Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
Richard Longnecker, Department of Microbiology and Immunology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA.
FUNDING
This research was supported by AI076183 (RL) and AI137267 (RL) from the National Institute of Allergy and Infectious Diseases, IRG-15-173-21 (JC) from the American Cancer Society and the Third Coast Center for AIDS Research pilot award (JC).
Conflict of interest. None declared.
REFERENCES
- Aaron PA, Jamklang M, Uhrig JPet al.. The blood–brain barrier internalises Cryptococcus neoformans via the EphA2-tyrosine kinase receptor. Cell Microbiol. 2018;20:e12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arii J, Goto H, Suenaga Tet al.. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature. 2010;467:859–62. [DOI] [PubMed] [Google Scholar]
- Babcock GJ, Decker LL, Volk Met al.. EBV persistence in memory B cells in vivo. Immunity. 1998;9:395–404. [DOI] [PubMed] [Google Scholar]
- Backovic M, Jardetzky TS, Longnecker R. Hydrophobic residues that form putative fusion loops of Epstein–Barr virus glycoprotein B are critical for fusion activity. J Virol. 2007;81:9596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M, Longnecker R, Jardetzky TS. Structure of a trimeric variant of the Epstein–Barr virus glycoprotein B. P Natl Acad Sci USA. 2009;106:2880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borza CM, Hutt-Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein–Barr virus. Nat Med. 2002;8:594–9. [DOI] [PubMed] [Google Scholar]
- Borza CM, Morgan AJ, Turk SMet al.. Use of gHgL for attachment of Epstein–Barr virus to epithelial cells compromises infection. J Virol. 2004;78:5007–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke AP, Yen TS, Shekitka KMet al.. Lymphoepithelial carcinoma of the stomach with Epstein–Barr virus demonstrated by polymerase chain reaction. Modern Pathol. 1990;3:377–80. [PubMed] [Google Scholar]
- Burkitt DP. Observations on the geography of malignant lymphoma. East Afr Med J. 1961;38:511–4. [PubMed] [Google Scholar]
- Burkitt D, O'Conor GT. Malignant lymphoma in African children. I. A clinical syndrome. Cancer. 1961;14:258–69. [DOI] [PubMed] [Google Scholar]
- Chen J, Jardetzky TS, Longnecker R. The large groove found in the gH/gL structure is an important functional domain for Epstein–Barr virus fusion. J Virol. 2013;87:3620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Rowe CL, Jardetzky TSet al.. The KGD motif of Epstein–Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio. 2012;3:e00290–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sathiyamoorthy K, Zhang Xet al.. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat Microbiol. 2018;3:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang X, Jardetzky TSet al.. The Epstein–Barr virus (EBV) glycoprotein B cytoplasmic C-terminal tail domain regulates the energy requirement for EBV-induced membrane fusion. J Virol. 2014;88:11686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshenko N, Liu W, Satlin LMet al.. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J Biol Chem. 2005;280:31116–25. [DOI] [PubMed] [Google Scholar]
- Chesnokova LS, Hutt-Fletcher LM. Fusion of Epstein–Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol. 2011;85:13214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. Fusion of epithelial cells by Epstein–Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. P Natl Acad Sci USA. 2009;106:20464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W, Song X, Yang Xet al.. Neuropilin-1 promotes epithelial-to-mesenchymal transition by stimulating nuclear factor-kappa B and is associated with poor prognosis in human oral squamous cell carcinoma. PLoS One. 2014;9:e101931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman CB, Wohlford EM, Smith NAet al.. Epstein–Barr virus type 2 latently infects T cells, inducing an atypical activation characterized by expression of lymphotactic cytokines. J Virol. 2015;89:2301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Jackson JO, Jardetzky TSet al.. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9:369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paschale M, Clerici P. Serological diagnosis of Epstein–Barr virus infection: problems and solutions. World J Virol. 2012;1:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Chakraborty S, Bandyopadhyay Cet al.. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS Pathog. 2013;9:e1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1:702–3. [DOI] [PubMed] [Google Scholar]
- Fingeroth JD, Weis JJ, Tedder TFet al.. Epstein–Barr virus receptor of human B lymphocytes is the C3d receptor CR2. P Natl Acad Sci USA. 1984;81:4510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JR, Atanasiu D, Saw WTet al.. Functional fluorescent protein insertions in herpes simplex virus gB report on gB conformation before and after execution of membrane fusion. PLoS Pathog. 2014;10:e1004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Xiao S, Xiao Yet al.. MYH9 is an essential factor for porcine reproductive and respiratory syndrome virus infection. Sci Rep. 2016;6:25120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia NJ, Chen J, Longnecker R. Modulation of Epstein–Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio. 2013;4:e00571–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues HJ, Rubinchikova YE, Dipersio CMet al.. Integrin alphaVbeta3 binds to the RGD motif of glycoprotein B of Kaposi's sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J Virol. 2008;82:1570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghez D, Lepelletier Y, Lambert Set al.. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J Virol. 2006;80:6844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–32. [DOI] [PubMed] [Google Scholar]
- Grove J, Marsh M. The cell biology of receptor-mediated virus entry. J Cell Biol. 2011;195:1071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunven P, Klein G, Henle Get al.. Epstein–Barr virus in Burkitt's lymphoma and nasopharyngeal carcinoma. Antibodies to EBV associated membrane and viral capsid antigens in Burkitt lymphoma patients. Nature. 1970;228:1053–6. [DOI] [PubMed] [Google Scholar]
- Hahn AS, Kaufmann JK, Wies Eet al.. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat Med. 2012;18:961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt W, Sugden B. Replication of Epstein–Barr viral DNA. C SH Perspect Biol. 2013;5:a013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henle W, Ho JH, Henle Get al.. Nasopharyngeal carcinoma: significance of changes in Epstein–Barr virus-related antibody patterns following therapy. Int J Cancer. 1977;20:663–72. [DOI] [PubMed] [Google Scholar]
- Imai S, Nishikawa J, Takada K. Cell-to-cell contact as an efficient mode of Epstein–Barr virus infection of diverse human epithelial cells. J Virol. 1998;72:4371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe Y, Sugimoto K, Yang Let al.. Epstein–Barr virus infection of human natural killer cell lines and peripheral blood natural killer cells. Cancer Res. 2004;64:2167–74. [DOI] [PubMed] [Google Scholar]
- Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–56. [DOI] [PubMed] [Google Scholar]
- Kirschner AN, Omerovic J, Popov Bet al.. Soluble Epstein–Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J Virol. 2006;80:9444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan HH, Sharma-Walia N, Streblow DNet al.. Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J Virol. 2006;80:1167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of pEpstein–Barr virus in vivo. J Virol. 2005;79:1296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Bouttier M, Vassy Ret al.. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood. 2009;113:5176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Turk SM, Hutt-Fletcher LM. The Epstein–Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J Virol. 1995;69:3987–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesing JB, Di Fiore S, Ritter Ket al.. Epstein–Barr virus BDLF2-BMRF2 complex affects cellular morphology. J Gen Virol. 2009;90:1440–9. [DOI] [PubMed] [Google Scholar]
- Longnecker R, Kieff E, Cohen JI.. Fields Viorology. Philadelphia, PA: Lippincott Williams & Wilkins, 2013. [Google Scholar]
- Lun SW, Cheung ST, Lo KW. Cancer stem-like cells in Epstein–Barr virus-associated nasopharyngeal carcinoma. Chin J Cancer. 2014;33:529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Zeisel MB, Xiao Fet al.. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura H, Kirschner AN, Longnecker Ret al.. Crystal structure of the Epstein–Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. P Natl Acad Sci USA. 2010;107:22641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane MP, Longnecker R. Cell-surface expression of a mutated Epstein–Barr virus glycoprotein B allows fusion independent of other viral proteins. P Natl Acad Sci USA. 2004;101:17474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller N, Hutt-Fletcher LM. Epstein–Barr virus enters B cells and epithelial cells by different routes. J Virol. 1992;66:3409–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohl BS, Sathiyamoorthy K, Jardetzky TSet al.. The conserved disulfide bond within domain II of Epstein–Barr virus gH has divergent roles in membrane fusion with epithelial cells and B cells. J Virol. 2014;88:13570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth SJ, Lake CM, Borza CMet al.. Epstein–Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol. 2000;74:6324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53:810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen MM, Haan KM, Longnecker Ret al.. Structure of the Epstein–Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell. 2002;9:375–85. [DOI] [PubMed] [Google Scholar]
- Murphy G, Pfeiffer R, Camargo MCet al.. Meta-analysis shows that prevalence of Epstein–Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009;137:824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR, Cooper NR. Early events in the infection of human B lymphocytes by Epstein–Barr virus: the internalization process. Virology. 1984;132:186–98. [DOI] [PubMed] [Google Scholar]
- Nemerow GR, Mold C, Schwend VKet al.. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein–Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J Virol. 1987;61:1416–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhierl B, Feederle R, Hammerschmidt Wet al.. Glycoprotein gp110 of Epstein–Barr virus determines viral tropism and efficiency of infection. P Natl Acad Sci USA. 2002;99:15036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonoyama M, Pagano JS. Homology between Epstein–Barr virus DNA and viral DNA from Burkitt's lymphoma and nasopharyngeal carcinoma determined by DNA–DNA reassociation kinetics. Nature. 1973;242:44–7. [DOI] [PubMed] [Google Scholar]
- Ogembo JG, Kannan L, Ghiran Iet al.. Human complement receptor type 1/CD35 is an Epstein–Barr Virus receptor. Cell Rep. 2013;3:371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang HB, Braun GB, Friman Tet al.. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat Commun. 2014;5:4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Son AI, Zhou R. Roles of EphA2 in development and disease. Genes. 2013;4:334–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate AE, Reimer JJ, Jardetzky TSet al.. Mapping regions of Epstein–Barr virus (EBV) glycoprotein B (gB) important for fusion function with gH/gL. Virology. 2011;413:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate AE, Smajlovic J, Jardetzky TSet al.. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein–Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J Virol. 2009;83:7678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathiyamoorthy K, Hu YX, Mohl BSet al.. Structural basis for Epstein–Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nat Commun. 2016;7:13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathiyamoorthy K, Jiang J, Hu YXet al.. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog. 2014;10:e1004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathiyamoorthy K, Jiang J, Mohl BSet al.. Inhibition of EBV-mediated membrane fusion by anti-gHgL antibodies. P Natl Acad Sci USA. 2017;114:E8703–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz Q, Vieira JM, Howard Bet al.. Neuropilin 1 and 2 control cranial gangliogenesis and axon guidance through neural crest cells. Development. 2008;135:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamaa AA, Zyada MM, Wagner Met al.. The significance of Epstein Barr virus (EBV) & DNA topoisomerase II alpha (DNA-Topo II alpha) immunoreactivity in normal oral mucosa, oral epithelial dysplasia (OED) and oral squamous cell carcinoma (OSCC). Diagn Pathol. 2008;3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang WF, Chen XMet al.. Expression of EphA2 and VEGF in squamous cell carcinoma of the tongue: correlation with the angiogenesis and clinical outcome. Oral Oncol. 2008;44:1110–7. [DOI] [PubMed] [Google Scholar]
- Shaw PL, Kirschner AN, Jardetzky TSet al.. Characteristics of Epstein–Barr virus envelope protein gp42. Virus Genes. 2010;40:307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She Y, Nong X, Zhang Met al.. Epstein–Barr virus infection and oral squamous cell carcinoma risk: a meta-analysis. PLoS One. 2017;12:e0186860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinevici N, O'Sullivan J. Oral cancer: deregulated molecular events and their use as biomarkers. Oral Oncol. 2016;61:12–8. [DOI] [PubMed] [Google Scholar]
- Sixbey JW, Vesterinen EH, Nedrud JGet al.. Replication of Epstein–Barr virus in human epithelial cells infected in vitro. Nature. 1983;306:480–3. [DOI] [PubMed] [Google Scholar]
- Snijder J, Ortego MS, Weidle Cet al.. An antibody targeting the fusion machinery neutralizes dual-tropic infection and defines a site of vulnerability on Epstein–Barr virus. Immunity. 2018;48:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorem J, Jardetzky TS, Longnecker R. Cleavage and secretion of Epstein–Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J Virol. 2009;83:6664–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorem J, Longnecker R. Cleavage of Epstein–Barr virus glycoprotein B is required for full function in cell–cell fusion with both epithelial and B cells. J Gen Virol. 2009;90:591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs MK, Armitage RJ, Comeau MRet al.. The extracellular domain of the Epstein–Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J Virol. 1996;70:5557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PL, Nemerow GR. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 2007;15:500–7. [DOI] [PubMed] [Google Scholar]
- Sun Y, Qi Y, Liu Cet al.. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J Virol. 2014;88:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M, Solis NV, Lionakis MSet al.. EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat Microbiol. 2018;3:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S, Tsuji T, Amagai Tet al.. Specific cell surface labels in the visual centers of Xenopus laevis tadpole identified using monoclonal antibodies. Dev Biol. 1987;122:90–100. [DOI] [PubMed] [Google Scholar]
- TerBush AA, Hafkamp F, Lee HJet al.. A Kaposi's sarcoma-associated herpesvirus infection mechanism is independent of integrins alpha3beta1, alphaVbeta3, and alphaVbeta5. J Virol. 2018;92:e00803–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CM, Deng W, Yip YLet al.. Epstein–Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin J Cancer. 2014;33:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CM, Yip YL, Lo KWet al.. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. P Natl Acad Sci USA. 2012;109:E3473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CM, Zhang G, Seto Eet al.. Epstein–Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int J Cancer. 2010;127:1570–83. [DOI] [PubMed] [Google Scholar]
- Tugizov SM, Berline JW, Palefsky JM. Epstein–Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat Med. 2003;9:307–14. [DOI] [PubMed] [Google Scholar]
- Tugizov SM, Herrera R, Palefsky JM. Epstein–Barr virus transcytosis through polarized oral epithelial cells. J Virol. 2013;87:8179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia SM, Hutt-Fletcher LM. Important but differential roles for actin in trafficking of Epstein–Barr virus in B cells and epithelial cells. J Virol. 2012;86:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RSet al.. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hutt-Fletcher LM. Epstein–Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J Virol. 1998;72:158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Zhang H, Zhang JPet al.. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun. 2015;6:6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf H, zur Hausen H, Becker V. EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nat New Biol. 1973;244:245–7. [DOI] [PubMed] [Google Scholar]
- Wu L, Borza CM, Hutt-Fletcher LM. Mutations of Epstein–Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J Virol. 2005;79:10923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Palefsky JM, Herrera Ret al.. Characterization of the Epstein–Barr virus glycoprotein BMRF-2. Virology. 2007;359:382–96. [DOI] [PubMed] [Google Scholar]
- Yao QY, Rickinson AB, Epstein MA. A re-examination of the Epstein–Barr virus carrier state in healthy seropositive individuals. Int J Cancer. 1985;35:35–42. [DOI] [PubMed] [Google Scholar]
- Yip YL, Pang PS, Deng Wet al.. Efficient immortalization of primary nasopharyngeal epithelial cells for EBV infection study. PLoS One. 2013;8:e78395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary IC. How neuropilin-1 regulates receptor tyrosine kinase signalling: the knowns and known unknowns. Biochem Soc Trans. 2011;39:1583–91. [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Y, Wang HBet al.. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat Microbiol. 2018;3:164–71. [DOI] [PubMed] [Google Scholar]
- zur Hausen H, Schulte-Holthausen H, Klein Get al.. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature. 1970;228:1056–8. [DOI] [PubMed] [Google Scholar]