

Abstract
Objective
Type I sialidosis (ST‐1) is a rare autosomal recessive inherited disorder. To date, there has been no study on ST‐1 patients in mainland China.
Methods
We reported in detail the cases of five Chinese ST‐1 patients from two centers, and summarized all worldwide cases. Then, we compared the differences between Chinese and foreign patients.
Results
A total of 77 genetically confirmed ST‐1 patients were identified: 12 from mainland China, 23 from Taiwan, 10 from other Asian regions, and 32 from European and American regions. The mean age of onset was 16.0 ± 6.7 years; the most common symptoms were myoclonus seizures (96.0%), followed by ataxia (94.3%), and blurred vision (67.2%). Compared to other groups, the onset age of patients from mainland China was much younger (10.8 ± 2.7 years). The incidence of visual impairment was lower in patients from other Asian regions than in patients from mainland China and Taiwan (28.6% vs. 81.8%–100%). Cherry‐red spots were less frequent in the Taiwanese patients than in patients from other regions (27.3% vs. 55.2%–90.0%). Furthermore, 48 different mutation types were identified. Chinese mainland and Taiwanese patients were more likely to carry the c.544A > G mutation (75% and 100%, respectively) than the patients from other regions (only 0%–10.0%). Approximately 50% of Chinese mainland patients carried the c.239C > T mutation, a much higher proportion than that found in the other populations. In addition, although the brain MRI of most patients was normal, 18F‐FDG‐PET analysis could reveal cerebellar and occipital lobe hypometabolism.
Interpretation
ST‐1 patients in different regions are likely to have different mutation types; environmental factors may influence clinical manifestations. Larger studies enrolling more patients are required.
Introduction
Sialidosis has been gradually recognized since it was named in the 1970s. 1 , 2 , 3 At present, we have clearly realized that the disease is an important autosomal recessive inherited lysosomal storage disorder (LSD), characterized by a deficiency in α‐N‐acetyl neuraminidase that results from a mutation in the neuraminidase gene (NEU1). 4 , 5 The impaired neuraminidase activity prevents oligosaccharides and glycoproteins from getting rid of the terminal sialic acid molecules, leading sialic acid‐rich macromolecules to aggregate in neurons and multiple organs. 4 , 6 , 7 For 40 years, sialidosis has always been classified into two types on the basis of phenotype and onset age. 8 Compared with type II sialidosis (ST‐2), type I (ST‐1) has a milder clinical course, usually affects adolescents or adults and is the focus of our study.
Currently, there is only one obsolete epidemiological study specializing in the Australian population for reference, with the prevalence of sialidosis estimated at 1 in 4.2 million live births. 9 Furthermore, ST‐1 is rarer than ST‐2, with only hundreds of patients reported in the world according to the literature. The clinical spectrum of sialidosis is expanding and overlaps with that of many other LSDs, and establishing a diagnosis purely based on clinical manifestations is impossible. 10 , 11 In addition, the increased excretion pattern of urinary oligosaccharides and bound sialic acid in sialidosis is also not specific or sensitive and may be best as a screening tool. 10 , 12 , 13 , 14 Usually, researchers diagnose sialidosis by detecting the activity of neuraminidase in cultured fibroblasts or leucocytes 13 , 15 , 16 , 17 ; however, this method is not accurate when simply considering this single enzyme because there is a secondary combined deficiency of neuraminidase in galactosialidosis, 5 leading to many misdiagnoses in previous literature. 18 , 19 , 20 , 21 The instability of enzymes in vitro is another drawback. 22 Theoretically, the severity of the phenotype correlated with the residual activity of neuraminidase, but uncertainty remains, the enzyme activity of some ST‐1 patients with late onset and chronic course is too low to be detected, 10 or severe infantile ST‐2 patients have a relatively higher level of reserve enzyme activity. 23 Hence, we should proceed from the root cause and make accurate diagnoses on the basis of the NEU1 gene.
By using gene sequencing technology, researchers are expected to aggregate information on mutation types in this sporadic and rare disease, build molecular models, and establish databases that can predict phenotypes through genotypes. 22 , 24 , 25 To date, approximately 50 mutations in the NEU1 gene have been reported in patients with sialidosis, and the number is still increasing. 6 Here, we introduce in detail five Chinese mainland ST‐1 patients from two different tertiary epilepsy centers, and we summarize all the genetically confirmed cases worldwide. Then, we determine the differences between Chinese and foreign patients. We wish to enhance the awareness of clinicians and to promote the diagnosis and treatment of the disease. To the best of our knowledge, our case series is the largest case group in mainland China to date.
Patients and Methods
Clinical analysis of five illustrative cases
We identified five genetically confirmed ST‐1 patients. Blood‐based gene sequencing was performed by a third‐party company with the technology of whole‐exon sequencing (WES) on HiSeq X10. The average depth of the WES ranged from 104.43X to 124.02X. The 20X coverage of WES ranged from 94.88% to 99.60%. The transcript number of the NEU1 gene was NM_000434.3.
Patients 1–3 came from Beijing Tiantan Hospital, and the other two patients came from Peking Union Medical College Hospital. All patients gave written informed consent for participation and written consent to permit the publication of clinical details.
Patient 1 underwent 18‐fluoro‐deoxyglucose (18F‐FDG) positron emission tomography (PET) scans during clinical evaluations. The PET procedure and data analysis were performed according to a previous study. 26 Details are described in the Supplementary Material.
Comparisons to previous reports of ST‐1 patients from different regions or with different genotypes
We conducted searches on the literature retrieval platforms WanFang Data (http://www.wanfangdata.com.cn/), CNKI (https://www.cnki.net/), and PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) for articles up to October 2019 by using the title/abstract keyword “sialidosis.” We selected original studies or case (series) reports that included genetically confirmed ST‐1 patients from 1996. Reference screening was conducted by two experienced neurological doctors, Tao‐Ran Li and Rui‐Juan Lv.
Articles on ST‐1 patients without genetic tests, on ST‐2 patients, and not written in English or Chinese were excluded. Researchers in Taiwan have made great contributions to sialidosis research, but several of their studies could not be included in our analysis. As the Taiwanese authors said, there were overlaps among the ST‐1 patients enrolled in these literatures, 27 , 28 , 29 and we cannot know which patients were repeatedly included. We speculated that the 11 patients in the recent literature 30 overlapped with those of previous reports 28 , 29 ; therefore, only three genetically proven Taiwanese patients were included in our analysis.
We summarized the selected cases and compared the clinical and genetic characteristics among patients from the Chinese mainland, Taiwan, other Asian regions, and European and American regions. In addition, we also compared patients with different genotypes.
Statistics analysis
We subgrouped cases from the literature review based on their ethnicities and genotypes. A one‐way ANOVA was performed for group comparison of onset age, and the Games–Howell test was used for the post hoc analysis because of heteroscedasticity. For categorical variables, Fisher's exact test or the chi‐squared test was used for group comparison. A P < 0.05 was considered significant. We performed all analyses using SPSS 13.0 software (SPSS, Chicago, IL).
Results
Clinical descriptions
The cases of the five Chinese mainland patients are described as follows. We provided a detailed description of patient 1 and a simple description of the rest in the Supplementary Material because of their similar examination results and because of spatial limitations. There is also a detailed and comparative description in Table 1. All patients were right‐handed and were born at term to nonconsanguineous Chinese mainland Han parents, with no abnormal antenatal or postnatal issues of note, and all reached developmental milestones appropriately. All patients had mutations in the NEU1 gene. Their past medical histories and family histories of seizures, ataxia, or other neurological disorders, were all unremarkable.
Table 1.
Clinical characteristics of Chinese mainland type I sialidosis patients in our study.
| Patient 1, this study | Patient 2, this study | Patient 3, this study | Patient 4, this study | Patient 5, this study | |
|---|---|---|---|---|---|
| Age/gender | 17/M | 14/F | 31/M | 20/F | 19/M |
| Past history | Easily tired after exercise; bad sports performance | Bad sports performance | Unremarkable | Bad sports performance | Unremarkable |
| Family history | Unremarkable | Unremarkable | Unremarkable | Unremarkable | Unremarkable |
| Symptom onset age | 12 years old | 12 years old | 17 years old | 10 years old | 10 years old |
| Initial symptoms | GTCS | Symmetric distal neuropathic pain | Myoclonus | A and falls | GTCS |
| Seizures | GTCS; MCS | GTCS; MCS | GTCS; MCS | GTCS; MCS | GTCS; MCS |
| Other symptoms | D; falls; A; hyperreflexia | CI; D; falls; A; bedridden; hyperreflexia | D; falls; A; hypertonia; hyperreflexia | D; bedridden | D; A; nystagmus; unequal pupils; hyperreflexia; ankle clonus |
| Visual acuity | Gradually declined | Gradually declined | Gradually declined | Gradually declined | Gradually declined |
| Aural acuity | Gradually declined; bilateral nervous deafness | Normal | Normal | Normal | Normal |
| Brain MRI | Normal | Widening of bilateral cerebellar sulcus | Normal | Normal | Mild atrophy of cerebellar vermis and enlargement of midbrain aqueduct |
| EMG | Bilateral F‐waves were not elicited | Normal | Normal | U | Neurogenic damage of extremities |
| BAEP | Normal | Normal | Normal | U | Abnormal waveform differentiation |
| VEP (P100) | Abnormal peak latency and amplitude | Abnormal peak latency and amplitude | Normal | U | Prolonged peak latency |
| SEP of upper limb | Huge potential | Huge potential | Huge potential | U | Huge potential |
| SEP of lower limb | Abnormal deep sensory conduction | Abnormal deep sensory conduction | Abnormal deep sensory conduction | U | Abnormal deep sensory conduction |
| Ocular fundus | CRS | Punctate opacity of binocular lens; CRS | CRS | Punctate opacity of binocular lens; CRS | CRS |
| Visual field | Diffuse defect of OU visual field | OS, defect except the lower quadrant of nasal field; OD, enlargement of physiological blind spot | Normal | Normal | Normal |
| OCT | Diffuse weakness of bilateral RNFL and GCC; hyperreflex of macular inner layer | Basically normal | Hyperreflex of macular inner layer | U | Hyperreflex of macular inner layer |
| Blood examination | Normal | Normal | Normal | Normal | Normal |
| CSF examination | Normal | Weakly positive of oligoclonal band | Normal | Normal | Normal |
| Mutation of NEU1 | c.803A > G; c.239C > T | c.544A > G; c.239C > T | c.544A > G; c.239C > T | c.838G > A; c.403C > T | c.1118T > C; c.544A > G |
| Transmission | Hereditary mutation | Hereditary mutation | Hereditary mutation | Hereditary mutation | U |
| Therapy | LEV, 750 mg/bid; VPA, 500 mg/bid; CZP, 0.5 mg/bid | LEV, 500 mg/bid to 750 mg/bid; CZP, 1 mg/tid | LEV, 500 mg/bid; CZP, 1 mg/qn | LEV, 500 mg/bid | U |
| Outcome | 2 m later: GTCS, zero; MCS and A, obviously improved | 6 m later: loss of self‐sufficiency; 2 y later: GTCS, 1–3/m; MCS, daily; A and visual function deteriorated even further | 2 y later: seizures, A and visual function deteriorated even further | Seizures and A improved | U |
F, female; M, male; U, unsure; GTCS, generalized tonic–clonic seizure; A, ataxia; MCS, myoclonus seizures; CI, cognitive impairment; D, dysarthria; MRI, magnetic resonance imaging; EMG, electromyography; BAEP, brainstem auditory evoked potential; VEP, visual evoked potential; SEP, somatosensory evoked potential; CRS, cherry red spot; OD, oculus dexter; OS, oculus sinister; OU, oculus unati; OCT, optical coherence tomography; RNFL, retina nerve fiber layer; GCC, ganglion cell complex; CSF, cerebrospinal fluid; LEV, levetiracetam; CZP, carbamazepine; VPA, valproic acid.
Patient 1. A 17‐year‐old man was easily fatigued after activities and had poor sports performance since childhood. He presented with typical nocturnal generalized tonic–clonic seizures (GTCSs) 5 years ago, 1–3 times per year, which were well controlled with levetiracetam (750 mg/bid) over the past 3 years. Four years ago, he developed walking instability and frequent falls, accompanied by ambiguous language. Three years ago, his limbs trembled involuntarily, several times per day. These syndromes have deteriorated gradually. On neurological examination, he had dysarthria, ataxia of the trunk and four limbs, an enhanced bilateral tendon reflex, and negative Babinski sign. Routine examinations of blood and cerebrospinal fluid, including onconeural and neuronal surface antibodies, were normal. Abdominal ultrasonography, heart rate variability, electromyography, and nerve conduction velocity all showed no significant abnormalities except F‐waves, which were not elicited bilaterally. The pure tone threshold examination indicated bilateral neuro‐deafness. The brainstem auditory evoked potential was normal, while the visual evoked potential (VEP) showed abnormal peak latency and amplitude of P100. The somatosensory evoked potential (SEP) showed a huge potential of bilateral N20‐P25 in the upper limb pathway (Fig. 1E) and abnormal deep sensory conduction peripherally in the lower limb pathway. The electroencephalogram (EEG) indicated frequent polyspike and polyspike‐slow waves in bilateral central, parietal, and midline regions, accompanying myoclonic seizures (Fig. 1B). Ophthalmological examination found that the best‐corrected visual acuity of the left eye was 50/100 and of the right was 60/100; a dark‐red, round, spotted lesion was detected in both macular regions, suggesting cherry‐red spots (Fig. 1C); the bilateral eye visual fields showed diffuse defects; the optical coherence tomography (OCT) indicated bilateral diffuse weakness of the retinal nerve fiber layer and ganglion cell complex, as well as the hyperreflexivity of the macular inner layer (Fig. 1D). Although the brain magnetic resonance imaging (MRI) was normal (Fig. 1A), the 18F‐FDG‐PET analysis revealed hypometabolism in the left cuneus and lingual gyrus, right inferior semilunar lobule of cerebellum, left cerebellar tonsil, and culmen of the cerebellum compared to controls (Fig. 2).
Figure 1.
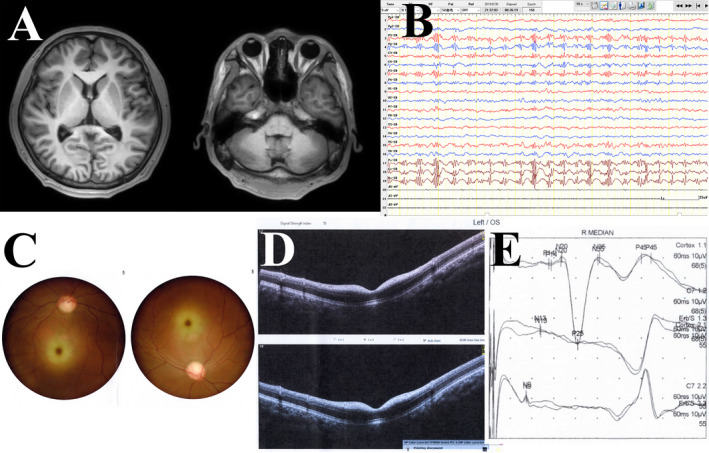
Examination results of patient 1. (A) Axial T1 brain magnetic resonance imaging was normal; (B) electroencephalogram showed bilateral polyspike and polyspike‐slow waves in the central, parietal, and midline regions; (C) fundus examination showed bilateral cherry‐red spots; (D) optical coherence tomography indicated bilateral diffuse weakness of the retinal nerve fiber layer and ganglion cell complex, as well as the hyperreflexivity of the macular inner layer; (E) somatosensory evoked potential showed a huge potential of bilateral N20‐P25 in the upper limb pathway.
Figure 2.
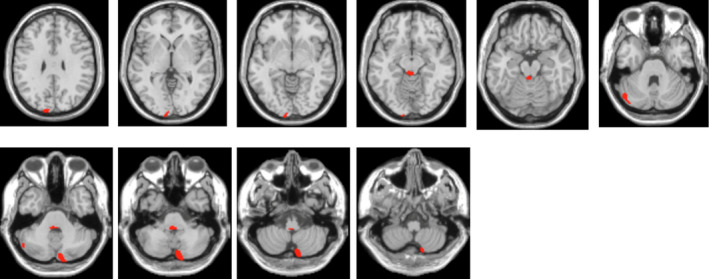
Regions displaying hypometabolism in patient 1 compared with controls. 18F‐FDG‐PET analysis revealed hypometabolism of the left cuneus and lingual gyrus, right inferior semilunar lobule of the cerebellum, left cerebellar tonsil, and culmen of the cerebellum.
The blood of the patient and his parents was examined by WES. Eventually, the patient was confirmed to have the heterozygous c.803A > G (p.Y268C) and c.239C > T (p.P80L) mutation in the NEU1 gene, inherited from his father and mother, respectively.
Clinical characteristics of ST‐1 patients
Eventually, we identified 35 articles according to the literature review (detailed information in the Supplementary Material). In addition to our 5 patients, another 72 ST‐1 patients from these articles were included for comparison. We collected clinical information as much as possible according to the description of the original literature, and we tried our best to ensure accuracy. The specific clinical characteristics of the 77 ST‐1 patients are summarized in Table 1 and the Supplementary Tables (Table S1 is a continuation of Table 1 and describes the other 7 mainland patients reported in the literature; Table S2 describes 65 patients from other regions). In Table 2, we summarized the data from all patients and compared the clinical data according to the origin of the patients.
Table 2.
Comparison of genetically confirmed ST‐1 patients from different ethnic groups.
| Clinical characteristics | Chinese mainland | Taiwan | Other Asian regions | European and American regions | Total |
|---|---|---|---|---|---|
| No. of cases | 12 | 23 | 10 | 32 | 77 |
| Ethnicity | The Han nationality | Taiwanese | East‐Asian, 1; Indian, 3; Korean, 1; Japanese, 5 | European, 27; American, 4; Ecuadorian, 1 | \ |
| Onset age (y) | 10.8 ± 2.7 (10 Ava) | 18.0 ± 6.6 (23 Ava)** | 15.8 ± 6.3 (9 Ava)* | 16.9 ± 6.7 (24 Ava)** | 16.0 ± 6.7 (66 Ava) |
| M/F | 7/5 | 17/6 | 6/4 | 9/18 (27 Ava) | 39/33 (72 Ava) |
| MCS | 100% (12, 12 Ava) | 100% (23, 23 Ava)# | 90.0% (9, 10 Ava)# | 93.3% (28, 30 Ava)# | 96.0% (72, 75 Ava) |
| Ataxia | 100% (9, 9 Ava) | 95.7% (22, 23 Ava)# | 100% (10, 10 Ava)# | 89.3% (25, 28 Ava)# | 94.3% (66, 70 Ava) |
| Blurred vision | 100% (11, 11 Ava) | 81.8% (18, 22 Ava)# | 28.6% (2, 7 Ava)** | 55.6% (10, 18 Ava)* | 67.2% (39, 58 Ava) |
| CRS | 72.7% (8, 11 Ava) | 27.3% (6, 22 Ava)* | 90.0% (9, 10 Ava)# | 55.2% (16, 29 Ava)# | 54.2% (39, 72 Ava) |
| VEP (abnormal P100) | 83.3% (5, 6 Ava) | 95.5% (21, 22 Ava)# | 100% (2, 2 Ava)# | 33.3% (3, 9 Ava)# | 79.5% (31, 39 Ava) |
| SEP (GCP) | 100% (4, 4 Ava) | 100% (22, 22 Ava)# | 100% (2, 2 Ava)# | 100% (7, 7 Ava)# | 100% (35, 35 Ava) |
| Brain MRI | 25.0% (2, 8 Ava) | 42.1% (8, 19 Ava)# | 42.9% (3, 7 Ava)# | 37.5% (6, 16 Ava)# | 38.8% (19, 49 Ava) |
| NEU1 mutation, c.544A > G | 75.0% (9, 12 Ava) | 100% (23, 23 Ava)* | 10.0% (1, 10 Ava)** | 0% (0, 32 Ava)** | 42.9% (33, 77 Ava) |
| NEU1 mutation, c.239C > T | 50.0% (6, 12 Ava) | 13.0% (3, 23 Ava)* | 10.0% (1, 10 Ava)# | 0% (0, 32 Ava)** | 13.0% (10, 77 Ava) |
ST‐1, Type I sialidosis; Ava, available; M, male; F, female; MCS, myoclonus seizures; CRS, cherry red spot; VEP, visual evoked potential; SEP, somatosensory evoked potential; GCP, giant cortical potential; MRI, magnetic resonance imaging. European: German, 3; Dutchman, 3; Greek, 1; French, 3; Turkey, 3; Czech, 1; Italian, 9; Spaniard, 1; Portuguese, 3. Compared with Chinese mainland patients, *P < 0.05, **P < 0.01, # P > 0.05.
Of the 77 patients, 12 were from mainland China, 23 were from Taiwan, 10 were from other Asian regions outside China, mainly Japan (5) and India (3), and 32 were from European and American regions, mainly from Europe (27). According to the information presently available, the proportion of males was approximately 54.2% (72 available); the mean age of onset was 16.0 ± 6.7 years (66 available). The most common symptoms were myoclonic seizures (96.0%, 75 available), followed by ataxia (94.3%, 70 available), and blurred vision (67.2%, 58 available). Other manifestations were difficult to count due to limited information, such as GTCSs, dysarthria, dysphagia, hearing loss, or cognitive impairment. Notably, though with a limited number of patients, the available data showed that the proportion of giant cortical potentials in SEP reached an amazing 100% (35 available), and 79.5% (39 available) of patients presented abnormal VEP, while cherry‐red spots were reported in only 39 of 72 (54.2%) patients. Abnormalities on brain MRI were found in 19 of 49 (38.8%) patients, 16/19 (84.2%) showed brain atrophy, and the remaining three showed a widening of the bilateral cerebellar sulcus (patient 2), frontotemporal encephalomalacia, and cerebellar meningocele. 29 Because of autosomal recessive inheritance, each patient has two mutation sites (perhaps homozygosity) in the NEU1 gene, corresponding to two mutation “occurrences.” Consequently, we acquired 150 mutation “occurrences” in these 77 patients (we could obtain only one mutation site for patients 60, 64, 66, and 67 12 , 24 , 25 ). As shown in Figure 3, there were 8 mutation types in Chinese mainland patients, 6 types in Taiwanese patients, 14 types in other Asian patients, and 30 types in European and American patients. In total, 48 different mutation types were identified. The most frequent mutation type was c.544A > G, which occurred 51 times in 33 persons, followed by c.239C > T (10 times, 10 persons), c.679G > A (7 times, 7 persons), c.200G > T (6 times, 3 persons), and c.982G > A (4 times, 4 persons); other types occurred only 1–3 times.
Figure 3.
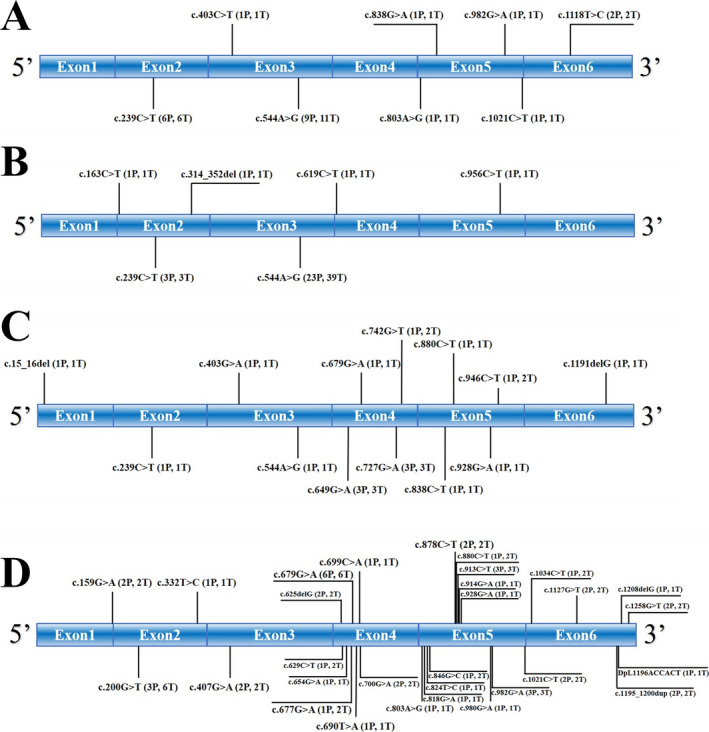
Mutation types of ST‐1 patients. (A–D) show the type and number of mutations in ST‐1 patients from the Chinese mainland, Taiwan, other Asian regions, and European and American regions, respectively. P means the number of patients; T means the mutation times. Each patient has two mutation sites (perhaps homozygosity) in the NEU1 gene, corresponding to two mutation “occurrences.” A mutation in the intron region of an Indian patient, g.2869A > T (1P, 1T), and a c.1258G > T (2P, 2T) mutation in the 3’‐UTR regions of two Americans patients are not marked in the figure.
Comparison of patients from different ethnic groups
As shown in Table 2, we divided patients into the abovementioned four groups according to their origins to assess whether regions and races have an impact on genotypes or phenotypes of ST‐1. We found that the onset age of patients from mainland China was significantly lower than that of the other three groups, and there was no significant difference among patients from other regions. There is no point in comparing the ratio of men to women for autosomal diseases. The major symptoms (myoclonus and ataxia) were similar among these four groups. Notably, compared with the astounding incidence rate of visual impairment in Chinese mainland and Taiwanese patients, 100% (11/11) and 81.8% (18/22), respectively, the incidence was obviously lower in patients from other Asian regions, only 28.6% (2/7). Cherry‐red spots were reported less frequently in Taiwanese patients than in the other three groups (27.3% vs. 55.2%–90.0%). All patients in the four groups had abnormal giant cortical potentials in SEP, while the VEP of patients from European and American regions tended to be less likely to be abnormal than that of patients from the other three groups (33.3% vs. 83.3%–100%). The probability of brain MRI abnormalities was between 25% and 42.9%, and there was no difference among groups. As shown in Figure 3 and Table 2, importantly, up to 9/12 (75%) Chinese mainland patients and 23/23 (100%) Taiwanese patients carried the c.544A > G mutation in the NEU1 gene, which was much higher than proportion found in the other Asian group and the European and American group (10.0% and 0%, respectively). In addition, the homozygous proportion of Taiwanese patients (16/23, 69.6%) was higher than that of Chinese mainland patients (2/9, 22.2%). Another concern was that Chinese mainland patients were also more likely to carry the c.239C > T (6/12, 50%) mutation than other groups (0%–13%).
Comparison of patients with different genotypes
To further understand the genotype–phenotype correlations, we divided patients according to their mutation types. As shown in Table 3, eight patients had the heterozygous c.544A > G and c.239C > T mutation and 18 had the homozygous c.544A > G mutation; all of them were from mainland China or Taiwan. The next high‐frequency genotypes were heterozygous c.649G > A and c.727G > A, homozygous c.200G > T and heterozygous c.679G > A, and c.913C > T. All three genotypes appeared three times; furthermore, the latter two belonged to familial aggregations, both coming from siblings of the same family. Four mutation types appeared two times, which are also shown in Table 3. The remaining 34 genotypes appeared only once, and the clinical details can be found in Table 1 and the Supplementary Tables.
Table 3.
Comparison of genetically confirmed ST‐1 patients with different genotypes.
| Mutation types | c.544A > G; c.239C > T | Homozygous c.544A > G | c.649G > A; c.727G > A | Homozygous c.200G > T | c.679G > A; c.913C > T | c.1195_1200dup; c.679G > A | c.159G > A; c.1127G > T | c.914G > A; c.625delG | c.700G > A; c.1021C > T |
|---|---|---|---|---|---|---|---|---|---|
| Ethnic regions (No.) | 8 | 18 | 3 | 3 | 3 | 2 | 2 | 2 | 2 |
| mainland China | 5 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Taiwan | 3 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| other Asian regions | 0 | 0 | 3a | 0 | 0 | 0 | 0 | 0 | 0 |
| European and American regions | 0 | 0 | 0 | 3b | 3b | 2b | 2b | 2a | 2a |
| Onset age (y) | 11.6 ± 3.1 (8 Ava) | 20.6 ± 6.3 (16 Ava)** | 21.3 ± 9.3 (3 Ava) | 23.3 ± 1.5 (3 Ava) | 27.3 ± 4.0 (3 Ava) | 10.5 ± 2.1 (2 Ava) | 16.0 ± 1.4 (2 Ava) | 17.0 ± 0 (2 Ava) | 26c |
| M/F | 4/4 | 14/4 | 2/1 | 1/2 | 1/2 | 0/2 | 0/2 | 0/2 | 1/0c |
| MCS | 100% (8, 8 Ava) | 100% (18, 18 Ava)# | 100% (3, 3 Ava) | 100% (3, 3 Ava) | 100% (3, 3 Ava) | 100% (2, 2 Ava) | 100% (2, 2 Ava) | 100% (2, 2 Ava) | 100% (2, 2 Ava) |
| Ataxia | 100% (6, 6 Ava) | 94.1% (16, 17 Ava)# | 100% (3, 3 Ava) | 100% (3, 3 Ava) | 100% (3, 3 Ava) | 50.0% (1, 2 Ava) | 100% (2, 2 Ava) | 100% (2, 2 Ava) | 100% (1, 1 Ava) |
| Blurred vision | 100% (8, 8 Ava) | 81.3% (13, 16 Ava)# | 50.0% (1, 2 Ava) | 0% (0, 3 Ava) | 0% (0, 2 Ava) | 100% (2, 2 Ava) | U | 0% (0, 2 Ava) | 100% (2, 2 Ava) |
| CRS | 75.0% (6, 8 Ava) | 6.3% (1, 16 Ava)** | 100% (3, 3 Ava) | 0% (0, 3 Ava) | 0% (0, 2 Ava) | 100% (2, 2 Ava) | 50.0% (1, 2 Ava) | 0% (0, 2 Ava) | 100% (2, 2 Ava) |
| VEP (abnormal P100) | 80.0% (4, 5 Ava) | 93.8% (15, 16 Ava)# | U | 0% (0, 3 Ava) | 0% (0, 3 Ava) | U | U | U | U |
| SEP (GCP) | 100% (5, 5 Ava) | 100% (15, 15 Ava)# | U | 100% (3, 3 Ava) | 100% (3, 3 Ava) | U | U | 100% (1, 1 Ava) | U |
| Brain MRI | 33.3% (1, 3 Ava) | 43.8% (7, 16 Ava)# | 0% (0, 2 Ava) | 33.3% (1, 3 Ava) | 0% (0, 3 Ava) | 0% (0, 2 Ava) | U | 100% (1, 1 Ava) | 100% (1, 1 Ava) |
ST‐1, Type I sialidosis; Ava, available; M, male; F, female; MCS, myoclonus seizures; CRS, cherry red spot; VEP, visual evoked potential; SEP, somatosensory evoked potential; GCP, giant cortical potential; MRI, magnetic resonance imaging; U, unsure. a, the patients came from different families; b, the patients were siblings. c, only one patient’s data can be acquired. Compared with the heterozygous c.544A > G and c.239C > T mutation patients, **P < 0.01, # P > 0.05.
Considering the limited sample size, the unavailability of some data and familial aggregations, we made statistical comparisons only of the two most frequent mutation types: the heterozygous c.544A > G and c.239C > T mutation and the homozygous c.544A > G mutation. We found differences in the onset age and incidence of cherry‐red spots; the onset age of the former genotype was 11.6 ± 3.1 years old, which was significantly lower than that of the latter (20.6 ± 6.3 years), while only one of the 16 homozygous c.544A > G mutation patients had cherry‐red spots, which was significantly lower than that of the patients with heterozygous c.544A > G and c.239C > T mutation (75.0%). We found no other differences, such as seizures or ataxia (Table 3).
Discussion
In this study, we included five ST‐1 patients from two tertiary epilepsy centers in mainland China. They have similar manifestations, examination results and mutation types (3/5 have the c.544A > G or c.239C > T mutation). Then, we explored previous literature, hoping to further determine the influence of regions and ethnicities on ST‐1. Finally, we included 12 patients from mainland China, 24 , 31 , 32 , 33 , 34 23 from Taiwan, 10 from other Asian regions, and 32 from Europe and America. We summarized their clinical characteristics and made a horizontal comparison. To the best of our knowledge, this study provides the largest number of patients from mainland China to date and includes all known genetically confirmed ST‐1 patients for the first time.
According to the present 77 ST‐1 patients, we found that there was no sex difference, and the clinical manifestations usually started in adolescence (16.0 ± 6.7 years). A similar conclusion was obtained in another study where the onset age was 15.5 ± 5.5 years. 35 Although there may be overlaps of the enrolled patients, three different studies suggested that the average age of onset in Taiwanese patients was 19.8 ± 6.2, 28 19.4 ± 6.1, 29 and 18.3 ± 9.5 years, 30 respectively, which was also similar to our result (18.0 ± 6.6 years). Moreover, we found that the mean onset age in the Caucasian population did not change after enlarging the sample size and enrolling African Americans in our study (16.9 ± 6.7 years) when compared with a previous study (16.7 ± 6.3 years). 35 Obviously, ST‐1 patients in mainland China were younger than all the other regions, only 10.8 ± 2.7 years, but the reasons for this observation are elusive. Although the onset age of the patients with heterozygous c.544A > G and c.239C > T mutation was lower than that of the patients with homozygous c.544A > G mutation, we considered that the result was also influenced by regional factors. Except for one patient whose onset age was 17 years, the onset age of the other nine patients from mainland China was 7–12 years, independent of genotypes. Further studies are needed to clarify this.
Consistent with previous studies, 28 , 29 , 30 , 35 almost all patients had myoclonus and ataxia. The incidence of epilepsy was between 72.5% and 83.3%, 28 , 29 , 30 , 35 mainly myoclonus seizures and GTCSs, while focal tonic seizures 35 or partial complex seizures 25 or only EEG discharge 13 can also exist. Notably, visual impairment in patients from other Asian regions (28.6%) was significantly lower than those observed in the Chinese mainland group and the Taiwanese group (100% and 81.8%, respectively); VEP abnormality in patients from European and American regions (33.3%) seem to be rare, significantly lower than those observed in the Chinese mainland group and the Taiwanese group (83.3% and 95.5%, respectively). In addition, visual impairment in patients from other Asian regions and VEP abnormality in patients from European and American regions were also significantly lower than previous studies of Taiwanese (75%–100% and 81.8%–94.1%, respectively). 28 , 29 , 30 Curiously, the proportion of visual impairment and VEP abnormality did not match that found in the other Asian population, and we speculated that this was related to the integrity of the patients' examinations, because many patients had completed only one of them, causing information deviations and statistical errors. Ahn et al. found that early‐onset patients are more likely to have visual impairment than late‐onset patients. 36 However, the Taiwanese patients in our study challenged that result; they had the highest average onset age and were also likely to have impairment. It was once thought that cherry‐red spots were inevitable in the course of ST‐1, 37 but now, we have realized that their observation is not sensitive or specific and just plays a suggestive role; these spots can disappear as the disease progresses 17 or emerge 20 years after the disease onset. 16 However, the presence of cherry‐red spots in progressive myoclonic epilepsy patients strongly suggests a diagnosis of sialidosis. 6 Contrary to the incidence in other regions (55.2%–72.7%) in our study or in a previous report of a Caucasian population (61.1%), 35 only 6 out of 22 Taiwanese patients have cherry‐red spots detected. The difference may be partially determined by the homozygous c.544A > G mutation, which is known to have higher residual enzymatic activity with similar half‐life and cause no obvious structural change in the lysosomal multienzyme complex. On the basis of the results, we supposed that late‐onset patients tend to be free of spots, probably due to the inclusion of Taiwanese patients. 36 Brain MRI is not necessary for the diagnosis because it is mostly normal or parts show nonspecific atrophy (32.7% in our study). However, similar to previous reports, 38 , 39 patient 1, with normal MRI, showed bilateral hypometabolism in the cerebellum and occipital lobe, indicating that 18F‐FDG‐PET is more sensitive for the detection of abnormalities than MRI. Furthermore, a complete ophthalmic examination, including the fundus, visual field, and OCT, is necessary for the comprehensive assessment of ST‐1, 16 , 34 , 40 and other examinations, such as EEG and SEP, are likely to be 100% abnormal. 28 , 29 , 30 , 35 Currently, there is no consensus on the diagnosis of ST‐1, but the following typical manifestations can be used to suspect the diagnosis. For those patients whose clinical presentation is consistent with adolescent onset of progressive myoclonic epilepsy, their detailed ophthalmic examination reveals cherry‐red spots, and clinicians should implement genetic tests to determine a definite diagnosis of ST‐1 as early as possible.
The five patients in our study showed some relatively rare signs, such as neuropathic pain (patient 2), cognitive impairment (patient 2), and deafness (patient 1), all of which have been reported previously. 17 , 23 , 41 Other atypical symptoms should also be noted, including scoliosis, 10 , 31 , 42 vertebral deformities, 10 back or hip pain, 10 , 14 syringomyelia, 10 and avascular osteonecrosis 14 , 43 ; an ST‐1 patient can even develop serious dysphagia and suffer from recurrent bronchopneumonia 44 or be misdiagnosed with conversion disorder. 14 Patient 5 had unequal pupils, but we are not sure whether this is related to the disease. These atypical symptoms further implied that NEU1 can indeed be mutated in the absence of typical clinical signs, further complicating the diagnosis of sialidosis.
Enzyme‐dependent diagnosis may be inappropriate, because its activity is likely to be affected by the sampling sites and experimental conditions, and the detection of neuraminidase alone is not specific. 4 , 5 It would also be impossible to make any accurate predictions as to phenotypes purely from biochemical evidence. 22 Most patients presented near‐zero activity for the defective enzyme, but not all of them manifested the most severe forms of the disease. 10 , 22 Thus, it is conceivable that environmental factors, including diet and different types of environmental exposure, in addition to neuraminidase mutation, may influence the penetrance of the disease or phenotype. 5 , 23 Currently, gene detection is essential for the diagnosis of sialidosis and is of great importance in the early diagnosis, early intervention, genetic counseling, prognosis prediction, and possible gene therapy in the future. We found substantial genetic heterogeneity among different regions. The c.544A > G and c.239C > T mutations were not found in the European and American population and were rarely seen in the other Asian population; however, they were the most common mutation types in mainland China (75% and 50%) and Taiwanese (100% and 13%) patients, indicating their same root and homology. The homozygous c.544A > G mutation of Taiwanese patients reached 69.6% (16/23), which may be due to small‐scale intermarriage limited to the island. The two common mutation types of Europeans and Americans were homozygous c.200G > T (3/32, 9.4%) and heterozygous c.679G > A and c.913C > T (3/32, 9.4%), both in three patients from two different families in Italy. 10 The heterozygous c.649G > A and c.727G > A mutation (3/10, 10.0%) is probably the most common type in Japanese patients, coming from three different families. 7 , 45 Other mutation types were rare and sporadic. We speculated that different countries or regions retain their own unique mutation types of ST‐1.
Our study also had some limitations. First, the retrospective analysis of the literature had the following inherent limitations: the diverse vocabularies could have led to a certain degree of interpretation, and the description of clinical features and typical examinations also differed from one article to another. Second, we tried our best to include as many genetically confirmed ST‐1 patients as possible, but there may be omissions, and importantly, articles written in languages other than English or Chinese were excluded, which may also cause statistical deviation. Third, the diagnosis of patient 59, 23 who developed characteristics of ST‐2 with coarse facies and dysmorphic features, was questionable, but we followed the original author's viewpoint. Fourth, patients 53 and 55 may be too young to show symptoms such as epilepsy or ataxia or others. 13 , 23 Fifth, we did not obtain all patients’ FDG‐PET image analysis data. Finally, since almost all our patients had poor prognosis, it remains challenging to provide treatment advice. Considering the shortcomings of our research and the limitations in this field, multicenter collaboration to include more subjects and long‐term follow‐up are needed in the future.
Author Contributions
Rui‐Juan Lv, Qun Wang, Li‐Ri Jin, Xiao‐Qiu Shao, and Yu‐Di Zhang provided the clinical data. Tao‐Ran Li acquired the literature data. Rui‐Juan Lv and Tao‐Ran Li drafted the manuscript. Qun Wang and Li‐Ri Jin critically revised the manuscript for important intellectual content.
Conflict of Interest
On behalf of all authors, the corresponding author confirms no conflict of interest.
Ethics Approval
The study was approved by the Medical Ethics Committee of Beijing Tiantan Hospital, Capital Medical University (No. XW20190056) and Medical Ethics Committee of Peking Union Medical College Hospital, Tsinghua University (No. PUMCH20190130), and was carried out in accordance with the Declaration of Helsinki. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Informed consent was obtained from all individual participants included in the study.
Supporting information
Table S1. Clinical data of the other 7 mainland type I sialidosis patients reported in previous references.
Table S2. Clinical data of the other 65 type I sialidosis patients from other regions reported in previous references.
Data S1. Brain 18F‐FDG‐PET studies of patient 1, Clinical descriptions of patient 2–5, and References included in the statistics.
Funding Information
This work was supported by grants from the Application Research of Capital Clinical Characteristics (No. Z181100001718082), the Beijing Dongcheng District Outstanding Talent Funding Project (No. 2019DCT‐M‐18) and the National Key R&D Program of China (2017YFC1307500).
Funding Statement
This work was funded by the Application Research of Capital Clinical Characteristics grant Z181100001718082; the Beijing Dongcheng District Outstanding Talent Funding Project grant 2019DCT‐M‐18; the National Key R&D Program of China grant 2017YFC1307500.
Contributor Information
Qun Wang, Email: wangqunneuro@163.com.
Li‐Ri Jin, Email: jinlirineuro@163.com.
Data Availability Statement
Data are available on reasonable request.
References
- 1. Cantz M, Gehler J, Spranger J. Mucolipidosis I: increased sialic acid content and deficiency of an alpha‐N‐acetylneuraminidase in cultured fibroblasts. Biochem Biophys Res Commun 1977;74:732–738. [DOI] [PubMed] [Google Scholar]
- 2. Durand P, Gatti R, Cavalieri S, et al. Sialidosis (mucolipidosis I). Helv Paediatr Acta 1977;32:391–400. [PubMed] [Google Scholar]
- 3. Sphranger J, Gehler J, Cantz M. Mucolipidosis I–a sialidosis. Am J Med Genet 1977;1:21–29. [DOI] [PubMed] [Google Scholar]
- 4. Franceschetti S, Canafoglia L. Sialidoses. Epileptic Disord 2016;18(S2):89–93. [DOI] [PubMed] [Google Scholar]
- 5. Khan A, Sergi C. Sialidosis: a review of morphology and molecular biology of a rare pediatric disorder. Diagnostics (Basel) 2018;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aravindhan A, Veerapandiyan A, Earley C, et al. Child neurology: type 1 sialidosis due to a novel mutation in NEU1 gene. Neurology 2018;90:622–624. [DOI] [PubMed] [Google Scholar]
- 7. Uchihara T, Ohashi K, Kitagawa M, et al. Sialidosis type I carrying V217M/G243R mutations in lysosomal sialidase: an autopsy study demonstrating terminal sialic acid in lysosomal lamellar inclusions and cerebellar dysplasia. Acta Neuropathol 2010;119:135–145. [DOI] [PubMed] [Google Scholar]
- 8. Lowden JA, O'Brien JS. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet 1979;31:1–18. [PMC free article] [PubMed] [Google Scholar]
- 9. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249–254. [DOI] [PubMed] [Google Scholar]
- 10. Canafoglia L, Robbiano A, Pareyson D, et al. Expanding sialidosis spectrum by genome‐wide screening: NEU1 mutations in adult‐onset myoclonus. Neurology 2014;82:2003–2006. [DOI] [PubMed] [Google Scholar]
- 11. Fernández‐Marmiesse A, Morey M, Pineda M, et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis 2014;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonten E, van der Spoel A, Fornerod M, et al. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev 1996;10:3156–3169. [DOI] [PubMed] [Google Scholar]
- 13. Mütze U, Bürger F, Hoffmann J, et al. Multigene panel next generation sequencing in a patient with cherry red macular spot: Identification of two novel mutations in NEU1 gene causing sialidosis type I associated with mild to unspecific biochemical and enzymatic findings. Mol Genet Metab Rep 2017;10:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schene IF, Kalinina Ayuso V, de Sain‐van der Velden M, et al. Pitfalls in diagnosing neuraminidase deficiency: psychosomatics and normal sialic acid excretion. JIMD Rep 2016;25:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Louboutin JP, Nogues B, Caillaud C, Elie B. Multimodality evoked potentials and EEG in a case of cherry red spot‐myoclonus syndrome and alpha‐neuraminidase deficiency (sialidosis type 1). Eur Neurol 1995;35:175–177. [DOI] [PubMed] [Google Scholar]
- 16. Michalewska Z, Gajos A, Michalewski J, et al. Spectral optical coherence tomography in a patient with type I sialidosis. Med Sci Monit 2011;17:CS129–CS131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Brien JS. The cherry red spot‐myoclonus syndrome: a newly recognized inherited lysosomal storage disease due to acid neuraminidase deficiency. Clin Genet 1978;14:55–60. [DOI] [PubMed] [Google Scholar]
- 18. Kleijer WJ, Hoogeveen A, Verheijen FW, et al. Prenatal diagnosis of sialidosis with combined neuraminidase and beta‐galactosidase deficiency. Clin Genet 1979;16:60–61. [DOI] [PubMed] [Google Scholar]
- 19. Matsuo T, Egawa I, Okada S, et al. Sialidosis type 2 in Japan. Clinical study in two siblings' cases and review of literature. J Neurol Sci 1983;58:45–55. [DOI] [PubMed] [Google Scholar]
- 20. Tsuji S, Yamada T, Tsutsumi A, Miyatake T. Neuraminidase deficiency and accumulation of sialic acid in lymphocytes in adult type sialidosis with partial beta‐galactosidase deficiency. Ann Neurol 1982;11:541–543. [DOI] [PubMed] [Google Scholar]
- 21. Tsvetkova IV, Petushkova NA, Zolotuchina TV, et al. Biochemical study of sialidosis type I in a Russian family. J Inherit Metab Dis 1987;10:18–23. [DOI] [PubMed] [Google Scholar]
- 22. Coutinho MF, Lacerda L, Macedo‐Ribeiro S, et al. Lysosomal multienzymatic complex‐related diseases: a genetic study among Portuguese patients. Clin Genet 2012;81:379–393. [DOI] [PubMed] [Google Scholar]
- 23. Bonten EJ, Arts WF, Beck M, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet 2000;9:2715–2725. [DOI] [PubMed] [Google Scholar]
- 24. Lukong KE, Elsliger MA, Chang Y, et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum Mol Genet 2000;9:1075–1085. [DOI] [PubMed] [Google Scholar]
- 25. Seyrantepe V, Poupetova H, Froissart R, et al. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat 2003;22:343–352. [DOI] [PubMed] [Google Scholar]
- 26. Navarro V, Kas A, Apartis E, et al. Motor cortex and hippocampus are the two main cortical targets in LGI1‐antibody encephalitis. Brain 2016;139(Pt 4):1079–1093. [DOI] [PubMed] [Google Scholar]
- 27. Chen CM, Lai SC, Chen IC, et al. First report of two Taiwanese siblings with sialidosis type I: a 10‐year follow‐up study. J Neurol Sci 2006;247:65–69. [DOI] [PubMed] [Google Scholar]
- 28. Huang YZ, Lai SC, Lu CS, et al. Abnormal cortical excitability with preserved brainstem and spinal reflexes in sialidosis type I. Clin Neurophysiol 2008;119:1042–1050. [DOI] [PubMed] [Google Scholar]
- 29. Lai SC, Chen RS, Wu Chou YH, et al. A longitudinal study of Taiwanese sialidosis type 1: an insight into the concept of cherry‐red spot myoclonus syndrome. Eur J Neurol 2009;16:912–919. [DOI] [PubMed] [Google Scholar]
- 30. Lu CS, Ng SH, Lai SC, et al. Cortical damage in the posterior visual pathway in patients with sialidosis type 1. Brain Imaging Behav 2017;11:214–223. [DOI] [PubMed] [Google Scholar]
- 31. Jing‐zi ZB, Chao Q, Su‐shan L, et al. Cherry‐red spot: a case report of sialidosis I and literature review. Chin J Clin Neurosci 2015;1:115–121. [Google Scholar]
- 32. Xuan LI, Ya‐Fang HU. Clinical application of next‐generation sequencing technology in diagnosis of hereditary ataxia. Master Dissertation of Nanjing Medical University, 2018.
- 33. Zhang J, Zhang YH, Chen JY, et al. Pathogenic gene variants and clinical phenotype features of 26 children with progressive myoclonic epilepsy. Zhonghua Er Ke Za Zhi 2019;57:458–464. [DOI] [PubMed] [Google Scholar]
- 34. Zou W, Wang X, Tian G. Fundus autofluorescence and optical coherence tomography of a macular cherry‐red spot in a case report of sialidosis. BMC Ophthalmol 2016;16:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fan SP, Lee NC, Lin CH. Clinical and electrophysiological characteristics of a type 1 sialidosis patient with a novel deletion mutation in NEU1 gene. J Formos Med Assoc 2020;119:406–412. [DOI] [PubMed] [Google Scholar]
- 36. Ahn JH, Kim AR, Lee C, et al. Type 1 sialidosis patient with a novel deletion mutation in the NEU1 gene: case report and literature review. Cerebellum 2019;18:659–664. [DOI] [PubMed] [Google Scholar]
- 37. Till JS, Roach ES, Burton BK. Sialidosis (neuraminidase deficiency) types I and II: neuro‐ophthalmic manifestations. J Clin Neuroophthalmol 1987;7:40–44. [PubMed] [Google Scholar]
- 38. Nishiyama Y, Sakamoto S, Katayama Y, et al. Neuroradiological findings on cerebral blood flow and metabolism of a case of adult onset sialidosis. Rinsho Shinkeigaku (Clin Neurol) 1997;37:982–986. [PubMed] [Google Scholar]
- 39. Sakazume Y, Tanaka M, Isobe I, et al. A case of middle‐aged onset sialidosis type I. Rinsho Shinkeigaku (Clin Neurol) 2004;44:541–544. [PubMed] [Google Scholar]
- 40. Wang IH, Lin TY, Kao ST. Optical coherence tomography features in a case of Type I sialidosis. Taiwan J Ophthalmol 2017;7:108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sekijima Y, Nakamura K, Kishida D, et al. Clinical and serial MRI findings of a sialidosis type I patient with a novel missense mutation in the NEU1 gene. Intern Med. 2013;52:119–124. [DOI] [PubMed] [Google Scholar]
- 42. Heroman JW, Rychwalski P, Barr CC. Cherry red spot in sialidosis (mucolipidosis type I). Arch Ophthalmol 2008;126:270–271. [DOI] [PubMed] [Google Scholar]
- 43. Urbanski G, Bekri S, Barth M, et al. A case of type I sialidosis with osteonecrosis revealing a new mutation in NEU1. J Inborn Errors Metab Screen 2014;2 10.1177/2326409814543468 [DOI] [Google Scholar]
- 44. Itoh K, Naganawa Y, Matsuzawa F, et al. Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes. J Hum Genet 2002;47:29–37. [DOI] [PubMed] [Google Scholar]
- 45. Naganawa Y, Itoh K, Shimmoto M, et al. Molecular and structural studies of Japanese patients with sialidosis type 1. J Hum Genet 2000;45:241–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical data of the other 7 mainland type I sialidosis patients reported in previous references.
Table S2. Clinical data of the other 65 type I sialidosis patients from other regions reported in previous references.
Data S1. Brain 18F‐FDG‐PET studies of patient 1, Clinical descriptions of patient 2–5, and References included in the statistics.
Data Availability Statement
Data are available on reasonable request.