

Abstract
Heterogeneous catalysts offer a cyclable platform for exploring efficient transformation systems, and their promising applications underpin a broad research interest. Covalent organic frameworks (COFs) are a class of crystalline porous networks that can integrate organic units into ordered skeletons and pores, offering an insoluble and robust platform for exploring heterogeneous catalysts. In this Outlook, we describe a conceptual scheme for designing catalytic COFs to promote various transformations. We summarize the general strategy for designing COFs to construct tailor-made skeletons and pores by emphasizing their structural uniqueness. We introduce different approaches to develop catalytic functions by sampling COFs into four regimes, i.e., skeletons, walls, pores, and systematically organized systems. We scrutinize their catalytic features and elucidate interplays with electrons, holes, and molecules by highlighting the key role of interface design in exploring catalytic COFs. We further envisage the key issues to be challenged, future research directions, and perspectives to show a full picture of designer heterogeneous catalysis based on COFs.
Short abstract
Exploring covalent organic frameworks to construct heterogeneous catalysts is summarized by introducing design principle, scrutinizing the current status, and evaluating challenges and opportunities.
Introduction
Organic transformation is key to the chemical industry and adds its irreplaceable values to the formation of an affluent society. However, most reactions are energy demanding, sharing a great part of world energy consumption. How to explore a catalyst that is stable, recyclable, and efficient is a key fundamental issue in chemistry.1,2 In this context, the heterogeneous catalyst is one of the most important transformation technologies in the chemical industry; it’s great potential underpins an increasing attention in developing new heterogeneous catalysts. Traditionally, inorganic and polymeric matrixes have been widely studied for loading catalysts to immobilize on the surface.3,4 However, these heterogeneous catalysts encounter problems of low efficiency as a result of site hiding, random and amorphous distribution, and leakage of active sites. Recently, a representative molecular platform, i.e., crystalline porous covalent organic frameworks (COFs), emerges as predesignable heterogeneous catalysts. They are stable and insoluble and allow for the full use of both the skeleton and pores to design various catalytic systems.5−7
COFs are a class of crystalline porous molecular frameworks that enable the covalent integration of organic units into ordered polymer backbones to create two- and three-dimensional (2D and 3D) structures with built-in pores. The growth of 2D and 3D polymer networks is guided by the topology diagram whereby the monomers with matched geometries are connected into polygonal backbones to construct different skeletons and specific pores (Figure 1).5−7 This design principle opens a chance to explore COFs as heterogeneous catalysts to promote chemical reactions. Compared to other crystalline frameworks and amorphous polymers, COFs are fascinating as they can merge a series of structural features including stability, porosity, designability, and tunability in one material. Owing to structural orderings, COFs allow for the precise installation of molecular catalysts to create a well-defined structure with a specific local interface including electron density, orientation, and spatial confinement. These characters are quite similar to those of homogeneous catalysts so that COFs can retain the possibility of designing reaction pathway and enhancing selectivity. COFs also feature a heterogeneous nature in which ordered skeletons facilitate charge carrier transport and open porous structures promote mass transport while their stability and insolubility allow for cycle use. Therefore, COF-based catalysts are unique in that they can merge the advantages of both homogeneous and heterogeneous catalysts into one material.
Figure 1.
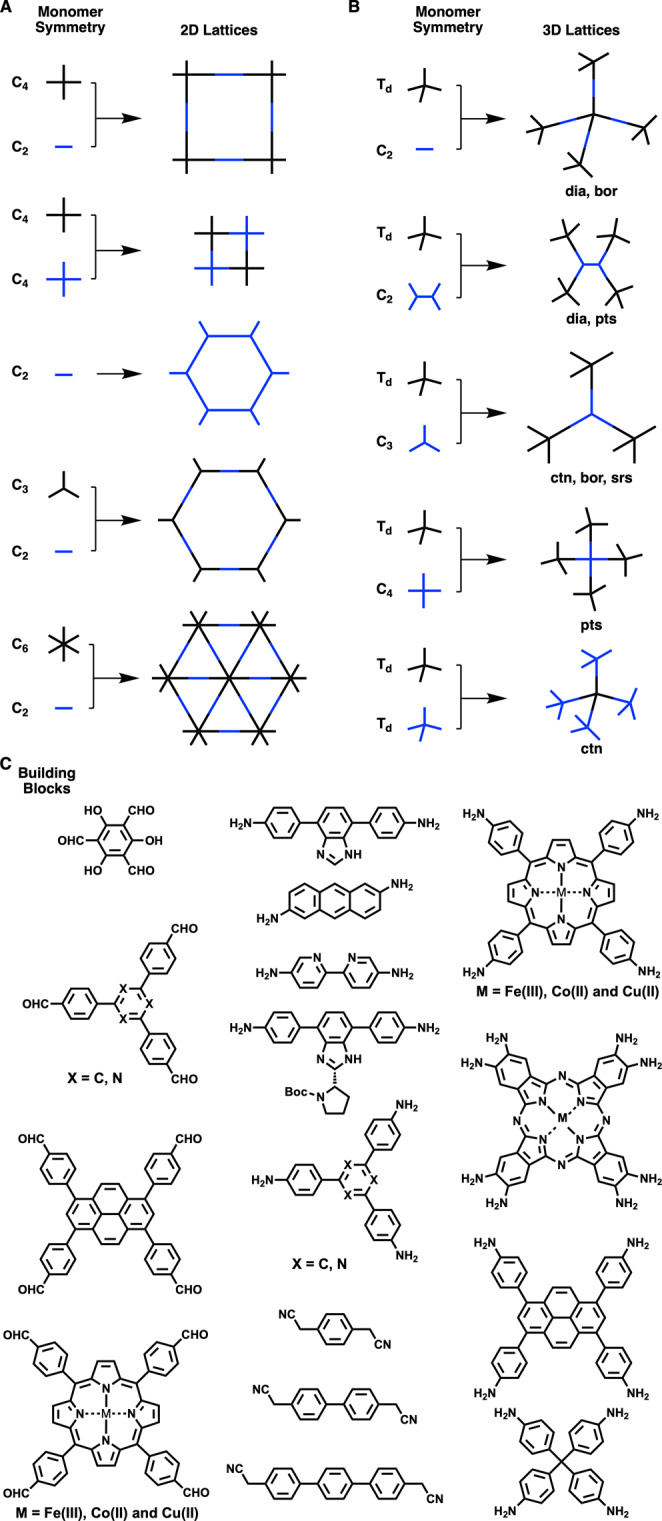
Basic topological diagrams for the design of (A) 2D and (B) 3D COFs. (C) Typical units for the synthesis of catalytic COFs.
Owing to the diversity of skeletons and pores, COFs can be developed using different chemistries to construct a diversity of heterogeneous catalysts (Figure 2). In this Outlook, we focus on scrutinizing catalytic functions of COFs, summarize design principle of different approaches, and disclose the origin of catalytic activity and selectivity by elucidating interactions with photon, exciton, electron, hole, ions, and molecules. We further outline the key fundamental issues to be addressed and point out future directions on exploring efficient, robust, and recyclable heterogeneous catalysts that are both important for basic research and applications.
Figure 2.
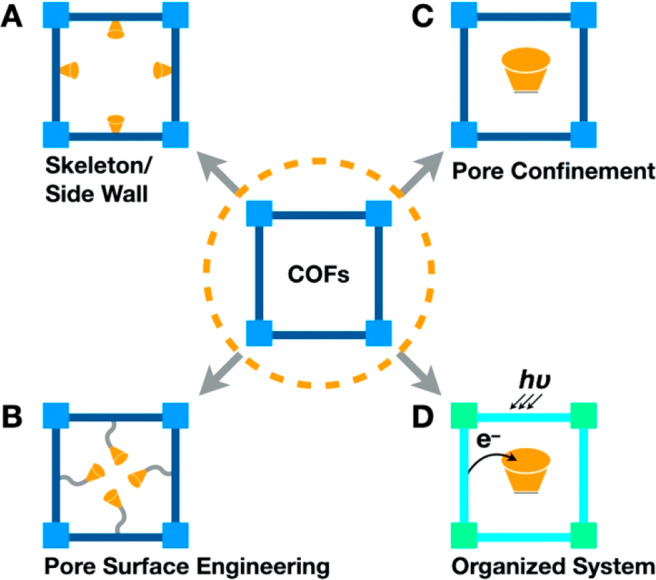
Designing COFs for heterogeneous catalysis based on (A) skeleton and side wall, (B) pore surface engineering, (C) pore confinement, and (D) systematically organized systems.
Design Principle
COFs are designed by a topology diagram whereby monomers are connected in a predetermined manner by covalent bonds to develop 2D or 3D skeletons and to form inherent pores.5−7 In 2D COFs, the 2D polymer networks stack to form layer skeletons and one-dimensional (1D) open channels in which the spatial separation between neighboring layers is controlled by π–π interactions and is usually 3–5 Å. This layered structure offers the chance to develop photocatalysts and electrocatalysts as the preorganized columnar π arrays provide pathways for facilitating exciton, electron, and hole transport that are key to photochemical and electrochemical processes (Figure 2A). On the other hand, the stacking structure would cover catalytic sites and leaves only those on the surface layers accessible to reactions; this reflects that 2D COFs is not suitable to directly integrate the catalytic sites to the π columns, especially exploring the focal point of the π backbones as catalytic centers should be avoided. Nevertheless, there is still a great possibility of developing the skeletons to serve as catalytic centers as π columns form pore walls in the 1D channels. For example, a general strategy is fixing catalysts to the side groups of monomers to construct 2D COFs, which would enable the integration of catalytic sites onto the pore walls (Figure 2B). This approach enables the pore surface engineering of various heterogeneous catalytic systems. For 3D COFs, the interpenetration structure impedes the direct use of backbones as catalytic centers, while the small pore size usually in the micropore range will also limit the scope of catalysts that can be integrated to the pore walls.8,9
The open pores of COFs allow for physical loading of various catalysts ranging from metal nanoparticles to enzymes, which construct catalytic systems by exploring confinement effects in the well-defined pores with precise local environments, as pore size, pore shape, and pore environment can be fully designed and controlled (Figure 2C). This constitutes a unique nanoreactor as each pore is independent from neighboring ones without mutual interference.
Chemical reactions usually involve multielectron process and this requires efficient transport of electrons to the reaction centers. COFs are extremely powerful to constitute such multielectron catalytic systems as the designability of both skeleton and pore enables a seamless connection of multiple processes into a systematic working package to allow a smooth electron flow. Therefore, COFs offer a platform for designing molecular systems in which various processes can be synergistically connected and interfaced for promoting reactions by reducing electron loss between steps (Figure 2D).
These considerations are based on the structural features and offer the fundamental concept in designing COFs-based heterogeneous catalysis. The design of catalytic COFs requires not only the design of the catalytic sites but the control of the interface as well. The interface design in COFs includes the electronic interface design associated with exciton migration, charge carrier separation and transfer, supramolecular interface design for setting the environment around catalytic centers, and pore interface for promoting mass transports. Especially, the electronic interface involves π units, polygonal topology, linkages, and molecular orbital energies of the frameworks, while the local interface treats the geometry, electronic and redox state, and spatial confinement of each catalytic site and the pore interface comprises pore size, shape, wall structure, and connectivity. These designs are distinct from those of molecular catalysts as COFs-based systems consist of more systematic control over a long distance and time range.
Compared to metal organic frameworks (MOFs),10−12 COFs are different in three aspects. (1) COFs are constructed with covalent bonds to attain enough stability for keeping crystalline porous structures in various solvents, including coordinative pyridine, dimethyl sulfoxide, and imidazole as well as boiling water, strong acid (concentrated HCl), and strong base (NaOH or KOH, 14 M).7 (2) COFs consist of a layered structure and 1D channels, which are sustained by intralayer π–π interactions; exfoliation is possible only under strong external power and yields few layer nanosheets, which retain the basic structural features of COFs. (3) The covalently linked catalytic sites hardly leak from the frameworks.
Catalytic Systems
Based on the above design principles, we have sampled COFs-based catalysts into four different structural origins, i.e., (1) skeleton design, (2) pore surface engineering, (3) pore confinement, and (4) systematic organization (Figure 2). These catalysts have been designed with different principles and applied for different types of transformations.
Catalysis Based on Backbones
COFs have been designed by exploring the skeleton π systems and the side walls to explore catalytic activity. For the skeleton π systems, the catalytic activity is based on the aligned π columns to enhance light absorption, exciton migration, and electron transport, which are key to the photochemical and electrochemical reactions. The direct use of residue units on the side walls and the postsynthetic metalation of coordinative sites on side walls can offer two different ways to explore specific interactions between the walls and reactants, which is key to catalysis on the side walls. For details, skeleton π systems have been developed for photocatalysis and electrocatalysts while the side walls have been developed for catalyzing various addition reactions.
Catalysts Based on π Skeletons
The first example for demonstrating photocatalysis based on π skeletons is a squaraine-linked CuP-SQ-COF (Figure 3A),13 which enables an extended π conjugation over the tetragonal lattice while the π stack forms layered CuP columns across the material. The extended π conjugation greatly extends a broad electronic absorption band from 300 to 700 nm while the π stack enhances the Q-band absorption to 2.63-fold that of monomeric CuP. From cyclic voltammetry, the CuP-SQ COF has a LUMO and HOMO level of −4.0 and −5.7 eV, respectively, yielding a band gap of 1.7 eV. Compared to the monomeric CuP, the HOMO level is increased by 0.3 eV, suggesting that the π clouds are more delocalized over the 2D skeletons. These results indicate a general strategy for extending π conjugation over the 2D COFs by using π conjugated linkages.
Figure 3.

Schematics of (A) CuP-SQ COF (A), (B) COF-366-Co, and (C) COF-367-M for catalysis on π skeletons.
Interestingly, the CuP-SQ COF harvests a broad range of visible light and serves as a photocatalyst for the activation of molecular oxygen to produce singlet oxygen. Remarkably, the CuP-SQ COF greatly enhances the efficiency by at least 20 times compared to monomeric CuP. As activation of molecular oxygen is driven by triplet-state energy transfer, this enhancement originates from an increased capability of CuP-SQ COF in producing the triplet excitation state owing to the ordered π arrays of COFs, while the 1D nanochannels of COFs may facilitate the adsorption of oxygen which shortens the distance to promote triplet energy transfer.
To take advantage of their light-harvesting functions, porphyrin and phthalocyanine units have been integrated into a series of porphyrin-co-phthalocyanine M1DPP-M2Pc-COFs and M1TPP-M2Pc-COFs.14 These COFs exhibit broad absorption bands up to 1350 nm and are active for activation of molecular oxygen. For example, ZnTPP-CuPc-COF produces singlet oxygen with an efficiency that is 1 order of magnitude higher compared to CuP-SQ COF.
Along this line of study, a C=C linked sp2 carbon TP COF with a LUMO energy of −3.23 eV and a band gap of 2.36 eV has been developed as artificial photosystem I.15 Interestingly, it promotes coenzyme regeneration and enhances the coenzyme-assisted synthesis of L-glutamate to achieve a high yield of 97% within 12 min.
As the π columns serve as ordered pathways for charge carrier transport,16−20 COFs are unique semiconductors with high carrier mobility. Based on this feature, COFs integrated with Co(II) porphyrin have been developed for catalyzing electroreduction of CO2 into CO (Figure 3B).21 The Co(II) porphyrin on the surface serves as a catalytic site while the porphyrin columns facilitate electron transport from the electrode to the catalytic site. An imine-linked COF-366-Co has been reported to perform CO2 reduction in water, while the Co(II) porphyrin content in the COFs can be synthetically tuned by mixing with different contents of copper(II) porphyrin as knot units in the polycondensation. The COF-367-Co exhibits a Faradaic efficiency of 91% and turnover number of up to 3 901 at an applied potential of −0.67 V (vs RHE). The turnover number is about 4.9-fold that of monomeric Co(II) porphyrin as the COF can promote electron transport to the catalytic cobalt(II) porphyrin sites. By decreasing the Co(II) porphyrin content to 1%, the resulting hybrid COF-367-Co (1%) (Figure 3C) achieves a greatly enhanced turnover number to reach 24 000. These results indicate that electrochemical catalysts can be designed by exploring the skeletons to install the π conduction path and catalytic sites.
Catalysts Based on Side Walls
Not only the skeletons but also the side walls are attractive for designing catalytic systems. As the aromatic building blocks stack in the framework, dense aromatic C–H units are aligned and extrude from the pore walls so that they are accessible to serve as catalytic sides. This possibility has been explored by designing an imine-linked pyrene anthracene Py-An-COF (Figure 4A),22 which promotes Diels–Alder reactions at room temperature under ambient pressure in water. This catalytic activity originates from the C–H···π interactions between the C–H units on the walls with the π systems of aromatic reactants. The C–H···π interactions reduce the entropy loss and activation energy. The heterogeneous catalyst with the π-electronic walls as catalytic bed shows a great potential for designing π walls for catalysis.
Figure 4.

Schematics of (A) Py-An COF, (B) Pd/COF-LZU1, (C) M/Salen-COF, (D) CCOF 4-M, and (E) Co-TpBpy COF and Ni-TpBpy COF for catalysis based on the side walls.
Coordination of metal species with the skeletons enables the designed synthesis of catalytic metal complexes on the pore walls. The C=N linkage has been demonstrated for the coordination with Pd(II) ions for the synthesis of Pd(II) complexed Pd/COF-LZU 1 (Figure 4B).23 The Pd(II) catalytic sites on the pore walls are accessible to reactants and substrates and exhibit excellent catalytic activity and recyclability for promoting Suzuki coupling reactions. An imine-linked Py-2,3-DHPh COF with a pyrene knot and catechol linker can coordinate with VO(acac)2 to form VO@Py-2,3-DHPh COF.24 Owing to the broad catalytic activity of V=O, the VO@Py-2,3-DHPh COF is promising for catalyzing various reactions.
Metallosalen complexes are well-established catalysts for promoting a diversity of different reactions. Exploring Salen units as a linker for polymerization with various knots to form Salen COFs enables the construction of Salen walls. Indeed, condensation of C3-symmetric salicyaldehyde-based monomers and ethylenediamine produces Salen-COF, which upon metalation with Cu(II), Ni(II), Zn(II), Co(II), and Mn(II) forms M/Salen COF (Figure 4C).25 The Salen ligands were periodically positioned at the walls of the hexagonal pores. Compared to the homogeneous counterparts, the similar catalytic activity of Co/Salen-COF is achieved in the Henry reaction while enhancing recyclability.
Chiral Salen-COF has been developed for asymmetric catalysis by integrating chiral metallosalen to the skeleton. An efficient one-step approach to synthesize a chiral Zn(Salen)-based imine-COF is based on a solvothermal condensation reaction of C3-symmetric trisalicylaldehydes with chiral 1,2-diaminocyclohexane in the presence of Zn(OAc)2·2H2O (Figure 4D).26 Incorporating hydrophobic tert-butyl groups onto the skeletons enhances chemical stability both in acidic and alkaline solutions. The postsynthetic exchange of Zn ions from the chiral Salen pockets with other metal species produces a series of heterogeneous metallosalen-based COFs containing a given ratio of hybrid Zn/M species (M = V, Mn, Fe, Cr, and Co). The metallosalen COFs exhibit excellent activity and selectivity in catalyzing asymmetric reactions. For example, CCOF 4-V(V) promotes cyanation of aldehydes with trimethylsilyl cyanide to achieve 89%–94% enantiomeric excess (ee). CCOF 4-Co facilitates the asymmetric Diels–Alder reaction of 1-amino-substiuted butadienes and acroleins in CHCl3 at room temp to reach ee values of 86%–96%. Both CCOF 4-Mn and 4-Fe are active in the epoxidation of alkenes, while CCOF 4-Cr promotes aminolysis of trans-stilbene oxide with different anilines, which is promising for synthesis of biologically important antiamino alcohols. Noticeably, CCOF 4-Cr-Mn with two different active metal centers catalyzes a sequential reaction to synthesize amino alcohols with 91% ee, in which epoxidation of alkene is catalyzed by the Mn moieties and the epoxide ring opening is consecutively promoted at the Cr centers.
2,2′-Bipyridine is a well-known bidentate ligand and has been developed for the synthesis of bipyridine COFs,24 which upon postsynthetic metalation produce metallo-bipyridine complexes (Figure 4E). For example, Co(II)-modified bipyridine Co-TpBpy COF has been developed as an electrocatalyst for oxygen evolution reaction (Figure 4E).27 This COF exhibits fast and stable water oxidation, while retaining 94% of its activity even after 1000 cycles with a turnover frequency of 0.23 s–1 and faradaic efficiency of 95%. A Ni(II)-bipyridine Ni-TpBpy COF has been reported as a synergistic catalyst for selective photoreduction of CO2 to CO (Figure 4E).28 The β-ketoenamine knots play a critical role in activation of CO2 and suppresses H2 evolution to achieve a rate of 4057 μ mol h–1 of CO in a 5-h reaction with a 96% selectivity over H2 generation. This result suggests the possibility of designing the knot and linker backbones to trigger a synergistic effect.
Catalysts Based on Pore Surface Engineering
Different from catalysts directly embedded on the walls, pore surface engineering enables the integration of catalytic sites into the channels without any coordinative monomers. Pore surface engineering explores three-component reactions systems with two knot or linker units to design and synthesize COFs with reactive sites on pore walls,29 which transform into catalytic sites via postsynthetic reactions. This strategy enables the integration of various molecular catalysts that cannot be introduced via direct polymerization.
We have explored pore surface engineering for the designed synthesis of catalytic COFs.30,31 Integrating chiral catalytic units into achiral COFs via pore surface engineering can produce chiral catalytic COFs. This method enables the introduction of chiral sites to the predetermined positions in the channels and controls the density of catalytic sites by tuning the content of three components in the polymerization, offering the possibility of control over the local interface of catalysts in the channels. More importantly, it can introduce chiral units at relatively low temperature that is critical for an asymmetric reaction; direct polymerization of chiral units under solvothermal conditions over a long reaction period and at a high temperature cannot avoid the risk of undesired thermal racemization and greatly decreases the enantioselectivity of the resulting catalysts. This lowered selectivity will directly deteriorate the catalytic performance in terms of the ee value.
The three-component polymerization reaction based on the combination of the tris(4-amino)triphenylene benzene (TAPB) knot and 2,5-bis(propynyloxy)terephthaldehyde (BPTA) and 2,5-dimethoxyterephthaldehyde (DMTA) as linker units yields intermediate [HC≡C]x-TPB-DMTP-COFs (x = 0.17, 0.34, and 0.5), which upon click reaction with an azide derivative of S-pyrrolidine form chiral [(S)-Py]x-TPB-DMTP-COFs with a different density of chiral organocatalysts in the channels (Figure 5A).30 These COFs are unique in stability as the methoxy groups induce the electron resonance effect by the lone pairs of the oxygen atoms to soften the polarization of the C=N linkages and thus stabilize the framework to achieve a robust stability in strong acid (12 M HCl) and base (14 M NaOH).
Figure 5.

Schematics of (A) [(S)-Py]x-TPB-DMTP-COFa, (B) [Pyr]x-H2P-COFs, and (C) CCOFs for catalysis based on pore surface engineering.
The resulting chiral COFs served as heterogeneous catalysts to promote asymmetric Michael addition in water at 25 °C. The open pores can accumulate reactants from the water phase and the reaction takes place on the chiral organocatalytic centers. As a result, [(S)-Py]x-TPB-DMTP-COFs achieve 94% ee and 97/3 dr. Among the series, [(S)-Py]0.17-TPB-DMTP-COF exhibits the highest reactivity owing to the fact that dense catalytic sites cause steric hindrance and a slow reaction. The reaction requires a large enough space to proceed; a small pore [(S)-Py]0.25-H2P-COF (Figure 5B) with the same S-pyrrolidine units in the 2.0 nm pore can promote Michael reaction but with less selectivity of 50% ee and 70/30 dr.31 Compared to S-pyrrolidine molecular catalyst and other solid catalysts, the chiral [(S)-Py]x-TPB-DMTP-COFs increase reaction rates and achieve high enantioselectivity and diastereoselectivity while retaining reactivity after recycle. The combination of these characters is highly desirable for heterogeneous organocatalysis.
Different from the click reaction, the imidazole unit has been developed to append catalytic sites. Condensation of TAPB with eight aldehydes of 2-substituted-1H-benzo[d]imidazole backbone enables the pore surface engineering of hexagonal CCOFs with eight different chiral sites on the imidazole rings (Figure 5C).32 Among CCOFs, TAH-CCOF2 displays the best activity and enantioselectivity in the asymmetric amination of ethyl 2-oxocyclopentane-1-carboxylate with di-tert-butylazodicarboxylate to achieve 96% yield and 99% ee in dichloromethane at −78 °C. The excellent performance originates from the local interface where the multiple N–H sites trigger hydrogen-bonding interactions with reactants and the site bulkiness controls spatial preference so that the enantioselectivity and catalytic activity are improved simultaneously. TAH-CCOF2 shows a broad scope of substrates and can be reused at least seven times.
Catalysts Based on Pore Confinement
The topology diagram enables the design of not only skeletons but also pores. Especially COFs can precisely predetermine the pore shape, size, and environment, which are the most important structural parameters that control the interactions with guest molecules. The 1D channels of COFs are unique as they combine a series of distinct features. (1) They are discrete in size and possess a precise polygonal shape with clear edges and corners. The pore size is predesignable from micropores to mesopores. (2) They are open on the (100) facet, whereas guest molecules can access these 1D channels from the top or the bottom of the 2D layers. (3) They are spatially isolated and independent from each other, offering a platform for designing a segregated yet ordered nanospace. (4) The oriented 1D pores offer an ultimate pathway for mass transport. Combining all these features in one framework, COFs create an unlimited potential for developing pores to design functions that are difficult to access with other molecular frameworks and porous materials. The most striking feature of 1D channels is that their walls can be predesigned via polymerization and pore surface engineering to build a tailor-made interface. The well-defined interface paves the chemical basis to trigger and control different interplays and determines pore physiochemical properties, thereby becoming the primary issue to be considered while designing pores. By loading and confining catalysts within the pores, COFs have been developed for various interesting catalyses.
The 1D nanochannels owing to discrete size and shape enable the synthesis of well-defined metal nanoparticles and confine them within the pores. For example, integration of thioether groups to the pore walls produces Thio-COF, which enables the in situ synthesis of ultrafine Pt or Pd nanoparticles at a high loading content (Figure 6A).33 The particles are well-controlled in size within the range of 1.7 ± 0.2 nm; these nanoparticles are nearly monodisperse. The 1D channels with a discrete shape and size allow for precise size control of the nanoparticles while the thioether units in the channels serve as a ligand for surface passivation to stabilize the nanoparticles. Such a spatial alignment isolates nanoparticles and prevents the aggregation to form large particles. These Pt and Pd nanoparticles confined in the nanochannels of COFs serve as catalysts for the nitrophenol reduction and Suzuki–Miyaura coupling reaction under mild conditions. PtNPs@Thio-COF allows the fast reduction of 4-nitrophenol to 4-aminophenol with a full conversion in only 8 min. PdNPs@Thio-COF with 0.1 mol % Pd nanoparticles is active for catalysis of Suzuki–Miyaura coupling between various aryl halides and phenylboronic acid, affording quantitative yields in 3 h. More importantly, the stability of NPs@Thio-COFs enables recycling over six reaction runs. Metal nanoparticles in COFs offer an approach to well-defined catalysts and other applications.34−37 However, the pore confinement without binding sites on the walls might lack control over nanoparticle deposition and eventually give rises to surface attachment.
Figure 6.

Schematics of (A) NPs@Thio-COF, (B) PPS@COF-TpBpy-Cu(II), and (C) PVP@[SO3H]x-COF for catalysis via pore confinement.
Combining two or more catalytic centers on a substrate to work cooperatively in reactions is considered as the state-of-the-art heterogeneous catalysis. Active sites are usually spatially separated and difficult to cooperate, making the combination a great challenge. A COF-TpBpy (Figure 6B) has been synthesized to bear a bipyridine linker which coordinates with Cu(II) to form Lewis acid catalytic sites on the pore walls, while in situ polymerization of ethyldiphenyl(4-vinylphenyl)phosphonium bromide allows for the introduction of Br-counterions to the linear polymer chains.38 The Cu(II) complex on walls and Br– anions in the pores work cooperatively to promote the cycloaddition reaction of epoxide to CO2 and quantitatively produce cyclic carbonates.
A series of acid-functionalized COF-[SO3H]x (Figure 6C) has been synthesized by incorporating perfluoroalkyl chains with a terminal sulfonic acid group.39 Encapsulation of linear polyvinylpyrrolidone (PVP) in the channels of COF-[SO3H]x by in situ polymerization of 1-vinyl-2-pyrrolidone enables the creation of a pseudo solvent environment as N-methylpyrrolidinone (NMP) where the nitrogen atoms trigger hydrogen-bonding interactions with the reactant fructose to promote its conversion. Indeed, the resulting COF creates a supramolecular interface to enable the cooperation of pyrrolidone moieties with the sulfonic acid catalytic sites which achieve enhanced activity and selectivity. COF-[SO3H]0.17 completes the reaction within 30 min, which is 1.4 times better than the NMP solvent. Note that this catalytic activity ranks among the best systems.
Loading Pd NPs in COF LZU1 (20 nm thick) in a core–shell NH2-MIL-125 Ti-MOF/COF LZU1 hybrid yields Pd/TiMOF@LZU1 COF.40 The Pd NPs hybrid in the presence of hydrogen under visible light promotes styrene hydrogenation to produce ethylbenzene with 100% conversion and over 99% selectivity in 15 min. In this case, the core TiMOF is excited to generate electron and the shell LZU1 COF serves as a bridge to promote electron transfer from the core TiMOF to surface Pd NPs where styrene hydrogenation occurs. Notably, the Pd NPs hybrid is efficient for photocatalytic dehydrogenation of NH3BH3, showing a TOF of 147 mol min–1 mol–1 (Pd). Interestingly, the Pd/TiMOF@LZU1 COF enables the construction of a dual-chamber microreactor with one chamber for dehydrogenation and another one for hydrogenation, which are connected by a gas-permeable PDMS membrane separator. Upon visible irradiation, hydrogen generated in the bottom chamber passed through the PDMS membrane to the top chamber for styrene hydrogenation. At a flow rate of 12.5 μL min–1, the flow system achieves a 94% conversion, which corresponds to a resident time of 68 s, both of which are superior to those (23% conversion and 20 min) of bulk reactors.
Catalysts Based on Systematic Organization
COFs owing to the ordered π columns and built-in pores offer a systematic platform for designing complex catalytic systems to merge different processes and reactions into one transformation. Exploring sunlight to produce hydrogen from water is a highly interesting and challenging subject. Hydrogen evolution consists of a series of sequential processes from light harvesting to electron transfer and catalytic reduction. These individual processes can be separately realized with different molecules, but they must be merged into one molecular system. First, a photon can be harvested by the light-harvesting to generate an exciton. Second, the exciton can be split into an electron and a hole at a suitable interface. Third, the charges can be transported and transferred to the reaction center. Therefore, the system must merge a light-harvesting antennae, a donor–acceptor interface for exciton splitting, charge transport pathways, and an interface for electron transfer to the reaction center. An ultimate goal is to split water into hydrogen and oxygen via systematic organization of tailor-made modules and interfaces to merge different processes in a seamless way.
Azine-linked COFs possess π-conjugated skeletons and are synthesized by condensation of aldehyde and hydrazine.41 A series of azine-linked Nx-COFs (Figure 7A, Nx-COF, x = 0, 1, 2, and 3) has been synthesized to have different numbers of nitrogen atoms (0–3) as a knot (Figure 2).42 The resulting Nx-COFs absorb a wide range of photons from ultraviolet to visible regions up to 475 nm. Nx-COFs (5 mg) have been used to build a photocatalytic system for hydrogen evolution from water, with a Pt nanoparticle catalyst (5 mL, 8 wt % hexachloroplatinic acid aqueous solution) and triethanolamine donor (100 mL, 0.738 mmol) in a phosphate buffer solution at pH = 7 and 25 °C under a 300-W xenon light source (λ ≥ 420 nm). The Nx-COFs exhibit a steady evolution of hydrogen without any induction period and show a linear proportion between the hydrogen amount and irradiation time. Interestingly, the evolution rate is dependent on the number of nitrogen atom at the focal aryl unit. For example, as the number (x) of nitrogen atoms is increased from 0 to 1, 2, and 3, Nx-COFs enhance the hydrogen evolution rate from 23 to 90, 438, and 1703 μmol h–1 g–1. N3-COF is catalytically stable to continuously produce hydrogen under 48-h irradiation. The hydrogen evolution rate of N3-COF is more than 2-fold as high as those of representative catalysts, such as poly(triazine imide) (864 μmol h–1 g–1),43 melon (720 μmol h–1 g–1),43 and g-C3N4 (840 μmol h–1 g–1).44
Figure 7.

Schematics of (A) Nx-COF, (B) FS-COF, (C) sp2C-COFERDN for catalysis based on systematic organization.
In Nx-COFs, as the number of nitrogen atoms is increased from 0 to 1, 2, and 3, the stabilization energy of the anion radical species of Nx-COFs decreases from 0.00 to −0.19, – 0.39, and −0.45 eV. Thus, Nx-COFs upon photoexcitation are reduced by a sacrificial donor to generate anionic radicals that can transfer electrons to the Pt catalyst for hydrogen evolution. This study shows the key role of the knot structure in designing an interface for promoting water reduction.
As the reaction takes place in water, a hydrophilic COF is highly desired. Hydrophilic benzothiophene sulfone-based FS-COF (Figure 7B) has been explored for hydrogen evolution.45 FS-COF exhibits a band gap of 1.85 eV and can harvest a wide range of visible light up to 700 nm. FS-COF bearing sulfone units on pore walls is hydrophilic and improves water affinity. In the presence of Pt nanoparticle as a reaction center (hexa-chloroplatinic acid, 5 μL, 8 wt % aqueous solution) and ascorbic acid (aqueous solution, 0.1 M, 25 mL) as a sacrificial donor, FS-COF (5 mg) achieves a hydrogen evolution rate as high as 10.1 mmol h–1 g–1 and is stable upon 50-h continuous irradiation. Note that this rate is 22-fold as high as that of N3-COF (0.47 mmol h–1 g–1) under identical conditions and 9-fold as high as that of the amorphous counterpart (1.12 mmol h–1 g–1). FS-COF combines an ordered π structure, increased light harvesting capability, improved hydrophilicity, and small Pt nanoparticles in one system, leading to an exceptional activity. This study thus shows that the combination of extended π structures with a hydrophilic interface is key to electron flow from COFs to Pt nanoparticles to water.
A series of hexagonal g-C40N3-COF, g-C31N3-COF, and g-C37N3-COF with C=C linkages have been developed to possess an optical band gap of 2.36, 2.40, and 2.52 eV, respectively.46 These band gap values suggest that the hexagonal topology cannot extend the π conjugation across the lattice.47 In the presence of TEOA donor, 3 wt % Pt-modified g-C40N3-COF shows a hydrogen evolution rate of 129.8 μmol h–1, while g-C31N3-COF and g-C37N3-COF exhibit a much low rate of 27.1 and 19.8 μmol h–1, respectively. The high performance of g-C40N3-COF originates from its light-harvesting and charge separation abilities together with a high crystallinity.
The above COFs for hydrogen evolution from water are robust in stability, but they possess partially π-conjugated skeletons. Integrating full π conjugation into COFs may offer a new way to achieve systematic combination of different functions. To fulfill such a purpose, the tetragonal skeleton is preferred as the hexagonal structure limits the π delocalization over the lattice.
To ensure a full π conjugation over the 2D skeleton, we have developed a C=C bond-linked all sp2 carbon conjugated sp2c-COF.48−50 Different from other topologies and linkages, the C=C bond-linked tetragonal sp2c-COF consists of an all sp2 carbon lattice and enables progressed π transmission along the x and y directions. The sp2c-COF is stable in boiling water, concentrated HCl, and NaOH solution (14 M) after 1 week. It keeps crystalline porous structure after 1-year exposure to air and room light under ambient conditions. Therefore, sp2c-COF is robust enough to build a photocatalytic system to promote hydrogen evolution from water.
To promote exciton splitting, an electron deficient unit 3-ethylrhodanine (ERDN) is introduced to the periphery of sp2c-COFERDN to the construction of the donor–acceptor interface (Figure 7C).50 Unexpectedly, the integration of ERDN terminals exerts positive effects on improving π conjugation, extending absorbance to even 800 nm, decreasing the band gap to 1.85 eV, and lowering HOMO and LUMO levels. Indeed, sp2c-COFERDN enhances the photocurrent as a result of improved π conjugation and the donor–acceptor interface, which enable exciton migration and its splitting into a hole and an electron.
Pt nanoparticles with a size of 1–3 nm (3 wt %) were in situ generated in sp2c-COFERDN (50 mg), and the resulting system in aqueous triethanolamine solution (100 mL, 10 vol %) was irradiated with a 300-W xenon lamp (≥420 nm). The sp2c-COFERDN achieves an impressive hydrogen production rate of 2120 μmol h–1 g–1, which is far superior to those of sp2c-COF (1360 μmol h–1 g–1) and sp2c-CMP (amorphous analogue, 140 μmol h–1 g–1) while the imine-linked pyrene-COF cannot generate hydrogen. Noticeably, sp2c-COFERDN keeps the crystalline porous structure after 20-h continuous irradiation. The lowered HOMO level of sp2c-COF is suitable for catalyzing water oxidation into oxygen. When dispersed in water (100 mL) with AgNO3 as the electron acceptor (0.01 M), Co(NO3)2 as the cocatalyst (0.6 mg), and La2O3 as the pH buffer agent (0.2 g), sp2c-COF (50 mg) produces oxygen at a rate of 22 μmol h–1 g–1 upon irradiation.50 Therefore, sp2c-COF with suitable HOMO and LUMO levels upon integration with reduction and oxidation centers can develop into a photocatalyst that promotes both hydrogen and oxygen evolution.
The fully π-conjugated sp2c-COFs develop a three molecule mechanism to enable photocatalytic hydrogen production. (1) The conjugated π skeleton enables harvesting of visible light. (2) The columnar arrays provide π pathways for exciton migration, split, and charge transport. (3) Docking metal centers in channels or on the surface forms a proximate interface to decrease the distance of electron transfer to the reaction center. These functions are seamlessly merged and render the system able to produce hydrogen by sunlight.
Outlook
COFs offer an emerging molecular platform for designing stable heterogeneous catalysts and have greatly changed the research direction and enhanced the flexibility in exploring efficient heterogeneous catalytic systems. Exploration of COFs-based catalysts involves the design of various interfaces, which are key to control the interplays with exciton, electron, hole, molecules, and ions. These interactions control both kinetics and dynamics of the overall reactions and thus determine the catalytic activity, selectivity, and efficiency.
COFs are obtained as polycrystalline materials with surface areas that are comparable with other inorganic porous materials including zeolites, mesoporous silicas, and MOFs. Most COFs possess porous structures such as pore size and surface area that are the same as or close to their theoretical values of virtual single crystal structures. This fact suggests that although most COFs are not in a single crystal state, their structural integrity cannot be underestimated. In this context, the interface design of most COFs with high crystallinity and porosity enables an overall control of properties and functions. Clearly, we are currently not at a stage that a single or a few defects will determine the functions we are working on. With further studies on the defect formation process and mechanism in COFs, we anticipate a much clearer picture on this aspect. The interior defects of COFs are still unclear and need further exploration. We classify a material as a COF by its high crystallinity and porosity; this criterion should be identified throughout the community. Nevertheless, even now we still see reported “COFs” although they have limited crystallinity and porosity.
Owing to a rich diversity of building blocks, COFs have a great probability of developing nanoreactors that can greatly surpass the activity, selectivity, and durability of molecular catalysts. This requires a precise engineering of interfaces between walls and catalytic sites. To develop catalytic systems that are inaccessible with molecular catalysts is an interesting topic so that the confined space with a well-defined structure can be fully explored for novel catalysis. Using the channel walls as well as pore size and shape to create a unique local interface for each catalytic site as observed in enzymes is an interesting subject worthy of further exploration. Using different walls to integrate different catalysts is a challenging goal where the synergistic effects between walls may play a key role in catalyzing multistep transformations. These directions should bring us to a new level to design truly high-performance heterogeneous catalysts.
As evidenced by the light-driven hydrogen production from water, COFs are superior to other state-of-the-art artificial antennae and semiconductors in many aspects. Especially the structural designability and synthetic controllability of organic π ordering, component, sequence, and density are unreachable with other systems. Therefore, COFs are highly promising to organize both oxidation and reduction sides into one molecular system to create a predesignable yet seamless electron flow diagram so that photoenergy can be efficiently transformed into chemical energy. We anticipate that artificial photosynthesis is possible through a full design of interplays of COFs with photon, exciton, electron, hole, ions, and molecules.
Acknowledgments
This work is supported from the NSFC (Grants 51973039 and 21975049) and STCSM (Grant 18520744800). D.J. acknowledges the MOE Tier 1 grant (Grant R-143-000-A71-114) and NUS startup grant (Grant R-143-000-A28-133).
Author Contributions
J.G. and D.J. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Schlögl R. Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2015, 54, 3465–3520. 10.1002/anie.201410738. [DOI] [PubMed] [Google Scholar]
- Thomas J. M.; Thomas W. J.. Principles and Practice of Heterogeneous Catalysis; Wiley: Weinheim, Germany, 2015. [Google Scholar]
- Heitbaum M.; Glorius F.; Escher I. Asymmetric Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2006, 45, 4732–4762. 10.1002/anie.200504212. [DOI] [PubMed] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Côté A. P.; Benin A. I.; Ockwig N. W.; O’Keeffe M.; Matzger A. J.; Yaghi O. M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. 10.1126/science.1120411. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ding X.; Jiang D. Covalent Organic Frameworks. Chem. Soc. Rev. 2012, 41, 6010–6022. 10.1039/c2cs35157a. [DOI] [PubMed] [Google Scholar]
- Geng K.; He T.; Liu R.; Dalapati S.; Tan K. T.; Li Z.; Tao S.; Gong Y.; Jiang Q.; Jiang D. Covalent Organic Frameworks: Design, Synthesis, and Functions. Chem. Rev. 2020, 10.1021/acs.chemrev.9b00550. [DOI] [PubMed] [Google Scholar]
- Fang Q.; Gu S.; Zheng J.; Zhuang Z.; Qiu S.; Yan Y. 3D Microporous Base-Functionalized Covalent Organic Frameworks for Size-Selective Catalysis. Angew. Chem., Int. Ed. 2014, 53, 2878–2882. 10.1002/anie.201310500. [DOI] [PubMed] [Google Scholar]
- Li H.; Pan Q.; Ma Y.; Guan X.; Xue M.; Fang Q.; Yan Y.; Valtchev V.; Qiu S. Three-Dimensional Covalent Organic Frameworks with Dual Linkages for Bifunctional Cascade Catalysis. J. Am. Chem. Soc. 2016, 138, 14783–14788. 10.1021/jacs.6b09563. [DOI] [PubMed] [Google Scholar]
- Yang D.; Gates B. C. Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research. ACS Catal. 2019, 9, 1779–1798. 10.1021/acscatal.8b04515. [DOI] [Google Scholar]
- Dhakshinamoorthy A.; Li Z.; Garcia H. Catalysis and Photocatalysis by Metal Organic Frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. 10.1039/C8CS00256H. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy A.; Opanasenko M.; Cejka J.; Garcia H. Metal Organic Frameworks as Heterogeneous Catalysts for the Production of Fine Chemicals. Catal. Sci. Technol. 2013, 3, 2509–2540. 10.1039/c3cy00350g. [DOI] [Google Scholar]
- Nagai A.; Chen X.; Feng X.; Ding X.; Guo Z.; Jiang D. A Squaraine-Linked Mesoporous Covalent Organic Framework. Angew. Chem., Int. Ed. 2013, 52, 3770–3774. 10.1002/anie.201300256. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ding X.; Chen L.; Wu Y.; Liu L.; Addicoat M.; Irle S.; Dong Y.; Jiang D. Two-Dimensional Artificial Light-Harvesting Antennae with Predesigned High-Order Structure and Robust Photosensitising Activity. Sci. Rep. 2016, 6, 32944. 10.1038/srep32944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Liu H.; Wu C.; Zhang Z.; Pan Q.; Hu F.; Wang R.; Li P.; Huang X.; Li Z. Fully Conjugated Two-Dimensional sp2-Carbon Covalent Organic Frameworks as Artificial Photosystem I with High Efficiency. Angew. Chem., Int. Ed. 2019, 58, 5376–5381. 10.1002/anie.201901194. [DOI] [PubMed] [Google Scholar]
- Wan S.; Guo J.; Kim J.; Ihee H.; Jiang D. A Belt-Shaped, Blue Luminescent, and Semiconducting Covalent Organic Framework. Angew. Chem., Int. Ed. 2008, 47, 8826–8830. 10.1002/anie.200803826. [DOI] [PubMed] [Google Scholar]
- Feng X.; Liu L.; Honsho Y.; Saeki A.; Seki S.; Irle S.; Dong Y.; Nagai A.; Jiang D. High-Rate Charge-Carrier Transport in Porphyrin Covalent Organic Frameworks: Switching from Hole to Electron to Ambipolar Conduction. Angew. Chem., Int. Ed. 2012, 51, 2618–2622. 10.1002/anie.201106203. [DOI] [PubMed] [Google Scholar]
- Jin S.; Supur M.; Addicoat M.; Furukawa K.; Chen L.; Nakamura T.; Fukuzumi S.; Irle S.; Jiang D. Creation of Superheterojunction Polymers via Direct Polycondensation: Segregated and Bicontinuous Donor-Acceptor π-Columnar Arrays in Covalent Organic Frameworks for Long-Lived Charge Separation. J. Am. Chem. Soc. 2015, 137, 7817–7827. 10.1021/jacs.5b03553. [DOI] [PubMed] [Google Scholar]
- Ding X.; Guo J.; Feng X.; Honsho Y.; Guo J.; Seki S.; Maitarad P.; Saeki A.; Nagase S.; Jiang D. Synthesis of Metallophthalocyanine Covalent Organic Frameworks that Exhibit High Carrier Mobility and Photoconductivity. Angew. Chem., Int. Ed. 2011, 50, 1289–1293. 10.1002/anie.201005919. [DOI] [PubMed] [Google Scholar]
- Guo J.; Xu Y.; Jin S.; Chen L.; Kaji T.; Honsho Y.; Addicoat M. A.; Kim J.; Saeki A.; Ihee H.; Seki S.; Irle S.; Hiramoto M.; Gao J.; Jiang D. Conjugated Organic Framework with Three-Dimensionally Ordered Stable Structure and Delocalized π Clouds. Nat. Commun. 2013, 4, 2736. 10.1038/ncomms3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Diercks C. S.; Zhang Y. B.; Kornienko N.; Nichols E. M.; Zhao Y. B.; Paris A. R.; Kim D.; Yang P.; Yaghi O. M.; Chang C. J. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. 10.1126/science.aac8343. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Xu H.; Chen X.; Gao J.; Jiang D. A π-Electronic Covalent Organic Framework Catalyst: π-Walls as Catalytic Beds for Diels-Alder Reactions under Ambient Conditions. Chem. Commun. 2015, 51, 10096–10098. 10.1039/C5CC03457D. [DOI] [PubMed] [Google Scholar]
- Ding S. Y.; Gao J.; Wang Q.; Zhang Y.; Song W. G.; Su C. Y.; Wang W. Construction of Covalent Organic Framework for Catalysis: Pd/COF-LZU1 in Suzuki-Miyaura Coupling Reaction. J. Am. Chem. Soc. 2011, 133, 19816–19822. 10.1021/ja206846p. [DOI] [PubMed] [Google Scholar]
- Chen X.; Huang N.; Gao J.; Xu H.; Xu F.; Jiang D. Towards Covalent Organic Frameworks with Predesignable and Aligned Open Docking Sites. Chem. Commun. 2014, 50, 6161–6163. 10.1039/C4CC01825G. [DOI] [PubMed] [Google Scholar]
- Li L.-H.; Feng X.-L.; Cui X.-H.; Ma Y.-X.; Ding S.-Y.; Wang W. Salen-Based Covalent Organic Framework. J. Am. Chem. Soc. 2017, 139, 6042–6045. 10.1021/jacs.7b01523. [DOI] [PubMed] [Google Scholar]
- Han X.; Xia Q.; Huang J.; Liu Y.; Tan C.; Cui Y. Chiral Covalent Organic Frameworks with High Chemical Stability for Heterogeneous Asymmetric Catalysis. J. Am. Chem. Soc. 2017, 139, 8693–8697. 10.1021/jacs.7b04008. [DOI] [PubMed] [Google Scholar]
- Aiyappa H. B.; Thote J.; Shinde D. B.; Banerjee R.; Kurungot S. Cobalt-Modified Covalent Organic Framework as a Robust Water Oxidation Electrocatalyst. Chem. Mater. 2016, 28, 4375–4379. 10.1021/acs.chemmater.6b01370. [DOI] [Google Scholar]
- Zhong W.; Sa R.; Li L.; He Y.; Li L.; Bi J.; Zhuang Z.; Yu Y.; Zou Z. A Covalent Organic Framework Bearing Single Ni Sites as a Synergistic Photocatalyst for Selective Photoreduction of CO2 to CO. J. Am. Chem. Soc. 2019, 141, 7615–7621. 10.1021/jacs.9b02997. [DOI] [PubMed] [Google Scholar]
- Nagai A.; Guo Z.; Feng X.; Jin S.; Chen X.; Ding X.; Jiang D. Pore Surface Engineering in Covalent Organic Frameworks. Nat. Commun. 2011, 2, 536. 10.1038/ncomms1542. [DOI] [PubMed] [Google Scholar]
- Xu H.; Gao J.; Jiang D. Stable, Crystalline, Porous, Covalent Organic Frameworks as a Platform for Chiral Organocatalysts. Nat. Chem. 2015, 7, 905. 10.1038/nchem.2352. [DOI] [PubMed] [Google Scholar]
- Xu H.; Chen X.; Gao J.; Lin J.; Addicoat M.; Irle S.; Jiang D. Catalytic Covalent Organic Frameworks via Pore Surface Engineering. Chem. Commun. 2014, 50, 1292–1294. 10.1039/C3CC48813F. [DOI] [PubMed] [Google Scholar]
- Wang L.-K.; Zhou J.-J.; Lan Y.-B.; Ding S.-Y.; Yu W.; Wang W. Divergent Synthesis of Chiral Covalent Organic Frameworks. Angew. Chem., Int. Ed. 2019, 58, 9443–9447. 10.1002/anie.201903534. [DOI] [PubMed] [Google Scholar]
- Lu S.; Hu Y.; Wan S.; McCaffrey R.; Jin Y.; Gu H.; Zhang W. Synthesis of Ultrafine and Highly Dispersed Metal Nanoparticles Confined in a Thioether-Containing Covalent Organic Framework and Their Catalytic Applications. J. Am. Chem. Soc. 2017, 139, 17082–17088. 10.1021/jacs.7b07918. [DOI] [PubMed] [Google Scholar]
- Kalidindi S. B.; Oh H.; Hirscher M.; Esken D.; Wiktor C.; Turner S.; Van Tendeloo G.; Fischer R. A. Metal@COFs: Covalent Organic Frameworks as Templates for Pd Nanoparticles and Hydrogen Storage Properties of Pd@COF-102 Hybrid Material. Chem. - Eur. J. 2012, 18, 10848–10856. 10.1002/chem.201201340. [DOI] [PubMed] [Google Scholar]
- Ma H.-C.; Kan J.-L.; Chen G.-J.; Chen C.-X.; Dong Y.-B. Pd NPs-Loaded Homochiral Covalent Organic Framework for Heterogeneous Asymmetric Catalysis. Chem. Mater. 2017, 29, 6518–6524. 10.1021/acs.chemmater.7b02131. [DOI] [Google Scholar]
- Pachfule P.; Panda M. K.; Kandambeth S.; Shivaprasad S. M.; Díaz D. D.; Banerjee R. Multifunctional and Robust Covalent Organic Framework-Nanoparticle Hybrids. J. Mater. Chem. A 2014, 2, 7944–7952. 10.1039/C4TA00284A. [DOI] [Google Scholar]
- Kamiya K.; Kamai R.; Hashimoto K.; Nakanishi S. Platinum-Modified Covalent Triazine Frameworks Hybridized with Carbon Nanoparticles as Methanol-Tolerant Oxygen Reduction Electrocatalysts. Nat. Commun. 2014, 5, 5040. 10.1038/ncomms6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Aguila B.; Perman J.; Nguyen N.; Ma S. Flexibility Matters: Cooperative Active Sites in Covalent Organic Framework and Threaded Ionic Polymer. J. Am. Chem. Soc. 2016, 138, 15790–15796. 10.1021/jacs.6b10629. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Tang Y.; Aguila B.; Wang S.; Xiao F.-S.; Thallapally P. K.; Al-Enizi A. M.; Nafady A.; Ma S. Reaction Environment Modification in Covalent Organic Frameworks for Catalytic Performance Enhance. Angew. Chem., Int. Ed. 2019, 58, 8670–8675. 10.1002/anie.201900029. [DOI] [PubMed] [Google Scholar]
- Dalapati S.; Jin S.; Gao J.; Xu Y.; Nagai A.; Jiang D. An Azine-Linked Covalent Organic Framework. J. Am. Chem. Soc. 2013, 135, 17310–17313. 10.1021/ja4103293. [DOI] [PubMed] [Google Scholar]
- Sun E.; Jang S.; Yim S.-J.; Ye L.; Kim D.-P. Metal Doped Core-Shell Metal-Organic Framework@Covalent Organic Frameworks (MOFs@COFs) Hybrids as a Novel Photocatalytic Platform. Adv. Funct. Mater. 2018, 28, 1707110. 10.1002/adfm.201707110. [DOI] [Google Scholar]
- Vyas V. S.; Haase F.; Stegbauer L.; Savasci G.; Podjaski F.; Ochsenfeld C.; Lotsch B. V. A Tunable Azine Covalent Organic Framework Platform for Visible Light-Induced Hydrogen Generation. Nat. Commun. 2015, 6, 8508. 10.1038/ncomms9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinghammer K.; Tuffy B.; Mesch M. B.; Wirnhier E.; Martineau C.; Taulelle F.; Schnick W.; Senker J.; Lotsch B. V. Triazine-Based Carbon Nitrides for Visible-Light-Driven Hydrogen Evolution. Angew. Chem., Int. Ed. 2013, 52, 2435–2439. 10.1002/anie.201206817. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen X.; Takanabe K.; Maeda K.; Domen K.; Epping J. D.; Fu X.; Antonietti M.; Wang X. Synthesis of a Carbon Nitride Structure for Visible-Light Catalysis by Copolymerization. Angew. Chem., Int. Ed. 2010, 49, 441–444. 10.1002/anie.200903886. [DOI] [PubMed] [Google Scholar]
- Wang X.; Chen L.; Chong S. Y.; Little M. A.; Wu Y.; Zhu W.; Clowes R.; Yan Y.; Zwijnenburg M. A.; Sprick R. S.; Cooper A. I. Sulfone-Containing Covalent Organic Frameworks for Photocatalytic Hydrogen Evolution from Water. Nat. Chem. 2018, 10, 1180–1189. 10.1038/s41557-018-0141-5. [DOI] [PubMed] [Google Scholar]
- Jin E.; Asada M.; Xu Q.; Dalapati S.; Addicoat M. A.; Brady M. A.; Xu H.; Nakamura T.; Heine T.; Chen Q.; Jiang D. Two-Dimensional sp2 Carbon-Conjugated Covalent Organic Frameworks. Science 2017, 357, 673–676. 10.1126/science.aan0202. [DOI] [PubMed] [Google Scholar]
- Bi S.; Yang C.; Zhang W.; Xu J.; Liu L.; Wu D.; Wang X.; Han Y.; Liang Q.; Zhang F. Two-Dimensional Semiconducting Covalent Organic Frameworks via Condensation at Arylmethyl Carbon Atoms. Nat. Commun. 2019, 10, 2467. 10.1038/s41467-019-10504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin E.; Geng K.; Lee K. H.; Jiang W.; Li J.; Jiang Q.; Irle S.; Jiang D. Topology-Templated Synthesis of Crystalline Porous Covalent Organic Frameworks. Angew. Chem., Int. Ed. 2020, 10.1002/anie.202004728. [DOI] [PubMed] [Google Scholar]
- Jin E.; Li J.; Geng K.; Jiang Q.; Xu H.; Xu Q.; Jiang D. Designed Synthesis of Stable Light-Emitting Two-Dimensional sp2 Carbon-Conjugated Covalent Organic Frameworks. Nat. Commun. 2018, 9, 4143. 10.1038/s41467-018-06719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin E.; Lan Z.; Jiang Q.; Geng K.; Li G.; Wang X.; Jiang D. 2D sp2 Carbon-Conjugated Covalent Organic Frameworks for Photocatalytic Hydrogen Production from Water. Chem. 2019, 5, 1632–1647. 10.1016/j.chempr.2019.04.015. [DOI] [Google Scholar]