

Abstract
Objective
The aim of the present study was to compare the levels of serum neurofilament light chain (sNfL) in frontotemporal lobar degeneration (FTLD) patients of different clinical subtypes (bvFTD, PPA, and FTLD‐MND) and with or without the C9orf72 repeat expansion, and to correlate sNfL levels to disease progression, assessed by the brain atrophy rate and survival time.
Methods
The sNfL levels were determined from 78 FTLD patients (C9orf72 repeat expansion carriers [n = 26] and non‐carriers [n = 52]) with Single Molecule Array (SIMOA). The progression of brain atrophy was evaluated using repeated T1‐weighted MRI scans and the survival time from medical records.
Results
In the total FTLD cohort, sNfL levels were significantly higher in C9orf72 repeat expansion carriers compared to non‐carriers. Considering clinical phenotypes, sNfL levels were higher in the C9orf72 repeat expansion carriers than in the non‐carriers in bvFTD and PPA groups. Furthermore, sNfL levels were the highest in the FTLD‐MND group (median 105 pg/mL) and the lowest in the bvFTD group (median 27 pg/mL). Higher sNfL levels significantly correlated with frontal cortical atrophy rate and subcortical grey matter atrophy rate. The higher sNfL levels also associated with shorter survival time.
Interpretation
Our results indicate that the C9orf72 repeat expansion carriers show elevated sNFL levels compared to non‐carriers and that the levels differ among different clinical phenotypes of FTLD. Higher sNfL levels correlated with a shorter survival time and cortical and subcortical atrophy rates. Thus, sNfL could prove as a potential prognostic biomarker in FTLD.
Introduction
Frontotemporal lobar degeneration (FTLD) is a neuropathological umbrella term including various clinical, genetic, and histopathological subtypes. The most common clinical phenotype is the behavioral variant frontotemporal dementia (bvFTD). Other main phenotypes consist of primary progressive aphasias (PPA) that further are divided into non‐fluent primary progressive aphasia (nfvPPA), semantic variant primary progressive aphasia (svPPA), and logopenic variant primary progressive aphasia (lvPPA). 1 , 2 Of predisposing genetic causes of familial FTLD, the C9orf72 hexanucleotide repeat expansion is the most common in different populations. 3 , 4 The C9orf72 repeat expansion is associated with TDP‐43 neuropathology. 5 This mutation is also associated with amyotrophic lateral sclerosis (ALS) and especially with the FTLD clinical phenotype concomitantly presenting with motoneuron disease (FTD‐ALS or FTD‐MND). 6 , 7 , 8
Due to diagnostic challenges and variability in disease progression, specific diagnostic and prognostic biomarkers are needed for FTLD. Also, considering future therapeutic trials, appropriate biomarkers are essential for determining the adequate time to start the treatment and for evaluation of treatment effects.
Neurofilaments are intracellular intermediate filaments present in the central and peripheral nervous systems, which form assemblies that maintain the shape and diameter of neuronal axons, thus regulating the response of the neurons to different stimuli. 9 Upon axonal damage, neurofilaments can leak out of the neurons to the extracellular space, and eventually end up in blood. Neurofilament light chain (NfL) is a subunit of the intracellular neurofilament assemblies, and its elevated concentrations in blood and cerebrospinal fluid (CSF) have been associated with neuroinflammation and axonal damage. 10 , 11 Elevated levels of CSF and serum NfL (sNfL) have been reported in several neurodegenerative conditions, such as FTLD, Alzheimer’s disease, (AD) and multiple sclerosis (MS), and also after head trauma and ischemic stroke, suggesting that it is a non‐specific biomarker for these conditions. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 In FTLD, CSF and serum NfL levels have been shown to vary between distinct clinical phenotypes and higher concentrations have been associated with a more aggressive disease progression and greater rate of brain atrophy. 15 , 22 , 23 Previous reports indicate that CSF and serum NfL concentrations correlate well with each other, proposing that sNfL may represent a potential minimally invasive biomarker for early diagnostic and prognostic purposes. 18
In this study, the aim was to analyze the sNfL levels in FTLD patients having different clinical phenotypes with and without the C9orf72 repeat expansion using the ultrasensitive SIMOA assay. Carriers and non‐carriers of the C9orf72 repeat expansion were analyzed separately since carriers are associated with TDP‐43 pathology and the certainty of the FTLD diagnosis is considered definite. Given that sNfL is a non‐specific biomarker for neuronal damage, we defined the characteristic levels of sNfL in different clinical phenotypes and genotypes of FTLD. Furthermore, we examined the prognostic value of sNfL by measuring how the sNfL levels correlate to brain atrophy rates and survival time in FTLD patients.
Materials and Methods
Patients
We examined a total of 78 subjects with FTLD including patients with a diagnosis of bvFTD (n = 54), PPA (n = 19), and FTD‐MND (n = 5). The study was conducted and patients were recruited at Kuopio University Hospital, Finland. The diagnoses of neurodegenerative diseases were set by an experienced neurologist specialized in memory disorders according to the most recent standard criteria 1 , 2 . For bvFTD, only patients fulfilling the clinical diagnostic criteria of probable (n = 35) or definite (n = 19) bvFTD were included. Patients were excluded if they had obvious other brain disorders or trauma that could affect cognitive functions, such as stroke or tumor. To evaluate disease progression, time from the FTLD diagnosis to death was calculated for those patients that died in the course of the study (N = 44).
The ethics committee of the Northern Savo Hospital District approved the research protocol in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants prior to enrollment.
Laboratory analyses
Genomic DNA was extracted from venous blood samples using QIAamp DNA blood mini extraction kit (QIAGEN). The blood sample was obtained in the early diagnostic phase. The presence of the C9orf72 repeat expansion was detected using Repeat‐Primed PCR (RP‐PCR) 3 . Patients were considered positive for the C9orf72 repeat expansion if the number of repeats were ≥ 30 and negative if the number of repeats was < 30. SNfL concentration and the genetic status of C9orf72 repeat expansion were determined from all patients; 26 were carriers and 52 non‐carriers for the repeat expansion. Mutations in the other FTLD‐associated genes were not systematically screened because the prevalence of these mutations is very low in Finnish patients. 24 , 25 , 26
Frozen (−80˚C) serum samples were thawed in room temperature, mixed, centrifuged (10,000g, 5 min, at room temperature), and transferred to a 96‐well plate. The sNfL was quantified using a NfL Advantage kit (REF#: 102258) for the Quanterix single molecule array (SIMOA, Lexington, MA, USA) according to the manufacturer’s instructions. 27 All measured values were within the calibration range. The mean intra‐assay CV was 4.6% and the inter‐assay CV was 13.9%.
MRI acquisition and quantification
The MRI examinations were performed with 3.0 T (n = 17) or 1.5 T (n = 24) scanners in the early diagnostic phase. Three‐dimensional T1‐weighted gradient echo sequences were acquired with the slice thickness varying from 1 to 1.5 mm. An appropriate cross‐sectional 3D T1‐weighted MRI scan was available for 41 patients, of whom 15 were C9orf72 repeat expansion carriers and 26 non‐carriers. Longitudinal data from repeated scans were available for 11 patients. There were six C9orf72 repeat expansion carriers and five non‐carriers having repeated scans. The time between the scans varied from 8 to 42 months, with a mean of 23 months.
The scans were preprocessed and analyzed using Freesurfer version 6.0.0 image analysis suite. The precise pipeline is demonstrated in http://surfer.nmr.mgh.harvard.edu. In short, processing includes motion correction and averaging of multiple volumetric T1‐weighted images, removal of non‐brain tissue using a hybrid watershed/surface deformation procedure, automated Talairach transformation, segmentation of the subcortical white matter and deep grey matter volumetric structures, intensity normalization, tessellation of the grey matter white matter boundary, automated topology correction, and surface deformation following intensity gradients to optimally place the grey/white and grey/cerebrospinal fluid borders at the location where the greatest shift in intensity defines the transition to the other tissue class. Cortices were parcellated with Desikan atlas, and thicknesses of cerebral lobes were merged as defined in the original publication. 28 Subcortical segmentation was performed using probabilistic atlas. 29 Freesurfer morphometric procedures have been demonstrated to show good test–retest reliability across scanner manufacturers and field strengths, 30 and thus we permitted the use of heterogeneous field strength.
To extract reliable volume and thickness estimates, images were automatically processed with the longitudinal stream 31 in FreeSurfer. Specifically, an unbiased within‐subject template space and image is created using robust, inverse consistent registration. 32 Several processing steps, such as skull stripping, Talairach transformations, atlas registration as well as spherical surface maps and parcellations, are then initialized with common information from the within‐subject template, significantly increasing reliability and statistical power. 31
Cortical measures are presented as a mean of cortical thicknesses. Subcortical grey matter structures of interest are presented as volumes. Volumes were corrected for total intracranial volume by simple division. Brain atrophy rates were calculated by subtracting baseline MRI measures from the follow‐up scan measures and dividing the remainder by baseline. Atrophy rates were then annualized using the time between scans:
Statistical analysis
Mean scores of demographic and neuropsychological data were compared across the genetic groups (C9orf72 repeat expansion carriers vs. non‐carriers) with t‐test. Chi‐square analysis was used for differences in gender distribution. Differences in sNfL levels between carriers and non‐carriers of the C9orf72 repeat expansion and between bvFTD and PPA were analyzed with the Mann–Whitney U‐test.
Given the statistically significant correlation between sNfL and age (P = 0.026), regression analyses using age as a covariate were conducted. For regression analyses, sNfL values were log‐transformed to deal with skewed data.
The difference between sNfL levels according to C9orf72 genetic status and clinical phenotype was evaluated using a multivariate linear regression model with age as a covariate.
A comparison of cortical thickness between the two genetic groups was performed with a t‐test and correlation between sNfL levels and cortical thickness with Pearson's correlation. Correction for multiple comparisons was performed with FDR < 0.05 correction.
Association of sNfL and C9orf72 genetic status with survival after disease onset was assessed using a multivariate cox regression model. The regression model was adjusted for age at disease onset. All tests were two‐sided and the standard statistical significance level was set at P < 0.05. Data were analyzed using SPSS 25.
Results
The patients carrying the C9orf72 repeat expansion were on average 5.3 years younger than those not carrying the repeat expansion (P = 0.008) (Table 1). The groups did not differ in gender distribution or the clinical severity of the disease as measured by MMSE and ADCS‐ADL tests (Table 1).
Table 1.
Demographic and clinical data.
| C9orf72 pos | C9orf72 neg | Total | P value | |
|---|---|---|---|---|
| N (%) | 26 (34) | 52 (67) | 78 | |
| Gender, female n (%) | 13 (50) | 29 (56) | 42 (54) | 0.630 |
| Age (SD) | 61.9 (9.4) | 67.2 (7.6) | 65.5 (8.6) | 0.008 |
| Serum NfL, pg/mL, mean (SD) | 78 (115) | 40 (35) | 53 (74) | 0.039 |
| MMSE (SD) | 22 (5.7) | 24 (3.6) | 23 (4.7) | 0.302 |
| ADCS‐ADL (SD) | 58 (17) | 55 (15) | 57 (15) | 0.267 |
| bvFTD, n (%) | 19 (36) | 35 (65) | 54 (100) | 0.739 |
| PPA, n (%) | 5 (26) | 14 (74) | 19 (100) | |
| FTLD‐MND, n (%) | 2 (40) | 3 (60) | 5 (100) |
ADCS‐ADL, Alzheimer’s Disease Co‐operative Study‐Activities of Daily Living; bvFTD, Behavioral Variant Frontotemporal Dementia; FTLD‐MND, Frontotemporal Lobar Degeneration with Motoneuron disease; MMSE, Mini Mental State Examination; NfL, Neurofilament light chain; PPA, Primary Progressive Aphasia.
Serum NfL levels are higher in C9orf72 repeat expansion carriers than in non‐carriers
In the total FTLD cohort, SIMOA‐based analysis of the sNfL levels indicated that sNfL concentration median was 10 pg/mL higher in the C9orf72 repeat expansion carriers compared to non‐carriers (median 40 pg/mL vs. 30 pg/mL, pMWU = 0.039). In the bvFTD group, sNfL levels did not differ between the carriers and non‐carriers (median 33 pg/mL vs. 27 pg/mL, pMWU = 0.24). In contrast, in the PPA group, sNfL concentration was significantly higher in C9orf72 repeat expansion carriers than in the non‐carriers (median 87 pg/mL vs. 37 pg/mL, pMWU = 0.019). Also, in the FTLD‐MND group, C9orf72 expansion carriers had higher levels of sNfL compared to non‐carriers, but the sample size was insufficient for reliable statistical comparison (median 106 pg/mL vs. 54 pg/mL).
Since the C9orf72 repeat expansion carriers were significantly younger and the sNfL levels showed a strong correlation with age (RP = 0.22, P = 0.026), we performed similar comparisons as above with multivariate linear regression analyses using age at the time of blood sample as a covariate, to take the effect of age into account. After the correction, the C9orf72 repeat expansion carriers in the total group had a higher concentration of sNfL to non‐carriers (β = 0.35, P = 0.002). Moreover, the C9orf72 repeat expansion carriers had higher sNfL concentrations in the bvFTD group (β = 0.30, P = 0.039) as well as in the PPA group (β = 0.59, P = 0.008) as compared to the non‐carriers (Fig. 1).
Figure 1.
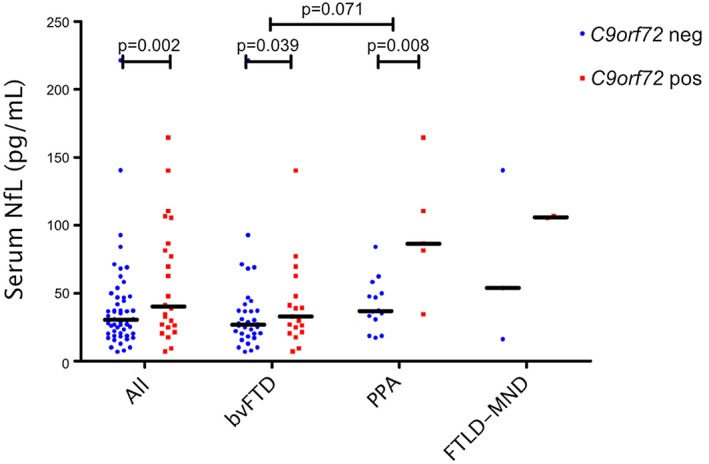
SNfL concentrations of FTLD patients displaying different clinical phenotypes and grouped according to the C9orf72 repeat expansion genetic status. Blue dots represent repeat expansion non‐carriers (C9orf72 neg) and red squares carriers (C9orf72 pos). Black horizontal lines indicate a group median. The P values displayed stem from the linear regression model with sNfL log‐transformed and age as a covariate. Statistical testing among the FTLD‐MND group and other groups was not done due to insufficient sample size.
Serum NfL levels in patients with different FTLD clinical phenotypes
We observed that the highest sNfL levels were present in the FTLD‐MND group (median 105 pg/mL), the second highest in the PPA group (median 47 pg/mL) and the lowest in the bvFTD group (median 27 pg/mL). The difference between the bvFTD and PPA groups was statistically significant (pMWU = 0.017). Statistical difference between FTLD‐MND and the two other groups was not assessed due to insufficient sample size in the FTLD‐MND group.
In the regression model using age as a covariate, the PPA groups had higher sNfL levels compared to bvFTD, but the difference was not statistically significant (β = 0.21, P = 0.071) (Fig. 1).
Serum NfL levels associated with shorter survival time
A total of 44 patients died during the follow‐up time of the study. Fifteen of them were C9orf72 repeat expansion carriers (58 % of all carriers) and 29 non‐carriers (56 % of all non‐carriers). The mean time from diagnosis to death was 4.4 (SD 3.5) years for carriers and 5.6 (SD 4.2) years for non‐carriers (P = 0.32). Higher sNfL levels associated with shorter survival time (β = −0.47, P = 0.002), in both carriers (β = −0.57, P = 0.035) and non‐carriers (β=−0.38, P = 0.044) of the C9orf72 repeat expansion. When dividing the whole cohort in three equally sized subgroups according to sNfL levels (low < 26.1 pg/mL, moderate 26.1–44.4 pg/mL, and high> 44.4 pg/mL), the survival was significantly shorter in the groups with the highest sNfL levels (HR 4.7, CI 95% 2.1–10.5, P < 0.001) and moderate sNfL levels (HR 2.4, CI 95% 1.0–5.6, P = 0.047) compared to the group with the lowest sNfL levels (P < 0.001) (Fig. 2A). Carriers of the C9orf72 repeat expansion showed a trend of shorter survival compared to non‐carriers; however, the difference was not statistically significant (HR 1.8, CI 95% 0.93–3.5, P = 0.080) (Fig. 2B).
Figure 2.
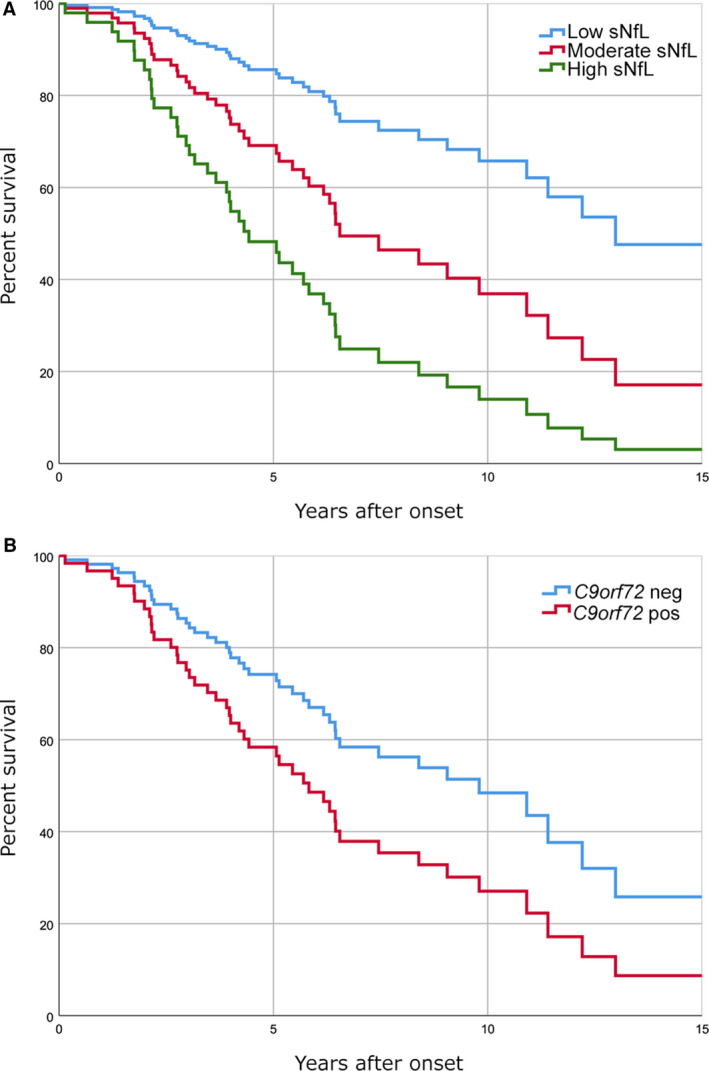
Survival rates after the onset of the disease in FTLD patients according to sNfL levels and C9orf72 genetic status. (A) Graph showing that the group with higher sNfL levels had a lower survival rate than those with low or moderate sNfL levels. The groups were formulated by dividing the whole FTLD cohort into three equally sized (n = 26) groups according to their sNfL levels (low < 26.1 pg/mL, moderate 26.1–44.4 pg/mL, and high> 44.4 pg/mL). Differences in survival among groups were statistically significant (P < 0.001) (B) Graph showing survival rates of the C9orf72 repeat expansion carriers (n = 26) and non‐carriers (n = 52). The difference in survival rate showed a trend of shorter survival of C9orf72 carriers compared to non‐carriers; however, the difference was not statistically significant (P = 0.080).
Association of serum NfL levels to cortical thickness and subcortical volume
First, the differences from baseline MRI scans in cortical thickness and subcortical volume between the C9orf72 repeat expansion carriers and non‐carriers were evaluated cross‐sectionally at the time of the diagnosis. There were no significant differences neither in the cortical thickness nor in the subcortical structure volumes between these groups. Interestingly, in a cross‐sectional correlation analysis on baseline MRI scans, a negative correlation between mean cortical thickness and sNfL concentration was found (RP = −0.26, P = 0.048).
Next, we tested whether the sNfL concentration correlated with brain atrophy rates, that is, the progression of atrophy based on repeated MRI measures (Table 2). The overall cortical atrophy rate was found to significantly correlate with elevated sNfL levels (Rp = −0.64, P = 0.018). Of separate brain lobes, only frontal lobe atrophy rate was observed to correlate with a higher sNfL concentration (RP = −0.72, P = 0.006). Furthermore, when correlating subcortical grey matter structures, there was a significant correlation between sNfL levels and the atrophy rate of the thalamus (Rp = −0.61, P = 0.024), caudate (Rp = −0.62, P = 0.022), pallidum (Rp = −0.73, P = 0.005), and putamen (Rp = −0.70, P = 0.008). Also, atrophy of the overall subcortical grey matter correlated with sNfL levels (Rp=−0.68, P = 0.011). All the statistically significant correlations between longitudinal MRI measures and sNfL levels survived the FDR < 0.05 correction for multiple comparisons.
Table 2.
Correlation of sNfL levels to cortical and subcortical grey matter atrophy rates.
| Mean atrophy rate, % (SD) | Corr. with sNfL levels (Rp) | P value | |
|---|---|---|---|
| Cortical Grey Matter | −1.9 (2.7) | −0.64 | 0.018 |
| Frontal | −3.0 (3.6) | −0.72 | 0.006 |
| Temporal | −1.3 (2.3) | −0.38 | 0.122 |
| Parietal | −1.1 (2.9) | −0.48 | 0.069 |
| Occipital | 1.3 (6.0) | −0.21 | 0.264 |
| Cingulate | −2.8 (3.6) | −0.36 | 0.142 |
| Subcortical Grey Matter | −4.1 (3.9) | −0.68 | 0.011 |
| Thalamus | −5.5 (5.3) | −0.61 | 0.024 |
| Caudate | −4.3 (5.2) | −0.62 | 0.022 |
| Putamen | −2.8 (3.6) | −0.70 | 0.008 |
| Pallidum | −1.5 (3.7) | −0.73 | 0.005 |
Discussion
The sNfL levels in FTLD patients with bvFTD, PPA, and FTLD‐MND phenotypes were higher in the C9orf72 repeat expansion carriers compared to non‐carriers. In addition, the sNfL levels were the highest in patients with FTLD‐MND and the lowest in bvFTD patients. Also, the elevated sNfL levels were proven to correlate with higher cortical and subcortical atrophy rates. Moreover, higher sNfL levels also associated with a shorter survival time. Altogether, our data suggest that higher sNfL levels associate with the C9orf72 repeat expansion and worse disease prognosis.
Our results are in line with previous findings. Previously, higher levels of CSF and serum NfL have been reported in FTLD patients carrying the C9orf72 repeat expansion compared to non‐carriers. 22 , 23 , 33 Also, after the onset of symptoms, sNfL levels do not change over time and remain elevated in sporadic FTLD patients, as well as FTLD patients with the C9orf72 repeat expansion. 20 , 23 Previous studies have also shown, that genetic mutations causing TDP‐43 neuropathology (C9orf72 and progranulin) tend to higher NfL levels and faster rate of neurodegeneration compared to tau pathology. 5 , 15 , 22 , 34 Notably, in our cohort, higher sNfL levels were strongly associated with atrophy of the frontal cortex and subcortical grey matter structures, as seen also in previous studies. 20 , 22 One previous study observed a similar association between elevated sNfL and brain atrophy among bvFTD patients, but not with patients with AD. 20 This could be explained by the degeneration of abundant neural connections originating from subcortical grey matter structures, as more prominent degeneration of these structures is seen in FTLD compared to AD. 35 Also, pathology‐specific analyses are needed to unveil whether the elevated NfL levels are only associated with certain underlying pathologies or certain areas of the cerebrum or both.
Elevated CSF and blood NfL levels compared to healthy controls, and sNfL levels’ association to disease severity and progression have also been found in several other neurodegenerative conditions involving axonal damage. 12 , 13 , 14 , 19 , 36 , 37 , 38 , 39 These results indicate that NfL measured from blood has poor differential diagnostic value among neurodegenerative diseases. 40 Due to the lack of specificity of the elevated sNfL levels between different neurodegenerative diseases, the diagnostic potential of sNfL may be restricted to discriminating neurodegenerative conditions from their non‐neurodegenerative mimics. For example, disparate sNfL levels could be used to differentiate FTLD from psychiatric conditions, as suggested by our previous study. 41 However, according to the present and previous findings, sNfL levels show great clinical potential as a non‐specific prognostic tool in various neurodegenerative conditions, including FTLD. 16 , 23 , 42
We also evaluated the differences in sNfL levels between distinct clinical phenotypes of FTLD. Previously, similar evaluations have been made with CSF‐derived NfL 33 , 34 and also with sNfL. 22 Our results with sNfL are in accordance with the previous studies and indicate that FTLD‐MND patients harbor the highest sNfL levels, patients with PPA the second highest, and bvFTD patients the lowest levels. The difference in the levels of sNfL between C9orf72 repeat expansion carriers and non‐carriers was apparent in patients manifesting with PPA and bvFTD clinical phenotypes. Also, the FTLD‐MND group showed a similar trend that the C9orf72 repeat expansion carriers had higher sNfL levels as compared to non‐carriers. However, the sample size of this group of patients was insufficient for statistical testing.
The greatest strength of our study is the large patient cohort considering the number of the C9orf72 repeat expansion carriers and non‐carriers with different clinical phenotypes of FTLD. All the patients were diagnosed according to the revised diagnostic criteria by an experienced neurologist specialized in memory disorders. A limitation in the present study is that healthy controls or asymptomatic C9orf72 repeat expansion carriers were not available for evaluating the increase in sNfL levels between patients and controls or between pre‐symptomatic and symptomatic C9orf72 repeat expansion carriers.
In conclusion, we observed that C9orf72 repeat expansion carriers are characterized by higher sNfL levels as compared to non‐carriers and that the sNfL levels differed among different clinical phenotypes of FTLD, with the highest levels detected in FTLD‐MND, the second highest in PPA, and the lowest levels in bvFTD patients. Higher sNfL levels also correlated with a shorter survival time as well as more severe cortical and subcortical atrophy rates. Therefore, sNfL levels could be considered as a biomarker of disease progression and as a potential minimally invasive prognostic tool in FTLD.
Authors’ Contributions
AC, KK, ES, AH, and AMR contributed to the study concept and design. AC, KK, ES, AH, AMR, AK, OJ, PH, AKH, and SKH were involved in data acquisition and analysis. All authors contributed to drafting the manuscript and accepted the final manuscript.
Conflicts of Interest
Dr. Haapasalo reports grants from Academy of Finland, during the conduct of the study. Other authors have no conflicts of interest to be reported.
Acknowledgments
This study has received funding from Finnish Medical Foundation, Finnish Alzheimer’s Disease Research Society, Olvi Foundation, Maire Taponen Foundation, Päivikki and Sakari Sohlberg Foundation, Finnish Cultural Foundation, Maud Kuistila Memorial Foundation, University of Oulu, Kuopio University Hospital and Academy of Finland (no. 315459, AH; no. 315460, AMR). This study is part of the FinFTD network research activities.
Funding Information
This study has received funding from Finnish Medical Foundation, Finnish Alzheimer’s Disease Research Society, Olvi Foundation, Maire Taponen Foundation, Päivikki and Sakari Sohlberg Foundation, Finnish Cultural Foundation, Maud Kuistila Memorial Foundation, University of Oulu, Kuopio University Hospital and Academy of Finland (no. 315459, AH; no. 315460, AMR). This study is part of the FinFTD network research activities.
Funding Statement
This work was funded by Maud Kuistilan Muistosäätiö grant ; Suomen Kulttuurirahasto grant ; Oulun Yliopisto grant ; OLVI‐Säätiö grant ; Maire Taponen Foundation grant ; Päivikki ja Sakari Sohlbergin Säätiö grant ; Academy of Finland grant ; Suomen Lääketieteen Säätiö grant ; Kuopion Yliopistollinen Sairaala grant ; Finnish Alzheimer's Disease Research Society grant .
References
- 1. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia [Internet]. Brain 2011;134:2456–2477. Available from https://academic.oup.com/brain/article‐lookup/doi/10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐Linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahoney CJ, Beck J, Rohrer JD, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features [Internet]. Brain 2012;135(Pt 3):736–750. Available from: http://brain.oxfordjournals.org/content/135/3/736.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper‐Knock J, Shaw PJ, Kirby J. The widening spectrum of C9ORF72‐related disease; Genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol 2014;127:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ng ASL, Rademakers R, Miller BL. Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Ann NY Acad Sci 2015;1338:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yao NY, Braedersz CP, Lin YC, et al. Elasticity in lonically cross‐linked neurofilament networks. Biophys J 2010;98:2147–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018;14:577–589. [DOI] [PubMed] [Google Scholar]
- 11. Peng L, Bi C, Xia D, et al. Increased cerebrospinal fluid neurofilament light chain in central nervous system inflammatory demyelinating disease. Mult Scler Relat Disord 2019;30:123–128. [DOI] [PubMed] [Google Scholar]
- 12. Skillbäck T, Farahmand B, Bartlett JW, et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology 2014;83:1945–1953. [DOI] [PubMed] [Google Scholar]
- 13. Skillbäck T, Mattsson N, Blennow K, Zetterberg H. Cerebrospinal fluid neurofilament light concentration in motor neuron disease and frontotemporal dementia predicts survival. Amyotroph Lateral Scler Front Degener 2017;18:397–403. [DOI] [PubMed] [Google Scholar]
- 14. Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016;73:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med 2019;25:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alcolea D, Vilaplana E, Suárez‐Calvet M, et al. CSF sAPPβ, YKL‐40, and neurofilament light in frontotemporal lobar degeneration. Neurology 2017;89:178–188. [DOI] [PubMed] [Google Scholar]
- 18. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 2016. 3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Disanto G, Barro C, Benkert P, et al. Serum Neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol 2017;81:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steinacker P, Anderl‐Straub S, Diehl‐Schmid J, et al. Serum neurofilament light chain in behavioral variant frontotemporal dementia. Neurology 2018;91:e1390–e1401. [DOI] [PubMed] [Google Scholar]
- 21. Onatsu J, Vanninen R, Jäkälä P, et al. Neurofilament light chain concentration correlates with infarct volume but not prognosis in acute ischemic stroke. J Stroke Cerebrovasc Dis 2019;28:2242–2249. [DOI] [PubMed] [Google Scholar]
- 22. Rohrer JD, Woollacott IOC, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia [Internet]. Neurology 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Ende EL, Meeter LH, Poos JM, et al. Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study [Internet]. Lancet Neurol 2019;18:1103–1111. Available from: https://www.sciencedirect.com/science/article/pii/S1474442219303540?via%3Dihub#fig1. [DOI] [PubMed] [Google Scholar]
- 24. Kaivorinne A‐L, Krüger J, Kuivaniemi K, et al. Role of MAPT mutations and haplotype in frontotemporal lobar degeneration in Northern Finland [Internet]. BMC Neurol 2008;8:48 Available from: http://www.biomedcentral.com/1471‐2377/8/48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaivorinne AL, Krüger J, Udd B, et al. Mutations in CHMP2B are not a cause of frontotemporal lobar degeneration in Finnish patients. Eur J Neurol 2010;17:1393–1395. [DOI] [PubMed] [Google Scholar]
- 26. Krüger J, Kaivorinne AL, Udd B, et al. Low prevalence of progranulin mutations in Finnish patients with frontotemporal lobar degeneration. Eur J Neurol 2009;16:27–30. [DOI] [PubMed] [Google Scholar]
- 27. Rissin DM, Kan CW, Campbell TG, et al. Single‐molecule enzyme‐linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol 2010;28:595–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 29. Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355. [DOI] [PubMed] [Google Scholar]
- 30. Han X, Jovicich J, Salat D, et al. Reliability of MRI‐derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. NeuroImage 2006;32:180–194. [DOI] [PubMed] [Google Scholar]
- 31. Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within‐subject template estimation for unbiased longitudinal image analysis. NeuroImage 2012;61:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reuter M, Rosas HD, Fischl B. Highly accurate inverse consistent registration: a robust approach. NeuroImage 2010;53:1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meeter LHH, Gendron TF, Sias AC, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol 2018;5:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Landqvist Waldö M, Frizell Santillo A, Passant U, et al. Cerebrospinal fluid neurofilament light chain protein levels in subtypes of frontotemporal dementia. BMC Neurol 2013;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bertoux M, O’Callaghan C, Flanagan E, et al. Fronto‐striatal atrophy in behavioral variant frontotemporal dementia and alzheimer’s disease. Front Neurol 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016;87:12–20. [DOI] [PubMed] [Google Scholar]
- 37. Mattsson N, Insel PS, Palmqvist S, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol Med 2016;8:1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pijnenburg YAL, Verwey NA, van der Flier WM, et al. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimer’s Dement Diagnosis, Assess Dis Monit 2015;1:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Malmeström C, Haghighi S, Rosengren L, et al. Neurofilament light protein and glial fibrillary acidic protein as biological markers in MS. Neurology 2003;61:1720–1725. [DOI] [PubMed] [Google Scholar]
- 40. Zhao Y, Xin Y, Meng S, et al. Neurofilament light chain protein in neurodegenerative dementia: A systematic review and network meta‐analysis. Neurosci Biobehav Rev 2019;102:123–138. [DOI] [PubMed] [Google Scholar]
- 41. Katisko K, Cajanus A, Jääskeläinen O, et al. Serum neurofilament light chain is a discriminative biomarker between frontotemporal lobar degeneration and primary psychiatric disorders [Internet]. J Neurol 2019. Available from: http://link.springer.com/10.1007/s00415‐019‐09567‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barro C, Benkert P, Disanto G, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018;141(8):2382–2391. [DOI] [PubMed] [Google Scholar]