

Abstract
Nitrogenases are responsible for biological nitrogen fixation, a crucial step in the biogeochemical nitrogen cycle. These enzymes utilize a two-component protein system and a series of iron–sulfur clusters to perform this reaction, culminating at the FeMco active site (M = Mo, V, Fe), which is capable of binding and reducing N2 to 2NH3. In this review, we summarize how different spectroscopic approaches have shed light on various aspects of these enzymes, including their structure, mechanism, alternative reactivity, and maturation. Synthetic model chemistry and theory have also played significant roles in developing our present understanding of these systems and are discussed in the context of their contributions to interpreting the nature of nitrogenases. Despite years of significant progress, there is still much to be learned from these enzymes through spectroscopic means, and we highlight where further spectroscopic investigations are needed.
1. Introduction
The conversion of atmospheric dinitrogen (N2) to bioavailable ammonia (NH3) is essential for life on earth and is a critical step in the biogeochemical nitrogen cycle. While N2 is abundant in the atmosphere, it is largely inert and must be fixed in a form that can be utilized by organisms for incorporation into amino acids, the building blocks of proteins, and nucleic acids, the building block of deoxyribonucleic acid (DNA). The fixation of nitrogen can occur in three ways (1) biologically through the catalytic action of the nitrogenase (N2ase) family of enzymes, (2) industrially through the Haber–Bosch process,1−4 or (3) to a much lesser extent within the earth’s atmosphere via lightning induced chemical conversion.5 Prior to the development of the Haber–Bosch process about 100 years ago, N2ases provided the primary source of bioavailable ammonia.6 Today, it is estimated that roughly equal portions of ammonia come from these two important processes.7−9 Despite decades of intense research, however, the mechanism of nitrogen reduction by either the industrial or the biological system has yet to be fully elucidated.10−12 This is a topic of intense ongoing scientific interest, as a fundamental understanding of N2 reduction would form a foundation to guide rational catalytic design and provide a path toward more renewable, sustainable energy solutions. This is further highlighted by the ongoing discussions of ammonia as a renewable carbon-free fuel.13
The mechanism of biological N2 reduction is of particular interest as N2ases operate at ambient temperatures and pressures and therefore may provide a means to decentralize fertilizer production.14 N2ases are found in bacteria and archaea, and no known examples occur in eukaryotes.15−17 There are just three isozymes of N2ase, which are delineated by the elemental composition of their cofactors: the Mo-dependent, the V-dependent, and the Fe-only N2ases.18−21 Each isozyme is encoded by a unique set of genes.16,22 However, they are all two component proteins,19,23,24 with a [Fe4S4] cluster containing iron protein (FeP) that serves as the reductase25−29 and a catalytic protein designated MFe (where M = Mo, V, or Fe for the Mo-, V- and Fe-forms, respectively). Within the catalytic protein, there are two metal cofactors, the 8Fe-7S P-cluster (which serves an electron transfer role) and the FeMco cluster (where M = Mo, V, or Fe), also known simply as FeMoco in MoFe, FeVco in VFe protein, and FeFeco in the iron-only FeFe protein. These cofactors are perhaps the most complex known in nature, being comprised of a [MoFe7S9C] cluster in the case of Mo and a [VFe7S9C(CO3)] cluster in the case of V.30−33 The full atomic composition of the catalytic cluster in Fe-only N2ase is yet to be revealed. When referring generally to any of the three clusters, we will use the term FeMco.
By far, the most studied N2ase has been the Mo-dependent form (Mo N2ase), and this will also be the dominant focus of the review. The often dubbed “alternative N2ases” are discussed in section 5. Mo N2ases have been studied since the 1960s, when the first reports for extracted N2ase from Azotobacter vinelandii and Clostridium pasteurianum were made.34−36 In the decades that followed, major advances have been made in obtaining insight into the structure, mechanism, and maturation of both this enzyme and the alternative V and Fe N2ases, which have been well summarized in previous reviews.11,27,29,37−59
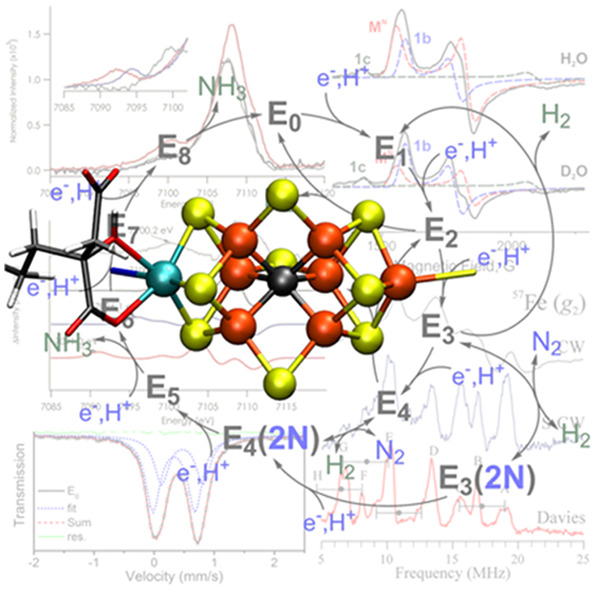
Figure 1 depicts the crystal structure of MoFe together with FeP.60 FeP is the site of ATP hydrolysis. It is generally believed that turnover is initiated by the binding of two molecules of ATP to reduced FeP, which then enables the binding of FeP to MoFe.60 The binding of FeP is proposed to induce a conformational change in the MoFe protein, allowing for an electron to be transferred from the all-ferrous P-cluster (PN) to FeMoco.61,62 Subsequently, an electron is then transferred from FeP to the P-cluster, regenerating the all-ferrous state PN.61 This somewhat nonintuitive order of events has been referred to as a “deficit spending mechanism”.42,62,63 This cycle then repeats, in total 8 times (Figure 2), delivering an electron together with a proton at each step, to give an overall stoichiometry of:64−66
Within the framework of the Lowe–Thorneley (LT) cycle (Figure 2), the resting state of MoFe protein is referred to as E0, and the subsequent En states refer to the number of electrons and protons that have been transferred to the catalytic cluster.
Figure 1.

Mo N2ase, demonstrating the binding orientation of FeP (green) relative to MoFe (blue). The MoFe α-subunit is displayed in dark blue, and the β-subunit is light blue. The two identical subunits of FeP are colored in light and dark green, respectively. Stick models of the M- (aka FeMoco) and P-clusters of MoFe, as well as the [Fe4S4] cluster of FeP (denoted as “F”), are provided detailing the coordinating residues of all three clusters, as well as the nomenclature of the Fe and S positions of FeMoco (PDB 4WZB).60
Figure 2.
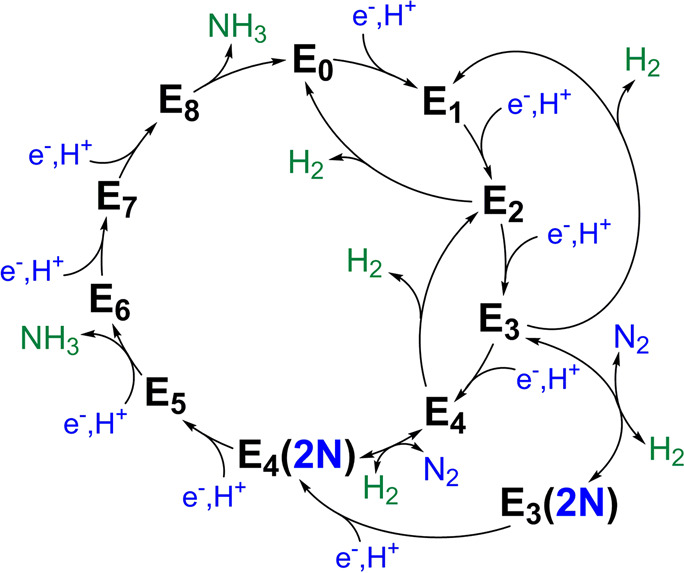
Lowe–Thorneley cycle describing the proposed intermediates of biological N2 reduction as catalyzed by Mo N2ase.64 While very hypothetical at the time of its publication, subsequent studies have substantiated the necessity of producing a stoichiometric quantity of H2 with each N2 released.67 Further aspects of the cycle, such as the release of NH3 from E5 and E8 (rather than from E7 and E8) remain debated.
To ultimately understand the mechanism of biological N2 reduction, one would like to understand how each of the metal clusters shown in Figure 1 functions during the reaction and in particular how the catalytic FeMoco cluster converts substrates into products. A full understanding of the mechanism requires knowing where the electrons and protons go at each step of the cycle and how the electronic and geometric structure of the catalytic active site is transformed. There has been tremendous progress toward this goal in recent years, and spectroscopy, particularly EPR based methods, has played a key role in current mechanistic proposals.11,39,41
Despite these recent advances, however, there is still much to learn about how N2ases function: on the level of the entire microbe, the processes of biological energy transduction, the protein–protein interactions, and the transformation of the inorganic cofactors during the reaction. There is also intense interest in understanding the biosynthesis of the metallocofactors.25,47,51
In the context of this review, we focus on presenting our understanding of N2ases from the perspective of inorganic chemists, concentrating on the geometric and electronic structure of the metal cofactors. As a variety of spectroscopic techniques have played key roles in helping to define the geometric and electronic properties of the cofactors, we first review the methods that have been essential in shaping our present understanding of N2ases. Many of these methods are also readily complemented by theory, and we briefly introduce the application of computational chemistry in N2ase research with the aim of demonstrating how it has been employed to complement spectroscopic studies. Model chemistry has also been crucial in informing spectroscopic methods, and hence we also review the contributions of molecular models as vital spectroscopic benchmarks. We then elaborate on the present understanding of each of the metal cofactors of N2ases, focusing on the Mo-dependent form of the enzyme as it has been the most intensely studied. This is followed by a discussion of the alternative N2ases. Finally, in closing, we discuss how our understanding of cofactor biosynthesis has been aided by spectroscopic studies.
2. Spectroscopic Methods
2.1. Electron Paramagnetic Resonance (EPR) Spectroscopy
As a technique, electron paramagnetic resonance (EPR) spectroscopy68,69 can be applied to study paramagnetic centers possessing one or more unpaired electrons. To investigate paramagnetic centers with half-integer, Kramers spin states (S = 1/2, 3/2, etc.), the more routine (and most commonly available) perpendicular-mode EPR spectrometer may be used. Detection of integer, non-Kramers spin states (S = 1, 2, etc.) requires less common and often significantly more challenging techniques and/or different instrumentation.70−75 The following sections 2.1.1–2.1.6) are intended to provide a very brief overview of EPR and its more advanced derivatives in the context of their application to N2ases.
As mentioned in the Introduction, the LT cycle of Mo N2ase dictates that each subsequent “En” intermediate is preceded by the addition of an electron and proton to the metal cluster (Figure 2) and therefore alternates between half-integer (n = even) and integer (n = odd) spin states. However, due to the side reactions which produce H2 during turnover, the accumulation of individual intermediates En>0 is exceptionally challenging experimentally, and some distribution of intermediates will inevitably exist. This is problematic for bulk techniques such as Mössbauer spectroscopy and most X-ray methods, where deconvoluting the contributions from each intermediate is a nontrivial process. EPR spectroscopy, however, has the unique advantage of spin state selectivity, and consequentially 6 of the 11 proposed intermediates illustrated in Figure 2 [E0, E2, E4, E4(2N), E6, and E8] have Kramers spin states and are approachable by “traditional” EPR methodologies. Additionally, the potential for each of these states to exhibit differing Kramers spins (i.e., S = 1/2 vs 3/2) or separable g-tensors allows for even further selectivity in samples containing a mixture of species. For example, in Mo N2ase, E0 is S = 3/2 and E4 is S = 1/2, allowing the two species to be readily distinguished by their differing EPR spectra. As a result, the contributions of EPR and its more advanced techniques76−79 to our current understanding of N2ases and specifically the catalytic intermediates of Mo N2ase have been unsurpassed.38,77,78,80
2.1.1. Basics of EPR Spectroscopy
The observed transitions of an EPR spectrum are characteristic of the electronic structure of the paramagnetic site and are described in terms of a g-tensor (or matrix).68,69 The three components of g (g = [g1/x, g2/y, g3/z]) can deviate from the free-electron value ge = 2.00232... due to a multitude of interactions, including spin–orbit coupling.81 In turn, information pertaining to the molecule’s orientation and coordination environment in the magnetic field can be gained from interpretation of the EPR spectrum. In measuring an EPR spectrum, the magnetic field is swept while a microwave field of incident energy is fixed. In the magnetic field, a spin-manifold’s degeneracy is lifted by the Zeeman effect82 and resonant transitions (Δms = ± 1) occur when the microwave photon energy matches the energy separation of the magnetic sublevels [for g = 2, resonance occurs at ∼3400 G for X-band frequencies (∼9.5 GHz) and at ∼12,500 G for Q-band frequencies (∼35 GHz)]. Similar to NMR spectroscopy, utilization of higher frequency microwave sources results in increased resolution of the g-tensor.
Biological samples, particularly those of transient intermediate states, are often not amenable to single-crystal studies and are prepared as frozen solutions. The EPR spectrum of a frozen solution is referred to as a “powder” spectrum, as it produces the same spectrum one would obtain for an infinitely large set of randomly oriented microcrystalline sites.69 Powder EPR spectra may take three general forms: isotropic, axial, or rhombic (Figure 3). Continuous-wave (CW) EPR spectroscopy yields a derivative of the EPR absorption spectrum due to the field-modulation detection methodology employed to improve the signal-to-noise ratio. In cases where a rapid adiabatic regime is entered, however, CW methods will produce the EPR absorption spectrum.83,84 All microwave pulse-detected EPR spectra will have an absorption line-shape, as these are based on integration (or other digitization) of the pulse echo intensity as a function of the swept magnetic field.79
Figure 3.

Representative EPR spectral line-shapes.
In cases where crystals can be obtained, single-crystal EPR can be measured by collecting spectra for a series of crystal orientations relative to the applied static magnetic field in the resonator cavity. This allows for a more intimate deconvolution of the EPR spectrum into its magnetically independent resonances and the orientation of the principal magnetic axes of the g-tensor to be determined relative to the structure. This technique has been successfully applied to the resting E0 state MoFe protein, where up to four distinct resonances have been observed, implying four magnetically (or at least orientationally) unique FeMoco clusters are contained per unit cell of the crystal.85,86 Additionally, in MoFe extracted from Azotobacter vinelandii, the gz = 4.31 component has been found to deviate from the principal approximate C3v axis of FeMoco by just 4.5°, which has led to the proposal that Mo is key in producing the high axial rhombicity (E/D = 0.053) observed in this otherwise relatively geometrically symmetric paramagnetic center.86
For an S = 1/2 system, the presence of a magnetic field will split the ground state into two sublevels with ms = ±1/2. However, additional spin-manifolds are present for systems with S ≥ 1, and the possible EPR spectra can be more complex. First, the electron-spin degeneracy may be lifted even in the absence of an external magnetic field through zero-field splitting (ZFS).87,88 ZFS is described in terms of two parameters: the axial parameter D, and the rhombic distortion E/D. For systems with large zero-field splitting, most laboratory spectrometers (such as X-band and Q-band) do not operate at sufficiently high frequencies to observe transitions between spin-manifold pairs (e.g., a transition from ms = −1/2 to ms = −3/2), and are instead limited to transitions within a single spin-manifold. For example, many high-spin Fe3+ (S = 5/2) sites have zero-field splittings large enough to sufficiently split the spin-manifolds such that only a single spin-manifold is probed, and the sign of D (±) determines the energy ordering of the spin-manifolds.87 This is also the case for all known S ≥ 1 species of N2ase.
2.1.2. ENDOR: Observed Transitions
While significant insight into the electronic structure of iron–sulfur clusters may be gained through the CW-EPR investigation of Kramers high-spin signals, much of its utility as applied to N2ases has been in the distinct fingerprinting of unique species, particularly those formed during turnover.40 Instead, much of the finer chemical understanding we have has come from advanced EPR techniques. In particular, electron nuclear double resonance (ENDOR) spectroscopy has been a central tool for the study of Mo N2ase, and the work of Brian M. Hoffman and co-workers has overwhelmingly established the potential of ENDOR spectroscopy to yield key information.11,38−40,77,78,80,89
The broad EPR line widths observed for both Mo N2ase and numerous other metalloproteins often forbids direct observation of small electron–nuclear hyperfine splittings when using CW-EPR. ENDOR, in its most simple terms, is “EPR-detected NMR”, and is capable of resolving small hyperfine coupling constants (A) buried under broad EPR line widths. To perform the experiment, an EPR manifold is first saturated by the incident microwave energy at a single magnetic field position and then further irradiated with radio frequency (RF) energy. When the incident RF energy matches that of available hyperfine resonances, the EPR absorption is perturbed slightly, resulting in an ENDOR transition. In this experiment, the magnetic field is not swept, and the observed spectrum is not dependent on the electronic Zeeman interaction, obeying the nuclear spin Hamiltonian:
| 1 |
The first term is the nuclear Zeeman interaction, where gn is the nuclear g-value of the NMR-active isotope, βn is the Bohr magneton, and I is the nuclear spin. The complete orientation dependent hyperfine tensor, A, also depends on the electronic spin, S, and nuclear spin I. The magnitude of the hyperfine interaction has no dependence on the external magnetic field, and therefore, unlike the g-tensor, hyperfine splittings do not gain higher resolution in the EPR or ENDOR spectra at higher microwave frequencies. However, the nuclear Larmor frequencies (νn) of various nuclei gain separation at higher magnetic fields (higher applied microwave frequencies), potentially separating overlapping nuclei. For this reason, Q-band has become the generally desired frequency for ENDOR measurements for its degree of separation of Larmor frequencies at g = 2, ∼12 000 G, νn(1H) = 53.2 MHz, νn(14N) = 3.84 MHz.
Two different splitting patterns can be observed in ENDOR (Figure 4). The first occurs in the “weak-field” limit, in which half the internal field (A/2) is smaller than the external field. In this case, the ENDOR transitions appear centered at their corresponding vn split by the hyperfine coupling constant A. The second pattern arises under the “strong-field” limit, which occurs when A/2 is greater than the Larmor frequency. Here, the ENDOR transitions are instead centered around A/2 and split by twice the Larmor frequency. This is simply summarized by the first-order equation:
| 2 |
ENDOR of nuclei with I ≥ 1 may also contain an additional splitting arising from nuclear quadrupole interaction, P (eq 1). The asymmetry of the quadrupolar nuclei and its interaction with an unequal charge distribution results in an internal electric field gradient.90−92 This quadrupole interaction is ever present for nuclei of I ≥ 1, even in the absence of an external magnetic field. The quadrupole splitting will further split the hyperfine pattern. In the case of Mo N2ase, only the 14N nuclei has a resolvable quadrupole splitting and has been studied.93,94
Figure 4.

General splitting diagram of expected hyperfine ENDOR transitions in both the weak and strong field limits.
2.1.3. ENDOR: Extracting Information
The ENDOR experiment provides considerably more information than just the hyperfine coupling constants. Because the hyperfine coupling interaction is related to spin delocalization, it can allow for determination of structure, ligand identity, covalency, mechanism studies, and the measurement of metal valency.77,78,95−97 The hyperfine coupling A observed at a single magnetic field position is only a partial component of the complete hyperfine tensor, A, with respect to the complete electronic g-tensor, g. The complete hyperfine tensor and its respective orientation is best determined in single-crystal studies, allowing for the most precision. However, as noted before, most studies of N2ases involve “powder”, randomly oriented (i.e., frozen solution) EPR samples. EPR spectra with g-anisotropy yield orientation selection such that at each resonance position of the EPR spectrum, a unique subset of orientations of the powder pattern are selected.98−100 This can be a powerful tool, allowing for complete hyperfine tensors to be determined from two-dimensional field-frequency ENDOR patterns, demonstrated in Figure 5. Inspections of the edges or extremes of the absorption pattern yield “single-crystal-like” positions, where the ENDOR reports only information along either the g1 or g3 axis. At these positions, the number of uniquely coupled nuclei may be counted. Subsequent analysis and simulations allow for the refinement of the tensors and their orientations by utilizing Euler’s angles to relate the orientations A and g.
Figure 5.
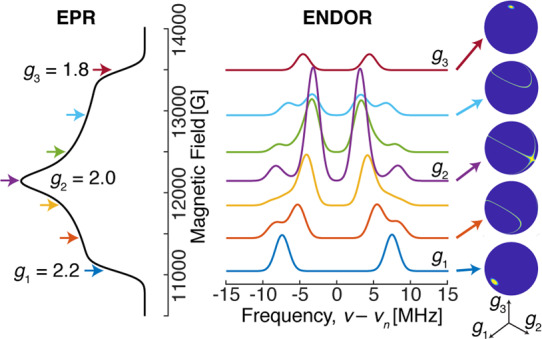
Example field-frequency pattern of a Q-band ENDOR pattern (middle) collected along a rhombic EPR spectrum (left). The spheres on the right highlight the unique subset of molecular orientations observed in the ENDOR spectrum at each selected magnetic field position, as indicated by arrows.
It is possible to decompose A into two separate components, namely local hyperfine interactions (Aloc) and anisotropic dipolar interactions (T): A = Aloc + T.95 Numerous interactions are contained in the Aloc term, including covalent bonding interactions and the isotropic hyperfine coupling contribution, aiso = [A1 + A2 + A3]/3. The term aiso is a measure of s orbital spin density, as only s-orbital electrons have any electron density probability positioned at the nucleus. The ratio of the observed aiso to the intrinsic isotropic hyperfine coupling constant of a given nucleus, a0, gives an estimate of the s-orbital spin density.69 Various mechanisms such as (i) spin polarization from electron–electron exchange interactions or (ii) hyperconjugation may contribute to apparent s-orbital density in systems that do not classically otherwise possess s-orbital density (i.e., organic π-radicals and transition metal centers such as 55Mn and 51V).95 In-depth analyses of Aloc have the potential to provide insight into the valency (or spin state) of metal ions, the degree of ligand covalency and refine the coordination sphere description of paramagnetic metallocenters.96,101
Because both p and d orbitals have nodes at the nucleus, their hyperfine contributions are formally anisotropic and they contribute to the local component of the dipolar tensor, T. This is only a first approximation and should be taken with a grain of salt; determination of the true value requires the integration of all electrons (and their splitting constants) from the full many-electron wave function.97 The nonlocal portion of the dipolar tensor is useful for determining distances and other geometric parameters such as angles. However, this is limited in range, typically up to ∼6–7 Å.
While the signs of individual hyperfine interactions (positive or negative) are difficult to experimentally determine, they can provide additional insight into the electronic and bonding structure of a paramagnetic center. For metallocenters, the unpaired electrons at the metal can share spin density with both ligands and nearby “through-space” nuclei. As a result, the sign of the hyperfine interaction can be either positive or negative depending on the bonding and spin distribution.95 While coupled systems such as iron–sulfur clusters have an overall positive spin density, individual iron sites within a cluster can have either positive or negative hyperfine signs, depending on their electronic coupling.96,102
As mentioned before, nuclei possessing a spin of I ≥ 1 exhibit an additional quadrupole interaction resulting from the electrostatic interaction of the valence electrons in their bonding environment.90−92 The quadrupole tensor yields significant insight into the local charge balance of the valence electrons of the probed atom, allowing for clear assignments of bond order and hybridization (e.g., sp, sp2 vs sp3).91,103,104
2.1.4. ENDOR: Experimental and Instrumental Concerns
The simplicity of the ENDOR experiment allows for measurements to be obtained using a variety of methods. ENDOR may be collected using either continuous-wave105 or pulsed microwave radiation106 while holding the magnetic field constant and varying the applied RF. The applied RF range is only limited by instrumentation (i.e., RF synthesizer and amplifiers) and the bandwidth of the sample cavity or resonator used. This makes ENDOR a broad-range technique that is able to selectively detect a variety of nuclei closely related to the paramagnetic center. Further use of isotopic sample or substrate enrichment (2H, 13C, 15N, etc.) allows for the additional deconvolution of signals. Furthermore, differing metallocofactors or intermediate states may give rise to different, separable EPR spectra. Separable EPR spectra allow ENDOR to be potentially selective for a single given species within a heterogeneous sample, as it only monitors signals associated with the selected EPR resonance.
ENDOR spectra can be collected by either a linear sweep of the RF105 or randomly hopping107 across a chosen RF range. Swept ENDOR often suffers from poorer line resolution and sweep “artifacts”108 which, compared to randomly hopped collected ENDOR, make the spectra more difficult to evaluate (Figure 6).109 By randomly hopping the frequency (stochastic collection) of the “pulsed” RF field in the CW-ENDOR experiment, an ENDOR spectrum free of artifacts associated with nuclear relaxation, which can broaden and possibly distort ENDOR line shapes, may be obtained (Figure 6).110
Figure 6.
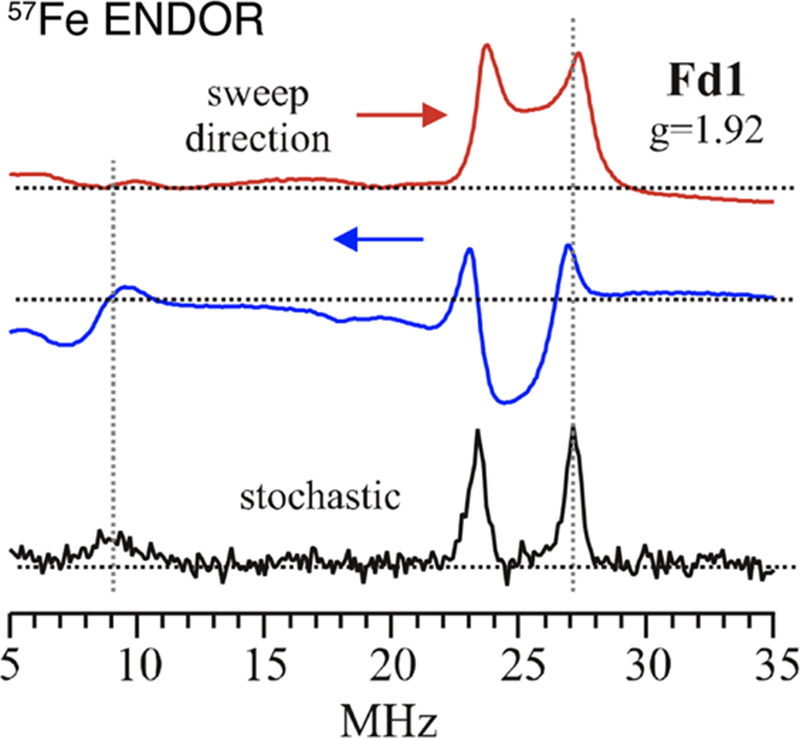
Example of RF swept CW ENDOR exhibiting “sweep artifacts” for the 57Fe ENDOR response of a [Fe2S2]1+ ferredoxin cluster from Aquifex aeolicus. The stochastically hopped RF rids the spectrum of sweep artifacts and improves line shapes in CW-ENDOR. Adapted with permission from ref (102). Copyright 2015 American Chemical Society.
The Mims111 and Davies112 sequences, named after their developers, are the two primary pulse sequences used in the majority of pulsed ENDOR studies. One can find a wide variety of other pulse sequences and their applicable techniques detailed by Schweiger and Jeschke.79 The Mims ENDOR technique uses the simple three microwave pulse stimulated-echo sequence,79 π/2 – τ – π/2 – TRF – π/2 – τ – echo, where the microwave pulses are represented by π/2, the time between applied pulses is τ, and a long RF pulse is applied during TRF. The microwave pulses, π/2, are typically tens of nanoseconds long, with τ on the order of hundreds of nanoseconds. The RF pulse, TRF, is typically quite long, on the order of microseconds. Because Mims ENDOR utilizes a stimulated-echo pulse sequence which drives side-bands into the excited EPR profile, the resulting ENDOR spectra possess “blind-spots” in the hyperfine response as a function of τ. The Mims ENDOR technique is primarily used for resolving small hyperfine couplings, generally less than 10 MHz.
The Davies ENDOR technique may be applied to any size hyperfine coupling but particularly excels at larger couplings (>5 MHz). The pulse sequence employs an initial preparation π microwave pulse to invert the electron spin, followed by an RF pulse, TRF. The excitation and mixing of nuclear manifolds are then detected by a “Hahn” pulse-echo detection sequence, forming the complete Davies ENDOR pulse sequence: π – TRF – π/2 – τ – π – τ – echo. The lengths of these pulses are similar to those in the Mims sequence. Unlike Mims ENDOR, however, the Davies ENDOR technique does not suffer from periodic blind-spots. Instead, it results in a single hole centered at either the nuclear Larmor frequency νn or A/2.
To overcome the limitations of techniques in evaluating relative hyperfine coupling signs,113 Peter Doan has developed the Pulsed ENDOR Saturation and Recovery (PESTRE) protocol, which allows for the easy readout of absolute hyperfine signs from a single hyperfine transition.114 This multistage technique simply establishes (i) a spin–echo baseline (BSL), (ii) saturates the ENDOR response, and (iii) monitors the dynamic recovery level (δDRL) of the spin–echo back to baseline postsaturation. The behavior of the δDRL response, with some applied logic regarding the ENDOR transition being probed and the sign of gn of the nuclei, allows for direct determination of the hyperfine sign. This technique has proven invaluable in 57Fe ENDOR studies of N2ase, as well as other iron–sulfur proteins and model complexes.114−122
Additional ENDOR methods have been applied to the study of Mo N2ase, such as the “pump–probe” triple-ENDOR technique and various other pulse sequences.76,79,117,123 These techniques and their application to Mo N2ase will be discussed in the context of the applied studies below.
2.1.5. Time-Domain EPR Experiments
Time-domain microwave pulse EPR experiments are frequently employed as complementary methods to ENDOR. Techniques such as electron spin echo envelope modulation (ESEEM) spectroscopy124,125 do not utilize an applied RF field but instead employ only microwave pulses to detect the NMR transitions. Generally, ESEEM is limited to the detection of smaller couplings (A < 10 MHz).
ESEEM has proven useful in studying Mo N2ase under unique circumstances. Typical ENDOR and ESEEM spectra are collected on Kramers spin systems, where the Larmor frequencies of coupled nuclei have appreciable magnetic strength and separability (i.e., 14N vs 1H). However, not all N2ase intermediates are Kramers states, and some intermediates with S > 1 non-Kramers states with significantly large negative zero-field splittings exhibit EPR intensity at low-fields.126 Non-Kramers ESEEM (and ENDOR) may be collected at these low fields, providing a possible magnetic characterization technique for these unique non-Kramers systems.126−128 In other studies, ESEEM (and ENDOR) spectroscopy excluded a possible exchangeable 14N atom as the identity of the central light atom of FeMoco.33,93,129,130 Additionally, the observation of weak 13C coupling by ESEEM spectroscopy has been proposed to arise from the central atom (discussed in section 4.1).33,93,129,130
The hyperfine sublevel correlation (HYSCORE) experiment is a four-pulse 2D ESEEM experiment that aims to alleviate detection problems associated with spectrometer dead time by effectively transferring nuclear spin coherence from one electron spin manifold to the other. The result of this transfer is a prolonged modulation decay signal, which increases the time available for detection. This two-dimensional experiment permits the correlation of hyperfine and quadrupole transitions, which often simplifies the assignments of features in multinuclear systems. Although this technique has not been commonly employed for the spectroscopic characterization of Mo N2ase, the reader is referred to the nonexhaustive list of references102,124,131,132 in which HYSCORE has been applied to other iron–sulfur proteins.
2.1.6. Special Topic: Cryoreduction and Cryoannealing
Utilization of high-energy sources such as X-rays or γ-energy photons for molecular samples frequently results in so-called “radiation damage”. However, despite its negative connotation, this can be utilized as an advantageous process.133,134 Such radiation is capable of generating high concentrations of nonspecific solvent (and some solute) radicals in frozen solutions at low temperatures (77 K). These radicals can be further used to generate and stabilize nonequilibrium redox states of metallocenters under cryogenic conditions. Cryoreduction allows for the generation of a new EPR-active center that is one-electron reduced and reflects the global structure and chemistry of the native site, opening up the possibility of advanced EPR characterization methods.135
Below the freezing point of the solvent, the global structure of a protein is restricted and large-scale movement is not possible. However, small changes such as proton transfer and other small conformational distortions may still be feasible, lending the potential to study the reactions and their kinetics through cryoannealing. This annealing process involves warming the sample to a desired temperature for a period of time to allow the sample and system to “relax” and equilibrate, followed by returning the sample to 77 K. In this stepwise fashion, the chemistry (e.g., H atom transfer, substrate bond cleavage, etc.) of trapped intermediates can be monitored by EPR spectroscopy, as well as other spectroscopic methods.70,135−137
2.2. Vibrational Spectroscopy
2.2.1. Infrared Spectroscopy
Infrared (IR) spectroscopy is an extremely widespread technique which utilizes electromagnetic radiation in the range of ∼20–14 000 cm–1 to probe the normal vibrational, translational, and rotational modes of molecular motion. In bioinorganic systems, the mid-IR region of 400–4000 cm–1 is utilized to probe vibrational modes involving both metals and their corresponding ligands. Generally, a change in dipole moment of the system is required to absorb infrared radiation, and the energy of this absorbed radiation corresponds to a unique vibrational mode. Given N atoms, the number of possible vibrational modes of a nonlinear molecule is calculated as 3N-6. Protein systems naturally have many atoms, which in turn results in an outstandingly high number of possible vibrational modes. As a result, proteins typically have very broad and intense spectra in energetic regions involving the excitation of amide vibrational modes, particularly below 1700–1800 cm–1. Because modes directly involving soft metal–ligand bonds (i.e., Fe–S, Mo–S, etc.) are usually low in energy and intensity, they are invariably obscured by modes involving the surrounding protein. However, IR is still particularly useful in detecting the binding and activation of substrates which produce modes outside of this amide window (>1700 cm–1 in D2O). More specifically, IR has been applied quite successfully to investigate the binding of small molecules such as CO and CN– in the FeMoco cluster through observation of how the energy of bound C≡O and C≡N– stretching modes are modulated.138−145 This topic is discussed in detail in sections 4.10 and 4.11.
2.2.2. Raman and Resonance Raman (RR)
Raman spectroscopy provides another method of investigating the vibrational modes of a system. This technique involves the excitation of electrons into a virtual energy state by absorption of an incoming photon, followed by relaxation back to various vibrational sublevels of the ground state accompanied by the emission of a photon in an inelastic relaxation process. The resulting energy difference between the incident photon and observed emitted photons corresponds to the energy of these vibrational modes. The ability to observe such vibrational modes is dependent on whether or not the interaction of the external electric field (from an incident photon source) with the electron cloud of the sample is capable of inducing an instantaneous dipole moment. Hence, the Raman effect results in a different set of selection rules than IR, requiring that a change in polarizability occurs with the vibration of the molecule.146 Conceptually, this means that there must be a change in the average electron distribution in the vibrationally excited state of the molecule relative to the ground state in order for the vibration to be Raman active. For more detailed information regarding the process of Raman spectroscopy, please refer to refs (147,148). For an example of the Raman vs IR active modes in CO2, as well as examples of Raman spectroscopy applied in bioinorganic chemistry, please refer to ref (149).
Resonance Raman (RR) spectroscopy is a particularly powerful derivative of Raman spectroscopy in bioinorganic chemistry. Standard Raman spectroscopy uses a nonselective incident photon energy to excite the system into a virtual excited state. Meanwhile, in RR, the incident photon energy is selected to match that of a particular electronic transition, and as a result the vibrational modes involving bonds that undergo changes in length and force constant during this electronic excitation are significantly enhanced. As electronic transitions involving metal-to-ligand or ligand-to-metal charge transfer (MLCT or LMCT) are often significantly lower in energy than those involving the protein backbone, incident photon energies in RR can be chosen to selectively enhance vibrational modes involving metal centers in proteins. UV–vis/nIR spectroscopy is commonly employed in tandem to determine the necessary incident photon energy for such selective excitations.
Raman, and particularly RR, have been used considerably to investigate FeP and similar model chemistry.150,151 However, few studies have been performed investigating either intact MoFe152 or isolated FeMoco,153 and to date there has been no report of RR on VFe or FeFe.
In addition to standard RR, impulsive coherent vibrational spectroscopy (ICVS) [also known as femtosecond pump–probe spectroscopy (FPPS)] has been successfully employed to study the vibrational modes of MoFe and extracted FeMoco.152,154 ICVS makes use of an ultrashort broadband pump pulse with an energy in resonance with an electronic excitation (typically in the visible range) followed by a weaker probe pulse. The transient absorption of the probe pulse is then measured as a function of the probe pulse delay using differential transmission, and the resulting complex oscillating signal can be treated using Fourier transform (FT) to produce a spectrum similar to that observed in RR. For more detailed information on ICVS and its application in biological systems, please see refs (155−157). ICVS is particularly useful for measuring low-frequency vibrational dynamics and holds several distinct advantages over other vibrational techniques such as IR and NRVS. As discussed in section 2.2.1, low energy IR features of the N2ase cofactors are typically completely obscured by features from the surrounding protein due to their low intensity. By exploiting a pump pulse corresponding to a LMCT or MLCT transition, features involving the metallocofactors can be selectively enhanced. Additionally, ICVS does not require the cryogenic temperatures or synchrotron light source needed for NRVS.
2.2.3. Nuclear Resonance Vibrational Spectroscopy (NRVS)
Nuclear resonance vibrational spectroscopy (NRVS) is a synchrotron-based vibrational spectroscopic technique capable of selectively observing modes involving the displacement of Mössbauer active nuclei.158−160 This technique is particularly useful in the context of N2ases, as 57Fe is a Mössbauer active isotope which is resonant at 14.4125 keV. The phenomenon of NRVS arises from changes in vibrational states which are coincident with the excitation of the nucleus. In dilute frozen solutions, the energy of photons emitted by excited Mössbauer-active nuclei can only deviate from the elastic line by discrete energies corresponding to normal-mode frequencies. These modes can be activated by scanning the incident energy relative to the elastic line. The resulting fluorescence-detected absorption spectrum is composed of peaks that are displaced from the recoilless elastic line by vibrational quanta exclusively involving the Mössbauer nucleus. In this way, NRVS can exclusively provide the entire set of vibrational modes involving the Mössbauer-active nuclei of interest in a given sample.161 In the context of N2ases, this refers to modes involving 57Fe. Furthermore, the intensity of the observed mode is directly related to the degree of displacement of the nuclei, adding a unique quantitative component. Using enriched 57Fe MoFe protein, as well as various mutants, significant insight into the electronic and vibronic structure of the Fe centers of Mo N2ase and possible reaction intermediates for several alternative substrates has been acquired.162−167 However, this technique is yet to be applied to the alternative N2ases.
2.3. Optical Spectroscopy
2.3.1. UV–vis
Ultraviolet, visible, and near-infrared (UV–vis/nIR) absorption spectroscopies utilize electromagnetic radiation in range of 100–2000 nm (5000–100 000 cm–1) to probe excited electronic states. The ability of a system to absorb photon radiation is dependent upon the strength of the electric dipole moment for the corresponding electronic transition. The visible spectra of isolated MoFe and FeP were first reported in 1966.23 Characteristic of larger iron–sulfur clusters, the UV–vis/nIR spectra of both MFe (M = Mo, V, Fe) and FeP are generally very broad, intense, and not particularly featured due to the large number of electronic excited states possible.168,169 The region of 10 000–30 000 cm–1 has generally been assigned to MLCT and LMCT type excitations involving the FeMco and P-clusters. Despite the broadness of these spectra, some changes are known to occur upon oxidation/reduction of these metal clusters (such as an increase in intensity ∼430 nm upon oxidation of MoFe), which has allowed UV–vis/nIR to be used in time-resolved experiments.62,170
2.3.2. Circular Dichroism (CD)
Circular dichroism (CD) spectroscopy depends on the differential absorption of left and right circularly polarized light (eq 3):
| 3 |
In biology, this technique is commonly employed to determine the asymmetry of the spatial structure of a system.171−174 It is often used in conjunction with conventional absorption spectroscopy and can greatly aid the assignment of excited electronic states. CD spectroscopy is applicable to all optically active systems, that is to say, those which are able to rotate the plane of polarized light.175 This requires that the system possesses chirality, meaning that mirror images of the chromophore and its environment are nonsuperimposable. This has made CD useful in bioinorganic chemistry, where metal centers often exhibit relatively low symmetry. The ability to differentially absorb left and right circularly polarized light depends primarily on the strength of the electric and magnetic dipoles of a given transition, where both are required for a given electronic transition to be allowed. As electronic excitations involving transition metals generally have weak electric dipole moments, CD spectra of such systems are usually dominated by transitions which are magnetic dipole allowed (which, unlike electronic dipole transitions, require no change in parity). A consequence of this different set of selection rules is that classic absorption and CD spectra can be compared to determine the electronically and magnetically allowed transitions of a transition metal complex, allowing detailed assignments to be made.
2.3.3. Magnetic Circular Dichroism (MCD)
Similar to CD spectroscopy, magnetic circular dichroism (MCD) spectroscopy also measures the difference between left and right circularly polarized light (referred to as LCP and RCP). However, MCD additionally has an applied magnetic field in parallel to the path of light, resulting in a Faraday effect which induces optical activity in all substances rather than just those with chirality.176,177 While UV–vis/nIR spectroscopy requires an electronic transition to have a nonzero electronic dipole moment in at least one direction (x, y, or z) to gain intensity, MCD spectroscopy requires that a transition is nonzero in two orthogonal directions. The resulting selection rules for MCD are Δml = +1 for LCP and Δml = −1 for RCP light, while Δms = 0.178 An important consequence of these selection rules is that some form of degeneracy in angular momentum, whether orbital or spin in origin, is necessary to gain MCD intensity. In low symmetry systems, orbital angular momentum is quenched. In these cases, only systems with spin degeneracy are able to make a significant contribution to the spectrum. This has made MCD particularly powerful in selectively providing insight into the electronic and geometric structural properties in transition metal complexes and metalloenzymes. For a more detailed description of the origins and formulation of MCD, the reader is directed to refs (179−181).
As mentioned, N2ases generally have very broad absorption spectra in the range of ∼10 000–30 000 cm–1 (∼1000–333 nm) in their resting state, where a lack of resolution has generally limited the usefulness of UV–vis/nIR spectroscopy in understanding the electronic structure of either N2ase component. In contrast, the first reports of MCD on Mo N2ase revealed that these proteins have rich and complex underlying spectra.182−185 The inherent complexity in the electronic and magnetic structure of the FeMco clusters has limited the ability to fully assign the electronic transitions found in their MCD spectra. Interpreting such spectra first requires a sound theoretical approach, which has remained elusive due to the complexity in modeling the electronic coupling of the eight metal centers of the FeMco clusters.96,169,186 However, MCD has been successfully applied to fingerprint changes in the paramagnetic metal clusters of these proteins, using spectral changes to relate changes which occur at the paramagnetic metal centers of either MFe or FeP proteins. This approach has been particularly useful in analyzing the pathways of FeMoco and P-cluster maturation.187−189
2.4. 57Fe Mössbauer Spectroscopy
The Mössbauer effect, discovered by Rudolf Mössbauer,190 involves the absorption and recoilless resonant emission of γ-radiation by 57Fe nuclei (or one of the 79 other Mössbauer-active nuclei). As a detailed description of Mössbauer spectroscopy is beyond the scope of this review, we will instead present a more general description of the technique and focus on the information content accessible through 57Fe Mössbauer spectroscopy as it relates to the study of N2ases. For more comprehensive reviews of 57Fe Mössbauer as applied to iron–sulfur systems, please see refs (191−193). The source of γ-radiation used in 57Fe Mössbauer spectroscopy is 57Co, which decays via electron capture (K-capture), emitting an electron neutrino and yielding 57Fe in an excited I = 5/2 nuclear spin state. This excited state may relax either directly to the I = 1/2 ground state or via an I = 3/2 excited state which then further relaxes to I = 1/2. It is this second process (decay of the I = 3/2 nuclear excited state) which produces the characteristic photons at ∼14.4 keV used in 57Fe Mössbauer spectroscopy (Figure 7). In a sample, the reverse process is observed, where the incident photons ∼14.4 keV are absorbed, exciting 57Fe nuclei from the I = 1/2 ground state to the I = 3/2 first excited state. The incident energy is modulated through acceleration of the source via the Doppler effect, and absorption is measured via transmission through the sample. As the Mössbauer effect requires recoilless absorption, the sample must either be in the solid state or a frozen solution.
Figure 7.

(a) An overview of the emission and absorption processes employed in 57Fe Mössbauer spectroscopy, as well as (moving left to right) the origin of the parameters δ, ΔEQ, and the magnetic hyperfine interaction. The simulated spectrum (b) is the result of the combined isomer shift and quadrupole splitting for a single unique Fe site. Meanwhile, the simulated spectrum (c) is the additional result of the magnetic hyperfine interaction, in which the zero-field splitting parameter D is much larger than the magnetic hyperfine interaction A (as is the case in Mo N2ase and more generally in iron–sulfur clusters). Energy splittings are not to scale.
The nuclear transition from the I = 1/2 ground state to the I = 3/2 first excited state is dependent on the 1s electron density at the nucleus, which is shielded by p and d electrons. Thus, the energy of this transition, the isomer shift δ, reported relative to a standard (typically α-Fe) in units of mm/s, is sensitive to the oxidation state, geometry and ligand environment of the absorbing 57Fe nucleus. Additionally, the nonspherical charge distribution of the I = 3/2 nuclear excited state generates a quadrupole moment that interacts with the electric field gradient at the 57Fe nucleus, producing a quadrupole doublet (Figure 7). As the electric field gradient is affected by the population of the 3d manifold, as well as the asymmetry of ligand charge distribution about the nucleus, the splitting of the quadrupole doublet |ΔEQ| is sensitive to the oxidation state and ligand set of the absorbing 57Fe.
In biological iron–sulfur clusters, the iron centers nearly always exist in a pseudotetrahedral coordination environment of sulfide and thiolate (or other weak field) ligands, resulting in locally high spin electronic configurations. Given the near-uniformity of the ligand coordination sphere around the Fe centers, the isomer shifts observed by Mössbauer spectroscopy can be readily used to determine Fe oxidation states using the equation δ(x) = [1.43 – 0.4x] mm/s (where x is the formal Fe oxidation state).194 This empirical correlation has been found to be consistent across both biological and synthetic iron–sulfur clusters (at or near 80 K). In fully localized iron–sulfur clusters, each Fe center gives rise to a separate quadrupole doublet, while in fully delocalized clusters the Mössbauer spectra afford an average of the individual Fe centers. In systems containing large iron–sulfur clusters or several different iron–sulfur clusters, the weighted signal average of multiple overlapping subspectra is observed because Mössbauer probes all 57Fe present in a sample. While deconvolution of such samples can be challenging, this limitation can be partly overcome through selective 57Fe labeling of specific clusters. Given that the natural abundance of 57Fe is only ca. 2%, 57Fe Mössbauer of biological samples invariably requires some amount of 57Fe enrichment to generate appreciable signal-to-noise ratios. In systems with multiple iron–sulfur clusters, such as N2ases, the ability to selectively 57Fe-label can be used to enhance the Mössbauer signal of one cluster over another.
An alternative method for deconvoluting overlapping Mössbauer signals is through magnetic Mössbauer measurements. In the presence of a magnetic field, the degenerate nuclear spin manifolds of 57Fe are further split into mI = ±1/2 and mI = ±3/2 levels, resulting in six transitions rather than the two observed in the absence of a magnetic field (Figure 7, right). Not only can magnetic splitting allow deconvolution of signals which otherwise overlap at zero field, the nature of the magnetic splitting as a function of magnetic field can also reveal a wealth of information through assignment of the magnetic hyperfine interactions. Specifically, the magnetic hyperfine splitting tensors (A) can be used to reveal spin density distributions and coupling interactions between Fe centers in larger iron–sulfur clusters. Thus, magnetic 57Fe Mössbauer spectroscopy can examine individual Fe centers within larger clusters, describing their spin density and electronic coupling interactions with other Fe centers within the cluster to form a more complete picture of the electronic structure. The detailed analysis of such measurements has been performed for the E0 state of Mo N2ase195 but is yet to be applied to either of the alternative N2ases or any of the currently established intermediates.
2.5. X-ray Spectroscopy
X-ray spectroscopy has played an important role in our understanding of the metallocofactors in N2ases for more than four decades. In 1978, Cramer et al. reported the first Mo extended X-ray absorption fine structure (EXAFS) spectrum of Mo N2ase,196,197 and ever since this technique has been extensively employed in the study of N2ases.163,166,196−210 On the basis of these data, they proposed that FeMoco could either be a linear Fe–Mo–Fe unit or a MoFe3 cubane-like structure. While ultimately crystal structures have revealed a much more complex cofactor,33,60,211,212 the early EXAFS already captured the important metrical parameters of the cofactor and provided great inspiration for bioinspired model chemistry (as described in section 3). In the following years, X-ray spectroscopy has continued to play a major role in our understanding of N2ases, and numerous examples of its utility will be referenced in the subsequent sections. Here, we briefly describe the information content of the primary X-ray spectroscopic methods that have been applied in N2ase research. We refer the reader to refs (213−218) for a more in-depth discussion of the methods.
Both X-ray absorption (XAS) and X-ray emission spectroscopies (XES) have been applied to study N2ases. Figure 8 provides a summary of both the XAS edges and XES lines and their nomenclature. The primary advantage of X-ray based spectroscopies is that they provide an element selective, isotope independent probe of both geometric and electronic structure. XAS is the measurement of the absorption of X-rays as a function of energy. An XAS spectrum is commonly divided into two regions: the X-ray absorption near edge structure or XANES region (which includes the pre-edge and edge) and the EXAFS region (Figure 9). For biological samples, XAS K-edges (excitations of 1s core electrons) are most often utilized. For instance, Fe and Mo K-edge XAS have been used extensively for studies of Mo N2ase. However, there are also limited reports of Mo L-edge XAS and even S and Se K-edge XAS.219−221
Figure 8.
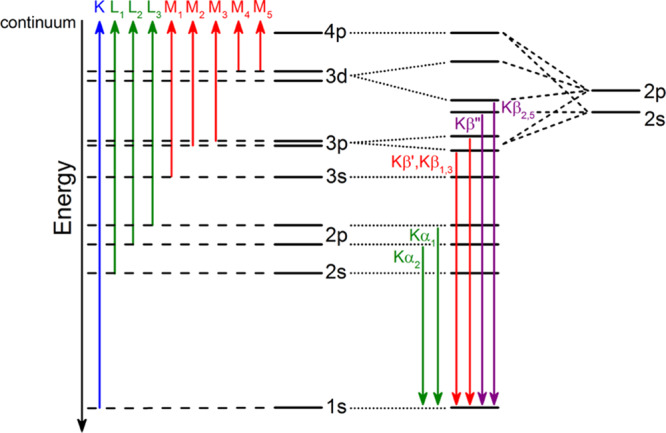
Nomenclature and origins of transitions contributing to (left) XAS excitations and (right) XES 1s core–hole relaxation processes.
Figure 9.

Fe K-edge XAS spectrum of Mo N2ase, demonstrating the pre-edge feature (lime), edge (cyan), and EXAFS (green) regions.
The XANES region can be further broken down into two regions, the pre-edge and the rising edge (highlighted in lime and cyan in Figure 9). In spectra of metals with a partially filled d-shell, such as Fe3+/2+ or Mo3+, the pre-edge corresponds to a 1s to nd transition, which is formally dipole forbidden.222 However, intensity can still be gained through p/d mixing, which imparts dipole allowed character to this feature.223−225 This results in relatively intense pre-edges for most iron–sulfur clusters due to the pseudotetrahedral coordination environment.207,226 The energy of the pre-edge reflects both the average oxidation state as well as the strength of the ligand field. More oxidized metals generally have higher energy pre-edges, while the pre-edges of more reduced metals are usually shifted to lower energy. The rising edge energy is often used as an indicator of oxidation state, with a higher energy edge reflecting a more oxidized metal site. Edges typically shift by ∼1 eV per one unit change in oxidation state. However, the complexity of iron–sulfur electronic structures have shown that these simple correlations break down, and the edge position will also be affected by the extent of delocalization between the iron sites.136,227,228
The EXAFS region provides information regarding the identity of the surrounding atoms and their distance from the photoabsorber. In the context of N2ase research, EXAFS has been vital in providing M–L distance parameters, which were crucial in the earliest structural determination of FeMoco and continue to play a role in the identification and description of intermediates.163,166,196−209 However, a major limitation of EXAFS is that the extracted distances reflect the average for all photoabsorbers of a given type. This means that in the context of FeMoco, while one obtains unique structural information from the perspective of the Mo EXAFS, the information from the Fe EXAFS is limited. We note that this is, of course, not just an issue for the EXAFS, but for the XAS edges and XES as well.
XES involves monitoring the fluorescent processes that occur after the ionization of a core electron. For a 1s core ionization, 2p-to-1s (Kα), 3p-to-1s (Kβ), and valence-to-1s (VtC, aka Kβ′′, and Kβ2,5) transitions are all possible, with the probability decreasing by 3 orders of magnitude across this series (Figure 10). The intense Kα emission lines are dominated by the metal 2p spin orbit coupling and as such provide little new chemical information. However, by combining high resolution Kα-detected emission with X-ray absorption in a two-dimensional experiment, one can obtain XAS edges with significantly higher resolution.213,229−231 Such experiments are known as high-energy resolution fluorescence detected (HERFD) XAS and have been applied to Mo N2ase at the Mo K-edge, Fe K-edge, and most recently at the Se K-edge for selenium substituted N2ases.137,220,232
Figure 10.
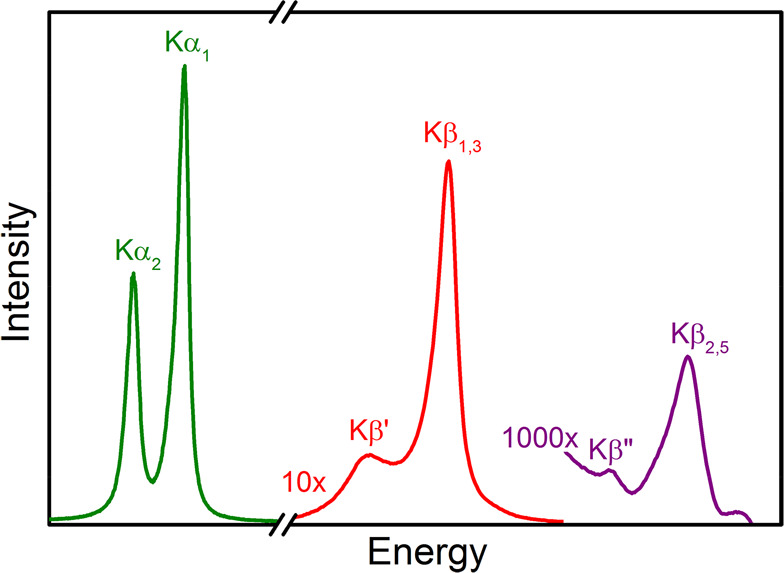
An XES spectrum demonstrating the fine structure of the K lines. The Kα1,2 features are approximately 10× more intense than the Kβ′ and Kβ1,3 features, and 1000× more so than the Kβ′′ and Kβ2,5 features.
At higher fluorescence energies, the Kβ emission lines appear (Figure 10, red). Because of the presence of a 3p core–hole in the final state, the spectra are dominated by 3p–3d exchange, which splits the Kβ emission into Kβ1,3 and Kβ′ spectral features. The splitting of these two features reflects the number of unpaired d-electrons; however, this simple picture is further modulated by covalency, making it challenging to extract quantitative information from the Kβ emission lines of iron–sulfur clusters. Nonetheless, clear differences have been observed between FeMoco and the P-clusters, suggesting a role for the central carbide in modulating the electronic structure.232
The highest in energy, and by far weakest in intensity, are the VtC emission features (Figure 10, purple). These are further divided into the Kβ2,5 and Kβ′′ features, corresponding to ligand np-to-metal 1s and ligand ns-to-metal 1s transitions, respectively. The VtC XES features gain intensity through a small amount of metal np character mixed into the ligand valence orbitals. This spectral region was key in establishing the presence of a central carbide in FeMoco based on the presence of a carbide 2s to iron 1s spectral feature.30 Subsequent VtC XES has also been important in understanding the biosynthesis of N2ases and in establishing the presence of a central carbide in FeVco.31
2.6. Theory and its Correlation with Spectroscopy
Spectroscopic methods can provide a bounty of information regarding the geometric and electronic structures of the iron–sulfur clusters found in N2ases. Theory can provide complementary information and in many cases further insight through a more complete description of these properties. However, theory is first and foremost limited by how well it describes a system of interest, making the direct calculation of geometric and spectroscopic parameters critical not only in aiding spectral interpretation but also in evaluating the applicability of the employed computational method. While ligand-field theory provides a simple and useful semiempirical approach to understanding the electronic structure of transition metal ions, quantum chemistry allows a less empirical, more direct way of calculating and understanding electronic structure.
In quantum chemistry, the starting point is the many-particle molecular wave function described by the Schrödinger equation. When combined with relativistic corrections, quantum theory is capable of describing the entire field of chemistry. However, approximations are required to the Schrödinger equation for it to be directly applied to molecular systems. Modern quantum chemistry has evolved into two main branches: (i) correlated wave function theory, where the electronic wave function is systematically expanded in terms of antisymmetric Slater determinants233 and (ii) density functional theory (DFT), where the Schrödinger equation is reformulated in terms of equations using the electron density.234 Both correlated wave function theory and DFT approaches have their advantages and caveats; however, due to current cost/accuracy trade-offs, quantum chemical calculations of the electronic structure of the metallocofactors of N2ases have almost exclusively been based on broken symmetry DFT (BS-DFT) approaches. The largest drawback of BS-DFT approaches is their ineptitude in describing pure spin states, and it is not clear from the outset how successful the single-determinant DFT strategy should be for systems as complex as FeMoco and the P-cluster. Additionally, DFT comes in many flavors (functionals) and the functional choice is not obvious. For iron–sulfur compounds, it has been found that nonhybrid functionals or hybrid functionals with low HF exchange typically predict molecular structures in much better agreement with experiment than popular hybrid functionals with 20% or higher HF exchange (e.g., B3LYP).235,236 This has recently been discussed in the literature by Raugei et al.,237 Cao et al.,238 and Thorhallsson et al.,239 where a comparison of multiple functionals show that only nonhybrid functionals and TPSSh produce resting state structures of FeMoco that can be considered in good agreement with the high-resolution crystal structure.33 The functional dependency seen in the molecular structure with little doubt carries over to the calculation of spectroscopic properties.
Recent work by Garnet Chan and co-workers has presented a multireference wave function (via a DMRG-CASSCF approach) description of the low-energy spectrum of the nitrogenase P-cluster in three oxidation states.240 This approach allows for the direct calculation of the pure spin states involved, and the calculations were able to predict the ground-state spin state of the different redox states of the P-cluster and give a description of the nature of the low-lying spin states. While it is still concerning that these calculations lack dynamical correlation, the fact that large active-space multireference calculations are becoming possible for such large iron–sulfur clusters is encouraging and paves the way for a robust treatment of the electronic structure and spectroscopic properties of these complex clusters.
2.6.1. Electronic Structure of Iron–Sulfur Clusters
Iron–sulfur clusters, including those found in N2ases, incorporate Fe ions almost exclusively in Fe2+ and Fe3+ oxidation states. The weak-field nature of the sulfide ligands combined with tetrahedral coordination of the Fe ions gives rise to high-spin electronic structures at the individual metal, i.e., an S = 5/2 spin state for Fe3+ and S = 2 for Fe2+. The large number of unpaired electrons per Fe ion combined with the presence of multiple Fe ions in each cluster makes such electronic structures highly challenging to calculate. Iron–sulfur clusters form highly covalent metal–ligand bonds with sulfides and thiolate groups and contain strong spin-polarization effects. As shown by molecular orbital (MO) calculations from Noodleman, iron–sulfur systems display an “inverted level scheme” where the majority (α) Fe 3d spin levels are lower in energy than the occupied sulfur 3p levels, and the minority (β) Fe 3d lie higher in energy than both.96 The minority spin levels are either empty, in the case of a single Fe3+ ion, or populated by one electron, in the case of an Fe2+ ion. This MO scheme suggests a very different environment for majority vs minority electrons. Studies from the Solomon group using photoelectron spectroscopy, polarized absorption, and MCD have supported the validity of this scheme.241−244
As a consequence of spin polarization and strong covalent metal–ligand bonds, iron–sulfur systems show strong spin coupling in which the ground spin states are almost always antiferromagnetically coupled low-spin states. Spin coupling of transition metal ions is often well described by the phenomenological Heisenberg–Dirac–van Vleck Hamiltonian, defined (in one of several ways) as
| 4 |
where ŜA and ŜB are the local spin operators of spin site A and spin site B. The exchange coupling parameter J is the strength of the interaction and its sign meaning antiferromagnetic (negative) or ferromagnetic (positive) interaction for this form of the equation. Heisenberg spin coupling results in a ladder of spin states from |SA + SB| to |SA – SB|, where SA and SB are the local spins on each site.245
Spin coupling of metal ions usually results in antiferromagnetic alignment of spins. This is due to the presence of superexchange,246 where the metal ion d-orbitals on each site interact via the bridging ligand orbitals, favoring antiferromagnetic alignment and low spin states. However, another phenomenon commonly encountered in iron–sulfur clusters favors ferromagnetic alignment of spins. This is called double exchange or resonance delocalization and results from direct overlap of metal d-orbitals.96,247−249 While superexchange almost always dominates in mixed valent iron–sulfur dimers,250−252 double exchange can become a competing factor in larger clusters.
A modified Heisenberg double exchange (HDE) Hamiltonian250−252 has been used to incorporate these effects:
| 5 |
where B is the double-exchange parameter.96,249 Pioneering work by Noodleman186,253 has utilized spin-polarized BS-DFT and the HDE Hamiltonian to study the electronic structure of iron–sulfur clusters in proteins ranging from the dimeric iron–sulfur clusters in ferredoxins and Rieske proteins, to the tetrameric clusters in ferredoxins and high potential iron–sulfur proteins (HIPIPs), to the large FeMoco130,254−256 and P-clusters257 of nitrogenase (with very complex spin coupling). The HDE model has provided a basis for interpretation of EPR, 57Fe Mössbauer, ENDOR, and NMR spectra in [Fe2S2], [Fe3S4], and [Fe4S4] clusters.258−265 As BS-DFT uses an unrestricted single-determinantal approach, it cannot describe the pure spin states of the spin coupled iron–sulfur clusters; rather, the BS-DFT states are weighted averages of the pure spin states. The energies of the broken symmetry states can, however, be used to parametrize a system-specific HDE Hamiltonian from which the energy spectrum of the pure spin states can be derived.
While the work by Noodleman demonstrated the general applicability of BS-DFT and the HDE Hamiltonian in iron–sulfur clusters, recent work by Chan and Neese has demonstrated the failure of the Heisenberg double exchange Hamiltonian to describe the low-energy spectra of these clusters.169 This was accomplished via large active-space CASSCF calculations using the DMRG approximation to the full-CI problem. Even a simple homovalent Fe dimer showed deviation from the Heisenberg spin ladder, evidence of non-Heisenberg behavior. Furthermore, the mixed-valence Fe2+–Fe3+ dimer gave rise to an unusually dense energy spectrum, at odds with the discrete spectrum predicted by the Heisenberg double exchange Hamiltonian. Clearly, the electronic structures of even the simplest iron–sulfur clusters are deeply complicated, and the clusters incorporated by nitrogenase pose an even greater challenge for theory.
2.6.2. Calculation of Absorption and Emission Spectra
The direct calculation of absorption and emission spectra from quantum chemistry involves the calculation of excited electronic states, following the calculation of the ground-state wave function. Within DFT, the most popular approach involves time-dependent DFT (TDDFT) where the transition energy is directly calculated via the time-dependent Kohn–Sham equations245 (often simplified further via the Tamm–Dancoff approximation). The intensity is calculated via transition dipole and quadrupole moments that are calculated as analytical derivatives of the total energy;245 for metal K-edge XAS, quadrupole contributions have been shown to be important for quantitative accuracy.266,267
TDDFT is, in many ways, an attractive approach to excited states as the equations can be easily solved for many transitions. The computation of K-edge XAS from TDDFT has been particularly successful for transition metal complex chemistry, both at metal and ligand edges, and has been applied to iron–sulfur clusters including FeMoco and FeVco for Fe and Mo K-edge XAS and even Mo L-edge XAS spectra.219,228,232,268 Absolute calculated XAS transition energies are usually systematically underestimated compared to experiment due to the flawed DFT potential near the nucleus and the approximate relativistic treatment. However, relative transition energies are usually the target of such calculations, and constant energy shifts can be readily applied to a specific computational method via empirical calibration.266 Calculations of transition metal X-ray emission spectra from a simpler one-electron DFT approach (core transition energies approximated as orbital energy differences) have also been quite successful,269 particularly for the valence-to-core region, and have been used to interpret experimental Fe XES spectra of the MoFe and VFe proteins.30,31
The disadvantages of TDDFT include the almost complete neglect of the multiplet structure because in TDDFT only particle–hole single excitations can be described and, therefore, electronic states arising from double excitations are not treated. Orbital relaxation effects are also not treated, leading to overestimation of transition energies. As the long-range behavior of most density functionals is incorrect (due to self-interaction error), this has severe consequences for the calculation of charge-transfer and Rydberg states; as a result, the energies of charge-transfer states are typically underestimated. While range-separated hybrid functionals have seen some success in alleviating the problems associated with long-range behavior and charge-transfer states, many problems remain.245 It is here that ab initio wave function based methods for the calculation of X-ray spectra show great promise.270 However, applications to iron–sulfur clusters have yet to be realized.
2.6.3. Calculation of Vibrational Spectroscopies
Theoretical vibrational spectra are relatively straightforward in quantum chemistry. Starting from the lowest energy point on a potential energy surface, where the first derivative of the energy is zero, the second derivative of the energy with respect to nuclear coordinates can be calculated to provide harmonic force constants, which can further be used to derive harmonic vibrational frequencies. While the calculation of anharmonic contributions remains a difficult task, harmonic vibrational frequencies from DFT protocols often agree surprisingly well with experimental fundamental frequencies (although often due to error cancellation).245 Additionally, empirical scaling factors (from calibration studies) are sometimes applied to calculated harmonic frequencies, which simultaneously correct for anharmonicity contributions and errors from the DFT method and model deficiencies.
Theoretical vibrational spectroscopy for large molecules and clusters is almost completely dominated by DFT, as correlated wave function theory methods are generally very expensive computationally and their first and second derivatives are quite complicated. DFT approaches have been quite successful for computing accurate molecular geometries, even for the iron–sulfur clusters of N2ases, and calculated vibrational frequencies have been found to be reasonably accurate as well.
IR intensities are easily calculated from the eigenvectors of the Hessian matrix and the derivative of the electric dipole integrals with respect to the nuclear coordinates, while Raman intensities require the derivative of the polarizability tensor.245 Calculations of 57Fe NRVS spectra are also relatively straightforward, requiring the calculation of the Hessian to give harmonic frequencies. The observable vibrational modes must involve displacement of the resonant 57Fe nucleus, and the NRVS intensity of each vibrational mode is proportional to the kinetic energy contribution from the 57Fe nucleus. Thus, the simple way to obtain computed NRVS spectra involves calculation of a Hessian from which both the harmonic vibrational frequencies and normal mode composition factors can be derived, which is then used to calculate the partial vibrational density of states (PVDOS). Additionally, Petrenko et al. have presented a first-principles approach to NRVS spectra where single- and double-phonon contributions to the NRVS signal and orientational dependence aspects are discussed.271
Calculations of vibrational spectra will likely continue to be performed primarily with DFT approaches. Of concern, however, is the functional dependency of the results and the use of empirical scaling factors that may even be different for different vibrational energy regions. These scaling factors are intended to account for both the systematic error arising from the functional but also for possible anharmonicity. The functional dependency and requirement of scaling factors thus requires careful calibration studies.
2.6.4. Calculation of Mössbauer Spectroscopic Parameters
The computation of Mössbauer properties from quantum mechanics is relatively straightforward. The nonmagnetic Mössbauer spectrum primarily depends on two properties: the isomer shift and the quadrupole splitting. Both of these properties can be derived from the ground-state electron density alone, and the modern theoretical approach has been to calculate electron densities at the Fe nucleus via DFT.
2.6.4.1. Isomer Shift (δ)
Isomer shifts are derived through linear correlation plots (calibration curves) of computed electron densities vs the experimental δ for a set of Fe compounds. Although electron density is highly sensitive to the potential at the nucleus (which is dependent on the method used, relativistic effects, and basis sets), these effects are systematic and can be effectively incorporated into the linear fit parameters. This means, however, that the fit parameters (slope and intercept) depend strongly on the methodology used to generate the calibration (the electronic structure method, e.g., density functional, basis sets, relativistic treatment) and thus in practice a calibration must be made for the computational protocol that will be applied to the specific system of interest.
Generally, these calibration curves are quite robust with regards to coordination number, oxidation state, etc., and should be universal for each protocol. Many calibration studies have appeared in recent years,254,272−275 including one specific to iron–sulfur compounds that highlighted the importance of accurate geometries when calculating isomer shifts of iron–sulfur compounds (nonhybrid functionals giving more accurate Fe–S and Fe–Fe distances).274 However, it has sometimes been pointed out that separate calibrations are required for a given class of compounds if the calibration set includes few such complexes, e.g., β-diketiminate compounds276 or iron–sulfur compounds.277 An accuracy of about 0.1 mm/s for absolute isomer shifts can generally be expected, although in practice it is advisible to focus on relative isomer shifts between compounds, as exemplified in a recent computational study on the isomer shifts of FeMoco.277
2.6.4.2. Quadrupole Splitting (ΔEQ)
Quadrupole splitting, ΔEQ, arises due to the interaction of the quadrupole moment of the 57Fe nucleus (the excited nuclear state, S = 3/2 here) with the electric field gradient at the nuclear position, determined by the molecular Fe ion and its environment. Calculation of the electric field gradient is straightforward in quantum chemistry codes and can be calculated as a simple expectation value.245 As the electric field gradient reflects the asymmetry of the electron distribution around the nucleus, it can be more sensitive to ligand-field effects. Good agreement between calculated and experimental quadrupole splittings have been obtained in systematic studies on a range of Fe complexes, but there is also a pronounced dependence on the density functional used and the implicit solvation model.278,279 The lack of explicit solvent molecules and crystal or protein environment in many calculations also likely plays a role in the disagreement between reported computed and experimental values.
3. Characterizing N2ases through Model Chemistry
Synthetic models of protein active sites offer well-defined systems that can serve as spectroscopic benchmarks to compare against protein data, allowing further insight into the biological system to be gained. There has been a long history of synthetic structural and reactivity studies aimed at better understanding the complexities of the iron–sulfur cofactors of nitrogenases, and several other reviews have explored these synthetic systems.280−282 Here, we will highlight those studies most relevant to the spectroscopic characterization of nitrogenase while also offering a historical perspective on the development of nitrogenase model chemistry.
3.1. F-Cluster
The F-cluster of FeP is a rather unusual example of an [Fe4S4]n+ cubane, a ubiquitous workhorse in biological electron transfer chemistry. While the [Fe4S4]2+/1+ couple is believed to be the relevant redox couple in the delivery of reducing equivalents to the MoFe protein, the [Fe4S4]0 redox level was also found to be biologically accessible (section 4.1).283 Additionally, both of these reduced states have been found to exist in two different spin ground states. In the case of the [Fe4S4]1+ redox state, the F-cluster demonstrates both the S = 1/2 state, the “conventional” spin ground state for [Fe4S4]1+ clusters, and a less commonly encountered S = 3/2 spin ground state. It was observed that the relative ratio of the two spin states could be controlled through the use of solvent additives.284 Urea yields a majority S = 3/2 state, while ethylene glycol shifts the population to a majority S = 1/2 state. It has been hypothesized that the relatively open nature of the F-cluster active site facilitates solvent access to the [Fe4S4]1+ cubane, although the exact nature of solvent effects on the protein–cluster interactions and how those effects preferentially stabilize one spin state over the other remains unclear.
The “conventional” S = 1/2 ground state can be easily understood using classical Heisenberg–Dirac van Vleck coupling, with a ferromagnetically coupled mixed-valent pair of Fe centers antiferromagnetically coupled to two ferrous Fe centers, resulting in the overall doublet ground state. The S = 3/2 state is not as simply rationalized, and instead requires invoking double exchange, as the classical Heisenberg exchange model fails to address electronic delocalization (see section 2.6). When the resonance (or delocalization) term B is introduced, the degeneracy of the Heisenberg spin ladder is lifted, and it is possible to stabilize the S = 3/2 state below the S = 1/2 state. The observation of S > 1/2 ground states in [Fe4S4]1+ cubanes has been reported both in biological and synthetic systems.285−287
A systematic study of the magnetic properties and spin ground states of 14 synthetic [Fe4S4]1+ clusters with varying terminal thiolate ligands using 57Fe Mössbauer, EPR, and magnetization measurements was reported by Holm and co-workers.287 In crystalline form, three classes of ground states were observed for the cubanes investigated: (i) pure S = 1/2 or 3/2, (ii) physical mixtures of S = 1/2 and 3/2, and (iii) spin admixtures (quantum mixing of spin states) of S = 1/2 and 3/2. As opposed to a physical mixture of the two spin states, experimental data (Mössbauer, EPR, magnetic susceptibility) for spin admixtures exhibit features similar to those indicative of S = 1/2 and 3/2 spin states but cannot be adequately fit or simulated as a convolution of any combination of distinct S = 1/2 and 3/2 states. While different thiolate ligands were observed to result in stabilization of different ground spin states (e.g., the ground state of (Et4N)3[Fe4S4(S-p-C6H4Br)4] is pure S = 1/2 while that of (Et4N)3[Fe4S4(SCy)4] is pure S = 3/2), changes in crystal packing and electrostatic interactions independent of thiolate identity also appeared to affect the electronic structure. For example, (Me4N)3[Fe4S4(SPh)4]•2MeCN exhibits a pure S = 3/2 state, (Et4N)3[Fe4S4(SPh)4], presented a physical mixture of S = 1/2 and 3/2, and (Et3NMe)3[Fe4S4(SPh)4] was classified as having an admixed ground state. Additionally, all complexes exhibited physical mixtures of S = 1/2 and S = 3/2 spin ground states when dissolved in dimethylformamide, with the ratio of the two states dependent on the identity of the coordinating thiolate ligand. Although an in-depth analysis of cluster structure and magnetic properties was performed, no direct correlation could be elucidated between structural distortions and spin ground states. These studies demonstrate that relatively small interactions can seriously perturb the ground state in [Fe4S4]1+ cubanes, and the exact mechanism remains poorly understood. Additionally, it is unclear whether or not the ground state has a significant effect on enzymatic activity.
Beyond the conventional [Fe4S4]2+/1+ redox couple, the [Fe4S4]0 level has also been accessible in the F-cluster through reduction with either Ti3+-citrate or the more biologically relevant flavodoxin hydroquinone, although the electronic structure of the resultant [Fe4S4]0 state was found to differ depending on the reductant (see section 4.1). This has made [Fe4S4]0 clusters an interesting synthetic target.
The first reported synthetic [Fe4S4]0 cluster was synthesized using terminal cyanide ligands.288 The UV–vis spectrum of the [Fe4S4(CN)4]4– cluster revealed an absorption band at 518 nm, consistent with the feature observed at 510 nm for the Ti3+-citrate reduced F-cluster.289 Additionally, the 57Fe Mössbauer spectrum revealed large isomer shifts (0.65 mm/s), consistent with the all-ferrous oxidation state assignment. While a spin ground state was not assigned, the Mössbauer spectrum was less consistent with an S = 4 ground state compared to that of the Ti3+ citrate reduced F-cluster.70 Several years later, a more stable carbene-supported [Fe4S4]0 cubane was reported290 whose electronic structure was found to be remarkably similar to the Ti3+-citrate reduced F-cluster, with both all-ferrous cubanes demonstrating an S = 4 spin state. Through 57Fe Mössbauer, EPR, and structural analysis in combination with DFT calculations, the S = 4 ground state was assigned as arising from spontaneous cluster distortion, breaking the formal Td symmetry to produce a 3:1 site differentiated geometry.291 This structural analysis was then applied to the crystal structure of the Ti3+-citrate reduced F-cluster and the unique Fe atom was assigned.
3.2. P-Clusters
Synthetic structural mimics of the P-cluster exhibiting the unusual fused double cubane motif with a μ6-S bridging the two cubane halves were first reported by the group of Holm.292,293 These heterometal iron–sulfur clusters were accessed from nucleophilic attack by hydrosulfide on the all-ferrous edge-bridged double cubane precursors (Figure 11, compound I). Through analogous reactions with hydroselenide,294 it was determined that the μ6-sulfide originates from one of the cubane-bridging μ4-S’s of the precursor, while the attacking hydrosulfide/hydroselenide and the second μ4-S are the sources of the μ2-S/Se bridges. Though the incorporation of heterometals and μ2-sulfide bridges deviate from the all-Fe P-cluster with its cysteine bridges, the structural topology remains consistent. Additionally, the reduced nature of the Fe centers (the synthetic vanadium containing cluster in particular, which is formally in the all-ferrous state) is consistent with the all-ferrous assignment of the PN-state of the P-cluster. Apart from zero-field 57Fe Mössbauer studies, however, spectroscopic characterization of these complexes is rather underdeveloped.
Figure 11.

Syntheses of selected structural models of the PN-cluster in the reduced state (I and II) and FeMoco (III and IV).
In addition to the heterometal clusters of Holm, Tatsumi has reported the synthesis of all-Fe clusters exhibiting the same PN-cluster topology through a self-assembly reaction.295,296 In nonpolar solvent, reaction of Fe precursor, thiol/thiourea ligands, and sulfur results in isolation of the P-cluster analogue [Fe8S7]4+. These clusters formally contain a 6Fe2+–2Fe3+ oxidation state distribution, equivalent to the P2+ (Pox) state, although they maintain the PN structural topology (see section 4.2). Tatsumi also demonstrated that these structures could be generated through a cubane condensation route (albeit in low yield) through reaction of [Fe4S4]4+ cubanes with phosphine (Figure 11, compound II).29757Fe Mössbauer analysis of these clusters revealed two quadrupole doublets in a 3:1 ratio, with isomer shifts consistent with the formal oxidation state assignment of 6Fe2+–2Fe3+. These [Fe8S7]4+ clusters are EPR silent, and magnetic data suggests an S = 0 ground state with antiferromagnetic coupling of the Fe centers, consistent with both PN and Pox. Through spectroscopic analysis, together with DFT studies, the two Fe3+ centers were assigned to the terminal positions, and antiferromagnetically coupled to each other as well as the three nearest Fe2+ centers.298,299
3.3. FeMoco: Structure
Starting in the late 1970s, Mo/Fe/S clusters were being synthesized as potential models of the nitrogenase active site when very little was known about the FeMoco structure. Throughout the 1980s, both Holm and Coucouvanis were synthesizing [MoFe3S4] cubanes, as well as larger Mo/Fe/S clusters, based on interpretations of the early Mo and Fe EXAFS data.196−198
Once the first crystal structure of FeMoco revealed the larger cluster framework (to which Fe EXAFS data had alluded), synthesis of larger iron–sulfur clusters intensified. The fused double-cubane clusters of Holm mentioned above, while exhibiting the structural topology of the PN cluster, also appeared as reasonable first approximations of FeMoco (and by extension FeVco and FeFeco). The synthetic analogues of FeMoco that best reproduce the overall structural topology were reported by Tatsumi.300,301 Supported by bulky thiolate ligands, these [Fe8S7] clusters replicate the fused double-cubane motif of FeMoco with an interstitial μ6-S and three “belt” μ2-thiolates substituting the μ6-C and μ2-S’s of the protein active site (Figure 11, compound III). These clusters could be viewed as PN cluster models in which the open face has been closed by a bridging thiolate. EPR measurements reveal an S = 1/2 ground state, indicative of antiferromagnetic coupling of the Fe centers.
More recently, Tatsumi and Ohki have reported the synthesis of an asymmetric double-cuboidal MoFe5S9 cluster from Cp*MoS3– through a templated reaction with FeCl2 and (Et4N)(SH) (Figure 11, compound IV).302 While still lacking two iron centers and a carbide, this is the first larger (>4Fe) iron–sulfur cluster to replicate the longitudinal asymmetry of FeMoco with Mo and Fe at opposite ends of the cluster. EPR of this complex has revealed an S = 1/2 ground state.
In 2002, the identification of an unspecified interstitial light atom missing from the 1992 crystal structure of FeMoco led to attempts to synthesize iron–sulfur clusters with various light atom incorporations (Figure 12). Successes include an oxo-containing [Fe8S6O] cluster from Tatsumi303 (V), an imido-containing [Fe4S3NtBu] cubane from Lee (VI), and more recently a heterometal-imido [WFe3S3NSiMe3] cubane304 (VII) and a heterometal-nitrido [WFe3S3N]2 edge-bridge double cubane305 (VIII) from Holm and Chen. The oxo-cluster reported by Tatsumi was prepared through a similar self-assembly reaction to that described in Figure 11 for the analogous all-sulfide complex with the addition of water and use of a mixed thiolate-alkoxide Fe dimer precursor. The reaction produces a mixture of the alkoxide substituted all-sulfide complex and the oxo-containing product, both of which crystallize from solution and must be manually separated. In this cluster, the interstitial oxo ligand asymmetrically bridges four of the six “belt” irons, with the two remaining Fe’s supported by metal–arene interactions from the bridging terphenylthiolate ligands. Although spectroscopic characterization of Tatsumi’s oxo-cluster (as well as the W imido and nitrido cubanes) is lacking, the effects of light atom incorporation in iron–sulfur clusters were probed in Lee’s imido cubanes using XAS.306
Figure 12.

Synthetic iron–sulfur clusters demonstrating light-atom oxo (V), imido (VI–VII), and nitrido (VIII) ligand incorporations.
To date, no synthetic weak-field iron–sulfur cluster has been synthesized that incorporates a μ6-carbide. Several iron carbonyl carbide clusters are known, but the strong field CO ligands stabilize locally low spin Fe0 and Fe1+ centers in contrast to the high spin Fe2+ and Fe3+ centers observed in FeMoco and FeVco. However, these iron carbonyl carbide complexes have proven instrumental in assigning the identity of the interstitial light atom of FeMoco as a carbide through VtC XES and have also provided insight into how the carbide modulates the electronic structure of the cluster.30,307,308 Recently, Rauchfuss reported the first sulfur substitution in the [Fe6C(CO)x] core, a promising first step toward a carbide-containing synthetic FeMoco model.309
The heterometal [MFe3S4] cubanes of Holm have proven to be the most spectroscopically relevant to the study of the electronic structure of FeMoco. These cubanes have not only provided high fidelity structural models of one-half of the FeMoco and FeVco clusters but have proven to be excellent electronic structural models as demonstrated through 57Fe Mössbauer, EPR, and X-ray spectroscopic studies as well as DFT calculations.232,268,286 Comparison of FeMoco to the [MoFe3S4] using high resolution Mo K-edge X-ray spectroscopy resulted in the assignment of a spin-coupled Mo3+ (Figure 13).268 More recently, Mo L-edge XAS measurements further confirmed the Mo3+ oxidation state assignment of FeMoco,219 and Fe and Mo X-ray magnetic circular dichroism (XMCD) studies of the [MoFe3S4] cubane support the non-Hund electronic structure assignment at Mo.310
Figure 13.

(a) High energy resolution fluorescence detected (HERFD) Fe K-edge XAS comparing the experimental (left) and calculated (right) pre-edge features of MoFe and VFe proteins, as well as the Mo and V cubanes and extracted FeMoco. (b) Schematic of the coupling schemes in [MFe3S4]3+/2+ clusters (M = Mo, V), comparing the Mo and V cubanes to the heterometal half of the FeMoco and FeVco as determined through DFT calculations. Reproduced in part with permission from ref (232). Copyright 2017 The Royal Chemistry Society under Creative Commons Attribution 3.0 Unported License https://creativecommons.org/licenses/by/3.0/.
3.4. FeMoco: Reactivity and Intermediates
Since the first report of an N2-bound transition metal complex,900 understanding N2 activation using molecular systems has developed into a thriving area of research. Early work by Chatt and Hidai on stoichiometric N2 reduction by phosphine-supported Mo and W complexes led to the development of the so-called Chatt cycle311,312 (now often referred to as the distal mechanism, Figure 14). The Chatt cycle details the activation of N2 through end-on binding to a low-valent metal center. Successive addition of protons and electrons results in protonation of the distal N of bound N2, weakening the N–N bond, and concomitantly strengthening the M–N bond until the N–N interaction is completely ruptured, resulting in the release of NH3 and generation of a metal nitride. Further protonation and reduction steps afford the second equivalent of NH3 and regeneration of the M–N2 complex.
Figure 14.

Outline of the potential catalytic mechanisms for homogeneous N2 reduction to NH3.
Following the pioneering work of Chatt and Hidai, the first molecular N2 reduction catalyst was reported by the group of Schrock.313,314 This system, which cycles through Mo3+/Mo6+, allowed for isolation and direct spectroscopic (IR, NMR) characterization of a number of catalytic intermediates, suggesting the system followed a Chatt-like mechanism. The demonstration of a catalytic Mo-based model system contributed to the early assumption that the unique heterometal, Mo (or V or Fe), was the site of N2 activation and reduction. Earlier work by Coucouvanis and co-workers on the catalytic activity of heterometal cubane clusters for non-N2 nitrogenase substrates (hydrazine, diazene, acetylene) also seemed to suggest the primary role of the heterometal site in these synthetic complexes315−322 and DFT calculations are in agreement.323 However, 1H ENDOR characterization of the Schrock Mo3+–hydride complex116 revealed a near axial hyperfine coupling tensor, ruling out terminal hydrides as the source of the signals observed in the E4(4H) state of Mo N2ase and casting doubt on the direct role of Mo in N2 reduction. For a detailed discussion of the E4(4H) state described in Figure 2, please refer to section 4.5. While other molecular Mo-based catalytic systems for N2 reduction have since emerged,324 spectroscopic characterization of relevance to N2ases is scant.
Peters reported the first molecular Fe system for catalytic N2 reduction,325 further turning attention from Mo to Fe as the site of N2 reduction in FeMoco. Through expansive development of a tris-phosphino-borane system, a large number of catalytic (and related noncatalytic) intermediates relevant to N2 activation by molecular systems have been isolated and studied by a suite of spectroscopic techniques, including 57Fe Mössbauer, EPR, X-ray, and IR spectroscopies.122,326−330 To date, this system has afforded the largest collection of molecular Fe N2 reduction intermediates from both distal and alternating pathways. These complexes offer insight into the potential mechanism of N2ase but more importantly yield discrete spectroscopic fingerprints to help identify potential biological intermediates. Here we will focus on a few specific complexes as they relate to the spectroscopic characterization of nitrogenase intermediates, particularly the E4(4H) intermediate and its hydrides.
Supported by a tripodal phosphine ligand, Peters reported the reaction of a bridging nitrido diferrous complex (Figure 15, compound IX) with H2 to give a bridging hydrido imido complex (X) that could then be oxidized to the mixed-valent hydrido imido complex (XI).331,332 Extensive ENDOR studies on this S = 1/2 system revealed that for both the bridging hydride and the imido proton the 1H dipole coupling tensor (T) approximates a fully rhombic interaction, consistent with the initial assignment of the E41H resonances as arising from bridging hydrides.331
Figure 15.

(left) Synthesis of a mixed-valent bridging hydrido imido diiron complex reported by Peters. (right) The stochastic-field modulation-detected 1H ENDOR field frequency pattern of XI, with experimental spectra in black and simulated spectra of the bridging hydride, 1Hμ, in blue. Reproduced with permission from ref (331). Copyright 2012 American Chemical Society.
The Peters group also extended their ligand framework to a bis-chelating disilylide system to support bimetallic complexes that bind both N2 and hydrides, complexes that can be viewed as models of associative N2 binding to the E4 state in nitrogenase (Figure 16).333 ENDOR studies on the mixed-valent N2 dihydride complex again demonstrated a rhombic dipolar coupling tensor (T) as diagnostic for [Fe–H–Fe] bridging hydrides and showed that an anchor model could be used to determine the bridging hydride’s orientation within the molecular framework (Figure 16).120 This same approach was later used to assign the geometric orientation of the two hydrides in the E4(4H) state of nitrogenase (see section 4.5). Additionally, the 14N quadrupole coupling constant was found to offer a measure of charge transfer between Fe and N and thus could potentially be used to gauge N2 activation in biological intermediates.
Figure 16.
A mixed-valent diiron N2 dihydride complex as a potential model of an associative N2 binding to the E4 state of nitrogenase. Reproduced with permission from ref (120). Copyright 2018 American Chemical Society.
ENDOR spectroscopy was also used to determine the chemical identity of a mononuclear Fe–N2 reduction intermediate (Figure 17).122 Low temperature protonation of the catalytically competent XII with excess acid HBArF4·2Et2O results in formation of a new S = 1/2 species tentatively assigned as the FeNNH2 hydrazido species (XIII), but other intermediates XIII′-A-E could not be ruled out. 1H ENDOR spectroscopy revealed two distinct resonances, eliminating symmetrically bound intermediate XIII′-B and diazenido XIII′-E as possibilities. The observed maximal hyperfine coupling (18 MHz) was significantly smaller than those typically observed for metal hydrides (ca. 40 MHz), ruling out XIII′-C and XIII′-D. Diazene isomer XIII′-A was eliminated by the short Fe–N distance determined from EXAFS, leaving the hydrazido formulation for XIII in which the N–NH2 is asymmetrically bound. Analysis of the 1H hyperfine tensors for both resonances resulted in assignment of a bent Fe–N–NH2 fragment of approximately 150°. Analogous noncatalytic intermediates were later structurally characterized using the silyl variant of the ligand,329 further supporting the FeNNH2 assignment. While the formally one-electron oxidized analogue [P3SiFeNNH2]1+ exhibits a near linear (176°) Fe–N–N fragment, the isoelectronic methylated analogue P3SiFeNNMe2 demonstrates a bent (158°) Fe–N–N fragment, consistent with the ENDOR- and DFT-assigned geometry of XIII.
Figure 17.

Proposed structure of N2 reduction intermediate XIII with alternate structures XIII′-A through XIII′-E. Geometric orientation of the −NNH2 fragment relative to Fe as determined through ENDOR spectroscopy. Reproduced with permission from ref (122). Copyright 2015 American Chemical Society.
The Peters group has also successfully extended the reactivity of their system to other transition metals, including Co,334 Ru,335 and Os,335 perhaps demonstrating intriguing possibilities for exploring engineered forms of nitrogenase.
The Peters system has allowed access to a large library of Fe-bound N2 reduction intermediates which have been well-characterized by a host of spectroscopic methods. However, the local electronic structures at Fe are often low-spin due to the strong-field effect of the phosphine ligands, which is in contrast to the weak-field ligand environment found in nitrogenases. In contrast, the Holland group has leveraged a β-diketiminate supported platform to synthesize models of substrate-bound nitrogenase intermediates that almost exclusively exhibit local high-spin electronic structures at the Fe centers. While a catalytically competent N2 reduction system has thus far not been reported, the Holland group has synthesized a number of complexes incorporating reduced nitrogenase substrate intermediates such as styrene and phenylhydrazine, modeling propargyl alcohol and N2 reduction intermediates, respectively. 1H ENDOR studies336 on the Fe–styrene complex (Figure 18, compound XIV) revealed distinct dipolar couplings for the two terminal vinylic H’s of the bound styrene, suggesting the measurements to be quite sensitive to the small crystallographically observed tilt of the alkene plane from normal to the Fe–C–C plane (Figure 19). The geometric distortion and corresponding disparate dipolar couplings contrast with the identical dipolar couplings observed for the propargyl alcohol intermediate, suggesting negligible tilt of the alkene fragment in the case of the protein intermediate.
Figure 18.

Various β-diketiminate supported Fe complexes of relevance to the spectroscopic characterization of nitrogenase intermediates.336−339,341−344
Figure 19.

Crystal structure of β-diketiminate-supported Fe styrene complex. ENDOR studies of 1H/2H substituted variants reveal two distinct dipolar couplings for the terminal vinylic H’s of the bound styrene, suggesting sensitivity of the ENDOR technique to the structurally observed tilt of the styrene fragment. Reproduced with permission from ref (336). Copyright 2015 The Royal Society of Chemistry under Creative Commons Attribution 3.0 Unported License https://creativecommons.org/licenses/by/3.0/.
The Holland group has also reported the synthesis of a three-coordinate terminal Fe1+ hydride complex, a rare structural motif stabilized by a bulky β-diketiminate ligand (Figure 18, compound XV). The terminal nature of the hydride was determined both in the solid state and in solution. ENDOR analysis338 reveals a near axial dipolar coupling tensor (T), similar to the Schrock Mo3+ terminal hydride complex. Together with the ENDOR studies on the bridging hydride complexes reported by Peters, these results further support the assignment of bridging hydrides in E4(4H).
The same β-diketiminate system has been used to prepare low-coordinate [Fe2S]n+ complexes incorporating a bridging sulfide that can bind substrates bridging between the two metals, with nitrogenase-relevant substrates including hydride, hydrazine, and phenylhydrazine. ENDOR analysis of the phenylhydrazine complex (Figure 18, compound XVI) revealed the characteristic hyperfine tensor values for a bound hydrazido ligand as well as the tensor orientations relative to the molecular frame.337 DFT calculations reproduced the experimental results well, demonstrating the utility of a combined ENDOR/DFT approach to identify nitrogenous intermediates.
Additionally, Holland has reported the cooperative reversible cleavage of N2 by 4 equiv of Fe1+ precursor to yield an Fe4 bis-nitrido complex (Figure 18 compound XVII).339 A less bulky β-diketiminate (with methyl rather than isopropyl substituents) was crucial for the formation of the bis-nitrido complex, as increased steric bulk on the ligand only resulted in N2 activation but no cleavage. Together with several other Fe–N2 complexes, compound XVII was studied via Fe VtC XES,340 demonstrating the sensitivity of this technique to the degree of N2 activation. Continuing with the smaller β-diketiminate ligand, the Holland group has reported an unusual [Fe4S3] cluster containing a three-coordinate [FeS3] fragment that can reductively cleave N2H4 to yield a [Fe4S3(NH2)] amido complex (Figure 18, compound XVIII) that has been well characterized by IR, NMR, Mössbauer, and X-ray absorption spectroscopies supported by DFT calculations.341 In addition, Holland has reported N2 binding to an Fe terphenyl–dithiolate complex supported by an Fe–arene interaction (Figure 18, compound XIX) that on further reduction and protonation yields substoichiometric amounts of N2H4 and NH3.342,343
More recently, a diiron dihydride/dideuteride complex was investigated using NRVS344 to probe the Fe–H/D stretching modes as models of the E4(4H) intermediate in nitrogenase (Figure 18, compound XX). The NRVS study revealed Fe–H/D–Fe vibrational modes that extended in energy from approximately 1430 to 400 cm–1. The higher energy vibrations were assigned as in- and out-of-phase hydride stretches perpendicular and parallel to the Fe–Fe vector, while the lower energy vibrations were assigned as Fe–H–Fe wagging modes (deformations of the Fe(μ-H)2Fe core). The modes were well-reproduced using DFT calculations.
4. Mo N2ase
Mo N2ase is by far the most studied of the N2ase systems. Of the three known N2ases, it is the only system present in all identified nitrogen-fixing organisms. As mentioned earlier, this two-protein system contains three metallocofactors, with the F-cluster belonging to FeP, while FeMoco and the P-cluster are found within MoFe. In this section, we will explore the spectroscopic works that have contributed to our current understanding of these metallocofactors, the intermediates that are formed during native catalysis, and finally the interactions of alternative substrates.
4.1. FeP and the F-Cluster
The reducing iron protein of N2ases, generally referred to here as FeP (but also as NifH, see section 6), serves not only as the sole reductase for the complementing catalytic MoFe/VFe/FeFe protein under turnover conditions but also a host of additional roles in the maturation of the FeMoco and P-clusters.345 Herein, we will focus on the spectroscopic characterization of this protein in the Mo-dependent N2ase and in its three well-researched oxidation states. The corresponding FeP of V and Fe N2ases (VnfH and AnfH, respectively) are discussed in section 5.4, while the additional roles of NifH in metallocofactor maturation are discussed in section 6.
It is well established from crystallography that FeP is a homodimer containing a [Fe4S4] cluster symmetrically bound at the interface between the two subunits (Figure 1).346 As isolated in the presence of dithionite (DT), the iron–sulfur cluster rests in the [Fe4S4]1+ oxidation state, producing a solvent-dependent S = 1/2 and S = 3/2 mixed-ground state, as demonstrated by EPR, 57Fe Mössbauer, and VTVH-MCD spectroscopy.284,347,348 While the physiological redox couple of FeP is generally accepted as [Fe4S4]2+/1+(which we will refer to as FePox and FePred, respectively),349−351 the primary physiological reductant of FeP, flavodoxin, is capable of further reducing FeP to the all-ferrous [Fe4S4]0 state (super-reduced FeP, which we will refer to as FePsred).283,352,353 This has generated interest in studying FeP across all three oxidation states, and considerable efforts have been put forth in characterizing and understanding the nature of the all-ferrous form.206,354−357
FeP must first bind 2 ATP358 in order to bind MoFe and transfer an electron to the P-cluster (Figure 20). The [Fe4S4]2+/1+ redox couple is associated with a significant change in the UV–vis spectrum around 430 nm,359 which has served as a critical feature for tracking the oxidation state of the cluster in a number of kinetic studies.349,360−364 Both FePox and FePred are capable of binding ADP/ATP, and through inhibition studies it has been proposed that ADP binds FePox more effectively, while ATP more strongly binds to FePred.360,365 Binding of either of these nucleotides results in a significant reduction in the midpoint potential of the [Fe4S4]2+/1+ redox couple, shifting from −295 mV to < −400 mV.37,366,367 Interestingly, significant conformational changes in both the protein and [Fe4S4]2+ cluster have only been observed in ADP/ATP binding of FePox as evidenced by CD and 57Fe Mössbauer spectroscopies.182,284,368−370 While no major changes have been observed via CD for the binding of ATP to FePred, conformational changes associated with the [Fe4S4]1+ cluster have been observed via EPR spectroscopy (through changes in the relative ratios of S = 1/2 and S = 3/2 components) and 57Fe Mössbauer spectroscopy.371
Figure 20.
FeP cycle of Mo N2ase with the commonly used nonphysiological and physiological reductants, DT and flavodoxin (FldHQ). Adapted with permission from ref (348). Copyright 2016 American Chemical Society.
4.1.1. FePox
FePox exhibits a diamagnetic S = 0 ground state, appearing as a moderately distorted [Fe4S4]2+ cluster with a symmetry reminiscent of ferrodoxins, such as those of Bacillus thermoproteolyticus and Clostridium acidurici.206,28457Fe Mössbauer studies have characterized two Fe-sites in FePox, present in a 3:1 ratio with ΔEQ = 1.22, 0.83 mm/s and δ = 0.45, 0.44 mm/s, respectively, at 4.2 K. Recent spatially resolved anomalous dispersion (SpReAD) analysis has proposed full valence delocalization between the Fe centers of FePox, with each Fe-center appearing as Fe2.5+.356 Upon binding of either ADP or ATP, a significant decrease in the quadrupole splitting (ΔEQ = 0.91, 0.58 mm/s) of these two components is observed; these changes are observed for at least three (and likely all four) Fe centers, making them indicative of a conformational change in the protein induced by nucleotide binding as opposed to binding at one of the Fe sites.284 This has been supported by both UV–vis and CD spectroscopies, where significant changes between 300 and 500 nm are found, particularly at 365 nm (Figure 21).182,370
Figure 21.

UV–visible (left) and circular dichroism (right) spectra of oxidized iron protein of molybdenum nitrogenase from A. vinelandii in the presence (dashed line) and absence (solid line) of MgATP.370 Reproduced with permission from ref (370). Copyright 1995 American Chemical Society.
It has been suggested, however, on the basis of RR spectroscopy, that such drastic changes may actually arise from dissociation of the homodimer and formation of two [Fe2S2]2+ clusters as degradation products in FePox(ATP)2.150 This was proposed based on the appearance of several additional vibrational frequencies (289, 391 cm–1) following addition of MgATP to FePox, which were consistent with a portion of the sample being converted to [Fe2S2]2+. It is notable that in previous 57Fe Mössbauer studies, enzymatic oxidation (accomplished by the addition of MoFe to FePred(MgATP)2 in the absence of a sacrificial reductant) resulted in the appearance of an additional signal consistent with a [Fe2S2]2+ cluster that accounted for ∼30% of FeP.284 Similar results were also found in the Leu127 deletion variant of FeP isolated from Azotobacter vinelandii, which was proposed to be an analogue of the MgATP-bound state of wild-type FeP151 but has since been found (via small-angle X-ray scattering (SAXS) measurements372) to have an extremely different, and quite open, structural conformation. Here, UV–vis, RR, and EPR spectroscopies were used together to demonstrate that upon oxidation with thionine to the S = 0 state a portion of [Fe2S2]2+ is also formed as a minor species, which could be greatly enhanced by the presence of glycerol.151
It has since been demonstrated using XAS, and again CD spectroscopy, that the original changes in the CD spectrum do not arise from degradation of FePox upon MgATP binding, and instead from conformational changes in the protein as originally proposed.369 This is consistent with early 57Fe Mössbauer studies, which showed that the [Fe4S4]2+ cluster undergoes significant electronic rearrangement with nucleotide binding.284 Therefore, [Fe2S2]2+ likely forms from degradation of FePox and is not a catalytically relevant state.151 Perhaps most importantly, the inconsistent appearance of [Fe2S2]2+ over varying experimental conditions is an important reminder of the sensitivity of FeP and the caution with which one must proceed in investigating these systems.
Another intriguing feature of the iron protein is its ability to decompose DT and spontaneously oxidize.182 This process depends on DT or a decomposition byproduct and is accelerated by MgATP and does not affect the activity of the protein. Although it displays similar spectroscopic properties to dye-oxidized protein, a lower midpoint potential was found for the self-oxidized iron protein in absence of nucleotide.370,373 However, when bound to MgATP, the midpoint potential of the iron protein is independent of the oxidation method. The mechanism of self-oxidation is still not understood and might not be physiologically relevant. Regardless, it needs to be taken into account in kinetics studies, as it significantly influences nitrogenase reactions performed in vitro with DT.374
4.1.2. FePred
As mentioned earlier, FePred contains the [Fe4S4]1+ cluster, which has been found to produce an S = 1/2, 3/2 admixture. The ratio observed in this admixture depends heavily on the presence of additional possible ligands in solution, such as ethylene glycol and urea, as well as on the binding of ADP or ATP to the protein.284,347,348,366,371,375 Early studies identified an S = 1/2 species via EPR spectroscopy, appearing at g = [2.05, 1.94, 1.88] associated with isolated FePred;366,376,377 however, spin integration of this signal yielded just 0.3 spins/4Fe, considerably lower than the 1 spin/4Fe anticipated for an S = 1/2 [Fe4S4]1+ cluster.359,375,376 Additional studies demonstrated this ratio could be modulated by the presence of either ethylene glycol378 or urea (Figure 22),375 either raising it to 1 spin/4Fe or virtually eliminating the S = 1/2 signal. This effect was later found to correspond inversely with the appearance of a rhombic S = 3/2 signal with negative zero field splitting (D ∼ −2.5 cm–1, E/D ∼ 0.22) with lower field inflections appearing around g = 5.79 and 5.15 (assigned to g1 of the ms = 3/2 and 1/2 manifolds, respectively).284
Figure 22.

X-band EPR spectrum of the resting FeP in (a) 50 mM Tris, 2.5 mM DT, (b) 50% (v/v) ethylene glycol, 25 mM Tris, 2 mM DT, and (c) 0.4 M urea, 50 mM Tris, 2 mM DT. All samples are pH 7.5. The respective insets are provided to emphasize the g ∼ 5 region. Adapted with permission from ref (284). Copyright 1985 American Society for Biochemistry and Molecular Biology.
57Fe Mössbauer measurements (Figure 23) investigating FePred under these various conditions were performed, resulting in a series of spectra favoring one spin state or the other, all of which could be fit with two components in a 1:1 ratio. FePred prepared in 50% ethylene glycol (∼90% S = 1/2) produced signals with δ = 0.59, 0.53 and ΔEQ = 1.60, 0.98, while the presence of 0.4 M urea (∼85% S = 3/2) resulted in δ = 0.54, 0.54 and a dramatic decrease in the quadrupole splitting, particularly of the first component (ΔEQ = 1.20, 0.80).284 These relative compositions were also later found to be in good agreement with VTVH-MCD studies.348
Figure 23.

Zero-field, 50 K 57Fe Mössbauer spectra of the resting FeP in (a) 50% (v/v) ethylene glycol, 25 mM Tris, 2 mM DT, (b) 50 mM Tris, 2.5 mM DT, and (c) 0.4 M urea, 50 mM Tris, 2 mM DT. All samples are pH 7.5. The solid line through (a) represents the least-squares fit described in the text. Adapted with permission from ref (284). Copyright 1985 American Society for Biochemistry and Molecular Biology.
The 57Fe Mössbauer spectra of the near-pure S = 1/2 and 3/2 ground states were further used as components to deconvolute the spectra of nucleotide-bound FePred(ADP)x (x < 2) and FePred(ATP)2. Here, ADP binding did not appear to perturb the ground-state composition relative to unbound FePred (∼45% S = 1/2, ∼55% S = 3/2, while ATP binding resulted in a bias toward the S = 1/2 state (∼60% S = 1/2, ∼40% S = 3/2).371 Further EPR measurements confirmed that ADP binding does not perturb FePred, although an additional broad signal at g = 4.8 appeared, purportedly due to sample heterogeneity. Meanwhile, formation of FePred(ATP)2 produced a new signal at g = 4.3, which was assigned to the ms = 3/2 sublevel of a fully rhombic S = 5/2 species. The S = 1/2 signal of FePred(ATP)2 also appeared to shift to a considerably more axial spectrum (g = [2.04, 1.90, 1.87]).367,371 Structural characterization of FePred and FePred(ATP)2 via EXAFS has evidenced that there are no major perturbations in the geometry of the [Fe4S4]1+ cluster upon ATP binding;379 therefore, these combined results provide strong evidence that the electronic structure perturbations observed in [Fe4S4]1+ upon ATP binding arise from conformational changes in the surrounding protein.
1H NMR has also played an important role in understanding the changes in electronic structure and geometry of FePred that occur during nucleotide binding.380,381 In particular, the cysteinyl protons of the [Fe4S4]1+ coordinating residues give rise to four features with distinct chemical shifts and spin–lattice relaxation times: in A. vinelandii, these components are referred to as A (49 ppm,<1 ms), B (23 ppm, 5 ms), C (17 ppm, 25 ms), and D (14 ppm, 17 ms). Integration of A:B:C:D yields a ratio of 4:4:3:1.381 This has resulted in the assignment of A and B to protons at the β-CH2 position of cysteine, and C and D to those at α-CH position, agreeing with the expected total of 12 protons. Binding of ATP to produce FePred(ATP)2 was found to produce a series of shifts in these signals,381 implying that ATP binding results in some reorientation of the [Fe4S4]1+-ligating cysteines relative to the magnetic moment of the cluster. These shifts are even more pronounced in FePred(ADP)2,381 further indicating that there are also conformational changes that may occur upon hydrolysis of ATP to ADP + Pi.
4.1.3. FePsred
While most in vitro studies of N2ases have used DT as a sacrificial reductant, the physiological reductants (e.g., flavodoxin hydroquinone, FldHQ) are capable of reducing FeP to the all-ferrous FePsred.283 The ability of FeP to form [Fe4S4]0, first reported in 1994 using methyl viologen (MV) as reductant,352 generated considerable interest over the next several decades regarding both the possible biological relevance and unique electronic structure of this state. Although kinetic studies have ultimately shown that FePsred is not a physiologically relevant state,349,351 the spectroscopic studies characterizing this state have provided important insight into the properties of [Fe4S4]0 systems.
A variety of reducing agents have been employed to generate the all-ferrous state besides MV and Fld, including Ti3+-citrate, 5‘-deazariboflavin, formadine sulfinic acid (FSA), bisaquo(1,4,9,12-tetraazacyclopentadecanyl)-Cr2+ (Cr2+-TACpD), Cr2+-EDTA, Eu2+-DTPA, or Eu2+-EGTA.185,206,382,383 It can also be generated by radiolytic reduction of FePred.70 Formation of [Fe4S4]0 by strong reducing methods (including Ti3+-citrate, 5‘-deazariboflavin, Cr2+-TACpD, Cr2+-EDTA, Eu2+-DTPA, and Eu2+-EGTA) is indicated by the appearance of a new absorption feature centered at ∼520 nm and the loss of any corresponding EPR signal corresponding to FePred and a new pinkish appearance (Figure 24).185,352,382,383 Interestingly, a similar color change from brown to red was observed in the super-reduced activator of 2-hydroxyglutaryl-CoA dehydratase from Acidaminococcus fermentans, with a new shoulder around 500 nm appearing in the UV–vis spectrum.384
Figure 24.

UV–vis spectra of the iron protein of molybdenum nitrogenase from A. vinelandii in the FePred (solid line) and the FePsred (dashed line) states. Reproduced with permission from ref (383). Copyright 1995 American Chemical Society.
57Fe Mössbauer, EPR, XAS, and VTVH-MCD spectroscopies have been used to characterize the Ti3+-citrate generated FePsred.70,185,289,355 The 57Fe Mössbauer spectra of this species was simulated with four doublets with an equivalent isomer shift of δ = 0.68 mm/s and varying quadrupole splitting (ΔEQ = 1.25, 1.40, 1.75, and 3.08 mm/s), all consistent with the presence of 4Fe2+ and in agreement with more recent anomalous dispersion experiments.356 Measurement of the parallel-mode EPR revealed an axial signal with an inflection at g = 16.4, indicative of an S = 4 species; this assignment was further strengthened by analysis of low-field (0.01 T) magnetic Mössbauer measurements. VTVH-MCD measurements were also in agreement with these findings, where fits of the MCD magnetization curves recorded at 490 nm indicated an S = 4 species.185 Further investigation of the magnetic properties of FePsred revealed a negative, fully rhombic zero-field splitting (D = −0.75 cm–1, E/D = 0.33).70 EXAFS measurements revealed the [Fe4S4]0 cluster distorts significantly into a tetragonally compressed structure, in which fitting requires splitting of both the Fe–S and Fe–Fe shells into two scattering contributions.206,355
Interestingly, neither of the key spectroscopic features of FePsred discussed above (520 nm feature in UV–vis, g = 16.4 signal in parallel mode EPR) are observed when FePsred is generated under weaker reducing conditions. Early reports of the formation of FePsred came from microcoulometric measurements of the titration of FePred with MV (reporting a midpoint potential of −460 mV vs NHE) coupled with the disappearance of the S = 1/2 EPR signal of FePred.352 This relatively high reduction potential for a [Fe4S4]1+/0 redox couple, combined with the fact that DT (−500 to −660 mV vs NHE, depending on the exact conditions385) does not generate FePsred, appeared quite confounding. While a later UV–vis spectroelectrochemical study using Cr2+-EDTA as a mediator measured a −790 mV potential for the [4Fe–4S]1+/0 couple (see inset, Figure 24),383 it has since been shown that [4Fe–4S]0 can be generated in vitro by FldHQ from A. vinelandii (Em = −515 mV).49,386 These more softly reduced forms of FePsred were found to likely have an S = 0 ground state based on magnetic susceptibility measurements using the Evans NMR method,283 rather than the S = 4 found when produced using harsher reductants. This suggested that the strong chemical reductants used to measure the redox potential may induce a conformational change that modulates the redox potential. These suspicions have been further supported by DFT-based studies, which found that protein conformation determines both the final spin state of [4Fe–4S]0 in Fesred and the potential of the [4Fe–4S]1+/0 redox couple.357 Intriguingly, the reduction of FePred to FePsred was ∼20% inhibited by MgATP binding, and 40% by MgADP, which is not the case for the reduction of FePox to FePred.283
The discovery of FePsred implied that FeP could transfer two electrons to the catalytic subunit of nitrogenase upon hydrolysis of two molecules of ATP,352 and therefore only 4 FeP cycles would be necessary for the reduction of one N2 molecule by N2ase, in contrast with the eight cycles required by the LT cycle (Figure 2). This hypothesis is supported by an ATP/e– ratio of 1:1 when Ti3+-citrate or FldHQ is used as a reductant with various MoFe:Fe ratios, whereas it is always above 2 with DT.386 This ratio is closer to in vivo studies of whole cells of Azotobacter chroococcum, which have claimed as little as 4–5 ATP are necessary to fix one molecule of N2.387 However, more recent studies have demonstrated ATP/e– ratios of ∼2 for reduction of a variety of substrates by Mo N2ase.349 FldHQ is only capable of single electron donation (FldHQ → FldSQ, SQ = semiquinone) under the experimental conditions in which FePsred is formed, suggesting that FeP would require binding of two FldHQ for complete reduction of the cluster.49,289 As nucleotide binding may lower the reduction potential of the [4Fe–4S]1+/0 redox couple, it is possible formation of FePsred via FldHQ is only possible in vivo under high-activity conditions, i.e., low MoFe/Fe and ADP/ATP ratios and high concentrations of FldHQ.283
4.2. The P-Cluster
57Fe Mössbauer spectroscopy has been instrumental in forming our current understanding of both FeMoco and the P-cluster of MoFe. The first studies to apply this technique to Mo N2ase were performed by Lang and co-workers as early as 1970, who studied 57Fe enriched MoFe protein from Klebsiella pneumoniae in the presence/absence of an applied magnetic field.388,389 On the basis of the differences between spectra in the presence/absence of reductant, these early studies correctly deduced that the Fe atoms in the protein form spin-coupled pairs. Two Mössbauer signals named “M4” and “M5”, which displayed isomer shifts of δ = 0.65 mm/s, ΔE = 3.05 mm/s and δ = 0.6 mm/s, ΔE = 0.8 mm/s, were described as “Fe2+with unusually strong π-bonding” and “Coupled Fe3+pairs with zero net spin. Possibly Fe2+”, respectively. These signals were found to remain during turnover in the presence of N2 but were replaced by several other signals (M1 and M2) in the presence of thionine.388,389 In retrospect, these two sets of signals would later be found to correspond to the resting and two-electron oxidized forms of the P-cluster (P0 and P2+, but commonly referred to as PN and POX).
Shortly following these initial studies, Münck, Zimmerman, and co-workers applied 57Fe Mössbauer and EPR spectroscopies in a series of cross-correlative investigations of MoFe extracted from Azotobacter vinelandii.390,391 Their approach allowed them not only to establish that 30(±2) Fe were present in MoFe but also to deconvolute the 57Fe Mössbauer spectra of resting MoFe into three main components with different relative total absorptions: (1) D/∼42%/S = 0, (2) Fe2+/∼13%/S = 0 or integer, and (3) M/∼40%/S = 3/2 where the D label was given to the main diamagnetic component, Fe2+ to the reduced rubredoxin-like signals (yet also diamagnetic), and M to the paramagnetic signals (now known as FeMoco), respectively. Spin-coupling was proposed as the only way to explain both the diamagnetism and a 3:1 ratio of D and Fe2+, which gave rise to the proposal of four diamagnetic [Fe4S4] clusters in MoFe. They were additionally responsible for dubbing these unique [Fe4S4] clusters as “P-clusters”, as they appeared to be directly bound to the protein (and hence “protein-clusters”).391
Further studies into the thionine oxidized forms of MoFe were performed, and it was found that four oxidations could occur before loss of the S = 3/2 EPR signal associated with FeMoco was observed. This behavior was attributed to the one-electron oxidation of each of the P-clusters, and the four-electron oxidized state of MoFe was labeled POX.391 While this state would theoretically generate a noninteger spin at each cluster and therefore an easily observed signal in the EPR spectrum, no new EPR signals were observed in POX, making this assignment a subject of debate over the next decade.392−395 VTVH-MCD studies performed by Johnson et al. on thionine-oxidized MoFe protein purported the spin state of POX to be consistent with either a S = 7/2 or S = 5/2 ground state,392 and magnetic susceptibility studies via NMR published by Orme-Johnson and co-workers found the effective magnetic moment of POX to be larger than the resting state of FeMoco, likely S = 5/2.393
In 1987, Münck, Orme-Johnson, and co-workers performed a series of 57Fe Mössbauer measurements using MoFe containing selectively 57Fe-enriched P-clusters (P[57Fe]M[56Fe]), allowing for more highly resolved spectra of the P-clusters to be obtained (Figure 25).394 This study showed that a component S, previously disregarded as a contaminant, both correlated with the other two signals assigned to the P-clusters (components D and Fe2+) and also disappeared upon oxidation. These new spectra revealed the ratio between components Fe2+:D:S as 4:10:2, forcing a revision of the assignment of the P-clusters as two types ([D3Fe2+] and [D2Fe2+S]) rather than four identical clusters.394 In the same year, Hagen and co-workers presented another proposal for the nature of the P-clusters based on the results of EPR studies using excess amounts of solid thionine to oxidize MoFe.395 In these additionally oxidized samples, new features at g = 10.4, 5.8, and 5.5 were observed using X-band EPR consistent with an S = 7/2 species. Further quantification of this species indicated a 1:1 ratio with FeMoco, at odds with the 2:1 stoichiometry of P:FeMoco suggested by Münck and Orme-Johnson.391,394 Two explanations were proposed to account for these observations: (1) there are two [Fe4S4] P-clusters in each half of the protein, both with S = 0, and oxidation of the P-clusters results in two types of oxidized P-clusters, or (2) each P-cluster consists of 8 Fe (S = 0), and that two-electron oxidation (via small amounts of thionine) produces P2+ with S = 0 (explaining the EPR-silence of POX).395 In this second explanation, use of solid thionine in sample preparation is proposed to result in continued oxidation of MoFe protein, producing EPR silent MOX and a new S = 7/2 signal from P3+. While the solid thionine experiments were repeated by Münck and Orme-Johnson,396 the disagreement over how to interpret the EPR and Mössbauer data and the actual nature of the P-clusters themselves, remained unresolved in the literature for several years. In 1992, Münck and Orme-Johnson performed non-Kramers integer-spin EPR on the thionine-oxidized POX sample, revealing an S = 3 or S = 4 non-Kramers system, and identifying POX as doubly oxidized P2+ (Figure 26, bottom).397 Furthermore, the P-clusters could now be firmly identified as two 8Fe-clusters per protein instead of four [Fe4S4] clusters, aided by the new 2.7398 and 2.2 Å399 crystal structures which proposed the P-cluster was formed from two [Fe4S4] clusters connected through two bridging cysteines. With the P-cluster established as an 8Fe cluster with S = 0 and the oxidation state of POX clarified as P2+, the S = 7/2 signal observed by Hagen and co-workers most likely corresponded with their proposed P3+ state.395
Figure 25.
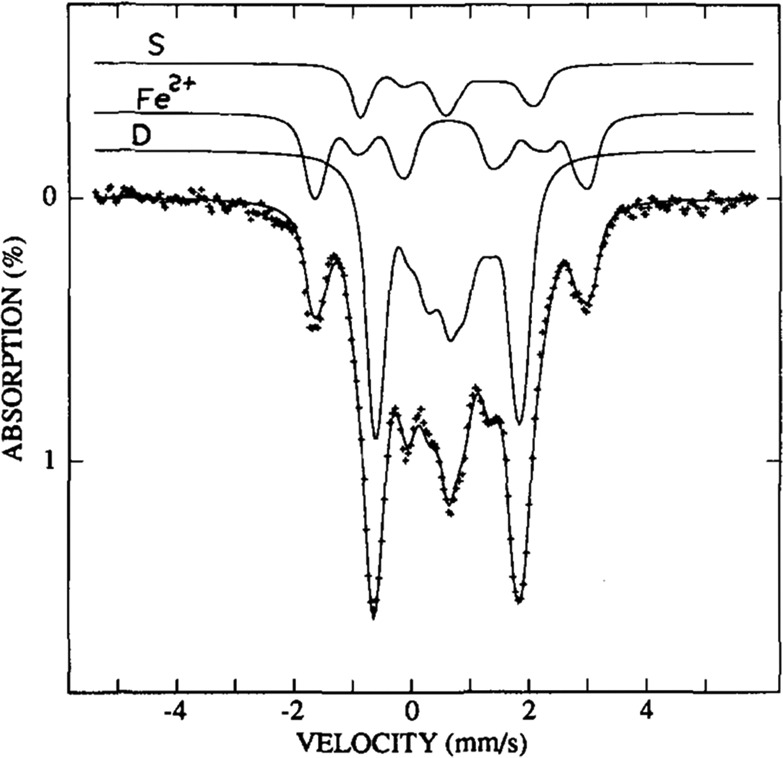
57Fe Mössbauer spectrum of selectively 57Fe-enriched MoFe (P[57Fe]M[56Fe]) recorded at 4.2 K, 6 T. The solid lines overlaid with the experimental data (dots) represents the sum of the spectral simulations of the individual fit components. Reproduced from with permission from ref (394). Copyright 1987 American Society for Biochemistry and Molecular Biology.
Figure 26.

(top) (a) Perpendicular mode X-band EPR of MoFe which has been oxidized using 2 equiv of thionine, with expansions emphasizing (b) the low-field, and (c) g ∼ 2 regions of the spectra.400 Spectra were collected at 15 K. (bottom) Low-field parallel mode X-band EPR spectra of MoFe titrated with thionine to a potential of −325 mV (vs SHE).397 Reproduced with permission from refs (397,400). Copyright 1993, 1992, respectively, American Chemical Society.
In 1993, separate works by Hales and Hagen reinvestigated the oxidized states of the P-cluster using EPR.400,401 Using again thionine as a reductant, Tittsworth and Hales elucidated at last the P1+ state, demonstrating it produces both S = 1/2 (g = [2.06, 1.95, 1.82]) and S = 5/2 (geff = [6.67, 5.30, ?]) signals (Figure 26, top).400 Meanwhile, Pierik and Hagen et al., employed a variety of redox dyes to measure the redox potentials between the P0, P2+, P3+, and a series of new irreversibly oxidized Psuperox species with possible spin states of S = 4, S = 7/2, S = 9/2, etc. (Figure 27).401
Figure 27.

Redox states of the P-cluster with observed spin states and redox potentials (vs NHE) indicated.401,402
In 1997, a higher resolution crystal structure of both the PN and POX (P2+) oxidation states of MoFe protein from A. vinelandii became available, clarifying the molecular structure of the P-cluster.403 The resting PN state was found to be a [Fe8S7] cluster (rather than [Fe8S8]) featuring a hexacoordinate sulfur that bridges two [Fe4S3] cubane-like clusters. Meanwhile, the P2+ structure features binding of the β-188Ser side chain to Fe6 with the backbone amide of α-88Cys bound to Fe5 (Figure 28). A more recent crystallographic study by Tezcan and co-workers of P2+ in MoFe extracted from Gluconacetobacter diazotrophicus (natively containing an Ala residue in place of the P-cluster-binding Ser) has found O-coordination from a Tyr residue to Fe8 (rather than the Fe6–Ser coordination found in A. vinelandii).404 The authors took this as a possible indication of the relevance of P2+ to catalysis and that an oxygenic ligand may act as a redox switch, and a recent mutation study of A. vinelandii revealed the presence of an oxygenic ligand is likely be crucial to the redox stability of the P-cluster.405
Figure 28.

Geometries of the P-cluster in PN, P1+m and P2+ states of MoFe extracted from A. vinelandii according to crystallographic studies (PDBs 3MIN, 6CDK, and 2MIN, respectively).403,406 Upon formation of P1+, Fe6 is coordinated by β-188Ser, while further oxidation to P2+ results in coordination of Fe5 by the amido backbone of α-88Cys.
While the spectroscopy of the P-cluster has been dominated by Mössbauer and EPR studies, several other methods have helped shape our current understanding of the properties of this redox center, including XAS, MCD, and theoretical studies.
Hodgson and co-workers conducted Fe K-edge XAS studies in 1998 on resting and two-electron oxidized forms of the apo-MoFe protein (lacking FeMoco).203 While a shift in the energetic position of the rising edge in XAS is traditionally seen as a good indicator of metal oxidation state, this was not observed when comparing PN and POX. The pre-edges and rising edges were mostly identical between the two redox states, with a small decrease in pre-edge intensity and an increase in white-line intensity (around 7126 eV) being observed when moving from POX→ PN. While the effect of redox state changes on the Fe K-edges of iron–sulfur compounds remains poorly understood, it is possible these changes are dependent on the mixed-valent nature of the Fe centers being oxidized or reduced.137,207,228
An MCD study by Hales and co-workers found the P1+ state could be stabilized in the ΔnifB-β-188Ser→Cys mutant of A. vinelandii.407 The MCD spectrum of this P1+ state was found to closely resemble the spectrum of [Fe4S4]1+ clusters, and it was hypothesized that the P-cluster could be viewed as two coupled 4Fe clusters, capable of donating either one or two electrons, depending on circumstances. Because the ΔnifB-β-188Cys mutant lacks an oxygenic ligand, it is unclear whether this mutant P1+ state resembles the native one. The later crystal structure of native P1+, solved for the first time in 2018, in fact demonstrates binding of β-188Ser to Fe6, and lacks the coordination of backbone amide nitrogen of α-88Cys to Fe5 seen in P2+(Figure 28).406
The first theoretical studies of the electronic structure of the P-cluster were performed by Mouesca and co-workers, focusing on reproducing 57Fe hyperfine couplings and spin states of the P2+ state.257 As most density functional theory studies of N2ase have focused on the FeMoco active site, very few have included the P-cluster. The study by Lancaster et al.,30 featured calculations of Fe VtC XES spectra of the P-cluster in order to understand interstitial atom effects on the spectra of FeMoco, leading to the interstitial carbide assignment. Similarly, 57Fe Mössbauer calculations of the P-cluster were performed where a comparison of the P-cluster and FeMoco isomer shifts was used to clarify the redox state of FeMoco.277 The unambiguous redox state assignment of the PN state via the Mössbauer isomer shifts thus proved a highly useful reference.
More recently, Ryde and co-workers performed QM/MM calculations investigating the P-cluster in redox states P0, P1+, P2+, and P3+, giving detailed insight into the protonation states and conformations of these redox states.408 An extensive exploration of the broken symmetry states of the P-cluster was undertaken, filling an important gap in the literature. A broken symmetry state, referred to as BSb11, was found to be lowest in energy and in best agreement with X-ray data.408 This state features an antiferromagnetic coupling arrangement of the local spins that is reminiscent of the BS7 arrangement in FeMoco.254
State-of-the-art multiconfigurational ab initio calculations have also been applied to the P-cluster by Chan and co-workers using the density matrix renormalization group (DMRG) approach.240 These calculations allow the direct calculation of the low-lying spin and electronic states of the P-cluster via a flexible multiconfigurational wave function (active space of 73–77 orbitals), avoiding the single-determinant BS- DFT approach where pure spin states are not accessible. The study gives a detailed description of the nature of the low-lying electronic states of the P0 state as well as the P1+ and P2+ states; however, the accuracy of the calculations is unfortunately unknown due to lack of dynamic correlation in these calculations. These DMRG calculations also are in disagreement with the DFT study by Ryde and co-workers regarding the nature of the lowest energy state of the P1+ and P2+ states.408
Much has been learned about the P-cluster over the course of nearly five decades of spectroscopic studies. The molecular structures of the early oxidized states are now established, while some uncertainty remains about the origin of the multiple spin states observed in the oxidized states P1+, P2+, and P3+ and the electronic structures that contribute to them. Future studies focusing on the electronic structure of these redox states will be important, and new theoretical methodology will likely be crucial to this goal. What also remains unclear is the precise nature of the redox processes of the P-cluster that occur during turnover conditions. According to the deficit-spending model, once FeP is bound, the P-cluster reduces FeMoco to form P1+ and E1 and is then promptly reduced back to PN by FeP.62 Is the P1+ state formed during these conditions the same as that encountered experimentally via EPR (showing mixture of S = 1/2 and S = 5/2 spin states) or X-ray diffraction (showing serine-binding to Fe6)? Additionally, the relevance of the P2+ state during N2ase catalysis is also unclear. As two electrons can be removed from PN at approximately the same potential, it seems conceivable that this could play a role during catalysis.
4.3. E0
In the resting state of MoFe (E0), the P-cluster is in an all-ferrous S = 0 state409 and FeMoco can be identified by a characteristic S = 3/2 EPR signal.390,410 Because its initial identification as an iron-rich molybdenum-containing cluster (initially with an 8:1 Fe:Mo ratio411), FeMoco has provided a puzzle for spectroscopists. Early EXAFS studies came up with a range of proposals for the possible structures of FeMoco,196−198 but it was not until the first crystal structure was reported in 1992 that the unprecedented complexity of this unique cofactor could be appreciated.412 Interestingly, the initial crystal structure revealed that the irons were all three coordinate, as at the time the resolution of the structure was not of sufficient to reveal the presence of a central atom. A decade later, a 1.16 Å resolution crystal structure revealed the presence of a light atom, however, a definitive assignment could not be made,212 and in the years that followed both experimentalists and theoreticians struggled to determine the identity of the central atom, with carbide, nitride, or oxide all being possible.93,129,130,164,413−423
In 2011, the full atomic composition of FeMoco was finally revealed by a combination of VtC XES and high-resolution crystallography, which provided experimental evidence for the presence of a central carbide (Figure 29).30,33 In 2014, Mo Kα-detected HERFD XAS revealed that the Mo is best described as Mo3+, indicating that the possible oxidation state distributions that could be consistent with the S = 3/2 EPR signal, were either [Mo3+6Fe3+1Fe2+]3–, [Mo3+4Fe3+3Fe2+]1–, or [Mo3+2Fe3+5Fe2+]1+.268 On the basis of 57Fe Mössbauer studies, combined with DFT calculations, a [Mo3+4Fe3+3Fe2+]1– distribution is favored.195,277 We also note that this assignment is favored by SpReAD analysis of crystals of MoFe protein and a QM/MM geometric comparison with the crystal structure.424,425 This was a particularly important finding, as knowledge of the total charge of the cluster is fundamental to any informed calculations of the mechanism. Additionally, this assignment shed light into why the oxidation state of Mo appears invariant during turnover when probed using either ENDOR or X-ray based methods,137,426 as redox activity at Mo during e– accumulation would require accessing Mo2+. It is of interest to note that in order to achieve the S = 3/2 total spin of FeMoco, the Mo is proposed to be in an unusual non-Hund electronic configuration in which Fe–Mo bond interactions stabilize this abnormal local spin configuration.268 Recently, this assignment was further supported by Mo L-edge XMCD studies of a [MoFe3S4] cubane cluster, in which experimental support for a non-Hund configuration at the Mo was provided for the first time.310
Figure 29.
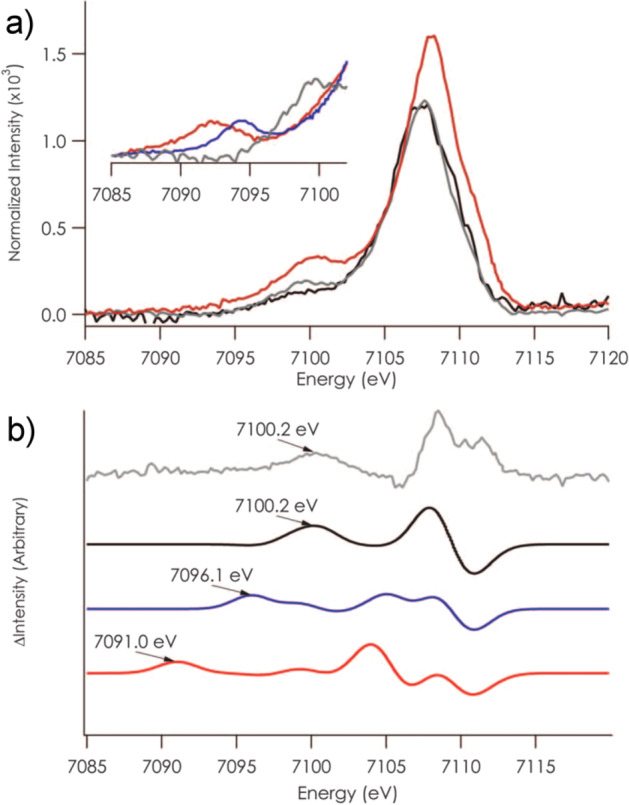
(a) comparison of the Fe VtC XES spectrum of extracted FeMoco (red), MoFe (gray), and P-cluster only ΔnifB (black). The inset region compares Fe2O3 (red), Fe3N (blue), and MoFe (gray). (b) Experimental difference VtC XES spectrum of MoFe–P-cluster (gray) compared with equivalent DFT calculated difference spectra using C4– (black), N3– (blue), and O2– (red) as the interstitial light atom of FeMoco. Reproduced with permission from ref (30). Copyright 2011 American Association for the Advancement of Science.
Despite knowledge of the total charge on the FeMoco cluster, a remaining open question has been the exact distribution of metal oxidation states within the cluster. Recently, Se Kα-HERFD XAS studies of selenium substituted MoFe protein provided experimental evidence for charge localization in FeMoco.220 Specifically, these studies showed that the S2B position (bridging Fe2 and Fe6) is electronically distinct from the S3A/5A position (bridging the Fe4–Fe5/Fe3–7). Through correlation with calculations, it is proposed that the Fe2–Fe6 pair is best described as an antiferromagnetically coupled diferric pair, while the Fe3/4/5/7 sites exhibit more localized ferrous character, with an average oxidation state of +2.25–2.5 for the Fe3/4/5/7 face. We point out that SpReAD studies have suggested that Fe1/3/7 are locally ferrous, with the remaining sites being more oxidized.425 On the basis of the Se HERFD XAS results and inspection of the crystal structure, it is suggested that additional electrostatic and hydrogen bonding interactions at the S3A and S5A bridge positions help stabilize the localization of charge.220 These findings thus suggest an important role of the protein environment in tuning the electronic structure of FeMoco. A QM/MM broken symmetry study reveals a more delocalized picture with mixed-valence delocalized pairs of the belt irons in the three lowest-lying electronic states.424
The pioneering theoretical studies on FeMoco by Noodleman and co-workers were the first to discuss in detail the complexity of the electronic structure of FeMoco by BS-DFT calculations and identified the many possible broken symmetry states in FeMoco (labeled from 1 through 10).254,255 Once an interstitial atom was included in the calculations, it became clear that the BS7 class of solutions (that maximize antiferromagnetic coupling) was lowest in energy according to multiple studies (regardless of interstitial atom and charge state assumed).30,130,422,427,428 A broken symmetry state labeled BS7–235 (Fe2, Fe3, and Fe5 being spin-down), features Fe6–Fe7 and Fe2–Fe3 as mixed-valence delocalized pairs and the geometry of this state is in best agreement with the crystal structure geometry and reproduces trends in the Mo–Fe and Fe–Fe distances. However, the calculations also predict the “spin isomeric” solutions BS7–346 and BS7–247 (where the mixed-valence pairs swap places) to be almost energetically equivalent (within 1 kcal/mol of each other). Thus, the possible presence of three low-lying electronic states in E0 (according to BS-DFT calculations) may complicate the question of localized vs delocalized nature on individual irons, as would vibronic coupling (currently not accounted for in calculations). Further, we emphasize that while BS-DFT has shown great utility in our understanding of FeMoco, it is clear that DFT methods are insufficient for properly describing the spin couplings in even simple mixed valent iron–sulfur dimers.168,169 Hence at present, the electronic complexity of FeMoco makes a rigorous theoretical description of the electronic structure and magnetic coupling prohibitive.
4.4. E1
The E1 state is proposed to arise from the first electron/proton transfer to the FeMoco cluster,64 and, like all other intermediates described by the Lowe–Thorneley cycle in Figure 2, cannot presently be generated as a pure species. However, significant populations of E1 can be generated during turnover under low electron flux. These conditions require both the absence of N2 and a large ratio of MoFe:FeP (with published reports ranging from 50:1 to 5:1).137,195,202,209 Large ratios of MoFe:FeP result in a slower rate of coordination of FeP to MoFe and, in turn, a slower flux of electrons to the FeMoco cluster. When appropriately balanced, the electron flux to FeMoco is slower than the rate of H2 production from E2, and as a result only the E0 and E1 states are significantly populated. While E1 itself is presently inaccessible to spin-selective techniques, such as EPR, due to its non-Kramers spin state, EPR can still be used to determine the ratio of E1 to E0 in a given turnover sample by quantitation of the remaining S = 3/2 signal of E0 relative to a resting state sample. In this way, contributions of E0 may be deconvoluted from a mixed species sample measured using a bulk spectroscopic technique (such as 57Fe Mössbauer, XAS, or XES) to generate “pure” spectra of E1. Beyond this point, bulk spectroscopic techniques still face challenges due to the large number of unique metal centers in the Mo N2ase enzyme (one Mo and 15 or 16 Fe33,429). Therefore, while changes in coordination, oxidation state, or spin state of Mo should be readily accessible when employing Mo-specific spectroscopic methods, any changes arising at a given Fe center may be obscured by a background of up to 15 additional Fe. This has effectively forced studies of E1 to push the boundaries of technique sensitivity and data quality.
In the earliest studies of this intermediate, Mo and Fe K-edge EXAFS reported significant contractions in the average Mo–Fe, Mo–O/N, and short Fe–Fe distances.202 Later, 57Fe Mössbauer spectroscopic studies of MoFe containing selectively 57Fe enriched FeMoco also observed small, but significant, changes in the observed average isomer shift upon reduction.195 Together, these studies implied that FeMoco was indeed the site of reduction/protonation in the E1 state and that the Mo site may undergo significant changes in its formation in terms of either oxidation state or coordination.
At the time of these studies, the resting E0 state was believed to contain Mo4+.195,430 Since this time, measurements of the E0 state using Mo Kα-HERFD and Mo L2,3-edge XAS have evidenced that Mo formally exists as Mo3+ in the resting state.219,268 Therefore, reduction of Mo when forming the E1 state would produce a formal Mo2+. More recent studies using Mo and Fe K-edge XAS and 57Fe Mössbauer have now demonstrated that E1 is the result of an Fe centered reduction137 and that Mo remains both invariant in valency and coordination in this state.137,209 The Mo and Fe K-edge EXAFS of E1 have also since been revised, demonstrating that the previously reported bond contractions were artifacts arising from an unphysical treatment of the data. Instead, the first coordination sphere of Mo remains effectively invariant, while a mild expansion of the average Fe–S distance is found in combination with a small contraction of the average short Fe–Fe distances (assigned as predominately arising from the FeMoco cluster); these changes are consistent with reduction based on comparison with numerous model complexes and other biological iron–sulfur clusters.60,203,346,356,431−439
Considering the large number of atoms involved in the FeMoco cluster (eight metal centers, nine sulfurs, a central carbide, not to mention coordinating homocitrate, histidine, and cysteine ligands) and the possibility of several total spin states (as well as 35 broken symmetry solutions), the active space for possible identities of the E1 state is extremely large. Several computational studies have undertaken the task of locating the site of protonation in E1,440,441 and the S2B site (Figure 1) has generally been found to be the most energetically favorable site of protonation (with S3A protonation disfavored by the protein environment). Further studies comparing the average changes in EXAFS-determined bond distances with those of a series of E1 computational models have also supported the S2B and S5A belt sulfide positions as the most likely candidates for protonation.209 This is perhaps not surprising, as some studies have proposed activation of both of these belt sulfide positions to occur in the E4(4H) state.121,239 BS-DFT studies have furthermore found E1 models with an MS = 2 BS state lowest in energy,209,440 tentatively suggesting a spin state of S = 2, and that the additional electron predominately resides on a relatively localized Fe in the Mo subcubane of the FeMoco cluster (in agreement with Fe K-edge XAS studies137).209
4.5. E4
The E4 intermediate, formed by the accumulation of 4[e–/H+] in MoFe, was first proposed by Lowe and Thorneley as the critical intermediate for binding of N2 and subsequent N–N bond activation (Figure 2).49,361 The half-integer spin state of this intermediate has made it susceptible to magnetic spectroscopic techniques, and in the 35 or so years since the proposal of its existence,64 EPR spectroscopy and its advanced counterpart, ENDOR spectroscopy, have played central roles in elucidating the chemical characteristics of E4.121,442 This has in turn provided invaluable insight into the nature of biological N2 activation. The E4 intermediate has been coined the “Janus” intermediate by Hoffman and co-workers, as it sits at the transition point.40 At this stage, nitrogenase may either return back to the E2 state via release of H2136,443 or it may continue forward to reduce N2 with an additional 4[e–/H+].
The S = 1/2 EPR spectrum of the E4 intermediate was first identified in the α-70Val→Ile mutant of (Azotobacter vinelandii) Mo N2ase (Figure 30).442 Prior to this study, the α-70Val residue of the MoFe protein had been targeted for mutations (α-70Val→Ala) to enlarge the substrate binding pocket at the Fe2/3/6/7 face of the FeMoco cluster (Figure 31), allowing for the reduction of larger alkyne substrates.89,444,445 The binding of large alkyne substrates to this multi-iron face of the cofactor naturally led to complementing experiments attempting to “close off” this substrate binding face. The α-70Val→Ile mutant was found to limit the reduction of all substrates at the catalytic cluster, with the exception of protons.442 A newly trapped intermediate formed under argon atmosphere during high electron flux turnover (using 1:4 MoFe:FeP) was readily identified by a unique S = 1/2 EPR signal. This same S = 1/2 intermediate was later observed in the wild-type protein also, supporting that subsequent spectroscopic studies of the α-70Val→Ile mutant have been representative of the wild-type intermediate.67
Figure 30.

X-band EPR spectra of the α-70Val→Ile MoFe variant in the resting (top) and turnover (bottom) states in the presence of an Ar atmosphere. Adapted with permission from ref (442). Copyright 2005 American Chemical Society.
Figure 31.
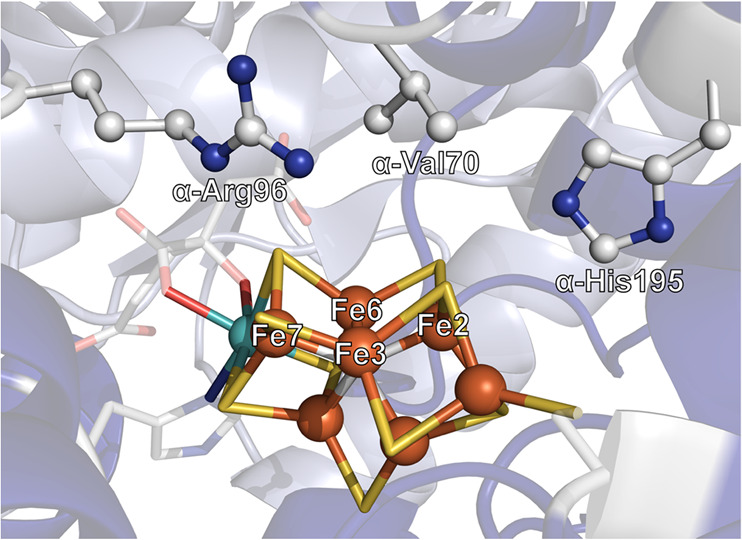
Depiction of the Fe2/3/6/7 face of FeMoco and the neighboring secondary coordination sphere amino acid side chains. Produced using the 3U7Q high resolution crystal structure of MoFe.33
While this new intermediate could be observed by EPR spectroscopy, its exact identity could not yet be assigned. The first 1H Q-band CW-ENDOR study of this intermediate revealed two strongly coupled hydrogen (H±) signals identified at the “single-crystal-like” positions of the ENDOR field-frequency pattern, Figure 32.442 The large 33 and 25 MHz 1H couplings observed are significantly larger than those previously found for the protons of acetylene when bound to Mo N2ase (18 MHz maximum).446 Through thorough analysis, the 1H hyperfine tensors were determined and recognized to be both nearly identical in their principal component values and near ideally rhombic in character. The resultant anisotropic dipolar component (T = A – aiso) of the hyperfine tensor was also nearly ideally rhombic (T = [t, 0, −t]), a case that had been predicted for bridging hydrides.447 Experimental support in the form of biomimetic hydride complexes has since followed120,331 and are discussed in greater detail in section 3.4. Most importantly, these results provided strong experimental support for the assignment of bridging hydride motifs in the E4 state. Given that the position of this intermediate on the LT cycle (Figure 2) is prior to N2 binding, it will be referred to as the E4(4H) state.
Figure 32.

Comparison of the 1H field-frequency pattern of the α-70Val→Ile turnover intermediate collected by standard CW RF swept ENDOR spectroscopy (left) and field-modulated CW stochastic ENDOR spectroscopy (right). Reproduced with permission from refs (121, 442) Copyright 2005, 2019, respectively, American Chemical Society.
Most recently, the 1H ENDOR of the E4(4H) intermediate has been revisited121 using field-modulated stochastic CW-ENDOR spectroscopy to provide extremely high resolution data.107,121Figure 32 highlights the comparison between the original Q-band CW-RF swept ENDOR and the increased resolution of the 1H ENDOR spectra achieved by the claimed improved instrumentation.121 By randomly hopping the frequency of the “pulsed” RF field in the CW-ENDOR experiment, the stochastic method rids the ENDOR spectrum of nuclear relaxation sweep artifacts that broaden and possibly distort the ENDOR lineshapes (Figure 6), as discussed in section 2.1.110
The newly acquired 1H ENDOR data definitively demonstrates that each of the hydrides have rhombic dipolar tensors, consistent with the bridging hydride assignment made previously.5 Furthermore, the higher resolution of the data has allowed for more accurate simulations of the 2D field-frequency ENDOR pattern, which has provided more precise information regarding the relative orientations of the hyperfine tensors (Figure 32). Specifically, the dipolar tensors of the hydrides are coaxial but permuted by a rotation that gives them orthogonal character. This reveals crucial information regarding their spatial arrangement and places key constraints on possible candidates for the identity of the intermediate. Using complementary BS-DFT calculations on models of various possible E4 intermediates, Hoffman and co-workers have investigated the possibility of both bridging and terminal hydride combinations with protonation at various sulfur sites.121 While several of these models were calculated to be too close in energy to clearly distinguish (Figure 33), the experimentally determined hyperfine tensors of E4 have served as critical criteria in evaluating model feasibility, allowing those involving protonation of the interstitial carbide to be confidently disregarded. Evaluation of the point dipolar hyperfine tensor for an E4(4H) intermediate with two bridging hydrides parallel to one another on the Fe2/3/6/7 face of FeMoco best reproduce the experimental data (Figure 33).121 Questions remain about the validity of the point-dipole model for such a complex system as hydride-bound FeMoco.
Figure 33.
Lowest-energy structures of the Janus intermediate as seen in recent density functional theory computations from Hoeke and co-workers.121 Relative energies ΔE are provided in kJ/mol calculated at the BP86 level, as reported in ref (237), and hydride bond distances are indicated in Å. Experimental 1H ENDOR spectroscopy only supports bridging hydride structures as shown in E4(4H)(a) and E4(4H)(c). Evaluation of 1H hyperfine tensors favor parallel bridging hydrides (c). Reproduced with permission from ref (121). Copyright 2019 American Chemical Society.
4.5.1. Classification of the Dihydride Intermediate in the LT Scheme
While the trapped intermediate with two bound hydrides described in section 4.5 has been assigned to the E4 state, the initial studies could not eliminate the possibility of it belonging to the E2 state (Figure 2), which also has a noninteger spin state. In 2007, a fundamental kinetic and EPR spectroscopic study by Lukoyanov et al. was able to formally assign the S = 1/2 EPR signal of the dihydride trapped intermediate to the E4 state by exploiting that E4 does not revert to E0 by microreversibility but rather by a side reaction which yields two molecules of H2.136 Annealing of the dihydride intermediate revealed the immediate loss of the S = 1/2 signal and the appearance of two new S = 3/2 signals which correlated with those observed during previous EPR experiments of turnover Mo N2ase (Figure 34).448 Further annealing resulted in the appearance of the S = 3/2 signal of the E0 state, demonstrating that relaxation of the dihydride species proceeded through a novel S = 3/2 intermediate, assigned as E2. Additionally, identical experiments performed in D2O yielded significant kinetic isotope effects for the production H2. Importantly, this cryoannealing study provided strong evidence that the dihydride species was the result of the accumulation of 4[e–/H+] equivalents, and therefore the E4 state.
Figure 34.
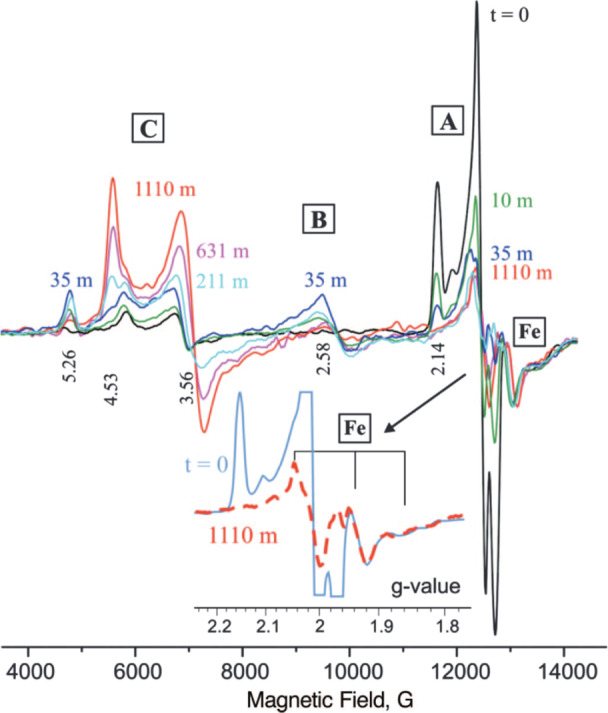
Q-band EPR spectra collected of α-70Val→Ile MoFe freeze-quenched during turnover (1.25:1 MoFe:FeP) and subsequently cryoannealed for the indicated periods of time at 253 K. The inset is an expansion of the g = 2 region showing the unchanged regions of the FeP S = 1/2 signal and the amount of reduced Fe-protein is indeed unchanging during step-annealing. Signal A corresponds to E4, B to E2, and C to resting state E0. Reproduced with permission from ref (136). Copyright 2007 National Academy of Sciences.
4.5.2. Electronics of E4(4H)
4.5.2.1. 95Mo ENDOR
The 95Mo ENDOR of the E0 resting state MoFe exhibits a small 95Mo hyperfine coupling (Figure 35). Measured at the low-field signal (g1′ = 4.5), the observed hyperfine splitting of α-70Val→Ile variant is A′ = 9.96 MHz. However, to make one-to-one comparisons with the E4 state, a fictitious S′ = 1/2 must be corrected for the true S = 3/2 spin state: A = (ge/g1′)A′. With this correction, the resulting isotropic 95Mo coupling is ∼7 MHz for the E0 state of the α-70Val→Ile Mo N2ase variant. It is notable that this is larger than the ∼6 MHz isotropic coupling observed for the wild-type Mo N2ase,430 indicating that there are some structural and/or electronic perturbations by the mutation detectable at the Mo center.426 However, this is not large enough to be affiliated with a change in oxidation state, unsurprisingly as reduction of FeMoco at Mo would imply the formation of Mo2+.
Figure 35.
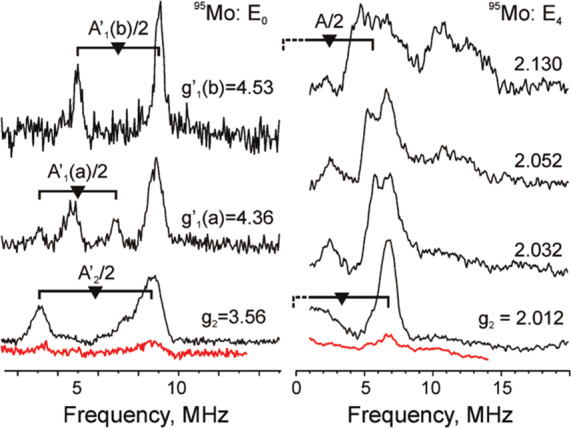
Davies 95Mo-ENDOR spectra of resting state (left) and CW 95Mo-ENDOR of E4 (right) for 95Mo-enriched (black) and unenriched (red) α-70Val→Ile MoFe. Reproduced with permission from ref (426). Copyright 2010 American Chemical Society.
The E4(4H) intermediate of the α-70Val→Ile N2ase variant exhibits an isotropic coupling of 4 MHz. The further decrease in isotropic hyperfine coupling is not supportive of reduction of the Mo center. Hoffman and co-workers have argued that the effective spin projection coefficient (K) of the Mo center is too small to be involved based on the assumption that the intrinsic isotropic hyperfine coupling for the Mo centers is aiso ≥ 100 MHz.426 However, the more recently assigned local S = 1/2 spin configuration of the Mo3+ center268 may complicate such simple spin-projection coefficient estimates. Given that no other available Mo3+ hyperfine measurements exist, intrinsic hyperfine couplings may not be accurately estimated, especially given the unusual proposed electronic structure of FeMoco.
While Hoffman and co-workers have concluded from the 95Mo ENDOR that Mo is not a site for hydride binding,426 further investigation of the molybdenum hyperfine couplings and electronic structure is still required to fully understand its small magnitude and to calibrate the relative scale of the observed changes in hyperfine couplings. While the 95Mo ENDOR seems to rule out reduction at Mo in E4(4H), it does show that the Mo site is somewhat sensitive to changes of the FeMoco cluster during turnover, perhaps through hydrogen bonding interactions with homocitrate.426,430
4.5.2.2. 57Fe ENDOR
Hoffman and co-workers have previously characterized the 57Fe hyperfine couplings of the E0 state of nitrogenase, along with various substrate-bound forms.101,430,449−451 These initial studies established an “electron inventory” methodology101,451 that can be used to determine the redox level of FeMoco,451 and Hoffman and co-workers further applied these methods to E4(4H).117
The complex ENDOR spectra collected for the E4(4H) state presented up to seven 57Fe hyperfine doublets that needed to be individually assigned (Figure 36). TRIPLE ENDOR, a “pump–probe” technique, was used to deconvolute many of the overlapping signals by assigning ν± transition pairs from TRIPLE difference spectra (a subtraction of “pump on” – “pump–off” spectra). While Davies pulsed ENDOR and TRIPLE ENDOR were able to identify five of the seven irons of FeMoco, these techniques were unable to yield the absolute sign of the hyperfine constants, which is necessary for the sum of the isotropic hyperfine coupling constants to be accurately determined. Therefore, the PESTRE technique114 briefly discussed in section 2.1, was employed to individually select 57Fe ν+ or ν– transitions, yielding the sign of the hyperfine by monitoring the electron–nuclear response in a multisequence protocol. This protocol allowed for the signs of 57Fe hyperfine constants for five of the seven observed Fe to be determined. Further use of an extension of the PESTRE technique, raw data (RD)-PESTRE was able to reveal that two irons possess nearly identical hyperfine couplings but opposite signs117 (Figure 36), bringing the total number of individual irons assigned in the E4(4H) state to six out of seven. Pairing the magnitude of the hyperfine couplings and the absolute hyperfine sign determined by the PESTRE technique, the most complete electronic picture of an isolated nitrogenase intermediate is obtained.
Figure 36.

(a) Triple Davies ENDOR spectra (top) with the 9.60 MHz RF “pump probe” on and off. The difference of the “pump off” and “pump on” spectra is shown at the bottom where the pump frequency is a negative peak and corresponding ν+/– features are positive. (b) Random-hop Davies pulsed (bottom) 57Fe ENDOR spectra collected at g = 2.01 for E4(4H) trapped in the α-70Val→Ile mutant. Individual peaks are labeled A–H in the Davies spectrum. Goalposts indicate 2ν (∼3.4–3.5 MHz) and the positions of the complementary oppositely signed transition pairs; color indicates the sign of the hyperfine coupling as either positive (red) or negative (blue). (c) PESTRE traces of the ν+ (trace A) and the ν– (trace C) of a single 57Fe ENDOR doublet. The sequence axis exhibits a schematic of the PESTRE protocol showing Stage I (RF off, BSL); Stage II (RF on, ENDOR signal); Stage III (RF off, DRL). (d) RD-PESTRE technique applied to determine the signs of hyperfine couplings at g = 2.01. An expanded region of the Davies spectrum is shown (right), along with the difference between dynamic reference level (DRL) and baseline (BSL) spin echoes, referred to as DRLδ (bottom). Reproduced with permission from ref (117). Copyright 2011 American Chemical Society.
With the assignment that the Mo center is not further reduced at the E4(4H) state, a complete “electron inventory” of the four reducing electrons is accounted for in the assignment of two bridging hydrides. Because these reducing equivalents at the E4(4H) state are stored in two hydrides, the FeMoco metallic core maintains the same redox level as E0. In fact, the isotropic 57Fe couplings observed for E4 are remarkably similar to the E0 state (MN) as characterized by Mössbauer spectroscopy.195 This raises an interesting question regarding the location of these hydrides on the cluster and the sensitivity of the metal-ion hyperfine couplings in determining hydride binding locations. The small reduction in the 95Mo hyperfine coupling from 7 to 4 MHz has been deemed too small to support hydride binding; however, the relative K cannot be well established without thorough 95Mo3+ hyperfine measurements of the unique electronic environment within FeMoco and appropriate model clusters. Furthermore, no dramatic changes are observed in the individual 57Fe couplings, meaning these are also not particularly diagnostic for hydride binding. In our view, the selective application of this logic could dismiss a possible role of the Mo center during catalysis, albeit the site directed mutagenesis performed by Dean and crystal structures by both Einsle and Rees strongly support that the Fe2/3/6/7 face is the site of substrate binding.89,442,444,445,452−454
4.5.3. Reductive Elimination and the “Janus” Intermediate
The limiting stoichiometry of biological nitrogen fixation via Mo N2ase has long required an additional molecule of H2 to be produced in addition to two molecules of ammonia.64 Whether this was an experimental limitation of the assays employed or an integral part of the mechanism of N2 activation by FeMoco was a subject of intense study spanning several decades.11,40 It has now been demonstrated that production of one H2 per two NH3 during turnover (and thus the limiting 8e– stoichiometry of Figure 2) is the result of the reductive elimination of the two bridging hydride species in the E4 state upon binding of N2, forming the E4(2N) intermediate (Figure 2).67 The proposed process has been extensively discussed and reviewed elsewhere.11,40,455 Briefly, upon binding of N2 the two bound hydrides of E4 terminate to form one molecule of H2, taking with it 2 of the 4 accumulated reducing equivalents. The remaining 2 reducing equivalents stay on the cluster and are now available to activate N2. This may occur through the formal reduction of a single iron site, e.g., Fe2+ → Fe0, or through two iron sites to bind and activate N2. The remaining 2 proton equivalents are still present and available to form a possible N2H2 substrate-bound intermediate at the E4 stage: E4(2N2H). This state has reversibility, where increases in the partial pressure of H2 release N2, producing the forward “reductive elimination” and reverse “oxidation addition” pathway (Figure 37).
Figure 37.
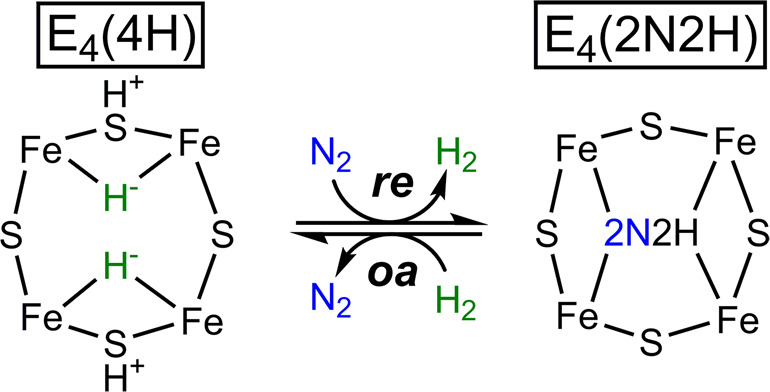
Representation of the E4 intermediates observed during turnover, only showing the Fe2/3/6/7 face of FeMoco to demonstrate their relationship through reductive elimination (re), and oxidative addition (oa).
4.5.4. Identifying the E4(2N2H) Intermediate
Upon turnover of the wild-type protein under variable partial pressures of N2 and H2, the reductive elimination step itself may be identified and studied. Wild-type Mo N2ase trapped during turnover with very low partial pressures of N2 yields majority E4(4H), while increased N2 partial pressures produce a new intermediate, also with an S = 1/2 signal (g = [2.09, 1.99, 1.97]) (Figure 38).67,456,457 By systematically varying the partial pressures of N2 and H2, Hoffman and co-workers clearly demonstrate that an equilibrium between the E4(4H) and a new “E4(2N2H)” states exists.458 This intermediate has a similar g-tensor to the hydrazine- and diazene-derived reaction intermediate of N2ase, as previously characterized by 15N and 1H ENDOR spectroscopy.459 This state shows a single 15N ENDOR signal but unlike the hydrazine/diazene reaction intermediates, no exchangeable 1H signals were found. E4(2N2H) was also found to relax back to E0 via the E2 intermediate step.
Figure 38.
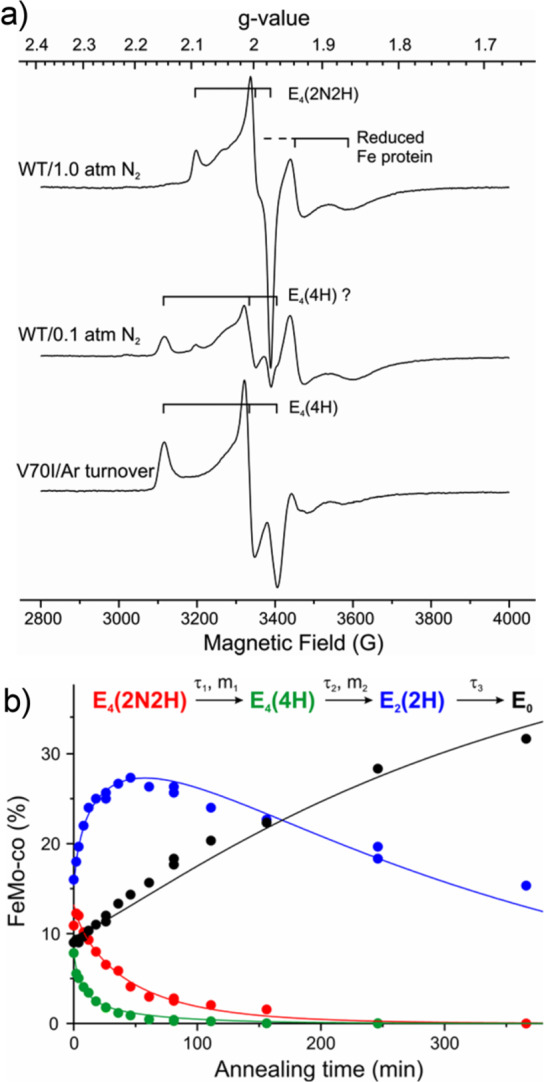
(a) Turnover of wild-type nitrogenase under 1 atm N2 and 0.1 atm N2, top and middle, respectively, compared to turnover of the α-70Val→Ile variant (bottom). The 0.1 atm N2 wild-type and α-70Val→Ile variant turnover samples exhibit the same E4(4H) signal corresponding to an E4 intermediate without N2 binding. Under 1.0 atm N2, the E4(4H) signal is absent and the signal for the E4(2N2H) is observed. (b) Time course kinetics of the observed EPR signals for cryoannealing of an E4 trapped turnover sample of wild-type nitrogenase. Incorporation of the E4(2N2H) intermediate signal to the kinetics shows it is coupled to the E4(4H) state and an equilibrium exists between the two E4 states. Reproduced with permission from ref (67). Copyright 2016 American Chemical Society.
On the basis of these studies, it was proposed that oxidative addition of H2 to the E4(2N2H) intermediate drives the equilibrium back to E4(4H). Further investigations of the kinetics found the loss of E4(2N2H) was coupled with the formation of E4(4H) (Figure 38), confirming the reversibility of the two states and supporting the reductive elimination/oxidative addition mechanisms. Further analysis of this equilibrium revealed that the reductive elimination step is near thermoneutral (ΔG0 = −2 kcal/mol) compared to the highly endergonic process of dinitrogen hydrogenation in the gas phase (ΔG0 = +50 kcal/mol).67
4.5.5. Photoinduced Reductive Elimination from E4(4H)
Spectroscopic support of both the thermodynamically and kinetically reversible reductive elimination of H2 from the E4 state of nitrogenase came in the form of in situ photolysis measurements of H2 reductive elimination.460It is well established that photolysis of mononuclear transition metal cis-dihydrides results in a reduction in the metal’s oxidation state,461−465 as suggested in the reductive elimination mechanism.67,460 The in situ photolysis of the E4 state at 12 K with 450 nm light results in a dramatic loss of the E4(4H) state (g = 2.15, 2.007, 1.965]) and the growth of a new species (g = [2.098, 2.0, 1.956]) (Figure 39), which we will refer to as E4(2H)*.460,466 The absence of low-field, high-spin features upon photolysis did not support an electronic structure similar to the S = 3/2 spin states of E2 (section 4.6) or E0, implying that the photolyzed product had not relaxed to one of these states. Q-band 1H stochastic CW-ENDOR measurements confirmed the reductive elimination of the hydrides by the significant 3-fold decrease of the strongly coupled 1H signals (Figure 39). Only a decrease in the signals arising from E4(4H) was observed, and the absence of any new signals implied that E4(2H)* was not a new hydride isomer or a rotationally hindered bound H2 molecule.460 While the latter will yield an ENDOR response at 2 K,118 a freely rotating H2 molecule will not due to the proton exchange of the rotation and the antisymmetric nuclear wave function of the 1-D rotor problem for a set of the identical (1H) protons. However, D2 substitution yields a symmetric nuclear wave function for the same 1-D rotor problem and therefore an ENDOR signal.467 The preparation of the E4 intermediate in D2O solvent yields D2 upon reductive elimination, even by photolysis (albeit with a significant kinetic isotope effect).460 However, no new 2H ENDOR signals are observed either, further indicating that a molecule of H2 or D2 is not bound to E4(2H)*. Subsequent cryoannealing measurements demonstrated that E4(4H) had lost a molecule of H2 through reductive elimination, and the remaining cluster is doubly reduced and contains the two protons from the original four. The E4 state could be regenerated by the oxidative addition of H2 to the cryoannealed photolysis E4(2H)* state at temperatures above 175 K.460
Figure 39.
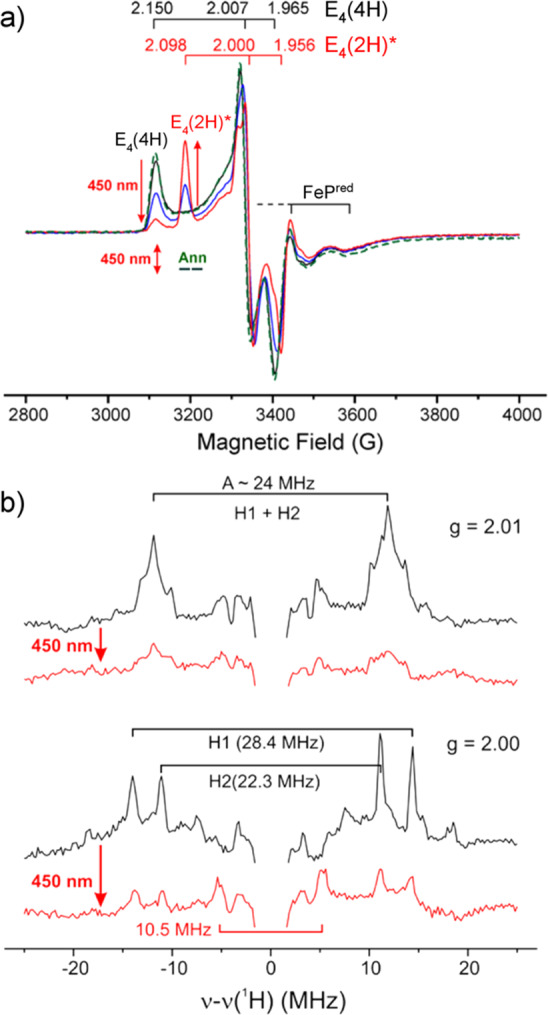
(a) X-band CW EPR spectra of in situ photolysis of E4(4H) trapped α-70Val→Ile MoFe, showing the rise of a new photolysis product, S, or also called E4(2H)* and the return to the E4(4H) state upon cryoannealing at 217 K for 2 min (Ann, green dashed line). (b) 1H CW stochastic ENDOR spectra of E4(4H) trapped nitrogenase before and after photolysis.460 Reproduced with permission from refs (117, 460). Copyright 2016 American Chemical Society.
It is important to note that while Hoffman and co-workers favor a parallel dihydride like structure of the E4 state,121 there is no precedent for parallel transition metal bound hydrides to undergo reductive elimination through either thermal or photochemical means. This makes the E4 state of nitrogenase a unique case, unsupported by current organometallic chemistry. As Hoffman and co-workers have previously suggested,121 it is entirely possible that the E4 structure elucidated at 2 K represents a single dihydride confirmation and that multiple confirmations are thermally accessible at ambient temperatures.
Subsequent kinetic analysis of the photolysis-cryoannealing experiment revealed a significant lag phase for the generation of the E4(2H)* state, implying the occurrence of one or more intermediate species and thus that photoinduced reductive elimination and release of H2 proceeds in a multistep fashion.466,468 Low temperature photolysis and cryoannealing experiments revealed a new intermediate (referred to as “B”) with a distinct S = 1/2 spectrum exhibiting significantly less g-anisotropy than either the E4 or E4(2H)* states (Figure 40). Intermediate B also appeared photoactive, and prolonged photolysis results in accumulation of the E4(2H)* state.
Figure 40.
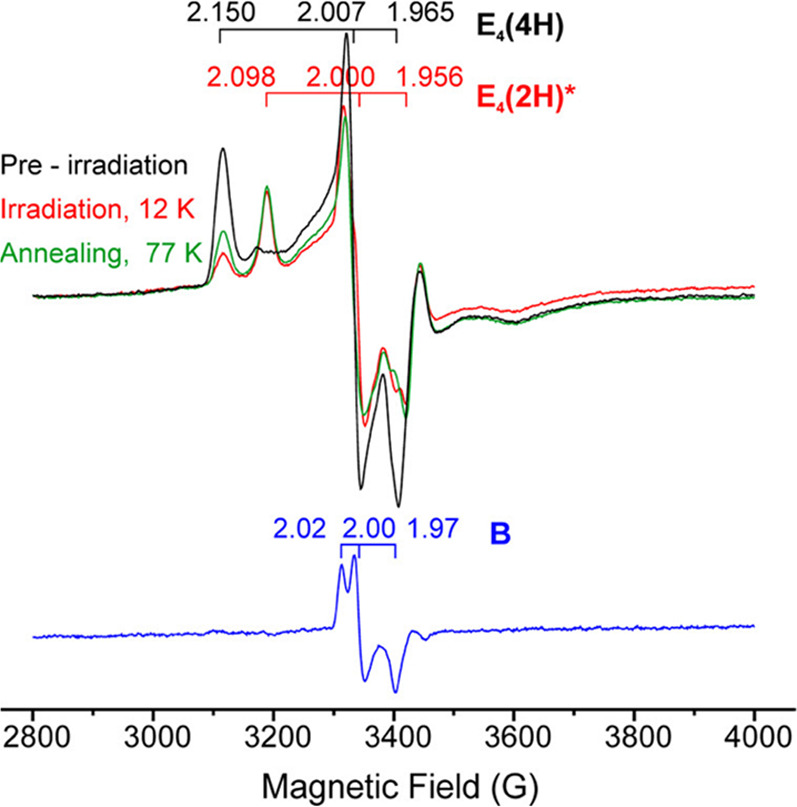
X-Band CW EPR spectra of the in situ photolysis of E4(4H) (black) to E4(2H)* (red) in the α-70Val→Ile MoFe variant. Partial recovery of E4(4H) at 77 K (green) is caused by the relaxation of the minor intermediate referred to as B, visualized by the subtraction–elimination spectrum (blue) generated from a combination of the three other spectra. Reproduced with permission from ref (466). Copyright 2017 American Chemical Society.
With support from DFT calculations, intermediate B has been proposed as an H2-bound complex that forms on the ground potential energy surface from the reductive elimination of two hydrides to generate H2 and therefore has also been referred to as E4(H2;2H).466,468 Although intermediate B possesses a unique EPR signal and is of significant interest for further characterization, the maximum 20% yield relative to E4(4H) and complete overlap of its EPR spectrum with those of E4(4H) and E4(2H)* has precluded its selective characterization by techniques such as ENDOR. Although the assignment of B as a H2-bound complex is supported by its photolability, there is presently no direct spectroscopic evidence to support its assignment as E4(H2;2H). The characterization of this putative H2-bound intermediate would be the “Denali”469 of ENDOR experiments, which some have called the “Everest of enzymes”.39
Overall, these photolysis studies have resulted in a proposed ground-state potential energy surface (Figure 41) for the N2/H2 equilibrium described in section 4.5.3 and Figure 37. On this surface, reductive elimination of H2 from E4(4H) results in the formation of E4(2H:H2) (intermediate B), which is unable to release H2 due to the unfavorable energy of E4(2H)*. However, upon binding of N2, release of H2 from E4(2H:H2) becomes energetically favorable, resulting in formation of the E4(2N2H) intermediate. The E4(4H) + N2 ↔ E4(2N2H) + H2 equilibrium is a thermally neutral process, in which N2 binds in a single, reversible step along with reductive elimination of H2. If FeMoco were capable of thermally releasing H2 by reductive elimination, then it should also undergo oxidative addition in the presence of D2/absence of N2, which has not been observed.49
Figure 41.

A putative ground-state potential energy diagram of the proposed intermediates resulting from photoinduced reductive elimination with and without N2.449 Transient EPR studies of the photolysis process suggest isomerization at an early stage into a new conformer, which has been interpreted as Fe–S bond cleavage (detailed by Lukoyanov et al. in ref (468). The relative positions of H+ on S in E4(4H), E4(H2;2H), and E4(2H)* are unknown.
4.6. E2
While E2 precedes the E4 intermediate, the E2 state was not spectroscopically established until several years following E4.136,443 Because of the continuous delivery of electrons and protons to the cofactor, trapping of the E2 intermediate in large quantities following two delivered equivalents of [e–/H+] has not been feasible. However, through trapping the S = 1/2 E4 intermediate (g = [2.14, 2.00, 1.96]), the E2 intermediate could be observed by annealing of E4 back to E0 through the appearance of two unique high-spin S = 3/2 EPR signals, referred to as 1b and 1c, at g(1b) = [4.21, 3.76, ?] and g(1c) = [4.69, ∼3.20, ?] (Figure 42).416 Kinetic traces of these EPR signals supported an E4(4H) (S = 1/2) → E2 (S = 3/2) → E0 (S = 3/2) relaxation process, and a strong kinetic isotope effect was found when repeated in the presence of D2O.
Figure 42.
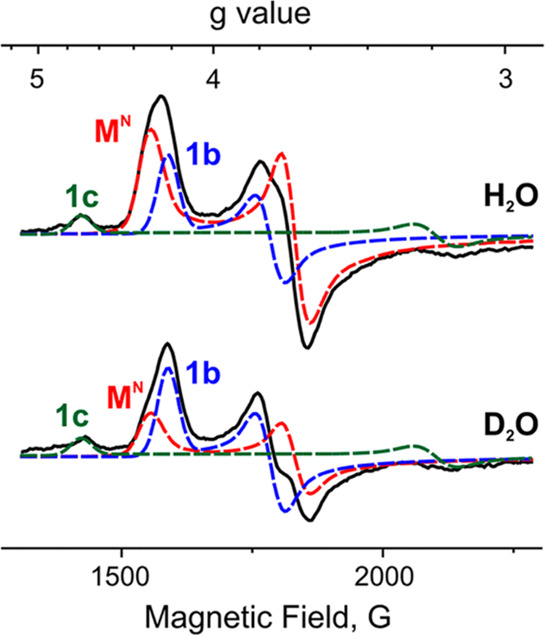
Low-field X-band EPR spectra of Mo N2ase trapped during high-flux (1.2:1 MoFe:FeP) turnover conditions under Ar in H2O (top) and D2O (bottom) buffer solutions. Spectra collected at 3.8 K. Reproduced with permission from ref (443). Copyright 2014 American Chemical Society.
Although earlier observations of the 1b and 1c EPR signals of reduced FeMoco were made in samples trapped during turnover, there was significant difficulty in assigning individual signals to specific intermediate states due to a lack of understanding of the kinetics of the reaction.448 Because of the large accumulation of 1b and possibly a misunderstanding of the rates, 1b was attributed to E3, which must formally be an integer-spin state.448
Though the E2 state has accumulated 2 equiv of [e–/H+], it is unknown directly from the EPR signal whether it possesses a hydride or not. 1,2H ENDOR spectroscopy technically can directly characterizing bound hydrides, but the relatively low accumulations of the E2 intermediate signals 1b and 1c have made ENDOR spectroscopic approaches challenging. Recognizing the photolability of a coordinated hydride to FeMoco (section 4.5), Hoffman and co-workers monitored the transformation of the various EPR signals of the E2 state during photolysis and subsequent cryoannealing.470 In situ photolysis led to an observed decrease of the 1b EPR signal and an increase in an EPR signal identical to 1c (called 1c*), making the conversion 1b → 1c* a single kinetic step. Furthermore, this conversion exhibited a kinetic isotope effect of ∼3, suggesting that hydrogens were involved in the rate limiting step of the reaction. Cryoannealing of the photolyzed product found the photolytically generated 1c* to be stable up to 77 K, relaxing back to 1b around 145 K. However, the natively trapped 1c state requires temperatures ≳230 K to relax, much higher than those needed for the relaxation for 1c*. The methods of generating 1c and 1c* likely produce slight differences in their active-site structures, yielding different thermal relaxation properties. Ultimately, the ability of 1c/1c* to relax to 1b indicates that photolysis yields only a hydride isomer, and there is no hydride eliminated through a hydride protonation mechanism.
4.7. Beyond E4: Reactions of N2H4 and N2H2
Beyond the E4 state, considerable work remains. However, several intermediates beyond this state have been characterized through the use of hydrazine and diazenes as substrates. Early on, hydrazine (N2H4) was found to be a substrate for Mo N2ase.471 However, the slow rate of its reaction suggested it may be too bulky to access the active site. The hydrazine to ammonia reaction is a simpler two-electron reduction than the 8-electron reduction of dinitrogen to ammonia. Hydrazine has additionally been discovered as a product of Mo N2ase under N2 turnover conditions if the reaction is acid-quenched472 and has been discovered as a minor reaction product for V N2ase.473,474 Together, these observations suggest that hydrazine could be a reaction intermediate on the N2 reduction pathway to ammonia and that hydrazine as a substrate might enter the latter phase of the N2 reduction cycle according to the proposed alternating pathway (where each nitrogen atom of dinitrogen is protonated in an alternating fashion, Figure 14).
Substituted diazenes such as methyldiazene and dimethyldiazene have also been revealed as substrates of Mo N2ase,456,475 and diazene itself has been proposed to be a possible reaction intermediate in the alternating mechanism of N2 reduction.11 When N2ase is frozen under N2 turnover conditions, an apparent EPR-silent state has often been found, creating difficulties for Kramers type EPR experiments.89 Hoffman and co-workers have thus explored trapping EPR-active intermediates using hydrazine and substituted diazenes as substrates as a way of indirectly studying N2 reaction intermediates. As these substrates react only slowly with the native form of the enzyme, mutant forms of the enzyme have been employed. As mentioned in section 4.5, the α-70Val→Ala mutation opens up space above the Fe2/3/6/7 face of FeMoco (shown in Figure 31), allowing for access of bulkier substrates.444,476 This mutation was found to result in considerably faster kinetics for hydrazine as a substrate.477 Another mutation used in these experiments is the α-195His→Gln mutation, where the suggested proton donor α-195His is mutated into a nonactive glutamine residue that reduces the capacity for N2 reduction by >95%.478−480
In 2005, Hoffman and co-workers described an EPR-ENDOR study of a hydrazine reaction intermediate for the α-70Val→Ala/α-195His→Gln mutant.481 The X-band EPR spectrum featured g-values of 2.09, 2.01, and 1.93, consistent with an S = 1/2 reaction intermediate. Isotope-labeled 15N2H4 and 14N2H4 substrates used in Q-band 15N Mims ENDOR experiments revealed that a single 1.5 MHz 15N ENDOR signal, suggestive of either symmetric substrate binding or the presence of a bound reaction product after N–N bond cleavage ([−NH2] or [−NH3]). A follow-up study in the same year compared the EPR and ENDOR spectra for the hydrazine intermediate in the α-70Val→Ala/α-195His→Gln mutant, a methyldiazene intermediate in the α-195His→Gln mutant, and a N2 reaction intermediate in the wild-type enzyme.456 All three substrates produced EPR signals consistent with S = 1/2 spin states with mildly varying g-values. The 15N hyperfine splitting at g1 = 2.08 was also different: 0.9 MHz for N2 in the wild-type, 1.5 MHz for methyldiazene in the α-195His→Gln mutant, and 1.9 MHz for the α-70Val→Ala/α-195His→Gln mutant. Furthermore, 1H CW-ENDOR revealed exchangeable protons with 1H hyperfine splittings at g1 of 8–9 MHz for methyldiazene and hydrazine intermediates, while none was observed for the N2 reduction intermediate.482 This was interpreted as different substrates resulting in different reaction intermediates, although methyldiazene and hydrazine intermediates were considerably more alike than the N2 reduction intermediate. The methyldiazene intermediate was explored in more detail in 2006, where 14N/15N isotope labeling was used to label each nitrogen atom in the substrate.483 When the substrate with terminal nitrogen labeled as 15N (H15N=N–CH3) was used, a 15N ENDOR signal appeared while no signal appeared when the other nitrogen was 15N labeled. This strongly suggests the terminal nitrogen atom to bind to the cofactor. 13C and 2H labeling experiments came to the same conclusion as no differences in 13C ENDOR could be seen when the carbon was 13C labeled nor were there differences in the 2H ENDOR when the carbon-bound hydrogens were labeled as 2H. The 1H ENDOR signal seen was found to be exchangeable. All in all, these results could be interpreted as the reaction intermediate consisting of at least an [NHx] fragment bound to the cofactor, derived from a terminally bound methyldiazene intermediate. In 2007, diazene was used directly as a substrate for N2ase via in situ formation by decomposition of azodiformate.459 Via the same type of EPR and ENDOR experiments with 15N/14N labeling on the α-70Val→Ala/α-195His→Gln mutant, it was demonstrated that hydrazine and diazene produce a common reaction intermediate under turnover conditions that contains at least an [NHx] fragment with [−NH], [−NH2], [−NH3], [−N2H2], and [−N2H3] as possibilities. In 2011, the nature of the intermediate was further clarified when it was revealed that if methyldiazene is used as a substrate for the same α-70Val→Ala/α-195His→Gln mutant (instead of the α-195His→Gln mutant used in the 2005 study) then the EPR and ENDOR spectra (see Figure 43) are identical for methyldiazene, hydrazine and diazene.132 Furthermore, use of 15N labeling on methyldiazene showed that the N–N bond is likely cleaved in this intermediate and 14N/15N HYSCORE shows no signal from a weakly coupled 15N. This allowed further constraints on the nature of the intermediate (named I) as [−NH], [−NH2], and [−NH3].
Figure 43.

Comparison of the Q-band 15N ReMims ENDOR spectrum of the α-70Val→Ala/α-195His→Gln MoFe double variant trapped during turnover (2:1 MoFe:FeP) with 15N2H4 (black), 15N2H2 (red), and 15NH=N–CH3 (blue, labeled “15MD” for methyldiazene). Adapted with permission from ref (132). Copyright 2011 American Chemical Society.
In a 2012 study, the EPR samples containing the S = 1/2 intermediate resulting from turnover in the presence of methyldiazene, hydrazine, or diazene were found to contain another intermediate that was EPR silent.126 This intermediate, named H, was found to be an integer-spin non-Kramers doublet according to Q-band CW-EPR (Figure 44) and non-Kramers ESEEM spectroscopy. The spin state was proposed to be S = 2. The ESEEM spectra show the absence of modulation of a second 14N in H–14N=14N–CH3 or 2H from H–14N=14N–C2H3, suggesting cleavage of the N–N bond and identifying intermediate H as another [−NHx] fragment. From the ESEEM spectra, 14N quadrupole couplings could also be deduced, giving e2qQ = 3.04 MHz and η = 0.47. As the possible NH3 candidate would have approximately C3v symmetry and an axial quadrupole tensor (η ∼ 0),484 intermediate H was deduced to contain an [−NH2] group with rhombic quadrupole character.126 Furthermore, intermediate H being an [−NH2] species was proposed to correspond to the E7 state of the LT cycle (Figure 2). As intermediate I has S = 1/2 and also contains an [NHX] group (i.e., cleaved N–N bond) was then proposed to correspond to the E8 state, i.e., an [−NH3] group.
Figure 44.

Q-Band CW-EPR absorption spectra of the α-70Val→Ala/α-195His→Gln double mutant MoFe variant trapped in the resting state and during turnover in the presence of 14N2H4, as indicated.126 [4Fe-4S] denotes signal arising from FeP. Adapted with permission from ref (126). Copyright 2012 National Academy of Sciences.
4.8. Acetylene Reduction
All three N2ases are capable of reducing acetylene, and for 50 years this property has been used routinely for in vitro assays of N2ase activity.485 Acetylene is likely the best-studied among N2ase substrates. While N2 is a competitive inhibitor of acetylene reduction, acetylene is a noncompetitive inhibitor of N2 reduction.486−488 Reduction of acetylene to ethylene requires only two electrons, and early studies established that ethylene is released from Mo N2ase in the E3 and E4 states.489 The reduction of acetylene to ethylene by N2ase is stereoselective: reduction of C2H2 in D2O, or C2D2 in H2O, predominantly produces cis-CHDCHD.485,486 It was found that this reaction proceeds by the addition of two protons to the same side of bound C2H2, and a mechanism involving an enzyme-bound η2-vinyl intermediate has been proposed.490
Kinetic studies of acetylene reduction by Mo N2ase showed that MoFe contains two binding sites for acetylene with varying affinity,491−493 a hypothesis which was supported by DFT calculations of a [MoFe7S9N]0 cluster model of FeMoco in which each acetylene binds to opposite sides of the cofactor, the first to the E1 state and the second at E2.494 The Km reported for the low-affinity binding site was found to be similar to that of the α-69Gly→Ser variant of MoFe, suggesting that the Fe2/3/6/7 face of FeMoco (capped by α-70Val, Figure 31) is the high-affinity acetylene binding site.492,495 Importantly, the EPR spectrum of α-69Gly→Ser MoFe was identical to that of native MoFe.495 The fact that N2 reduction is not altered in this “acetylene-resistant” variant prompted the hypothesis that N2 shares the same location as the low-affinity C2H2 binding site. Fe N2ase also seems to include two binding sites for acetylene but not V N2ase.53 Interestingly, the VFe residue corresponding to α-69Gly in MoFe is actually a leucine, and the Km for acetylene binding is 10-fold higher than for Mo N2ase, suggesting only the low-affinity C2H2 binding site exists in V N2ase.
EXAFS studies have presented no changes in the metallocofactor structures of Mo N2ase upon incubation with C2H2.496 The EPR spectrum of Mo N2ase in the resting E0 state is similarly unperturbed,497,498 with the rhombicity of the S = 3/2 signal slightly decreasing in Mo N2ase of Klebsiella pneumoniae upon incubation with acetylene.497−499 However, 57Fe ENDOR revealed a shift in highest- and lowest-energy doublets and 1H ENDOR showed an increase in intensity from unresolved proton resonances, suggesting acetylene interacts weakly with the cofactor at E0.499 DFT calculations on a [MoFe7S9N]0 analogue of FeMoco suggested weak and reversible binding of acetylene was already possible at the resting state.494
Under turnover conditions, the S = 3/2 signal observed by EPR decreases up to 90% without the appearance of any new paramagnetic species. A similar trend has been observed with V N2ase: upon incubation with C2H2 under turnover conditions, both S = 1/2 and S = 3/2 EPR signals related to FeVco decrease without the appearance of any new signals.500 In contrast, a weak axial S = 1/2 species with g values of 2.125 and 2.000 has been observed upon incubation of K. pneumoniae Mo N2ase with ethylene under turnover conditions.491,501
4.8.1. Variants
Major advances in the characterization of the binding mode of acetylene on FeMoco have come from kinetic and spectroscopic studies using variants of MoFe with mutations in side chains surrounding the cofactor, such as α-70Val, α-195His, and α-96Arg. The side chains of these three residues all perturb the environment around the Fe2/3/6/7 face of FeMoco (Figure 31).
4.8.1.1. α-70Val
α-70Val has been suggested to be involved in substrate binding, and a MoFe variant substituted at this position allowed the first identification of an E4 intermediate (see section 4.5).442 The substitution of α-70Val with a residue possessing a longer side chain, such as isoleucine, decreases N2 and C2H2 reduction activities of MoFe, while H2 evolution activity remains at the same level, implying a major role of this residue in substrate binding at FeMoco.502
4.8.1.2. α-195His
Histidine 195 is strictly conserved among N2ases. This residue participates in one of three NH-S hydrogen bonds at the cofactor.398 The EPR spectrum of MoFe whose α-195His residue has been substituted with a glutamine residue (α-195His→Gln MoFe) looks similar to that of unsubstituted MoFe, although a slight rhombic shift can be observed with geff = [4.33, 3.77, 2.00] for wild-type MoFe which shift to geff = [4.36, 3.64, 2.00] in α-195His→Gln MoFe.480,503 Thus, this substitution does not alter the spin state of FeMoco. Crystallographic studies have revealed that α-195His→Gln MoFe has the same structure as MoFe, where the hydrogen bonding interaction between α-195His and the belt sulfide of FeMoco is replaced by a similar interaction between the glutamine and this S atom.504 This modified enzyme still binds N2 but cannot reduce it; however, it does reduce C2H2 at similar rates to wild-type MoFe.480 N2 inhibits both C2H2 and H+ reduction, suggesting that N2 and C2H2 compete for occupancy of the same active site.
Under turnover conditions, the incubation of α-195His→Gln MoFe led to three new S = 1/2 EPR signals replacing the resting-state signal.505 A series of new features are observed in the g ∼2 region, with g-values of 2.12, 2.00, 1.98, and 1.95, and a minor shoulder at g = 1.97. Turnover experiments performed at different temperatures allowed features at g = 2.12, g = 2.00, and g = 1.97 to be attributed to three different species. All three species were present in samples trapped during low electron flux turnover, and modifying the electron flux only resulted in a change in the overall amplitude of the signals, not of their ratios, implying that these species are not precursors to one another. The signal at g = 1.97 (termed SEPR3) could not be further resolved using Q-band EPR, and its origin still remains unclear.446 The feature at g = [2.12, 1.98, 1.95] is part of a rhombic signal, designated SEPR1 (Figure 45). Because of the deviation of giso from that of a free electron, this signal has been attributed to an unpaired electron on FeMoco. The remaining signal at g = 2.00, termed SEPR2, is reminiscent of a radical species, exhibiting a small g-anisotropy caused by minor spin–orbit coupling. Q-Band EPR generated better-resolved features for this signal than the X-band EPR experiments, allowing the full S = 1/2 species to be characterized with g = [2.007, 2.000, 1.992] (Figure 45).446
Figure 45.

Q-Band EPR spectrum recorded at 2 K of the α-195His→Gln MoFe variant trapped during turnover in the presence of acetylene. Reproduced with permission from ref (446). Copyright 2000 American Chemical Society.
EPR experiments using deuterium and 13C-labeled acetylene were utilized to determine which of the SEPR signals were associated with substrate interactions.446,505 Upon incubation with C2D2, the line widths of SEPR1 narrow relative to those observed with C2H2, which was attributed to the smaller nuclear gyromagnetic moment of deuterons. Likewise, incubation with 13C2H2 led to a broadening of the SEPR1 features. Taken together, these results suggest that SEPR1 can be attributed to a C2H2-derived species bound to FeMoco under turnover conditions in an n-even state of the Lowe–Thorneley cycle (Figure 2). These results were supported by further Q-band 13C ENDOR experiments on α-195His→Gln MoFe incubated with 13C2H2 under turnover conditions, which indicated the presence of one or several fragments derived from 13C2H2 bound to FeMoco.446 The appearance of three separate 13C hyperfine couplings of decreasing strength (aiso(C1) = 2.5 MHz; aiso (C2) = 2.3 MHz; A(C3) ≤ 0.5 MHz) evidenced that there is a minimum of two C2Hx molecules interacting with the FeMoco cluster in the observed state. 1H ENDOR measurements on H2O/D2O samples revealed only nonexchangeable proton signals and no signal arising from solvent protons, further supporting the attribution of SEPR1 to the binding of C2H2-derived compounds to FeMoco.
Furthermore, a signal similar to SEPR1 with g = [2.12, 1.998, 1.987] could be observed when incubating α-195His→Gln MoFe with ethylene under turnover conditions, suggesting that this signal should be attributed to ethylene bound to FeMoco.501 The amplitude of this signal also displayed a sigmoidal behavior as a function of C2H2 concentration at low concentrations, implying cooperative binding of two C2H2-derived molecules to FeMoco.504 Meanwhile, turnover with C2D2 or 13C2H2 did not induce significant changes in the SEPR2 signal, suggesting that the corresponding species is not an intermediate radical of reduced acetylene but likely a carboxylate-centered radical species, either from the homocitrate or from glutamate or aspartate residues, which is generated during turnover.446,504,505 However, the SEPR2 signal found in the α-195His→Gln variant is broadened upon 57Fe enrichment, indicating the signal of minimal g-anisotropy is associated with the FeMoco center.451 Further characterization is therefore required for a clear assignment of both signals.
It has been suggested that C2H2 binds to two Fe atoms of FeMoco as a bridging dianion.446,506 However, Q-band CW and Mims pulsed 1,2H-ENDOR studies finally identified SEPR1 as a product complex, likely corresponding to FeMoco bound to ethylene via a single Fe atom, thereby forming a ferracycle.451 Interpretation of 57Fe ENDOR studies allowed the determination of the electronic state of the SEPR1 species, which was found to be similar to that of FeMoco in the loCO EPR active species (see section 4.10).
The ability to detect a C2H2-induced EPR signal in α-195His→Gln MoFe and not wild-type MoFe may be due to a higher binding affinity for either ethylene or an intermediate in this variant. In such a case, the intermediate species would not reach sufficient concentrations for detection in wild-type via EPR. It is also possible that the product is released in another EPR-silent state.495
4.8.1.3. α-96Arg
Arginine 96 is involved in hydrogen bonding with the same Fe2/3/6/7 face of FeMoco as α-195His. MoFe variants in which α-96Arg was substituted with alanine, leucine, glutamine, or histidine residues were shown to have similar EPR spectra to wild-type MoFe, with a prominent S = 3/2 signal with geff = [4.26, 3.67, 2.00], suggesting that the ground-state electronic properties of FeMoco are unperturbed in the variants.507 However, substitution at α-96Arg was found to change the catalytic properties of MoFe: the Michaelis constant (Km) for C2H2 reduction by α-96Arg→Leu MoFe was 3-fold lower than that for MoFe, and the variant catalyzed 3-fold more trans-ethylene formation during C2D2 reduction.490 Incubation of α-96Arg→Leu MoFe with acetylene induced modifications in the EPR spectra, with the appearance of a new S = 3/2 species with geff = [4.50, 3.50, 2.00].507 These changes were not seen upon incubation with N2 or C2H4. Similar changes were observed in the EPR spectra of the Ala, Gln, and His variants, with minor differences in effective g-values and line shapes. Interestingly, substitution with Lys did not induce modifications in the spectrum compared with native MoFe.
Incubation of α-96Arg→Leu with increasing concentrations of C2H2 resulted in a subsequent decrease of the resting state S = 3/2 signal at geff = [4.26, 3.67, 2.00] while increasing the signal at g = 4.50, suggesting that incubation with C2H2 alters the electronic properties of FeMoco in α-96Arg→Leu MoFe. Surprisingly, 13C ENDOR analyses of α-96Arg→Leu MoFe incubated with 13C2H2 did not reveal any spectral modifications, implying that the FeMoco modifications are not caused by direct binding of C2H2 to the cofactor.
The recent elucidation of the crystal structure of α-96Arg→Gln MoFe showed C2H2 trapped near FeMoco, positioned in a binding pocket surround by side chains of α-96Arg→Gln, α-70Val, and α-98Asn residues at a distance of 4 Å from the Fe2/3/6/7 face of FeMoco.508 The stabilization of C2H2 at the active site was suggested to result from an increase in the size of the binding pocket following substitution of the bulky Arg side chain.
4.9. Propargyl Alcohol Reduction
When it was discovered that modification of the Fe2/3/6/7 face at the α-70Val site could control substrate size acceptance of nitrogenase,476 the first experiments investigated the α-70Val→Ala modification to “open up” this substrate binding site to larger substrates such as propargyl alcohol (HC≡C–CH2OH), which can be reduced by 2e– to form allyl alcohol:444,477
Trapping α-70Val→Ala Mo N2ase during turnover in the presence of propargyl alcohol results in nearly the complete disappearance of the S = 3/2 resting-state EPR signal of E0, producing a nearly axial S = 1/2 signal (g = [2.123, 1.998, 1.986]), indicating a high occupancy of the trapped intermediate.444 Substrate concentration dependence studies indicate that propargyl alcohol binds in a single event, unlike the multiple substrate binding intermediate of acetylene.504
An alternate version of the Mims ENDOR sequence, Re-Mims ENDOR,123 was utilized to observed the small hyperfine couplings and eliminate the “blind-spots” of the traditional Mims ENDOR approach and collect distortion-free spectra. The 13C ENDOR of the uniformly 13C labeled propargyl alcohol collected at the “single-crystal like” field position yielded three 13C hyperfine couplings (Figure 46, left), and the evaluation and simulation of the full 2D field frequency ENDOR pattern yielded the presence of three carbons with decreasing isotropic hyperfine couplings: aiso(C3) = 3.7 MHz, aiso(C2) = 1.1 MHz and aiso(C1) = 0.4 MHz.
Figure 46.

(top, left) Q-band Mims and re-Mims pulsed 13C ENDOR and (right) Q-band 1,2H ENDOR collected at g = 1.989 (∼g3) of the S = 1/2 signal which appears during turnover of the α-70Val→Ala MoFe variant in the presence of propargyl alcohol. The time between applied pulses (τ) used in collection of each spectrum is indicated for the 13C ENDOR, as well as aiso for each unique observed C. For the 1,2H ENDOR, τ = 488 ns. (bottom, left) Proposed structure of the observed activated propargyl alcohol reduction intermediate. Unique aiso values are indicated in parentheses for 1Ha and 1Hb, and superscripts “S” and “C” indicate solvent-derived and nonexchangeable protons, respectively. Adapted with permission from ref (445). Copyright 2004 American Chemical Society.
The 13C hyperfine couplings alone are informative enough to reveal the structure of the bound substrate; however, further structural insight was gained through H/D labeling of the propargyl alcohol and trapping in either H2O or D2O for study using 1,2H ENDOR. Quantitative stochastic field-modulated 1H CW-ENDOR was employed for the large 1H proton couplings (Figure 46, top right) and 2H Mims (Figure 46, bottom right) for the smaller deuterium couplings of the isotopically labeled samples. The large 1Ha ∼ 20 MHz coupled signal of propargyl alcohol in 1H2O decreases by one-half in D2O solvent. The same halved signal is observed for the d-propargyl alcohol trapped intermediate in 1H2O. The large 1Ha ∼ 20 MHz signal is completely absent in d-propargyl alcohol samples prepared in D2O. The 2H Mims ENDOR yields complementary results, where the 2Ha signal decreases by one-half in samples prepared using either 1H2O/d-propargyl alcohol or D2O/propargyl alcohol and is completely absent in samples prepared in 1H2O/propargyl alcohol. This immediately indicated that two strongly coupled protons (∼20 MHz) were present in the propargyl alcohol intermediate, with one deriving from substrate and the other from solvent during reduction. The equivalency of the substrate proton and solvent derived couplings required that they be symmetrically equivalent. Additionally, another solvent derived proton coupling was observed with a coupling of ∼6 MHz. When the 13C and 1,2H ENDOR results are combined together, a comprehensive structure of the bound propargyl alcohol intermediate is generated (Figure 46, bottom left).445
The trapped propargyl alcohol intermediate has also been studied using 57Fe NRVS along with Fe and Mo K-edge XAS.163 Nearly superimposable Mo K-edge XANES and EXAFS spectra of resting-state (E0) and propargyl alcohol turnover samples indicated no oxidation state changes or alterations in the first coordination sphere around Mo. However, slight variations in the Mo EXAFS corresponding to Mo–Fe scattering interactions were observed, implying possible perturbation of Mo–Fe scattering distance due to changes at the iron sites ∼5.1 Å away (Fe2/3/4).
The Fe K-edge XAS of the bound propargyl alcohol intermediate exhibit only small changes relative to the resting state. However, as substrate binding to one or two irons affects only a small fraction of the 15 total irons observed in the sample, the changes are not expected to be large. Therefore, the lack of changes in the XAS at the Mo is more conclusive than at the Fe. The Fe EXAFS collected to a relatively high k-value (>17 Å) yielded excellent data to resolve scattering interactions (resolving strength of ΔR ∼0.10 Å). The fitted Fe EXAFS of resting-state and propargyl alcohol-bound N2ase show slight distortions consistent with substrate binding to iron. The EXAFS model favored by George et al.163 proposes a distortion in the average coordination sphere of Fe that would result in an increase in the average Fe-carbide distance by up to 0.35 Å upon substrate binding. NRVS, along with normal-mode analysis, concludes that the FeMoco core remains intact and there is no evidence of iron-sulfide bond breaking.
4.10. CO Reduction
Binding of CO in N2ases has been a topic of interest since the first publications reporting the inhibitory effect of CO on the N2 reducing capabilities of MoFe.509,510 Since then, numerous studies investigating the interaction of CO with N2ases, both as an inhibitor and substrate, have been undertaken,141,487,511−517 with the first spectroscopic study of this system reported in 1979.492 The primary spectroscopies used to investigate the species formed upon CO binding have been EPR and FT-IR. Both are powerful techniques for somewhat orthogonal reasons. EPR and EPR-based techniques such as ENDOR are predominately used to study Kramers systems and are only applicable to integer spin systems under special circumstances. This has allowed EPR to selectively look at the half-integer spin species formed under CO binding conditions without the convolution and interference of integer spin species and in turn provide detailed information regarding the electronic and geometric structures of a specific subset of species. However, the EPR observable CO-bound species reported to date only account for a fraction of the total FeMoco present.504,518 In contrast, FT-IR is a spin-insensitive technique which can be used to observe the modulation of the C≡O stretching frequency upon binding. This theoretically enables FT-IR to observe all possible bound forms of CO in MoFe, regardless of cluster oxidation state.
4.10.1. EPR Species hiCO and loCO, and hi(5)CO
The first unique, identifiable CO-bound species in MoFe come from a report of Davis and co-workers, in which two unique S = 1/2 signals were observed while exposing MoFe to CO during turnover. The first species, referred to as “hi-CO” was observed when a stoichiometric excess of CO was present, while “lo-CO” appeared when CO was provided in a substoichiometric ratio (Figure 47).492
Figure 47.

X-Band EPR spectra at ∼g = 2 of MoFe from Clostridium pasteurianum under turnover (1:3.4 MoFe:FeP) in the presence of (a) 0.3 atm CO (excess), (b) 0.00125 atm CO (substoichiometric), and (c) no CO. Adapted with permission from ref (492). Copyright 1979 American Chemical Society.
ENDOR has since provided significant insight into the nature of these two species.101,519−521 Foremost, ENDOR has been used to demonstrate that both species correspond to CO bound forms of FeMoco,519 as well as provide additional information regarding the nature of their binding to the cluster. From 13C ENDOR, hiCO was proposed to arise from two CO terminally bound to the FeMoco cluster, while the loCO species is best described as a single CO bound to FeMoco in a bridging fashion forming an Fe–C–Fe bond.521 This hypothesis regarding loCO closely matches the XRD structure of the purported loCO bound form of MoFe published over 15 years later,452 further highlighting the powerful insight ENDOR can provide regarding electronic and structural properties. It is worthwhile to note that these ENDOR studies were performed using an ethylene glycol (EG) quenching method, in which the turnover of MoFe in the presence of FeP was halted by the addition of ethylene glycol to a final concentration of 40–50% by volume rather than by the more traditional freeze quenching method. In this way, it was also found that removal of the CO headspace from an EG quenched hiCO sample resulted in the appearance of the loCO S = 1/2 signal and that exchanging the headspace back to CO resulted in the reconstitution of the hiCO signal.518 These experiments implied that hiCO and loCO existed in the same oxidation state. These results were later supported by photolysis experiments which showed the hiCO signal could be photolyzed to loCO at low temperatures and that annealing resulted in reformation of hiCO with an activation barrier of 0.93 kcal/mol.522
At higher e– flux conditions, a third independent EPR signal referred to as “hi(5)CO” has also been reported to form after 10 min reaction time.518 This S = 3/2 signal requires an excess of CO to form and does not appear to interconvert with either of the more well-known loCO and hiCO signals. In addition, the signal is correlated with the disappearance of the S = 3/2 signal of the E0 state and is also somewhat invariant to mutations modifying the secondary coordination environment of the FeMoco cluster.523
4.10.2. IR Species of hiCO and loCO
As mentioned, FT-IR should be capable of revealing not only the singly and doubly CO bound species which are responsible for loCO, hiCO, and hi(5)CO, but also all of those which are EPR silent. To this end, a variety of CO stretching features have been found under excess and substoichiometric CO conditions.138
4.10.2.1. loCO Conditions
LoCO conditions produced a single feature at 1904 cm–1, assumingly correlated with the hiCO peak at 1906 cm–1. Further studies have shown that this single loCO feature is correlated with a second slower forming feature at 1715 cm–1, which appears at time scales >200 s.139,141 This has led to the hypothesis that an initial terminally bound CO is formed under limiting CO conditions, which interconverts to a bridging form at longer time scales.
4.10.2.2. hiCO Conditions
In an original report, hiCO conditions produced four unique features at 1958, 1936, 1906, and 1880 cm–1 (Figure 48).138 Time-resolved measurements found the 1906 cm–1 feature to appear only transiently, maximizing at 7 s, while the peaks at 1936 and 1958 cm–1 continued to grow. This, combined with the observation of a single feature at 1904 cm–1 under loCO conditions, led to the hypothesis that this feature arose from a single CO bound state of the FeMoco cluster, which is transiently converted to a two CO bound form in the presence of excess CO. The relative energy of all of these features also led to the proposal that the CO species contributing to these features are all terminally bound, contrary to the proposed bridging state of loCO observed via 13C ENDOR.521
Figure 48.

Transient infrared spectra of MoFe from K. pneumoniae during high e– flux turnover (1:3.4 MoFe:FeP) in the presence of (a) excess (0.5 mM) 12CO, (b) excess (0.5 mM) 13CO, (c) substoichiometric (∼0.06 mM) 12CO, and (d) in the absence of CO. Adapted with permission from ref (138). Copyright 1997 American Chemical Society.
Photolysis studies have been particularly instrumental in correlating a number of the features observed under hiCO conditions, and three unique species (hiCO1, hiCO2, and hiCO3) have been proposed along with their corresponding photolyzed forms (loCO1, loCO2, and loCO3).142,143 The hiCO1 species is assigned to the 1958 cm–1 peak (1973 cm–1 at low temperatures), the decay of which is concomitant with the formation of a feature at 1711 cm–1 (loCO1), as well as a decay at 1679 cm–1. As the 1679 cm–1 stretch is well buried in the amide bands of the protein, which generate a large background, this feature would be indiscernible by absorption measurements. The peak at 1711 cm–1 is also assumed to correspond to a purported slowly developing (>200 s), long-lived feature at 1715 cm–1.138,139,141 Annealing of the photolysis-generated loCO1 species at 70 K was also found to result in reformation of hiCO1, and the activation energy for this event was found to be 0.88 kcal/mol, similar to the 0.93 kcal/mol observed for the interconversion of the EPR observed loCO and hiCO species.138,522 Next, a “hiCO2” species has been assigned based on photolysis of the α-195His→Asn and α-195His→Gln variants trapped under hiCO conditions. In the α-195His→Asn mutant, which alters the stabilizing interaction of the S2B position, decreased intensity was observed at 1938 and 1858 cm–1 upon photolysis, concomitant with increases in intensity at 1780 and 1760 cm–1 (loCO2). The α-195His→Gln variant showed a similar decrease in intensity of the 1932 cm–1 feature, with a small increase in intensity at 1748 cm–1. While this photolysis event was not initially observed in wild-type hiCO, samples which had been stored several months under liquid N2 displayed further decreases in intensity at 1938 cm–1, which corresponded with an increase in intensity at 1784 cm–1. This observation is similar to those made for the variants. Lastly, a less reversible “hiCO3” species was found in α-195His→Gln, where a decrease in intensity at 1938 and 1911 cm–1 corresponds to an increase at 1921 cm–1 (loCO3).142,143
DFT has also been used to investigate a series of potential binding modes of CO to FeMoco524−526 and has been primarily correlated to experimental studies through comparisons with IR and NRVS spectra.142,143,166 From these correlations, it has been proposed that the aforementioned “hiCO3” species is consistent with a model in which Fe2 and Fe6 each bind a CO terminally, and the S2B position is protonated.143,166 Meanwhile, hiCO1 is proposed to be a nearly identical species, except that the Fe2-bound CO is protonated to form a formyl group.166
4.10.3. Bridging Observations from EPR and IR
From the considerable body of work regarding CO binding in Mo N2ase, the observation of various species is primarily dependent on three variables, namely the ratio of CO:MoFe, the electron flux (controlled by the ratio of MoFe:FeP), and time. While the modulation of CO concentration has been instrumental for all comparisons of species of “loCO” and “hiCO”, only a select few studies have raised concerns about the influence of e– flux on the observed species.492,518 This is particularly crucial in drawing comparisons between the body of FT-IR and EPR studies which form our current picture of CO binding in MoFe. Furthermore, few have also investigated the transient nature of these species, which is dependent on a variety of factors including both the absolute and relative concentrations of MoFe and FeP, the concentration of MgATP, and reductant, as well as the presence or absence of a MgATP regeneration system. Lastly, a considerable number of studies have utilized chemical quenching (predominately EG, but also trehalose) as a method to halt turnover in the presence of CO.142,143,166,518−522,527 While this technique does indeed stabilize the loCO and hiCO EPR species developed during turnover, it is still uncertain exactly how this method impacts the formation of other nonobservable species, and therefore the direct comparison of results obtained from freeze quenching and chemical quenching should be made with caution.
Despite the breadth of conditions, some correlations between the EPR and FT-IR can be drawn. Reiterating, three unique EPR signals have been found corresponding to CO binding, two of which appear regardless of e– flux and a third which requires high e– flux conditions (at least 1:1 MoFe:FeP). Although the majority of studies have been performed using EG quenching, all three signals have also been observed following freeze quenching.492,523 Studies under loCO conditions have observed the appearance of the loCO EPR signal only under moderate to high electron flux conditions, and samples trapped through either EG quenching or freeze quenching have shown the appearance of this signal at time scales between 12 s and 15 min.492,518 Stopped-flow (SF) FT-IR studies have evidenced two features that can form under loCO conditions at 1904 and 1715 cm–1, where the first is seen to maximize and decay after 7 s while under high-flux turnover in the absence of a MgATP regeneration system, while the second requires several minutes to form.138,139,141 The 13C ENDOR of the loCO state favors an Fe–C–Fe bridged state, which the 1715 cm–1 feature would be consistent with.521 However, the observation that the loCO EPR signal forms within 12 s under similar electron flux conditions to the transient 1904 cm–1 feature raises further questions.492
The direct observation by FT-IR of a minimum of three additional features under turnover in the hiCO condition (with several further additional features implicated by photolysis) significantly complicates their correlation to the two observed hiCO EPR signals. First, the hi(5)CO signal has been reported to occur only with at least intermediate electron flux conditions (4:1 MoFe:FeP) and at time scales of >6 min.518,523 However, no detailed information regarding the transient behavior of hi(5)CO has been reported, and it is therefore unclear whether this species may appear at short time scales under these conditions. SF FT-IR studies have been performed at both 4:1 and 1:3.4 MoFe:FeP conditions and have consistently reported the same four CO-dependent features in wild-type MoFe at 1958, 1938, 1906, and 1880 cm–1.138,145 Photolysis studies have implicated that 1958 and 1938/1906 cm–1 correlate with two different species of hiCO, where hiCO1 at 1958/1679 cm–1 is converted to loCO1 at 1711 cm–1 upon photolysis, and similarly hiCO3 at 1938/1906 cm–1 is converted to loCO3 at 1921 cm1.142,143 One interpretation of these two species may be that both bind two CO but are in two different oxidation states. The similar interconversion activation energies of the IR observed loCO1/hiCO1138 and EPR observed loCO/hiCO522 is suggestive that these species correspond with one another. If this were true, then hiCO1 would likely represent an EPR-active species with one terminally bound CO and one bridging or formyl-like CO, while hiCO3 represents an EPR-silent species with two terminally bound CO. However, this hypothesis of hiCO1 is partly at odds with the 13C ENDOR, in which the hyperfine tensors (A) of both CO in hiCO are isotropic.521 This would not be expected if either a bridging or formyl-like CO were present. Additionally, the similar transient profiles of the features attributed to hiCO1 and hiCO3 from SF FT-IR imply these two species form independently from one another, and the lack of low e– flux SF FT-IR studies (or at least lower than those in which hi(5)CO is observed) precludes the definitive assignment of either hiCO FT-IR species with either of the EPR active hiCO species. Furthermore, the weak CO-dependent feature at 1880 cm–1 is unaccounted for in these considerations.
Significant progress has been made in our understanding of the interaction of CO with Mo N2ase. However, several open questions remain. First, no species have been observed, by either EPR or SF FT-IR, at time scales <1 s.138,145,492 Meanwhile it is known that CO inhibition of substrates such as azide and acetylene happen at significantly shorter time scales.499 Therefore, the mechanism by which CO inhibits N2 and other substrate reactivity remains elusive. Second, while it is clear that MoFe must undergo turnover to bind CO, it is still unknown to which state initial CO binding occurs (i.e., E1 or E2, etc.). As CO does not inhibit H2 reduction, it can be assumed that MoFe is capable of continuing to cycle through at least the E0–E2 states while maintaining bound CO. This is further complicated by the observation that CO can be reduced to simple hydrocarbons by MoFe over longer periods of time. Therefore, it is unclear whether some of the longer-lived intermediates and states actually represent partially activated CO species such as formyl-bound FeMoco cluster. Lastly, the relationships between species individually characterized by either EPR or FT-IR remain unclear, as well as the exact relationships of these species to the known single CO-bound crystal structure (PDB 4TKZ), which has implicated the displacement of the bridging S2B sulfide (Figure 1) by CO.452 To this end, great insight could be gained by a cross-correlative, time-resolved study which utilizes these orthogonal spectroscopic techniques to disentangle the myriad of speciation generated during CO inhibition and activation.
4.11. Cyanide
Similar to CO, HCN has been reported to act as both an electron-flux inhibitor and substrate of N2ases,511,528,529 and some N2 fixing bacteria such as Azotobacter vinelandii have even been found to proliferate in the presence of cyanide-containing medium, producing methane and ammonia through six-electron reduction.530 It has also been an important focus in ATP-independent reduction studies, as, like N2, complete reduction requires six electrons.531 The majority of spectroscopic studies have focused on the interaction of cyanide with extracted FeMoco in varying oxidation states and have utilized a wide variety of methods including (but not limited to): XAS,201,496 UV–vis,53219F NMR,496 FT-IR,140 and EPR.140 These combined studies have evidenced that CN– binding in the extracted cofactor is dependent on oxidation state, with Mo being the more favorable binding site in the more reduced states of the cluster201,496,532 and Fe in the more oxidized states.140 However, very few studies have spectroscopically probed the interactions of cyanide with the enzymes themselves.490,498,507 Several important kinetic observations have been made in the study of cyanide reduction by Mo N2ase. The first is that unlike N2, the presence of “infinite” concentrations of cyanide can nearly completely suppress H2 production during turnover.529 This implies that cyanide reduction does not follow the same reaction mechanism as N2. Second, it was observed that as electron flux was increased, reduction of cyanide became less favorable than H2 production.529 Although the 1:1 MoFe:FeP electron flux conditions (which have been proposed to populate E4) were still found to favor cyanide reduction, these results imply that cyanide not only binds to an earlier intermediate state En than N2, but also that binding is less favorable in higher En states. This, along with studies demonstrating that cyanide is a noncompetitive inhibitor of N2, led to the proposal that cyanide binds to the E0 state.529,533 However, EPR studies since this time have not indicated cyanide is capable of binding to the resting state of wild-type MoFe.498,507
CW-EPR was used to investigate the α-96Arg→Leu variant of Mo N2ase and its interaction with cyanide.490 This mutation effectively reduces the steric bulk around the Fe2/3/6/7 face of the FeMoco cluster (Figure 31). Under resting conditions, the addition of considerable quantities of cyanide (starting with ∼24× excess) resulted in the disappearance of the S = 3/2 E0 spectrum, and replacement with two new S = 3/2 signals with g1 components at g = 4.37 and 4.06. These new signals completely replace E0 in the presence of ∼240× excess cyanide. Further Q-band Mims 13C ENDOR measurements were performed, which found a single aiso = 0.42 MHz, comparable with that observed in CO inhibition studies and strong evidence for direct binding of cyanide to one of the Fe of FeMoco.507 These results demonstrated that while the oxidation state of E0 is sufficient to bind cyanide (albeit with apparently low affinity, possibly due to steric interactions) and the Fe2/3/6/7 face appears to be a likely binding site, the interaction is prevented in the E0 state of wild-type MoFe due to steric interactions with the surrounding amino acid side chains.
Much is still to be discovered regarding the interaction of cyanide with N2ases. In particular, the En state to which it binds in wild-type Mo N2ase as well as the exact location and mode of binding are yet to be determined. Additionally, any spectroscopic characterization of the interaction of cyanide with the alternative nitrogenases V and Fe N2ase is yet to be produced.
5. Alternative N2ases
While all diazotrophs described to-date produce Mo N2ase, some additionally possess systems allowing for the production of so-called alternative N2ases: V N2ase21 and Fe N2ase.19 These are the products from distinct gene clusters whose expression is stimulated under Mo-limiting conditions. All structural genes retain high similarity throughout the N2ase family, with crucial residues being strictly conserved, such as the cysteine and the histidine coordinating the cofactor. However, variations have still been observed in sequences neighboring both the P-cluster and FeVco or FeFeco,534 and, despite recent kinetic studies proposing N2ases all follow a universal reductive-elimination/oxidative-addition mechanism for N2 binding and activation,535 there are still large differences in catalytic efficiency and reactivity with alternative substrates.536,537
5.1. V N2ase
V N2ase displays different reactivity than Mo N2ase, reducing N2, H+, and C2H2 less efficiently while simultaneously much more efficiently reducing substrates such as CO.20,512,538 In the presence of N2, the H2 evolution activity of V N2ase is higher than that of Mo N2ase, with a ratio NH3/H2 of 0.9 instead of 2.3.500 Recent studies have evidenced that this does not necessarily arise from differences in the mechanism of N2 activation by these enzymes535 but likely from differing efficiency in proton and electron transfer to N2 as a substrate. However, in contrast to Mo N2ase, V N2ase can bind CO in the resting state.539 During turnover it is also capable of reducing CO to various hydrocarbons with an 800-fold greater efficiency than Mo N2ase.538 ATP is hydrolyzed at similar rates by Mo or V N2ases in the presence of CO during turnover; however, while H2 evolution by V N2ase is reduced by about one-third, it is unaffected in Mo N2ase.538 Additionally, not only methane, ethylene, and ethane have been identified as products of CO reduction by V N2ase but also higher-order alkanes including propene, propane, butene, and butane.512 These differences in reactivity could either be due to variations in the protein scaffolds surrounding the active site, or to discrepancies in the cofactors themselves.540
5.1.1. Similarities and Differences between FeMoco and FeVco
Quantification of Fe, S, V, and Mo in both the MoFe and VFe proteins, as well as extracted FeMoco and FeVco, have suggested that the cofactors of these proteins share similar cluster compositions.20,541 The relative total absorption intensities reported in the 57Fe Mössbauer spectra of VFe have also indicated this protein possesses the same Fe composition as MoFe, with two 7Fe-containing FeVco cofactors and two 8Fe-containing P-clusters.542 However, differences in the UV–vis, MCD, and resting state EPR spectra of MoFe and VFe20,184,543 (not to mention observably different colors in the extracted cofactors541) indicate discrepancies in their electronic properties.
The crystallographic structure of VFe has only recently become available,32 and structural studies of the metallocofactors of VFe have relied heavily on the use of X-ray spectroscopy and its correlations with both model complex chemistry and MoFe.31,200,544−546 From the first EXAFS studies of VFe, there were strong indications that the FeVco and FeMoco are structurally homologous. V and Fe K-edge EXAFS indicated V–S and V–Fe distances at 2.35 and ∼2.63–2.73 Å,544−546 similar to those observed for the average Mo–S and Mo–Fe distances. The Fe EXAFS of VFe also produced average Fe–S, short Fe–Fe, and long Fe–Fe distances at 2.24, ∼2.65, and 3.68 Å, respectively, typical of prismanes and again very similar to MoFe.200,544,545 More recent studies using Fe VtC XES studies of both VFe and extracted FeVco have also identified that this cluster, like FeMoco, contains an interstitial carbide at its heart. This was identified by the presence of the same Kβ′′ feature ∼7100 eV in both VFe and MoFe (Figure 49), which is dominantly the result of fluorescent emission from the 2s atomic orbital of carbon in a μ6-coordinated carbide state.30,31,539 Slight differences in the Kβ2,5 region ∼7100 eV were also observed, which could be caused by a shift of the sulfur 3p orbitals to higher energies in FeVco relative to FeMoco, consistent with a greater complement of reduced iron in the former.31 The subtle modulations in the Fe VtC XES were well reproduced by DFT calculations, assuming [MoFe7S9C] and [VFe7S9C] clusters for MoFe and VFe, respectively. It is of interest to note that this was prior to the identification of a bridging carbonate in the crystal structure of FeVco32 and suggests that either the VtC is not sensitive to the replacement of a sulfide by carbonate and/or that perhaps differences exist between the solution and single crystal structures. Spectroscopic validation for the presence of a bridging carbonate in FeVco is presently still needed.
Figure 49.
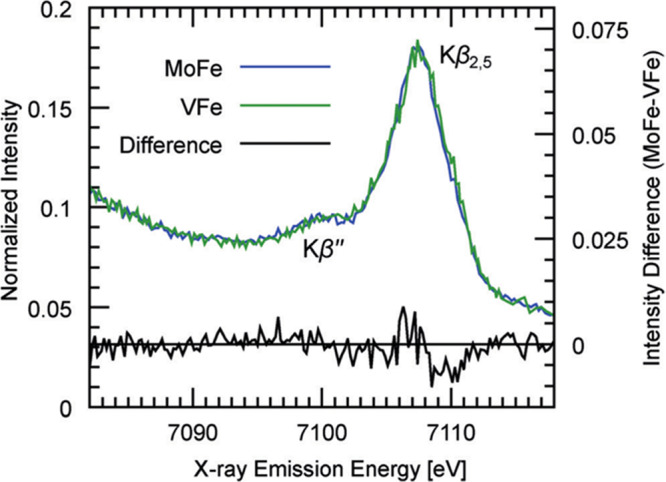
Fe Kβ VtC XES spectra of MoFe (blue) and VFe (green) proteins from A. vinelandii. The difference spectrum is shown in black. The similarity between both spectra in the Kβ″ energy indicates that FeVco also contains an interstitial carbon. Reproduced with permission from ref (31). Copyright 2015 John Wiley and Sons.
Electronically, the FeVco and FeMoco clusters also appear quite similar. Vanadium K-edge XAS measurements of the VFe protein have been used to approximate the oxidation state of V in FeVco through comparisons with the [Me4N][V3+Fe3S4Cl3(DMF)3] heterocubane.546 The similar position of the rising edge observed in VFe and this heterocubane (approximately 3 eV higher in energy than metallic vanadium) implied that the V of VFe exhibit an oxidation state between V2+ and V4+, most likely V3+.547 Both also exhibited a small pre-edge feature that was assigned to a 1s → 3d transition which is quadrupole-allowed but dipole-forbidden in octahedral symmetry.546 Perhaps not surprisingly, these results imply V and Mo are in the same oxidation state in their respective clusters, and similarly coordinated (though this then indicates that the d-electron configuration differs, d3 for Mo vs d2 for V).268,546
Further Fe Kα- and Kβ-HERFD XAS measurements of MoFe and VFe, as well as Mo and V-containing cubane analogues, have revealed that the dominant pre-edge features of the two proteins are nearly superimposable (Figure 13).232 Both also exhibit an additional pre-edge feature, which has been attributed to a metal-to-metal charge transfer (MMCT) transition from the Fe 1s to the low-lying unoccupied orbitals either the 4d of Mo3+ or 3d of V3+. However, this second pre-edge feature is considerably less intense in VFe, suggesting poorer overlap between the Fe donor and V acceptor orbitals.232 Additionally, while the position of the rising edge of the Fe XAS appears near identical in MoFe and VFe, VFe exhibits a considerably more intense white-line feature. The intensity of this feature has been found to correlate with Fe oxidation state in other iron–sulfur clusters (including reduced P-clusters and FeMoco), increasing upon reduction.137,203,228 Taken together with the changes observed in the Fe VtC XES, the average valency of the Fe centers of FeVco appears lower than that of FeMoco, likely exhibiting a total valency of V3+3Fe3+4Fe2+.232 The greater reduced complement of ferrous iron in the resting form of FeVco could explain why FeVco is able to bind CO in the resting state, while in the case of FeMoco, the cluster must be reduced to bind CO.539
As shown in Figure 50, CO binding in the VFe resting-state produces a new rhombic, S = 1/2 signal (g = [2.09, 1.99, 1.91]), analogous to the one observed for MoFe in the loCO state (see section 4.10).539 Incubation of apo-VFe (lacking FeVco) with CO did not lead to any modulation of the EPR spectrum, demonstrating that this new CO-induced signal arises from binding to FeVco.548 Interestingly, the intensity of this new EPR signal correlated with the potential of the reductant used (DT, Eu2+-EGTA, or Eu2+-DTPA), suggesting a redox dependence of CO binding to the VFe protein.539 Incubation under turnover conditions led to a decrease in the intensity of this EPR signal and the production of C2H4 and C2H6, demonstrating reductive coupling. When the CO pressure was increased to 2.6 atm in the presence of Eu2+-DTPA, an additional EPR signal arose (g = [2.13, 2.01, 1.97]), resulting in a “hiCO” spectrum with presumably multiple bound CO molecules bound to FeVco.548 Interestingly, a similar signal appeared when VFe was incubated with CO under low electron flux turnover conditions. However, GC-MS analyses after VFe turnover with labeled substrates revealed that only the CO moiety in the loCO conformation was catalytically competent and that the extra CO molecules in the hiCO state were not involved in C–C coupling. Consistently, intensity of the loCO EPR signal correlated with the decrease of N2 reduction in inhibition studies, whereas the signal corresponding to additional CO binding appeared only once N2 reduction was abolished. Hence, given that the inhibition of N2 reduction in V N2ase by CO may be distinctly different than that observed in Mo N2ase, it is possible that CO and N2 could compete for the same binding site in VFe, while additional molecules of CO would bind independently.
Figure 50.

Experimental (top traces) and simulated (bottom traces) EPR spectra of CO-bound VFe of A. vinelandii. (a) loCO, (b) hiCO, and (c) difference spectrum. Reproduced with permission from ref (548). Copyright 2018 John Wiley and Sons.
5.1.2. The P-Clusters of VFe
Despite decades of study, the structure and oxidation state of the P-clusters of V N2ase remains a subject of controversy. In contrast to resting-state Mo N2ase, whose P-clusters are all-ferrous and therefore diamagnetic, results reported for V N2ase have not been as clear-cut. 57Fe Mössbauer measurements of 57Fe-enriched VFe in the DT-reduced state have identified a diamagnetic component consisting of three quadrupole doublets, characteristic of P-clusters in the resting PN state.542 The observed ΔEQ and δ parameters for the Fe2+, D, and S iron sites are very similar to those reported for the P-clusters of Mo N2ase (see section 4.2),390,391 and calculations of the relative contributions of these components to the total spectrum confirmed that the iron composition of the P-clusters of VFe is similar to that of MoFe.542 Just over half of the total iron content in DT-reduced VFe was found to be attributable to the P-clusters in a diamagnetic state, implying that all EPR signals detected could be attributed to FeVco. This is also consistent with earlier low-temperature MCD studies which identified the P-clusters in V N2ase as diamagnetic in the DT-reduced enzyme and paramagnetic in the thionine-oxidized enzyme.184
However, EPR studies have suggested that the P-clusters have different electronic or structural properties in V N2ase (Figure 51).500 When oxidized using indigo disulfonate (IDS), the P-clusters of VFe remain EPR silent.500 In contrast, for MoFe, a strong signal is observed in parallel-mode EPR which has been associated with the two-electron oxidized state of the P-cluster (P2+), with an apparent g1 value at 11.8 in Azotobacter vinelandii and 16.0 in Gluconacetobacter diazotrophicus, as discussed in section 4.2.397,401,404 Furthermore, an S = 1/2 EPR active species with g = [2.03, 1.92, 1.92] which integrates to 0.9–1.0 spin per dimer has been observed in DT-reduced forms of both wild-type VFe and apo-VFe purified from the ΔnifB strain of A. vinelandii.20,500,543,54957Fe Mössbauer experiments performed on holo-V N2ase had previously attributed this signal to FeVco,542 which is naturally absent in VFe purified from ΔnifBA. vinelandii.404 An analogous S = 1/2 signal (g = [2.06, 1.95, 1.82]) has been described in partially oxidized Mo N2ase and attributed to the P-cluster in the P1+ state.400 Therefore, this S = 1/2 signal in V N2ase may also arise from a one-electron oxidized form of the P-clusters of VFe. Fe K-edge XAS studies of apo-VFe have corroborated this observation, as the rising edge of apo-VFe purified from ΔnifBA. vinelandii is shifted to slightly higher energies compared to apo-MoFe, indicating a slightly more oxidized P-cluster.550 The P-clusters of V N2ase could then resemble the FeS clusters present in Mo N2ase purified from ΔnifHA. vinelandii, precursors of the mature P-clusters.550 In both Mo and V N2ases, the iron protein has been identified as an essential player of P-cluster maturation (see section 6.5).550−552
Figure 51.
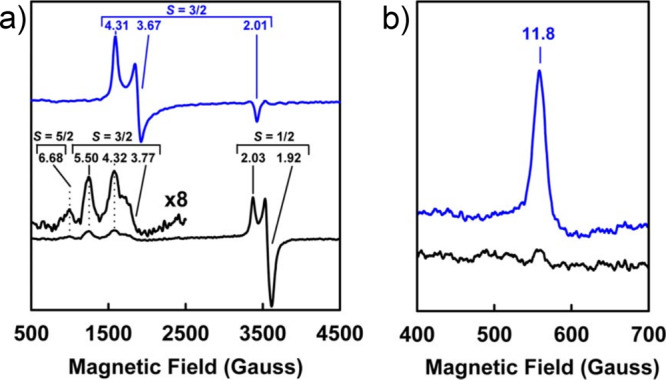
EPR spectra of MoFe (blue) and VFe (black), (a) perpendicular mode, both proteins DT-reduced; (b) parallel mode, both proteins IDS-oxidized. The signal observed for MoFe in (b) has been affiliated with the P2+ state of the cluster, as discussed in section 4.2. Spectra recorded at 15 K, 50 mW. Adapted with permission from ref (500). Copyright 2009 National Academy of Sciences.
5.1.3. FeVco
While the S = 1/2 signal is considered more debatable, all EPR studies have identified features consistent with an S = 3/2 ground state of FeVco, both in VFe and as an extracted cofactor (Figure 52).170,541 In VFe from Azotobacter chroococcum, several S = 3/2 signals are observed, with possible corresponding inflections at g = 5.6, 4.35, 3.77.170 Similar features at g = 5.5, 4.32, 3.77 have also been observed in VFe extracted from A. vinelandii, which may correspond to two different S = 3/2 species with varying rhombicity.20,500,543,549,553 Additional signals have also been found with inflections at g = 5.74 and 5.40 attributed to an highly rhombic S = 3/2 spin system with negative zero-field splitting (D = −0.74 cm–1).312 These values appear to differ slightly depending on the method of purification: Hales and co-workers initially reported g = 5.80 and 5.40,20,553 then 5.68, 5.45 (D = −0.74 cm–1, E/D = 0.29), which shifted to 5.71, 5.42 (E/D = 0.28) following reduction.554 Close inspection of these spectra clearly show at least two rhombic S = 3/2 are present in all of these cases.
Figure 52.
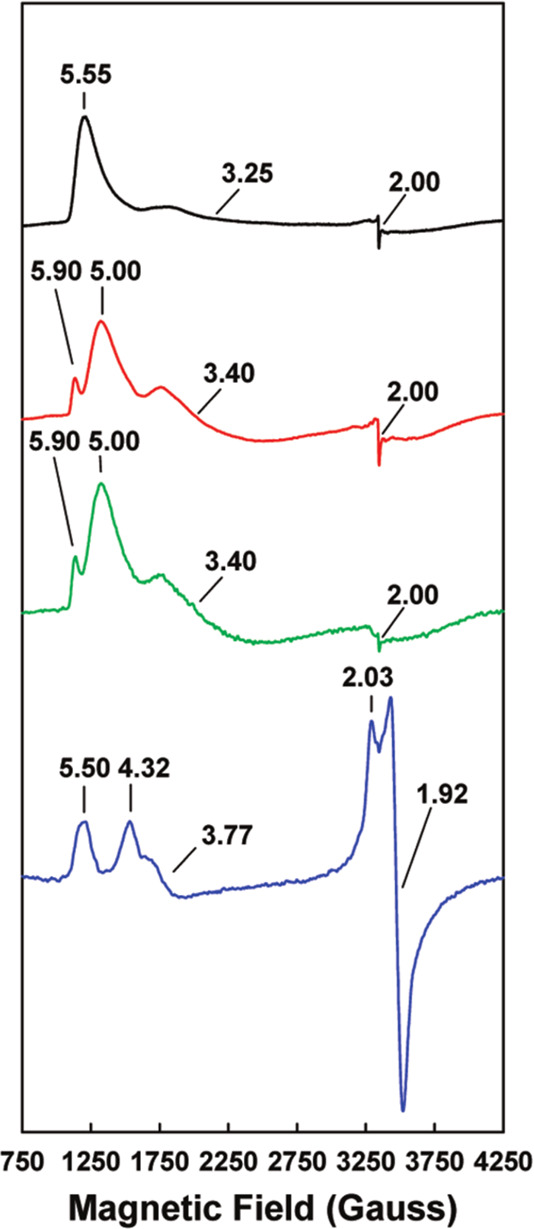
EPR spectra of FeVco extracted in N-methylformamide (NMF) (black), extracted in NMF in the presence of 10 mM thiophenol (red), extracted in NMF in the presence of 10 mM 1,4-benzene dithiol (green), and in native VFe (blue). Apparent g values are indicated. Reproduced with permission from ref (541). Copyright 2010 American Chemical Society.
When looking closely at difference spectra of as-isolated minus enzymatically reduced VFe, the features at geff = [6.7, 5.3, 4.3] come out quite clearly: the former two seem to be associated with an excited state and were assigned to the excited doublet of an S = 5/2 spin system (D = −1.6 cm–1, rhombicity E/D = 0.029). Similar signals at g = 6.67 and 5.30 were observed in partially oxidized MoFe, suggesting that these signals correspond to P-clusters in the P1+ state. It should be noted that the MoFe g = 6.67 signal is narrower, suggesting a different environment of the P-clusters.554
5.1.4. Oxidized Forms of V N2ase
Oxidation of V N2ase by thionine was found to be reversible when performed under strictly anoxic conditions. The UV–visible spectrum of thionine-oxidized V N2ase resembles the one of V N2ase oxidized under air, where the shoulder at 380 nm characteristic of the reduced N2ase disappears and is replaced with shoulders near 330 and 420 nm.170 These shoulders were less marked in the spectrum of V N2ase from A. vinelandii after oxidation by ferricyanide (see Figure 53) than in the thionine-oxidized V N2ase of A. chroococcum.170,549
Figure 53.
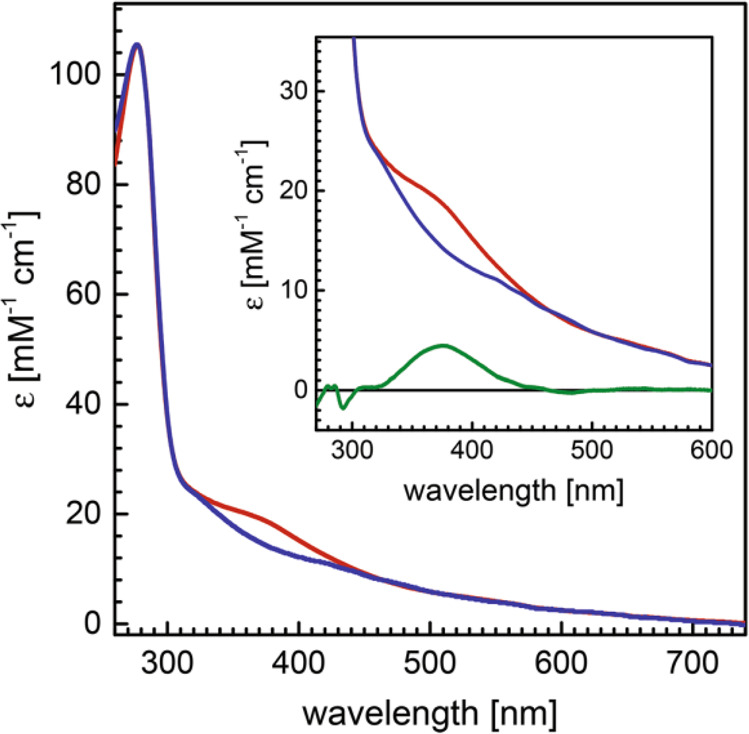
UV–visible spectra of VFe from A. vinelandii, in the reduced (red) and ferricyanide oxidized (blue) states. The inset shows the disappearance of the shoulder at 375 nm (difference spectrum, green). Adapted with permission from ref (549). Copyright 2017 Springer Berlin Heidelberg.
Early V K-edge XAS experiments demonstrated that features in the pre-edge and rising edge were similar for thionine oxidized, DT-reduced, and turnover (electron flux ∼1:1 VFe:FeP) V N2ase, suggesting that no major change occurs in the local environment of vanadium upon either oxidation or reduction of the enzyme. This was corroborated by EXAFS analysis, which demonstrated essentially no changes in the first shell coordination distances between all three samples.555 The relative intensity of the pre-edge feature and comparison with model complexes pointed toward a distorted octahedral coordination environment for vanadium in all states. Fourier transforms of the V EXAFS measurements indicated that distances of first-shell atoms to V, with V–S and V–Fe of approximately 2.3 and 2.75 Å respectively, were similar in both the thionine-oxidized and DT-reduced samples. In contrast, Fe EXAFS revealed that small changes occur in the Fe–S and short Fe–Fe distances between the reduced and oxidized forms of VFe. Upon oxidation by thionine, the average Fe–S distance decreased from 2.32 to 2.29 Å, and the long Fe–Fe interaction increased from 3.78 to 3.74 Å; meanwhile, the average short Fe–Fe distance increased slightly from 2.67 to 2.69 Å.200 These slight changes in bond lengths suggest that no major cluster reorganization occurs upon oxidation, but instead minor distortions consistent with the oxidation of an iron–sulfur cluster have occurred.
5.1.5. Lessons Learned from Hybrid Enzymes
Much can be learned about the influence of the cofactor construction vs protein environment by studying hybrid enzymes: either Mo N2ase artificially maturated with FeVco, or V N2ase artificially maturated with FeMoco. These studies have provided mixed results, with varying substrate reducing capabilities depending on organism and preparation method.540,541,550,556−558 While the product profiles for many of the common substrates/inhibitors of N2ases (N2, C2H2, CO) have been well explored, thus far EPR has remained the sole tool used to spectroscopically characterize these hybrids.
The EPR spectrum of the FeMoco-containing VFe hybrid generated by the reconstitution of apo-VFe with NMF-extracted FeMoco displays a broad S = 3/2 signal with geff = [4.65, 3.49, ?], decreasing in intensity upon turnover.557 The rhombicity of this signal (E/D = 0.093) is considerably higher than that of FeMoco (E/D = 0.053), indicating the protein scaffold imparts greater rhombicity in the cluster; however, it is still well below that of FeVco (E/D = 0.29). It should be noted that the orientation of FeMoco in the active site pocket of V N2ase may not be well-defined, as suggested by the broadness of the S = 3/2 signal. Subsequent studies of FeMoco-containing VFe generated in vivo (through the suppression of the nifDK genes responsible for generating MoFe) produced a similar EPR signal with inflections at g = 4.48 and 3.46.558 However, an additional inflection at g = 5.36 was also observed, implying an additional S = 3/2 species was present. This is similar to the EPR spectrum of native VFe, in which two unique S = 3/2 species with varying rhombicities are observed (see section 5.1.3).
MoFe reconstituted with NMF-extracted FeVco appears less efficient in both N2 and acetylene reduction, although it is capable of producing ethane from acetylene.540,541 This FeVco-containing MoFe hybrid exhibits broad inflections around g = 5.50, 4.32, and 3.72, again implying the presence of at least two S = 3/2 species with varying rhombicity as also observed in both native VFe and FeMoco-substituted VFe.541 Interestingly, substitution of FeMoco into VFe significantly bolsters the amount of hydrocarbon product observed during turnover in the presence of CO.558 This is in contrast to FeVco-substituted MoFe, which produces a substoichiometric amount of reduced hydrocarbon product from CO, similar to native MoFe.540 These results imply the protein scaffold plays a significantly greater role in CO reducing ability than the cluster. On the other hand, electron allocation in N2 reduction by the FeMoco-substituted VFe appears more similar to that of MoFe than VFe, suggesting the identity of the cluster may play a larger role in N2 reduction.557 Taken together, these studies have demonstrated that both cofactor and protein scaffold are important for tuning the activity of N2ases, with the magnitude of the roles of these two factors varying depending on substrate. However, it seems that for either MoFe or VFe the native cofactor in its respective protein scaffold is required for optimal efficiency in N2, H+, or C2H2 reduction.
5.1.6. A Late Turnover Intermediate?
Recently, following isolation using a particular set of reductant conditions, V N2ase was isolated in a new form described as a turnover state, whose crystal structure revealed a light atom replacing a belt sulfide.454 The EPR spectrum of this new species displayed the same signals as the resting state, albeit with significantly greater intensities relative to the S = 1/2 feature at g ∼ 2. This species was proposed to correspond to either the E6 or E8 state of N2ase, with a nitride ligand bound to two iron atoms.454 QM/MM calculations have since hinted this ligand may instead be a hydroxo group, possibly originating from a deprotonated water bound to the cofactor.559 These studies underline the role played by belt sulfides and neighboring iron atoms in ligand binding, already highlighted by works on inhibitors (see CO, section 4.10).
In conclusion, spectroscopic analyses on V N2ase have revealed strong similarities to Mo N2ase, consistent with the proposition of a universal mechanism for N2 reduction by N2ases.535 Further spectroscopic studies, notably investigating the turnover states of VFe, will be important for shedding light on the impact of the heterometal on the mechanism of N2 reduction, especially the reductive elimination of H2 and the binding of N2.
5.2. Fe N2ase
Fe N2ase was the last of the three known N2ases to be discovered, first in Azotobacter vinelandii three decades ago, and a few years later in Rhodobacter capsulatus and Rhodospirillum rubrum.19,560,561 It has been characterized as a heterometal-free N2ase, insofar as the metallocofactors do not contain any metals besides Fe.562,563 So far, little is known about the mechanism of this enzyme, and no kinetic model comparable to the Lowe–Thorneley model has been proposed; however, recent assays of N2, H+ and C2H2 reduction by Fe N2ase of A. vinelandii suggested that this enzyme follows a similar reductive elimination/oxidative addition pathway to Mo N2ase, with an “eight-electron” stoichiometry.535,564
Genetic analyses have led to the hypothesis that the catalytic cofactor of Fe N2ase would be analogous to FeMoco and FeVco, with the heterometal being replaced by an iron atom. In particular, the nifB and nifV genes are required for Fe N2ase activity, which suggests that FeFeco contains a hexavalent carbide and binds a homocitrate molecule (see section 6 for more details regarding the nifB and nifV). Likewise, the cysteine and histidine residues that coordinate the catalytic cofactor in Mo and V N2ases are also conserved in Fe N2ase.565 Production of H2 by Fe N2ase is much higher than either Mo or V N2ase, and it is less inhibited by N2. Under high N2 pressure, Mo N2ase produces NH3:H2 in a 2:1 ratio; this goes up 2:7.5 in Fe N2ase.563 Fe N2ase also reduces acetylene in a pH-dependent fashion, with up to 5.5% of acetylene reduced to ethane at pH 6.6.563 Another striking difference is its capacity to produce methane (CH4) from carbon dioxide (CO2), which has been evidenced in several diazotrophic bacterial species in vitro and shown to be catalyzed by Fe N2ase driven by its corresponding FeP and ATP hydrolysis.569 The production of hybrid enzymes consisting of Fe N2ase maturated with FeMoco or the contrary show further similarities between FeMoco and FeFeco.328 FeMoco-containing Fe N2ase is capable of fixing N2, as well as reducing C2H2 in C2H4,566,567 and Mo N2ase reconstituted with FeFeco can reduce acetylene to ethylene.568
5.2.1. FeFeco
In contrast to FeMoco and FeVco, the DT-reduced FeFeco of Fe N2ase has an integer spin ground state, likely S = 0. This may arise from the antiferromagnetic coupling of the Fe atoms in the cofactor. An EPR spectrum (Figure 54B) of the reduced protein (measured at 4 K, 20 mW) has been shown to produce a weak resonance in the g = 1.9–2.1 region, which has not been found to correlate with catalytic activity;563 in other cases, however, no signal has been observed.561 No S = 3/2 signals are observable, and a signal observed at g = 5.44 signal was attributed to either an altered FeFe cofactor or a cofactor breakdown product.562,56357Fe Mössbauer measurements confirm these results, where both the FeFeco and P-clusters of DT-reduced Fe N2ase are characterized as having a diamagnetic ground state.204
Figure 54.

EPR spectra of the MoFe (A) and FeFe proteins from R. capsulatus (B, reduced; C, oxidized). Spectra (A,B) collected at 4 K, 20 mW; spectrum (C) collected at 16 K, 100 mW. Adapted with permission from ref (563). Copyright 1996 John Wiley and Sons.
Similar to MoFe and VFe, 57Fe Mössbauer measurements of FeFe performed at 77 K revealed three main spectral components: a ferrous impurity, the P-cluster, and FeFeco.204 The similar integrated absorption area of FeFeco and the P-cluster supported that the FeFe cofactor contains eight iron atoms. The FeFeco component of the spectrum could be fit using two doublets of equivalent intensity with δ = 0.37/0.42 mm/s and ΔEQ = 0.74/1.00 mm/s. These parameters are very similar to those of FeMoco and FeVco, implying a similar electronic structure. Combining the average isomer shift of ∼0.4 mm/s with its diamagnetic ground state and even number of Fe atoms is suggestive that FeFeco contains an even number of ferrous and ferric sites.204
Further EXAFS studies highlighted the structural similarities between FeFeco and the catalytic sites of Mo and V N2ases.204 Fourier transforms of Fe K-edge EXAFS showed that the average 2.6 and 3.7 Å short/long Fe–Fe distances that were previously observed in FeMoco and FeVco are also retained in FeFeco. The 12 Fe–Fe scatterers at ∼3.7 Å associated with the diagonals of the quadratic faces are present in FeFeco. The EXAFS analysis also revealed a short Fe–S scattering path at 2.3 Å present in FeFeco and in a [Fe6S6Cl6]3– prismane model, similar to that observed in FeMoco and FeVco. Simulations based on the spectra showed that the only possible arrangement of Fe atoms in FeFeco is the trigonal prismatic arrangement of FeMoco.204 Taken together, these results suggest that the structure of FeFeco is homologous to FeMoco and FeVco, with the heterometal exchanged for an iron atom.
5.2.2. Fe N2ase: Oxidized and Turnover States
In contrast to the DT-reduced enzyme, EPR-active species are observable in Fe N2ase under both oxidative and native turnover conditions.563 Oxidation using ferricyanide results in the appearance of four unique signals (Figure 54C): (1) a weak resonance at g = 6.50, which was originally suggested to belong to an S = 9/2 species, (2) a strong signal at g ∼ 4.30, which may correspond with adventitious Fe3+, and (3) two overlapping S = 1/2 signals, the first rhombic and narrow with g = [2.00, 1.98, 1.96] the second relatively broad, with ∼g = [2.27, 2.06, ?] (and only detectable below 20 K). This first S = 1/2 signal forms irreversibly upon oxidation and therefore has been attributed to a damaged cluster. The second was found to be transient, i.e., disappearing upon reduction or further oxidation, implying the corresponding species could go through two redox steps.563 This signal could either arise from an atypical P-cluster or from an oxidized state of FeFeco.
Under turnover conditions in the presence of N2, C2H2, or even simply H+, a new S = 1/2 EPR signal, with g = 1.96, 1.92, and 1.77, was detected.563,570 In contrast, no signal has been detected with Mo N2ase under the same experimental conditions.570 As the strongest signal appears in the presence of dinitrogen, it may represent a reduced state of the FeFe protein.
5.2.3. The P-clusters of Fe N2ase
Like in Mo and V N2ase, the P-clusters of Fe N2ase are diamagnetic in the DT-reduced state.204 In contrast to V N2ase, no S = 1/2 EPR signal was detected in the as-isolated enzyme, suggesting no oxidized P-cluster is present. 57Fe Mössbauer studies have presented similar parameters for the components of P-clusters in both Fe and Mo N2ase, suggesting both proteins bear effectively identical P-clusters. The iron composition of Fe N2ase P-clusters is similar to those in Mo and V N2ases, with each cluster containing two Fe2+, five D, and one S sites (section 4.2).542 Fe K-edge EXAFS measurements have also confirmed the similarities between the P-clusters of Fe and Mo N2ases.204
5.2.4. Zn in Fe N2ase
The detection of high amounts of Zn, 1–2 ions per protein, presented the question whether Zn was involved in the cofactors of Fe N2ase.563 Zn K-edge EXAFS measurements did not show any 2.6 Å scatterers, which would otherwise correspond to a Zn–Fe interaction, and revealed strong Zn–N features (2.02 Å) as well as Zn–S ligands (2.33 Å).204 The Zn atoms detected in Fe N2ase are therefore not part of the cofactors and are likely coordinated by cysteine residues elsewhere in the enzyme.
5.3. W N2ase
A small number of studies have reported the isolation and spectroscopic characterization of a W-incorporated N2ase from both Azotobacter vinelandii and Rhodobacter capsulatus.571,572 W N2ase is actually the nif-encoded Mo N2ase in which the Mo atom of FeMoco has been replaced by W. Studying such an artificial N2ase, in which the presence of a non-native metal modulates activity, could help in answering some critical questions about the mechanism of substrate reduction.
W N2ase isolated from A. vinelandii contained as much Mo as W.571 In contrast, W N2ase isolated from R. capsulatus was found to contain one W atom and a negligible amount of Mo (0.01 per protein), and the iron quantification was consistent with this protein containing only one P-cluster and one FeWco.572 No N2 activity could be detected, and acetylene reduction activity was found to be 1000-fold lower than that of Mo N2ase, with 1 nmol ethylene formed per min per mg of protein. However, this enzyme still exhibited high rates of H2 evolution under an argon atmosphere, corresponding to 25% of the H2 evolution activity of native Mo N2ase. Additionally, acetylene was not found to inhibit this hydrogen evolution activity.
Two major S = 3/2 and S = 1/2 signals were observed in the EPR spectrum of W N2ase of R. capsulatus (Figure 55). The S = 3/2 signal resembles that of FeMoco in Mo N2ase of this organism (geff = [4.29, 3.67, 2.01]). The small feature at g = 3.67 in the EPR spectrum of W N2ase has therefore been attributed to a small contamination of Mo N2ase maturated with FeMoco, consistent with the detection of 0.01 atoms of Mo per tetramer. The rhombic S = 3/2 signal of FeWco has observed g values at 4.19 and 3.93, the putative third geff component likely masked by the prominent S = 1/2 signal (Figure 55). These values imply a slightly smaller rhombicity of FeWco in comparison with FeMoco. The intensity of this signal was found to be most intense at 4 K and decreased at higher temperatures, indicating positive zero-field splitting. Under turnover conditions, the EPR intensity of this S = 3/2 is only decreased by 20%, implying that FeWco is either not as easily enzymatically reduced as FeMoco or that the reduced states of the cluster are more susceptible to H2 production. No decrease in the amplitude of the W N2ase-specific EPR signals was observed for the protein isolated from A. vinelandii under turnover conditions, suggesting that FeWco was not reduced.571
Figure 55.

EPR spectrum of WFe from R. capsulatus under turnover conditions (thereby removing contributions from contaminating MoFe). Adapted with permission from ref (572). Copyright 2003 American Chemical Society.
Further spectroscopic characterization, particularly by X-ray spectroscopy, could shed light on the specificities of W N2ase and the redox properties of FeWco, possibly explaining the major differences observed in catalysis for this enzyme compared to Mo N2ase.
5.4. The Iron Protein of Alternative Nitrogenases
The FeP reductase components of V and Fe N2ases (termed VnfH and AnfH, respectively) were isolated later19,20 and have been far less characterized than their Mo N2ase counterpart (NifH) discussed in section 4.1. However, most studies tend to show that all reductases share a similar structure and function, which is consistent with their high sequence similarity; vnfH genes share ∼90% identity with nifH genes, while anfH genes show ∼60% identity with both vnfH and nifH genes.53 Interestingly, cross-reactivity is observed for components of Mo and V N2ases, whereas Fe N2ase specifically requires AnfH for activity.573 Accordingly, recent elucidation of the crystal structure of ADP-bound VnfH showed a high degree of structural conservation with NifH.574 UV–vis spectra of VnfH also showed a high similarity to NifH, with the reduced protein spectrum displaying a pronounced shoulder near 360 nm while the oxidized spectrum showed pronounced shoulders at 320 and 400 nm.575 IDS-oxidized VnfH is EPR-silent, while the superreduced form displays a g = 16.4 signal in parallel mode measurements, as also observed in NifH.576 The EPR spectra of reduced VnfH and AnfH are also similar to that of reduced NifH, displaying a mixture of S = 1/2 and S = 3/2 signals originating from the [Fe4S4]+ cluster. Furthermore, binding of MgATP induces a shift from a rhombic to an axial signal in the cluster of VnfH, consistent with a change in the conformation of the active center upon nucleotide binding.20,577 However, the intensities of both signals are smaller in NifH than in the other iron proteins, which may indicate a difference in the cluster oxidation state; also, the ratios between the S = 1/2 and S = 3/2 signals are slightly higher for VnfH and AnfH than for NifH.577 All EPR spectra of the reduced proteins became identical upon mixing with 50% glycerol.577 Likewise, all reduced reductases displayed pre-edge features of similar intensity at the Fe K-edge, implying a shared coordination geometry of the Fe atoms. However, for NifH, both the pre-edge feature and rising edge were shifted toward higher energy by 0.2 eV, suggesting that the Fe atoms in NifH are more ferric than in the other iron proteins. EXAFS analyses confirmed that the clusters of all reductases share a common structure. Yet, whereas the clusters of all three reductases could be modeled by two stacked [Fe2S2] rhomboids offset by 90°, only the cluster of NifH can be represented by a model with two unequal [Fe2S2] rhomboids which are bent to a greater degree out-of-plane relative to each other. In the first “universal” model, every Fe atom in the cluster participates in a tetrahedron, with one Fe neighbor at ∼2.50 Å and two more at ∼2.70 Å. In the second “NifH-specific” model, the iron–sulfur distances are decreased and the Fe sites are not equivalent.
In conclusion, while sequence analyses and cross-reactivity experiments highlight similarities between NifH and VnfH and differences with AnfH, spectroscopic studies have shown that the clusters of VnfH and AnfH share analogous geometry and electronic structures and differ only slightly from NifH. Further spectroscopic experiments with nucleotide-bound proteins and structural characterization of AnfH are necessary to reconcile those divergences.
6. Cluster Biosynthesis
As highlighted extensively in this review, the complex mechanism of biological nitrogen fixation has been a spectroscopic playground for well over half a century. For almost as long, so too have the mechanisms of nitrogenase metallocluster biosynthesis and component protein maturation. In this subsection, the processes involved in nitrogenase metallocluster biosynthesis will be described with a focus on Mo N2ase and emphasis on the insights revealed by spectroscopy. A more detailed review of N2ase metallocluster biosynthesis and insertion can be found in the review from Rubio and co-workers,578 also in this series, and this subject has been covered in several recent reviews.25,47,51,579
Before proceeding, we would like to bridge the protein nomenclature used in sections 1–5 with the gene nomenclature of N2ases that is commonly employed in biochemistry and more suitable for discussing the topic of cluster biosynthesis. Most of the genes required for synthesis of MoFe are located in the nif (nitrogen fixation) regulon (a type of gene cluster), while those specifically associated with VFe and FeFe are located in the vnf (vanadium-dependent nitrogen fixation) and anf (alternative nitrogen fixation) regulons. The specific proteins are then encoded by genes with names like nifA, nifB, etc., and the proteins produced from these genes are named NifA, NifB, etc. Specifically, in the nif regulon, the genes encoding FeP and the two subunits of MoFe are nifH, nifD, and nifK, respectively, and their protein counterparts are NifH, NifD, and NifK, respectively. nifDK then refers to the pair of genes encoding MoFe or NifDK. Likewise, in the vnf regulon, we have vnfH coding for VnfH and vnfD, K and G coding for VnfDKG, and in the anf regulon, we have anfH coding for AnfH and anfD, K, and G coding for AnfDKG.
6.1. Overview
All three metalloclusters of nitrogenase (M-cluster, P-cluster, and the F-cluster of FeP) are synthesized initially from free iron (Fe3+ and Fe2+) and sulfide, which is liberated via cysteine desulfuration by the proteins NifS and NifU.580,581 Iron–sulfur clusters can be assembled on NifU582 and transferred to NifH,583 NifB,584 and NifDK581 (also VnfDKG and AnfDKG) either directly or via an alternative scaffold, NifIscA.585 The mature P-cluster is formed from [Fe4S4] clusters, presumably also synthesized by NifSU, in a process requiring the action of NifH550 and possibly NifZ586 (a potential chaperone protein) and additional accessory proteins.587 NifB accepts three [Fe4S4] clusters, two are the substrates for cofactor biosynthesis, while the third is required for the catalytic function.588 NifB is a radical S-adenosylmethionine (SAM) enzyme that provides a methyl group from which the central carbide is formed,589,590 donates an additional sulfide,591 and fuses the two substrate [Fe4S4] clusters together with the carbide and sulfide into a precursor of FeMoco (as well as FeVco and FeFeco), known as the NifB cofactor592 (or the L-cluster). This precursor cluster is transferred to NifEN (or VnfEN), possibly via an intermediate cluster binding protein NifX.593 In NifEN, the L-cluster is converted to the final M-cluster via the action of NifH by replacing one Fe ion with Mo and adding homocitrate.594 Homocitrate is synthesized by NifV,595 and Mo is thought to be transferred to NifH from NifQ along with homocitrate.596 Transfer of mature FeMoco to NifDK is also thought to require the action of NifH597 and possibly a carrier protein, NifY or NafY.598
6.2. The Assembly of [Fe4S4] Clusters on NifU
It has long been known that NifS and NifU are essential for the function of both FeP and MoFe.580,581,599,600 NifS was elegantly determined to be a pyridoxal P dependent cysteine desulfurase601,602 capable of reconstituting FeP from l-cysteine and Fe2+.603 NifU, produced heterologously in Escherichia coli, was demonstrated to be a homodimer with one [Fe2S2] cluster per monomer, bound to its central domain.604 Through a combination of UV–vis, RR, EPR, and VTVH-MCD spectroscopies, the [Fe2S2] cluster was demonstrated to be cysteine coordinated but with distinct properties from other well-characterized classes of [Fe2S2] ferredoxins.604 The central domain of NifU has sequence homology to the bacterioferritin-associated E. coli [Fe2S2] ferredoxin.605,606 The cluster redox potential was determined to be −254 mV vs SHE by UV–vis spectroelectrochemical titration, suggesting a possible role in electron transfer. Subsequently, NifU was shown to bind Fe3+ in its N-terminal domain.607 The UV–vis and RR spectroscopic properties of the Fe3+ ion were shown to be very similar to those of rubredoxin. The Fe3+ bound to the N-terminal domain could be transformed into labile [Fe2S2] clusters by the cysteine desulfurase action of NifS.608 These clusters presented UV–vis and RR spectra indicative of complete cysteine coordination and a cluster geometry similar to that of human ferrochelatase. NifU and NifS are capable of directly activating apo-nifH (FeP),583 suggesting that the [Fe2S2] clusters formed on NifU must somehow go on to form [Fe4S4] clusters either in NifU itself or NifH. NifU was also shown to be capable of assembling labile [Fe4S4] clusters both in its N-terminal IscU-like domain and its C-terminal NfuA-like domain.582 Using Mössbauer spectroscopy, the accumulation of 57Fe labeled [Fe4S4] clusters could be observed in the C-terminal domain, while [Fe2S2] clusters accumulated in the N-terminal domain, followed by reductive coupling to yield a [Fe4S4] cluster bridging both subunits of the NifU dimer. Both the [Fe4S4] clusters in the C-terminal and N-terminal domains were capable of activating apo-NifH.
The capacity of NifSU to transfer [Fe4S4] clusters to NifB was suggested as ΔnifNSU strains of Klebsiella pneumoniae were unable to accumulate the intermediate NifB cofactor, while accumulation was still observed for ΔnifN strains (Figure 56).584 However, nifB overexpression could rescue the levels of NifB cofactor, suggesting that alternative iron–sulfur cluster synthesis pathways can also play a role. Importantly, the levels of active NifB do not appear to be altered in ΔnifNSU, indicating that the SAM cluster of NifB is not provided by NifSU. It is unknown whether NifSU is required for assembly of the permanent [Fe4S4] cluster (the O-cluster) in NifEN. The P-cluster in NifDK likely originates from [Fe4S4] clusters synthesized by NifSU because ΔnifSUAzotobacter vinelandii strains show reduced MoFe protein activity that cannot be recovered upon addition of FeMo cofactor.580 However, this may also be related to the importance of active NifH for P-cluster maturation. Thus, direct demonstration of the requirement of NifSU for insertion of the O- and P-clusters is still lacking.
Figure 56.

Initial stages of nitrogenase cluster biosynthesis. NifU (green ovals) assembles [Fe4S4] clusters in both its N-terminal (NT) and C-terminal (CT) domains using iron, presumably from an iron donor protein, and sulfur donated by NifS catalyzed cysteine desulfuration. NifU is likely to be the primary donor of [Fe4S4] clusters to NifH (orange) NifB (blue), the O-cluster in NifEN (yellow/pink), and the P-cluster precursors of NifDK (green/purple).
6.3. Synthesis of the 8Fe Precursor of FeMoco on NifB
The fundamental role of NifB in nitrogenase cofactor biosynthesis has long been recognized;600 however, spectroscopic studies of NifB were lacking for many years due to the instability of the purified protein. In 1994, Shah and co-workers were able to purify a FeMoco precursor, termed NifB-co, which accumulated in strains of Klebsiella pneumoniae lacking NifN.592 At that time, NifB-co appeared EPR silent and was, therefore, suggested to be diamagnetic, in contrast to isolated FeMoco, which exhibits an S = 3/2 signal similar to that observed in MoFe.410 It was subsequently shown using radioactive 55Fe and 35S labeling of NifB-co that the Fe and S from NifB-co were incorporated into FeMoco via NifEN and an additional protein, called “gamma”.609 Comparisons of the XAS and NRVS measurements of NifB-co bound to the putative chaperone protein NifX with FeMoco bound to NafY suggested that both cofactors contain 6Fe cores with a light interstitial atom,164 implying that the light interstitial atom was inserted by NifB. The presence of additional “capping” metals could not be confirmed; however, the authors suggested that an 8Fe structure was unlikely.164 Later, 57Fe Mössbauer spectroscopy on NifB-co bound to NifX demonstrated that this cluster is, in fact, composed of 8 Fe, with two spectroscopically distinct classes of Fe ions in a 3:1 ratio.610 These data also agreed with the conclusion that the cluster is diamagnetic. An alternative intermediate cofactor, the so-called “VK-cluster”, can be extracted from NifEN.593 This cluster was suggested also to possess eight Fe ions, but exhibits an S = 1/2 EPR signal both in the reduced and oxidized states. Possibly, slight modification of the 8Fe core structure leads to significant changes in the electronic structure of NifB-co, forming the VK-cluster.
A fusion protein between NifB and NifN allowed the purification and EPR spectroscopic characterization of active NifB.611 This study demonstrated that NifB contains at least two distinct [Fe4S4] clusters, one corresponding to the SAM cluster and the other that could be converted to an 8Fe precursor in a SAM-dependent mechanism (Figure 57). The same fusion protein was used to demonstrate that NifB inserted a central carbide ion into the 8Fe precursor589 via initial transfer to a [Fe4S4] cluster bridging sulfide.590 NifB homologues from two methanogenic nitrogen-fixing organisms (Methanosarcina acetivorans and Methanobacterium thermoautotrophicum) were identified to be much more stable than NifB from A. vinelandii and thus amenable to detailed analysis of the synthesis of NifB-co (also called the L-cluster).612 The three [Fe4S4] clusters bound to NifB from a separate methanogen (Methanocaldococcus infernus) were characterized by EPR in wild-type and mutant forms of NifB in which the SAM cluster, one of the auxiliary (K) clusters, or both the SAM cluster and K-cluster were disrupted. EPR spectroscopy revealed the presence of a [Fe4S4] cluster in the double cluster mutant, confirming the presence of three [Fe4S4] clusters in NifB.588 ESEEM measurements (discussed briefly in section 2.1) suggested the presence of proteinaceous nitrogen ligands bound to both of the K-clusters. However, in a subsequent study on the NifB of M. acetivorans, nitrogen ligation was found for only one K-cluster and that this was lost upon fusion of the K-clusters into the L-cluster.613 Furthermore, it was found that only the presence of the SAM cluster and nitrogen ligated auxiliary cluster were required for SAM activity and formation of methanethiol, the product of methyl transfer to cluster-bound sulfide. The final piece of the NifB puzzle was the origin of the ninth sulfur in the [8Fe-9S–C] L-cluster. This was recently elegantly demonstrated to arise from sulfite.591 The NifB-bound cluster in the absence of added sulfite, named the L*-cluster, was shown to be very similar in structure and electronic properties to the L-cluster.614
Figure 57.

FeMoco biosynthesis on NifB and NifEN. NifB catalyzes the formation of a carbide using S-adenosylmethionine (SAM), which inserts between the two K-clusters, displacing two μ-3 sulfides into “belt” positions forming the so-called L*-cluster. The L*-cluster then picks up an additional “belt” sulfide from a sulfite anion, giving the final product of NifB, the so-called L-cluster (also known as NifB-co). The L-cluster is then transferred to NifEN. Upon NifEN, a capping Fe is removed and replaced with a Mo and a homocitrate molecule is bound, both events requiring the action of NifH, to give the final FeMoco cluster (also known as FeMoco).
6.4. Processing of the L-cluster by NifEN
The fundamental role of NifEN as a scaffold for binding of FeMoco precursors was first suggested based on its high level of sequence homology with NifDK.615 NifEN was purified and found to possess an iron–sulfur cluster based on its UV–vis spectrum,616 which was later characterized by EPR, RR, and VTVH-MCD spectroscopies,189 and a redox midpoint potential of −350 mV vs SHE was determined. Transfer of the L-cluster from NifB to NifEN may occur directly or via alternative cofactor binding proteins such as NifX.593 A FeMoco precursor with a fairly isotropic EPR spectrum (g = 1.92) in the oxidized state was discovered on ΔnifHDK NifEN, which contains the interstitial carbide617 and which could be fully converted to a FeMoco-like species capable of activating MoFe purified from a ΔnifB strain of A. vinelandii (ΔnifB MoFe) using NifH, molybdate, homocitrate, and ATP.594
The ΔnifHDK NifEN bound cofactor was analyzed using Fe K-edge XAS and, by analysis of the EXAFS region, found to possess an average of 3.8 short-range Fe–Fe distances (3.3 at 2.66 Å and 0.5 at 2.88 Å) and 1.6 long-range Fe–Fe distances (0.8 at 3.68 Å and 0.8 at 3.80 Å).618 These data best correlate with an 8Fe cluster model. Furthermore, both the NifEN-bound FeMoco precursor and FeMoco itself exhibit a second pre-edge feature at 7113.6 eV that has been assigned as a metal–metal charge-transfer feature.618 The intensity of this pre-edge feature has been correlated with the heterometal identity in FeMoco and FeVco, with the former exhibiting more intense MMCT transitions. The weaker MMCT observed in the pre-edge data of the NifEN-bound FeMoco precursor thus provides further support for the 8Fe cluster model. Additionally, we note that ΔnifB MoFe and ΔnifB NifEN, both of which lack the FeMoco and FeMoco-like cofactors, also lack this second pre-edge feature, only exhibiting a single peak at 7112.2 eV.618 This may suggest that the structural modifications due to the presence a central carbide may allow for more facile MMCT in FeMoco and FeMoco-like clusters relative to typical iron–sulfur clusters. More recently, valence-to-core Fe Kβ X-ray emission spectroscopy showed the presence of an interstitial carbide in the NifEN-bound precursor.617 By comparing ΔnifHDK NifEN (containing the L-cluster) and ΔnifB NifEN (lacking the L-cluster), a Kβ′′ feature at 7099.7 eV was identified, which could only arise from demotion of an electron from a carbon-ligand-centered valence molecular orbital to the 1s core–hole.
Ultimately, the crystal structure of precursor-bound NifEN was solved, revealing the FeMoco precursor to be an [Fe8S9] cluster bound close to the surface of the NifE subunit, close to the O-cluster.619 Extraction of the 8Fe cofactor into NMF allowed this precursor to be spectroscopically characterized in the absence of the [Fe4S4] O-cluster.620 Thus, an interesting S = 1/2 EPR signal with g = 1.97 and 1.83 was observed for the reduced state of the cluster, while a signal at g = 1.93 was observed in the oxidized state. Fe K-edge XAS revealed very similar properties of the isolated 8Fe precursor to that of NMF-extracted FeMoco but with EXAFS that supported an 8 Fe model for the cluster.620 VTVH-MCD was used to compare the electronic structures of NifEN, NifEN bound with the 8Fe precursor, and NifEN bound with FeMoco.621 The permanent [Fe4S4] clusters of NifEN were found to have properties similar to those of typical [Fe4S4] clusters, although they were present in a mixed spin state with contributions from an S > 1/2 state. Both precursors were found to have similar electronic structures to FeMoco in MoFe, and FeMoco in NifEN was found to be in an S = 3/2 state, the same as in MoFe.
Cofactor processing on NifEN produces only small changes in the Fe K-edge EXAFS, going from two long Fe–Fe scatterers at 3.68 and 3.80 Å in the unprocessed precursor to one at 3.70 Å in the processed precursor, suggesting slight shortening of the Fe–Fe distances and possibly an increased symmetry in the cluster.622 The NifEN precursor, prior to processing with molybdate, produces no Mo K-edge X-ray absorption spectrum, while a spectrum very similar to that of MoFe is produced after processing. Small discrepancies are observed between processed NifEN and MoFe, including a slight shift (∼0.6 eV) of the absorption edge to higher energy and a small difference in white-line intensity.622 The Mo K-edge EXAFS of the NifEN precursor were best fit by assuming a reduced number of Mo–S and Mo–Fe scatterers compared with MoFe and three statistically disordered Mo–O/N scatterers at 2.13 Å or two types of Mo–O/N scatterers at 2.00 and 2.17 Å.622 Taken together, the Fe and Mo EXAFS data suggested that the NifEN bound precursor is very similar to FeMoco in MoFe but with slightly longer bonds.
At the same time, a ΔnifHDK NifEN form with a bound FeMoco-precursor containing Mo was purified and characterized.623 When measured using EPR, this precursor produced an isotropic g = 1.95 signal, similar to that observed by Hu and co-workers for their molybdenum-free NifEN-bound precursor.623 This suggested that this form of NifEN contained the same 8Fe FeMoco precursor but also with Mo bound at an additional site. Mo K-edge XAS suggested that Mo bound in this form had the same oxidation state as Mo in MoFe, but a very different ligand environment.624 The Mo K-edge of MoFe has additional structure in the white-line region around 20020 eV, while the Mo K-edge in NifEN has a shoulder at 20008 eV. Mo EXAFS analysis proposed a coordination environment with 3 Fe at a distance of 2.68 Å and 3 S at a distance of 2.35 Å but showed no evidence for O/N scatterers, suggesting a lack of homocitrate. However, the Mo was suggested not to be bound in a FeMoco precursor but in a separate [Mo-3Fe-4S] cluster. This was based on (i) the fact that the Mo K-edges of NifEN and MoFe suggest different Mo environments,624 (ii) NifEN had not interacted with NifH, which is required for the final steps of FeMoco synthesis,594 (iii) the NifEN-bound precursor can be extracted and shown to contain no Mo,593 and (iv) some preparations of NifEN may contain a [3Fe-4S] cluster,189,594 which could potentially bind Mo. However, it is not clear whether this mode of Mo binding is involved in the physiological incorporation of Mo into FeMoco on NifEN.
Purified NifEN (presumably with a bound FeMoco precursor) was found to be catalytically active for acetylene (∼50 nmol min–1 mg–1) and azide (∼70 nmol min–1 mg–1) reduction but not nitrogen reduction.625 Interestingly, these activities are only 1–2 orders of magnitude lower than in MoFe (∼2000 nmol min–1 mg–1 for acetylene and ∼600 nmol min–1 mg–1 for azide). The interactions of these substrates with the bound cluster were studied using EPR spectroscopy, demonstrating that acetylene produced a novel isotropic g = 2.02 signal during turnover conditions, while azide caused a slight perturbation to the normal S = 1/2 EPR signal originating from the NifEN bound precursor under both turnover and nonturnover conditions. These observations are in line with the sequence and structural homology of NifEN and NifDK.
Subsequently, Yoshizawa and co-workers showed that increasing the amount of DT present during maturation of the NifEN bound precursor allowed the complete maturation of the L-cluster to FeMoco, highlighting the crucial role of the reductant in this process.626 The final precursor obtained with this approach displayed an EPR spectrum similar to that of FeMoco in the reduced state and lacked the isotropic g = 1.92 signal characteristic of the partially maturated precursor. Furthermore, Mo K-edge XAS measurements demonstrated this final precursor was very similar to FeMoco in MoFe, where the EXAFS now more clearly defined the Mo–O (one at 2.12 Å and two at 2.24 Å), Mo–S (three at 2.37 Å), and Mo–Fe (two at 2.71 Å and one at 2.90 Å) distances. It was further demonstrated that the same maturation process could be used with V or Fe to generate FeVco and FeFeco on NifEN, at least partially.627
Omitting homocitrate from the maturation procedure led to the isolation of a form of the precursor on NifEN containing Mo but lacking homocitrate.628 This cluster had very similar properties to that of the precursor containing homocitrate but with subtle differences in the EPR and XAS spectra. The Mo K-edge was slightly shifted (<1 eV), and EXAFS revealed a more relaxed ligand sphere around Mo. Particularly interesting was the apparent inability of this cluster, lacking homocitrate, to be transferred to MoFe, suggesting homocitrate is crucial either for cluster transfer or in retaining the transferred cluster in MoFe. This is in contrast, however, with a previous study that identified a form of MoFe containing an unusual FeMoco cluster, proposed to contain Mo but lack homocitrate.629
6.5. P-Cluster Maturation and the Role of NifH
The P-cluster of NifDK is the initial entry point for electrons coming from NifH and plays a role in transferring electrons to the M-cluster in a complex series of events summarized as the deficit spending mechanism of electron transfer.62 Key to the function of the P-cluster is its ability to switch between different structural conformations depending on its redox state. This ability is related to the fact that it is composed of two [Fe4S4] clusters (P*-clusters) that have been fused together with the removal of one bridging μ3 sulfide ligand. Synthesis of these two [Fe4S4] clusters is probably carried out by NifSU.580 The maturation of the two P*-clusters into the P-cluster appears to be carried out by NifH, although a mechanism for this is not yet clear.
It was noted by Ribbe and co-workers that the P-clusters in MoFe purified from ΔnifB and ΔnifH strains were spectroscopically distinct.188,439,551 While DT-reduced ΔnifH MoFe exhibits a dominant axial EPR spectrum around g = 2, DT-reduced ΔnifB MoFe has a much lower proportion of this signal (Figure 58B, comparing 1 and 3).551 Similar EPR signals were observed much earlier in ΔnifH MoFe from A. vinelandii(629,630) and ΔnifB MoFe from Klebsiella pneumoniae.631 Gavini and co-workers reported that the signal integrated only to 0.1–0.3 spins per tetrameric protein.630 Ma and co-workers did not report the EPR spin quantitation of their signals, but observed that reconstitution of ΔnifH MoFe with a form of FeMoco lacking homocitrate, referred to as the MoFe cluster, reduced the intensity of this signal.629 However, the intensity of the signal dramatically increased to two spins per tetramer upon incubation with NifH, ATP, and DT. Hawkes and co-workers attributed their signal to a contaminant protein as the intensity of the signal varied between batches and appeared to diminish during protein purification.631 Additionally, Ribbe and co-workers observed a g = 11.8 signal in parallel mode EPR, arising from an integer spin ground state of the P2+ oxidation state of the P-cluster in ΔnifB MoFe when oxidized by IDS, as shown in Figure 58.551 IDS-oxidized ΔnifH MoFe completely lacks this signal. Subtle differences in the UV–vis spectra of the clusters were also observed (Figure 59), and while ΔnifH MoFe was shown to be incapable of oxidizing NifH, it did appear to facilitate ATP hydrolysis by NifH.
Figure 58.

Parallel (A) and perpendicular (B)-mode EPR spectra of DT-reduced and IDS-oxidized ΔnifB (1 and 2) and ΔnifH (3 and 4). Sample concentrations were 10 mg/mL. Reproduced with permission from ref (551). Copyright 2002 American Society for Biochemistry and Molecular Biology.
Figure 59.
UV–vis spectra of purified ΔnifB (solid line) and ΔnifH (dotted line) MoFe. Samples concentrations were 10 mg/mL. Reproduced with permission from ref (551). Copyright 2002 American Society for Biochemistry and Molecular Biology.
Fe K-edge XAS of ΔnifB MoFe and ΔnifH MoFe also differ significantly,439 particularly in the EXAFS region, where two distinct scatterers corresponding to Fe–S and Fe–Fe paths are observed for ΔnifH MoFe, while for ΔnifB MoFe the intensity of the Fe–Fe scattering path is greatly diminished; this was suggested to arise from phase-cancellation of the individual Fe–Fe scattering paths in ΔnifB and is also found to occur in MoFe.203 Thus, the authors concluded that their EXAFS data agreed with an 8Fe structure. Lastly, comparison of VTVH-MCD spectra of ΔnifB MoFe and ΔnifH MoFe support the notion that the former contains the mature 8Fe P-cluster, while the latter contains pairs of [Fe4S4] clusters, albeit with an unusually high spin contribution to the ground state.188
Hu and co-workers demonstrated that ΔnifH MoFe of A. vinelandii could be treated with NifH and ATP to recover some activity.550 However, this did not appear to be associated with any changes in the EPR or X-ray spectroscopic properties of the P-cluster. This suggests that while P-cluster maturation requires the activity of NifH, this alone is insufficient and other factors are needed. One of these factors appears to be NifZ. The ΔnifBZ strain of A. vinelandii produces a form of MoFe in which one of the P-clusters in the tetrameric MoFe protein has been maturated while the other has not.586 EPR spectroscopy revealed contributions from both the axial spectrum centered around g = 2 under reducing conditions (indicative of the immature P-cluster) and g = 11.8 in parallel mode under oxidizing conditions (indicative of the maturated P-cluster) in ΔnifBZ MoFe. Upon incubation with purified NifZ, NifH, and ATP, the S = 1/2 signal around g = 2 spectrum was lost and the signal intensity of the parallel mode feature increased approximately 2-fold, indicating that the second P-cluster was maturated. This process was also followed by VTVH-MCD.187
Later, it was shown that the immature P-clusters in ΔnifH MoFe could be completely maturated upon addition of large amounts of NifH, ATP, and DT.632 This was followed by the disappearance of the S = 1/2 EPR signal around g = 2 and appearance of the integer spin EPR signal at g = 11.8. Maturation was also followed by changes in XAS and EXAFS, showing a shift in the Fe K-edge and disappearance of the Fe–Fe scattering contribution at 2.4 Å. Further evidence for a reductive coupling mechanism of P-cluster formation came from the nonenzymatic maturation of the P-cluster precursors on ΔnifH MoFe to mature P-clusters using titanium citrate as a reductant.633 VTVH-MCD measurements suggested that titanium citrate generated all-ferrous [Fe4S4]0 clusters, which underwent fusion via loss of a bridging sulfide ligand followed by reoxidation to mature P-clusters. This process was later shown to be biphasic, where the formation of the first P-cluster in one-half of the tetrameric MoFe was rapid (<5 min), while the formation of the second P-cluster was slow (<2 h) and stimulated by NifZ.634
While it is still unclear what the role of NifZ is in this whole process, it has been suggested that formation of the first P-cluster in the MoFe tetramer induces a conformational change that increases the separation between the two [Fe4S4] clusters in the other half of the tetramer that should form the second P-cluster (Figure 60). Somehow, the interaction of NifZ with MoFe facilitates the binding of NifH, which then induces a conformational change that repositions the two [Fe4S4] clusters, allowing their reductive coupling. However, a recent study by Jimenez-Vicente and co-workers demonstrated that both P-clusters could be maturated in vitro in the absence of NifZ, questioning its essential role.635 It is suggested instead that NifZ simply speeds up the process. Structural characterization of the P-clusters in ΔnifH MoFe and ΔnifBZ MoFe would help to answer these questions.
Figure 60.

Final stages of nitrogenase cluster biosynthesis. The P-clusters on NifDK are formed from two canonical [Fe4S4] cluster precursors (P* clusters) by removing a sulfide from one cluster and fusing the two clusters together through the action of NifH and possibly also NifW and NifZ. Originally, the P-clusters were thought to be assembled one at a time, with only formation of the second cluster requiring NifZ. However, recently this has been disputed.635 Transfer of FeMoco from NifEN to NifDK requires the action of NifH and possibly other factors.
6.6. Transfer of FeMoco to MoFe: Yet Another Job for NifH
An early study of the MoFe protein formed from the ΔnifH strain of Azotobacter vinelandii showed that this strain could not synthesize FeMoco. As a result, ΔnifH MoFe lacked FeMoco and had no activity, but the activity could be reconstituted simply by adding isolated FeMoco to cell free extracts.636 However, activities as high as wild-type MoFe could not be achieved, and the efficiency of ΔnifH MoFe reconstitution appeared to depend on FeMoco concentration.637 Thus, it was established that restoration of full activity required preincubation of ΔnifH MoFe with NifH, ATP and FeMoco, demonstrating a requirement for NifH before or during FeMoco insertion. Later, it was shown that ΔnifH MoFe preincubated with NifH and ATP and repurified could be maturated with FeMoco, suggesting that MoFe was converted into a form in which the FeMoco binding site became accessible.597 This coincided with the binding of an additional subunit named gamma, later identified as NafY.598 This subunit was also found bound to ΔnifB MoFe.638 In this case, the action of NifH had already been performed and MoFe was ready to accept FeMoco. Interestingly, it was shown that the apo-form of NifH, in which the [Fe4S4] cluster was removed using chelating agents, was still capable of promoting FeMoco synthesis and insertion into MoFe,639 while mutants deficient in ATP binding were not.640
6.7. Spectroscopic Properties of Additional Maturases
In the model diazotroph Azotobacter vinelandii, as well as in other bacteria, there are a host of additional maturases required for MoFe maturation, and while some have well characterized functions, others are still not understood. NifM functions as a peptidyl proline cis–trans isomerase for NifH. Heterologous production of NifH in Escherichia coli and Saccharomyces cerevisiae (yeast) could be achieved in the absence of NifM but with enhanced yields in E. coli in its presence.641 However, only NifH produced in the presence of NifM supported acetylene reduction by NifDK, suggesting that the action of NifM was essential for putting NifH in an active conformation.642 Astonishingly, a variant of NifH in which a proline was mutated to serine was shown to support NifM-independent cell growth in A. vinelandii.643 However, this raises the question of why organisms would waste resources synthesizing NifM if a simple point mutation is sufficient. More recently, NifM was shown to be required for expression of active NifH in yeast mitochondria.644 Preparations of NifH from yeast mitochondria lacking NifM were colorless, suggesting a lack of the [Fe4S4] cluster. However, the precise role of the peptidyl proline isomerization carried out by NifM is still not clear. It is unknown whether ΔnifM NifH from native diazotrophs contains a correctly assembled [Fe4S4] cluster with the same properties as wild-type NifH or whether ΔnifM NifH can interact with MoFe and bind and hydrolyze ATP. In particular, spectroscopic characterization of the [Fe4S4] cluster in ΔnifM NifH by EPR, 57Fe Mössbauer, and MCD spectroscopies could provide useful information.
NifQ appears to be important for incorporation of Mo into MoFe nitrogenase. ΔnifQ strains of Klebsiella pneumoniae are defective in nitrogen fixation unless grown at elevated Mo concentrations645 or by addition of cysteine.646 NifQ was also found to serve as a Mo donor for FeMoco biosynthesis.647 On the basis of EPR spectral properties, NifQ was proposed to bind a [MoFe3S4] cluster. The EPR spectrum of the as-isolated state exhibited an isotropic S = 1/2 feature reminiscent of [Fe3S4] cluster spectra, while the DT-reduced spectrum contained a high spin feature, which was suggested to be related to the presence of Mo. Comparison of field swept ESEEM spectra of FeMoco and DT-reduced NifQ suggested that the high spin feature in NifQ originated from an S = 3/2 species. The suggested [MoFe3S4] cluster structure in NifQ was supported by Mo and Fe K-edge XAS, which showed similar absorption edges to FeMoco but with a slight contribution from a species similar to molybdate.208 EXAFS revealed Mo–S and Mo–Fe scattering distances of 2.34 and 2.71 Å, respectively, as well as Fe–S and Fe–Fe distances of 2.27 and 2.71 Å, respectively, consistent with a [MoFe3S4] cluster structure. In addition, a second Mo binding site including S and O ligands was suggested with a single Mo–S scattering distance of 2.23 Å, and one long and one short Mo–O distance of 2.12 and 1.73 Å. This was suggested to originate from a partially incorporated molybdate species and that incorporation of Mo into a sulfide coordination environment facilitates transfer to FeMoco in some way. However, Mo is thought to be transferred to FeMoco via formation of a complex with homocitrate on NifH.596 Therefore, the exact route by which Mo ends up on FeMoco is still an open question, which could possibly be resolved with time-resolved spectroscopic studies of metal transfer reactions between the various components.
NifV is well established as the enzyme responsible for homocitrate synthesis through the condensation of acetyl coenzyme A and α-ketoglutarate.595 It is closely related to a number of other acetyl CoA acetyltransferase enzymes, including homocitrate synthase from Schizosaccharomyces pombe and Thermus thermophilus.457,648,649 These enzymes usually have a divalent metal cation (possibly Mg2+) ligated by two conserved histidine and one conserved glutamate ligands. The remaining coordination sites available on the metal ion are involved in coordinating hydroxyl groups in the substrate and product, while catalysis proceeds via deprotonation of the acetyl group of acetyl CoA followed by nucleophilic attack of the carbonyl group in α-ketoglutarate and release of CoA as a leaving group. A similar mechanism should apply for NifV, however, it is not known whether this enzyme binds divalent metal cations and, if so, whether it prefers a particular ion. It was noted that NifV is oxygen labile, but the reason for this was not identified.595
The exact role of NifW remains unclear and has been variously proposed as being involved in homocitrate processing650 and insertion,28 protection of MoFe from oxygen damage,651 or even in P-cluster maturation.587,635
NifX appears to be involved in transferring partially assembled FeMoco precursors between NifB and NifEN or between NifEN and NifDK. This role of the cofactor chaperone has been suggested based on the high sequence similarity of NifX to the C-terminus of bacterial NifB.65255Fe labeled NifB-co was used to show accumulation of 55Fe on NifX,653 and 99Mo was also shown to accumulate on NifX.654 The structure of the cluster bound to NifX was studied using EPR spectroscopy593 as well as XAS/NRVS.164 In one case, NifX was found to bind both the proposed end product of NifB (NifB-co or L-cluster) and also the cluster bound initially by NifEN (VK-cluster). While DT-reduced NifX/NifB-co is EPR silent, DT-reduced NifX/VK-cluster gives an axial g = 1.95 spectrum indicative of an S = 1/2 cluster. Interestingly, the oxidized cluster also exhibits an S = 1/2 EPR spectrum with axial symmetry (g = 2.02 and 1.89). Clearly, these two forms of the precursor have very distinct properties. NifX bound NifB-co was studied in detail by XAS and found to contain a 6Fe core with average Fe–S distances of 2.26 Å and a mixture of long (3.74 Å) and short (2.66 Å) Fe–Fe distances.164 A single Fe-X (X = C, N, O) distance of 2.04 Å was observed compared with a distance of 2.00 Å in NafY bound FeMoco. At the time, the nature of the interstitial light atom was not yet elucidated, but this observation provided support for the notion that it was inserted by NifB. The NRVS data exhibited Fe–S stretching modes at 270, 315, 385, and 408 cm–1 as well as lower energy cluster “breathing” modes, similar to those observed for FeMoco. Combined simulation of the spectra using an [Fe6S9X] structure accounted for the majority of the features and fit the experimental data better than 7Fe or 8Fe models. However, it was noted that a 7Fe structure could not be ruled out. It is entirely possible that the NifX-bound species represents a breakdown product of the L-cluster lacking one or both of the capping Fe ions.
6.8. Differences in Cofactor Maturation for VFe and FeFe
As mentioned in section 5, many diazotrophs produce nitrogenases with alternative cofactors in which Mo is substituted with either V or Fe.53 These alternative N2ases are not simply Mo N2ase with a substituted heterometal but are entirely different proteins which rely on different sets of genes for expression. In Azotobacter vinelandii, the genes encoding all three enzymes are present,19 and thus, in the absence of Mo, V N2ase is exclusively produced, while in the absence of both Mo and V, Fe N2ase is produced. The structural genes (vnfDKG and anfDKG) for these N2ases are not the only things that differ. Additionally, each alternative N2ase has its own Fe-protein (encoded by vnfH and anfH)19,20 and several maturase components that are also specialized. While the early stages of maturation from [Fe4S4] cluster synthesis to [Fe8S9C] cluster synthesis are conserved, further cofactor processing is carried out by VnfEN in the case of FeVco.655 VnfX, a homologue of NifX, has been shown to bind NifB-co and a precursor of FeVco and could, therefore, be a chaperone for these clusters.656 In the case of FeFeco, it is not clear whether NifEN or VnfEN are required, but an AnfEN has not been identified.
Interestingly, the P-clusters of V N2ase display spectral properties very similar to those of Mo N2ase prior to P-cluster maturation, indicating that the P-clusters in V-nitrogenase may be separate [Fe4S4] clusters.550 However, these observations conflict with the recent crystal structure of the V-nitrogenase, which clearly shows the same P-cluster structure as Mo N2ase (as discussed in section 5.1.2).32
6.9. Outlook on Iron–Sulfur Cluster Assembly in N2ases
N2ase cofactor assembly and insertion is a complex process requiring the involvement of a host of proteins, and a variety of spectroscopic methods have been critical to provide insight into many key processes. In particular, the synthesis of [Fe4S4] clusters on NifU,582 the conversion of [Fe4S4] clusters into the L-cluster containing the interstitial carbide582,589 and additional sulfur ligand,591 and the final maturation of the L-cluster to FeMoco594 can now be considered understood. However, even here a number of key questions remain, particularly concerning how precursor clusters are transferred from one maturase to the next and how such a process is regulated. A number of studies indicate direct interactions between many of the components,587,657−659 but the key question is whether this is regulated such that only proteins containing fully assembled clusters are able to transfer their product to the next maturase. An astounding piece of the maturase machinery is NifH,345 which is capable of facilitating reduction, heterometal atom insertion and homocitrate attachment to the precursor cluster on NifEN,596 maturation of the P-clusters on NifDK,550 and insertion of the final FeMoco cluster into NifDK.597 In addition, NifH serves its fundamental role as the N2ase reductase FeP, the only direct electron transfer partner capable of facilitating high levels of N2 reduction.345 In the case of NifH, further experiments to study the spectroscopic properties of NifH bound complexes should provide insight into how NifH is capable of undertaking so many different tasks and what the common themes are. The roles of many of the other maturase components are much less well understood. However, as many of the components may interact with cluster intermediates or other metals, the spectroscopic methods detailed within this review could be further used to investigate their functions and interactions with target proteins. Lastly, while the maturation of Mo nitrogenase is quite well understood, the same cannot be said for the alternative nitrogenases, where many open questions remain. Because of the sequence and structural homology,534 much of the process is likely to follow a similar mechanism.
7. Summary and Outlook
In this review, we have discussed how a wide variety of spectroscopic methods have been applied to understand the structure, mechanism, alternative reactivity, and biosynthesis of nitrogenases. Our current understanding of nitrogenases has also been greatly assisted by parallel spectroscopic studies of related model complexes as well as theoretical studies of both models and the enzyme systems themselves. While some spectroscopic techniques have shone particularly brightly in studying certain aspects of these enzymes and their reaction mechanisms (such as the application of EPR and ENDOR in the E4 state121,442), in most cases our deepest insights have come from the application of multiple orthogonal spectroscopic techniques.
The large number of Fe atoms in MoFe/VFe/FeFe have made these enzymes particularly challenging to study using bulk spectroscopic methods such as Mössbauer, NRVS, and Fe K-edge X-ray spectroscopies. Despite this, these techniques have provided insight into the nature of the geometric and electronic structure of FeMoco through combinations of selective 57Fe enrichment of either FeMoco or the P-clusters (in the case of 57Fe Mössbauer and 57Fe NRVS), very careful data processing, and correlation with model complexes and theory. Meanwhile, EPR and its more advanced derivatives, such as ENDOR and ESEEM, have been overwhelmingly successful in the investigation of intermediate Kramers species due to their high degree of selectivity and sensitivity, which has allowed for individual species within mixtures to be characterized. In contrast, the information obtained from MCD has been more limited, due to the fact that the method is not selective for spin (both Kramers and non-Kramers paramagnetic centers are observed) and the interpretation of MCD of iron sulfur clusters is largely limited to fingerprinting in the absence of a spin-Hamiltonian formulation that can appropriately describe the magnetic coupling within the catalytic cofactor. Despite these limitations, MCD has still been useful in characterizing major changes that occur in cluster formation.
While major strides in our understanding of N2ases have been made since the first isolations of the MoFe and FeP enzymes ∼55 years ago, there is still much to be uncovered. Particularly, most of the non-Kramers states of the LT catalytic cycle of Mo N2ase are still minimally characterized (if at all), and much remains to be gained in the investigation of E3 and E5 as well as further characterization of E7. Additionally, although recent reports have demonstrated that V and Fe N2ases follow the same limiting stoichiometry as Mo N2ase,535 there is little to no spectroscopic characterization of the intermediates formed in the V-dependent and Fe-only enzymes during either H+ or N2 catalysis. Major questions also remain regarding the nature of the intermediates formed in all three of these enzymes during turnover with alternative carbon-based substrates such as CO and CN–. Furthermore, the roles of many of the maturase components which aid in the formation and insertion of the metallocofactors of N2ases are still not well understood.
Of central importance to all three enzymes is the question of belt sulfide mobility, particularly for the S2B position, which has now been implicated as labile in crystallographic studies of both MoFe and VFe.452−454 To truly take advantage of the insight these purported intermediate structures represent, a bridge, in the form of single-crystal spectroscopic studies, must be built to connect with observed solution-phase species. Even more monumental, the challenge of developing methods capable of clearly detecting the presence or absence of a single belt sulfide in intermediates which cannot be crystallized remains. It is clear that N2ases will provide a playground for spectroscopic studies for years to come.
Acknowledgments
We acknowledge the Max Planck Society for funding. C.V.S., S.D., and J.B. also acknowledge the DFG SPP 1927 “Iron–Sulfur for Life” (projects DE 1877/1-1 and BI 2198/1-1) for funding. Additionally, R.B. thanks the Icelandic Research Fund grant no. 162880051 and C.V.S. the IMPRS-RECHARGE for further funding. L.D. acknowledges the Peter und Traudl Engelhorn Stiftung for funding. The authors would also like to thank Prof. Brian M. Hoffman for helpful discussions during the preparation of the manuscript.
Glossary
Abbreviations Used
- 12-c-4
12-crown-4 or 1,4,7,10-tetraoxacyclododecane
- 1H
hydrogen
- 2H
deuterium
- A
hyperfine coupling constant
- A
complete hyperfine coupling tensor
- ADP
adenosine diphosphate
- aiso
isotropic hyperfine coupling contribution
- Ala
alanine
- Aloc
local hyperfine interaction tensor
- AnfH
Fe-protein of Fe nitrogenase
- Arg
arginine
- Asn
asparagine
- ATP
adenosine triphosphate
- B
magnetic field
- B
the double exchange parameter
- βn
Bohr magneton
- BS
broken symmetry
- BSL
spin–echo baseline
- Bu
butyl
- CASSCF
complete active space self-consistent field
- CD
circular dichroism
- CI
configuration interaction
- CN
cyanide
- CO
carbon monoxide
- CoA
coenzyme A
- Cp
cyclopentadienyl
- CW
continuous wave
- Cys
cysteine
- D
axial zero-field splitting parameter
- δ
isomer shift
- δDRL
dynamic recovery level
- ΔEQ
quadrupole splitting parameter
- DFT
density functional theory
- DMF
dimethylformamide
- DMRG
density matrix renormalization group
- DNA
deoxyribonucleic acid
- DT
sodium dithionite
- DTPA
diethylenetriamine pentaacetate
- E/D
rhombicity parameter
- EDTA
ethylenediaminetetraacetic acid
- EG
ethylene glycol
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- ESEEM
electron spin echo envelope modulation
- Et
ethyl
- EXAFS
extended X-ray absorption fine structure
- F-cluster
Fe4S4 cluster of the Fe protein
- FeFe
N2-reducing protein component of Fe nitrogenase
- FeFeco
FeFe cofactor
- FeMco
N2-reducing catalytic cofactor of nitrogenases
- FeMoco
FeMo cofactor
- FeP
Fe-protein reductase
- FePox
one-electron oxidized state of the Fe protein
- FePred
resting oxidation state of the Fe protein
- FePsred
all-ferrous super-reduced oxidation state of the Fe protein
- FeVco
FeV cofactor
- Fld
flavodoxin
- FPPS
femtosecond pump–probe spectroscopy
- FSA
5′-deazariboflavin, formadine sulfinic acid
- FT
Fourier transform
- g
g-tensor
- g
complete electronic g-tensor
- GC-MS
gas chromatography–mass spectrometry
- Gln
glutamine
- HBArF4·2Et2O
Brookhart’s acid, bis(diethylether)oxonium tetrakis(bis(3,5-trifluoromethyl)phenyl)borate
- HDE
Heisenberg double exchange
- HERFD
high-energy resolution fluorescence detection
- HF
Hartree–Fock
- HIPIP
high potential iron protein
- His
histidine
- HQ
hydroquinone
- HYSCORE
hyperfine sublevel correlation
- I
nuclear spin
- ICVS
impulsive coherent vibrational spectroscopy
- IDS
indigo disulfonate
- IR
infrared spectroscopy
- K
effective spin projection coefficient
- Km
the Michaelis constant
- LCP
left circularly polarized
- Leu
leucine
- LMCT
ligand-to-metal charge transfer
- LT
Lowe–Thorneley
- M-cluster
N2-reducing catalytic cluster of nitrogenases
- MCD
magnetic circular dichroism
- Me
methyl
- MeCN
acetonitrile
- ml
magnetic quantum number
- MLCT
metal-to-ligand charge transfer
- MMCT
metal-to-metal charge transfer
- MN
the resting state of FeMoco
- MO
molecular orbital
- MoFe
N2-reducing protein component of Mo nitrogenase
- Mox
the one electron oxidized state of FeMoco
- ms
spin quantum number
- MV
methyl viologen
- N2ase
nitrogenase
- NHE
normal hydrogen electrode
- NifH
Fe-protein of Mo nitrogenase
- nIR
near-infrared
- NMF
N-Methylformamide
- NMR
nuclear magnetic resonance
- νn
Larmor frequency
- NRVS
nuclear resonance vibrational spectroscopy
- P-cluster
the electron-transfer metallocofactor of MFe
- P1+
one-electron oxidized state of the P-cluster
- P2+
two-electron oxidized state of the P-cluster
- P3+
three-electron oxidized state of the P-cluster
- PESTRE
pulsed ENDOR saturation and recovery
- Ph
phenyl
- Pi
phosphate
- PN
as-isolated, all-ferrous resting state of the P-cluster
- POX
two-electron oxidized state of the P-cluster
- PVDOS
partial vibrational density of states
- QM/MM
quantum mechanics/molecular mechanics
- RCP
right circularly polarized
- RD-PESTRE
raw data PESTRE
- RF
radio frequency
- RR
resonance Raman
- S
electronic spin
- SAM
S-adenosylmethionine
- SAXS
small-angle X-ray scattering
- Ser
serine
- SF
stopped-flow
- SHE
standard hydrogen electrode
- SpReAD
spatially resolved anomalous dispersion
- SQ
semiquinone
- T
anisotropic dipolar interaction tensor
- TACpD
bisaquo(1,4,9,12-tetraazacyclopentadecane)
- tBu
tert-butyl
- Td
tetrahedral
- TDDFT
time-dependent density functional theory
- TRF
radio frequency pulse
- UV
ultraviolet
- Val
valine
- VFe
N2-reducing protein component of V nitrogenase
- Vis
visible
- VnfH
Fe-protein of V nitrogenase
- VtC
valence-to-1s electronic transition
- VTVH
variable temperature, variable field
- WFe
W-containing nitrogenase
- XANES
X-ray absorption near-edge spectroscopy
- XAS
X-ray absorption spectroscopy
- XES
X-ray emission spectroscopy
- XMCD
X-ray magnetic circular dichroism
- ZFS
zero field splitting
Biographies
Casey Van Stappen completed his B.Sc. (2011) and M.Sc. (2013) degrees in Chemistry at the University of Minnesota, Duluth, under the supervision of Prof. Viktor Nemykin. Following several years of postgraduate study at the University of Michigan, Ann Arbor, under the advisement of Profs. Nicolai Lehnert and Vincent Pecoraro, he transferred his studies to the Max Planck Institute for Chemical Energy Conversion (2015) as a member of the IMPRS-RECHARGE, and completed his Ph.D. (2019) in Chemistry via enrollment at Ruhr-Universität Bochum. He is currently a postdoctoral research associate at the Max Planck Institute for Chemical Energy Conversion. His research interests lie in combining spectroscopic and computational methods to investigate small molecule activation, particularly in biological and biologically inspired systems.
Laure Decamps completed her B.Sc. (2008) and M.Sc. (2010) degrees in Biology at the Paris-Diderot University, France, before carrying out her Ph.D. (2014) on radical SAM enzymes with Dr. Olivier Berteau at the French National Institute for Agricultural Research (INRA) in Jouy-en-Josas and the Paris-Sud University. She then was a postdoctoral researcher in the group of Prof. Oliver Einsle at the University of Freiburg, Germany, where she focused on the crystallization of proteins involved in nitrogen fixation. Now a research group leader in the Department of Inorganic Spectroscopy at the Max Planck Institute for Chemical Energy Conversion, her research focuses on the biology and biochemistry of nitrogenases and their maturation.
George E. Cutsail III earned his B.Sc. of Chemistry (2010) at the University of Maryland, Baltimore County, as part of the Meyerhoff Scholars Program. He earned his Ph.D. (2014) as an NSF Graduate Research Fellow at Northwestern University. There, he studied advanced EPR spectroscopic techniques, particularly ENDOR spectroscopy applied to metalloproteins and biomimetic complexes, with Prof. Brian M. Hoffman. George then moved to Germany for his postdoctoral research in X-ray spectroscopy at the Max Planck Institute for Chemical Energy Conversion (MPI-CEC) under the advisement of Prof. Serena DeBeer. During his postdoc, George was awarded an Alexander von Humboldt Fellowship. Currently, George is an independent Max Planck Research Group Leader at the MPI CEC, employing advanced paramagnetic spectroscopic techniques to understand fundamental chemical energy processes.
Ragnar Björnsson received B.Sc. and M.Sc. degrees from the University of Iceland, Iceland, and a Ph.D. from the University of St. Andrews, UK, supervised by Prof. Michael Bühl. He was a postdoctoral researcher with Prof. Frank Neese and Prof. Serena DeBeer at the Max Planck Institute for Chemical Energy Conversion and later a research fellow at the University of Iceland. He is currently a group leader in the Department of Inorganic Spectroscopy at the Max Planck Institute for Chemical Energy Conversion. His research interests are in theoretical inorganic and bioinorganic chemistry with a special focus on theoretical modelling of structure and spectroscopic properties of metalloenzymes, including nitrogenase.
Justin T. Henthorn earned his B.Sc. in Chemistry and B.A. in Economics (2010) at Ohio University as a Barry M. Goldwater and Ernest F. Hollings scholar. He earned his Ph.D. (2016) as an NSF Graduate Research Fellow at the California Institute of Technology. There he studied the multiproton, multielectron chemistry of π-bound Mo–quinonoid complexes with Prof. Theodor Agapie. Justin then moved to Germany for his postdoctoral research in X-ray spectroscopy at the Max Planck Institute for Chemical Energy Conversion as an Alexander von Humboldt Fellow under the advisement of Prof. Serena DeBeer. His postdoctoral work has focused on developing selenium as a selective X-ray spectroscopic probe of the FeMoco electronic structure.
James A. Birrell obtained his undergraduate degree in Natural Sciences in 2008 and his Ph.D. degree in Biochemistry in 2012, both from the University of Cambridge (UK). During his doctoral studies, he carried out functional and spectroscopic characterization of mitochondrial complex I under the supervision of Prof. Judy Hirst at the MRC Mitochondrial Biology Unit. He then moved to the Max Planck Institute for Chemical Energy Conversion (MPI-CEC) for a postdoctoral fellowship with Prof. Wolfgang Lubitz from 2013 until 2017. Since December 2017, he is a group leader in Prof. Serena DeBeer’s Department of Inorganic Spectroscopy, also at the MPI-CEC. As a biochemist, his research mostly focuses on producing metalloenzymes relevant to energy conversion reactions, and studying them with spectroscopic methods.
Serena DeBeer is a Professor and Director at the Max Planck Institute for Chemical Energy Conversion in Mülheim an der Ruhr, Germany. She is also an Adjunct Professor in the Department of Chemistry and Chemical Biology at Cornell University, an honorary faculty member at Ruhr University in Bochum, and the group leader of the PINK Beamline at the Energy Materials In-Situ Laboratory at Helmholtz Zentrum in Berlin. She received her B.Sc. in Chemistry at Southwestern University in 1995 and her Ph.D. from Stanford University in 2002. From 2002 to 2009, she was a staff scientist at the Stanford Synchrotron Radiation Laboratory, before moving to a faculty position at Cornell. She is the recipient of a European Research Council Synergy Award (2019), the American Chemical Society Inorganic Chemistry Lectureship Award (2016), the Society of Biological Inorganic Chemistry Early Career Award (2015), a European Research Council Consolidator Award (2013), a Kavli Fellowship (2012), and an Alfred P. Sloan Research Fellowship (2011). Research in the DeBeer group is focused on the development and application of advanced X-ray spectroscopic tools for understanding mechanisms in biological, homogeneous, and heterogeneous catalysis. Her research group has been active in the field of nitrogenase research for nearly a decade.
The authors declare no competing financial interest.
References
- Haber F. Über die Darstellung des Ammoniaks aus Stickstoff und Wasserstoff. Naturwissenschaften 1922, 10, 1041–1049. 10.1007/BF01565394. [DOI] [Google Scholar]
- Haber F. Neue Arbeitsweisen - Wissenschaft und Wirtschaft nach dem Kriege. Naturwissenschaften 1923, 11, 753–756. 10.1007/BF01551097. [DOI] [Google Scholar]
- Appl M. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH, 2006. [Google Scholar]
- Schlögl R. Catalytic Synthesis of Ammonia-a ″Never-Ending Story″?. Angew. Chem., Int. Ed. 2003, 42, 2004–2008. 10.1002/anie.200301553. [DOI] [PubMed] [Google Scholar]
- Tuck A. F. Production of Nitrogen Oxides by Lightning Discharges. Q. J. R. Meteorol. Soc. 1976, 102, 749–755. 10.1002/qj.49710243404. [DOI] [Google Scholar]
- Boyd E. S.; Anbar A. D.; Miller S.; Hamilton T. L.; Lavin M.; Peters J. W. A Late Methanogen Origin for Molybdenum-dependent Nitrogenase. Geobiology 2011, 9, 221–232. 10.1111/j.1472-4669.2011.00278.x. [DOI] [PubMed] [Google Scholar]
- Canfield D. E.; Glazer A. N.; Falkowski P. G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- Gruber N.; Galloway J. N. An Earth-system Perspective of the Global Nitrogen Cycle. Nature 2008, 451, 293–296. 10.1038/nature06592. [DOI] [PubMed] [Google Scholar]
- Thamdrup B. New Pathways and Processes in the Global Nitrogen Cycle. Annu. Rev. Eco. Evol. Syst. 2012, 43, 407–428. 10.1146/annurev-ecolsys-102710-145048. [DOI] [Google Scholar]
- Kandemir T.; Schuster M. E.; Senyshyn A.; Behrens M.; Schlögl R. The Haber-Bosch Process Revisited: On The Real Structure and Stability of ″Ammonia Iron″ Under Working Conditions. Angew. Chem., Int. Ed. 2013, 52, 12723–12726. 10.1002/anie.201305812. [DOI] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062. 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M. The Mechanism for Nitrogenase Including All Steps. Phys. Chem. Chem. Phys. 2019, 21, 15747–15759. 10.1039/C9CP02073J. [DOI] [PubMed] [Google Scholar]
- Service R.Ammonia—A Renewable Fuel Made From Sun, Air, and Water—Could Power the Globe Without Carbon. Science 2018, 10.1126/science.aau7489. [DOI] [Google Scholar]
- Liu C.; Sakimoto K. K.; Colon B. C.; Silver P. A.; Nocera D. G. Ambient Nitrogen Reduction Cycle Using a Hybrid Inorganic-Biological System. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6450–6455. 10.1073/pnas.1706371114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd E. S.; Peters J. W. New Insights into the Evolutionary History of Biological Nitrogen Fixation. Front. Microbiol. 2013, 4, 201. 10.3389/fmicb.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos P. C.; Fang Z.; Mason S. W.; Setubal J. C.; Dixon R. Distribution of Nitrogen Fixation and Nitrogenase-like Sequences Amongst Microbial Genomes. BMC Genomics 2012, 13, 162. 10.1186/1471-2164-13-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond J.; Siefert J. L.; Staples C. R.; Blankenship R. E. The Natural History of Nitrogen Fixation. Mol. Biol. Evol. 2004, 21, 541–554. 10.1093/molbev/msh047. [DOI] [PubMed] [Google Scholar]
- Bortels H. Molybdän als Katalysator bei der Biologischen Stickstoffbindung. Arch. Microbiol. 1930, 1, 333–342. 10.1007/BF00510471. [DOI] [Google Scholar]
- Chisnell J. R.; Premakumar R.; Bishop P. E. Purification of a Second Alternative Nitrogenase from a nifHDK Deletion Strain of Azotobacter vinelandii. J. Bacteriol. 1988, 170, 27–33. 10.1128/JB.170.1.27-33.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales B. J.; Case E. E.; Morningstar J. E.; Dzeda M. F.; Mauterer L. A. Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25, 7251–7255. 10.1021/bi00371a001. [DOI] [PubMed] [Google Scholar]
- Robson R. L.; Eady R. R.; Richardson T. H.; Miller R. W.; Hawkins M.; Postgate J. R. The Alternative Nitrogenase of Azotobacter chroococcum is a Vanadium Enzyme. Nature 1986, 322, 388–390. 10.1038/322388a0. [DOI] [Google Scholar]
- Hamilton T. L.; Jacobson M.; Ludwig M.; Boyd E. S.; Bryant D. A.; Dean D. R.; Peters J. W. Differential Accumulation of nif Structural Gene mRNA in Azotobacter vinelandii. J. Bacteriol. 2011, 193, 4534–4536. 10.1128/JB.05100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulen W. A.; LeComte J. R. The Nitrogenase System from Azotobacter: Two-Enzyme Requirement for N2 Reduction, ATP-dependent H2 evolution, and ATP Hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 1966, 56, 979–986. 10.1073/pnas.56.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson L. E. Components of Cell-free Extracts of Clostridium pasteurianum Required for ATP-dependent H2 Evolution from Dithionite and for N2 Fixation. Biochim. Biophys. Acta, Gen. Subj. 1966, 127, 18–25. 10.1016/0304-4165(66)90470-3. [DOI] [PubMed] [Google Scholar]
- Dean D. R.; Bolin J. T.; Zheng L. Nitrogenase Metalloclusters: Structures, Organization, and Synthesis. J. Bacteriol. 1993, 175, 6737–6744. 10.1128/JB.175.21.6737-6744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman R. V.; Burris R. H. Nitrogenase and Nitrogenase Reductase Associate and Dissociate with Each Catalytic Cycle. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 2699–2702. 10.1073/pnas.75.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. Nitrogenase: a Nucleotide-Dependent Molecular Switch. Annu. Rev. Biochem. 1994, 63, 235–264. 10.1146/annurev.bi.63.070194.001315. [DOI] [PubMed] [Google Scholar]
- Kim J.; Rees D. C. Nitrogenase and Biological Nitrogen Fixation. Biochemistry 1994, 33, 389–397. 10.1021/bi00168a001. [DOI] [PubMed] [Google Scholar]
- Winter H. C.; Burris R. H. Nitrogenase. Annu. Rev. Biochem. 1976, 45, 409–426. 10.1146/annurev.bi.45.070176.002205. [DOI] [PubMed] [Google Scholar]
- Lancaster K. M.; Roemelt M.; Ettenhuber P.; Hu Y.; Ribbe M. W.; Neese F.; Bergmann U.; DeBeer S. X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees J. A.; Bjornsson R.; Schlesier J.; Sippel D.; Einsle O.; DeBeer S. The Fe-V Cofactor of Vanadium Nitrogenase Contains an Interstitial Carbon Atom. Angew. Chem., Int. Ed. 2015, 54, 13249–13252. 10.1002/anie.201505930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippel D.; Einsle O. The Structure of Vanadium Nitrogenase Reveals an Unusual Bridging Ligand. Nat. Chem. Biol. 2017, 13, 956–961. 10.1038/nchembio.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulen W. A.; Burns R. C.; LeComte J. R. Nitrogen Fixation: Cell-free System with Extracts of Azotobacter. Biochem. Biophys. Res. Commun. 1964, 17, 265–271. 10.1016/0006-291X(64)90395-X. [DOI] [PubMed] [Google Scholar]
- Carnahan J. E.; Mortenson L. E.; Mover H. F.; Castle J. E. Nitrogen Fixation in Cell-free Extracts of Clostridium pasteurianum. Biochim. Biophys. Acta 1960, 38, 188–189. 10.1016/0006-3002(60)91225-7. [DOI] [PubMed] [Google Scholar]
- Schneider K. C.; Bradbeer C.; Singh R. N.; Wang L. C.; Wilson P. W.; Burris R. H. Nitrogen Fixation by Cell-Free Preparations from Microorganisms. Proc. Natl. Acad. Sci. U. S. A. 1960, 46, 726–733. 10.1073/pnas.46.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge H. L.; Tezcan F. A.. Electron Transfer in Nitrogenase. Chem. Rev. 2020, 10.1021/acs.chemrev.9b00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos P. C.; Igarashi R. Y.; Lee H.-I.; Hoffman B. M.; Seefeldt L. C.; Dean D. R. Substrate Interactions with the Nitrogenase Active Site. Acc. Chem. Res. 2005, 38, 208–214. 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- Hoffman B. M.; Dean D. R.; Seefeldt L. C. Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation. Acc. Chem. Res. 2009, 42, 609–619. 10.1021/ar8002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M.; Lukoyanov D.; Dean D. R.; Seefeldt L. C. Nitrogenase: A Draft Mechanism. Acc. Chem. Res. 2013, 46, 587–595. 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Decoding the Nitrogenase Mechanism: The Homologue Approach. Acc. Chem. Res. 2010, 43, 475–484. 10.1021/ar900254x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Peters J. W.; Raugei S.; Beratan D. N.; Antony E.; Dean D. R. Energy Transduction in Nitrogenase. Acc. Chem. Res. 2018, 51, 2179–2186. 10.1021/acs.accounts.8b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickerman N. S.; Ribbe M. W.; Hu Y. Nitrogenase Cofactor Assembly: An Elemental Inventory. Acc. Chem. Res. 2017, 50, 2834–2841. 10.1021/acs.accounts.7b00417. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Dean D. R. Role of Nucleotides in Nitrogenase Catalysis. Acc. Chem. Res. 1997, 30, 260–266. 10.1021/ar960260e. [DOI] [Google Scholar]
- Hu Y.; Ribbe M. W. Biosynthesis of the Metalloclusters of Nitrogenases. Annu. Rev. Biochem. 2016, 85, 455–483. 10.1146/annurev-biochem-060614-034108. [DOI] [PubMed] [Google Scholar]
- Mortenson L. E.; Thorneley R. N. F. Structure and Function of Nitrogenase. Annu. Rev. Biochem. 1979, 48, 387–418. 10.1146/annurev.bi.48.070179.002131. [DOI] [PubMed] [Google Scholar]
- Rubio L. M.; Ludden P. W. Biosynthesis of the Iron-Molybdenum Cofactor of Nitrogenase. Annu. Rev. Microbiol. 2008, 62, 93–111. 10.1146/annurev.micro.62.081307.162737. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Dean D. R. Mechanism of Mo-Dependent Nitrogenase. Annu. Rev. Biochem. 2009, 78, 701–722. 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess B. K.; Lowe D. J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3012. 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- Dos Santos P. C.; Dean D. R.; Hu Y.; Ribbe M. W. Formation and Insertion of the Nitrogenase Iron-Molybdenum Cofactor. Chem. Rev. 2004, 104, 1159–1173. 10.1021/cr020608l. [DOI] [PubMed] [Google Scholar]
- Ribbe M. W.; Hu Y.; Hodgson K. O.; Hedman B. Biosynthesis of Nitrogenase Metalloclusters. Chem. Rev. 2014, 114, 4063–4080. 10.1021/cr400463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess B. K. The Iron-Molybdenum Cofactor of Nitrogenase. Chem. Rev. 1990, 90, 1377–1406. 10.1021/cr00106a002. [DOI] [Google Scholar]
- Eady R. R. Structure-Function Relationships of Alternative Nitrogenases. Chem. Rev. 1996, 96, 3013–3030. 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. Structural Basis of Biological Nitrogen Fixation. Chem. Rev. 1996, 96, 2965–2982. 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Biosynthesis of Nitrogenase FeMoco. Coord. Chem. Rev. 2011, 255, 1218–1224. 10.1016/j.ccr.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eady R. R. Current Status of Structure Function Relationships of Vanadium Nitrogenase. Coord. Chem. Rev. 2003, 237, 23–30. 10.1016/S0010-8545(02)00248-5. [DOI] [Google Scholar]
- Igarashi R. Y.; Seefeldt L. C. Nitrogen Fixation: The Mechanism of the Mo Dependent Nitrogenase. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 351–384. 10.1080/10409230391036766. [DOI] [PubMed] [Google Scholar]
- Rehder D. Vanadium Nitrogenase. J. Inorg. Biochem. 2000, 80, 133–136. 10.1016/S0162-0134(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Tezcan F. A.; Haynes C. A.; Walton M. Y.; Andrade S.; Einsle O.; Howard J. B. Structural Basis of Biological Nitrogen Fixation. Philos. Trans. R. Soc., A 2005, 363, 971–984. 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- Tezcan F. A.; Kaiser J. T.; Mustafi D.; Walton M. Y.; Howard J. B.; Rees D. C. Nitrogenase Complexes: Multiple Docking Sites for a Nucleotide Switch Protein. Science 2005, 309, 1377–1380. 10.1126/science.1115653. [DOI] [PubMed] [Google Scholar]
- Danyal K.; Mayweather D.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Conformational Gating of Electron Transfer from the Nitrogenase Fe Protein to MoFe Protein. J. Am. Chem. Soc. 2010, 132, 6894–6895. 10.1021/ja101737f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danyal K.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Electron Transfer within Nitrogenase: Evidence for a Deficit-Spending Mechanism. Biochemistry 2011, 50, 9255–9263. 10.1021/bi201003a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval S.; Danyal K.; Shaw S.; Lytle A. K.; Dean D. R.; Hoffman B. M.; Antony E.; Seefeldt L. C. Electron Transfer Precedes ATP Hydrolysis during Nitrogenase Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16414–16419. 10.1073/pnas.1311218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D. J.; Thorneley R. N. F. The Mechanism of Klebsiella pneumoniae Nitrogenase Action. Pre-steady-state Kinetics of H2 Formation. Biochem. J. 1984, 224, 877–886. 10.1042/bj2240877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Lowe D. J. The Mechanism of Klebsiella pneumoniae Nitrogenase Action. Pre-steady-state Kinetics of an Enzyme-bound Intermediate in N2 Reduction and of NH3 formation. Biochem. J. 1984, 224, 887–894. 10.1042/bj2240887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eady R. R.; Leigh G. J. Metals in the Nitrogenases. J. Chem. Soc., Dalton Trans. 1994, 2739–2747. 10.1039/dt9940002739. [DOI] [Google Scholar]
- Lukoyanov D.; Khadka N.; Yang Z. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Reductive Elimination of H2 Activates Nitrogenase to Reduce the N≡N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc. 2016, 138, 10674–10683. 10.1021/jacs.6b06362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil J. A.; Bolton J. R.. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, 2nd ed.; Wiley-Interscience: Hoboken, N.J., 2007. [Google Scholar]
- Abragam A.; Bleaney B.. Electron Paramagnetic Resonance of Transition Ions; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Yoo S. J.; Angove H. C.; Burgess B. K.; Hendrich M. P.; Münck E. Mössbauer and Integer-Spin EPR Studies and Spin-Coupling Analysis of the [4Fe-4S]0 Cluster of the Fe Protein from Azotobacter vinelandii Nitrogenase. J. Am. Chem. Soc. 1999, 121, 2534–2545. 10.1021/ja9837405. [DOI] [Google Scholar]
- Hendrich M. P.; Debrunner P. G. Integer-spin Electron Paramagnetic Resonance of Iron Proteins. Biophys. J. 1989, 56, 489–506. 10.1016/S0006-3495(89)82696-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telser J.; Krzystek J.; Ozarowski A. High-frequency and High-field Electron Paramagnetic Resonance (HFEPR): A New Spectroscopic Tool for Bioinorganic Chemistry. JBIC, J. Biol. Inorg. Chem. 2014, 19, 297–318. 10.1007/s00775-013-1084-3. [DOI] [PubMed] [Google Scholar]
- Krzystek J.; England J.; Ray K.; Ozarowski A.; Smirnov D.; Que L. Jr.; Telser J. Determination by High-Frequency and -Field EPR of Zero-Field Splitting in Iron(IV) Oxo Complexes: Implications for Intermediates in Nonheme Iron Enzymes. Inorg. Chem. 2008, 47, 3483–3485. 10.1021/ic800411c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzystek J.; Park J. H.; Meisel M. W.; Hitchman M. A.; Stratemeier H.; Brunel L. C.; Telser J. EPR Spectra from ″EPR-silent″ Species: High-Frequency and High-field EPR Spectroscopy of Pseudotetrahedral Complexes of Nickel(II). Inorg. Chem. 2002, 41, 4478–4487. 10.1021/ic020198j. [DOI] [PubMed] [Google Scholar]
- Juarez-Garcia C.; Hendrich M. P.; Holman T. R.; Que L.; Münck E. Combined Mössbauer and EPR Studies of the S = 3 State of an Exchange-Coupled FeIIICuII Complex: Test for Quantitative EPR Analysis of Integer Spin Systems. J. Am. Chem. Soc. 1991, 113, 518–525. 10.1021/ja00002a020. [DOI] [Google Scholar]
- Gemperle C.; Schweiger A. Pulsed Electron Nuclear Double-Resonance Methodology. Chem. Rev. 1991, 91, 1481–1505. 10.1021/cr00007a011. [DOI] [Google Scholar]
- Hoffman B. M. Electron Nuclear Double Resonance (ENDOR) of Metalloenzymes. Acc. Chem. Res. 1991, 24, 164–170. 10.1021/ar00006a002. [DOI] [Google Scholar]
- Hoffman B. M. ENDOR of Metalloenzymes. Acc. Chem. Res. 2003, 36, 522–529. 10.1021/ar0202565. [DOI] [PubMed] [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- DeRose V. J.; Hoffman B. M. Protein Structure and Mechanism Studied by Electron Nuclear Double Resonance Spectroscopy. Methods Enzymol. 1995, 246, 554–589. 10.1016/0076-6879(95)46025-X. [DOI] [PubMed] [Google Scholar]
- Gast P.; Groenen E. J. J. EPR Interactions—g-Anisotropy. eMagRes. 2016, 5, 1435–1444. 10.1002/9780470034590.emrstm1500. [DOI] [Google Scholar]
- Zeeman P. On the Influence of Magnetism on the Nature of The Light Emitted by a Substance (Part II). Astrophys. J. 1897, 5, 332. 10.1086/140355. [DOI] [Google Scholar]
- Mailer C.; Taylor C. P. S. Rapid Adiabatic Passage EPR of Ferricytochrome c: Signal Enhancement and Determination of Spin-Lattice Relaxation Time. Biochim. Biophys. Acta, Protein Struct. 1973, 322, 195–203. 10.1016/0005-2795(73)90293-6. [DOI] [PubMed] [Google Scholar]
- Mailer C.; Hoffman B. M. Tumbling of an Adsorbed Nitroxide Using Rapid Adiabatic Passage. J. Phys. Chem. 1976, 80, 842–846. 10.1021/j100549a015. [DOI] [Google Scholar]
- Gurbiel R. J.; Bolin J. T.; Ronco A. E.; Mortenson L.; Hoffman B. M. Single-Crystal EPR and ENDOR Study of Nitrogenase from Clostridium pasteurianum. J. Magn. Reson. 1991, 91, 227–240. 10.1016/0022-2364(91)90188-Y. [DOI] [Google Scholar]
- Spatzal T.; Einsle O.; Andrade S. L. A. Analysis of the Magnetic Properties of Nitrogenase FeMo Cofactor by Single-Crystal EPR Spectroscopy. Angew. Chem., Int. Ed. 2013, 52, 10116–10119. 10.1002/anie.201303000. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–349. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- Boca R. Zero-field splitting in Metal Complexes. Coord. Chem. Rev. 2004, 248, 757–815. 10.1016/j.ccr.2004.03.001. [DOI] [Google Scholar]
- Barney B. M.; Lee H.-I.; Dos Santos P. C.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Breaking the N2 Triple Bond: Insights into the Nitrogenase Mechanism. Dalton Trans. 2006, 2277–2284. 10.1039/b517633f. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Goldfarb D. EPR Interactions - Nuclear Quadrupole Couplings. eMagRes. 2017, 6, 495–509. 10.1002/9780470034590.emrstm1504. [DOI] [Google Scholar]
- Townes C. H.; Dailey B. P. Determination of Electronic Structure of Molecules from Nuclear Quadrupole Effects. J. Chem. Phys. 1949, 17, 782–796. 10.1063/1.1747400. [DOI] [Google Scholar]
- Lucken E. A. C.Nuclear Quadrupole Coupling Constants; Academic Press: New York, 1969. [Google Scholar]
- Yang T. C.; Maeser N. K.; Laryukhin M.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. The Interstitial Atom of the Nitrogenase FeMo-cofactor: ENDOR and ESEEM Evidence That it is Not a Nitrogen. J. Am. Chem. Soc. 2005, 127, 12804–12805. 10.1021/ja0552489. [DOI] [PubMed] [Google Scholar]
- True A. E.; Mclean P.; Nelson M. J.; Orme-Johnson W. H.; Hoffman B. M. Comparison of Wild-Type and nifV Mutant Molybdenum-Iron Proteins of Nitrogenase from Klebsiella pneumoniae by ENDOR Spectroscopy. J. Am. Chem. Soc. 1990, 112, 651–657. 10.1021/ja00158a024. [DOI] [Google Scholar]
- Carrington A.; McLachlan A. D.. Introduction to Magnetic Resonance with Applications to Chemistry and Chemical Physics; Harper & Row: New York, 1967. [Google Scholar]
- Noodleman L.; Peng C. Y.; Case D. A.; Mouesca J. M. Orbital Interactions, Electron Delocalization and Spin Coupling in Iron-Sulfur Clusters. Coord. Chem. Rev. 1995, 144, 199–244. 10.1016/0010-8545(95)07011-L. [DOI] [Google Scholar]
- Harmer J. R. Hyperfine Spectroscopy—ENDOR. eMagRes. 2016, 5, 1493–1514. 10.1002/9780470034590.emrstm1515. [DOI] [Google Scholar]
- Hoffman B. M.; Martinsen J.; Venters R. A. General Theory of Polycrystalline ENDOR Patterns. g and Hyperfine Tensors of Arbitrary Symmetry and Relative Orientation. J. Magn. Reson. 1984, 59, 110–123. 10.1016/0022-2364(84)90287-7. [DOI] [Google Scholar]
- Hoffman B. M.; Venters R. A.; Martinsen J. General Theory of Polycrystalline ENDOR Patterns. Effects of Finite EPR and ENDOR Component Linewidths. J. Magn. Reson. 1985, 62, 537–542. 10.1016/0022-2364(85)90225-2. [DOI] [Google Scholar]
- Doan P. E. The Past, Present, and Future of Orientation-Selected ENDOR Analysis: Solving the Challenges of Dipolar-Coupled Nuclei. Paramagnetic Resonance of Metallobiomolecules 2003, 858, 55–81. 10.1021/bk-2003-0858.ch003. [DOI] [Google Scholar]
- Lee H.-I.; Hales B. J.; Hoffman B. M. Metal-Ion Valencies of the FeMo Cofactor in CO-Inhibited and Resting State Nitrogenase by 57Fe Q-Band ENDOR. J. Am. Chem. Soc. 1997, 119, 11395–11400. 10.1021/ja971508d. [DOI] [Google Scholar]
- Cutsail G. E. III; Telser J.; Hoffman B. M. Advanced Paramagnetic Resonance Spectroscopies of Iron-Sulfur Proteins: Electron Nuclear Double Resonance (ENDOR) and Electron Spin Echo Envelope Modulation (ESEEM). Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 1370–1394. 10.1016/j.bbamcr.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townes C. H.; Dailey B. P. Nuclear Quadrupole Coupling and Ionic Character of Molecules. Phys. Rev. 1950, 78, 346–347. [Google Scholar]
- Townes C. H.; Dailey B. P. Nuclear Quadrupole Effects and Electronic Structure of Molecules in the Solid State. J. Chem. Phys. 1952, 20, 35–40. 10.1063/1.1700192. [DOI] [Google Scholar]
- Roberts J. E.; Cline J. F.; Lum V.; Gray H. B.; Freeman H.; Peisach J.; Reinhammar B.; Hoffman B. M. Comparative ENDOR Study of Six Blue Copper Proteins. J. Am. Chem. Soc. 1984, 106, 5324–5330. 10.1021/ja00330a048. [DOI] [Google Scholar]
- Davoust C. E.; Doan P. E.; Hoffman B. M. Q-Band Pulsed Electron Spin-Echo Spectrometer and Its Application to ENDOR and ESEEM. J. Magn. Reson., Ser. A 1996, 119, 38–44. 10.1006/jmra.1996.0049. [DOI] [Google Scholar]
- Brüggemann W.; Niklas J. R. Stochastic ENDOR. J. Magn. Reson., Ser. A 1994, 108, 25–29. 10.1006/jmra.1994.1084. [DOI] [Google Scholar]
- Feher G. Electron Nuclear Double Resonance (ENDOR) Experiments. Physica 1958, 24, S80–S87. 10.1016/S0031-8914(58)80506-6. [DOI] [Google Scholar]
- Epel B.; Arieli D.; Baute D.; Goldfarb D. Improving W-band Pulsed ENDOR Sensitivity-Random Acquisition and Pulsed Special TRIPLE. J. Magn. Reson. 2003, 164, 78–83. 10.1016/S1090-7807(03)00191-5. [DOI] [PubMed] [Google Scholar]
- Doan P. E.; Gurbiel R. J.; Hoffman B. M. The Ups and Downs of Feher-Style ENDOR. Appl. Magn. Reson. 2007, 31, 649–663. 10.1007/BF03166608. [DOI] [Google Scholar]
- Mims W. B. Pulsed ENDOR Experiments. Proc. R. Soc. A 1965, 283, 452–457. 10.1098/rspa.1965.0034. [DOI] [Google Scholar]
- Davies E. R. A New Pulse ENDOR Technique. Phys. Lett. A 1974, 47, 1–2. 10.1016/0375-9601(74)90078-4. [DOI] [Google Scholar]
- Epel B.; Niklas J.; Antonkine M. L.; Lubitz W. Absolute Signs of Hyperfine Coupling Constants as Determined by Pulse ENDOR of Polarized Radical Pairs. Appl. Magn. Reson. 2006, 30, 311–327. 10.1007/BF03166203. [DOI] [Google Scholar]
- Doan P. E. Combining Steady-state and Dynamic Methods for Determining Absolute Signs of Hyperfine Interactions: Pulsed ENDOR Saturation and Recovery (PESTRE). J. Magn. Reson. 2011, 208, 76–86. 10.1016/j.jmr.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutsail G. E. III; Stein B. W.; Subedi D.; Smith J. M.; Kirk M. L.; Hoffman B. M. EPR, ENDOR, and Electronic Structure Studies of the Jahn-Teller Distortion in an FeV Nitride. J. Am. Chem. Soc. 2014, 136, 12323–12336. 10.1021/ja505403j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney R. A.; Hetterscheid D. G. H.; Hanna B. S.; Schrock R. R.; Hoffman B. M. Formation of {[HIPTN3N]Mo(III)H}− by Heterolytic Cleavage of H2 as Established by EPR and ENDOR Spectroscopy. Inorg. Chem. 2010, 49, 704–713. 10.1021/ic902006v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan P. E.; Telser J.; Barney B. M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. 57Fe ENDOR Spectroscopy and ’Electron Inventory’ Analysis of the Nitrogenase E4 Intermediate Suggest the Metal-Ion Core of FeMo-Cofactor Cycles Through Only One Redox Couple. J. Am. Chem. Soc. 2011, 133, 17329–17340. 10.1021/ja205304t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Kinney R. A.; Hoffman B. M.; Peters J. C. A Nonclassical Dihydrogen Adduct of S = 1/2 Fe(I). J. Am. Chem. Soc. 2011, 133, 16366–16369. 10.1021/ja207003m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutsail G. E. III; Doan P. E.; Hoffman B. M.; Meyer J.; Telser J. EPR and 57Fe ENDOR Investigation of 2Fe Ferredoxins from Aquifex aeolicus. JBIC, J. Biol. Inorg. Chem. 2012, 17, 1137–1150. 10.1007/s00775-012-0927-7. [DOI] [PubMed] [Google Scholar]
- Yang H.; Rittle J.; Marts A. R.; Peters J. C.; Hoffman B. M. ENDOR Characterization of (N2)FeII(μ-H)2FeI(N2)−: A Spectroscopic Model for N2 Binding by the di-μ-Hydrido Nitrogenase Janus Intermediate. Inorg. Chem. 2018, 57, 12323–12330. 10.1021/acs.inorgchem.8b02021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeke V.; Tociu L.; Case D. A.; Seefeldt L. C.; Raugei S.; Hoffman B. M. High-Resolution ENDOR Spectroscopy Combined with Quantum Chemical Calculations Reveals the Structure of Nitrogenase Janus Intermediate E4(4H). J. Am. Chem. Soc. 2019, 141, 11984–11996. 10.1021/jacs.9b04474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.; Cutsail G. E. III; Rittle J.; Connor B. A.; Gunderson W. A.; Zhang L.; Hoffman B. M.; Peters J. C. Characterization of an Fe≡N-NH2 Intermediate Relevant to Catalytic N2 Reduction to NH3. J. Am. Chem. Soc. 2015, 137, 7803–7809. 10.1021/jacs.5b03432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan P. E.; Hoffman B. M. Making Hyperfine Selection in Mims ENDOR independent of deadtime. Chem. Phys. Lett. 1997, 269, 208–214. 10.1016/S0009-2614(97)00293-5. [DOI] [Google Scholar]
- Deligiannakis Y.; Louloudi M.; Hadjiliadis N. Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy as a Tool to Investigate the Coordination Environment of Metal Centers. Coord. Chem. Rev. 2000, 204, 1–112. 10.1016/S0010-8545(99)00218-0. [DOI] [Google Scholar]
- Van Doorslaer S. Hyperfine Spectroscopy: ESEEM. eMagRes. 2017, 6, 51–70. 10.1002/9780470034590.emrstm1517. [DOI] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Barney B. M.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Unification of Reaction Pathway and Kinetic Scheme for N2 Reduction Catalyzed by Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5583–5587. 10.1073/pnas.1202197109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B. M. ENDOR and ESEEM of a Non-Kramers Doublet in an Integer-Spin System. J. Phys. Chem. 1994, 98, 11657–11665. 10.1021/j100096a006. [DOI] [Google Scholar]
- Song R.; Doan P. E.; Gurbiel R. J.; Sturgeon B. E.; Hoffman B. M. Non-Kramers ENDOR and ESEEM of the S = 2 Ferrous Ion of [Fe(II)EDTA]2–. J. Magn. Reson. 1999, 141, 291–300. 10.1006/jmre.1999.1887. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Benton P. M. C.; Laryukhin M.; Igarashi R. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. The Interstitial Atom of the Nitrogenase FeMo-Cofactor: ENDOR and ESEEM Show It Is Not an Exchangeable Nitrogen. J. Am. Chem. Soc. 2003, 125, 5604–5605. 10.1021/ja034383n. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Pelmenschikov V.; Maeser N.; Laryukhin M.; Yang T. C.; Noodleman L.; Dean D. R.; Case D. A.; Seefeldt L. C.; Hoffman B. M. Testing if the Interstitial Atom, X, of the Nitrogenase Molybdenum-Iron Cofactor Is N or C: ENDOR, ESEEM, and DFT Studies of the S = 3/2 Resting State in Multiple Environments. Inorg. Chem. 2007, 46, 11437–11439. 10.1021/ic7018814. [DOI] [PubMed] [Google Scholar]
- Harmer J.; Mitrikas G.; Schweiger A. Advanced Pulse EPR Methods for the Characterization of Metalloproteins. Biol. Magn. Reson. 2009, 28, 13–61. 10.1007/978-0-387-84856-3_2. [DOI] [Google Scholar]
- Lukoyanov D.; Dikanov S. A.; Yang Z. Y.; Barney B. M.; Samoilova R. I.; Narasimhulu K. V.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. ENDOR/HYSCORE Studies of the Common Intermediate Trapped during Nitrogenase Reduction of N2H2, CH3N2H, and N2H4 Support an Alternating Reaction Pathway for N2 Reduction. J. Am. Chem. Soc. 2011, 133, 11655–11664. 10.1021/ja2036018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. J.; Fu J.; Guo Y.; Drury O. B.; Friedrich S.; Rauchfuss T.; Volkers P. I.; Peters J. C.; Scott V.; Brown S. D.; et al. X-ray Photochemistry in Iron Complexes from Fe(0) to Fe(IV) - Can a Bug Become a Feature?. Inorg. Chim. Acta 2008, 361, 1157–1165. 10.1016/j.ica.2007.10.039. [DOI] [Google Scholar]
- Stumpf V.; Gokhberg K.; Cederbaum L. S. The Role of Metal Ions in X-ray-induced Photochemistry. Nat. Chem. 2016, 8, 237–241. 10.1038/nchem.2429. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Khadka N.; Yang Z. Y.; Fielding A. J.; Lukoyanov D.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Exploring Electron/Proton Transfer and Conformational Changes in the Nitrogenase MoFe Protein and FeMo-cofactor Through Cryoreduction/EPR Measurements. Isr. J. Chem. 2016, 56, 841–851. 10.1002/ijch.201600026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Barney B. M.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Connecting Nitrogenase Intermediates with the Kinetic Scheme for N2 Reduction by a Relaxation Protocol and Identification of the N2 Binding State. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1451–1455. 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stappen C.; Davydov R.; Yang Z. Y.; Fan R.; Guo Y.; Bill E.; Seefeldt L. C.; Hoffman B. M.; DeBeer S. Spectroscopic Description of the E1 State of Mo Nitrogenase Based on Mo and Fe X-ray Absorption and Mössbauer Studies. Inorg. Chem. 2019, 58, 12365–12376. 10.1021/acs.inorgchem.9b01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. J.; Ashby G. A.; Wharton C. W.; Thorneley R. N. F. Time-Resolved Binding of Carbon Monoxide to Nitrogenase Monitored by Stopped-Flow Infrared Spectroscopy. J. Am. Chem. Soc. 1997, 119, 6450–6451. 10.1021/ja971088s. [DOI] [Google Scholar]
- Thorneley R. N. F.; George S. J. In Prokaryotic Nitrogen Fixation: A Model System for Analysis of a Biological Process; Triplett E. W., Ed.; Horizon Scientific Press: Wyndham, UK, 2000. [Google Scholar]
- Pickett C. J.; Vincent K. A.; Ibrahim S. K.; Gormal C. A.; Smith B. E.; Fairhurst S. A.; Best S. P. Synergic Binding of Carbon Monoxide and Cynaide to the FeMo Cofactor of Nitrogenase: Relic Chemistry of an Ancient Enzyme?. Chem. - Eur. J. 2004, 10, 4770–4776. 10.1002/chem.200400382. [DOI] [PubMed] [Google Scholar]
- Tolland J. D.; Thorneley R. N. F. Stopped-flow Fourier Transform Infrared Spectroscopy Allows Continuous Monitoring of Azide Reduction, Carbon Monoxide Inhibition, and ATP Hydrolysis by Nitrogenase. Biochemistry 2005, 44, 9520–9527. 10.1021/bi050453m. [DOI] [PubMed] [Google Scholar]
- Yan L.; Dapper C. H.; George S. J.; Wang H.; Mitra D.; Dong W.; Newton W. E.; Cramer S. P. Photolysis of Hi-CO Nitrogenase - Observation of a Plethora of Distinct CO Species Using Infrared Spectroscopy. Eur. J. Inorg. Chem. 2011, 2011, 2064–2074. 10.1002/ejic.201100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Pelmenschikov V.; Dapper C. H.; Scott A. D.; Newton W. E.; Cramer S. P. IR-Monitored Photolysis of CO-Inhibited Nitrogenase: A Major EPR-Silent Species with Coupled Terminal CO Ligands. Chem. - Eur. J. 2012, 18, 16349–16357. 10.1002/chem.201202072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paengnakorn P.; Ash P. A.; Shaw S.; Danyal K.; Chen T.; Dean D. R.; Seefeldt L. C.; Vincent K. A. Infrared Spectroscopy of the Nitrogenase MoFe Protein Under Electrochemical Control: Potential-triggered CO Binding. Chem. Sci. 2017, 8, 1500–1505. 10.1039/C6SC02860H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.-Y.; Seefeldt L. C.; Dean D. R.; Cramer S. P.; George S. J. Steric Control of the Hi-CO MoFe Nitrogenase Complex Revealed by Stopped-Flow Infrared Spectroscopy. Angew. Chem., Int. Ed. 2011, 50, 272–275. 10.1002/anie.201005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A. B. Resonance Raman Intensity Analysis of Excited-State Dynamics. Acc. Chem. Res. 1997, 30, 519–527. 10.1021/ar960240c. [DOI] [Google Scholar]
- McCreery R. L.Raman Spectroscopy for Chemical Analysis; Wiley-Interscience, 2000. [Google Scholar]
- Gardiner D. J.; Graves P. R.; Bowley J. J.; Gerrard D. L.; Louden J. D.; Turrell G.. Practical Raman Spectroscopy; Springer-Verlag: Berlin, 1989. [Google Scholar]
- Spiro T. G.; Czernuszewicz R. S. In Physical Methods in Bioinorganic Chemistry. Spectroscopy and Magnetism; Que L., Jr., Ed.; University Science Books: Sausalito, CA, 2000. [Google Scholar]
- Fu W.; Morgan V.; Mortenson L. E.; Johnson M. K. Resonance Raman Studies of the [4Fe-4S] to [2Fe-2S] Cluster Conversion in the Iron Protein of Nitrogenase. FEBS Lett. 1991, 284, 165–168. 10.1016/0014-5793(91)80676-T. [DOI] [PubMed] [Google Scholar]
- Sen S.; Igarashi R.; Smith A.; Johnson M. K.; Seefeldt L. C.; Peters J. W. A Conformational Mimic of the MgATP-Bound ″On State″ of the Nitrogenase Iron Protein. Biochemistry 2004, 43, 1787–1797. 10.1021/bi0358465. [DOI] [PubMed] [Google Scholar]
- Maiuri M.; Delfino I.; Cerullo G.; Manzoni C.; Pelmenschikov V.; Guo Y.; Wang H.; Gee L. B.; Dapper C. H.; Newton W. E.; et al. Low Frequency Dynamics of the Nitrogenase MoFe Protein via Femtosecond Pump Probe Spectroscopy - Observation of a Candidate Promoting Vibration. J. Inorg. Biochem. 2015, 153, 128–135. 10.1016/j.jinorgbio.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko L. A.; Roschupkina O. S.; Sadkov A. P.; Marakushev S. A.; Mikhailov G. M.; Borod’ko Yu. G. Spectroscopic Investigation of FeMo-Cofactor. Coenzyme A as one of the probable components of an Active Site of Nitrogenase. Biochem. Biophys. Res. Commun. 1980, 96, 1384–1392. 10.1016/0006-291X(80)90104-7. [DOI] [PubMed] [Google Scholar]
- Delfino I.; Cerullo G.; Cannistraro S.; Manzoni C.; Polli D.; Dapper C.; Newton W. E.; Guo Y.; Cramer S. P. Observation of Terahertz Vibrations in the Nitrogenase FeMo Cofactor by Femtosecond Pump-probe Spectroscopy. Angew. Chem., Int. Ed. 2010, 49, 3912–3915. 10.1002/anie.200906787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Wende T.; Kukura P. Principles and Applications of Broadband Impulsive Vibrational Spectroscopy. J. Phys. Chem. A 2015, 119, 9506–9517. 10.1021/acs.jpca.5b05948. [DOI] [PubMed] [Google Scholar]
- Karunakaran V.; Sun Y.; Benabbas A.; Champion P. M. Investigations of the Low Frequency Modes of Ferric Cytochrome c Using Vibrational Coherence Spectroscopy. J. Phys. Chem. B 2014, 118, 6062–6070. 10.1021/jp501298c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfseder B.; Seidner L.; Stock G.; Domcke W. Femtosecond Pump-probe Spectroscopy of Electron-transfer Systems: A Nonperturbative Approach. Chem. Phys. 1997, 217, 275–287. 10.1016/S0301-0104(97)00046-3. [DOI] [Google Scholar]
- Chumakov A. I.; Rüffer R.; Grünsteudel H.; Grünsteudel H. F.; Grübel G.; Metge J.; Leupold O.; Goodwin H. A. Energy Dependence of Nuclear Recoil Measured with Incoherent Nuclear Scattering of Synchrotron Radiation. Europhys. Lett. 1995, 30, 427–432. 10.1209/0295-5075/30/7/009. [DOI] [Google Scholar]
- Seto M.; Yoda Y.; Kikuta S.; Zhang X. W.; Ando M. Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett. 1995, 74, 3828–3831. 10.1103/PhysRevLett.74.3828. [DOI] [PubMed] [Google Scholar]
- Sturhahn W.; Toellner T. S.; Alp E. E.; Zhang X.; Ando M.; Yoda Y.; Kikuta S.; Seto M.; Kimball C. W.; Dabrowski B. Phonon Density of States Measured by Inelastic Nuclear Resonant Scattering. Phys. Rev. Lett. 1995, 74, 3832–3835. 10.1103/PhysRevLett.74.3832. [DOI] [PubMed] [Google Scholar]
- Sturhahn W. Nuclear Resonant Spectroscopy. J. Phys.: Condens. Matter 2004, 16, S497–S530. 10.1088/0953-8984/16/5/009. [DOI] [Google Scholar]
- Cramer S. P.; Xiao Y.; Wang H.; Guo Y.; Smith M. C. Nuclear Resonance Vibrational Spectroscopy (NRVS) of FeS Model Compounds, Fe-S Proteins, and Nitrogenase. Hyperfine Interact. 2007, 170, 47–54. 10.1007/s10751-006-9473-4. [DOI] [Google Scholar]
- George S. J.; Barney B. M.; Mitra D.; Igarashi R. Y.; Guo Y.; Dean D. R.; Cramer S. P.; Seefeldt L. C. EXAFS and NRVS Reveal a Conformational Distortion of the FeMo-cofactor in the MoFe Nitrogenase Propargyl Alcohol Complex. J. Inorg. Biochem. 2012, 112, 85–92. 10.1016/j.jinorgbio.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. J.; Igarashi R. Y.; Xiao Y.; Hernandez J. A.; Demuez M.; Zhao D.; Yoda Y.; Ludden P. W.; Rubio L. M.; Cramer S. P. Extended X-ray absorption Fine Structure and Nuclear Resonance Vibrational Spectroscopy Reveal that NifB-co, a FeMo-co Precursor, Comprises a 6Fe Core with an Interstitial Light Atom. J. Am. Chem. Soc. 2008, 130, 5673–5680. 10.1021/ja0755358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Brecht E.; Aznavour K.; Nix J. C.; Xiao Y.; Wang H.; George S. J.; Bau R.; Keable S.; Peters J. W.; et al. Nuclear Resonance Vibrational Spectroscopy (NRVS) of Rubredoxin and MoFe Protein Crystals. Hyperfine Interact. 2013, 222, 77–90. 10.1007/s10751-012-0643-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A. D.; Pelmenschikov V.; Guo Y.; Yan L.; Wang H.; George S. J.; Dapper C. H.; Newton W. E.; Yoda Y.; Tanaka Y.; et al. Structural Characterization of CO-Inhibited Mo-Nitrogenase by Combined Application of Nuclear Resonance Vibrational Spectroscopy, Extended X-ray absorption Fine Structure, and Density Functional Theory: New Insights into the Effects of CO Binding and the Role of the Interstitial Atom. J. Am. Chem. Soc. 2014, 136, 15942–15954. 10.1021/ja505720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Fisher K.; Smith M. C.; Newton W. E.; Case D. A.; George S. J.; Wang H.; Sturhahn W.; Alp E. E.; Zhao J.; et al. How Nitrogenase Shakes - Initial Information about P-cluster and FeMo-cofactor Normal Modes from Nuclear Resonance Vibrational Spectroscopy (NRVS). J. Am. Chem. Soc. 2006, 128, 7608–7612. 10.1021/ja0603655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilkuri V. G.; DeBeer S.; Neese F. Ligand Field Theory and Angular Overlap Model Based Analysis of the Electronic Structure of Homovalent Iron-Sulfur Dimers. Inorg. Chem. 2020, 59, 984–995. 10.1021/acs.inorgchem.9b00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Sivalingam K.; Neese F.; Chan G. K. Low-energy Spectrum of Iron-Sulfur Clusters Directly from Many-particle Quantum Mechanics. Nat. Chem. 2014, 6, 927–933. 10.1038/nchem.2041. [DOI] [PubMed] [Google Scholar]
- Eady R. R.; Robson R. L.; Richardson T. H.; Miller R. W.; Hawkins M. The Vanadium Nitrogenase of Azotobacter chroococcum. Purification and Properties of the VFe Protein. Biochem. J. 1987, 244, 197–207. 10.1042/bj2440197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L.; Wallace B. A. Protein Secondary Structure Analyses from Circular Dichroism Spectroscopy: Methods and Reference Databases. Biopolymers 2008, 89, 392–400. 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- Greenfield N. J. Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc. 2006, 1, 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K.; Berova N.; Woody R.. Circular Dichroism: Principles and Applications, 2nd ed.; Wiley-VCH, 2000. [Google Scholar]
- Banerjee B.; Misra G.; Ashraf M. T.. Chapter 2 - Circular Dichroism. Data Processing Handbook for Complex Biological Data Sources; Misra G., Ed.; Academic Press, 2019; Chapter 2, 10.1016/B978-0-12-816548-5.00002-2. [DOI] [Google Scholar]
- Cotton A. Recherches sur L’absorption et la Dispersion de la Lumière par les Milieux doués du Pouvoir Rotatoire. J. Phys. Theor. Appl. 1896, 5 (1), 237. 10.1051/jphystap:018960050023700. [DOI] [Google Scholar]
- Faraday M. XLIX. Experimental Researches in Electricity.—Nineteenth Series. London, Edinburgh, and Dublin Philos. Mag. J. Sci. 1846, 28, 294–317. 10.1080/14786444608645086. [DOI] [Google Scholar]
- Faraday M. I. I. Experimental Researches in Electricity.—Nineteenth Series. Philos. Trans. R. Soc. London 1846, 136, 1. 10.1098/rstl.1846.0001. [DOI] [Google Scholar]
- Neese F.; Solomon E. I. MCD C-Term Signs, Saturation Behavior, and Determination of Band Polarizations in Randomly Oriented Systems with Spin S ≥ 1/2. Applications to S = 1/2 and S = 5/2. Inorg. Chem. 1999, 38, 1847–1865. 10.1021/ic981264d. [DOI] [PubMed] [Google Scholar]
- Mason R.A Practical Guide to Magnetic Circular Dichroism Spectroscopy; John Wiley & Sons, Inc.: Hoboken, NJ, 2007. [Google Scholar]
- Stephens P. J. Magnetic Circular Dichroism. Annu. Rev. Phys. Chem. 1974, 25, 201–232. 10.1146/annurev.pc.25.100174.001221. [DOI] [Google Scholar]
- Stephens P. J. Magnetic Circular Dichroism. Adv. Chem. Phys. 2007, 35, 197–264. 10.1002/9780470142547.ch4. [DOI] [Google Scholar]
- Stephens P. J.; McKenna C. E.; Smith B. E.; Nguyen H. T.; McKenna M.-C.; Thomson A. J.; Devlin F.; Jones J. B. Circular Dichroism and Magnetic Circular Dichroism of Nitrogenase Proteins. Proc. Natl. Acad. Sci. U. S. A. 1979, 76, 2585–2589. 10.1073/pnas.76.6.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P. J.; McKenna C. E.; McKenna M. C.; Nguyen H. T.; Devlin F. Circular Dichroism and Magnetic Circular Dichroism of Reduced Molybdenum-Iron Protein of Azotobacter vinelandii Nitrogenase. Biochemistry 1981, 20, 2857–2864. 10.1021/bi00513a023. [DOI] [PubMed] [Google Scholar]
- Morningstar J. E.; Johnson M. K.; Case E. E.; Hales B. J. Characterization of the Metal-Clusters in the Nitrogenase Molybdenum-Iron and Vanadium-Iron Proteins of Azotobacter vinelandii Using Magnetic Circular-Dichroism Spectroscopy. Biochemistry 1987, 26, 1795–1800. 10.1021/bi00381a001. [DOI] [PubMed] [Google Scholar]
- Yoo S. J.; Angove H. C.; Burgess B. K.; Münck E.; Peterson J. Magnetic Circular Dichroism Study of the All-Ferrous [4Fe-4S] Cluster of the Fe-Protein of Azotobacter vinelandii Nitrogenase. J. Am. Chem. Soc. 1998, 120, 9704–9705. 10.1021/ja981867o. [DOI] [Google Scholar]
- Noodleman L.; Case D. A. Density-Functional Theory of Spin Polarization and Spin Coupling in Iron-Sulfur Clusters. Adv. Inorg. Chem. 1992, 38, 423–470. 10.1016/S0898-8838(08)60070-7. [DOI] [Google Scholar]
- Cotton M. S.; Rupnik K.; Broach R. B.; Hu Y.; Fay A. W.; Ribbe M. W.; Hales B. J. VTVH-MCD Study of the ΔnifBΔnifZ MoFe Protein from Azotobacter vinelandii. J. Am. Chem. Soc. 2009, 131, 4558–4559. 10.1021/ja807525m. [DOI] [PubMed] [Google Scholar]
- Broach R. B.; Rupnik K.; Hu Y.; Fay A. W.; Cotton M.; Ribbe M. W.; Hales B. J. Variable-Temperature, Variable-Field Magnetic Circular Dichroism Spectroscopic Study of the Metal Clusters in the ΔnifB and ΔnifH MoFe Proteins of Nitrogenase from Azotobacter vinelandii. Biochemistry 2006, 45, 15039–15048. 10.1021/bi061697p. [DOI] [PubMed] [Google Scholar]
- Goodwin P. J.; Agar J. N.; Roll J. T.; Roberts G. P.; Johnson M. K.; Dean D. R. The Azotobacter vinelandii NifEN Complex Contains Two Identical [4Fe-4S] Clusters. Biochemistry 1998, 37, 10420–10428. 10.1021/bi980435n. [DOI] [PubMed] [Google Scholar]
- Mössbauer R. L. Kernresonanzfluoreszenz von Gammastrahlung in Ir191. Eur. Phys. J. A 1958, 151, 124–143. 10.1007/BF01344210. [DOI] [Google Scholar]
- Beinert H.; Holm R. H.; Münck E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- Schunemann V.; Winkler H. Structure and Dynamics of Biomolecules Studied by Mössbauer Spectroscopy. Rep. Prog. Phys. 2000, 63, 263–353. 10.1088/0034-4885/63/3/202. [DOI] [Google Scholar]
- Pandelia M.-E.; Lanz N. D.; Booker S. J.; Krebs C. Mössbauer Spectroscopy of Fe/S Proteins. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 1395–1405. 10.1016/j.bbamcr.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Hoggins J. T.; Steinfink H. Empirical Bonding Relationships in Metal-Iron-Sulfide Compounds. Inorg. Chem. 1976, 15, 1682–1685. 10.1021/ic50161a041. [DOI] [Google Scholar]
- Yoo S. J.; Angove H. C.; Papaefthymiou V.; Burgess B. K.; Münck E. Mössbauer Study of the MoFe Protein of Nitrogenase from Azotobacter vinelandii Using Selective 57Fe Enrichment of the M-Centers. J. Am. Chem. Soc. 2000, 122, 4926–4936. 10.1021/ja000254k. [DOI] [Google Scholar]
- Cramer S. P.; Gillum W. O.; Hodgson K. O.; Mortenson L. E.; Stiefel E. I.; Chisnell J. R.; Brill W. J.; Shah V. K. The Molybdenum Site of Nitrogenase. 2. A Comparative Study of Mo-Fe Proteins and the Iron-Molybdenum Cofactor by X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 1978, 100, 3814–3819. 10.1021/ja00480a027. [DOI] [Google Scholar]
- Cramer S. P.; Hodgson K. O.; Gillum W. O.; Mortenson L. E. The Molybdenum Site of Nitrogenase. Preliminary Structural Evidence from X-Ray Absorption Spectroscopy. J. Am. Chem. Soc. 1978, 100, 3398–3407. 10.1021/ja00479a023. [DOI] [Google Scholar]
- Antonio M. R.; Teo B.-K.; Orme-Johnson W. H.; Nelson M. J.; Groh S. E.; Lindahl P. A.; Kauzlarich S. M.; Averill B. A. Iron EXAFS of the Iron-Molybdenum Cofactor of Nitrogenase. J. Am. Chem. Soc. 1982, 104, 4703–4705. 10.1021/ja00381a045. [DOI] [Google Scholar]
- Chen J.; Christiansen J.; Campobasso N.; Bolin J. T.; Tittsworth R. C.; Hales B. J.; Rehr J. J.; Cramer S. P. Verfeinerung eines Modells für den Nitrogenase-Mo-Fe-Cluster mit Einkristall-Mo- und -Fe-EXAFS. Angew. Chem. 1993, 105, 1661–1663. 10.1002/ange.19931051109. [DOI] [Google Scholar]
- Chen J.; Christiansen J.; Tittsworth R. C.; Hales B. J.; George S. J.; Coucouvanis D.; Cramer S. P. Iron EXAFS of Azotobacter vinalndii Nitrogenase Mo-Fe and V-Fe Proteins. J. Am. Chem. Soc. 1993, 115, 5509–5515. 10.1021/ja00066a019. [DOI] [Google Scholar]
- Liu H. I.; Filipponi A.; Gavini N.; Burgess B. K.; Hedman B.; Di Cicco A.; Natoli C. R.; Hodgson K. O. EXAFS Studies of FeMo-Cofactor and MoFe Protein: Direct Evidence for the Long-Range Mo-Fe-Fe Interaction and Cyanide Binding to the Mo in FeMo-Cofactor. J. Am. Chem. Soc. 1994, 116, 2418–2423. 10.1021/ja00085a022. [DOI] [Google Scholar]
- Christiansen J.; Tittsworth R. C.; Hales B. J.; Cramer S. P. Fe and Mo EXAFS of Azotobacter vinelandii Nitrogenase in Partially Oxidized and Singly Reduced Forms. J. Am. Chem. Soc. 1995, 117, 10017–10024. 10.1021/ja00145a012. [DOI] [Google Scholar]
- Musgrave K. B.; Liu H. I.; Ma L.; Burgess B. K.; Watt G.; Hedman B.; Hodgson K. O. EXAFS Studies on the PN and POX States of the P-clusters in Nitrogenase. JBIC, J. Biol. Inorg. Chem. 1998, 3, 344–352. 10.1007/s007750050243. [DOI] [Google Scholar]
- Krahn E.; Weiss B. J. R.; Kröckel M.; Groppe J.; Henkel G.; Cramer S. P.; Trautwein A. X.; Schneider K.; Müller A. The Fe-only Nitrogenase from Rhodobacter capsulatus: Identification of the Cofactor, an Unusual, High-nuclearity Iron-Sulfur Cluster, by Fe K-edge EXAFS and 57Fe Mössbauer Spectroscopy. JBIC, J. Biol. Inorg. Chem. 2002, 7, 37–45. 10.1007/s007750100263. [DOI] [PubMed] [Google Scholar]
- Strange R. W.; Eady R. R.; Lawson D.; Hasnain S. S. XAFS Studies of Nitrogenase: the MoFe and VFe Proteins and the Use of Crystallographic Coordinates in Three-dimensional EXAFS Data Analysis. J. Synchrotron Radiat. 2003, 10, 71–75. 10.1107/S0909049502017272. [DOI] [PubMed] [Google Scholar]
- Mitra D.; George S. J.; Guo Y.; Kamali S.; Keable S.; Peters J. W.; Pelmenschikov V.; Case D. A.; Cramer S. P. Characterization of [4Fe-4S] Cluster Vibrations and Structure in Nitrogenase Fe Protein at Three Oxidation Levels via Combined NRVS, EXAFS, and DFT Analyses. J. Am. Chem. Soc. 2013, 135, 2530–2543. 10.1021/ja307027n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J.; DeBeer S. The Role of X-ray Spectroscopy in Understanding the Geometric and Electronic Structure of Nitrogenase. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 1406–1415. 10.1016/j.bbamcr.2014.11.027. [DOI] [PubMed] [Google Scholar]
- George S. J.; Hernandez J. A.; Jimenez-Vicente E.; Echavarri-Erasun C.; Rubio L. M. EXAFS Reveals Two Mo Environments in the Nitrogenase Iron-Molybdenum Cofactor Biosynthetic Protein NifQ. Chem. Commun. 2016, 52, 11811–11814. 10.1039/C6CC06370E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stappen C.; Thorhallsson A. T.; Decamps L.; Bjornsson R.; DeBeer S. Resolving the Structure of the E1 State of Mo Nitrogenase through Mo and Fe K-edge EXAFS and QM/MM Calculations. Chem. Sci. 2019, 10, 9807–9821. 10.1039/C9SC02187F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flank A. M.; Weininger M.; Mortenson L. E.; Cramer S. P. Single Crystal EXAFS of Nitrogenase. J. Am. Chem. Soc. 1986, 108, 1049–1055. 10.1021/ja00265a034. [DOI] [Google Scholar]
- Kim J.; Woo D.; Rees D. C. X-ray Crystal Structure of the Nitrogenase Molybdenum-Iron Protein from Clostridium pasteurianum at 3.0-Å Resolution. Biochemistry 1993, 32, 7104–7115. 10.1021/bi00079a006. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. Nitrogenase MoFe-Protein at 1.16 Å Resolution: A Central Ligand in the FeMo-Cofactor. Science 2002, 297, 1696–1700. 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- Glatzel P.; Bergmann U. High resolution 1s core hole X-ray Spectroscopy in 3d transition Metal Complexes—Electronic and Structural information. Coord. Chem. Rev. 2005, 249, 65–95. 10.1016/j.ccr.2004.04.011. [DOI] [Google Scholar]
- Glatzel P.; Sikora M.; Smolentsev G.; Fernández-García M. Hard X-ray Photon-in Photon-out Spectroscopy. Catal. Today 2009, 145, 294–299. 10.1016/j.cattod.2008.10.049. [DOI] [Google Scholar]
- Penner-Hahn J. E. In Comprehensive Coordination Chemistry II. From Biology to Nanotechnology; McCleverty J. A.; Meyer T. J., Ed.; Elsevier: London, UK, 2003; Vol. 2. [Google Scholar]
- DeBeer S. In Methods in Enzymology; David S. S., Ed.; Elsevier, 2018; Vol. 599. [Google Scholar]
- Kowalska J. K.; Lima F. A.; Pollock C. J.; Rees J. A.; DeBeer S. A Practical Guide to High-resolution X-ray Spectroscopic Measurements and their Applications in Bioinorganic Chemistry. Isr. J. Chem. 2016, 56, 803–815. 10.1002/ijch.201600037. [DOI] [Google Scholar]
- Pollock C. J.; DeBeer S. Insights into the Geometric and Electronic Structure of Transition Metal Centers from Valence-to-Core X-ray Emission Spectroscopy. Acc. Chem. Res. 2015, 48, 2967–2975. 10.1021/acs.accounts.5b00309. [DOI] [PubMed] [Google Scholar]
- Bjornsson R.; Delgado-Jaime M. U.; Lima F. A.; Sippel D.; Schlesier J.; Weyhermüller T.; Einsle O.; Neese F.; DeBeer S. Molybdenum L-Edge XAS Spectra of MoFe Nitrogenase. Z. Anorg. Allg. Chem. 2015, 641, 65–71. 10.1002/zaac.201400446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henthorn J. T.; Arias R. J.; Koroidov S.; Kroll T.; Sokaras D.; Bergmann U.; Rees D. C.; DeBeer S. Localized Electronic Structure of Nitrogenase FeMoco Revealed by Selenium K-Edge High Resolution X-ray Absorption Spectroscopy. J. Am. Chem. Soc. 2019, 141, 13676–13688. 10.1021/jacs.9b06988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedman B.; Frank P.; Gheller S. F.; Roe A. L.; Newton W. E.; Hodgson K. O. New Structure Insights into the Iron-Molybdenum Cofactor from Azotobacter vinelandii Nitrogenase through Sulfur K and Molybdenum L X-ray Absorption Edge Studies. J. Am. Chem. Soc. 1988, 110, 3798–3805. 10.1021/ja00220a013. [DOI] [Google Scholar]
- Laporte O.; Meggers W. F. Some Rules of Spectral Structure*. J. Opt. Soc. Am. 1925, 11, 459–463. 10.1364/JOSA.11.000459. [DOI] [Google Scholar]
- Jørgensen C. K. Chemical Bonding Inferred from Visible and Ultraviolet Absorption Spectra. Solid State Phys. 1962, 13, 375–462. 10.1016/S0081-1947(08)60460-9. [DOI] [Google Scholar]
- Srivastava U. C.; Nigam H. L. X-ray Absorption Edge Spectrometry (XAES) as Applied to Coordination Chemistry. Coord. Chem. Rev. 1973, 9, 275–310. 10.1016/S0010-8545(00)82080-9. [DOI] [Google Scholar]
- Shulman G. R.; Yafet Y.; Eisenberger P.; Blumberg W. E. Observations and Interpretation of X-ray Absorption Edges in Iron Compounds and Proteins. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 1384–1388. 10.1073/pnas.73.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George G. N.; Pickering I. J.; Prince R. C.; Zhou Z. H.; Adams M. W. W. X-ray Absorption Spectroscopy of Pyrococcus furiosus Rubredoxin. JBIC, J. Biol. Inorg. Chem. 1996, 1, 226–230. 10.1007/s007750050047. [DOI] [Google Scholar]
- Lugo-Mas P.; Dey A.; Xu L.; Davin S. D.; Benedict J.; Kaminsky W.; Hodgson K. O.; Hedman B.; Solomon E. I.; Kovacs J. A. How Does Single Oxygen Atom Addition Affect the Properties of an Fe-Nitrile Hydratase Analogue? The Compensatory Role of the Unmodified Thiolate. J. Am. Chem. Soc. 2006, 128, 11211–11221. 10.1021/ja062706k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J. K.; Hahn A. W.; Albers A.; Schiewer C. E.; Bjornsson R.; Lima F. A.; Meyer F.; DeBeer S. X-ray Absorption and Emission Spectroscopic Studies of [L2Fe2S2]n Model Complexes: Implications for the Experimental Evaluation of Redox States in Iron-Sulfur Clusters. Inorg. Chem. 2016, 55, 4485–4497. 10.1021/acs.inorgchem.6b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen K.; Siddons D. P.; Hastings J. B.; Berman L. E. Elimination of the Inner-Shell Lifetime Broadening in X-Ray-Absorption Spectroscopy. Phys. Rev. Lett. 1991, 67, 2850–2853. 10.1103/PhysRevLett.67.2850. [DOI] [PubMed] [Google Scholar]
- Eisenberger P.; Platzman P. M.; Winick H. X-Ray Resonant Raman Scattering: Observation of Characteristic Radiation Narrower than the Lifetime Width. Phys. Rev. Lett. 1976, 36, 623–626. 10.1103/PhysRevLett.36.623. [DOI] [Google Scholar]
- Glatzel P.; Sikora M.; Fernández-García M. Resonant X-ray Spectroscopy to Study K Absorption Pre-edges in 3d Transition Metal Compounds. Eur. Phys. J.: Spec. Top. 2009, 169, 207–214. 10.1140/epjst/e2009-00994-7. [DOI] [Google Scholar]
- Rees J. A.; Bjornsson R.; Kowalska J. K.; Lima F. A.; Schlesier J.; Sippel D.; Weyhermüller T.; Einsle O.; Kovacs J. A.; DeBeer S. Comparative Electronic Structures of Nitrogenase FeMoco and FeVco. Dalton Trans. 2017, 46, 2445–2455. 10.1039/C7DT00128B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A.; Ostlund N. S.. Modern Quantum Chemistry; McGraw-Hill: New York, 1989. [Google Scholar]
- Koch W.; Holthausen M. C.. A Chemist’s Guide to Density Functional Theory; Wiley-VCH: Weinheim, 2000. [DOI] [PubMed] [Google Scholar]
- Szilagyi R. K.; Winslow M. A. On the Accuracy of Density Functional Theory for Iron-Sulfur Clusters. J. Comput. Chem. 2006, 27, 1385–1397. 10.1002/jcc.20449. [DOI] [PubMed] [Google Scholar]
- Harris T. V.; Szilagyi R. K. Iron-Sulfur Bond Covalency from Electronic Structure Calculations for Classical Iron-Sulfur Clusters. J. Comput. Chem. 2014, 35, 540–552. 10.1002/jcc.23518. [DOI] [PubMed] [Google Scholar]
- Raugei S.; Seefeldt L. C.; Hoffman B. M. Critical Computational Analysis Illuminates the Reductive-elimination Mechanism that Activates Nitrogenase for N2 Reduction. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E10521–E10530. 10.1073/pnas.1810211115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Ryde U. Extremely Large Differences in DFT Energies for Nitrogenase Models. Phys. Chem. Chem. Phys. 2019, 21, 2480–2488. 10.1039/C8CP06930A. [DOI] [PubMed] [Google Scholar]
- Thorhallsson A. T.; Benediktsson B.; Bjornsson R. A Model for Dinitrogen Binding in the E4 State of Nitrogenase. Chem. Sci. 2019, 10, 11110–11124. 10.1039/C9SC03610E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Guo S.; Sun Q.; Chan G. K.-L. Electronic landscape of the P-cluster of Nitrogenase as revealed through many-Electron quantum wavefunctions. Nat. Chem. 2019, 11, 1026–1033. 10.1038/s41557-019-0337-3. [DOI] [PubMed] [Google Scholar]
- Butcher K. D.; Didziulis S. V.; Briat B.; Solomon E. I. Variable Photon Energy Photoelectron Spectroscopy on FeCl4–: An Unusual Electronic Structure for High-Spin d5 Complexes. J. Am. Chem. Soc. 1990, 112, 2231–2242. 10.1021/ja00162a024. [DOI] [Google Scholar]
- Gebhard M. S.; Deaton J. C.; Koch S. A.; Millar M.; Solomon E. I. Single-Crystal Spectral Studies of Fe(SR)4– [R = 2,3,5,6,-(Me)4C6H]: The Electronic Structure of the Ferric Tetrathiolate Active Site. J. Am. Chem. Soc. 1990, 112, 2217–2231. 10.1021/ja00162a023. [DOI] [Google Scholar]
- Gebhard M. S.; Koch S. A.; Millar M.; Devlin F. J.; Stephens P. J.; Solomon E. I. Single-Crystal Spectroscopic Studies of Fe(SR)42- (R = 2-(Ph)C6H4): Electronic Structure of the Ferrous Site in Rubredoxin. J. Am. Chem. Soc. 1991, 113, 1640–1649. 10.1021/ja00005a030. [DOI] [Google Scholar]
- Butcher K. D.; Gebhard M. S.; Solomon E. I. Variable-Energy Photoelectron Spectroscopic Comparison of the Bonding in Ferric Sulfide and Ferric Chloride: An Alternative Description of the Near-IR-Visible Spin-Forbidden Transitions in High-Spin d5 Complexes. Inorg. Chem. 1990, 29, 2067–2074. 10.1021/ic00336a008. [DOI] [Google Scholar]
- Neese F. Prediction of Molecular Properties and Molecular Spectroscopy with Density Functional Theory: From Fundamental Theory to Exchange-Coupling. Coord. Chem. Rev. 2009, 253, 526–563. 10.1016/j.ccr.2008.05.014. [DOI] [Google Scholar]
- Malrieu J. P.; Caballol R.; Calzado C. J.; de Graaf C.; Guihéry N. Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. 10.1021/cr300500z. [DOI] [PubMed] [Google Scholar]
- Zener C. Interaction Between the d-Shells in the Transition Metals. II. Ferromagnetic Compounds of Manganese with Perovskite Structure. Phys. Rev. 1951, 82, 403–405. 10.1103/PhysRev.82.403. [DOI] [Google Scholar]
- Anderson P. W.; Hasegawa H. Considerations on Double Exchange. Phys. Rev. 1955, 100, 675–681. 10.1103/PhysRev.100.675. [DOI] [Google Scholar]
- Noodleman L.; Case D. A.; Mouesca J.-M.; Lamotte B. Valence Electron Delocalization in Polynuclear Iron-Sulfur Clusters. JBIC, J. Biol. Inorg. Chem. 1996, 1, 177–182. 10.1007/s007750050039. [DOI] [Google Scholar]
- Girerd J.-J. Electron Transfer Between Magnetic Ions in Mixed Valence Binuclear Systems. J. Chem. Phys. 1983, 79, 1766–1775. 10.1063/1.446021. [DOI] [Google Scholar]
- Noodleman L.; Baerends E. J. Electronic Structure, Magnetic Properties, ESR, and Optical Spectra for 2-Fe Ferredoxin Models by LCAO-Xα Valence Bond Theory. J. Am. Chem. Soc. 1984, 106, 2316–2327. 10.1021/ja00320a017. [DOI] [Google Scholar]
- Noodleman L.; Davidson E. R. Ligand Spin Polarization and Antiferromagnetic Coupling in Transition Metal Dimers. Chem. Phys. 1986, 109, 131–143. 10.1016/0301-0104(86)80192-6. [DOI] [Google Scholar]
- Noodleman L. Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers. J. Chem. Phys. 1981, 74, 5737–5743. 10.1063/1.440939. [DOI] [Google Scholar]
- Lovell T.; Li J.; Liu T.; Case D. A.; Noodleman L. FeMo Cofactor of Nitrogenase: A Density Functional Study of States MN, MOX, MR, and MI. J. Am. Chem. Soc. 2001, 123, 12392–12410. 10.1021/ja011860y. [DOI] [PubMed] [Google Scholar]
- Lovell T.; Liu T.; Case D. A.; Noodleman L. Structural, Spectroscopic, and Redox Consequences of a Central Ligand in the FeMoco of Nitrogenase: A Density Functional Theoretical Study. J. Am. Chem. Soc. 2003, 125, 8377–8383. 10.1021/ja0301572. [DOI] [PubMed] [Google Scholar]
- Lovell T.; Torres R. A.; Han W.-G.; Liu T.; Case D. A.; Noodleman L. Metal Substitution in the Active Site of Nitrogenase MFe7S9 (M = Mo4+, V3+, Fe3+). Inorg. Chem. 2002, 41, 5744–5753. 10.1021/ic020474u. [DOI] [PubMed] [Google Scholar]
- Mouesca J.-M.; Noodleman L.; Case D. A. Analysis of the 57Fe Hyperfine Coupling Constants and Spin States in Nitrogenase P-Clusters. Inorg. Chem. 1994, 33, 4819–4830. 10.1021/ic00100a004. [DOI] [Google Scholar]
- Gibson J. F.; Hall D. O.; Thornley J. H. M.; Whatley F. R. The Iron Complex in Spinach Ferredoxin. Proc. Natl. Acad. Sci. U. S. A. 1966, 56, 987–990. 10.1073/pnas.56.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius G.; Lamotte B. Single-Crystal ENDOR Study of a 57Fe-Enriched Iron Sulfur [Fe4S4]3+ Cluster. J. Am. Chem. Soc. 1989, 111, 2464–2469. 10.1021/ja00189a015. [DOI] [Google Scholar]
- Brintzinger H.; Palmer G.; Sands R. H. On the Ligand field of Iron in Ferredoxin from Spinach Chloroplasts and Related Nonheme Iron Enzymes. Proc. Natl. Acad. Sci. U. S. A. 1966, 55, 397–404. 10.1073/pnas.55.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Briganti F.; Luchinat C.; Scozzafava A.; Sola M. Proton NMR Spectroscopy and the Electronic Structure of the High Potential Iron-Sulfur Protein from Chromatium vinosum. J. Am. Chem. Soc. 1991, 113, 1237–1245. 10.1021/ja00004a025. [DOI] [Google Scholar]
- Papaefthymiou V.; Millar M. M.; Münck E. Mössbauer and EPR Studies of a Synthetic Analog for the Iron-Sulfur Fe4S4 Core of Oxidized and Reduced High-Potential Iron Proteins. Inorg. Chem. 1986, 25, 3010–3014. 10.1021/ic00237a017. [DOI] [Google Scholar]
- Kappl R.; Ebelshäuser M.; Hannemann F.; Bernhardt R.; Hüttermann J. Probing Electronic and Structural Properties of the Reduced [2Fe–2S] Cluster by Orientation-Selective 1H ENDOR Spectroscopy: Adrenodoxin versus Rieske Iron-Sulfur Protein. Appl. Magn. Reson. 2006, 30, 427–459. 10.1007/BF03166211. [DOI] [Google Scholar]
- Banci L.; Bertini I.; Briganti F.; Luchinat C.; Scozzafava A.; Oliver M. V. 1H NMR Spectra of Oxidized High-Potential Iron-Sulfur Protein (HiPIP) from Rhodocyclus gelatinosus. A Model for Oxidized HiPIPs. Inorg. Chem. 1991, 30, 4517–4524. 10.1021/ic00024a011. [DOI] [Google Scholar]
- Mouesca J. M.; Lamotte B.; Rius G. Comparison Between Spin Population Distributions in Two Different [Fe4S4]3+ Clusters by Proton ENDOR in Single Crystals of a Synthetic Model Compound. J. Inorg. Biochem. 1991, 43, 251. 10.1016/0162-0134(91)84241-Z. [DOI] [Google Scholar]
- DeBeer George S.; Petrenko T.; Neese F. Prediction of Iron K-edge Absorption Spectra Using Time-Dependent Density Functional Theory. J. Phys. Chem. A 2008, 112, 12936–12943. 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]
- Bernadotte S.; Atkins A. J.; Jacob C. R. Origin-independent Calculation of Quadrupole Intensities in X-ray Spectroscopy. J. Chem. Phys. 2012, 137, 204106. 10.1063/1.4766359. [DOI] [PubMed] [Google Scholar]
- Bjornsson R.; Lima F. A.; Spatzal T.; Weyhermüller T.; Glatzel P.; Bill E.; Einsle O.; Neese F.; DeBeer S. Identification of a Spin-coupled Mo(III) in the Nitrogenase Iron-Molybdenum Cofactor. Chem. Sci. 2014, 5, 3096–3103. 10.1039/C4SC00337C. [DOI] [Google Scholar]
- Lee N.; Petrenko T.; Bergmann U.; Neese F.; DeBeer S. Probing Valence Orbital Composition with Iron Kβ X-ray Emission Spectroscopy. J. Am. Chem. Soc. 2010, 132, 9715–9727. 10.1021/ja101281e. [DOI] [PubMed] [Google Scholar]
- Chantzis A.; Kowalska J. K.; Maganas D.; DeBeer S.; Neese F. Ab Initio Wave Function-Based Determination of Element Specific Shifts for the Efficient Calculation of X-ray Absorption Spectra of Main Group Elements and First Row Transition Metals. J. Chem. Theory Comput. 2018, 14, 3686–3702. 10.1021/acs.jctc.8b00249. [DOI] [PubMed] [Google Scholar]
- Petrenko T.; Sturhahn W.; Neese F. First-principles Calculation of Nuclear Resonance Vibrational Spectra. Hyperfine Interact. 2007, 175, 165–174. 10.1007/s10751-008-9600-5. [DOI] [Google Scholar]
- Neese F. Prediction and Interpretation of the 57Fe Isomer Shift in Mössbauer Spectra by Density Functional Theory. Inorg. Chim. Acta 2002, 337, 181–192. 10.1016/S0020-1693(02)01031-9. [DOI] [Google Scholar]
- Römelt M.; Ye S.; Neese F. Calibration of Modern Density Functional Theory Methods for the Prediction of 57Fe Mössbauer Isomer Shifts: Meta-GGA and Double-Hybrid Functionals. Inorg. Chem. 2009, 48, 784–785. 10.1021/ic801535v. [DOI] [PubMed] [Google Scholar]
- Sandala G. M.; Hopmann K. H.; Ghosh A.; Noodleman L. Calibration of DFT Functionals for the Prediction of 57Fe Mössbauer Spectral Parameters in Iron-Nitrosyl and Iron-Sulfur Complexes: Accurate Geometries Prove Essential. J. Chem. Theory Comput. 2011, 7, 3232–3247. 10.1021/ct200187d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell T.; Han W.-G.; Liu T.; Noodleman L. A Structural Model for the High-Valent Intermediate Q of Methane Monooxygenase from Broken-Symmetry Density Functional and Electrostatics Calculations. J. Am. Chem. Soc. 2002, 124, 5890–5894. 10.1021/ja0121282. [DOI] [PubMed] [Google Scholar]
- McWilliams S. F.; Brennan-Wydra E.; MacLeod K. C.; Holland P. L. Density Functional Calculations for Prediction of 57Fe Mössbauer Isomer Shifts and Quadrupole Splittings in β-Diketiminate Complexes. ACS Omega 2017, 2, 2594–2606. 10.1021/acsomega.7b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsson R.; Neese F.; DeBeer S. Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem. 2017, 56, 1470–1477. 10.1021/acs.inorgchem.6b02540. [DOI] [PubMed] [Google Scholar]
- Sinnecker S.; Slep L. D.; Bill E.; Neese F. Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in 57Fe Mössbauer Spectra. Inorg. Chem. 2005, 44, 2245–2254. 10.1021/ic048609e. [DOI] [PubMed] [Google Scholar]
- Pápai M.; Vankó G. On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory. J. Chem. Theory Comput. 2013, 9, 5004–5020. 10.1021/ct4007585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickerman N. S.; Tanifuji K.; Hu Y.; Ribbe M. W. Synthetic Analogues of Nitrogenase Metallocofactors: Challenges and Developments. Chem. - Eur. J. 2017, 23, 12425–12432. 10.1002/chem.201702496. [DOI] [PubMed] [Google Scholar]
- Lee S. C.; Lo W.; Holm R. H. Developments in the Biomimetic Chemistry of Cubane-Type and Higher Nuclearity Iron–Sulfur Clusters. Chem. Rev. 2014, 114, 3579–3600. 10.1021/cr4004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čorić I.; Holland P. L. Insight into the Iron-Molybdenum Cofactor of Nitrogenase from Synthetic Iron Complexes with Sulfur, Carbon, and Hydride Ligands. J. Am. Chem. Soc. 2016, 138, 7200–7211. 10.1021/jacs.6b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery T. J.; Wilson P. E.; Zhang B.; Bunker J.; Harrison R. G.; Nyborg A. C.; Thiriot D.; Watt G. D. Flavodoxin Hydroquinone Reduces Azotobacter vinelandii Fe Protein to the All-ferrous Redox State with a S = 0 Spin State. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17131–17136. 10.1073/pnas.0603223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P. A.; Day E. P.; Kent T. A.; Orme-Johnson W. H.; Münck E. Mössbauer, EPR, and Magnetization Studies of the Azotobacter vinelandii Fe Protein - Evidence for a [4Fe-4S]1+ Cluster with Spin S = 3/2. J. Biol. Chem. 1985, 260, 11160–11173. [PubMed] [Google Scholar]
- Carney M. J.; Holm R. H.; Papaefthymiou G. C.; Frankel R. B. Demonstration of Alternative Spin States in Clusters Containing the Biologically Relevant [Fe4S4]1+ Core. J. Am. Chem. Soc. 1986, 108, 3519–3521. 10.1021/ja00272a064. [DOI] [Google Scholar]
- Carney M. J.; Kovacs J. A.; Zhang Y.-P.; Papaefthymiou G. C.; Spartalian K.; Frankel R. B.; Holm R. H. Comparative Electronic Properties of Vanadium-Iron-Sulfur and Molybdenum-Iron-Sulfur Clusters Containing Isoelectronic Cubane-Type [VFe3S4]2+ and [MoFe3S4]3+ Cores. Inorg. Chem. 1987, 26, 719–724. 10.1021/ic00252a016. [DOI] [Google Scholar]
- Carney M. J.; Papaefthymiou G. C.; Spartalian K.; Frankel R. B.; Holm R. H. Ground Spin State Variability in [Fe4S4(SR)4]3-. Synthetic Analogues of the Reduced Clusters in Ferredoxins and Other Iron-Sulfur Proteins: Cases of Extreme Sensitivity of Electronic State and Structure to Extrinsic Factors. J. Am. Chem. Soc. 1988, 110, 6084–6095. 10.1021/ja00226a025. [DOI] [PubMed] [Google Scholar]
- Scott T. A.; Berlinguette C. P.; Holm R. H.; Zhou H.-C. Initial Synthesis and Structure of an All-ferrous Analogue of the Fully Reduced [Fe4S4]0 Cluster of the Nitrogenase Iron Protein. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9741–9744. 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angove H. C.; Yoo S. J.; Burgess B. K.; Münck E. Mössbauer and EPR Evidence for an All-Ferrous Fe4S4 Cluster with S = 4 in the Fe Protein of Nitrogenase. J. Am. Chem. Soc. 1997, 119, 8730–8731. 10.1021/ja9712837. [DOI] [Google Scholar]
- Deng L.; Holm R. H. Stabilization of Fully Reduced Iron–Sulfur Clusters by Carbene Ligation: The [FenSn]0 Oxidation Levels (n = 4, 8). J. Am. Chem. Soc. 2008, 130, 9878–9886. 10.1021/ja802111w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti M.; Deng L.; Holm R. H.; Münck E.; Bominaar E. L. Mössbauer, Electron Paramagnetic Resonance, and Theoretical Studies of a Carbene-Based All-Ferrous Fe4S4 Cluster: Electronic Origin and Structural Identification of the Unique Spectroscopic Site. Inorg. Chem. 2009, 48, 2735–2747. 10.1021/ic802192w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J.-L.; Zhou H.-C.; Holm R. H. Vanadium–Iron–Sulfur Clusters Containing the Cubane-type [VFe3S4] Core Unit: Synthesis of a Cluster with the Topology of the PN Cluster of Nitrogenase. Inorg. Chem. 2003, 42, 4624–4631. 10.1021/ic0301369. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zuo J.-L.; Zhou H.-C.; Holm R. H. Rearrangement of Symmetrical Dicubane Clusters into Topological Analogues of the P Cluster of Nitrogenase: Nature’s Choice?. J. Am. Chem. Soc. 2002, 124, 14292–14293. 10.1021/ja0279702. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Holm R. H. Synthesis of a Molecular Mo2Fe6S9 Cluster with the Topology of the PN Cluster of Nitrogenase by Rearrangement of an Edge-Bridged Mo2Fe6S8 Double Cubane. J. Am. Chem. Soc. 2003, 125, 3910–3920. 10.1021/ja0214633. [DOI] [PubMed] [Google Scholar]
- Ohki Y.; Sunada Y.; Honda M.; Katada M.; Tatsumi K. Synthesis of the P-Cluster Inorganic Core of Nitrogenases. J. Am. Chem. Soc. 2003, 125, 4052–4053. 10.1021/ja029383m. [DOI] [PubMed] [Google Scholar]
- Ohki Y.; Imada M.; Murata A.; Sunada Y.; Ohta S.; Honda M.; Sasamori T.; Tokitoh N.; Katada M.; Tatsumi K. Synthesis, Structures, and Electronic Properties of [8Fe-7S] Cluster Complexes Modeling the Nitrogenase P-Cluster. J. Am. Chem. Soc. 2009, 131, 13168–13178. 10.1021/ja9055036. [DOI] [PubMed] [Google Scholar]
- Ohki Y.; Tanifuji K.; Yamada N.; Cramer R. E.; Tatsumi K. Formation of a Nitrogenase P-cluster [Fe8S7] Core via Reductive Fusion of Two All-Ferric [Fe4S4] Clusters. Chem. - Asian J. 2012, 7, 2222–2224. 10.1002/asia.201200568. [DOI] [PubMed] [Google Scholar]
- Shoji M.; Saito T.; Takeda R.; Kitagawa Y.; Kawakami T.; Yamanaka S.; Okumura M.; Yamaguchi K. Assignments of the Mössbauer Spectra of an Inorganic [8Fe–7S] Complex Based on the First-principle Calculations. Chem. Phys. Lett. 2007, 446, 228–232. 10.1016/j.cplett.2007.08.049. [DOI] [Google Scholar]
- Shoji M.; Koizumi K.; Kitagawa Y.; Yamanaka S.; Okumura M.; Yamaguchi K.; Ohki Y.; Sunada Y.; Honda M.; Tatsumi K. Theory of Chemical Bonds in Metalloenzymes V: Hybrid-DFT Studies of the inorganic [8Fe–7S] core. Int. J. Quantum Chem. 2006, 106, 3288–3302. 10.1002/qua.21201. [DOI] [Google Scholar]
- Ohki Y.; Ikagawa Y.; Tatsumi K. Synthesis of New [8Fe-7S] Clusters: A Topological Link Between the Core Structures of P-Cluster, FeMo-co, and FeFe-co of Nitrogenases. J. Am. Chem. Soc. 2007, 129, 10457–10465. 10.1021/ja072256b. [DOI] [PubMed] [Google Scholar]
- Hashimoto T.; Ohki Y.; Tatsumi K. Synthesis of Coordinatively Unsaturated Mesityliron Thiolate Complexes and Their Reactions with Elemental Sulfur. Inorg. Chem. 2010, 49, 6102–6109. 10.1021/ic100692v. [DOI] [PubMed] [Google Scholar]
- Tanifuji K.; Sickerman N.; Lee C. C.; Nagasawa T.; Miyazaki K.; Ohki Y.; Tatsumi K.; Hu Y.; Ribbe M. W. Structure and Reactivity of an Asymmetric Synthetic Mimic of Nitrogenase Cofactor. Angew. Chem., Int. Ed. 2016, 55, 15633–15636. 10.1002/anie.201608806. [DOI] [PubMed] [Google Scholar]
- Ohta S.; Ohki Y.; Hashimoto T.; Cramer R. E.; Tatsumi K. A Nitrogenase Cluster Model [Fe8S6O] with an Oxygen Unsymmetrically Bridging Two Proto-Fe4S3 Cubes: Relevancy to the Substrate Binding Mode of the FeMo Cofactor. Inorg. Chem. 2012, 51, 11217–11219. 10.1021/ic301348f. [DOI] [PubMed] [Google Scholar]
- Xu G.; Wang Z.; Ling R.; Zhou J.; Chen X.-D.; Holm R. H. Ligand Metathesis as Rational Strategy for the Synthesis of Cubane-type Heteroleptic Iron–Sulfur Clusters Relevant to the FeMo Cofactor. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5089–5092. 10.1073/pnas.1801025115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Zhou J.; Wang Z.; Holm R. H.; Chen X.-D. Controlled Incorporation of Nitrides into W-Fe-S Clusters. Angew. Chem. 2019, 131, 16621–16625. 10.1002/ange.201908968. [DOI] [PubMed] [Google Scholar]
- Pollock C. J.; Tan L. L.; Zhang W.; Lancaster K. M.; Lee S. C.; DeBeer S. Light-Atom Influences on the Electronic Structures of Iron–Sulfur Clusters. Inorg. Chem. 2014, 53, 2591–2597. 10.1021/ic402944r. [DOI] [PubMed] [Google Scholar]
- Delgado-Jaime M. U.; Dible B. R.; Chiang K. P.; Brennessel W. W.; Bergmann U.; Holland P. L.; DeBeer S. Identification of a Single Light Atom within a Multinuclear Metal Cluster Using Valence-to-Core X-ray Emission Spectroscopy. Inorg. Chem. 2011, 50, 10709–10717. 10.1021/ic201173j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGale J.; Cutsail G. E. III; Joseph C.; Rose M. J.; DeBeer S. Spectroscopic X-ray and Mössbauer Characterization of M6 and M5 Iron(Molybdenum)-Carbonyl Carbide Clusters: High Carbide-Iron Covalency Enhances Local Iron Site Electron Density Despite Cluster Oxidation. Inorg. Chem. 2019, 58, 12918–12932. 10.1021/acs.inorgchem.9b01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Rauchfuss T. B.; Woods T. J. Iron Carbide–Sulfide Carbonyl Clusters. Inorg. Chem. 2019, 58, 8271–8274. 10.1021/acs.inorgchem.9b01231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J. K.; Henthorn J. T.; Van Stappen C.; Trncik C.; Einsle O.; Keavney D.; DeBeer S. X-ray Magnetic Circular Dichroism Spectroscopy Applied to Nitrogenase and Related Models: Experimental Evidence for a Spin-Coupled Molybdenum(III) Center. Angew. Chem., Int. Ed. 2019, 58, 9373–9377. 10.1002/anie.201901899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen A. D.; Senoff C. V. Nitrogenpentammineruthenium(II) complexes. Chem. Commun. 1965, 621. 10.1039/c19650000621. [DOI] [Google Scholar]
- Chatt J.; Dilworth J. R.; Richards R. L. Recent Advances in the Chemistry of Nitrogen Fixation. Chem. Rev. 1978, 78, 589–625. 10.1021/cr60316a001. [DOI] [Google Scholar]
- Hidai M.; Mizobe Y. Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev. 1995, 95, 1115–1133. 10.1021/cr00036a008. [DOI] [Google Scholar]
- Yandulov D. V.; Schrock R. R. Reduction of Dinitrogen to Ammonia at a Well-Protected Reaction Site in a Molybdenum Triamidoamine Complex. J. Am. Chem. Soc. 2002, 124, 6252–6253. 10.1021/ja020186x. [DOI] [PubMed] [Google Scholar]
- Yandulov D. V.; Schrock R. R. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- Coucouvanis D.; Mosier P. E.; Demadis K. D.; Patton S.; Malinak S. M.; Kim C. G.; Tyson M. A. The Catalytic Reduction of Hydrazine to Ammonia by the MoFe3S4 Cubanes and Implications Regarding the Function of Nitrogenase. Evidence for Direct Involvement of the Molybdenum Atom in Substrate Reduction. J. Am. Chem. Soc. 1993, 115, 12193–12194. 10.1021/ja00078a079. [DOI] [Google Scholar]
- Demadis K. D.; Coucouvanis D. Syntheses and Structural Characterization of a New Class of Double Cubanes That Contain MoFe3S4 Subunits and Molybdenum-Coordinated, Bridging Mercapto-Carboxylate Ligands. Effective Catalysts for the Reduction of Hydrazine to Ammonia. Inorg. Chem. 1994, 33, 4195–4197. 10.1021/ic00097a001. [DOI] [Google Scholar]
- Laughlin L. J.; Coucouvanis D. Use of [MoFe3S4]3+ Single Cubanes in the Catalytic Reduction of Acetylene to Ethylene and Ethane. Identification of Molybdenum and Iron Atoms as Catalytic Sites during Substrate Reduction and Implications for Nitrogenase Action. J. Am. Chem. Soc. 1995, 117, 3118–3125. 10.1021/ja00116a016. [DOI] [Google Scholar]
- Malinak S. M.; Demadis K. D.; Coucouvanis D. Catalytic Reduction of Hydrazine to Ammonia by the VFe3S4 Cubanes. Further Evidence for the Direct Involvement of the Heterometal in the Reduction of Nitrogenase Substrates and Possible Relevance to the Vanadium Nitrogenases. J. Am. Chem. Soc. 1995, 117, 3126–3133. 10.1021/ja00116a017. [DOI] [Google Scholar]
- Coucouvanis D. Functional Analogs for the Reduction of Certain Nitrogenase Substrates. Are Multiple Sites Within the Fe/Mo/S Active Center Involved in the 6e– Reduction of N2?. JBIC, J. Biol. Inorg. Chem. 1996, 1, 594–600. 10.1007/s007750050098. [DOI] [Google Scholar]
- Coucouvanis D.; Demadis K. D.; Malinak S. M.; Mosier P. E.; Tyson M. A.; Laughlin L. J. Catalytic and Stoichiometric Multielectron Reduction of Hydrazine to Ammonia and Acetylene to Ethylene with Clusters that Contain the MFe3S4 Cores (M = Mo, V). Relevance to the Function of Nitrogenase. J. Mol. Catal. A: Chem. 1996, 107, 123–135. 10.1016/1381-1169(95)00168-9. [DOI] [Google Scholar]
- Demadis K. D.; Malinak S. M.; Coucouvanis D. Catalytic Reduction of Hydrazine to Ammonia with MoFe3S4–Polycarboxylate Clusters. Possible Relevance Regarding the Function of the Molybdenum-Coordinated Homocitrate in Nitrogenase. Inorg. Chem. 1996, 35, 4038–4046. 10.1021/ic960098b. [DOI] [PubMed] [Google Scholar]
- Malinak S. M.; Simeonov A. M.; Mosier P. E.; McKenna C. E.; Coucouvanis D. Catalytic Reduction of cis-Dimethyldiazene by the [MoFe3S4]3+ Clusters. The Four-Electron Reduction of a N = N Bond by a Nitrogenase-Relevant Cluster and Implications for the Function of Nitrogenase. J. Am. Chem. Soc. 1997, 119, 1662–1667. 10.1021/ja963475s. [DOI] [Google Scholar]
- Thorhallsson A. T.; Bjornsson R. Computational Mechanistic Study of [MoFe3S4] Cubanes for Catalytic Reduction of Nitrogenase Substrates. Inorg. Chem. 2019, 58, 1886–1894. 10.1021/acs.inorgchem.8b02669. [DOI] [PubMed] [Google Scholar]
- Arashiba K.; Miyake Y.; Nishibayashi Y. A Molybdenum Complex Bearing PNP-type pincer Ligands Leads to the Catalytic Reduction of Dinitrogen into Ammonia. Nat. Chem. 2011, 3, 120–125. 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]
- Anderson J. S.; Rittle J.; Peters J. C. Catalytic Conversion of Nitrogen to Ammonia by an Iron Model Complex. Nature 2013, 501, 84–87. 10.1038/nature12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo T. J.; Thompson N. B.; Peters J. C. A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc. 2016, 138, 5341–5450. 10.1021/jacs.6b01706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Peters J. C. An Fe-N2 Complex That Generates Hydrazine and Ammonia via Fe=NNH2: Demonstrating a Hybrid Distal-to-Alternating Pathway for N2 Reduction. J. Am. Chem. Soc. 2016, 138, 4243–4248. 10.1021/jacs.6b01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Peters J. C. Proton-Coupled Reduction of an Iron Cyanide Complex to Methane and Ammonia. Angew. Chem., Int. Ed. 2016, 55, 12262–12265. 10.1002/anie.201606366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Peters J. C. N–H Bond Dissociation Enthalpies and Facile H Atom Transfers for Early Intermediates of Fe–N2 and Fe–CN Reductions. J. Am. Chem. Soc. 2017, 139, 3161–3170. 10.1021/jacs.6b12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson N. B.; Green M. T.; Peters J. C. Nitrogen Fixation via a Terminal Fe(IV) Nitride. J. Am. Chem. Soc. 2017, 139, 15312–15315. 10.1021/jacs.7b09364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney R. A.; Saouma C. T.; Peters J. C.; Hoffman B. M. Modeling the Signatures of Hydrides in Metalloenzymes: ENDOR Analysis of a Di-iron Fe(μ-NH)(μ-H)Fe Core. J. Am. Chem. Soc. 2012, 134, 12637–12647. 10.1021/ja303739g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S. D.; Mehn M. P.; Peters J. C. Heterolytic H2 Activation Mediated by Low-Coordinate L3Fe-(μ-N)-FeL3 Complexes to Generate Fe(μ-NH)(μ-H)Fe Species. J. Am. Chem. Soc. 2005, 127, 13146–13147. 10.1021/ja0544509. [DOI] [PubMed] [Google Scholar]
- Rittle J.; McCrory C. C. L.; Peters J. C. A 106-Fold Enhancement in N2-Binding Affinity of an Fe2(μ-H)2 Core upon Reduction to a Mixed-Valence FeIIFeI State. J. Am. Chem. Soc. 2014, 136, 13853–13862. 10.1021/ja507217v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo T. J.; Thompson N. B.; Suess D. L. M.; Ung G.; Peters J. C. Evaluating Molecular Cobalt Complexes for the Conversion of N2 to NH3. Inorg. Chem. 2015, 54, 9256–9262. 10.1021/acs.inorgchem.5b00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo J. Jr.; Peters J. C. Catalytic Nitrogen-to-Ammonia Conversion by Osmium and Ruthenium Complexes. J. Am. Chem. Soc. 2017, 139, 16105–16108. 10.1021/jacs.7b10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horitani M.; Grubel K.; McWilliams S. F.; Stubbert B. D.; Mercado B. Q.; Yu Y.; Gurubasavaraj P. M.; Lees N. S.; Holland P. L.; Hoffman B. M. ENDOR Characterization of an Iron-alkene Complex Provides Insight into a Corresponding Organometallic Intermediate of Nitrogenase. Chem. Sci. 2017, 8, 5941–5948. 10.1039/C7SC01602F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees N. S.; McNaughton R. L.; Gregory W. V.; Holland P. L.; Hoffman B. M. ENDOR Characterization of a Synthetic Diiron Hydrazido Complex as a Model for Nitrogenase Intermediates. J. Am. Chem. Soc. 2008, 130, 546–555. 10.1021/ja073934x. [DOI] [PubMed] [Google Scholar]
- Chiang K. P.; Scarborough C. C.; Horitani M.; Lees N. S.; Ding K.; Dugan T. R.; Brennessel W. W.; Bill E.; Hoffman B. M.; Holland P. L. Characterization of the Fe-H Bond in a Three-Coordinate Terminal Hydride Complex of Iron(I). Angew. Chem., Int. Ed. 2012, 51, 3658–3662. 10.1002/anie.201109204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock C. J.; Grubel K.; Holland P. L.; DeBeer S. Experimentally Quantifying Small-molecule Bond Activation using Valence-to-Core X-ray Emission Spectroscopy. J. Am. Chem. Soc. 2013, 135, 11803–11808. 10.1021/ja3116247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosha D. E.; Chilkuri V. G.; Van Stappen C.; Bill E.; Mercado B. Q.; DeBeer S.; Neese F.; Holland P. L. Planar Three-coordinate Iron Sulfide in a Synthetic [4Fe-3S] Cluster with Biomimetic Reactivity. Nat. Chem. 2019, 11, 1019–1025. 10.1038/s41557-019-0341-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čorić I.; Mercado B. Q.; Bill E.; Vinyard D. J.; Holland P. L. Binding of Dinitrogen to an Iron–Sulfur–Carbon Site. Nature 2015, 526, 96–99. 10.1038/nature15246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speelman A. L.; Čorić I.; Van Stappen C.; DeBeer S.; Mercado B. Q.; Holland P. L. Nitrogenase-Relevant Reactivity of a Synthetic Iron-Sulfur-Carbon Site. J. Am. Chem. Soc. 2019, 141, 13148–13157. 10.1021/jacs.9b05353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Gee L. B.; Wang H.; MacLeod K. C.; McWilliams S. F.; Skubi K. L.; Cramer S. P.; Holland P. L. High-Frequency Fe-H Vibrations in a Bridging Hydride Complex Characterized by NRVS and DFT. Angew. Chem., Int. Ed. 2018, 57, 9367–9371. 10.1002/anie.201804601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasniewski A. J.; Sickerman N. S.; Hu Y.; Ribbe M. W. The Fe Protein: An Unsung Hero of Nitrogenase. Inorganics 2018, 6, 25. 10.3390/inorganics6010025. [DOI] [Google Scholar]
- Georgiadis M. M.; Komiya H.; Chakrabarti P.; Woo D.; Kornuc J. J.; Rees D. C. Crystallographic Structure of the Nitrogenase Iron Protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. 10.1126/science.1529353. [DOI] [PubMed] [Google Scholar]
- Hagen W. R.; Eady R. R.; Dunham W. R.; Haaker H. A Novel S = 3/2 EPR Signal Associated with Native Fe-proteins of Nitrogenase. FEBS Lett. 1985, 189, 250–254. 10.1016/0014-5793(85)81033-4. [DOI] [PubMed] [Google Scholar]
- Onate Y. A.; Finnegan M. G.; Hales B. J.; Johnson M. K. Variable Temperature Magnetic Circular Dichroism Studies of Reduced Nitrogenase Iron Proteins and [4Fe-4S]+ Synthetic Analog Clusters. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1993, 1164, 113–123. 10.1016/0167-4838(93)90237-L. [DOI] [PubMed] [Google Scholar]
- Yang Z.-Y.; Ledbetter R.; Shaw S.; Pence N.; Tokmina-Lukaszewska M.; Eilers B.; Guo Q.; Pokhrel N.; Cash V. L.; Dean D. R.; et al. Evidence That the Pi Release Event Is the Rate-Limiting Step in the Nitrogenase Catalytic Cycle. Biochemistry 2016, 55, 3625–3635. 10.1021/acs.biochem.6b00421. [DOI] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Dean D. R. Electron transfer in Nitrogenase Catalysis. Curr. Opin. Chem. Biol. 2012, 16, 19–25. 10.1016/j.cbpa.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D.; Watt G. D. Nucleotide-Assisted [Fe4S4] Redox State Interconversions of the Azotobacter vinelandii Fe Protein and Their Relevance to Nitrogenase Catalysis. Biochemistry 2013, 52, 4791–4799. 10.1021/bi301547b. [DOI] [PubMed] [Google Scholar]
- Watt G. D.; Reddy K. R. N. Formation of an All Ferrous Fe4S4 Cluster in the Iron Protein Component of Azotobacter vinelandii Nitrogenase. J. Inorg. Biochem. 1994, 53, 281–294. 10.1016/0162-0134(94)85115-8. [DOI] [Google Scholar]
- Nyborg A. C.; Johnson J. L.; Gunn A.; Watt G. D. Evidence for a Two-Electron Transfer Using the All-Ferrous Fe Protein during Nitrogenase Catalysis. J. Biol. Chem. 2000, 275, 39307–39312. 10.1074/jbc.M007069200. [DOI] [PubMed] [Google Scholar]
- Angove H. C.; Yoo S. J.; Münck E.; Burgess B. K. An All-ferrous State of the Fe Protein of Nitrogenase. Interaction with Nucleotides and Electron Transfer to the MoFe Protein. J. Biol. Chem. 1998, 273, 26330–26337. 10.1074/jbc.273.41.26330. [DOI] [PubMed] [Google Scholar]
- Musgrave K. B.; Angove H. C.; Burgess B. K.; Hedman B.; Hodgson K. O. All-Ferrous Titanium(III) Citrate Reduced Fe Protein of Nitrogenase: An XAS Study of Electronic and Metrical Structure. J. Am. Chem. Soc. 1998, 120, 5325–5326. 10.1021/ja980598z. [DOI] [Google Scholar]
- Wenke B. B.; Spatzal T.; Rees D. C. Site-Specific Oxidation State Assignments of the Irons in the [4Fe:4S]2+/1+/0 States of the Nitrogenase Fe-Protein. Angew. Chem., Int. Ed. 2019, 58, 3894–3897. 10.1002/anie.201813966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.-L.; Perrin B. S. Jr.; Niu S.; Huang Q.; Ichiye T. Protein Dynamics and the All-ferrous [Fe4S4] Cluster in the Nitrogenase Iron Protein. Protein Sci. 2016, 25, 12–18. 10.1002/pro.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman R. V.; Orme-Johnson W. H.; Burris R. H. Role of Magnesium Adenosine 5′-Triphosphate in the Hydrogen Evolution Reaction Catalyzed by Nitrogenase from Azotobacter vinelandii. Biochemistry 1980, 19, 2333–2342. 10.1021/bi00552a009. [DOI] [PubMed] [Google Scholar]
- Anderson G. L.; Howard J. B. Reactions with the Oxidized Iron Protein of Azotobacter vinelandii Nitrogenase: Formation of a 2Fe Center. Biochemistry 1984, 23, 2118–2122. 10.1021/bi00305a002. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Lowe D. J. Nitrogenase of Klebsiella pneumoniae. Kinetics of the Dissociation of Oxidized Iron Protein from Molybdenum-Iron Protein: Identification of the Rate-limiting Step for Substrate Reduction. Biochem. J. 1983, 215, 393–403. 10.1042/bj2150393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D. J.; Thorneley R. N. F. The Mechanism of Klebsiella pneumoniae Nitrogenase action. The Determination of Rate Constants Required for the Simulation of the Kinetics of N2 Reduction and H2 Evolution. Biochem. J. 1984, 224, 895–901. 10.1042/bj2240895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T. A.; Fairhurst S.; Lowe D. J.; Watmough N. J.; Eady R. R. Electron Transfer and Half-reactivity in Nitrogenase. Biochem. Soc. Trans. 2011, 39, 201. 10.1042/BST0390201. [DOI] [PubMed] [Google Scholar]
- Danyal K.; Shaw S.; Page T. R.; Duval S.; Horitani M.; Marts A. R.; Lukoyanov D.; Dean D. R.; Raugei S.; Hoffman B. M.; et al. Negative Cooperativity in the Nitrogenase Fe Protein Electron Delivery Cycle. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E5783. 10.1073/pnas.1613089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.; Lowe D. J.; Tavares P.; Pereira A. S.; Huynh B. H.; Edmondson D.; Newton W. E. Conformations Generated During Turnover of the Azotobacter vinelandii Nitrogenase MoFe Protein and Their Relationship to Physiological Function. J. Inorg. Biochem. 2007, 101, 1649. 10.1016/j.jinorgbio.2007.07.037. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Lanzilotta W. N.; Mortenson L. E.; Watt G. D.; Seefeldt L. C. Evidence for a Central Role of Lysine 15 of Azotobacter vinelandii Nitrogenase Iron Protein in Nucleotide Binding and Protein Conformational Changes. J. Biol. Chem. 1995, 270, 13112–13117. 10.1074/jbc.270.22.13112. [DOI] [PubMed] [Google Scholar]
- Zumft W. G.; Mortenson L. E.; Palmer G. Electron-Paramagnetic-Resonance Studies on Nitrogenase. Investigation of the Oxidation-Reduction Behaviour of Azoferredoxin and Molybdoferredoxin with Potentiometric and Rapid-Freeze Techniques. Eur. J. Biochem. 1974, 46, 525–535. 10.1111/j.1432-1033.1974.tb03646.x. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Seefeldt L. C. The [4Fe-4S] Cluster Domain of the Nitrogenase Iron Protein Facilitates Conformational Changes Required for the Cooperative Binding of Two Nucleotides. Biochemistry 1996, 35, 15654–15662. 10.1021/bi961886f. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Stephens P. J.; Eran H.; Luo G.-M.; Zhang F. X.; Matai D.; Nguyen H. T. In Advances in Nitrogen Fixation Research; Veeger C., Newton W. E., Eds.; Springer: The Hague, Netherlands, 1983; Vol. 4. [Google Scholar]
- Ryle M. J.; Lanzilotta W. N.; Seefeldt L. C.; Scarrow R. C.; Jensen G. M. Circular Dichroism and X-ray Spectroscopies of Azotobacter vinelandii Nitrogenase Iron Protein. MgATP and MgADP Induced Protein Conformational Changes Affecting the [4Fe-4S] Cluster and Characterization of a [2Fe-2S] Form. J. Biol. Chem. 1996, 271, 1551–1557. 10.1074/jbc.271.3.1551. [DOI] [PubMed] [Google Scholar]
- Watt G. D.; Wang Z.-C.; Knotts R. R. Redox Reactions of and Nucleotide Binding to the Iron Protein of Azotobacter vinelandii. Biochemistry 1986, 25, 8156–8162. 10.1021/bi00373a005. [DOI] [Google Scholar]
- Lindahl P. A.; Gorelick N. J.; Münck E.; Orme-Johnson W. H. EPR and Mössbauer Studies of Nucleotide-bound Nitrogenase Iron Protein from Azotobacter vinelandii. J. Biol. Chem. 1987, 262, 14945–14953. [PubMed] [Google Scholar]
- Sarma R.; Mulder D. W.; Brecht E.; Szilagyi R. K.; Seefeldt L. C.; Tsuruta H.; Peters J. W. Probing the MgATP-Bound Conformation of the Nitrogenase Fe Protein by Solution Small-Angle X-ray Scattering. Biochemistry 2007, 46, 14058–14066. 10.1021/bi700446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordewener J.; Haaker H.; Veeger C. Binding of MgATP to the Nitrogenase Proteins from Azotobacter vinelandii. Eur. J. Biochem. 1983, 132, 47–54. 10.1111/j.1432-1033.1983.tb07323.x. [DOI] [PubMed] [Google Scholar]
- Duyvis M. G.Kinetics of Nitrogenase from Azotobacter vinelandii. Wageningen Agricultural University, May 1997. [Google Scholar]
- Zumft W. G.; Palmer G.; Mortenson L. E. Electron Paramagnetic Resonance Studies on Nitrogenase. II. Interaction of Adenosine 5′-Triphosphate with Azoferredoxin. Biochim. Biophys. Acta, Bioenerg. 1973, 292, 413–421. 10.1016/0005-2728(73)90047-9. [DOI] [PubMed] [Google Scholar]
- Palmer G.; Multani J. S.; Cretney W. C.; Zumft W. G.; Mortenson L. E. Electron Paramagnetic Resonance Studies on Nitrogenase. I. The Properties of Molybdoferredoxin and Azoferrodoxin. Arch. Biochem. Biophys. 1972, 153, 325–332. 10.1016/0003-9861(72)90452-3. [DOI] [PubMed] [Google Scholar]
- Orme-Johnson W. H.; Hamilton W. D.; Jones T. L.; Tso M. Y.; Burris R. H.; Shah V. K.; Brill W. J. Electron Paramagnetic Resonance of Nitrogenase and Nitrogenase Components from Clostridium pasteurianum W5 and Azotobacter vinelandii OP. Proc. Natl. Acad. Sci. U. S. A. 1972, 69, 3142–3145. 10.1073/pnas.69.11.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaksma A.; Haaker H.; Veeger C. Fully Active Fe-protein of the Nitrogenase from Azotobacter vinelandii Contains at least Eight Iron Atoms and Eight Sulphide Atoms per Molecule. Eur. J. Biochem. 1983, 133, 71–76. 10.1111/j.1432-1033.1983.tb07430.x. [DOI] [PubMed] [Google Scholar]
- Lindahl P. A.; Teo B.-K.; Orme-Johnson W. H. EXAFS Studies of the Nitrogenase Iron Protein from Azotobactor vinelandii. Inorg. Chem. 1987, 26, 3912–3916. 10.1021/ic00270a018. [DOI] [PubMed] [Google Scholar]
- Meyer J.; Gaillard J.; Moulis J. M. Hydrogen-1 Nuclear Magnetic Resonance of the Nitrogenase Iron Protein (Cp2) from Clostridium pasteurianum. Biochemistry 1988, 27, 6150–6156. 10.1021/bi00416a048. [DOI] [PubMed] [Google Scholar]
- Lanzilotta W. N.; Holz R. C.; Seefeldt L. C. Proton NMR Investigation of the [4Fe-4S]1+ Cluster Environment of Nitrogenase Iron Protein from Azotobacter vinelandii: Defining Nucleotide-Induced Conformational Changes. Biochemistry 1995, 34, 15646–15653. 10.1021/bi00048a007. [DOI] [PubMed] [Google Scholar]
- Vincent K. A.; Tilley G. J.; Quammie N. C.; Streeter I.; Burgess B. K.; Cheesman M. R.; Armstrong F. A. Instantaneous, Stoichiometric Generation of Powerfully Reducing States of Protein Active Sites using Eu(II) and Polyaminocarboxylate Ligands. Chem. Commun. 2003, 2590–2591. 10.1039/b308188e. [DOI] [PubMed] [Google Scholar]
- Guo M.; Sulc F.; Ribbe M. W.; Farmer P. J.; Burgess B. K. Direct Assessment of the Reduction Potential of the [4Fe-4S]1+/0 Couple of the Fe Protein from Azotobacter vinelandii. J. Am. Chem. Soc. 2002, 124, 12100–12101. 10.1021/ja026478f. [DOI] [PubMed] [Google Scholar]
- Hans M.; Buckel W.; Bill E. Spectroscopic Evidence for an All-ferrous [4Fe-4S]0 Cluster in the Superreduced Activator of 2-Hydroxyglutaryl-CoA Dehydratase from Acidaminococcus fermentans. JBIC, J. Biol. Inorg. Chem. 2008, 13, 563–574. 10.1007/s00775-008-0345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew S. G. The Redox Potential of Dithionite and SO2– from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen plus Hydrogenase. Eur. J. Biochem. 1978, 85, 535–547. 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- Erickson J. A.; Nyborg A. C.; Johnson J. L.; Truscott S. M.; Gunn A.; Nordmeyer F. R.; Watt G. D. Enhanced Efficiency of ATP Hydrolysis during Nitrogenase Catalysis Utilizing Reductants That Form the All-Ferrous Redox State of the Fe Protein. Biochemistry 1999, 38, 14279–14285. 10.1021/bi991389+. [DOI] [PubMed] [Google Scholar]
- Dalton H.; Postgate J. R. Growth and Physiology of Azotobacter Chroococcum in Continuous Culture. J. Gen. Microbiol. 1969, 56, 307–319. 10.1099/00221287-56-3-307. [DOI] [PubMed] [Google Scholar]
- Kelly M.; Lang G. Evidence from Mössbauer Spectroscopy for the Role of Iron in Nitrogen Fixation. Biochim. Biophys. Acta, Bioenerg. 1970, 223, 86–104. 10.1016/0005-2728(70)90134-9. [DOI] [PubMed] [Google Scholar]
- Smith B. E.; Lang G. Mössbauer Spectroscopy of the Nitrogenase Proteins from Klebsiella pneumoniae. Structural Assignments and Mechanistic Conclusions. Biochem. J. 1974, 137, 169–180. 10.1042/bj1370169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münck E.; Rhodes H.; Orme-Johnson W. H.; Davis L. C.; Brill W. J.; Shah V. K. Nitrogenase. VIII. Mössbauer and EPR Spectroscopy. The MoFe Protein Component from Azotobacter vinelandii OP. Biochim. Biophys. Acta, Protein Struct. 1975, 400, 32–53. 10.1016/0005-2795(75)90124-5. [DOI] [PubMed] [Google Scholar]
- Zimmermann R.; Münck E.; Brill W. J.; Shah V. K.; Henzl M. T.; Rawlings J.; Orme-Johnson W. H. Nitrogenase X: Mössbauer and EPR Studies on Reversibly Oxidized MoFe Protein from Azotobacter vinelandii OP. Nature of the Iron Centers. Biochim. Biophys. Acta, Protein Struct. 1978, 537, 185–207. 10.1016/0005-2795(78)90504-4. [DOI] [PubMed] [Google Scholar]
- Johnson M. K.; Thomson A. J.; Robinson A. E.; Smith B. E. Characterization of the Paramagnetic Centres of the Molybdenum-Iron Protein of Nitrogenase from Klebsiella pneumoniae Using Low Temperature Magnetic Circular Dichroism Spectroscopy. Biochim. Biophys. Acta, Protein Struct. 1981, 671, 61–70. 10.1016/0005-2795(81)90094-5. [DOI] [Google Scholar]
- Smith J. P.; Emptage M. H.; Orme-Johnson W. H. Magnetic Susceptibility Studies of Native and Thionine-oxidized Molybdenum-Iron Protein from Azotobacter vinelandii Nitrogenase. J. Biol. Chem. 1982, 257, 2310–2313. [PubMed] [Google Scholar]
- McLean P. A.; Papaefthymiou V.; Orme-Johnson W. H.; Münck E. Isotopic Hybrids of Nitrogenase. Mössbauer Study of MoFe Protein With Selective 57Fe Enrichment of the P-cluster. J. Biol. Chem. 1987, 262, 12900–12903. [PubMed] [Google Scholar]
- Hagen W. R.; Wassink H.; Eady R. R.; Smith B. E.; Haaker H. Quantitative EPR of an S = 7/2 System in Thionine-oxidized MoFe Proteins of Nitrogenase. A Redefinition of the P-cluster Concept. Eur. J. Biochem. 1987, 169, 457–465. 10.1111/j.1432-1033.1987.tb13633.x. [DOI] [PubMed] [Google Scholar]
- Lindahl P. A.; Papaefthymiou V.; Orme-Johnson W. H.; Münck E. Mössbauer Studies of Solid Thionin-oxidized MoFe Protein of Nitrogenase. J. Biol. Chem. 1988, 263, 19412–19418. [PubMed] [Google Scholar]
- Surerus K. K.; Hendrich M. P.; Christie P. D.; Rottgardt D.; Orme-Johnson W. H.; Münck E. Mössbauer and Integer-Spin EPR of the Oxidized P-clusters of Nitrogenase: POX is a Non-Kramers System with a Nearly Degenerate Ground Doublet. J. Am. Chem. Soc. 1992, 114, 8579–8590. 10.1021/ja00048a034. [DOI] [Google Scholar]
- Kim J.; Rees D. C. Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 1992, 257, 1677–1682. 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]
- Chan M. K.; Kim J.; Rees D. C. The Nitrogenase FeMo-Cofactor and P-Cluster Pair: 2.2 Å Resolution Structures. Science 1993, 260, 792–794. 10.1126/science.8484118. [DOI] [PubMed] [Google Scholar]
- Tittsworth R. C.; Hales B. J. Detection of EPR Signals Assigned to the 1-equiv-oxidized P-Clusters of the Nitrogenase MoFe-Protein from Azotobacter vinelandii. J. Am. Chem. Soc. 1993, 115, 9763–9767. 10.1021/ja00074a050. [DOI] [Google Scholar]
- Pierik A. J.; Wassink H.; Haaker H.; Hagen W. R. Redox Properties and EPR Spectroscopy of the P Clusters of Azotobacter vinelandii MoFe Protein. Eur. J. Biochem. 1993, 212, 51–61. 10.1111/j.1432-1033.1993.tb17632.x. [DOI] [PubMed] [Google Scholar]
- Chan J. M.; Christiansen J.; Dean D. R.; Seefeldt L. C. Spectroscopic Evidence for Changes in the Redox State of the Nitrogenase P-Cluster During Turnover. Biochemistry 1999, 38, 5779–5785. 10.1021/bi982866b. [DOI] [PubMed] [Google Scholar]
- Peters J. W.; Stowell M. H. B.; Soltis M. S.; Finnegan M. G.; Johnson M. K.; Rees D. C. Redox-Dependent Structural Changes in the Nitrogenase P-Cluster. Biochemistry 1997, 36, 1181–1187. 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- Owens C. P.; Katz F. E. H.; Carter C. H.; Oswald V. F.; Tezcan F. A. Tyrosine-Coordinated P-cluster in G. diazotrophicus Nitrogenase: Evidence for the Importance of O-Based Ligands in Conformationally Gated Electron Transfer. J. Am. Chem. Soc. 2016, 138, 10124–10127. 10.1021/jacs.6b06783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge H. L.; Rittle J.; Williamson L. M.; Xu W. A.; Gagnon D. M.; Tezcan F. A. Redox-Dependent Metastability of the Nitrogenase P-Cluster. J. Am. Chem. Soc. 2019, 141, 10091–10098. 10.1021/jacs.9b04555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keable S. M.; Zadvornyy O. A.; Johnson L. E.; Ginovska B.; Rasmussen A. J.; Danyal K.; Eilers B. J.; Prussia G. A.; LeVan A. X.; Raugei S.; et al. Structural Characterization of the P1+ Intermediate State of the P-cluster of Nitrogenase. J. Biol. Chem. 2018, 293, 9629–9635. 10.1074/jbc.RA118.002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik K.; Hu Y.; Lee C. C.; Wiig J. A.; Ribbe M. W.; Hales B. J. P+ State of Nitrogenase P-cluster Exhibits Electronic Structure of a [Fe4S4]+ Cluster. J. Am. Chem. Soc. 2012, 134, 13749–13754. 10.1021/ja304077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; Börner M. C.; Bergmann J.; Caldararu O.; Ryde U. Geometry and Electronic Structure of the P-Cluster in Nitrogenase Studied by Combined Quantum Mechanical and Molecular Mechanical Calculations and Quantum Refinement. Inorg. Chem. 2019, 58, 9672–9690. 10.1021/acs.inorgchem.9b00400. [DOI] [PubMed] [Google Scholar]
- Morgan T. V.; Mortenson L. E.; McDonald J. W.; Watt G. D. Comparison of Redox and EPR Properties of the Molybdenum Iron Proteins of Clostridium pasteurianum and Azotobacter vinelandii Nitrogenases. J. Inorg. Biochem. 1988, 33, 111–120. 10.1016/0162-0134(88)80039-4. [DOI] [PubMed] [Google Scholar]
- Rawlings J.; Shah V. K.; Chisnell J. R.; Brill W. J.; Zimmerman R.; Münck E.; Orme-Johnson W. H. Novel Metal Cluser in the Iron-Molybdenum Cofactor of Nitrogenase. J. Biol. Chem. 1978, 253, 1001–1004. [PubMed] [Google Scholar]
- Shah V. K.; Brill W. J. Isolation of an Iron-Molybdenum Cofactor from Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 3249–3253. 10.1073/pnas.74.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn J.; Rees D. C. Crystallographic Structure and Functional Implications of the Nitrogenase Molybdenum–Iron Protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. 10.1038/360553a0. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. Modeling a Central Ligand in the Nitrogenase FeMo Cofactor. J. Am. Chem. Soc. 2003, 125, 1466–1467. 10.1021/ja029041g. [DOI] [PubMed] [Google Scholar]
- Vrajmasu V.; Münck E.; Bominaar E. L. Density Functional Study of the Electric Hyperfine Interactions and the Redox-Structural Correlations in the Cofactor of Nitrogenase. Analysis of General Trends in 57Fe Isomer Shifts. Inorg. Chem. 2003, 42, 5974–5988. 10.1021/ic0301371. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. Chemical Activity of the Nitrogenase FeMo Cofactor with a Central Nitrogen Ligand: Density Functional Study. J. Am. Chem. Soc. 2004, 126, 3920–3927. 10.1021/ja037792s. [DOI] [PubMed] [Google Scholar]
- Hinnemann B.; Nørskov J. K. Catalysis by Enzymes: the Biological Ammonia Synthesis. Top. Catal. 2006, 37, 55–70. 10.1007/s11244-006-0002-0. [DOI] [Google Scholar]
- Pelmenschikov V.; Case D. A.; Noodleman L. Ligand-Bound S = 1/2 FeMo-cofactor of Nitrogenase: Hyperfine Interaction Analysis and Implication for the Central Ligand X Identity. Inorg. Chem. 2008, 47, 6162–6172. 10.1021/ic7022743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Zhou Z.; Wan H.; Zhang Q. Enzymatic and Catalytic Reduction of Dinitrogen to Ammonia: Density Functional Theory Characterization of Alternative Molybdenum Active Sites. Int. J. Quantum Chem. 2005, 103, 344–353. 10.1002/qua.20492. [DOI] [Google Scholar]
- Dance I. The Correlation of Redox Potential, HOMO Energy, and Oxidation State in Metal Sulfide Clusters and Its Application to Determine the Redox Level of the FeMo-co Active-site Cluster of Nitrogenase. Inorg. Chem. 2006, 45, 5084–5091. 10.1021/ic060438l. [DOI] [PubMed] [Google Scholar]
- Dance I. The Consequences of an Interstitial N Atom in the FeMo Cofactor of Nitrogenase. Chem. Commun. 2003, 324–325. 10.1039/b211036a. [DOI] [PubMed] [Google Scholar]
- Huniar U.; Ahlrichs R.; Coucouvanis D. Density Functional Theory Calculations and Exploration of a Possible Mechanism of N2 Reduction by Nitrogenase. J. Am. Chem. Soc. 2004, 126, 2588–2601. 10.1021/ja030541z. [DOI] [PubMed] [Google Scholar]
- Harris T. V.; Szilagyi R. K. Comparative Assessment of the Composition and Charge State of Nitrogenase FeMo-cofactor. Inorg. Chem. 2011, 50, 4811–4824. 10.1021/ic102446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. W.; Szilagyi R. K. Exploring New Frontiers of Nitrogenase Structure and Mechanism. Curr. Opin. Chem. Biol. 2006, 10, 101–108. 10.1016/j.cbpa.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Benediktsson B.; Bjornsson R. QM/MM Study of the Nitrogenase MoFe Protein Resting State: Broken-Symmetry States, Protonation States, and QM Region Convergence in the FeMoco Active Site. Inorg. Chem. 2017, 56, 13417–13429. 10.1021/acs.inorgchem.7b02158. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Schlesier J.; Burger E.-M.; Sippel D.; Zhang L.; Andrade S. L. A.; Rees D. C.; Einsle O. Nitrogenase FeMoco Investigated by Spatially Resolved Anomalous Dispersion Refinement. Nat. Commun. 2016, 7, 10902–10907. 10.1038/ncomms10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z. Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Is Mo Involved in Hydride Binding by the Four-Electron Reduced (E4) Intermediate of the Nitrogenase MoFe Protein?. J. Am. Chem. Soc. 2010, 132, 2526–2527. 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kästner J.; Blöchl P. E. Ammonia Production at the FeMo Cofactor of Nitrogenase: Results from Density Functional Theory. J. Am. Chem. Soc. 2007, 129, 2998–3006. 10.1021/ja068618h. [DOI] [PubMed] [Google Scholar]
- Dance I. Electronic Dimensions of FeMo-co, the Active Site of Nitrogenase, and its Catalytic Intermediates. Inorg. Chem. 2011, 50, 178–192. 10.1021/ic1015884. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Kaiser J. T.; Meloni G.; Yang K.-Y.; Spatzal T.; Andrade S. L. A.; Einsle O.; Howard J. B.; Rees D. C. The Sixteenth Iron in the Nitrogenase MoFe Protein. Angew. Chem., Int. Ed. 2013, 52, 10529–10532. 10.1002/anie.201303877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venters R. A.; Nelson M. J.; McLean P. A.; True A. E.; Levy M. A.; Hoffman B. M.; Orme-Johnson W. H. ENDOR of the Resting State of Nitrogenase Molybdenum-Iron Proteins from Azotobacter vinelandii, Klebsiella pneumoniae, and Clostridium pasteurianum: 1H, 57Fe, 95Mo, and 33S Studies. J. Am. Chem. Soc. 1986, 108, 3487–3498. 10.1021/ja00272a054. [DOI] [Google Scholar]
- Bobrik M. A.; Laskowski E. J.; Johnson R. W.; Gillum W. O.; Berg J. M.; Hodgson K. O.; Holm R. H. Selenium Substitution in [Fe4S4(SR)4]2-: Synthesis and Comparative Properties of [Fe4X4(YC6H5)4]2- (X, Y = S, S) and the Structure of [(CH3)4N]2[Fe4Se4(SC6H5)4]. Inorg. Chem. 1978, 17, 1402–1410. 10.1021/ic50184a003. [DOI] [Google Scholar]
- Fomitchev D. V.; McLauchlan C. C.; Holm R. H. Heterometal Cubane-Type MFe3S4 Clusters (M= Mo, V) Trigonally Symmetrized with Hydrotris(pyrazolyl)borate(1-) and Tris(pyrazolyl)methanesulfonate(1-) Capping Ligands. Inorg. Chem. 2002, 41, 958–966. 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- Hauser C.; Bill E.; Holm R. H. Single- and Double-Cubane Clusters in the Multiple Oxidation States [VFe3S4]3+,2+,1+. Inorg. Chem. 2002, 41, 1615–1624. 10.1021/ic011011b. [DOI] [PubMed] [Google Scholar]
- Berg J. M.; Hodgson K. O.; Holm R. H. Crystal Structure of [(C2H5)4N]3[Fe4S4(SCH2Ph)4], a Reduced Ferredoxin Site Analog with a Nontetragonal Fe4S4 Core Structure in the Solid State. J. Am. Chem. Soc. 1979, 101, 4586–4593. 10.1021/ja00510a025. [DOI] [Google Scholar]
- Sharp C. R.; Duncan J. S.; Lee S. C. [Fe4S4]q Cubane Clusters (q = 4+, 3+, 2+) with Terminal Amide Ligands. Inorg. Chem. 2010, 49, 6697–6705. 10.1021/ic100742c. [DOI] [PubMed] [Google Scholar]
- Ohki Y.; Tanifuji K.; Yamada N.; Imada M.; Tajima T.; Tatsumi K. Synthetic Analogues of [Fe4S4(Cys)3(His)] in Hydrogenases and [Fe4S4(Cys)4] in HiPIP Derived from All-ferric [Fe4S4{N(SiMe3)2}4]. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 12635–12640. 10.1073/pnas.1106472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moula G.; Matsumoto T.; Miehlich M. E.; Meyer K.; Tatsumi K. Synthesis of an All-Ferric Cuboidal Iron-Sulfur Cluster [FeIII4S4(SAr)4]. Angew. Chem., Int. Ed. 2018, 57, 11594–11598. 10.1002/anie.201803679. [DOI] [PubMed] [Google Scholar]
- Strop P.; Takahara P. M.; Chiu H.-J.; Angove H. C.; Burgess B. K.; Rees D. C. Crystal Structure of the All-Ferrous [4Fe-4S]0 Form of the Nitrogenase Iron Protein from Azotobacter vinelandii. Biochemistry 2001, 40, 651–656. 10.1021/bi0016467. [DOI] [PubMed] [Google Scholar]
- Corbett M. C.; Hu Y.; Naderi F.; Ribbe M. W.; Hedman B.; Hodgson K. O. Comparison of Iron-Molybdenum Cofactor-deficient Nitrogenase MoFe Proteins by X-ray Absorption Spectroscopy. Implications for P-cluster Biosynthesis. J. Biol. Chem. 2004, 279, 28276–28282. 10.1074/jbc.M403156200. [DOI] [PubMed] [Google Scholar]
- Cao L.; Caldararu O.; Ryde U. Protonation and Reduction of the FeMo Cluster in Nitrogenase Studied by Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations. J. Chem. Theory Comput. 2018, 14, 6653–6678. 10.1021/acs.jctc.8b00778. [DOI] [PubMed] [Google Scholar]
- Dance I. Survey of the Geometric and Electronic Structures of the Key Hydrogenated Forms of FeMo-co, the Active Site of the Enzyme Nitrogenase: Principles of the Mechanistically Significant Coordination Chemistry. Inorganics 2019, 7, 8. 10.3390/inorganics7010008. [DOI] [Google Scholar]
- Igarashi R. Y.; Laryukhin M.; Dos Santos P. C.; Lee H.-I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Trapping H– Bound to the Nitrogenase FeMo-cofactor Active Site during H2 Evolution: Characterization by ENDOR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 6231–6241. 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Duval S.; Danyal K.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. A Confirmation of the Quench-Cryoannealing Relaxation Protocol for Identifying Reduction States of Freeze-Trapped Nitrogenase Intermediates. Inorg. Chem. 2014, 53, 3688–3693. 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton P. M. C.; Laryukhin M.; Mayer S. M.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Localization of a Substrate Binding Site on the FeMo-Cofactor in Nitrogenase: Trapping Propargyl Alcohol with an α-70-Substituted MoFe Protein. Biochemistry 2003, 42, 9102–9109. 10.1021/bi034595x. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Igarashi R. Y.; Laryukhin M.; Doan P. E.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. An Organometallic Intermediate during Alkyne Reduction by Nitrogenase. J. Am. Chem. Soc. 2004, 126, 9563–9569. 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- Lee H.-I.; Sørlie M.; Christiansen J.; Song R. T.; Dean D. R.; Hales B. J.; Hoffman B. M. Characterization of an Intermediate in the Reduction of Acetylene by the Nitrogenase α-Gln195 MoFe Protein by Q-band EPR and 13C, 1H ENDOR. J. Am. Chem. Soc. 2000, 122, 5582–5587. 10.1021/ja000542g. [DOI] [Google Scholar]
- Willems J.-P.; Lee H.-I.; Burdi D.; Doan P. E.; Stubbe J.; Hoffman B. M. Identification of the Protonated Oxygenic Ligands of Ribonucleotide Reductase Intermediate X by Q-Band 1,2H CW and Pulsed ENDOR. J. Am. Chem. Soc. 1997, 119, 9816–9824. 10.1021/ja9709942. [DOI] [Google Scholar]
- Fisher K.; Newton W. E.; Lowe D. J. Electron Paramagnetic Resonance Analysis of Different Azotobacter vinelandii Nitrogenase MoFe-Protein Conformations Generated During Enzyme Turnover: Evidence for S = 3/2 Spin States from Reduced MoFe-Protein Intermediate. Biochemistry 2001, 40, 3333–3339. 10.1021/bi0012686. [DOI] [PubMed] [Google Scholar]
- Hoffman B. M.; Venters R. A.; Roberts J. E.; Nelson M.; Orme-Johnson W. H. 57Fe ENDOR of the Nitrogenase MoFe Protein. J. Am. Chem. Soc. 1982, 104, 4711–4712. 10.1021/ja00381a050. [DOI] [Google Scholar]
- True A. E.; Nelson M. J.; Venters R. A.; Orme-Johnson W. H.; Hoffman B. M. 57Fe Hyperfine Coupling Tensors of the FeMo Cluster in Azotobacter vinelandii MoFe Protein: Determination by Polycrystalline ENDOR Spectroscopy. J. Am. Chem. Soc. 1988, 110, 1935–1943. 10.1021/ja00214a045. [DOI] [Google Scholar]
- Lee H.-I.; Sørlie M.; Christiansen J.; Yang T. C.; Shao J.; Dean D. R.; Hales B. J.; Hoffman B. M. Electron Inventory, Kinetic Assignment (En), Structure, and Bonding of Nitrogenase Turnover Intermediates with C2H2 and CO. J. Am. Chem. Soc. 2005, 127, 15880–15890. 10.1021/ja054078x. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Einsle O.; Howard J. B.; Rees D. C. Ligand Binding to the FeMo-cofactor: Structures of CO-bound and Reactivated Nitrogenase. Science 2014, 345, 1620–1623. 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatzal T.; Perez K. A.; Howard J. B.; Rees D. C. Catalysis-dependent Selenium Incorporation and Migration in the Nitrogenase Active Site Iron-Molybdenum Cofactor. eLife 2015, 4, e11620 10.7554/eLife.11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippel D.; Rohde M.; Netzer J.; Trncik C.; Gies J.; Grunau K.; Djurdjevic I.; Decamps L.; Andrade S. L. A.; Einsle O. A Bound Reaction Intermediate Sheds Light on the Mechanism of Nitrogenase. Science 2018, 359, 1484–1489. 10.1126/science.aar2765. [DOI] [PubMed] [Google Scholar]
- Yang Z.-Y.; Khadka N.; Lukoyanov D.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. On Reversible H2 Loss Upon N2 Binding to FeMo-cofactor of Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16327–16332. 10.1073/pnas.1315852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Yang T. C.; Igarashi R. Y.; Dos Santos P. C.; Laryukhin M.; Lee H.-I.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Intermediates Trapped During Nitrogenase Reduction of N≡N, CH3-N = NH, and H2N-NH2. J. Am. Chem. Soc. 2005, 127, 14960–14961. 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- Bulfer S. L.; Scott E. M.; Couture J.-F.; Pillus L.; Trievel R. C. Crystal Structure and Functional Analysis of Homocitrate Synthase, an Essential Enzyme in Lysine Biosynthesis. J. Biol. Chem. 2009, 284, 35769–35780. 10.1074/jbc.M109.046821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Yang Z.-Y.; Khadka N.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Identification of a Key Catalytic Intermediate Demonstrates that Nitrogenase Is Activated by the Reversible Exchange of N2 for H2. J. Am. Chem. Soc. 2015, 137, 3610–3615. 10.1021/jacs.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; McClead J.; Lukoyanov D.; Laryukhin M.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Diazene (HN = NH) Is a Substrate for Nitrogenase: Insights into the Pathway of N2 Reduction. Biochemistry 2007, 46, 6784–6794. 10.1021/bi062294s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Khadka N.; Dean D. R.; Raugei S.; Seefeldt L. C.; Hoffman B. M. Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride (Janus) State Involves a FeMo-cofactor-H2 Intermediate. Inorg. Chem. 2017, 56, 2233–2240. 10.1021/acs.inorgchem.6b02899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmann J.; Munhá R. F.; Fryzuk M. D. The Hydride Route to the Preparation of Dinitrogen Complexes. Chem. Commun. 2010, 46, 1013–1025. 10.1039/b922853e. [DOI] [PubMed] [Google Scholar]
- Dugan T. R.; Holland P. L. New Routes to Low-coordinate Iron Hydride Complexes: The Binuclear Oxidative Addition of H2. J. Organomet. Chem. 2009, 694, 2825–2830. 10.1016/j.jorganchem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz R. N. Metal dihydride Complexes: Photochemical Mechanisms for Reductive Elimination. Pure Appl. Chem. 1998, 70, 2211–2220. 10.1351/pac199870112211. [DOI] [Google Scholar]
- Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. Studies of Low-Coordinate Iron Dinitrogen Complexes. J. Am. Chem. Soc. 2006, 128, 756–769. 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Sadique A. R.; Smith J. M.; Dugan T. R.; Cowley R. E.; Brennessel W. W.; Flaschenriem C. J.; Bill E.; Cundari T. R.; Holland P. L. The Reactivity Patterns of Low-Coordinate Iron–Hydride Complexes. J. Am. Chem. Soc. 2008, 130, 6624–6638. 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov D.; Khadka N.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Reversible Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride State, the E4(4H) Janus Intermediate. J. Am. Chem. Soc. 2016, 138, 1320–1327. 10.1021/jacs.5b11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson W. A.; Suess D. L. M.; Fong H.; Wang X.; Hoffmann C. M.; Cutsail G. E. III; Peters J. C.; Hoffman B. M. Free H2 Rotation vs Jahn-Teller Constraints in the Nonclassical Trigonal (TPB)Co-H2 Complex. J. Am. Chem. Soc. 2014, 136, 14998–15009. 10.1021/ja508117h. [DOI] [PubMed] [Google Scholar]
- Lukoyanov D.; Krzyaniak M. D.; Dean D. R.; Wasielewski M. R.; Seefeldt L. C.; Hoffman B. M. Time-Resolved EPR Study of H2 Reductive Elimination from the Photoexcited Nitrogenase Janus E4(4H) Intermediate. J. Phys. Chem. B 2019, 123, 8823–8828. 10.1021/acs.jpcb.9b07776. [DOI] [PubMed] [Google Scholar]
- Newell M.; USGS Office of Communications, 2015.
- Lukoyanov D. A.; Khadka N.; Yang Z.-Y.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. Hydride Conformers of the Nitrogenase FeMo-cofactor Two-Electron Reduced State E2(2H), Assigned Using Cryogenic Intra Electron Paramagnetic Resonance Cavity Photolysis. Inorg. Chem. 2018, 57, 6847–6852. 10.1021/acs.inorgchem.8b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L. C. Hydrazine as a Substrate and Inhibitor of Azotobacter vinelandii Nitrogenase. Arch. Biochem. Biophys. 1980, 204, 270–276. 10.1016/0003-9861(80)90033-8. [DOI] [PubMed] [Google Scholar]
- Thorneley R. N. F.; Eady R. R.; Lowe D. J. Biological Nitrogen Fixation by way of an Enzyme-bound Dinitrogen-hydride Intermediate. Nature 1978, 272, 557–558. 10.1038/272557a0. [DOI] [Google Scholar]
- Dilworth M. J.; Eady R. R. Hydrazine is a Product of Dinitrogen Reduction by the Vanadium-Nitrogenase from Azotobacter chroococcum. Biochem. J. 1991, 277, 465–468. 10.1042/bj2770465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth M. J.; Eldridge M. E.; Eady R. R. Correction for Creatine Interference with the Direct Indophenol Measurement of NH3 in Steady-State Nitrogenase Assays. Anal. Biochem. 1992, 207, 6–10. 10.1016/0003-2697(92)90491-O. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Simeonov A. M.; Eran H.; Bravo-Leerabhandh M. Reduction of Cyclic and Acyclic Diazene Derivates by Azotobacter vinelandii Nitrogenase: Diazirine and trans-Dimethyldiazene. Biochemistry 1996, 35, 4502–4514. 10.1021/bi950964g. [DOI] [PubMed] [Google Scholar]
- Mayer S. M.; Niehaus W. G.; Dean D. R. Reduction of Short Chain Alkynes by a Nitrogenase α-70Ala-substituted MoFe Protein. J. Chem. Soc., Dalton Trans. 2002, 802–807. 10.1039/b107336b. [DOI] [Google Scholar]
- Barney B. M.; Igarashi R. Y.; Dos Santos P. C.; Dean D. R.; Seefeldt L. C. Substrate Interaction at an Iron-Sulfur Face of the FeMo-cofactor during Nitrogenase Catalysis. J. Biol. Chem. 2004, 279, 53621–53624. 10.1074/jbc.M410247200. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Kim C.-H.; Newton W. E. Azotobacter vinelandii Nitrogenases with Substitutions in the FeMo-Cofactor Environment of the MoFe Protein: Effects of Acetylene or Ethylene on Interactions with H+, HCN, and CN–. Biochemistry 2000, 39, 10855–10865. 10.1021/bi0001628. [DOI] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Newton W. E. Differential Effects on N2 Binding and Reduction, HD Formation, and Azide Reduction with α-195His- and α-191Gln-Substituted MoFe Proteins of Azotobacter vinelandii Nitrogenase. Biochemistry 2000, 39, 15570–15577. 10.1021/bi0017834. [DOI] [PubMed] [Google Scholar]
- Kim C.-H.; Newton W. E.; Dean D. R. Role of the MoFe Protein α-Subunit Histidine-195 Residue in FeMo-Cofactor Binding and Nitrogenase Catalysis. Biochemistry 1995, 34, 2798–2808. 10.1021/bi00009a008. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Laryukhin M.; Igarashi R. Y.; Lee H.-I.; Dos Santos P. C.; Yang T.-C.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Trapping a Hydrazine Reduction Intermediate on the Nitrogenase Active Site. Biochemistry 2005, 44, 8030–8037. 10.1021/bi0504409. [DOI] [PubMed] [Google Scholar]
- Barney B. M.; Lukoyanov D.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. A methyldiazene (HN = N-CH3)-derived Species Bound to the Nitrogenase Active-site FeMo Cofactor: Implications for Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17113–17118. 10.1073/pnas.0602130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barney B. M.; Lukoyanov D.; Igarashi R. Y.; Laryukhin M.; Yang T.-C.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Trapping an Intermediate of Dinitrogen (N2) Reduction on Nitrogenase. Biochemistry 2009, 48, 9094–9102. 10.1021/bi901092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt R. D.; Zimmermann J.-L.; Sauer K.; Klein M. P. Ammonia Binds to the Catalytic Manganese of the Oxygen-Evolving Complex of Photosystem II. Evidence by Electron Spin-Echo Envelope Modulation Spectroscopy. J. Am. Chem. Soc. 1989, 111, 3522–3532. 10.1021/ja00192a006. [DOI] [Google Scholar]
- Hardy R. W. F.; Holsten R. D.; Jackson E. K.; Burns R. C. The Acetylene-Ethylene Assay for N2 Fixation: Laboratory and Field Evaluation. Plant Physiol. 1968, 43, 1185–1207. 10.1104/pp.43.8.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth M. J. Acetylene Reduction by Nitrogen-fixing Preparations from Clostridium pasteurianum. Biochim. Biophys. Acta, Gen. Subj. 1966, 127, 285–294. 10.1016/0304-4165(66)90383-7. [DOI] [PubMed] [Google Scholar]
- Hwang J. C.; Chen C. H.; Burris R. H. Inhibition of Nitrogenase-catalyzed Reductions. Biochim. Biophys. Acta, Bioenerg. 1973, 292, 256–270. 10.1016/0005-2728(73)90270-3. [DOI] [PubMed] [Google Scholar]
- Rivera-Ortiz J. M.; Burris R. H. Interactions Among Substrates and Inhibitors of Nitrogenase. J. Bacteriol. 1975, 123, 537–545. 10.1128/JB.123.2.537-545.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D. J.; Fisher K.; Thorneley R. N. F. Klebsiella pneumoniae Nitrogenase. Mechanism of Acetylene Reduction and its Inhibition by Carbon Monoxide. Biochem. J. 1990, 272, 621–625. 10.1042/bj2720621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton P. M. C.; Christiansen J.; Dean D. R.; Seefeldt L. C. Stereospecificity of Acetylene Reduction Catalyzed by Nitrogenase. J. Am. Chem. Soc. 2001, 123, 1822–1827. 10.1021/ja003662x. [DOI] [PubMed] [Google Scholar]
- Lowe D. J.; Eady R. R.; Thorneley R. N. F. Electron-Paramagnetic-Resonance Studies on Nitrogenase of Klebsiella pneumoniae. Evidence for Acetylene- and Ethylene-Nitrogenase Transient Complexes. Biochem. J. 1978, 173, 277–290. 10.1042/bj1730277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L. C.; Henzl M. T.; Burris R. H.; Orme-Johnson W. H. Iron-Sulfur Clusters in the Molybdenum-Iron Protein Component of Nitrogenase. Electron Paramagnetic Resonance of the Carbon Monoxide Inhibited State. Biochemistry 1979, 18, 4860–4869. 10.1021/bi00589a014. [DOI] [PubMed] [Google Scholar]
- Shen J.; Dean D. R.; Newton W. E. Evidence for Multiple Substrate-Reduction Sites and Distinct Inhibitor-Binding Sites from an Altered Azotobacter vinelandii Nitrogenase MoFe Protein. Biochemistry 1997, 36, 4884–4894. 10.1021/bi9628578. [DOI] [PubMed] [Google Scholar]
- Kästner J.; Blöchl P. E. Model for Acetylene Reduction by Nitrogenase Derived from Density Functional Theory. Inorg. Chem. 2005, 44, 4568–4575. 10.1021/ic0500311. [DOI] [PubMed] [Google Scholar]
- Christiansen J.; Cash V. L.; Seefeldt L. C.; Dean D. R. Isolation and Characterization of an Acetylene-resistant Nitrogenase. J. Biol. Chem. 2000, 275, 11459–11464. 10.1074/jbc.275.15.11459. [DOI] [PubMed] [Google Scholar]
- Conradson S. D.; Burgess B. K.; Vaughn S. A.; Roe A. L.; Hedman B.; Hodgson K. O.; Holm R. H. Cyanide and Methylisocyanide Binding to the Isolated Iron-Molybdenum Cofactor of Nitrogenase. J. Biol. Chem. 1989, 264, 15967–15974. [PubMed] [Google Scholar]
- Smith B. E.; Lowe D. J.; Bray R. C. Studies by Electron Paramagnetic Resonance on the Catalytic Mechanism of Nitrogenase of Klebsiella pneumoniae. Biochem. J. 1973, 135, 331–341. 10.1042/bj1350331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes B. D.; Fisher K.; Lowe D. J. Nitrogenase of Klebsiella pneumoniae: Electron Nuclear Double Resonance (ENDOR) Studies on the Substrate Reduction Site. Biochem. J. 1994, 297, 261–264. 10.1042/bj2970261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean P. A.; True A.; Nelson M. J.; Lee H. I.; Hoffman B. M.; Orme-Johnson W. H. Effects of Substrates (Methyl Isocyanide, C2H2) and Inhibitor (CO) on Resting-state Wild-type and NifV–Klebsiella pneumoniae MoFe Proteins. J. Inorg. Biochem. 2003, 93, 18–32. 10.1016/S0162-0134(02)00580-9. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Unique Features of the Nitrogenase VFe Protein from Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 9209–9214. 10.1073/pnas.0904408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes T. R.; Lowe D. J.; Smith B. E. Nitrogenase from Klebsiella pneumoniae. An E.P.R. Signal Observed During Enzyme Turnover under Ethylene is Associated with the Iron Molybdenum Cofactor. Biochem. J. 1983, 211, 495–497. 10.1042/bj2110495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma R.; Barney B. M.; Keable S.; Dean D. R.; Seefeldt L. C.; Peters J. W. Insights into Substrate Binding at FeMo-cofactor in Nitrogenase from the Structure of an α-70Ile MoFe Protein Variant. J. Inorg. Biochem. 2010, 104, 385–389. 10.1016/j.jinorgbio.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose V. J.; Kim C.-H.; Newton W. E.; Dean D. R.; Hoffman B. M. Electron Spin Echo Envelope Modulation Spectroscopic Analysis of Altered Nitrogenase MoFe Proteins from Azotobacter vinelandii. Biochemistry 1995, 34, 2809–2814. 10.1021/bi00009a009. [DOI] [PubMed] [Google Scholar]
- Sørlie M.; Christiansen J.; Lemon B. J.; Peters J. W.; Dean D. R.; Hales B. J. Mechanistic Features and Structure of the Nitrogenase α-Gln195 MoFe Protein. Biochemistry 2001, 40, 1540–1549. 10.1021/bi0013997. [DOI] [PubMed] [Google Scholar]
- Sørlie M.; Christiansen J.; Dean D. R.; Hales B. J. Detection of a New Radical and FeMo-cofactor EPR Signal During Acetylene Reduction by the α-H195Q Mutant of Nitrogenase. J. Am. Chem. Soc. 1999, 121, 9457–9458. 10.1021/ja991599+. [DOI] [Google Scholar]
- Lee H.-I.; Cameron L. M.; Christiansen J.; Christie P. D.; Pollock R. C.; Song R.; Sørlie M.; Orme-Johnson W. H.; Dean D. R.; Hales B. J.; Hoffman B. M.. Q-Band ENDOR Studies of the Nitrogenase MoFe Protein under Turnover Conditions. Substrate-Inhibitor-Binding to and Metal-Ion Valencies of the FeMo-Cofactors. In Paramagnetic Resonance of Metallobiomolecules; ACS Symposium Series, American Chemical Society, 2003; Vol. 858, Chapter 8, pp 150–178. [Google Scholar]
- Benton P. M. C.; Mayer S. M.; Shao J.; Hoffman B. M.; Dean D. R.; Seefeldt L. C. Interaction of Acetylene and Cyanide with the Resting State of Nitrogenase α-96-Substituted MoFe Proteins. Biochemistry 2001, 40, 13816–13825. 10.1021/bi011571m. [DOI] [PubMed] [Google Scholar]
- Keable S. M.; Vertemara J.; Zadvornyy O. A.; Eilers B. J.; Danyal K.; Rasmussen A. J.; De Gioia L.; Zampella G.; Seefeldt L. C.; Peters J. W. Structural Characterization of the Nitrogenase Molybdenum-Iron Protein with the Substrate Acetylene Trapped Near the Active Site. J. Inorg. Biochem. 2018, 180, 129–134. 10.1016/j.jinorgbio.2017.12.008. [DOI] [PubMed] [Google Scholar]
- Lind C. J.; Wilson P. W. Mechanism of Biological Nitrogen Fixation. VIII. Carbon Monoxide as an Inhibitor for Nitrogen Fixation by Red Clover. J. Am. Chem. Soc. 1941, 63, 3511–3514. 10.1021/ja01857a073. [DOI] [Google Scholar]
- Wilson P. W.; Lind C. J. Carbon Monoxide Inhibition of Azotobacter in Microrespiration Experiments. J. Bacteriol. 1943, 45, 219–232. 10.1128/JB.45.3.219-232.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockshin A.; Burris R. H. Inhibitors of Nitrogen Fixation in Extracts from Clostridium pasteurianum. Biochim. Biophys. Acta, Gen. Subj. 1965, 111, 1–10. 10.1016/0304-4165(65)90466-6. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Lee C. C.; Ribbe M. W. Extending the Carbon Chain: Hydrocarbon Formation Catalyzed by Vanadium/Molybdenum Nitrogenases. Science 2011, 333, 753–755. 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Ribbe M. W. Nitrogenases-A Tale of Carbon Atom(s). Angew. Chem., Int. Ed. 2016, 55, 8216–8226. 10.1002/anie.201600010. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Catalytic Reduction of CN–, CO, and CO2 by Nitrogenase Cofactors in Lanthanide-Driven Reactions. Angew. Chem., Int. Ed. 2015, 54, 1219–1222. 10.1002/anie.201410412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Insights into Hydrocarbon Formation by Nitrogenase Cofactor Homologs. mBio 2015, 6, e00307-15 10.1128/mBio.00307-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller C. J.; Lee C. C.; Stiebritz M. T.; Rettberg L. A.; Hu Y. Strategies Towards Capturing Nitrogenase Substrates and Intermediates via Controlled Alteration of Electron Fluxes. Chem. - Eur. J. 2019, 25, 2389–2395. 10.1002/chem.201803735. [DOI] [PubMed] [Google Scholar]
- Yang Z.-Y.; Dean D. R.; Seefeldt L. C. Molybdenum Nitrogenase Catalyzes the Reduction and Coupling of CO to Form Hydrocarbons. J. Biol. Chem. 2011, 286, 19417–19421. 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron L. M.; Hales B. J. Investigation of CO Binding and Release from Mo-Nitrogenase during Catalytic Turnover. Biochemistry 1998, 37, 9449–9456. 10.1021/bi972667c. [DOI] [PubMed] [Google Scholar]
- Christie P. D.; Lee H.-I.; Cameron L. M.; Hales B. J.; Orme-Johnson W. H.; Hoffman B. M. Identification of the CO-Binding Cluster in Nitrogenase MoFe Protein by ENDOR of 57Fe Isotopomers. J. Am. Chem. Soc. 1996, 118, 8707–8709. 10.1021/ja961289g. [DOI] [Google Scholar]
- Pollock R. C.; Lee H.-I.; Cameron L. M.; DeRose V. J.; Hales B. J.; Orme-Johnson W. H.; Hoffman B. M. Investigation of CO Bound to Inhibited Forms of Nitrogenase MoFe Protein by 13C ENDOR. J. Am. Chem. Soc. 1995, 117, 8686–8687. 10.1021/ja00138a033. [DOI] [Google Scholar]
- Lee H.-I.; Cameron L. M.; Hales B. J.; Hoffman B. M. CO Binding to the FeMo Cofactor of CO-inhibited Nitrogenase: 13CO and 1H Q-band ENDOR Investigation. J. Am. Chem. Soc. 1997, 119, 10121–10126. 10.1021/ja9715096. [DOI] [Google Scholar]
- Maskos Z.; Hales B. J. Photo-lability of CO Bound to Mo-Nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2003, 93, 11–17. 10.1016/S0162-0134(02)00480-4. [DOI] [PubMed] [Google Scholar]
- Maskos Z.; Fisher K.; Sørlie M.; Newton W. E.; Hales B. J. Variant MoFe Proteins of Azotobacter vinelandii: Effects of Carbon Monoxide on Electron Paramagnetic Resonance Spectra Generated During Enzyme Turnover. JBIC, J. Biol. Inorg. Chem. 2005, 10, 394–406. 10.1007/s00775-005-0648-2. [DOI] [PubMed] [Google Scholar]
- Dance I. Calculated Vibrational Frequencies for FeMo-co, the Active Site of Nitrogenase, Bearing Hydrogen Atoms and Carbon Monoxide. Dalton Trans. 2011, 40, 6480–6489. 10.1039/c1dt10505a. [DOI] [PubMed] [Google Scholar]
- Dance I. Mechanisms of the S/CO/Se Interchange Reactions at FeMo-co, the Active Site Cluster of Nitrogenase. Dalton Trans. 2016, 45, 14285–14300. 10.1039/C6DT03159E. [DOI] [PubMed] [Google Scholar]
- Durrant M. C. An Atomic Level Model for the Interactions of Molybdenum Nitrogenase with Carbon Monoxide, Acetylene, and Ethylene. Biochemistry 2004, 43, 6030–6042. 10.1021/bi036300l. [DOI] [PubMed] [Google Scholar]
- Gee L. B.; Scott A. D.; Dapper C. H.; Newton W. E.; Cramer S. P. Is Trehalose an Effective Quenching Agent of Azotobacter vinelandii Mo-nitrogenase Turnover?. Inorg. Chim. Acta 2016, 453, 74–77. 10.1016/j.ica.2016.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K.; Dilworth M. J.; Newton W. E. Azotobacter vinelandii Vanadium Nitrogenase: Formaldehyde Is a Product of Catalyzed HCN Reduction, and Excess Ammonia Arises Directly from Catalyzed Azide Reduction. Biochemistry 2006, 45, 4190–4198. 10.1021/bi0514109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Burgess B. K.; Corbin J. L. Nitrogenase Reactivity: Cyanide as Substrate and Inhibitor. Biochemistry 1982, 21, 4393–4402. 10.1021/bi00261a031. [DOI] [PubMed] [Google Scholar]
- Koksunan S.; Vichitphan S.; Laopaiboon L.; Vichitphan K.; Han J. Growth and Cyanide Degradation of Azotobacter vinelandii in Cyanide-Containing Wastewater System. J. Microbiol. Biotechnol. 2013, 23, 572–578. 10.4014/jmb.1209.09026. [DOI] [PubMed] [Google Scholar]
- Roth L. E.; Tezcan F. A. ATP-Uncoupled, Six-Electron Photoreduction of Hydrogen Cyanide to Methane by the Molybdenum–Iron Protein. J. Am. Chem. Soc. 2012, 134, 8416–8419. 10.1021/ja303265m. [DOI] [PubMed] [Google Scholar]
- Cui Z.; Dunford A. J.; Durrant M. C.; Henderson R. A.; Smith B. E. Binding Sites of Nitrogenase: Kinetic and Theoretical Studies of Cyanide Binding to Extracted FeMo-Cofactor Derivatives. Inorg. Chem. 2003, 42, 6252–6264. 10.1021/ic030108q. [DOI] [PubMed] [Google Scholar]
- Lowe D. J.; Fisher K.; Thorneley R. N. F.; Vaughn S. A.; Burgess B. K. Kinetics and Mechanism of the Reaction of Cyanide with Molybdenum Nitrogenase from Azotobacter vinelandii. Biochemistry 1989, 28, 8460–8466. 10.1021/bi00447a028. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Kechris K. J.; Rees D. C.; Glazer A. N. Multiple Amino Acid Sequence Alignment Nitrogenase Component 1: Insights into Phylogenetics and Structure-Function Relationships. PLoS One 2013, 8, e72751 10.1371/journal.pone.0072751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D. F.; Lukoyanov D. A.; Kallas H.; Trncik C.; Yang Z.-Y.; Compton P.; Kelleher N.; Einsle O.; Dean D. R.; Hoffman B. M.; et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism To Achieve N2 Reduction. Biochemistry 2019, 58, 3293–3301. 10.1021/acs.biochem.9b00468. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Lee C. C.; Ribbe M. W. Vanadium Nitrogenase: A Two-Hit Wonder?. Dalton Trans. 2012, 41, 1118–1127. 10.1039/C1DT11535A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mus F.; Alleman A. B.; Pence N.; Seefeldt L. C.; Peters J. W. Exploring the Aternatives of Biological Nitrogen Fixation. Metallomics 2018, 10, 523–538. 10.1039/C8MT00038G. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Hu Y.; Ribbe M. W. Vanadium Nitrogenase Reduces CO. Science 2010, 329, 642. 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Fay A. W.; Weng T.-C.; Krest C. M.; Hedman B.; Hodgson K. O.; Hu Y.; Ribbe M. W. Uncoupling Binding of Substrate CO from Turnover by Vanadium Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 13845–13849. 10.1073/pnas.1519696112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Tanifuji K.; Newcomb M.; Liedtke J.; Hu Y.; Ribbe M. W. A Comparative Analysis of the CO-reducing Activities of MoFe Proteins Containing Mo- and V-Nitrogenase Cofactors. ChemBioChem 2018, 19, 649–653. 10.1002/cbic.201800035. [DOI] [PubMed] [Google Scholar]
- Fay A. W.; Blank M. A.; Lee C. C.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Characterization of Isolated Nitrogenase FeVco. J. Am. Chem. Soc. 2010, 132, 12612–12618. 10.1021/ja1019657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi N.; Moore V.; Lloyd S. G.; Hales B. J.; Huynh B. H. Mössbauer Characterization of the Metal Clusters in Azotobacter vinelandii Nitrogenase VFe Protein. J. Biol. Chem. 1994, 269, 20920–20924. [PubMed] [Google Scholar]
- Morningstar J. E.; Hales B. J. Electron Paramagnetic Resonance Study of the Vanadium-Iron Protein of Nitrogenase from Azotobacter vinelandii. J. Am. Chem. Soc. 1987, 109, 6854–6855. 10.1021/ja00256a051. [DOI] [Google Scholar]
- George G. N.; Coyle C. L.; Hales B. J.; Cramer S. P. X-Ray Absorption of Azotobacter vinelandii Vanadium Nitrogenase. J. Am. Chem. Soc. 1988, 110, 4057–4059. 10.1021/ja00220a066. [DOI] [Google Scholar]
- Harvey I.; Arber J. M.; Eady R. R.; Smith B. E.; Garner C. D.; Hasnain S. S. Iron K-edge X-ray-absorption Spectroscopy of the Iron-Vanadium Cofactor of the Vanadium Nitrogenase from Azotobacter chroococcum. Biochem. J. 1990, 266, 929–931. [PMC free article] [PubMed] [Google Scholar]
- Arber J. M.; Dobson B. R.; Eady R. R.; Stevens P.; Hasnain S. S.; Garner C. D.; Smith B. E. Vanadium K-edge X-ray Absorption Spectrum of the VFe Protein of the Vanadium Nitrogenase of Azotobacter chroococcum. Nature 1987, 325, 372–374. 10.1038/325372a0. [DOI] [Google Scholar]
- Wong J.; Lytle F. W.; Messmer R. P.; Maylotte D. H. K-edge absorption Spectra ofSelected Vanadium Compounds. Phys. Rev. B: Condens. Matter Mater. Phys. 1984, 30, 5596–5610. 10.1103/PhysRevB.30.5596. [DOI] [Google Scholar]
- Lee C. C.; Wilcoxen J.; Hiller C. J.; Britt R. D.; Hu Y. Evaluation of the Catalytic Relevance of the CO-Bound States of V-Nitrogenase. Angew. Chem., Int. Ed. 2018, 57, 3411–3414. 10.1002/anie.201800189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippel D.; Schlesier J.; Rohde M.; Trncik C.; Decamps L.; Djurdjevic I.; Spatzal T.; Andrade S. L.; Einsle O. Production and Isolation of Vanadium Nitrogenase from Azotobacter vinelandii by Molybdenum Depletion. JBIC, J. Biol. Inorg. Chem. 2017, 22, 161–168. 10.1007/s00775-016-1423-2. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Corbett M. C.; Fay A. W.; Webber J. A.; Hedman B.; Hodgson K. O.; Ribbe M. W. Nitrogenase Reactivity with P-cluster Variants. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13825–13830. 10.1073/pnas.0506967102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbe M. W.; Hu Y.; Guo M.; Schmid B.; Burgess B. K. The FeMoco-deficient MoFe Protein Produced by a nifH Deletion Strain of Azotobacter vinelandii Shows Unusual P-cluster Features. J. Biol. Chem. 2002, 277, 23469–23476. 10.1074/jbc.M202061200. [DOI] [PubMed] [Google Scholar]
- Chatterjee R.; Allen R. M.; Ludden P. W.; Shah V. K. In vitro Synthesis of the Iron-Molybdenum Cofactor and Maturation of the nif-Encoded Apodinitrogenase. Effect of Substitution of VnfH for NifH. J. Biol. Chem. 1997, 272, 21604–21608. 10.1074/jbc.272.34.21604. [DOI] [PubMed] [Google Scholar]
- Hales B. J.; True A. E.; Hoffman B. M. Detection of a New Signal in the EPR Spectrum of Vanadium Nitrogenase from Azotobacter vinelandii. J. Am. Chem. Soc. 1989, 111, 8519–8520. 10.1021/ja00204a035. [DOI] [Google Scholar]
- Tittsworth R. C.; Hales B. J. Oxidative Titration of the Nitrogenase VFe Protein from Azotobacter vinelandii: An Example of Redox-Gated Electron Flow. Biochemistry 1996, 35, 479–487. 10.1021/bi951430i. [DOI] [PubMed] [Google Scholar]
- Arber J. M.; Dobson B. R.; Eady R. R.; Hasnain S. S.; Garner C. D.; Matsushita T.; Nomura M.; Smith B. E. Vanadium K-edge X-ray-Absorption Spectroscopy of the Functioning and Thionine-Oxidized Forms of the VFe-protein of the Vanadium Nitrogenase from Azotobacter chroococcum. Biochem. J. 1989, 258, 733–737. 10.1042/bj2580733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Eady R. R.; Lowe D. J.; Gormal C. The Vanadium-Iron Protein of Vanadium Nitrogenase from Azotobacter chroococcum contains an Iron-Vanadium Cofactor. Biochem. J. 1988, 250, 299–302. 10.1042/bj2500299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore V. G.; Tittsworth R. C.; Hales B. J. Construction and Characterization of Hybrid Component 1 from V-Nitrogenase Containing FeMo Cofactor. J. Am. Chem. Soc. 1994, 116, 12101–12102. 10.1021/ja00105a080. [DOI] [Google Scholar]
- Rebelein J. G.; Lee C. C.; Newcomb M.; Hu Y.; Ribbe M. W. Characterization of an M-Cluster-Substituted Nitrogenase VFe Protein. mBio 2018, 9, e00310-18 10.1128/mBio.00310-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benediktsson B.; Thorhallsson A. Th.; Bjornsson R. QM/MM Calculations Reveal a Bridging Hydroxo Group in a Vanadium Nitrogenase Crystal Structure. Chem. Commun. 2018, 54, 7310–7313. 10.1039/C8CC03793K. [DOI] [PubMed] [Google Scholar]
- Lehman L. J.; Roberts G. P. Identification of an Alternative Nitrogenase System in Rhodospirillum rubrum. J. Bacteriol. 1991, 173, 5705–5711. 10.1128/JB.173.18.5705-5711.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K.; Müller A.; Schramm U.; Klipp W. Demonstration of a Molybdenum- and Vanadium-Independent Nitrogenase in a nifHDK-Deletion Mutant of Rhodobacter capsulatus. Eur. J. Biochem. 1991, 195, 653–661. 10.1111/j.1432-1033.1991.tb15750.x. [DOI] [PubMed] [Google Scholar]
- Müller A.; Schneider K.; Knuttel K.; Hagen W. R. EPR Spectroscopic Characterization of an ’Iron Only’ Nitrogenase. S = 3/2 Spectrum of Component 1 Isolated from Rhodobacter capsulatus. FEBS Lett. 1992, 303, 36–40. 10.1016/0014-5793(92)80472-S. [DOI] [PubMed] [Google Scholar]
- Schneider K.; Gollan U.; Dröttboom M.; Selsemeier-Voigt S.; Müller A. Comparative Biochemical Characterization of the Iron-Only Nitrogenase and the Molybdenum Nitrogenase from Rhodobacter capsulatus. Eur. J. Biochem. 1997, 244, 789–800. 10.1111/j.1432-1033.1997.t01-1-00789.x. [DOI] [PubMed] [Google Scholar]
- Harris D. F.; Lukoyanov D. A.; Shaw S.; Compton P.; Tokmina-Lukaszewska M.; Bothner B.; Kelleher N.; Dean D. R.; Hoffman B. M.; Seefeldt L. C. Mechanism of N2 Reduction Catalyzed by Fe-Nitrogenase Involves Reductive Elimination of H2. Biochemistry 2018, 57, 701–710. 10.1021/acs.biochem.7b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüddekopf K.; Hennecke S.; Liese U.; Kutsche M.; Klipp W. Characterization of anf Genes Specific for the Alternative Nitrogenase and Identification of nif Genes Required for both Nitrogenases in Rhodobacter capsulatus. Mol. Microbiol. 1993, 8, 673–684. 10.1111/j.1365-2958.1993.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Gollan U.; Schneider K.; Muller A.; Schuddekopf K.; Klipp W. Detection of the in vivo Incorporation of a Metal Cluster into a Protein. The FeMo Cofactor is Inserted into the FeFe Protein of the Alternative Nitrogenase of Rhodobacter capsulatus. Eur. J. Biochem. 1993, 215, 25–35. 10.1111/j.1432-1033.1993.tb18003.x. [DOI] [PubMed] [Google Scholar]
- Pau R. N.; Eldridge M. E.; Lowe D. J.; Mitchenall L. A.; Eady R. R. Molybdenum-Independent Nitrogenases of Azotobacter vinelandii: a Functional Species of Alternative Nitrogenase-3 Isolated from a Molybdenum-Tolerant Strain Contains an Iron-Molybdenum Cofactor. Biochem. J. 1993, 293, 101–107. 10.1042/bj2930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.; Lehman L.; Petrovich R.; Shah V. K.; Roberts G. P.; Ludden P. W. Purification and Characterization of the Alternative Nitrogenase from the Photosynthetic Bacterium Rhodospirillum rubrum. J. Bacteriol. 1996, 178, 1445–1450. 10.1128/JB.178.5.1445-1450.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Harris D. F.; Yu Z.; Fu Y.; Poudel S.; Ledbetter R. N.; Fixen K. R.; Yang Z. Y.; Boyd E. S.; Lidstrom M. E.; et al. A Pathway for Biological Methane Production using Bacterial Iron-Only Nitrogenase. Nat. Microbiol. 2018, 3, 281–286. 10.1038/s41564-017-0091-5. [DOI] [PubMed] [Google Scholar]
- Siemann S.; Schneider K.; Dröttboom M.; Müller A. The Fe-only Nitrogenase and the Mo Nitrogenase from Rhodobacter capsulatus. A Comparative Study on the Redox Properties of the Metal Clusters Present in the Dinitrogenase Components. Eur. J. Biochem. 2002, 269, 1650–1661. 10.1046/j.1432-1327.2002.02804.x. [DOI] [PubMed] [Google Scholar]
- Hales B. J.; Case E. E. Nitrogen Fixation by Azotobacter vinelandii in Tungsten-containing Medium. J. Biol. Chem. 1987, 262, 16205–16211. [PubMed] [Google Scholar]
- Siemann S.; Schneider K.; Oley M.; Müller A. Characterization of a Tungsten-Substituted Nitrogenase Isolated from Rhodobacter capsulatus. Biochemistry 2003, 42, 3846–3857. 10.1021/bi0270790. [DOI] [PubMed] [Google Scholar]
- Premakumar R.; Chisnell J. R.; Bishop P. E. A Comparison of the Three Dinitrogenase Reductases Expressed by Azotobacter vinelandii. Can. J. Microbiol. 1989, 35, 344–348. 10.1139/m89-053. [DOI] [Google Scholar]
- Rohde M.; Trncik C.; Sippel D.; Gerhardt S.; Einsle O. Crystal Structure of VnfH, the Iron Protein Component of Vanadium Nitrogenase. JBIC, J. Biol. Inorg. Chem. 2018, 23, 1049–1056. 10.1007/s00775-018-1602-4. [DOI] [PubMed] [Google Scholar]
- Eady R. R.; Richardson T. H.; Miller R. W.; Hawkins M.; Lowe D. J. The Vanadium Nitrogenase of Azotobacter chroococcum. Purification and Properties of the Fe Protein. Biochem. J. 1988, 256, 189–196. 10.1042/bj2560189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller C. J.; Stiebritz M. T.; Lee C. C.; Liedtke J.; Hu Y. Tuning Electron Flux through Nitrogenase with Methanogen Iron Protein Homologues. Chem. - Eur. J. 2017, 23, 16152–16156. 10.1002/chem.201704378. [DOI] [PubMed] [Google Scholar]
- Blank M. A.; Lee C. C.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Structural Models of the [Fe4S4] Clusters of Homologous Nitrogenase Fe Proteins. Inorg. Chem. 2011, 50, 7123–7128. 10.1021/ic200636k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burén S.; Jiménez-Vicente E.; Echavarri-Erasun C.; Rubio L. M.. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 10.1021/acs.chemrev.9b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. C.; Dos Santos P. C.; Dean D. R. NifU and NifS are Required for the Maturation of Nitrogenase and Cannot Replace the Function of isc-Gene Products in Azotobacter vinelandii. Biochem. Soc. Trans. 2005, 33, 90–93. 10.1042/BST0330090. [DOI] [PubMed] [Google Scholar]
- Jacobson M. R.; Brigle K. E.; Bennett L. T.; Setterquist R. A.; Wilson M. S.; Cash V. L.; Beynon J.; Newton W. E.; Dean D. R. Physical and Genetic Map of the Major nif Gene Cluster from Azotobacter vinelandii. J. Bacteriol. 1989, 171, 1017–1027. 10.1128/JB.171.2.1017-1027.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy C.; Dean D. The nifU, nifS and nifV Gene Products are Required for Activity of all Three Nitrogenases of Azotobacter vinelandii. Mol. Gen. Genet. 1992, 231, 494–498. 10.1007/BF00292722. [DOI] [PubMed] [Google Scholar]
- Smith A. D.; Jameson G. N.; Dos Santos P. C.; Agar J. N.; Naik S.; Krebs C.; Frazzon J.; Dean D. R.; Huynh B. H.; Johnson M. K. NifS-Mediated Assembly of [4Fe-4S] Clusters in the N- and C-Terminal Domains of the NifU Scaffold Protein. Biochemistry 2005, 44, 12955–12969. 10.1021/bi051257i. [DOI] [PubMed] [Google Scholar]
- Dos Santos P. C.; Smith A. D.; Frazzon J.; Cash V. L.; Johnson M. K.; Dean D. R. Iron-Sulfur Cluster Assembly. NifU-directed Activation of the Nitrogenase Fe Protein. J. Biol. Chem. 2004, 279, 19705–19711. 10.1074/jbc.M400278200. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Curatti L.; Rubio L. M. Evidence for nifU and nifS Participation in the Biosynthesis of the Iron-Molybdenum Cofactor of Nitrogenase. J. Biol. Chem. 2007, 282, 37016–37025. 10.1074/jbc.M708097200. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Agar J. N.; Smith A. D.; Frazzon J.; Dean D. R.; Huynh B. H.; Johnson M. K. IscA, an Alternate Scaffold for Fe-S Cluster Biosynthesis. Biochemistry 2001, 40, 14069–14080. 10.1021/bi015656z. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Fay A. W.; Lee C. C.; Ribbe M. W. P-cluster Maturation on Nitrogenase MoFe Protein. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10424–10429. 10.1073/pnas.0704297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Vicente E.; Yang Z.-Y.; Ray W. K.; Echavarri-Erasun C.; Cash V. L.; Rubio L. M.; Seefeldt L. C.; Dean D. R. Sequential and Differential Interaction of Assembly Factors during Nitrogenase MoFe Protein Maturation. J. Biol. Chem. 2018, 293, 9812–9823. 10.1074/jbc.RA118.002994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxen J.; Arragain S.; Scandurra A. A.; Jimenez-Vicente E.; Echavarri-Erasun C.; Pollmann S.; Britt R. D.; Rubio L. M. Electron Paramagnetic Resonance Characterization of Three Iron-Sulfur Clusters Present in the Nitrogenase Cofactor Maturase NifB from Methanocaldococcus infernus. J. Am. Chem. Soc. 2016, 138, 7468–7471. 10.1021/jacs.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Lee C. C.; Ribbe M. W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-cluster. Science 2012, 337, 1672–1675. 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Ribbe M. W. Refining the Pathway of Carbide Insertion into the Nitrogenase M-cluster. Nat. Commun. 2015, 6, 8034. 10.1038/ncomms9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanifuji K.; Lee C. C.; Sickerman N. S.; Tatsumi K.; Ohki Y.; Hu Y.; Ribbe M. W. Tracing the ’Ninth Sulfur’ of the Nitrogenase Cofactor via a Semi-synthetic Approach. Nat. Chem. 2018, 10, 568–572. 10.1038/s41557-018-0029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah V. K.; Allen J. R.; Spangler N. J.; Ludden P. W. In vitro Synthesis of the Iron-Molybdenum Cofactor of Nitrogenase. Purification and Characterization of NifB Cofactor, the Product of NIFB Protein. J. Biol. Chem. 1994, 269, 1154–1158. [PubMed] [Google Scholar]
- Hernandez J. A.; Igarashi R. Y.; Soboh B.; Curatti L.; Dean D. R.; Ludden P. W.; Rubio L. M. NifX and NifEN Exchange NifB Cofactor and the VK-cluster, a Newly Isolated Intermediate of the Iron-Molybdenum Cofactor Biosynthetic Pathway. Mol. Microbiol. 2007, 63, 177–192. 10.1111/j.1365-2958.2006.05514.x. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Fay A. W.; Ribbe M. W. Identification of a Nitrogenase FeMo Cofactor Precursor on NifEN Complex. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3236–3241. 10.1073/pnas.0409201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; White R. H.; Dean D. R. Purification of the Azotobacter vinelandii nifV-Encoded Homocitrate Synthase. J. Bacteriol. 1997, 179, 5963–5966. 10.1128/JB.179.18.5963-5966.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Corbett M. C.; Fay A. W.; Webber J. A.; Hodgson K. O.; Hedman B.; Ribbe M. W. Nitrogenase Fe Protein: A Molybdate/Homocitrate Insertase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17125–17130. 10.1073/pnas.0602651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R. M.; Homer M. J.; Chatterjee R.; Ludden P. W.; Roberts G. P.; Shah V. K. Dinitrogenase Reductase- and MgATP-dependent Maturation of Apodinitrogenase from Azotobacter vinelandii. J. Biol. Chem. 1993, 268, 23670–23674. [PubMed] [Google Scholar]
- Rubio L. M.; Rangaraj P.; Homer M. J.; Roberts G. P.; Ludden P. W. Cloning and Mutational Analysis of the γ Gene from Azotobacter vinelandii Defines a New Family of Proteins Capable of Metallocluster Binding and Protein Stabilization. J. Biol. Chem. 2002, 277, 14299–14305. 10.1074/jbc.M107289200. [DOI] [PubMed] [Google Scholar]
- Jacobson M. R.; Cash V. L.; Weiss M. C.; Laird N. F.; Newton W. E.; Dean D. R. Biochemical and Genetic Analysis of the nifUSVWZM Cluster from Azotobacter vinelandii. Mol. Gen. Genet. 1989, 219, 49–57. 10.1007/BF00261156. [DOI] [PubMed] [Google Scholar]
- Roberts G. P.; MacNeil T.; MacNeil D.; Brill W. J. Regulation and Characterization of Protein Products Coded by the nif (Nitrogen Fixation) Genes of Klebsiella pneumoniae. J. Bacteriol. 1978, 136, 267–279. 10.1128/JB.136.1.267-279.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; White R. H.; Cash V. L.; Dean D. R. Mechanism for the Desulfurization of L-Cysteine Catalyzed by the nifS Gene Product. Biochemistry 1994, 33, 4714–4720. 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- Zheng L.; White R. H.; Cash V. L.; Jack R. F.; Dean D. R. Cysteine Desulfurase Activity Indicates a Role for NIFS in Metallocluster Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2754–2758. 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L.; Dean D. R. Catalytic Formation of a Nitrogenase Iron-Sulfur Cluster. J. Biol. Chem. 1994, 269, 18723–18726. [PubMed] [Google Scholar]
- Fu W.; Jack R. F.; Morgan T. V.; Dean D. R.; Johnson M. K. nifU Gene Product from Azotobacter vinelandii Is a Homodimer That Contains Two Identical [2Fe-2S] Clusters. Biochemistry 1994, 33, 13455–13463. 10.1021/bi00249a034. [DOI] [PubMed] [Google Scholar]
- Garg R. P.; Vargo C. J.; Cui X.; Kurtz D. M. Jr. A [2Fe-2S] Protein Encoded by an Open Reading Frame Upstream of the Escherichia coli Bacterioferritin Gene. Biochemistry 1996, 35, 6297–6301. 10.1021/bi9600862. [DOI] [PubMed] [Google Scholar]
- Quail M. A.; Jordan P.; Grogan J. M.; Butt J. N.; Lutz M.; Thomson A. J.; Andrews S. C.; Guest J. R. Spectroscopic and Voltammetric Characterisation of the Bacterioferritin-Associated Ferredoxin of Escherichia coli. Biochem. Biophys. Res. Commun. 1996, 229, 635–642. 10.1006/bbrc.1996.1856. [DOI] [PubMed] [Google Scholar]
- Agar J. N.; Yuvaniyama P.; Jack R. F.; Cash V. L.; Smith A. D.; Dean D. R.; Johnson M. K. Modular Organization and Identification of a Mononuclear Iron-Binding Site within the NifU Protein. JBIC, J. Biol. Inorg. Chem. 2000, 5, 167–177. 10.1007/s007750050361. [DOI] [PubMed] [Google Scholar]
- Yuvaniyama P.; Agar J. N.; Cash V. L.; Johnson M. K.; Dean D. R. NifS-directed Assembly of a Transient [2Fe-2S] Cluster within the NifU Protein. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 599–604. 10.1073/pnas.97.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R. M.; Chatterjee R.; Ludden P. W.; Shah V. K. Incorporation of Iron and Sulfur from NifB Cofactor into the Iron-Molybdenum Cofactor of Dinitrogenase. J. Biol. Chem. 1995, 270, 26890–26896. 10.1074/jbc.270.45.26890. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Echavarri-Erasun C.; Demuez M.; Jiménez-Vicente E.; Bominaar E. L.; Rubio L. M. The Nitrogenase FeMo-Cofactor Precursor Formed by NifB Protein: A Diamagnetic Cluster Containing Eight Iron Atoms. Angew. Chem., Int. Ed. 2016, 55, 12764–12767. 10.1002/anie.201606447. [DOI] [PubMed] [Google Scholar]
- Wiig J. A.; Hu Y.; Ribbe M. W. NifEN-B Complex of Azotobacter vinelandii is Fully Functional in Nitrogenase FeMo Cofactor Assembly. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 8623–8627. 10.1073/pnas.1102773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Wiig J. A.; Lee C. C.; Hu Y. Identification and Characterization of Functional Homologs of Nitrogenase Cofactor Biosynthesis Protein NifB from Methanogens. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 14829–14833. 10.1073/pnas.1510409112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettberg L. A.; Wilcoxen J.; Lee C. C.; Stiebritz M. T.; Tanifuji K.; Britt R. D.; Hu Y. Probing the Coordination and Function of Fe4S4 Modules in Nitrogenase Assembly Protein NifB. Nat. Commun. 2018, 9, 2824. 10.1038/s41467-018-05272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasniewski A. J.; Wilcoxen J.; Tanifuji K.; Hedman B.; Hodgson K. O.; Britt R. D.; Hu Y.; Ribbe M. W. Spectroscopic Characterization of an Eight-Iron Nitrogenase Cofactor Precursor That Lacks the ″9th Sulfur″. Angew. Chem., Int. Ed. 2019, 58, 14703–14707. 10.1002/anie.201907593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigle K. E.; Weiss M. C.; Newton W. E.; Dean D. R. Products of the Iron-Molybdenum Cofactor-Specific Biosynthetic Genes, nifE and nifN, Are Structurally Homologous to the Products of the Nitrogenase Molybdenum-Iron Protein Genes, nifD and nifK. J. Bacteriol. 1987, 169, 1547–1553. 10.1128/JB.169.4.1547-1553.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paustian T. D.; Shah V. K.; Roberts G. P. Purification and Characterization of the nifN and nifE Gene Products from Azotobacter vinelandii Mutant UW45. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 6082–6086. 10.1073/pnas.86.16.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster K. M.; Hu Y.; Bergmann U.; Ribbe M. W.; DeBeer S. X-ray Spectroscopic Observation of an Interstitial Carbide in NifEN-Bound FeMoco Precursor. J. Am. Chem. Soc. 2013, 135, 610–612. 10.1021/ja309254g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett M. C.; Hu Y.; Fay A. W.; Ribbe M. W.; Hedman B.; Hodgson K. O. Structural Insights into a Protein-bound Iron-Molybdenum Cofactor Precursor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1238–1243. 10.1073/pnas.0507853103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. T.; Hu Y.; Wiig J. A.; Rees D. C.; Ribbe M. W. Structure of Precursor-Bound NifEN: A Nitrogenase FeMo Cofactor Maturase/Insertase. Science 2011, 331, 91–94. 10.1126/science.1196954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Blank M. A.; Lee C. C.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Spectroscopic Characterization of the Isolated Iron-Molybdenum Cofactor (FeMoco) Precursor from the Protein NifEN. Angew. Chem., Int. Ed. 2011, 50, 7787–7790. 10.1002/anie.201102724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik K.; Hu Y.; Fay A. W.; Ribbe M. W.; Hales B. J. Variable-temperature, Variable-field Magnetic Circular Dichroism Spectroscopic Study of NifEN-bound Precursor and ″FeMoco″. JBIC, J. Biol. Inorg. Chem. 2011, 16, 325–332. 10.1007/s00775-010-0728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Corbett M. C.; Fay A. W.; Webber J. A.; Hodgson K. O.; Hedman B.; Ribbe M. W. FeMo Cofactor Maturation on NifEN. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 17119–17124. 10.1073/pnas.0602647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboh B.; Igarashi R. Y.; Hernandez J. A.; Rubio L. M. Purification of a NifEN Protein Complex That Contains Bound Molybdenum and a FeMo-Co Precursor from an Azotobacter vinelandii ΔnifHDK Strain. J. Biol. Chem. 2006, 281, 36701–36709. 10.1074/jbc.M606820200. [DOI] [PubMed] [Google Scholar]
- George S. J.; Igarashi R. Y.; Piamonteze C.; Soboh B.; Cramer S. P.; Rubio L. M. Identification of a Mo-Fe-S Cluster on NifEN by Mo K-Edge Extended X-ray Absorption Fine Structure. J. Am. Chem. Soc. 2007, 129, 3060–3061. 10.1021/ja0663428. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Yoshizawa J. M.; Fay A. W.; Lee C. C.; Wiig J. A.; Ribbe M. W. Catalytic Activities of NifEN: Implications for Nitrogenase Evolution and Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 16962–16966. 10.1073/pnas.0907872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa J. M.; Blank M. A.; Fay A. W.; Lee C. C.; Wiig J. A.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Optimization of FeMoco Maturation on NifEN. J. Am. Chem. Soc. 2009, 131, 9321–9325. 10.1021/ja9035225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa J. M.; Fay A. W.; Lee C. C.; Hu Y.; Ribbe M. W. Insertion of Heterometals into the NifEN-associated Iron-Molybdenum Cofactor Precursor. JBIC, J. Biol. Inorg. Chem. 2010, 15, 421–428. 10.1007/s00775-009-0614-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay A. W.; Blank M. A.; Yoshizawa J. M.; Lee C. C.; Wiig J. A.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Formation of a Homocitrate-free Iron-Molybdenum Cluster on NifEN: Implications for the Role of Homocitrate in Nitrogenase Assembly. Dalton Trans. 2010, 39, 3124–3130. 10.1039/c000264j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Brosius M. A.; Burgess B. K. Construction of a Form of the MoFe Protein of Nitrogenase That Accepts Electrons from the Fe Protein but Does Not Reduce Substrate. J. Biol. Chem. 1996, 271, 10528–10532. 10.1074/jbc.271.18.10528. [DOI] [PubMed] [Google Scholar]
- Gavini N.; Ma L.; Watt G.; Burgess B. K. Purification and Characterization of a FeMo Cofactor-Deficient MoFe Protein. Biochemistry 1994, 33, 11842–11849. 10.1021/bi00205a021. [DOI] [PubMed] [Google Scholar]
- Hawkes T. R.; Smith B. E. Purification and Characterization of the Inactive MoFe Protein (NifB–Kp1) of the Nitrogenase from nifB Mutants of Klebsiella pneumoniae. Biochem. J. 1983, 209, 43–50. 10.1042/bj2090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; Blank M. A.; Fay A. W.; Yoshizawa J. M.; Hu Y.; Hodgson K. O.; Hedman B.; Ribbe M. W. Stepwise Formation of P-cluster in Nitrogenase MoFe Protein. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18474–18478. 10.1073/pnas.0909149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik K.; Lee C. C.; Wiig J. A.; Hu Y.; Ribbe M. W.; Hales B. J. Nonenzymatic Synthesis of the P-cluster in the Nitrogenase MoFe Protein: Evidence of the Involvement of All-Ferrous [Fe4S4]0 Intermediates. Biochemistry 2014, 53, 1108–1116. 10.1021/bi401699u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik K.; Lee C. C.; Hu Y.; Ribbe M. W.; Hales B. J. A VTVH MCD and EPR Spectroscopic Study of the Maturation of the “Second” Nitrogenase P-Cluster. Inorg. Chem. 2018, 57, 4719–4725. 10.1021/acs.inorgchem.8b00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Vicente E.; Yang Z.-Y.; Martin Del Campo J. S.; Cash V. L.; Seefeldt L. C.; Dean D. R. The NifZ Accessory Protein has an Equivalent Function in Maturation of both Nitrogenase MoFe Protein P-clusters. J. Biol. Chem. 2019, 294, 6204–6213. 10.1074/jbc.RA119.007905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A. C.; Dean D. R.; Burgess B. K. Iron-Molybdenum Cofactor Biosynthesis in Azotobacter vinelandii Requires the Iron Protein of Nitrogenase. J. Biol. Chem. 1987, 262, 14327–14332. [PubMed] [Google Scholar]
- Robinson A. C.; Chun T. W.; Li J. G.; Burgess B. K. Iron-Molybdenum Cofactor Insertion into the Apo-MoFe Protein of Nitrogenase Involves the Iron Protein-MgATP Complex. J. Biol. Chem. 1989, 264, 10088–10095. [PubMed] [Google Scholar]
- Paustian T. D.; Shah V. K.; Roberts G. P. Apodinitrogenase: Purification, Association with a 20-Kilodalton Protein, and Activation by the Iron-Molybdenum Cofactor in the Absence of Dinitrogenase Reductase. Biochemistry 1990, 29, 3515–3522. 10.1021/bi00466a014. [DOI] [PubMed] [Google Scholar]
- Rangaraj P.; Shah V. K.; Ludden P. W. ApoNifH Functions in Iron-Molybdenum Cofactor Synthesis and Apodinitrogenase Maturation. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 11250–11255. 10.1073/pnas.94.21.11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraj P.; Ryle M. J.; Lanzilotta W. N.; Ludden P. W.; Shah V. K. In vitro Biosynthesis of Iron-Molybdenum Cofactor and Maturation of the nif-encoded Apodinitrogenase. Effect of Substitution for NifH with Site-specifically Altered Forms of NifH. J. Biol. Chem. 1999, 274, 19778–19784. 10.1074/jbc.274.28.19778. [DOI] [PubMed] [Google Scholar]
- Berman J.; Gershoni J. M.; Zamir A. Expression of Nitrogen Fixation Genes in Foreign Hosts. Assembly of Nitrogenase Fe Protein in Escherichia coli and in yeast. J. Biol. Chem. 1985, 260, 5240–5243. [PubMed] [Google Scholar]
- Howard K. S.; McLean P. A.; Hansen F. B.; Lemley P. V.; Koblan K. S.; Orme-Johnson W. H. Klebsiella pneumoniae nifM Gene Product Is Required for Stabilization and Activation of Nitrogenase Iron Protein in Escherichia coli. J. Biol. Chem. 1986, 261, 772–778. [PubMed] [Google Scholar]
- Gavini N.; Tungtur S.; Pulakat L. Peptidyl-Prolyl cis/trans Isomerase-Independent Functional nifH Mutant of Azotobacter vinelandii. J. Bacteriol. 2006, 188, 6020–6025. 10.1128/JB.00379-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Torrejón G.; Jiménez-Vicente E.; Buesa J. M.; Hernandez J. A.; Verma H. K.; Rubio L. M. Expression of a Functional Oxygen-labile Nitrogenase Component in the Mitochondrial Matrix of Aerobically Grown Yeast. Nat. Commun. 2016, 7, 11426. 10.1038/ncomms11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperial J.; Ugalde R. A.; Shah V. K.; Brill W. J. Role of the nifQ Gene Product in the Incorporation of Molybdenum into Nitrogenase in Klebsiella pneumoniae. J. Bacteriol. 1984, 158, 187–194. 10.1128/JB.158.1.187-194.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde R. A.; Imperial J.; Shah V. K.; Brill W. J. Biosynthesis of the Iron-Molybdenum Cofactor and the Molybdenum Cofactor in Klebsiella pneumoniae: Effect of Sulfur Source. J. Bacteriol. 1985, 164, 1081–1087. 10.1128/JB.164.3.1081-1087.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J. A.; Curatti L.; Aznar C. P.; Perova Z.; Britt R. D.; Rubio L. M. Metal trafficking for Nitrogen Fixation: NifQ Donates Molybdenum to NifEN/NifH for the Biosynthesis of the Nitrogenase FeMo-cofactor. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11679–11684. 10.1073/pnas.0803576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T.; Tomita T.; Wulandari A. P.; Kuzuyama T.; Nishiyama M. Mechanism of Substrate Recognition and Insight into Feedback Inhibition of Homocitrate Synthase from Thermus thermophilus. J. Biol. Chem. 2010, 285, 4195–4205. 10.1074/jbc.M109.086330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfer S. L.; Scott E. M.; Pillus L.; Trievel R. C. Structural Basis for L-Lysine Feedback Inhibition of Homocitrate Synthase. J. Biol. Chem. 2010, 285, 10446–10453. 10.1074/jbc.M109.094383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masepohl B.; Angermüller S.; Hennecke S.; Hübner P.; Moreno-Vivian C.; Klipp W. Nucleotide Sequence and Genetic Analysis of the Rhodobacter capsulatus ORF6-nifUISVW Gene Region: Possible Role of NifW in Homocitrate Processing. Mol. Gen. Genet. 1993, 238, 369–382. 10.1007/BF00291996. [DOI] [PubMed] [Google Scholar]
- Kim S.; Burgess B. K. Evidence for the Direct Interaction of the nifW Gene Product with the MoFe Protein. J. Biol. Chem. 1996, 271, 9764–9770. 10.1074/jbc.271.16.9764. [DOI] [PubMed] [Google Scholar]
- Moreno-Vivian C.; Schmehl M.; Masepohl B.; Arnold W.; Klipp W. DNA Sequence and Genetic Analysis of the Rhodobacter capsulatus nifENX Gene Region: Homology Between NifX and NifB Suggests Involvement of NifX in Processing of the Iron-Molybdenum Cofactor. Mol. Gen. Genet. 1989, 216, 353. 10.1007/BF00334376. [DOI] [PubMed] [Google Scholar]
- Rangaraj P.; Rüttimann-Johnson C.; Shah V. K.; Ludden P. W. Accumulation of 55Fe-Labeled Precursors of the Iron-Molybdenum Cofactor of Nitrogenase on NifH and NifX of Azotobacter vinelandii. J. Biol. Chem. 2001, 276, 15968–15974. 10.1074/jbc.M100907200. [DOI] [PubMed] [Google Scholar]
- Rangaraj P.; Ludden P. W. Accumulation of 99Mo-containing Iron-Molybdenum Cofactor Precursors of Nitrogenase on NifNE, NifH, and NifX of Azotobacter vinelandii. J. Biol. Chem. 2002, 277, 40106–40111. 10.1074/jbc.M204581200. [DOI] [PubMed] [Google Scholar]
- Wolfinger E. D.; Bishop P. E. Nucleotide Sequence and Mutational Analysis of the vnfENX Region of Azotobacter vinelandii. J. Bacteriol. 1991, 173, 7565–7572. 10.1128/JB.173.23.7565-7572.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüttimann-Johnson C.; Staples C. R.; Rangaraj P.; Shah V. K.; Ludden P. W. A Vanadium and Iron Cluster Accumulates on VnfX during Iron-Vanadium-Cofactor Synthesis for the Vanadium Nitrogenase in Azotobacter vinelandii. J. Biol. Chem. 1999, 274, 18087–18092. 10.1074/jbc.274.25.18087. [DOI] [PubMed] [Google Scholar]
- White T. C.; Harris G. S.; Orme-Johnson W. H. Electrophoretic Studies on the Assembly of the Nitrogenase Molybdenum-Iron Protein from the Klebsiella pneumoniae nifD and nifK Gene Products. J. Biol. Chem. 1992, 267, 24007–24016. [PubMed] [Google Scholar]
- Homer M. J.; Paustian T. D.; Shah V. K.; Roberts G. P. The nifY Product of Klebsiella pneumoniae Is Associated with Apodinitrogenase and Dissociates upon Activation with the Iron-Molybdenum Cofactor. J. Bacteriol. 1993, 175, 4907–4910. 10.1128/JB.175.15.4907-4910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraj P.; Ryle M. J.; Lanzilotta W. N.; Goodwin P. J.; Dean D. R.; Shah V. K.; Ludden P. W. Inhibition of Iron-Molybdenum Cofactor Biosynthesis by L127Δ NifH and Evidence for a Complex Formation Between L127Δ NifH and NifNE. J. Biol. Chem. 1999, 274, 29413–29419. 10.1074/jbc.274.41.29413. [DOI] [PubMed] [Google Scholar]