

Abstract
Amyloid β-peptide, the principal component of characteristic cerebral plaques of Alzheimer’s disease (AD), is produced through intramembrane proteolysis of the amyloid precursor protein (APP) by γ-secretase. Despite the importance in the pathogenesis of AD, the mechanisms of intramembrane proteolysis and substrate processing by γ-secretase remain poorly understood. Here, complementary all-atom simulations using a robust Gaussian accelerated molecular dynamics (GaMD) method and biochemical experiments were combined to investigate substrate processing of wildtype and mutant APP by γ-secretase. The GaMD simulations captured spontaneous activation of γ-secretase, with hydrogen bonded catalytic aspartates and water poised for proteolysis of APP at the ε cleavage site. Furthermore, GaMD simulations revealed that familial AD mutations I45F and T48P enhanced the initial ε cleavage between residues Leu49–Val50, while M51F mutation shifted the ε cleavage site to the amide bond between Thr48–Leu49. Detailed analysis of the GaMD simulations allowed us to identify distinct low-energy conformational states of γ-secretase, different secondary structures of the wildtype and mutant APP substrate, and important active-site subpockets for catalytic function of the enzyme. The simulation findings were highly consistent with experimental analyses of APP proteolytic products using mass spectrometry and Western blotting. Taken together, the GaMD simulations and biochemical experiments have enabled us to elucidate the mechanisms of γ-secretase activation and substrate processing, which should facilitate rational computer-aided drug design targeting this functionally important enzyme.
Short abstract
Complementary accelerated molecular simulations, mass spectrometry, and Western blotting experiments have revealed spontaneous activation of γ-secretase bound by the wildtype amyloid precursor protein (APP) substrate and shift of the ε cleavage site in processing of mutant APP, yielding different APP intracellular domains (AICD50−99 and AICD49−99).
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by cerebral atrophy, beginning with areas of the brain involved in learning and memory. Deposition of 42-residue amyloid β-peptide (Aβ42) in the form of plaques is a defining pathological feature of AD and begins many years before onset of symptoms.1 For these reasons, Aβ42 has been a major target for the development of potential therapeutics2 as well as a key biomarker for AD.3 Aβ peptides are derived through proteolytic processing of the membrane-traversing amyloid precursor protein (APP), first by β-secretase outside the membrane, generating a membrane-bound 99-residue C-terminal fragment (C99), and then by γ-secretase within the membrane.4 γ-Secretase is a membrane-embedded aspartyl protease complex, with presenilin (PS1) as the catalytic component that carries out intramembrane proteolysis of >90 substrates, including APP and the Notch family of cell-surface receptors.5 Cleavage of the APP transmembrane (TM) domain by γ-secretase determines the length of Aβ peptides, the proportion of the hydrophobic TM domain retained in the Aβ product, and therefore the tendency of Aβ to aggregate into plaques.
Proteolysis of the APP TM domain by γ-secretase is complex.6 Initial endoproteolysis of C99 at the ε site generates 48- or 49-residue Aβ (Aβ48 or Aβ49) and corresponding APP intracellular domains (AICD49–99 or AICD50–99) (Figure S1).7 These initially formed Aβ peptides are then trimmed every 3–4 amino acids through a carboxypeptidase activity of γ-secretase along two pathways, Aβ48 → Aβ45 → Aβ42 → Aβ38 and Aβ49 → Aβ46 → Aβ43 → Aβ40,8,9 and this trimming is dictated by three active-site pockets that recognize substrate residues P1′, P2′, and P3′ (i.e., immediately C-terminal of the scissile amide bond).10 Mutations in the APP TM domain associated with early onset familial AD (FAD) can skew ε cleavage in favor of Aβ48 (i.e., to the pathological Aβ42 pathway).10,11 Alternatively, these mutations can be “pathway switchers”, affecting carboxypeptidase activity to switch from the Aβ40 pathway to the Aβ42 pathway.10
Little is known about the mechanism by which γ-secretase accomplishes intramembrane proteolysis. A substantial advance in understanding substrate recognition came recently with reports of cryoelectron microscopic (cryo-EM) structure determination of the γ-secretase complex bound to the Notch and APP substrates (Figure S1).12,13 The average resolutions of the Notch- and APP-bound γ-secretase structures were 2.7 and 2.6 Å, respectively, although lower resolutions were obtained for flexible protein regions. The cryo-EM structures were consistent with expectations from previous studies using small-molecule probes and mutagenesis. In both structures, the substrate TM assumed a helical conformation starting from the extracellular side and was surrounded by TM2, TM3, and TM5 of PS1. The helix ended just before entry into the enzyme active site, becoming first partially unwound and then fully extended into a β-strand toward the intracellular side. The substrate β-strand interacted with an antiparallel β-strand in the intracellular side of PS1 TM7, which in turn interacted with another β-strand from the enzyme TM6. This β-sheet motif was suggested to be essential for substrate recognition by the γ-secretase.12,13 While a tour de force for the field, stabilization of the substrate-enzyme complex required (1) mutation of one of the catalytic aspartates (Asp385) to alanine in PS1 (inactivating the enzyme) and (2) double cysteine mutagenesis and disulfide cross-linking between substrate and presenilin (with the potential for deviation from normal wildtype interactions).
Computational modeling, especially molecular dynamics (MD) simulation, has proven useful in understanding the structural dynamics of γ-secretase. Previous studies have provided valuable insights into the conformational changes,14−17 enzyme allosteric modulation,18 substrate binding,14,17,19−21 water distribution,14,15 lipid interactions15 and ligand binding of γ-secretase.22−24 In several of these studies, a putative active conformation was described for the substrate-free (apo) γ-secretase with the two catalytic aspartates moving to close proximity,14−16 but none has characterized the enzyme active state poised for proteolysis with both the water and peptide substrate. Hence, the dynamic mechanisms of enzyme activation and substrate processing by γ-secretase remained poorly understood.
Here, we present the first report of MD computational modeling of activation of APP-bound γ-secretase using the latest cryo-EM structures of substrate-bound enzyme. The enzyme and substrate were computationally restored to the wildtype. Extensive all-atom simulations using a novel and robust Gaussian accelerated molecular dynamics (GaMD) method were employed to capture the extremely slow motions underlying activation of γ-secretase for proteolysis of substrate within the cell membrane (kcat in proteoliposomes estimated at 1.9 h–1).25
GaMD is an enhanced sampling computational technique that works by adding a harmonic boost potential to smooth the biomolecular potential energy surface.26 GaMD greatly reduces energy barriers and accelerates biomolecular simulations by orders of magnitude.27 GaMD does not require predefined collective variables or reaction coordinates. Compared with the enhanced sampling methods that rely on careful selection of the collective variables, GaMD is of particular advantage for studying complex biological processes28 such as enzyme activation and substrate processing by γ-secretase. Moreover, because the boost potential follows a Gaussian distribution, biomolecular free energy profiles can be properly recovered through cumulant expansion to the second order.26 GaMD builds on the previous accelerated MD (aMD) method,29,30 but solves its energetic reweighting problem31 for free energy calculations of large biomolecules. GaMD has successfully revealed physical pathways and mechanisms of protein folding and ligand binding, which are consistent with experiments and long-time scale conventional MD simulations.26,32,33 It has also been applied to characterize protein–protein,34,35 protein–membrane,36 and protein-nucleic acid37,38 interactions. Therefore, GaMD was applied in this study for enhanced sampling of the γ-secretase complex, a well-known slow enzyme.39,40
Furthermore, the GaMD simulations were highly consistent with parallel mass spectrometry (MS) and Western blotting biochemical experiments on the processing of both wildtype and mutant APP substrates. Remarkably, one of the mutations (M51F) in APP shifted the substrate ε cleavage site to the amide bond between residue Thr48–Leu49, while another two mutations (I45F and T48P) enhanced the ε cleavage between Leu49–Val50 compared with the wildtype. The GaMD simulations and biochemical experiments together offered a deep atomic-level understanding of intramembrane proteolysis by γ-secretase.
Results
Activation of Computationally Restored Wildtype γ-Secretase Is Captured in GaMD Simulations
Our initial testing GaMD simulations using the earlier published cryo-EM structure of Notch-bound γ-secretase (Figure S1A)—with Asp385 computationally restored— showed that Asp257 rather than Asp385 should be protonated in the active site, as in this case the two aspartates were able to approach each other for catalysis (Table 1 and Figure S2). Further testing GaMD simulations using the cryo-EM structure of APP-bound γ-secretase (Figure S1B and Table 1) revealed an active conformation of the PS1 catalytic subunit with computationally restored Asp385, while the enzyme–substrate disulfide bond was kept (Figure S3 and Movie S1). Building upon these testing results, we proceeded to remove the artificial enzyme–substrate disulfide bond to completely restore the wildtype γ-secretase for further simulations (Table 1). During three 2-μs GaMD enhanced simulations, spontaneous activation of APP-bound γ-secretase was observed starting from its inactive cryo-EM conformation (Figures 1A and S4A and Movie S2). The activation was characterized by coordinated hydrogen bonding interactions between the active-site aspartates, APP, and a water molecule. Active site Asp257 and Asp385 moved closer to form a hydrogen bond between the protonated Asp257 and the carbonyl oxygen in Leu49 of the scissile amide bond in APP (Figure 1B). The two aspartates were ∼7 Å apart between their Cγ atoms. Water entered the enzyme active site from the intracellular side and formed hydrogen bonds with the aspartates. The hydrogen bonds with the catalytic aspartates activated the water needed for nucleophilic attack of the carbonyl carbon of the scissile amide bond in APP. The distance between the carbonyl carbon of Leu49 and water oxygen was ∼3.8 Å. This active conformation is well poised for ε cleavage of the amide bond between residues Leu49 and Val50 of APP.
Table 1. Summary of GaMD Simulations Performed on Different Systems of γ-Secretase Bound by the Notch and APP Substrates.
| enzyme | substrate | disulfide bonda | Natomsb | dimension (Å3) | simulation (ns) | ΔVavg (kcal/mol)c | σΔv (kcal/mol)d |
|---|---|---|---|---|---|---|---|
| D385A (Cryo-EM) | notch | present | 240,021 | 141 × 124 × 146 | 300 × 1 | 12.91 | 7.91 |
| D385-protonated | notch | present | 240,358 | 141 × 124 × 146 | 300 × 3 | 9.97 | 6.76 |
| D257-protonated | notch | present | 240,358 | 141 × 124 × 146 | 300 × 3 | 10.36 | 6.46 |
| D385A (Cryo-EM) | APP | present | 253,650 | 141 × 124 × 147 | 300 × 1 | 12.53 | 6.58 |
| wildtype | APP | present | 253,647 | 141 × 124 × 147 | 300 × 3 | 10.46 | 6.91 |
| wildtype | APP | absent | 241,351 | 141 × 124 × 147 | 2000 × 3 | 10.45 | 6.78 |
| wildtype | I45F APP | absent | 241,355 | 141 × 124 × 147 | 1100 × 3 | 10.30 | 6.79 |
| wildtype | T48P APP | absent | 241,348 | 141 × 124 × 147 | 1400 × 3 | 10.87 | 6.87 |
| wildtype | M51F APP | absent | 241,360 | 141 × 124 × 147 | 1500 × 3 | 10.08 | 7.38 |
The artificial disulfide bond between the N-terminus of APP and PS1 HL1 loop of the γ-secretase is kept (“present”) or removed (“absent”).
Natoms is the number of atoms in the simulation systems.
ΔVavg and
σΔV are the average and standard deviation of the GaMD boost potential, respectively.
Figure 1.

Conformational changes of the catalytic subunit presenilin (PS1) and APP substrate during activation of the computationally restored wildtype γ-secretase. (A) Comparison of the inactive cryo-EM structure (green) and wildtype active conformation of APP-bound PS1 (red). (B) The active site poised for proteolysis. Water entered the active site and formed hydrogen bonds with the catalytic aspartates, being ready for nucleophilic attack on the scissile amide bond between residues Leu49 and Val50 of APP for ε cleavage. (C–F) Conformational changes of (C) PS1 TM2, (D) PS1 TM6a, (E) the C-terminus of APP, (F) PS1 TM1 and PS1 TM8 during activation of γ-secretase. The extracellular end of TM2 moved outward by ∼2.5 Å in the active PS1 relative to the inactive cryo-EM structure. The PS1 TM6a moved upward by ∼2 Å compared to the cryo-EM structure. The C-terminal β-strand region of APP moved closer to interact with the PS1 TM6a helix. Residue Leu52 of APP moved by ∼6 Å toward nonpolar residues Val272, Leu270, and Ala275 in the enzyme TM6a. The intracellular ends of TM8 and TM1 moved from the cryo-EM structure by ∼4.5 Å and ∼3.5 Å, respectively.
Water molecules entered the active site of γ-secretase through a channel formed by the C-terminal β-strand of APP, the C-terminal β-strand of PS1 TM7 and the C-terminal loop region of TM6a, which was open to the intracellular solvent (Figure S5). The residues comprising this channel are listed in Table S1. In addition, water molecules visited another pocket adjacent to the active site. This pocket was located between the TM8 and TM9 in PS1 near the protein surface. Since the polar water molecules cannot diffuse into the hydrophobic lipid bilayer, they returned to the active site and navigated through the open channel to the intracellular solvent. This was reflected in the pathway of the water molecule that formed hydrogen bonds with catalytic aspartates for activation of the enzyme (Figure S5B).
RMSFs were calculated from GaMD simulations of the enzyme–substrate complex (Figure S6). In nicastrin, extracellular helices α1, α2, α4a, the C-terminal regions of α5, α12, α17, and TM domain exhibited high fluctuations with ∼3 Å RMSF (Figure S6A). The TM6 and Helix-8 of APH1 were also flexible during the simulations. In PS1, TM2 extracellular domain, TM6, and TM6a were flexible with ∼2.5–3 Å RMSF. Through structural clustering of GaMD simulation snapshots (see Methods), the top cluster was obtained as the representative wildtype active conformation of the enzyme. Relative to the cryo-EM structure, the extracellular end of TM2 moved outward by 2.5 ± 0.5 Å (Figure 1C), and TM6a moved upward by 2.0 ± 0.3 Å (Figure 1D). Conformational changes of these domains involved a significant number of PS1 FAD mutation sites, including Gln127, Arg128, Ser132, Pro264, Pro267, Arg269, Leu271, Val272, Glu273, and Thr274 (www.alzforum.org). Interestingly, His131 from TM2 and Cys263 from TM6a flipped their side chains. The N-terminal helix region of APP moved outward by 10.0 ± 2.0 Å during enzyme activation (Figure 1A), while the C-terminal β-strand of APP moved by 6.0 ± 1.0 Å to interact with the PS1 TM6a helix. In the process, APP residue Leu52 made new contacts with residues Val272 and Ala275 in TM6a of PS1 (Figure 1E). The movement was consistent with the previous finding that TM6a undergoes large conformational change upon substrate binding and plays a key role in activation of the enzyme.13 In addition, the intracellular ends of TM8 and TM1 moved by 4.5 ± 0.8 Å and 3.5 ± 0.5 Å, respectively (Figure 1F). Residues Ser104, Phe105, and Tyr106 in the N-terminal region of PS1 HL1 changed into a helical conformation during enzyme activation (Figure S7B,F). In summary, we have captured activation of computationally restored wildtype γ-secretase bound by wildtype APP in the GaMD simulations.
GaMD Simulations Correlated with Biochemical Experiments on Cleavage of Wildtype and Mutant APP
MS experiments were carried out to analyze AICD species (AICD49–99 and AICD50–99) generated in proteolysis of the wildtype APP and three mutants (I45F, T48P, and M51F) by γ-secretase assay (Figure 2A–D). For the wildtype APP, the MALDI-TOF analysis showed the presence of both AICD species, but the AICD50–99 species had relatively higher intensity than the AICD49–99 species (Figure 2A). The difference in the amount of AICD fragments suggested that the γ-secretase preferred ε cleavage between Leu49-Val50 to the cleavage between Thr48–Leu49 in the wildtype APP, as has been previously reported.10 Such experimental data correlated well with GaMD simulations with the wildtype APP substrate, during which the activated enzyme was poised to cleave wildtype APP between Leu49–Val50 (Figure 1B).
Figure 2.

Mass spectrometry and Western blotting of the APP intracellular domain (AICD) fragments and GaMD free energy profiles of wildtype and mutant APP-bound γ-secretase. (A–D) The intensity of different AICD fragments detected by mass spectrometry for (A) wildtype (AICD 50–99, expected mass: 6905.6 g/mol, observed mass: 6907.4 g/mol; AICD 49–99, expected mass: 7018.8 g/mol, observed mass: 7019.6 g/mol), (B) I45F (AICD 50–99, expected mass: 6905.6 g/mol, observed mass: 6905.4 g/mol; AICD 49–99, expected mass: 7018.8 g/mol, observed mass: 7019.8 g/mol), (C) T48P (AICD 50–99, expected mass: 6905.6 g/mol, observed mass: 6907.4 g/mol; AICD 49–99, expected mass: 7018.8 g/mol, observed mass: 7041.8 g/mol) and (D) M51F (AICD 49–99, expected mass: 7034.8 g/mol, observed mass: 7031.4 g/mol; AICD 48–99, expected mass: 7135.8 g/mol, observed mass: 7132.2 g/mol) APP substrate as cleaved by γ-secretase. (E–G) 2D free energy profiles of the Asp257:Cγ–Asp385:Cγ and Asp257:protonated O–Leu49:O distances calculated from GaMD simulations of (E) wildtype, (F) I45F, and (G) T48P APP substrate. (H) 2D free energy profile of the Asp257:Cγ–Asp385:Cγ and Asp257:protonated O–Thr48:O distances calculated from GaMD simulations of the M51F APP substrate. (I) The total amount of AICD species in γ-secretase determined in vitro by Western blotting using anti-Flag antibodies of γ-secretase.
During activation, the wildtype APP-bound γ-secretase also sampled “inhibited’ and “intermediate” low-energy states as identified from the GaMD reweighted free energy profile (Figure 2E). In the inhibited state, the catalytic aspartates moved very close to each other, with only ∼4 Å distance between the Cγ atoms, while the substrate was ∼6 Å away from the active site. This conformation could not accommodate water between the aspartates to form hydrogen bonds. A similar inhibited state of the enzyme was also observed in the dipeptidic inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT)-bound cryo-EM structure (PDB: 5FN2)41 (Figure S8). With the enzyme active site in the inhibited state, APP substrate moved away from the catalytic aspartates in the GaMD simulations. The carbonyl oxygen in Leu49 of APP was ∼6 Å from the protonated oxygen of Asp257 (Figure 2E). In the intermediate state, while the Cγ atoms of the catalytic aspartates were ∼6 Å apart, being similar to the active conformation, the catalytic Asp257 was still ∼7 Å away from the target carbonyl oxygen of Leu49 in the APP substrate (Figure 2E). The intermediate state adopted by γ-secretase may reflect its flexibility during activation and substrate processing and the well-known slow kinetics of the enzyme.
For the I45F and T48P mutants of APP substrate, the MS analysis showed a decreased amount of the AICD49–99 species from proteolysis of both mutants compared with the wildtype substrate, and AICD50–99 was the predominant AICD product (Figure 2B,C). Thus, ε cleavage between Leu49–Val50 was even more preferred for these two mutants than for the wildtype substrate. These results are consistent with recent findings that these two FAD mutations act as “pathway switchers” to increase the Aβ42/Aβ40 ratio, rather than shifting the ε cleavage site toward formation of Aβ48 and the Aβ42 pathway.10 In parallel with the experiments, further GaMD simulations were performed on γ-secretase bound by the I45F and T48P mutant APP substrates. The I45F mutant substrate-bound γ-secretase became activated during 1.1 μs GaMD simulations (Figure S4B). A low-energy conformation was observed in the I45F active state for which the distance between the Cγ atoms of Asp257 and Asp385 was ∼7 Å, while the Leu49 carbonyl oxygen and Asp257 protonated oxygen formed a hydrogen bond with ∼3 Å distance (Figure 2F). The boost potential was 10.30 ± 6.79 kcal/mol in the GaMD simulations of the I45F mutant substrate-bound enzyme, which was comparable to that of the wildtype system (10.45 ± 6.78 kcal/mol) (Table 1). However, the I45F mutant APP substrate-bound γ-secretase was activated within a shorter simulation time compared with the wildtype APP substrate-bound enzyme, with higher probability of conformations for the ε cleavage between Leu49–Val50 in APP (Figure S4A,B). The simulation findings agreed well with the experimental data. Analysis of AICD products by MALDI mass spectrometry revealed a higher peak intensity of AICD50–99 than AICD49–99 for the I45F mutant APP compared with the wildtype APP (Figure 2A,B). Another low-energy conformation of I45F APP substrate-bound enzyme was observed in the inhibited state (Figure 2F), being similar to the inhibitor DAPT-bound structure of γ-secretase (PDB: 5FN2).41
For the T48P mutant APP substrate-bound γ-secretase, activation was observed during one of three 1.5 μs GaMD simulations (Figure S4C). Low-energy conformations were identified from the free energy profile in the active, inhibited, and intermediate states (Figure 2G). In the T48P active state, the catalytic aspartates were positioned ∼7 Å apart (Cγ to Cγ), and the substrate Leu49 carbonyl oxygen aligned with the protonated oxygen of Asp257 to form a hydrogen bond. The boost potential was 10.87 ± 6.87 kcal/mol, which was also comparable to that of wildtype APP simulations (Table 1). The T48P mutant APP substrate-bound γ-secretase transitioned into the active state within a shorter simulation time compared to the wildtype system (Figure S4A,C). The T48P APP substrate mutant had a higher probability than the wildtype APP substrate of aligning the aspartates and water with the scissile amide bond between Leu49–Val50 in APP. This computational finding was again consistent with MALDI mass spectrometric analysis of AICD products: AICD50–99 intensity is higher than AICD49–99 for the T48P mutant substrate compared to that of the wildtype system (Figure 2A,C). The observed inhibited state (Figure 2G) was similar to that seen in the wildtype and I45F systems (Figure 2E,F) as well as the inhibitor DAPT-bound cryo-EM structure of γ-secretase (PDB: 5FN2).41 In the T48P intermediate state, the Cγ atoms of the catalytic aspartates were ∼6 Å apart, while the Leu49 carbonyl oxygen of APP substrate and the protonated oxygen of Asp257 were ∼7 Å apart (Figure 2G). The I45F and T48P mutant APP substrate-bound γ-secretase showed a similar structural flexibility as the wildtype system in the RMSFs calculated from GaMD simulations (Figure S6A). In both systems, extracellular helices α1, α2, α4a, the C-terminal regions of α5, α12, α17, and TM domain of nicastrin exhibited high fluctuations. The TM6 and helix-8 of APH-1, and the TM2 extracellular domain, TM6, and TM6a of PS1 were also flexible during the GaMD simulations (Figure S6C,D).
Shifted ε Cleavage Site of APP in the M51F Mutant
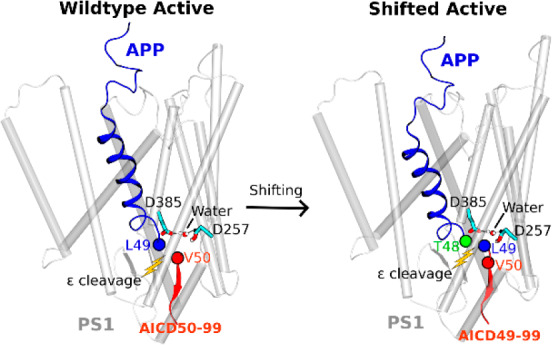
MS analysis of AICD products from the M51F mutant system revealed AICD49–99 as the major product, suggesting that the predominant ε cleavage site of M51F APP was between residues Thr48–Leu49 (Figure 2D). A low level of AICD48–99 was also detected, revealing that the M51F APP substrate was cleaved to a limited degree between Ile47–Thr48 (Figure 2D). This was consistent with previous studies that a Phe residue is not tolerated in the P2’ position of substrate or transition-state analogue inhibitors of γ-secretase.10,42 Thus, M51F mutation of the APP substrate shifted the ε cleavage site from Leu49-Val50 to Thr48-Leu49. Such a shift of ε cleavage was consistently observed in 1.5 μs GaMD simulations of the M51F mutant APP substrate bound to γ-secretase (Figure S4D and Movie S3). The protonated oxygen of PS1 Asp257 was hydrogen bonded to the carbonyl oxygen of Thr48, and the activated water molecule targeted the scissile amide bond between Thr48 and Leu49 in the M51F APP mutant for ε cleavage. In comparison, residue Thr48 in the wildtype APP maintained a distance of ∼8–9 Å between its carbonyl oxygen and the protonated oxygen of the PS1 Asp257 (Figure S4E). A distinct low-energy state was identified for the “shifted” conformation in the free energy profile of the M51F APP system (Figure 2H). In the shifted state, the Cγ atoms of the catalytic aspartates were ∼7 Å apart, and the carbonyl oxygen of APP substrate Thr48 and protonated oxygen of PS1 Asp257 formed a hydrogen bond with ∼3 Å distance. Moreover, in one of the three GaMD simulations, the ε cleavage site of M51F mutant APP was further shifted to the amide bond between Ile47–Thr48. The distance between the carbonyl oxygen of APP substrate residue Ile47 and the protonated oxygen of PS1 Asp257 became ∼3 Å (Figure S9). The Cγ atoms of the catalytic aspartates were ∼7 Å apart. This observation was consistent with the low level of AICD48–99 fragment detected by MS of AICD products of the M51F APP mutant (Figure 2D).
In addition to the MS experiments, the effect of APP mutations was investigated by detecting the total amount of Flag-tagged AICD species in in vitro γ-secretase assays by Western blotting using anti-Flag antibodies (Figure 2I). The AICD production increased substantially for the M51F mutant substrate compared to wildtype APP substrate, although we note that this increase was not apparent in a previous study.10 In contrast, ε proteolysis of the I45F and T48P mutants showed no drastic change in the total AICD level compared with wildtype APP substrate (Figure 2I). This was highly consistent with the GaMD simulations. In the systems with wildtype, I45F and T48P mutant APP substrate, the low-energy inhibited state was observed in the free energy profiles of γ-secretase, but not for the M51F mutant system (Figure 2E–H). Relative to the active low-energy minimum, the PMF free energy value of the inhibited state was 0.19, 0.29, and 1.80 kcal/mol for the wildtype, T48P and I45F APP systems, respectively. However, it is important to note that the GaMD simulations and free energy profiles were still not converged, especially for the T48P and M51F mutant systems, similarly for populations of the different enzyme conformational states. Nevertheless, the relative population probability of the inhibited to the active state could be estimated according to the Boltzmann distribution of the PMF values as roughly 0.74, 0.62, and 0.05 for the wildtype, T48P, and I45F APP systems, respectively.
Structural clustering was performed on GaMD simulations of M51F APP-bound γ-secretase, and the top cluster was identified as the shifted conformational state of the enzyme. Compared with the wildtype active conformation (Figure 3A), the extracellular end of TM2 in PS1 moved outward by 5.5 ± 0.5 Å (Figure 3B). The helix involving residues Thr124, Val125, Gly126, and Gln127 became disordered in this region (Figure 3B). The APP substrate moved downward by 4.0 ± 0.5 Å in the substrate binding channel of the enzyme (Figure 3A,C). In comparison, the catalytic aspartates and flanking regions of TM6 and TM7 moved less than APP (Figure 3C,E). Upon shifting of the ε cleavage site, local rearrangements of APP residues were required to establish the coordinated hydrogen bonding interactions at the active site (Figure 3C and Movie S3). Side chain flipping of the APP Thr48 residue led to formation of a hydrogen bond between its carbonyl oxygen and the PS1 Asp257 protonated oxygen (Figure S4D). Residue Leu49 initially facing the activated water between these catalytic aspartates flipped the side chain and moved downward by 4.0 ± 0.5 Å. The PS1 TM6 helix in the M51F shifted state moved toward the active site by 4.0 ± 0.6 Å relative to the wildtype active conformation (Figure 3A). Moreover, the TM6a helix tilted by 60° ± 5° and 6.0 ± 1.0 Å distance relative to the wildtype active conformation (Figure 3A,D). Meanwhile, the β-strand at the C-terminus of APP deformed to a turn as it moved away from the TM6a helix. APP Leu52, interacting with the nonpolar residues of TM6a in the wildtype active conformation, flipped its side chain and moved away from these residues by 6.0 ± 0.6 Å (Figure 3E). The intracellular domains of TM1 and TM8 in the M51F shifted state moved by 2.5 ± 0.3 Å compared with the wildtype active conformation (Figure 3F). PS1 FAD mutation sites Ala79, Val82, Ile83, Met84, Leu85, Pro88, Leu424, and Ala426 from TM1 and TM8 showed similar movements of their side chains. In addition, RMSF of the M51F mutant APP-bound γ-secretase calculated from GaMD simulations showed higher flexibility in TM2, TM6, and TM6a regions of PS1 (Figure S6B). This extra flexibility is consistent with the ability of the M51F mutant system to readjust the positioning of the substrate in the active site in shifting the ε cleavage site.
Figure 3.

Conformational changes of the catalytic subunit presenilin (PS1) and APP in the shifted active (M51F) states of γ-secretase compared with the active (wildtype) state. (A) Overview of the active (red) and shifted active (blue) conformations of APP-bound PS1. (B) The extracellular end of the PS1 TM2 moved outward by ∼5.5 Å in the shifted active (M51F) conformation relative to the active (wildtype) structure. Residues Thr124, Val125, Gly126, and Gln127 in this region lost the helical conformation in the shifted active state. (C) The active site poised to attack the scissile amide bond between residues Leu49 and Val50 in the active state (red) and between residues Thr48 and Leu49 in the shifted active state (blue) of APP for ε cleavage. Side chain flipping of the APP Thr48 residue led to formation of a hydrogen bond between its carbonyl oxygen and the PS1 Asp257 protonated oxygen. Residue Leu49 initially facing the center of two aspartates flipped to the other side with a downward movement of ∼4 Å. (D) The N-terminus of PS1 TM6 moved toward the active site by ∼4 Å and the TM6a helix tilted by ∼60°. (E) The β-strand at the C-terminus of APP substrate deformed to a turn as it moved away from TM6a in PS1. The APP Leu52 interacting with nonpolar residues in PS1 TM6a in the active conformation flipped its side chain and moved in the opposite direction by ∼6 Å. (F) The intracellular ends of TM1 and TM8 moved by ∼2.5 Å and ∼2.5 Å in PS1, respectively.
Changes in Secondary Structures of APP Substrate Mutants
Changes in secondary structures of the wildtype and mutant APP substrate in γ-secretase were monitored during the GaMD simulations (Figures 4, S10, and S11). Secondary structures of APP substrate in the active conformations of the wildtype, I45F and T48P mutant systems and the shifted active conformation of the M51F mutant system were compared using their top ranked structural clusters obtained from the corresponding simulations (Figure S12). For wildtype APP substrate, residues Gly29 to Val46 formed a helical conformation throughout the simulations except between residues 42 and 43 (Figure 4A). Residues Asn27–Lys28 fluctuated between turn and coil conformations during last ∼700 ns for activation, whereas Ile47–Leu49 fluctuated between helix and turn conformations throughout the simulation. The N-terminal region of APP substrate was very flexible and sampled turn and coil conformations. The C-terminal residues Leu52 to Lys55 primarily maintained an antiparallel β-sheet conformation. Residues Val50–Met51, immediately after the Leu49–Val50 ε cleavage site, formed a turn for a number of times that exposed this APP scissile amide bond to the enzyme aspartates and coordinated water for proteolysis.
Figure 4.

Time courses of the APP secondary structures in the (A) wildtype, (B) I45F, (C) T48P, and (D) M51F forms as bound to γ-secretase calculated from their representative GaMD simulations. Results of the other simulations are plotted in Figures S8 and S9.
The I45F and T48P mutants of APP substrate, which maintained ε cleavage between Leu49–Val50, showed similar secondary structures as the wildtype substrate, although unique features were also observed in each mutant. Both mutant substrates formed turns at residues Val50–Met51 despite fluctuations (Figure 4B,C) and adopted a β-sheet conformation at the C-terminus during simulations. However, only residues Ile31–Val46 formed a helix in the I45F mutant, with ∼2–3 residues toward the N-terminus losing the helical conformation (Figure 4B). The N-terminal region of I45F mutant APP substrate was thus more flexible than the wildtype substrate and bent over the HL1 loop of PS1 (Figures S7C,F and S13). For the I45F APP mutant substrate, multiple hydrogen bonds were formed between the N-terminal residues of APP and PS1 HL1. Residue Gln112 in the PS1 HL1 loop—mutated to Cys112 in the cryo-EM structure to generate a disulfide bond and restored in our simulation—formed three hydrogen bonds with residues Ser26, Asn27, and Lys28 of the APP N-terminus (Figure S13A). Two of these hydrogen bonds involved backbone atoms. In addition, the backbone N atom of Ile114 in PS1 HL1 formed a hydrogen bond with the backbone O atom of Lys28 of APP. These hydrogen bonds contributed to a parallel β-sheet between the PS1 HL1 and APP N-terminus as reflected in the secondary structure plots (Figures 4B, S10, S13A). In contrast, the N-terminal loop of wildtype APP was observed flexible without bending over the PS1 HL1 (Figure S7B,F). For the T48P mutant APP, residues Gly29–Ala42 formed a helical conformation, whereas residues Thr43–Ile47 fluctuated between the α-helix, 3-10 helix, and turn conformations (Figure 4C). Residues Ser104, Phe105, Tyr106, and Thr107 of the PS1 HL1 loop formed a helix in the T48P active conformation, similar to what was observed in the wildtype active state (Figure S7D,F).
For the M51F mutant APP, residues Ala30-Val46 formed a helical conformation during the GaMD simulations (Figure 4D). A longer turn appeared starting from residue Leu49 to Leu52 in M51F APP during the simulations (Figures 4D, 3E, and S12). In comparison, a turn was formed for only residues Val50–Met51 in the wildtype APP that exposed the Leu49–Val50 scissile amide bond for ε cleavage (Figures 4A and S12). The shift of this turn correlated with the shift of the ε cleavage site. The C-terminal β-strand became shorter in the M51F APP substrate mutant (Figure 4D) and even completely disappeared in the representative M51F shifted active conformational state (Figures S12 and 3E). As the M51F mutant APP substrate moved downward relative to PS1, its N-terminus formed more interactions with PS1 HL1 (Figure S7E,F). The backbone O and N atoms of Gly111 in HL1 often formed hydrogen bonds with the backbone atoms N of Glu22 and O of Phe20 in the APP substrate, respectively (Figure S7E,F and S12B). These hydrogen bonds resulted in a parallel β-sheet conformation (Figure 4D). The N-terminus of the T48P mutant APP was also found in proximity with the PS1 HL1 loop (Figure S7D,F). The PS1 HL1 loop—with high flexibility and multiple interactions with APP substrate—make it one of the most important regions of PS1 in the context of enzymatic function and Alzheimer’s disease pathogenesis.43,44 The PS1 HL1 has a large number of FAD mutation sites, including Phe105, Gly111, Leu113, Tyr115, and Gln127. Hence, our simulation findings were consistent with the literature regarding the importance of the HL1 loop.
The C-terminus of APP substrate-bound to the active wildtype conformation moved toward PS1 TM6a region by ∼6 Å during enzyme activation (Figure 1E). The C-terminus of APP maintained β-sheet conformations with the N-terminus of PS1 TM7 throughout the simulations. Hence, with the movement of C-terminus of APP, the N-terminus of PS1 TM7 also moved along by ∼6 Å (Figure S14). In contrast, I45F and T48P mutant APPs maintained the β-sheet conformations with the N-terminus of PS1 TM7 without the movement of the C-terminus. M51F mutant APP lost its interaction with the PS1 TM7 and hence losing the β-sheet conformations (Figures 4D and 3E).
Comparison of the S1′, S2′, and S3′ Active-Site Subpockets in the Wildtype and Mutant APP Substrate-Bound γ-Secretase
Representative active conformations of PS1 were identified as the top ranked structural clusters from the GaMD simulations of the wildtype, I45F, and T48P mutant systems and the shifted active conformation from the M51F system simulations. These conformations were aligned and compared for the enzyme active-site S1′, S2′, and S3′ subpockets that were occupied by APP substrate residues P1′, P2′, and P3′, respectively10 (Figure 5). In the active wildtype conformation, the S1′ subpocket occupied by P1′ residue (Val50) constituted residues mostly from TM6 and TM6a as listed in Table S2. The S3′ subpocket occupied by P3′ residue (Leu52) constituted residues mostly from TM6a and the C-terminus of PS1-NTF. The S1′ and S3′ subpockets were located on the same side with respect to APP (Figure 5A). In contrast, the S2′ subpocket occupied by P2′ residue (Met51) constituted residues mostly from TM8, TM8–TM9 loop, and the β-strand region of TM7 (Table S2).
Figure 5.
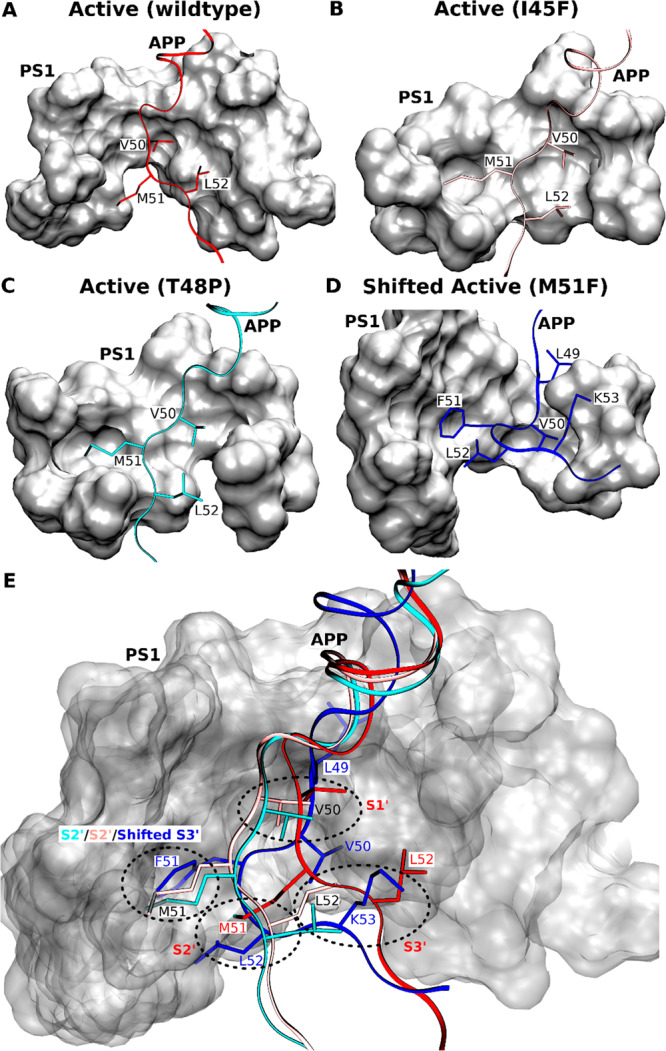
(A–D) Comparison of the locations of APP substrate residues P1′, P2′, and P3′ in the (A) wildtype active, (B) I45F active, (C) T48P active, and (D) shifted active M51F APP substrate-bound conformations of γ-secretase. (E) Comparison of the corresponding PS1 active-site S1′, S2′, and S3′ pockets in these different conformational states of γ-secretase.
In the I45F and T48P active conformations (Figure 5A,–C), the S1′ and S3′ subpockets occupied by the P1′ (Val50) and P3′ (Leu52) residues, respectively, embodied the same S1′ and S3′ subpockets of the wildtype active conformation. The S2′ pocket occupied by P2′ (Met51) of the I45F and T48P mutant APP substrate comprised residues from TM8, the TM8–TM9 loop, the β-strand region of TM7 and part of TM1. Notably, both the S1′ and S2′ subpockets involved the PAL motif (P433–A434–L435) in the TM9 N-terminal region that is considered important for substrate binding.45 This S2′ subpocket occupied by the APP mutants was located on the same side of substrate but ∼4 Å above the extended S2′ subpocket in the wildtype active conformation.
For the M51F APP mutant, the presence of a bulky residue Phe at the P2′ position induced local rearrangements and shifted the ε cleavage site. With the shift, Leu49, Val50, and Phe51 became the new P1′, P2′, and P3′ residues, respectively. The new P1′ residue occupied a distinct subpocket near to the S1′ subpocket in the wildtype active conformation (Figure 5A,D,E). In contrast, the new P2′ residue occupied a new subpocket in the space between the S1′ and S3′ subpockets of the wildtype active conformation of PS1. The new P3′ residue occupied the same extended pocket as S2′ subpocket in the I45F active and T48P active conformations (Figure 5B,C,E). Hence, the new subpocket occupied by the P3′ residue (F51) is termed “shifted S3′ subpocket” here and also involved the PAL motif (Figure 5D,E). Moreover, the L52 (P4′) and K53 (P5′) residues in the M51F shifted active conformation occupied what were the S2′ and S3′ subpockets in the wildtype active conformation of PS1, respectively (Figure 5D,E).
The location of the S2′ subpocket differed among the active conformations of the wildtype active, I45F active, and T48P active conformations. As the C-terminus of I45F and T48P mutant APP moved by ∼6 Å compared with the wildtype APP (Figure S14), the P2′ residue (Met51) of these mutants occupied a different S2′ subpocket (Figure 5). Because of the shift in the ε cleavage site, the C-terminus of M51F mutant APP lost interactions with the N-terminus of PS1 TM7 and PS1 TM6a. This resulted in large conformational tilting of PS1 TM6a helix in the M51F shifted conformation (Figure 3). Therefore, the conformational changes and molecular interactions of the APP with the γ-secretase provided important insights into the mechanisms of activation and substrate processing by the enzyme.
Discussion
The PS1-containing γ-secretase complex is a founding member of intramembrane-cleaving proteases (I-CLiPs) which carry out hydrolysis of substrate TM domains within the hydrophobic environment of the lipid bilayer.46 I-CLiPs also include the S2P metalloproteases, rhomboid serine proteases, and presenilin-like aspartyl proteases. Although microbial representatives of each of these other I-CLiP classes have been crystallized for high-resolution structure determination,47−50 visualizing the active state and elucidating the molecular mechanism of intramembrane proteolysis has been challenging. Only very recently has a rhomboid protease been studied through time-resolved X-ray crystallography to reveal how this serine protease hydrolyzes transmembrane substrates.51 Most recently, structures of the γ-secretase complex bound to Notch and APP substrates have been reported, providing critical insights into substrate recognition of γ-secretase.12,13 Nevertheless, mutations in the enzyme and substrate needed for stabilization of the substrate–enzyme complex precluded visualization of the active protease and raised the possibility of unnatural substrate interactions.
Using the latest cryo-EM structures, we have, for the first time, developed an all-atom MD model for activation of the APP substrate-bound γ-secretase poised for intramembrane proteolysis that is in excellent agreement with mass spectrometry and Western blotting biochemical experiments. Extensive simulations using a novel GaMD enhanced sampling method have captured spontaneous activation of γ-secretase in the presence of APP and water (Figure 6). The catalytic aspartates moved into close proximity, similar to previous simulation findings,14−16 although these studies were performed without the APP substrate bound to the γ-secretase active site. Previous studies suggested a putative active conformation of the apo γ-secretase but was unable to fully characterize the enzyme activation involving additional coordinated hydrogen bond interactions with the substrate. In the GaMD simulations, water molecules entered the active site, one of which coordinated with the two aspartates (Figure 6B and Movies S1 and S2). Moreover, Asp257 formed a hydrogen bond with the carbonyl oxygen of the scissile amide bond between APP residues Leu49–Val50. The activated water molecule was poised for nucleophilic attack on the backbone carbon atom of this activated Leu49–Val50 amide bond. While a number of regions of nicastrin, Aph-1, and Pen-2 displayed flexibility during simulations of the activated enzyme–substrate complex, the PS1 TM6a was the most noteworthy, as this region interacted directly with substrate near the cleavage site and appeared to play a role in enzyme activation. The wildtype enzyme–substrate complex additionally sampled the inhibited and intermediate conformational states, the former closely resembling the conformation of the DAPT inhibitor-bound γ-secretase.41 The current ∼2-μs GaMD simulation of γ-secretase with wildtype APP captured the enzyme activation for ε cleavage of APP between Leu49–Val50. The ε cleavage of wildtype APP between Thr48–Leu49 with lower probability, as detected by MS, would likely require a longer simulation time and more sufficient sampling.
Figure 6.

Summary of the (A) inactive cryo-EM, (B) active (wildtype), and (C) shifted active (M51F) conformational states of the APP substrate-bound γ-secretase. Distinct AICD products were generated from the wildtype and M51F mutant APP. The complementary simulations and experiments have revealed mechanisms of the γ-secretase activation and its ε cleavage of the APP substrate.
GaMD simulations on I45F and T48P APP substrate-bound γ-secretase revealed faster activation of PS1 for proteolysis at the ε cleavage site between Leu49–Val50 with these two FAD mutations compared to the complex with wildtype APP substrate. These observations were consistent with MS analysis of AICD proteolytic products: the two FAD mutant substrates were cleaved by γ-secretase with a greater AICD50–99/AICD49–99 ratio than was the wildtype substrate. Moreover, the M51F mutation resulted in dramatic conformational changes of APP (Figure 6C and Movie S3), setting up ε cleavage between Thr48–Leu49. These results were entirely consistent with the known incompatibility of Phe in the P2′ position.10 MS experimental results also showed the major AICD product generated by γ-secretase from the M51F mutant APP substrate was due to cleavage between Thr48–Leu49. Little or no cleavage occurred between Leu49–Val50. In addition, Western blotting revealed a substantial increase in the total AICD production in the in vitro γ-secretase assay for the M51F mutant APP substrate compared to the wildtype APP substrate. In contrast, I45F and T48P mutant APP-bound γ-secretase showed a similar amount of AICD production as the wildtype APP bound γ-secretase. This was in exceptional agreement with the GaMD simulation: the low-energy inhibited state was observed in the free energy profiles of the wildtype, I45F and T48P mutant APP bound γ-secretase, but absent in the M51F mutant APP system. These strong correlations between the GaMD simulations and biochemical experiments provided substantial validity to our dynamic model of γ-secretase. However, the GaMD simulations and calculated free energy profiles were still not converged, especially for the T48P and M51F mutant systems. It is exceedingly difficult to accomplish converged simulations on large and complex biomolecules such as γ-secretase even with GaMD enhanced sampling. Nevertheless, relatively low-energy conformational states could be still identified from the GaMD free energy profiles, e.g., the active, intermediate, and inhibited for the T48P APP system (Figure 2G). The same low-energy conformational states were also identified in the free energy profile calculated from GaMD simulations of the wildtype APP system (Figure 2E) and two of those states from simulations of the I45F APP system (Figure 2F).
The active-site S1′, S2′, and S3′ subpockets were visualized in the wildtype active, I45F active, T48P active, and M51F shifted active conformations of PS1 obtained from GaMD simulations. The protein residues (Figure 5 and Table S2) found in the S1′ and S3′ subpockets of the active wildtype, I45F, and T48P conformations were the same as those identified in a recent computational study by Hitzenberger et.al.24 However, the S2′ pocket of the wildtype active PS1 was identified in a distinct location that shifted by ∼4 Å toward the APP C-terminus from the previously described S2′ pocket.24 The subpocket described by Hitzenberger et al.,24 on the other hand, appeared to be the S2′ pocket in the I45F and T48P active conformations and the shifted S3′ subpocket for the M51F APP (Figure 5E). Shift of the S2′ subpocket from the wildtype active conformation to the I45F and T48P active conformations resulted from the simultaneous movements of the APP C-terminus and PS1 TM7 N-terminus toward the PS1 TM6a in order to maintain the β-sheet structure of this domain in the GaMD simulations (Figure S14). In comparison, the I45F and T48P APP-bound γ-secretase appeared to sample a smaller conformational space than the WT APP-bound enzyme in their free energy profiles as shown in Figure 2, suggesting that the mutant complexes underwent lower fluctuations with reduced conformational flexibility compared to the WT. Therefore, the GaMD simulations revealed a newly identified S2′ subpocket for wildtype APP, while the previously described S2′ subpocket24 was used as the S2′ for I45F and T48P APP as well as the shifted S3′ for the M51F APP (Figure 5).
In summary, we have combined all-atom GaMD simulations with MS and Western blotting experiments to probe the mechanisms of γ-secretase activation and its ε cleavage of the wildtype and mutant APP substrates. Extensive GaMD simulations using the latest cryo-EM structures of γ-secretase have captured spontaneous activation of the enzyme, for which the active-site Asp385 has been restored and the artificial enzyme–substrate disulfide bond has been removed. The active conformation is characterized by water-bridged hydrogen bonds between the two catalytic aspartates, one of which formed another hydrogen bond with the carbonyl oxygen of the target scissile amide bond for the ε cleavage of APP. Free energy calculations of the GaMD simulations also allowed us to identify distinct intermediate, inhibited, and shifted active conformational states of γ-secretase. The simulations predicted ε cleavage preferences of the wildtype and three mutants of APP that were highly consistent with MS and Western blotting experimental findings of the AICD species. The validated GaMD simulations were then used to interpret the experimental data at an atomistic level. Remarkably, the M51F mutation shifted the ε cleavage site of APP from the amide bond between Leu49–Val50 to the Thr48–Leu49 bond, generating predominantly the AICD49–99 fragment instead of the AICD50–99 as detected by MS. Finally, the GaMD simulations have systematically revealed the active-site S1′, S2′, and S3′ subpockets of γ-secretase that interact with the P1′, P2′, and P3′ residues in the wildtype and mutant APP. This provides an in-depth picture of the ε proteolytic cleavage of different APP substrates by γ-secretase. The GaMD method is apparently very well suited for the study of this extremely slow-acting membrane protease complex. In order to fully understand the functional mechanisms of γ-secretase, further simulation and experimental studies have been planned on the tripeptidase activity of the enzyme and effects of other FAD mutations in both the APP substrate and γ-secretase. These studies are expected to greatly facilitate rational drug design targeting γ-secretase for the AD therapeutic treatments.
Materials and Methods
Cloning
All mutations in C100 FLAG were introduced by site-directed mutagenesis (QuickChange Lightning Site Directed Mutagenesis kit, Agilent) in pET 22b vector. All constructs were verified by sequencing by ACGT.
C100-FLAG Substrate Purification
Escherichia coli BL21 cells were grown in LB media until the OD600 reached 0.6. Cells were induced with 0.5 mM IPTG and were grown post induction for 4 h. The cells were then pelleted by centrifugation and resuspended in 50 mM HEPES pH 8, 1% Triton X-100. The cells were lysed by a French press, and the lysate was incubated with anti-FLAG M2-agarose beads from SIGMA. Bound substrates were then eluted from the beads with 100 mM glycine pH 2.5, 0.25% NP-40 detergent and then neutralized with Tris HCl prior to being stored at −80 °C.
γ-Secretase Expression and Purification
γ-Secretase was expressed in HEK 395F cells by transfection with pMLINK vector containing all four components (presenilin-1, Pen-2, Aph-1, nicastrin) of γ-secretase complex (provided by Yigong Shi). For transfection, HEK 395F cells were grown in unsupplemented Freestyle 293 media (Life Technologies, 12338-018) until the cell density reached 2 × 106 cells/mL. 150 μg of vector was mixed with 450 μg of 25 kDa linear polyethylemimines (PEI) and incubated for 30 min at room temperature. The DNA–PEI mixtures were added to HEK cells, and cells were grown for 60 h. The cells were harvested, and γ-secretase was purified as described previously.10
In Vitro γ-Secretase Assay and Detection of AICD Species
γ-Secretase purification and assays were carried out as described previously.10 Briefly, 30 nM purified γ-secretase was dissolved into total brain lipid extract (Avanti) in 50 mM HEPES pH 7.0, 150 mM NaCl, 0.25% CHAPSO. The detergent/lipid/enzyme solution was mixed with SM-2 biobeads (Bio-Rad) for 2 h at 4 °C to remove the detergent. After removal of the bio beads, the proteoliposome solution was mixed with 3 mM recombinant C100 substrates to initiate the cleavage reaction. The reaction was carried out for 16 h at 37 °C. After 16 h, AICD-Flag products were isolated by immunoprecipitation with anti-FLAG M2 beads (SIGMA) in 10 mM MES pH 6.5, 10 mM NaCl, 0.05% DDM detergent overnight at 4 °C. AICD products were then eluted from the anti-FLAG beads with acetonitrile/water (1:1) with 0.1% trifluoroacetic acid. The elutes were run on a Bruker MALDI-TOF mass spectrometer.
Western Blotting
Samples from γ-secretase assays were run on 4–12% bis-tris gel and transferred into PVDF membrane. The membrane was treated with 5% dry milk in PBS Tween-20 for 1 h at ambient temperature. The membrane was then incubated with the anti-Flag M2 antibodies at 4 °C overnight. The membrane was washed three times with PBS Tween-20 and was incubated with antimouse secondary antibodies for 1 h. The membrane was washed and imaged for chemiluminescence.
Gaussian Accelerated Molecular Dynamics (GaMD)
GaMD is an enhanced sampling technique, in which a harmonic boost potential is added to smooth the potential energy surface and reduce the system energy barriers.26 GaMD is able to accelerate biomolecular simulations by orders of magnitude.27,33 GaMD does not need predefined collective variables. Moreover, because GaMD boost potential follows a Gaussian distribution, biomolecular free energy profiles can be properly recovered through cumulant expansion to the second order.26 GaMD has successfully overcome the energetic reweighting problem in free energy calculations that was encountered in the previous accelerated molecular dynamics (aMD) method29,31 for free energy calculations of large molecules. GaMD has been implemented in widely used software packages including AMBER,26,52 NAMD,32 and GENESIS.53 A brief summary of GaMD is provided here.
Consider a system with N atoms at positions 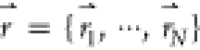 . When the system potential
. When the system potential 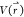 is lower than a reference energy E, the modified
potential
is lower than a reference energy E, the modified
potential 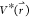 of the system is calculated as
of the system is calculated as
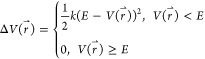 |
1 |
where k is the harmonic force constant. The two adjustable parameters E and k are automatically determined based on three enhanced sampling principles.26 The reference energy needs to be set in the following range:
| 2 |
where Vmax and Vmin are the system minimum and maximum potential
energies. To ensure that eq 2 is valid, k has to satisfy: 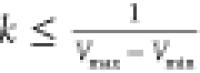 Let us define
Let us define 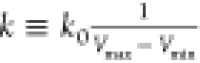 , then 0 < k0 ≤ 1. The standard deviation of ΔV needs
to be small enough (i.e., narrow distribution) to ensure proper energetic
reweighting:54 σΔV = k(E – Vavg) σV ≤ σ0 where Vavg and σV are the average and standard deviation of the system potential energies,
σΔV is the standard deviation
of ΔV with σ0 as a user-specified
upper limit (e.g., 10kBT) for proper reweighting. When E is set to the lower
bound E = Vmax, k0 can be calculated as
, then 0 < k0 ≤ 1. The standard deviation of ΔV needs
to be small enough (i.e., narrow distribution) to ensure proper energetic
reweighting:54 σΔV = k(E – Vavg) σV ≤ σ0 where Vavg and σV are the average and standard deviation of the system potential energies,
σΔV is the standard deviation
of ΔV with σ0 as a user-specified
upper limit (e.g., 10kBT) for proper reweighting. When E is set to the lower
bound E = Vmax, k0 can be calculated as
| 3 |
Alternatively,
when the threshold energy E is set to its upper bound E = Vmin + 1/k, 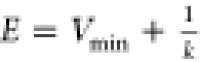 , k0 is set to
, k0 is set to
| 4 |
if k0″ is found to be between 0 and 1. Otherwise, k0 is calculated using eq 3.
Similar to aMD, GaMD provides schemes to add only the total potential boost ΔVP, only dihedral potential boost ΔVD, or the dual potential boost (both ΔVP and ΔVD). The dual-boost simulation generally provides higher acceleration than the other two types of simulations.55 The simulation parameters comprise of the threshold energy E for applying boost potential and the effective harmonic force constants, k0P and k0D for the total and dihedral potential boost, respectively.
Energetic Reweighting of GaMD Simulations
To calculate
the potential of mean force (PMF)56 from
GaMD simulations, the probability distribution along a reaction coordinate
is written as p*(A). Given the boost
potential 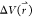 of each frame, p*(A) can be reweighted to recover the
canonical ensemble distribution, p(A), as
of each frame, p*(A) can be reweighted to recover the
canonical ensemble distribution, p(A), as
| 5 |
where M is the number of
bins, β = kBT and 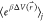 is the ensemble-averaged Boltzmann
factor
of
is the ensemble-averaged Boltzmann
factor
of 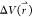 for simulation
frames found in the jth bin. The ensemble-averaged
reweighting factor can be
approximated using cumulant expansion:
for simulation
frames found in the jth bin. The ensemble-averaged
reweighting factor can be
approximated using cumulant expansion:
| 6 |
where the first two cumulants are given by
| 7 |
The boost potential obtained from GaMD simulations usually follows near-Gaussian distribution. Cumulant expansion to the second order thus provides a good approximation for computing the reweighting factor.26,54 The reweighted free energy F(A) = −kBT ln p(A) is calculated as
| 8 |
where F* (A) = −kBT ln p*(A) is the modified free energy obtained from GaMD simulation and FC is a constant.
System Setup
The earlier published cryo-EM structure of γ-secretase bound by Notch (PDB: 6IDF)12 was used for initial GaMD simulations. This system was used to optimize our simulation protocol, especially the protonation state of aspartates in the active site, which were assumed to be the same as γ-secretase processed various substrates. Another cryo-EM structure of γ-secretase bound by APP (PDB: 6IYC)13 was used to perform further GaMD simulations as per the optimized protocol. For the wildtype enzyme, residue Asp385 that was mutated to Ala at the active site in the cryo-EM structure was restored for setting up the simulation system. Similarly, the disulfide bond between Cys112 of PS1-Q112C and Cys24 of APP-V24C were removed, and the wildtype residues were restored for simulation setup. Five unresolved residues at the N-terminus of APP substrate C83 were added through homology modeling using SWISS-MODEL.57 All chain termini were capped with neutral groups, i.e., the acetyl group (ACE) for the N-terminus and methyl amide group (CT3) for C terminus. Protein residues were set to the standard CHARMM protonation states at neutral pH with the psfgen plugin in VMD.58 Then the complex was embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer with all overlapping lipid molecules removed using the Membrane plugin in VMD58 (Figure S1). The system charges were then neutralized at 0.15 M NaCl using the Solvate plugin in VMD.58 Periodic boundary conditions were applied on the simulation systems. The simulation systems of γ-secretase bound by APP are summarized in Table 1.
For APP mutant simulations systems, isoleucine, threonine,and methionine were mutated to phenylalanine, proline, and phenylalanine computationally at the 29th, 32nd, and 35th residue of APP substrate, respectively. These corresponded to I45F, T48P, and M51F mutations as per the numbering based on C99, the substrate that was cleaved to Aβ, although the actual substrate in the model was C83.
Simulation Protocol
The CHARMM36 parameter set59 was used for the protein and POPC lipids. Initial energy minimization and thermalization of the γ-secretase complex followed the same protocol as used in the previous GaMD simulations of membrane proteins.33,60 The simulation proceeded with equilibration of lipid tails. With all the other atoms fixed, the lipid tails were energy minimized for 1000 steps using the conjugate gradient algorithm and melted with constant number, volume, and temperature (NVT) run for 0.5 ns at 310 K. Each system was further equilibrated using constant number, pressure, and temperature (NPT) run at 1 atm and 310 K for 10 ns with 5 kcal (mol Å2)−1 harmonic position restraints applied to the protein. Further equilibration of the systems was performed using an NPT run at 1 atm and 310 K for 0.5 ns with all atoms unrestrained. Conventional MD simulation was performed on each system for 10 ns at 1 atm pressure and 310 K with a constant ratio constraint applied on the lipid bilayer in the X–Y plane. The GaMD simulations were carried out using AMBER 18.26,52 Dual-boost GaMD simulations were performed to study the substrate-bound γ-secretase complex (Table 1). In the GaMD simulations, the threshold energy E for adding boost potential was set to the lower bound, i.e., E = Vmax.26,32 The simulations included 50 ns equilibration after adding the boost potential and then multiple independent production runs lasting 1–2 μs with randomized initial atomic velocities. GaMD production simulation frames were saved every 0.2 ps for analysis.
Simulation Analysis
The VMD58 and CPPTRAJ61 tools were used for trajectory analysis. In particular, distance was calculated between the Cγ atoms of catalytic aspartate residues. Hydrogen bond distance was calculated between donor protonated oxygen atom of PS1 Asp257 and the acceptor carbonyl oxygen atom of APP substrate residue Leu49, Thr48, or Ile47. Root-mean-square fluctuations (RMSFs) were calculated for the protein residues, averaged over three independent GaMD simulations and color coded for schematic representation of each complex system. The CPPTRAJ was used to calculate the protein secondary structure plots. The PyReweighting toolkit54 was applied to reweight GaMD simulations for free energy calculations by combining all simulation trajectories for each system. A bin size of 1 Å was used for the PMF calculation of distances. The cutoff was set to 500 frames in each bin for calculating the 2D PMF profiles. Protein snapshots were taken every 1 ps for structural clustering. Clustering was performed on the GaMD simulations of wildtype, I45F, T48P, and M51F mutant APP bound γ-secretase based on the RMSD of PS1 using hierarchical agglomerative algorithm in CPPTRAJ61 generating ∼10 representative structural clusters for each system. The top structural cluster was identified as the representative active (wildtype) and shifted active conformational states of the wildtype and M51F mutant APP bound γ-secretase systems, respectively. The top structural cluster was also identified as the active (I45F and T48P) conformational state of the I45F and T48P mutant APP bound γ-secretase.
Acknowledgments
This work used supercomputing resources with allocation Award TG-MCB180049 through the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. ACI-1548562, and Project M2874 through the National Energy Research Scientific Computing Center (NERSC), which is a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231, and the Research Computing Cluster at the University of Kansas. This work was supported in part by the startup funding in the College of Liberal Arts and Sciences at the University of Kansas (Y.M.) and GM122894 from the National Institutes of Health (M.S.W.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00296.
The authors declare no competing financial interest.
Supplementary Material
References
- Selkoe D. J.; Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8 (6), 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F.; Lozupone M.; Logroscino G.; Imbimbo B. P. A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15 (2), 73–88. 10.1038/s41582-018-0116-6. [DOI] [PubMed] [Google Scholar]
- Molinuevo J. L.; Ayton S.; Batrla R.; Bednar M. M.; Bittner T.; Cummings J.; Fagan A. M.; Hampel H.; Mielke M. M.; Mikulskis A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136 (6), 821–853. 10.1007/s00401-018-1932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Kaether C.; Thinakaran G.; Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect. Med. 2012, 2 (5), a006270 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe M. S. Structure and Function of the gamma-Secretase Complex. Biochemistry 2019, 58 (27), 2953–2966. 10.1021/acs.biochem.9b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H.; Fukumori A.; Tagami S.; Okochi M. Making the final cut: pathogenic amyloid-beta peptide generation by gamma-secretase. Cell Stress 2018, 2 (11), 292–310. 10.15698/cst2018.11.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y.; Misonou H.; Sato T.; Dohmae N.; Takio K.; Ihara Y. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J. Biol. Chem. 2001, 276 (38), 35235–35238. 10.1074/jbc.C100357200. [DOI] [PubMed] [Google Scholar]
- Qi-Takahara Y.; Morishima-Kawashima M.; Tanimura Y.; Dolios G.; Hirotani N.; Horikoshi Y.; Kametani F.; Maeda M.; Saido T. C.; Wang R.; et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J. Neurosci. 2005, 25 (2), 436–445. 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami M.; Nagashima Y.; Sano Y.; Ishihara S.; Morishima-Kawashima M.; Funamoto S.; Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29 (41), 13042–13052. 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc D. M.; Montagna D. R.; Seghers M. C.; Wolfe M. S.; Selkoe D. J. The amyloid-beta forming tripeptide cleavage mechanism of gamma-secretase. eLife 2016, 5, e17578 10.7554/eLife.17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.; Dohmae N.; Qi Y.; Kakuda N.; Misonou H.; Mitsumori R.; Maruyama H.; Koo E. H.; Haass C.; Takio K.; et al. Potential link between amyloid beta-protein 42 and C-terminal fragment gamma 49–99 of beta-amyloid precursor protein. J. Biol. Chem. 2003, 278 (27), 24294–24301. 10.1074/jbc.M211161200. [DOI] [PubMed] [Google Scholar]
- Yang G. H.; Zhou R.; Zhou Q.; Guo X. F.; Yan C. Y.; Ke M.; Lei J. L.; Shi Y. G. Structural basis of Notch recognition by human gamma-secretase. Nature 2019, 565 (7738), 192–197. 10.1038/s41586-018-0813-8. [DOI] [PubMed] [Google Scholar]
- Zhou R.; Yang G. H.; Guo X. F.; Zhou Q.; Lei J. L.; Shi Y. G. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363 (6428), eaaw0930. 10.1126/science.aaw0930. [DOI] [PubMed] [Google Scholar]
- Hitzenberger M.; Zacharias M. Structural Modeling of gamma-Secretase A beta(n) Complex Formation and Substrate Processing. ACS Chem. Neurosci. 2019, 10 (3), 1826–1840. 10.1021/acschemneuro.8b00725. [DOI] [PubMed] [Google Scholar]
- Hitzenberger M.; Zacharias M. gamma-Secretase Studied by Atomistic Molecular Dynamics Simulations: Global Dynamics, Enzyme Activation, Water Distribution and Lipid Binding. Front. Chem. 2019, 6, 640. 10.3389/fchem.2018.00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo-Ortiz R.; Chavez-Garcia C.; Straub J. E.; Dominguez L. Characterizing the structural ensemble of gamma-secretase using a multiscale molecular dynamics approach. Chemical Science 2017, 8 (8), 5576–5584. 10.1039/C7SC00980A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somavarapu A. K.; Kepp K. P. Membrane dynamics of γ-secretase provides a molecular basis for β-amyloid binding and processing. ACS Chem. Neurosci. 2017, 8 (11), 2424–2436. 10.1021/acschemneuro.7b00208. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Feng Z.; Xie X. Q.; Bahar I. Allosteric Modulation of Intact gamma-Secretase Structural Dynamics. Biophys. J. 2017, 113 (12), 2634–2649. 10.1016/j.bpj.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz A.; Mylonas N.; Hogel P.; Silber M.; Heinel H.; Menig S.; Vogel A.; Feyrer H.; Huster D.; Luy B.; Langosch D.; Scharnagl C.; Muhle-Goll C.; Kamp F.; Steiner H. Modulating hinge flexibility in the APP transmembrane domain alters γ-secretase cleavage. Biophys. J. 2019, 116 (11), 2103–2120. 10.1016/j.bpj.2019.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehury B.; Tang N.; Kepp K. P. Molecular dynamics of C99-bound γ-secretase reveal two binding modes with distinct compactness, stability, and active-site retention: implications for Aβ production. Biochem. J. 2019, 476 (7), 1173–1189. 10.1042/BCJ20190023. [DOI] [PubMed] [Google Scholar]
- Götz A.; Högel P.; Silber M.; Chaitoglou I.; Luy B.; Muhle-Goll C.; Scharnagl C.; Langosch D. Increased H-Bond Stability Relates to Altered ε-Cleavage Efficiency and Aβ Levels in the I45T Familial Alzheimer’s Disease Mutant of APP. Sci. Rep. 2019, 9 (1), 1–12. 10.1038/s41598-019-41766-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsik N.; am Ende C. W.; Geoghegan K. F.; Nguyen C.; Mukherjee P.; Mente S.; Seneviratne U.; Johnson D. S.; Li Y.-M. Mapping the Binding Site of BMS-708163 on gamma-Secretase with Cleavable Photoprobes. Cell Chem. Biol. 2017, 24 (1), 3–8. 10.1016/j.chembiol.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit D.; Hitzenberger M.; Lismont S.; Zoltowska K. M.; Ryan N. S.; Mercken M.; Bischoff F.; Zacharias M.; Chávez-Gutiérrez L. Extracellular interface between APP and Nicastrin regulates Aβ length and response to γ-secretase modulators. EMBO J. 2019, 38 (12), e101494 10.15252/embj.2019101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzenberger M.; Zacharias M. Uncovering the Binding Mode of γ-Secretase Inhibitors. ACS Chem. Neurosci. 2019, 10 (8), 3398–3403. 10.1021/acschemneuro.9b00272. [DOI] [PubMed] [Google Scholar]
- Bolduc D. M.; Montagna D. R.; Gu Y.; Selkoe D. J.; Wolfe M. S. Nicastrin functions to sterically hinder gamma-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (5), E509–518. 10.1073/pnas.1512952113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Feher V. A.; McCammon J. A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11 (8), 3584–3595. 10.1021/acs.jctc.5b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y. Acceleration of Biomolecular Kinetics in Gaussian Accelerated Molecular Dynamics. J. Chem. Phys. 2018, 149 (7), 072308. 10.1063/1.5024217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Unconstrained enhanced sampling for free energy calculations of biomolecules: a review. Mol. Simul. 2016, 42 (13), 1046–1055. 10.1080/08927022.2015.1121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; Mongan J.; McCammon J. A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120 (24), 11919–11929. 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- Voter A. F. Hyperdynamics: Accelerated molecular dynamics of infrequent events. Phys. Rev. Lett. 1997, 78 (20), 3908–3911. 10.1103/PhysRevLett.78.3908. [DOI] [Google Scholar]
- Shen T. Y.; Hamelberg D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008, 129 (3), 034103. 10.1063/1.2944250. [DOI] [PubMed] [Google Scholar]
- Pang Y. T.; Miao Y.; Wang Y.; McCammon J. A. Gaussian Accelerated Molecular Dynamics in NAMD. J. Chem. Theory Comput. 2017, 13 (1), 9–19. 10.1021/acs.jctc.6b00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Gaussian Accelerated Molecular Dynamics: Theory, Implementation and Applications. Annu. Rep. Comput. Chem. 2017, 13, 231–278. 10.1016/bs.arcc.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Miao Y. Mechanistic Insights into Specific G Protein Interactions with Adenosine Receptors. J. Phys. Chem. B 2019, 123 (30), 6462–6473. 10.1021/acs.jpcb.9b04867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. Mechanism of the G-Protein Mimetic Nanobody Binding to a Muscarinic G-Protein-Coupled Receptor. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (12), 3036–3041. 10.1073/pnas.1800756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai A.; Wang J.; Miao Y. G-Protein-Coupled Receptor-Membrane Interactions Depend on the Receptor Activation State. J. Comput. Chem. 2020, 41, 460–471. 10.1002/jcc.26082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East K. W.; Newton J. C.; Morzan U. N.; Narkhede Y. B.; Acharya A.; Skeens E.; Jogl G.; Batista V. S.; Palermo G.; Lisi G. P. Allosteric Motions of the CRISPR-Cas9 HNH Nuclease Probed by NMR and Molecular Dynamics. J. Am. Chem. Soc. 2020, 142 (3), 1348–1358. 10.1021/jacs.9b10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci C. G.; Chen J. S.; Miao Y.; Jinek M.; Doudna J. A.; McCammon J. A.; Palermo G. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Cent. Sci. 2019, 5 (4), 651–662. 10.1021/acscentsci.9b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp F.; Winkler E.; Trambauer J.; Ebke A.; Fluhrer R.; Steiner H. Intramembrane proteolysis of β-amyloid precursor protein by γ-secretase is an unusually slow process. Biophys. J. 2015, 108 (5), 1229–1237. 10.1016/j.bpj.2014.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langosch D.; Scharnagl C.; Steiner H.; Lemberg M. Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics. Trends Biochem. Sci. 2015, 40 (6), 318–327. 10.1016/j.tibs.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Bai X.-c.; Rajendra E.; Yang G.; Shi Y.; Scheres S. H. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 2015, 4, e11182 10.7554/eLife.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler W. P.; Das C.; Wolfe M. S. Probing pockets S2-S4′ of the gamma-secretase active site with (hydroxyethyl)urea peptidomimetics. Bioorg. Med. Chem. Lett. 2004, 14 (8), 1935–1938. 10.1016/j.bmcl.2004.01.077. [DOI] [PubMed] [Google Scholar]
- Cai T.; Yonaga M.; Tomita T. Activation of γ-secretase trimming activity by topological changes of transmembrane domain 1 of presenilin 1. J. Neurosci. 2017, 37 (50), 12272–12280. 10.1523/JNEUROSCI.1628-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi-Niidome S.; Sasaki T.; Osawa S.; Sato T.; Morishima K.; Cai T.; Iwatsubo T.; Tomita T. Cooperative roles of hydrophilic loop 1 and the C-terminus of presenilin 1 in the substrate-gating mechanism of γ-secretase. J. Neurosci. 2015, 35 (6), 2646–2656. 10.1523/JNEUROSCI.3164-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C.; Takagi S.; Tomita T.; Iwatsubo T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci. 2008, 28 (24), 6264–6271. 10.1523/JNEUROSCI.1163-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe M. S. Intramembrane proteolysis. Chem. Rev. 2009, 109 (4), 1599–1612. 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Yan N.; Feng L.; Oberstein A.; Yan H.; Baker R. P.; Gu L.; Jeffrey P. D.; Urban S.; Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol. 2006, 13 (12), 1084–1091. 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- Feng L.; Yan H.; Wu Z.; Yan N.; Wang Z.; Jeffrey P. D.; Shi Y. Structure of a site-2 protease family intramembrane metalloprotease. Science 2007, 318 (5856), 1608–1612. 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- Li X.; Dang S.; Yan C.; Gong X.; Wang J.; Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature 2013, 493 (7430), 56–61. 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.; Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature 2006, 444 (7116), 179–180. 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Cho S.; Baker R. P.; Ji M.; Urban S. Ten catalytic snapshots of rhomboid intramembrane proteolysis from gate opening to peptide release. Nat. Struct. Mol. Biol. 2019, 26 (10), 910–918. 10.1038/s41594-019-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S. T. E.; Cheatham I.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.. et al. AMBER 18; University of California: San Francisco. 2018. [Google Scholar]
- Oshima H.; Re S.; Sugita Y. Replica-Exchange Umbrella Sampling Combined with Gaussian Accelerated Molecular Dynamics for Free-Energy Calculation of Biomolecules. J. Chem. Theory Comput. 2019, 15 (10), 5199–5208. 10.1021/acs.jctc.9b00761. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Sinko W.; Pierce L.; Bucher D.; Walker R. C.; McCammon J. A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10 (7), 2677–2689. 10.1021/ct500090q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelberg D.; de Oliveira C. A. F.; McCammon J. A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127 (15), 155102. 10.1063/1.2789432. [DOI] [PubMed] [Google Scholar]
- Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput. Phys. Commun. 1995, 91 (1–3), 275–282. 10.1016/0010-4655(95)00053-I. [DOI] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T; de Beer T. A P; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46 (W1), W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850 (5), 861–871. 10.1016/j.bbagen.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel K.; Miao Y.; McCammon J. A. Accelerated Molecular Dynamics Simulations of Ligand Binding to a Muscarinic G-protein Coupled Receptor. Q. Rev. Biophys. 2015, 48 (04), 479–487. 10.1017/S0033583515000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9 (7), 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.