

Abstract
Objective
The detection of prion seeding activity in CSF and olfactory mucosal brushings using real‐time quaking‐induced conversion assays allows highly accurate clinical diagnosis of sporadic Creutzfeldt–Jakob disease. To gauge transmission risks associated with these biospecimens and their testing, we have bioassayed prion infectivity levels in patients’ brain tissue, nasal brushings, and CSF, and assessed the pathogenicity of amplified products of real‐time quaking‐induced conversion assays seeded with Creutzfeldt–Jakob disease prions.
Methods
We obtained olfactory mucosal brushings and CSF from patients with a final diagnosis of sporadic Creutzfeldt–Jakob disease subtype MM1 (n = 3). Samples were inoculated intracerebrally into Tg66 transgenic mice that overexpress the homologous human 129M prion protein. The mice were evaluated for clinical, neuropathological, and biochemical evidence of prion infection.
Results
Patients’ brain tissue at 102 to 105 fold dilutions affected 47/48 Tg66 mice. In contrast, maximum acutely tolerable doses of insoluble pellets from their olfactory mucosa brushings caused evidence of prion disease in only 4/28 inoculated mice, and no effects were seen with 10‐fold dilutions. No clinical prion disease was observed in mice inoculated with antemortem CSF samples or prion‐seeded real‐time quaking‐induced conversion assay products.
Interpretation
Pellets from patients’ olfactory mucosa brushings had ≥10,000‐fold lower infectivity per unit volume than brain tissue, while CSF lacked detectable infectivity. Nonetheless, the results suggest that appropriate precautions may be warranted in surgical interventions involving the olfactory areas. The lack of pathogenic infectivity in the real‐time quaking‐induced conversion assay products provides evidence that the assay does not replicate biohazardous prions in vitro.
Introduction
Prion diseases are fatal, transmissible neurodegenerative disorders that include Creutzfeldt–Jakob disease (CJD), variably protease‐sensitive prionopathy, Gerstmann–Sträussler–Scheinker syndrome, and fatal familial insomnia in humans. Although most human prion disease cases are of sporadic or genetic origin, many cases have been initiated by infections through iatrogenic exposures of patients via tissue transplants, blood transfusions, cadaveric growth hormone inoculations, or contamination of surgical instruments. 1
The most common form of human prion disease is sporadic CJD (sCJD) with an incidence of 1–2 cases per million per year worldwide. In sCJD, the clinical and pathological phenotype are influenced by two major disease modifiers which include the polymorphism methionine (M)/valine (V) at codon 129 of PRNP and the glycotype of protease‐resistant prion protein (PrPres). Two main PrPres types (1 and 2), have been distinguished by the electrophoretic migration of PrPres unglycosylated fragment at 21 or 19 kDa, respectively. 2 The combination of these molecular factors results in six distinct molecular subtypes. 3 The most common is MM1, which accounts for about 70% of all sCJD cases.
The accuracy of antemortem diagnosis of sCJD has improved recently with the implementation of ultrasensitive real‐time quaking induced conversion (RT‐QuIC) assays of patients’ cerebrospinal fluid (CSF) 4 , 5 , 6 , 7 , 8 , 9 , 10 and nasal brushings of the olfactory mucosa (OM). 7 , 11 RT‐QuIC assays allow the detection of a prion disease‐specific biomarker by amplifying the presence of prion seeding activity in diagnostic specimens by many orders of magnitude in vitro. 4 , 12 The detection of prion seeding activity in CSF and OM specimens from sCJD patients raises two important practical issues: (1) Do CSF and OM specimens from sCJD patients contain biohazardous levels of prions? And (2), are the highly amplified products of sCJD‐seeded RT‐QuIC reactions infectious, pathogenic, and therefore, potentially hazardous to laboratory workers?
No transmission studies of OM specimens from sCJD patients have been reported. CSF samples have been inoculated into laboratory animals with divergent results. 13 , 14 , 15 Moreover, from these previous reports, it is unclear what proportion of the CSF specimens was collected antemortem versus postmortem. CSF samples collected postmortem would have much greater potential for contamination from brain parenchymal tissue, raising questions about origin of prion infectivity detected therein.
In the present study, we have tested OM and CSF samples from sCJD patients for their ability to transmit prion disease when inoculated intracerebrally into sCJD‐susceptible Tg66 transgenic mice that overexpress human prion protein. 16 In addition, we similarly tested the amplified products of sCJD‐seeded RT‐QuIC reactions.
Results
OM and brain inoculations
Insoluble pelleted material from individual OM brushings from three MM1 sCJD patients who were previously reported to be positive by RT‐QuIC, 7 , 11 and dilutions thereof, were inoculated intracerebrally into groups of six Tg66 transgenic mice as described in the Methods section. This humanized mouse model overexpresses human M129 PrPC (the normal cellular prion protein) by 8‐ to 16‐fold and expresses no murine PrPC. 16 One of six mice inoculated with OM pellets from Patient 2 (Table 1) showed typical clinical signs of prion disease leading to euthanasia at 331 days postinoculation (dpi). Prion disease in this mouse was confirmed by immunoblotting for proteinase K (PK)‐resistant PrP (Fig. 1A) as well as histological and RT‐QuIC analyses of brain tissue (Table 1, Figs. 1, 2). Analysis of the brain of the clinically ill mouse indicated that the overall pattern of abnormal PrP deposition closely resembled that of mice inoculated with brain tissue from the same patient (Figs. 1B‐D). Moreover, higher magnification comparisons indicated that spongiform changes in the preoptic area and brain stem were associated with abnormal PrP deposits in the mice receiving the OM and either relatively high (10‐2 dilution) or near end‐point (10‐5) doses of the brain inocula (Fig. 2). These observations provide evidence that the prions in Patient 2’s OM and brain specimens were of the same strain, and argue against the possibility that the disease in the single mouse inoculated with Patient 2’s OM specimen was due to inadvertent contamination of the OM specimen with a distinct laboratory strain. Second passage of the clinically affected sCJD OM‐inoculated Tg66 mouse into other Tg66 mice also confirmed that this mouse had a prion infection (Table 1).
Table 1.
1st and 2nd passages of sCJD OM samples in humanized Tg66 mice.
| 1st passages | |||||
|---|---|---|---|---|---|
| Donor | Inoculum dilution | Clinical prion disease (+/total) | Survival time dpi (mean +/‐ SD) | Neuropathology and PrP IHC (+/total tested) | RT‐QuIC (+/ total tested) |
| CJD Patient 2 | Neat | 1/6 |
Pos: 331 Neg: 531 +/‐ 0 |
1/6 1 | 1/6 1 |
| 10‐1 | 0/5 | 605 +/‐ 4 | 0/5 | 0/5 | |
| 10‐2 | 0/5 | 607 +/‐ 0 | 0/5 | 0/5 | |
| 10‐3 | 0/5 | 573 +/‐ 69 | NT | 0/4 | |
| CJD Patient 8 | Neat | 0/6 | 483 +/‐ 42 | 0/6 | 0/6 |
| 10‐1 | 0/6 | 632 +/‐ 51 | 0/4 | 0/2 | |
| 10‐2 | 0/5 | 638 +/‐ 42 | NT | 0/4 | |
| 10‐3 | 0/6 | 676 +/‐ 16 | NT | 0/2 | |
| Conc’d pool | 0/12 | 317 3 , 379 (n = 11) | 0/12 | 0/12 | |
| CJD Patient 11 | Conc’d pool | 2/4 2 |
Pos: 308,303 Neg/Pos: 267 4 Neg: 379 Acute: 1 (n = 7) |
3/3 1 | 3/4 1 |
| Non‐CJD | Neat | 0/5 | 493 +/‐40 | 0/4 | 0/5 |
| Uninoculated | not applicable | 0/8 |
317 (n = 4) 378 (n = 4) |
0/5 | 0/5 |
| 2nd passages | ||||
|---|---|---|---|---|
| Donor | Inoculum dilution | Clinical prion disease (+/total) | Survival time dpi (mean +/‐ SD) | Neuropathology and PrP IHC (+/total tested) |
|
sCJD OM‐inoculated Tg66 mouse brain |
10‐4 | 6/6 | 201 +/‐ 6 | 6/6 |
| 10‐5 | 6/6 | 277 +/‐ 57 | 6/6 | |
| 10‐6 | 0/6 | 378 +/‐ 0 | 0/3 | |
| TITER | 6.3 x 106 LD50/mg tissue | |||
| sCJD brain (human standard) 5 | 10‐4 | 6/6 | 199 +/‐ 6 | 5/5 |
| 10‐5 | 3/6 | 235 +/‐ 8 | 2/2 | |
| 10‐6 | 1/6 | 240 | 1/6 | |
| TITER | 2.9 × 106 LD50/mg tissue | |||
Dpi, days postinoculation; Conc’d, concentrated several fold (nominally) compared to the individual OM brushings; NT, not tested.
Same mouse or mice were positive for neuropathology and RT‐QuIC.
In this group, a total of 11 mice were inoculated, but only four survived beyond 1 day, as explained in the main text.
Euthanized with neurological disease partially consistent with prion disease, but negative by histology or RT‐QuIC.
Euthanized at 267 dpi due to trauma, before clinical signs, but positive for prion disease histology and RT‐QuIC.
This was a 1st passage of human sCJD brain as a positive control for the 2nd passage of the sCJD OM sample.
Figure 1.
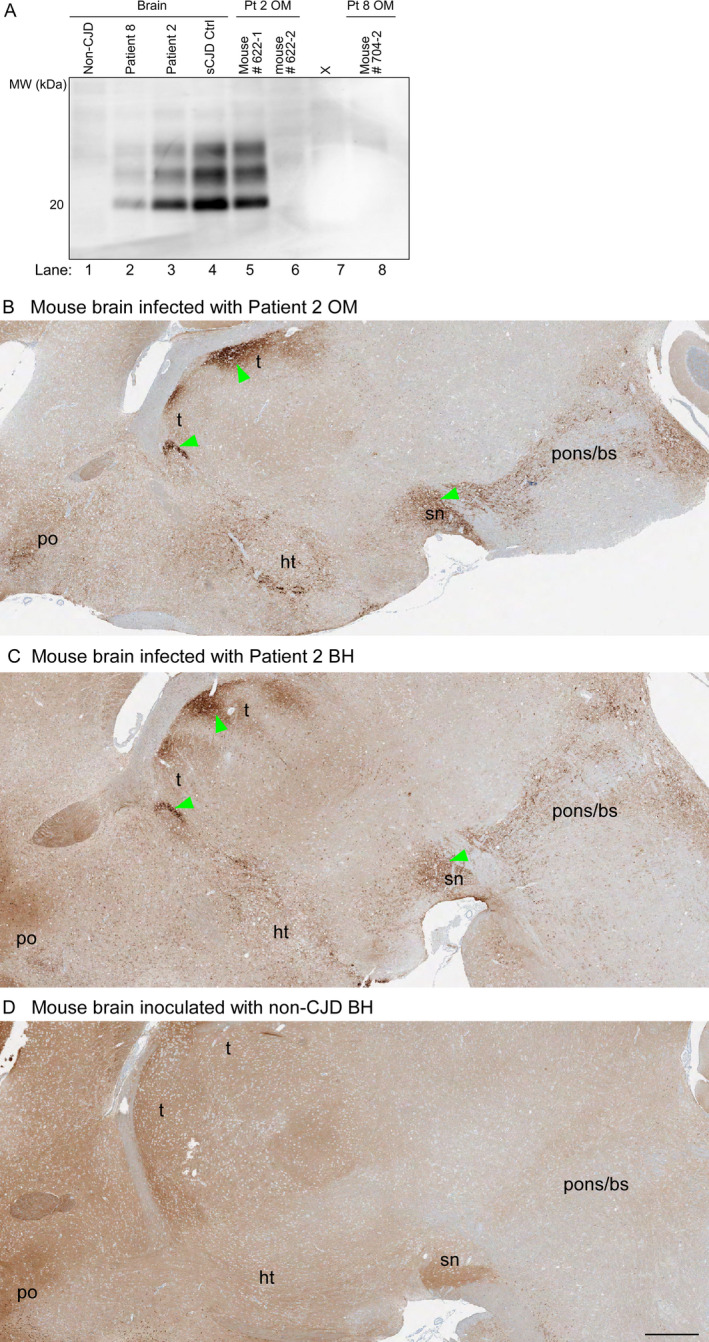
PrP analyses of inocula and brains from Tg66 mouse recipients. A, Immunoblots of PrPres in sCJD human brain inocula and brains of Tg66 mice inoculated with human brain or OM samples. Lanes 1–4: human brain homogenates from a non‐CJD control case and three MM1 sCJD cases (Patient 8, Patient 2, and a previously established positive control case (sCJD Ctrl). Lanes 5–6, 8: Tg66 mouse brain homogenates from individual animals inoculated with OM specimens from sCJD Patients 2 or 8 (Pt 2 and 8, respectively). Lane 7 contained an irrelevant sample. The position of a 20‐kDa molecular weight marker is shown on the left. B‐D, PrP immunohistochemical staining of brain sections from clinically affected Tg66 mice inoculated with OM (B) or brain homogenate (BH) (C) samples from sCJD Patient 2 (B, C) or a healthy mouse inoculated with healthy human (non‐prion disease) control BH (D). Brain regions: t = thalamus; po = preoptic area; ht = hypothalamus; sn = substantia nigra; pons/bs = pons/brain stem. B shows the single mouse that had clinical signs of prion disease following OM inoculation. Green arrows indicate examples of regions where abnormal PrP deposition was observed. Bar = 0.5 mm. PrP monoclonal antibody 3F4 was used as described in Methods
Figure 2.
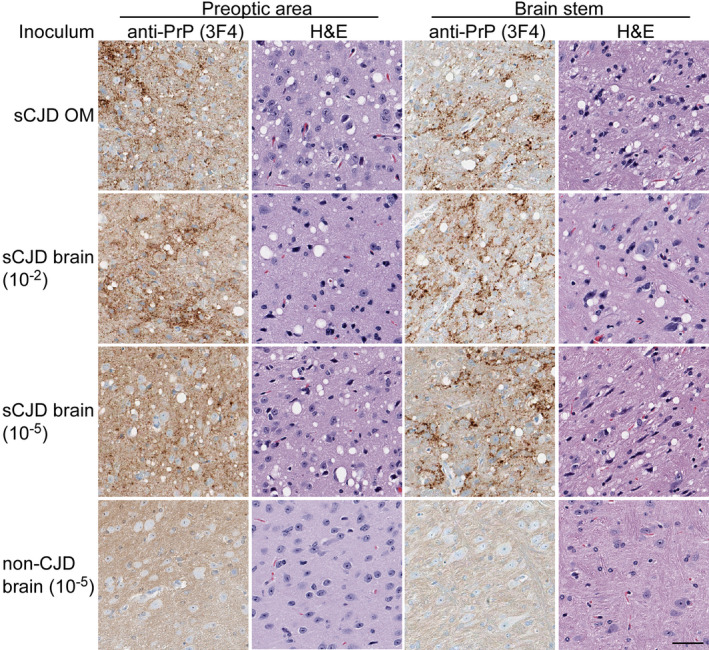
Higher magnification views of anti‐PrP or hematoxylin and eosin (H & E) staining of preoptic and brain stem areas from Tg66 mice inoculated with OM (top row) or brain homogenate (BH) samples from sCJD Patient 2 (second and third rows) or a nonprion disease control (bottom row). The OM inoculated mouse was euthanized at 331 days postinoculation (dpi). The second and third rows show stains from terminally ill mice inoculated with a relatively high (10‐2 dilution) or near end‐point (10‐5) doses of sCJD BH, at 180 and 294 dpi, respectively. Note that the dpi for the sCJD‐inoculated mouse represented in the third row is much closer to that of the OM‐inoculated mouse in the top row than the dpi of the mouse represented in the second row that received the higher dose of BH. Thus, this mouse is more closely matched in age and incubation time to the OM‐ and negative control‐inoculated mice (331 dpi) in the top and bottom rows, respectively. All panels have equivalent magnification; bar = 50 µm
None of the first‐passage mice inoculated with OM samples from Patient 8 showed any evidence of prion disease prior to being euthanized at means of 483–676 dpi, including samples from pools of three or four OM brushings (Table 1). Finally, although the inoculation of an OM pool from Patient 11 caused acute distress in seven of 11 inoculated mice, three of the four remaining mice ultimately showed clinical signs of the prion disease, which was confirmed by postmortem histological and RT‐QuIC analyses of brain tissue (Table 1). The unexpectedly acute distress postinoculation in these mice, causing death or requiring euthanasia within 24 h, is consistent with the presence of bacteria and/or endotoxin, which were likely concentrated by the sample pooling. Altogether, maximum acutely tolerable doses of sCJD OM samples and pools of samples caused evidence of prion disease in a total of 4/28 inoculated mice, and no effects were seen with ≥10‐fold dilutions of individual OM samples. This indicated that CJD infectivity levels in OMs collected from these sCJD patients were either slightly above or below the threshold of detection in the mouse bioassay.
By comparison, inoculation of brain tissue from two of these sCJD patients at 102 to 104 fold dilutions caused prion disease in all recipient mice (n = 32) and 105 fold dilutions required euthanasia of 10/11 mice (Table 2). This sensitivity for the Tg66 mice for MM1 sCJD brain tissue dilutions was similar to that reported for another humanized mouse model (Tg340) that has been used to detect infectivity in human sCJD plasma samples. 17 By comparing the estimated range of OM insoluble pellet volumes (20–100 µL) to the volume of solid brain tissue equivalents inoculated into the mice, we calculated that the OM pellets from these cases had at least 104 fold lower titers of sCJD prions than solid brain tissue. Also consistent with this conclusion was the observation that the incubation periods in the clinically affected OM‐inoculated mice (303–331 dpi) were similar to, or longer than, the means observed with the mice inoculated with 105 fold dilutions of brain tissue from the same sCJD patients (277–334 dpi; Tables 1, 2). Interestingly, although 106 fold dilutions of sCJD brain tissue samples from sCJD Patients 2, Patient 8, and our sCJD standard case did not cause clinical disease in any of the mice (collectively n = 12), 7/8 of those mice were positive by RT‐QuIC (Table 2), providing evidence that the RT‐QuIC assay is more sensitive for MM1 sCJD than the Tg66 mouse bioassay.
Table 2.
Inoculation of human sCJD brain samples into Tg66 mice.
| BH Donor | Inoculum dilution | Clinical prion disease (+/total) | Survival time dpi (mean +/‐ SD) | Neuropathology and PrP IHC (+/total tested) | RT‐QuIC (+/ total tested) |
|---|---|---|---|---|---|
| sCJD Patient 2 | 10‐2 | 5/5 | 175 +/‐7 | 2/2 | |
| 10‐3 | 5/5 | 189 +/‐2 | NT | ||
| 10‐4 | 6/6 | 220 +/‐18 | NT | ||
| 10‐5 | 5/5 | 334 +/‐111 | 2/2 | 1/1 | |
| 10‐6 | 0/5 | 569 +/‐35 | 0/5 | 3/4 | |
| TITER | 6.3 × 106 LD50/mg tissue | ||||
| sCJD Patient 8 | 10‐2 | 4/4 | 167 +/‐1 | 2/2 | |
| 10‐3 | 6/6 | 185 +/‐5 | NT | ||
| 10‐4 | 6/6 | 219 +/‐11 | NT | ||
| 10‐5 | 5/6 |
277 +/‐19 Neg: 525 |
2/3 | 2/2 | |
| 10‐6 | 0/6 | 539 +/‐74 | 2/5 | 4/4 | |
| TITER | 4.3 × 106 LD50/mg tissue | ||||
| sCJD standard | 10‐3 | 6/6 | 180 +/‐4 | 2/2 | |
| 10‐4 | 5/5 | 204 +/‐5 | |||
| 10‐5 | 2/5 |
Pos: 254, 269 Neg: 505 +/‐32 |
2/2 | 2/2 | |
| 10‐6 | 0/4 | 511 +/‐60 | 3/3 | 4/4 | |
| TITER | 1.6 × 106 LD50/mg tissue | ||||
|
Non‐CJD Control |
10‐2 | 0/7 | 414 +/‐110 | 0/4 | 0/5 |
NT: not tested.
Comparison of nasal mucus and OM brushings by RT‐QuIC
As an indication of the relative amounts of prion seeding activity in OM brushings versus nasal secretions obtained from the nasal vestibule of patients affected with sCJD (n = 13) and non‐CJD controls (n = 5), we tested both types of sample with RT‐QuIC. Based on our selection of a mean fluorescence threshold for positivity that is above the highest value from the non‐CJD controls, 11/13 sCJD OM brushings were RT‐QuIC positive within 4–42 h (average of 21.5 ± 13.7 h), whereas only 2/13 of the nasal mucus samples were positive, in this case between 26 and 38 h (average of 32.2 ± 8.6 h) (Fig. 3). We cannot exclude the possibility that these two nasal mucus samples were contaminated with OM material during the collection procedure. Nonetheless, overall, the results indicate that there was lower, and usually undetectable, RT‐QuIC seeding activity in nasal secretions compared to brushings of the OM specifically.
Figure 3.
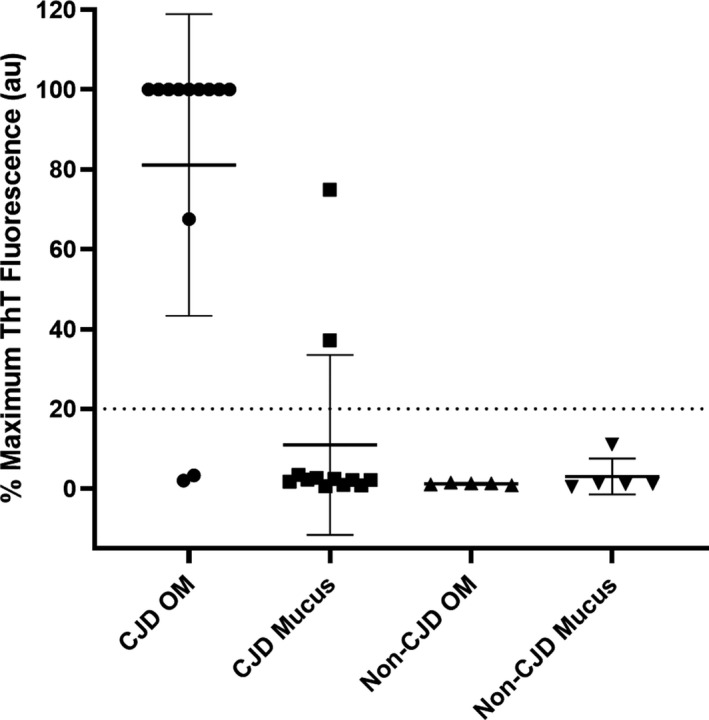
Comparison of sCJD and non‐CJD nasal mucus and OM brushings by RT‐QuIC. Average fluorescence maxima obtained within 55 h from quadruplicate reactions for each patient. Bars show the average +/− SD for type of case. The dashed line indicates a provisional positive fluorescence threshold selected to be above all data points for the non‐CJD controls yet below most of the CJD OM samples
CSF inoculations
We also inoculated Tg66 mice with 1:1 or 1:20 dilutions of RT‐QuIC‐positive CSF specimens from sCJD patients into the Tg66 mice. None of the inoculated mice showed clinical signs of prion disease over periods averaging 528–705 dpi (Table 3). Brains of most of these clinically negative mice were further tested for RT‐QuIC seeding activity, prion disease neuropathology, or both, and none had any consistent evidence of prion infection. These results indicated that, despite being positive in the more sensitive RT‐QuIC assay, CSF samples collected from these sCJD patients lacked prions that were detectable by intracerebral bioassay in Tg66 mice.
Table 3.
Inoculation of sCJD CSF samples into Tg66 mice.
| Donor | Inoculum dilution | Clinical prion disease (+/total) | Survival time dpi (mean +/‐ SD) | Neuropathology and PrP IHC (+/total tested) | RT‐QuIC (+/ total tested) |
|---|---|---|---|---|---|
| sCJD Patient 1 | 1:1 | 0/6 | 556 +/‐56 | 0/5 | 0/5 |
| 1:20 | 0/6 | 616 +/‐64 | NT | 0/4 | |
| sCJD Patient 2 | 1:1 | 0/6 | 547 +/‐74 | NT | 0/4 |
| 1:20 | 0/5 | 657 +/‐40 | NT | 0/3 | |
| sCJD Patient 8 | 1:1 | 0/5 | 528 +/‐89 | 0/2 | 0/4 |
| 1:20 | 0/5 | 587 +/‐13 | NT | 0/2 | |
| Non‐CJD | 1:1 | 0/6 | 550+/‐73 | 0/3 | 0/5 |
NT: not tested.
Inoculation of RT‐QuIC products
To address the issue of whether sCJD prions might be amplified in sCJD‐seeded RT‐QuIC reactions, we inoculated reaction products into Tg66 mice. We chose products from reactions seeded with hamster PrPC 90‐231 because this is the most commonly used substrate for human prion disease diagnostics with second generation RT‐QuIC conditions. Products from multiple reactions seeded with brain homogenates from sCJD Patients 2 and 8 were pooled and confirmed to contain the expected proteinase K (PK)‐resistant products (Fig. 4A and B). Aliquots of the RT‐QuIC products containing 5 or 0.5 µg of recombinant PrP were each inoculated intracerebrally into 5–6 mice. The final sCJD brain homogenate dilutions in these inocula were 2 × 10−5 or 2 × 10−6, respectively, which bracket the endpoint dilution of these brain homogenates with respect to their ability to cause terminal illness in mice (Table 2). None of the recipients showed clinical signs of prion disease prior to euthanasia at 410–665 dpi (Table 4) despite the fact that, without processing through RT‐QuIC reactions, 2 × 10‐5 dilutions of these same brain homogenates caused disease in a total of 11/12 inoculated mice with mean incubation periods of 334 +/‐111 (Patient 2) and 277 +/‐17 dpi (Patient 8) (Table 2). These results provided evidence that, if anything, sCJD lethal doses were reduced, rather than amplified, during the RT‐QuIC assays and/or the recovery of reaction products from the wells.
Figure 4.
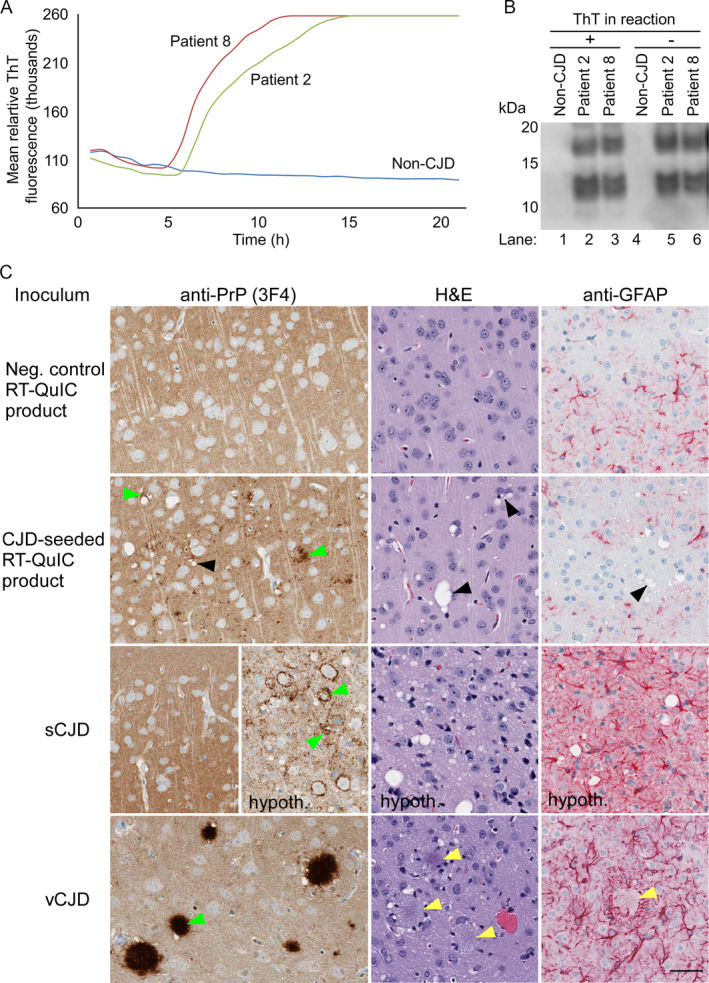
Analyses of RT‐QuIC products that were inoculated (A, B) and the resultant neuropathology (C). A. RT‐QuIC amplification of prion seeding activity in quadruplicate reactions seeded with 10‐3 dilutions of brain tissue from Patients 2 and 8, and a non‐CJD patient. The y axis indicates the average thioflavin T fluorescence of four replicate wells. B, Immunoblotting of proteinase K‐digested RT‐QuIC conversion products generated at the same time as the reactions in A. Lanes 1–3 are conversion products containing ThT while lanes 4–6 are products generated in the absence of ThT. C, Histological analyses of brain from Tg66 mice inoculated with RT‐QuIC reaction products seeded with BH from either a negative control patient without CJD (at 560 dpi; top row) or an MM1 sCJD Patient 8 (at 565 dpi; second row). For comparison, corresponding tissues from Tg66 mice inoculated with sCJD (180 dpi; third row) or variant CJD MM1 (vCJD; at 396 dpi; bottom row) are shown. All panels show sections of cerebral cortex, except for those labeled “hypoth” which are from the hypothalamus. The latter are shown because the hypothalamus was the most affected brain area while cerebral cortex was relatively spared, as is typical of sCJD‐inoculated Tg66 mice. The antibody or stain used for each column is shown on top. Green arrowheads show examples of abnormal PrP deposition. Black arrowheads show examples of vacuoles present in brain of mice inoculated with sCJD‐seeded RT‐QuIC product. Scale bar = 50 µm and applies to all panels
Table 4.
Inoculation of sCJD‐seeded RT‐QuIC products into Tg66 mice.
| RT‐QuIC product inoculum | Total PrP inoculated | Clinical prion disease (+/total) | Survival time dpi (mean +/‐ SD) | Atypical neuropathology and PrP IHC 1 (+/total tested) | RT‐QuIC (+/ total tested) |
|---|---|---|---|---|---|
| CJD Patient 2‐seeded | 5 µg | 0/5 | 644 +/‐ 7 | 4/4 | 2/2 |
| 0.5 µg | 0/6 | 554 +/‐104 | 2/3 | NT | |
| CJD Patient 8‐seeded | 5 µg | 0/5 | 560 +/‐ 62 | 2/2 | 1/1 |
| 0.5 µg | 0/6 | 624 +/‐53 | 6/6 | 3/3 | |
| Control Patient‐seeded | 2.5 µg | 0/4 | 526 +/‐36 | 0/4 | 0/4 |
NT: not tested. IHC: immunohistochemistry.
Although abnormal vacuolation and PrP deposition were observed in the brains designated as positive in this column, these features differed markedly from those seen with any bona fide prion disease in this mouse model.
Nonetheless, we also checked the brains of many of the inoculated mice for RT‐QuIC seeding activity or signs of subclinical pathology (Table 4). Not surprisingly, given our past findings with inoculations of other types of RT‐QuIC products, 18 , 19 and the fact that we inoculated several logs of in vitro seeding activity into the brain of each mouse, we detected RT‐QuIC seeding activity in the brains of all of the mice tested. Immunohistochemical analyses showed abnormal PrP staining and regional distribution that were markedly different from those seen in either uninoculated aged Tg66 mice or mice inoculated with either human sCJD or variant CJD (vCJD) brain homogenate (Fig. 4C). Abnormal PrP deposits observed in mice inoculated with sCJD‐seeded RT‐QuIC reaction product were restricted to the cerebral cortex. In contrast, sCJD‐infected mice had a combination of diffuse, perineuronal and small aggregate PrP deposits most prominent in the thalamus, hypothalamus, brainstem, substantia nigra, and preoptic area, and vCJD‐infected mice had widespread abnormal PrP plaques (Figs. 1, 2 and 4). Vacuolation was also observed in the RT‐QuIC product inoculated mice, however there was little evidence for astroglial responses that are typical of Tg66 mice infected with sCJD or vCJD (Fig. 4). Altogether, these results suggested that although the sCJD‐seeded RT‐QuIC products persisted long term after inoculation into the brain, and may have induced further aggregation of endogenous PrPC and vacuolation, they failed to elicit either clinical disease or astrogliosis. In contrast, based on previous determinations of PrPSc concentration in human MM1 sCJD brain tissue (~200 ng/mL 10% homogenate) 20 and our end‐point dilution bioassay titrations of MM1 sCJD brains in Tg66 mice (~5 × 106 LD50/ mg tissue; Table 2), we calculate that the lethal dose of MM1 sCJD PrPSc in brain is ~0.4 fg, or at least 10 billion‐fold more lethal per unit mass than the RT‐QuIC product that is seeded by it.
Discussion
Our current findings are relevant in the assessment of risks of tissue infectivity and sCJD transmission. As noted above, sCJD is well known to be transmissible following invasive iatrogenic exposures to prion infected tissues or neurosurgical procedures involving contaminated instruments. 1 Divergent conclusions about risks of sCJD associated with extraneural surgical procedures have also been reported, 21 and the strength of any such conclusions remain under debate. Importantly, other epidemiological studies have shown that health professionals in contact with CJD patients do not have enhanced occupational risk for CJD, suggesting that any occupational risks of sCJD infection via exposure in clinical settings may be negligible. 22
With respect to CSF collected by lumbar puncture from sCJD patients, our humanized mouse bioassays did not detect levels of sCJD prions capable of causing disease despite the finding that these CSF samples were positive for prion seeding activity using the more sensitive RT‐QuIC assay. In contrast, as noted above, earlier transmission studies using nonhuman primates or other experimental animals have sometimes detected sCJD infectivity in CSF 13 , 14 , 15 . Brown et al. ( 13 and Dr. Paul Brown, personal communication) reported that CSF samples from 27 CJD cases were inoculated into 120 animals (65 nonhuman primates, 40 Guinea‐pigs, and 15 other animal species) with successful transmissions for only four inocula (one Pan troglodytes, two Cebus capucinus, and one Cavia porcellus). It is likely that three of the four CSF samples that transmitted disease were collected postmortem (Dr. Paul Brown, personal communication). Postmortem degradation of brain tissues or exposure of the aspiration needle to brain tissue may have potentially contaminated CSF samples as has been suspected in comparisons of tau seeding activity in postmortem versus antemortem CSF specimens from tauopathy cases. 23 , 24 Draganescu and coworkers also reported successful transmission to Guinea pigs (Cavia porcellus) for six of seven CSF samples from CJD patients, 14 but the neuropathology of the diseased animals did not clearly show the characteristic spongiform changes of the cerebral grey matter, questioning the reliability of transmission. Finally, Tateishi and coworkers reported no transmission of an unspecified number of CSF samples from sCJD patients following intracerebral inoculation into mice. 15 Another important uncertainty is the relative sensitivity of species of nonhuman primates and our Tg66 mice to sCJD. In comparing animal bioassay models, a feature that might be expected to enhance the susceptibility of Tg66 mice relative to primates and wild‐type rodents is their overexpression of M129 human PrPC that is exactly sequence‐matched to our MM1 sCJD brain inocula. On the other hand, humans and other primates have much longer lifespans than rodents, giving more time for infections to progress to clinical disease. In any case, our side‐by‐side comparison of brain and CSF inocula in Tg66 mice indicated that CSF from living MM1 sCJD patients is at least several orders of magnitude less infectious than the same patients’ brain tissue. Moreover, our failure to cause disease by injecting relatively large volumes of sCJD CSF directly into the brain (typically the most efficient route) suggests that the transmission risks associated with incidental exposure to such CSF specimens may be negligible.
Consistent with our observations that sCJD OM samples often have 3–4 logs higher levels of prion seeding activity per µl than CSF samples, 11 our bioassays indicated that our OM samples, and concentrated pools thereof, had sCJD infectivity that caused prion disease in a small minority of the recipient mice. Thus, OM infectivity was only slightly above the detection limit of the bioassay, which involved inoculation directly into the brain. Similarly, barely detectable levels of sCJD infectivity have been detected in plasma samples of two of four MM1 sCJD patients bioassayed in the humanized Tg340 mice mentioned earlier. 17 Although our data indicate that there was higher infectivity in OM than CSF, the OM infectivity levels were again orders of magnitude lower than in brain tissue. Nonetheless, immunochemical analyses of the olfactory neuroepithelium of human CJD cases showed that the deposits of the pathological prion protein were unevenly distributed in the cilia. 25 Taken together, these findings suggest that the olfactory area of the nasal vault represents a site of potential risk, albeit relatively low, in mediating iatrogenic CJD transmission. Therefore, instruments used for nasal vault surgery should be considered for prion decontamination procedures or disposal. One study reported that people having experienced nose or throat surgery, without specifying the specific surgery of each subject, had three times the risk of sCJD compared to the general population. 26 Additional and more detailed studies are required to better estimate the risk of surgical procedures involving the nasal vault. A limitation of this study is that other individual MM1 sCJD cases, and cases with other sCJD strains 27 besides the most common MM1 sCJD subtype that we have evaluated here, might give different levels of infectivity in OM brushings or mucus. Further transmission studies should provide broader perspective on this issue.
The fact that pathogenic sCJD prions do not amplify in our RT‐QuIC reactions using recombinant hamster PrPC 90‐231 should help to allay fears of additional biohazard being generated in conducting these tests. However, we note that some RT‐QuIC assays for human prions use full‐length hamster, human, bank vole, or other recombinant PrPC substrate, 28 and one cannot conclude from our current data that RT‐QuIC products generated with other PrPC substrates or under other assay conditions are as safe as those generated with hamster PrPC 90‐231.
Methods
Ethics
This project was approved by the ethics committees at the University of Verona and the Istituto Superiore di Sanità (Italy), which is recognized by the Office for Human Research Protections of the U.S. Department of Health and Human Services. Informed consent for participation in research was obtained in accordance with the Declaration of Helsinki and the Additional Protocol to the Convention on Human Rights and Biomedicine, concerning Biomedical Research. All the sampling of OM and CSF was performed after written informed consent was obtained from each patient or the patient’s representative. The analyses of human specimens that were performed at the NIAID were performed under Exemption 11517 for the use of encoded samples from the NIH Office of Human Subjects Research Protections. Tg66 mice were used according to RML animal protocol number 2013‐022 approved by the RML Animal Care and Use Committee.
Brain tissue
The control sCJD stock brain homogenate (designated herein as the “positive standard”) was derived from a frozen brain sample of sCJD MM1 from a patient and was kindly provided by Robert Rohwer (University of Maryland, Baltimore, MD). Brain‐tissue specimens were obtained at autopsy from two patients with MM1 sporadic Creutzfeldt–Jakob disease at the University of Verona and were processed there for neuropathological examination and biochemical analyses. The brain tissue samples were aseptically diluted to 10% w/v into RT QuIC buffer (0.05 M Tris‐Cl pH 7.5, 0.15 M NaCl, 0.5% Triton X100, and 1X Roche Minitab protease inhibitors) and processed using a bead beater (Biospec Products) for 40 sec at full power, then centrifuged at 400× g for 2 min. The supernatants were removed, and 0.1 ml aliquots were frozen at −80 °C prior to further use. Just prior to inoculation, an aliquot was thawed and then sonicated for 2 min at high power in a cup horn (Misonix). Subsequent serial 10‐fold dilutions were aseptically made in phosphate buffered saline (PBS), pH 7.4, containing 2% fetal bovine serum (FBS) and held at room temperature until inoculation within 2 h.
OM specimens
OM samples were obtained from individuals with swabs, and insoluble material was collected as described previously. 7 , 11 The donors included normal control and probable sCJD patients at the time of sampling, with the sCJD diagnoses being later confirmed by postmortem neuropathological analysis of brain tissue. OM samples were stored at −80°C from a time shortly after harvest until further use. Resuspended pellets of both the sCJD OM samples, but not the controls, were positive by RT‐QuIC analysis as we have reported previously. 7 , 11 Individual OM brushings were thawed, centrifuged at 3220× g at 4°C for 10 min. Supernatants were removed, and the pellets resuspended in 600 µl of PBS with 2% FBS and cuphorn sonicated at full power for 2 min (designated ‘neat’ OM samples). Serial dilutions were performed into PBS with 2% FBS and held at room temperature until inoculation within 2 h.
Pooled OM samples were prepared by combining three or four replicate brushings, collected in a single session from an individual, with 1 ml of PBS that was used to rinse each of the source tubes. The combined brushings were vortexed and centrifuged at 3,220× g at 4 °C for 10 min. The supernatants were removed and the pellets resuspended in 300 µl of PBS with 2% FBS with vortexing and sonication prior to inoculation within 2 h.
Nasal mucus specimens
Nasal secretions and OM samples were collected from 13 patients affected with sCJD using a specifically designed flocked brush (FLOQbrush®, Copan Italia Spa, Brescia, Italy). Nasal mucus secretions were collected from the nasal vestibule, while the OM brushings were collected by insertion of the brush to the top of the nasal cavity where the OM is located. The samples were processed and stored at –80°C. 7 , 11 The samples were thawed at room temperature and prepared for RT‐QuIC as described below.
CSF specimens
CSF samples obtained by lumbar puncture stored at −80°C were thawed, diluted 1:1 with 2X PBS containing 4% FBS, sonicated in a cup horn for 2 min at full power, and held at room temperature until inoculation within 2 h.
RT‐QuIC products for inoculation
Brain tissue dilutions (10‐3) from sCJD Patients 2 and 8 were each used to seed parallel RT‐QuIC reactions with and without thioflavin T (ThT) as described below. Approximately 12 h after the ThT‐containing reactions achieved maximally positive fluorescence readings (at ~10‐12 h) the products from five replicate ThT‐free reactions were collected with scraping of the bottom of the wells, pooled, and frozen until inoculation as described below. The final dilution of sCJD brain tissue in the RT‐QuIC reaction product was 2 x 10‐5 and the total recombinant PrP was 100 µg/ml.
Tg66 mouse bioassays
The brain, OM, CSF, and RT‐QuIC product preparations described above were inoculated intracerebrally (IC) using a volume of 25 µl (pooled OM samples only) or 50 µl (all other samples) per mouse. After inoculation the mice were monitored repeatedly over the next 24 h for acute signs of distress, in which case they were euthanized. Such distress was only observed with some of the OM inoculations (Table 1). The mice were monitored for clinical signs of neurological disease, in particular progressive deterioration of the use of nestlet material, followed by poor grooming, myoclonus, ataxia, tremors, kyphosis, weight loss, and somnolence (albeit with hyperactivity upon stimulation). Mice were euthanized by CO2 asphyxiation upon loss of appetite or thirst. The time from first positive indication of disease to euthanasia was ~10–14 days. For mice that became sick with prion disease, the clinical progression was indistinguishable between OM‐ and brain‐inoculated CJD animals. Brains were excised and separated into sagittal halves. The half contralateral to the inoculation site was placed immediately into 50 ml 10% neutralized formalin for fixation and processed for histological analysis. The ipsilateral half was placed into a microtube and flash frozen for subsequent RT‐QuIC and immunoblotting analyses.
RT‐QuIC assays
RT‐QuIC testing of brain samples from Tg66 mice was done blinded. Reactions were seeded with 2 μl of 10‐3 brain tissue dilutions for the initial screening and 10‐3 to 10‐9 brain tissue dilutions for end‐point dilution analysis. The RT‐QuIC reaction mix was composed of 10 mM phosphate buffer (pH 7.4), 300 mM NaCl, 0.1 mg/ml N‐terminally truncated recombinant Syrian hamster PrP (residues 90–231; GenBank accession number K02234), 10 μM ThT, 1 mM ethylenediaminetetraacetic acid tetrasodium salt (EDTA), and 0.001% SDS. Ninety‐eight microliter of this reaction mix were loaded into each well of a black 96‐well plate with a clear bottom (Nunc) and seeded with brain tissue dilutions. The plate was sealed (plate sealer, Nalgene Nunc International), incubated at 55 °C in a BMG FLUOstar Omega plate reader with cycles of 1‐min shaking (700 rpm double orbital) and 1‐min rest. ThT fluorescence measurements (450 +/‐ 10 nm excitation and 480 +/‐ 10 nm emission; bottom read) were taken every 45 min. The fluorescence threshold for a positive result was calculated as the mean of all values from negative brain tissues plus three standard deviations. For prion seeding activity quantitation, end‐point dilution assays were performed with Spearman–Kärber analyses to estimate the seeding dose (±SE) giving ThT positivity in 50% of technical replicate wells (SD50) 12 . For RT‐QuIC testing of nasal mucous and OM specimens, we used a recombinant full‐length Syrian hamster PrP (residues 23–231) substrate and the reactions were incubated at 42°C for 55 h, using experimental conditions that were otherwise the same as those described for the brain sample assays above. Mean maximum fluorescence values from quadruplicate reactions were normalized between plates as percentages of means from a positive sCJD control on each plate. As a provisional basis for discriminating positive from negative samples, we selected a threshold above the highest mean obtained from non‐CJD controls as shown with a dashed line in Figure 3. As is true with all RT‐QuIC assays, such a threshold will be subject to adjustment as larger and more varied negative and positive control specimens are assayed in future studies.
Hematoxylin and Eosin (H&E) and Immunohistochemistry (IHC) staining
Tissues were processed and stained for H&E, anti‐GFAP, and anti‐PrP (b3F4) staining as previously described. 29
Author Contributions
Conceived the overall project: GJR, GZ, MP, and BC. Clinically evaluated patients and/or collected specimens: GZ, MP, LS, SF. Mouse bioassays: GJR, LDR, BR. Neuropathology of mice: BR, GJR. Gel analyses: AGH, LDR. RT‐QuIC assays: CDO, MB, MF, BRG. Manuscript preparation: BC, GZ, MP, GJR, BR, CDO, BRG, SM.
Conflict of Interest
BC is an inventor on the following patents related to RT‐QuIC technology: #2554996 (France, UK, Germany), #2179293 (Switzerland, Germany, France, UK, Ireland) and #8,216,788 (USA).
Funding Information
This work was funded in part by the Intramural Research Program of the NIAID (to BC) and Italian Ministry of Health grant RF 2013‐02354884 to GZ and MP.
Acknowledgments
We thank Drs. Karin Peterson, Ankit Srivastava, and Durbadal Ojha for initial in‐house review of this manuscript. We thank Nancy Kurtz and Lori Lubke for performing histological staining of brain tissue, Jeff Severson for animal husbandry, and Santina Castriciano (Copan Italia Spa) for donating flocked brushes. We deeply thank families of CJD patients for their generous donations.
Funding Statement
This work was funded by Division of Intramural Research, National Institute of Allergy and Infectious Diseases grant AI001086‐08; Italian Ministry of Health grant RF 2013‐02354884.
Contributor Information
Gianluigi Zanusso, Email: gianluigi.zanusso@univr.it.
Byron Caughey, Email: bcaughey@nih.gov.
References
- 1. Brown P, Brandel JP, Sato T, et al. Iatrogenic Creutzfeldt‐Jakob disease, final assessment. Emerging Infect Dis. 2012;18:901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt‐Jakob disease. Ann Neurol. 1996;39:767–778. [DOI] [PubMed] [Google Scholar]
- 3. Zerr I, Parchi P. Sporadic Creutzfeldt‐Jakob disease. Handb Clin Neurol. 2018;153:155–174. [DOI] [PubMed] [Google Scholar]
- 4. Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real‐time quaking‐induced conversion. Nat Med. 2011;17(2):175–178. [DOI] [PubMed] [Google Scholar]
- 5. McGuire LI, Peden AH, Orru CD, et al. RT‐QuIC analysis of cerebrospinal fluid in sporadic Creutzfeldt‐Jakob disease. Ann Neurol. 2012;72(2):278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orru CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT‐QuIC detection of human Creutzfeldt‐Jakob disease using cerebrospinal fluid. mBio 2015;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bongianni M, Orrù CD, Groveman BR, et al. Diagnosis of human prion disease using real‐time quaking‐induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol. 2017;74(2):1–8. [DOI] [PubMed] [Google Scholar]
- 8. Foutz A, Appleby BS, Hamlin C, et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann Neurol. 2017;81(1):79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Franceschini A, Baiardi S, Hughson AG, et al. High diagnostic value of second generation CSF RT‐QuIC across the wide spectrum of CJD prions. Sci Rep. 2017;7(1):10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Groveman BR, Orru CD, Hughson AG, et al. Extended and direct evaluation of RT‐QuIC assays for Creutzfeldt‐Jakob disease diagnosis. Ann Clin Transl Neurol. 2017;4(2):139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Orru CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt‐Jakob disease using nasal brushings. New Engl J Med. 2014;371(6):519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilham JM, Orrú CD, Bessen RA, et al. Rapid end‐point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 2010;6(12):e1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown P, Gibbs CJ Jr, Rodgers‐Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–529. [DOI] [PubMed] [Google Scholar]
- 14. Draganescu N, Girjabu E, Antipa C, et al. Creutzfeldt‐Jakob disease: experimental transmission in the guinea pig. Virologie. 1984;35(2):95–98. [PubMed] [Google Scholar]
- 15. Tateishi J, Sato Y, Koga M, et al. Experimental transmission of human subacute spongiform encephalopathy to small rodents. I. Clinical and histological observations. Acta Neuropathol. 1980;51(2):127–134. [DOI] [PubMed] [Google Scholar]
- 16. Race B, Meade‐White KD, Miller MW, et al. Susceptibilities of nonhuman primates to chronic wasting disease. Emerging Infect Dis. 2009;15(9):1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Douet JY, Zafar S, Perret‐Liaudet A, et al. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt‐Jakob disease. Emerging Infect Dis. 2014;20(1):114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kraus A, Raymond GJ, Race B, et al. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J Virol. 2017;91(21):e01276–e1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Groveman BR, Raymond GJ, Campbell KJ, et al. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog 2017;13(9):e1006623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim C, Haldiman T, Cohen Y, et al. Protease‐sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt‐Jakob disease are indicator of progression rate. PLoS Pathog 2011;7(9):e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Pedro CJ, Ruiz Tovar M, Ward H, et al. Sensitivity to biases of case‐control studies on medical procedures, particularly surgery and blood transfusion, and risk of Creutzfeldt‐Jakob disease. Neuroepidemiol. 2012;39(1):1–18. [DOI] [PubMed] [Google Scholar]
- 22. Alcalde‐Cabero E, Almazan‐Isla J, Brandel JP, et al. Health professions and risk of sporadic Creutzfeldt‐Jakob disease, 1965 to 2010. Euro Surveill. 2012;17(15). [PubMed] [Google Scholar]
- 23. Takeda S, Commins C, DeVos SL, et al. Seed‐competent HMW tau species accumulates in the cerebrospinal fluid of Alzheimer's disease mouse model and human patients. Ann Neurol. 2016;80(3):355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saijo E, Metrick MA 2nd, Koga S, et al. 4‐Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020;139:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zanusso G, Ferrari S, Cardone F, et al. Detection of pathologic prion protein in the olfactory epithelium in sporadic Creutzfeldt‐Jakob disease. N Engl J Med. 2003;348(8):711–719. [DOI] [PubMed] [Google Scholar]
- 26. Ward HJ, Everington D, Cousens SN, et al. Risk factors for sporadic Creutzfeldt‐Jakob disease. Ann Neurol. 2008;63(3):347–354. [DOI] [PubMed] [Google Scholar]
- 27. Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA. 2010;107(26):12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orrú CD, Groveman BR, Raymond LD, et al. Bank vole prion protein as an apparently universal substrate for RT‐QuIC‐based detection and discrimination of prion strains. PLOS Pathog. 2015;11(6):e1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Race B, Williams K, Chesebro B. Transmission studies of chronic wasting disease to transgenic mice overexpressing human prion protein using the RT‐QuIC assay. Vet Res. 2019;50(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]